Embed Size (px)
Citation preview

A comparison of algorithms for identification of specimens using DNA barcodes: examples
from gymnosperms
Damon P. Little and Dennis Wm. Stevenson
Cullman Program for Molecular Systematic StudiesThe New York Botanical Garden, Bronx, New York

Why is DNA barcoding useful?

Why is DNA barcoding useful?
(1) Non–specialists can identify specimens (e.g., customs inspectors, ethnobotanists).
(2) Morphologically deficient or incomplete specimens can be identified (e.g., powders).

application to conservation:
Cycadopsida:all 305 species are protected by CITES (Convention on International Trade in Endangered Species)
5 genera are appendix I
6 genera are appendix II* Cycas machonie

((GTGCTCGGGC and TCTCGCACTG) and not CGCCTCCCCT)
nrITS 2:
Encephalartos feroxLepidozamia hopei
CITES appendix ICITES appendix II
CGCCTCCCCT

selection of the barcode locus
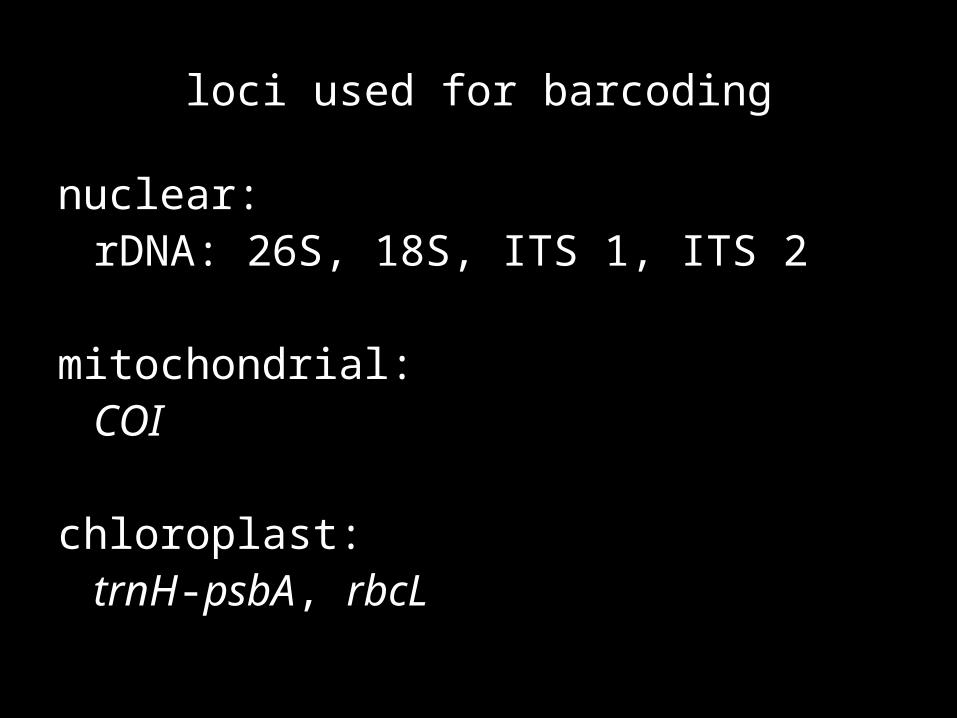
loci used for barcoding
nuclear: rDNA: 26S, 18S, ITS 1, ITS 2
mitochondrial:COI
chloroplast:trnH-psbA, rbcL

Consortium for the Barcode Of Life (CBOL)
cpDNA: matK, rpoC1, rpoB, YCF5, accD, ndhJ
Edinburgh (UK) => Podocarpus, Araucaria, Asterella, Anastrophyllum
Instituto de Biologia UNAM (Mexico) => Agave
Kew (UK) => Conostylis, Pinus, Equisetum, Dactylorhiza
National Biodiversity Institute (South Africa) => Encephalartos, Mimetes
Natural History Museum (Denmark) => Hordeum, Scalesia, Crocus
Natural History Museum (UK) => Tortella, Ptychomniaceae, Asplenium,
New York Botanical Garden (USA) => Elaphoglossum, Cupressus, Labordia
Universidad de los Andes (Colombia) => Lauraceae
University of Cape Town (South Africa) => Anastrophyllum, Bryum
Universidade Estadual de Feira de Santana (Brazil) => Laelia, Cattleya

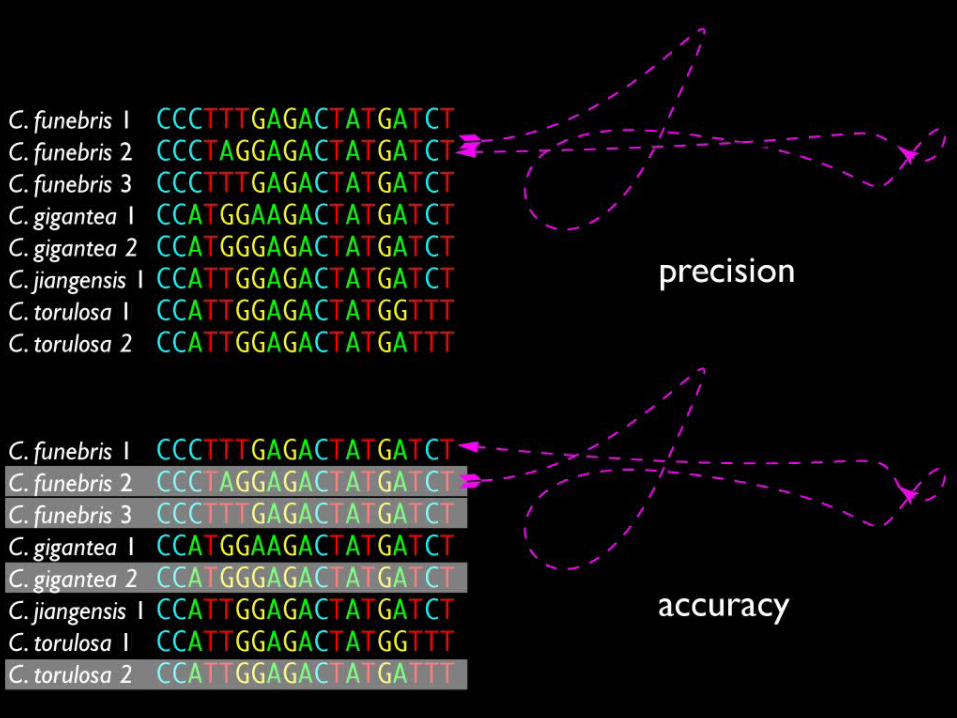
measuring precision and accuracy
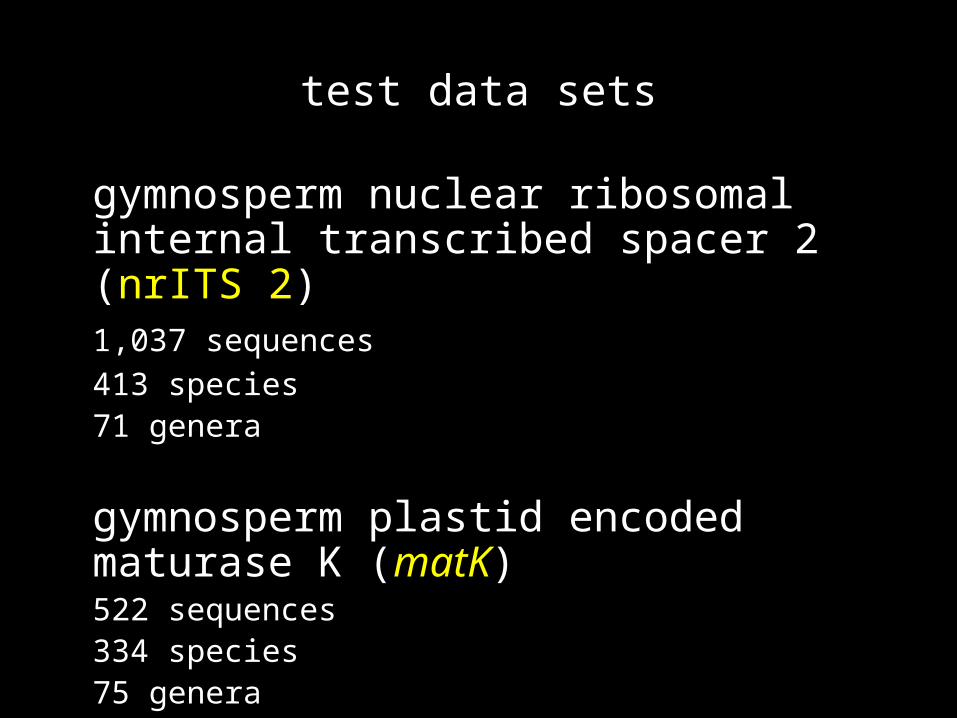
test data sets
gymnosperm nuclear ribosomal internal transcribed spacer 2 (nrITS 2)
1,037 sequences
413 species71 genera
gymnosperm plastid encoded maturase K (matK)
522 sequences334 species75 genera
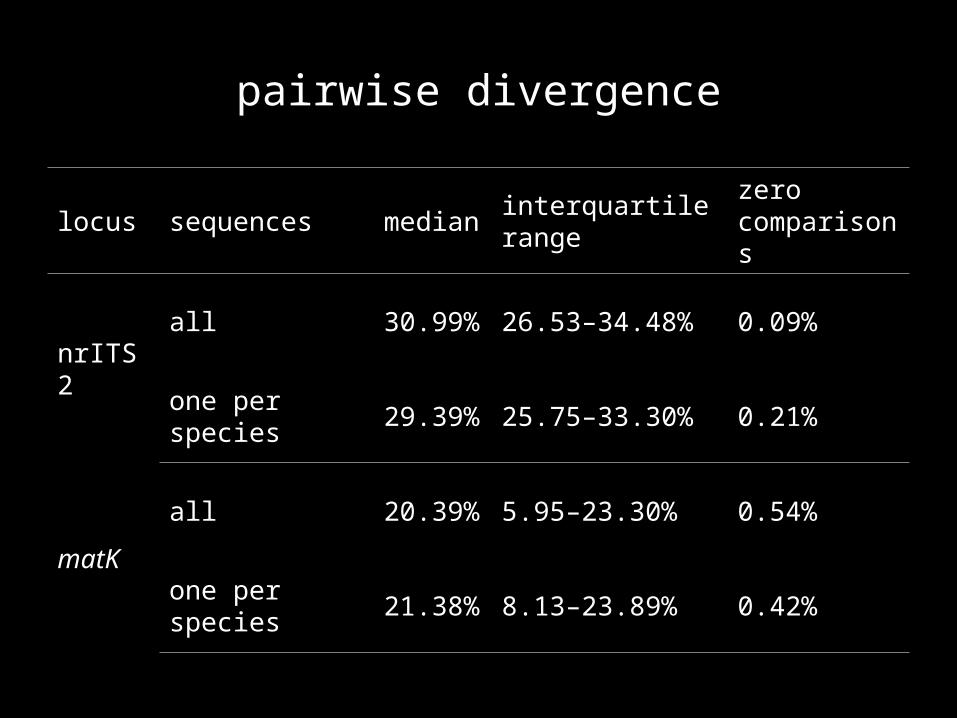
pairwise divergence
locus sequences median interquartile rangezero comparisons
nrITS 2
all 30.99% 26.53–34.48% 0.09%
one per species 29.39% 25.75–33.30% 0.21%
matK
all 20.39% 5.95–23.30% 0.54%
one per species 21.38% 8.13–23.89% 0.42%

hierarchical clustering
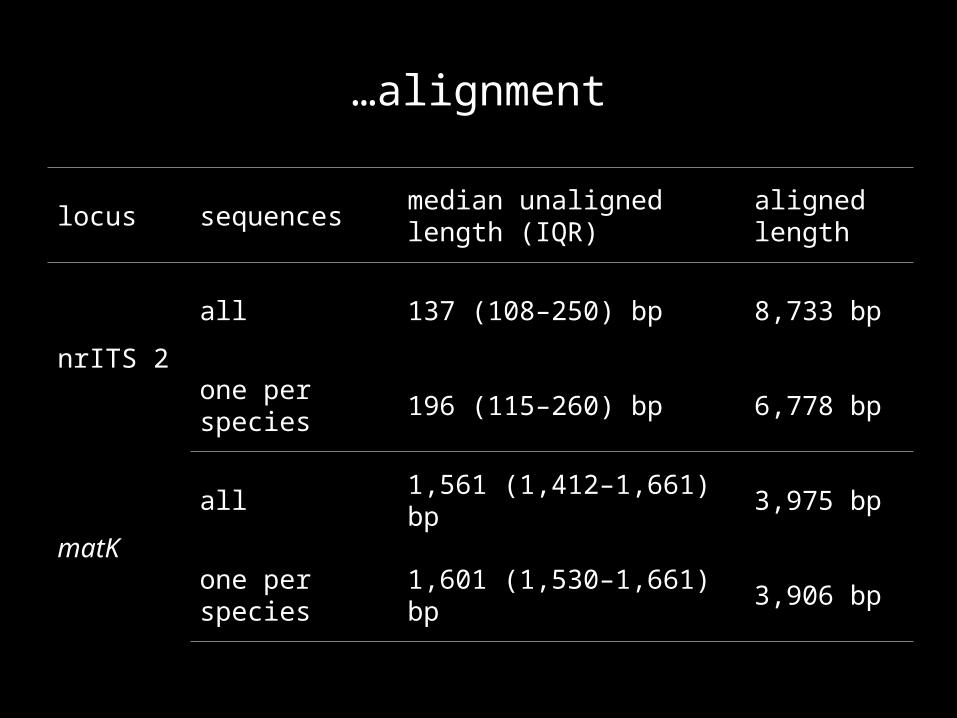
…alignment
locus sequencesmedian unaligned length (IQR)
aligned length
nrITS 2
all 137 (108–250) bp 8,733 bp
one per species 196 (115–260) bp 6,778 bp
matK
all 1,561 (1,412–1,661) bp 3,975 bp
one per species 1,601 (1,530–1,661) bp 3,906 bp
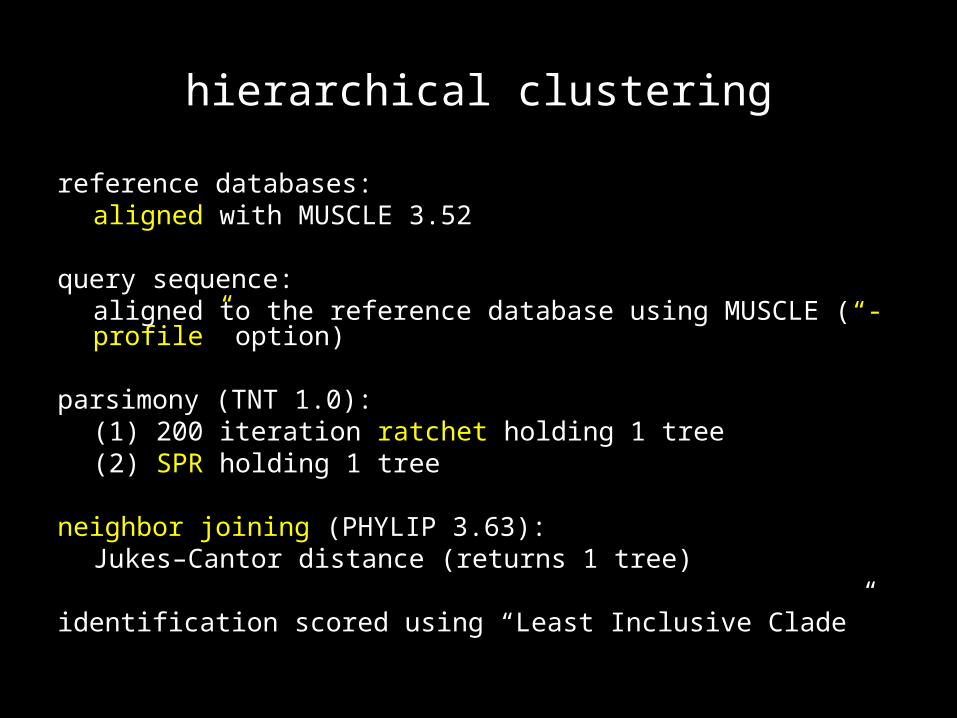
hierarchical clustering
reference databases:aligned with MUSCLE 3.52
query sequence:aligned to the reference database using MUSCLE (“-profile” option)
parsimony (TNT 1.0):(1) 200 iteration ratchet holding 1 tree(2) SPR holding 1 tree
neighbor joining (PHYLIP 3.63):Jukes–Cantor distance (returns 1 tree)
identification scored using “Least Inclusive Clade”
Will and Rubinoff (2004)...
identification ambiguity due to tree shape
Fitch (1971) optimization of group membership variables


Least Inclusive Clade
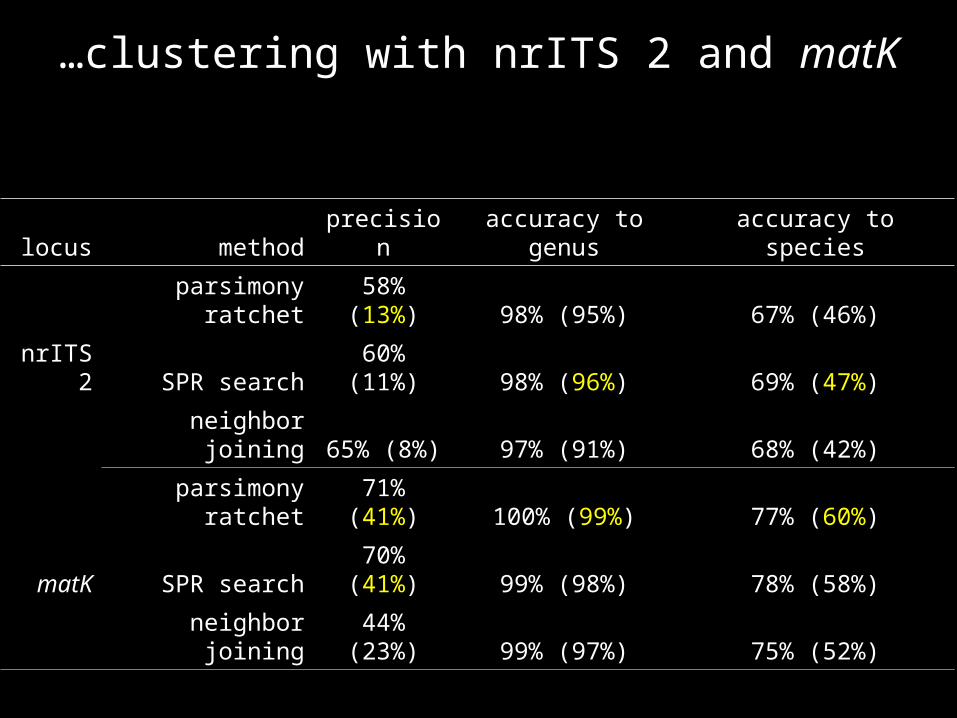
…clustering with nrITS 2 and matK
locus method precision accuracy to genus accuracy to species
parsimony ratchet 58% (13%) 98% (95%) 67% (46%)
nrITS 2 SPR search 60% (11%) 98% (96%) 69% (47%)
neighbor joining 65% (8%) 97% (91%) 68% (42%)
parsimony ratchet 71% (41%) 100% (99%) 77% (60%)
matK SPR search 70% (41%) 99% (98%) 78% (58%)
neighbor joining 44% (23%) 99% (97%) 75% (52%)
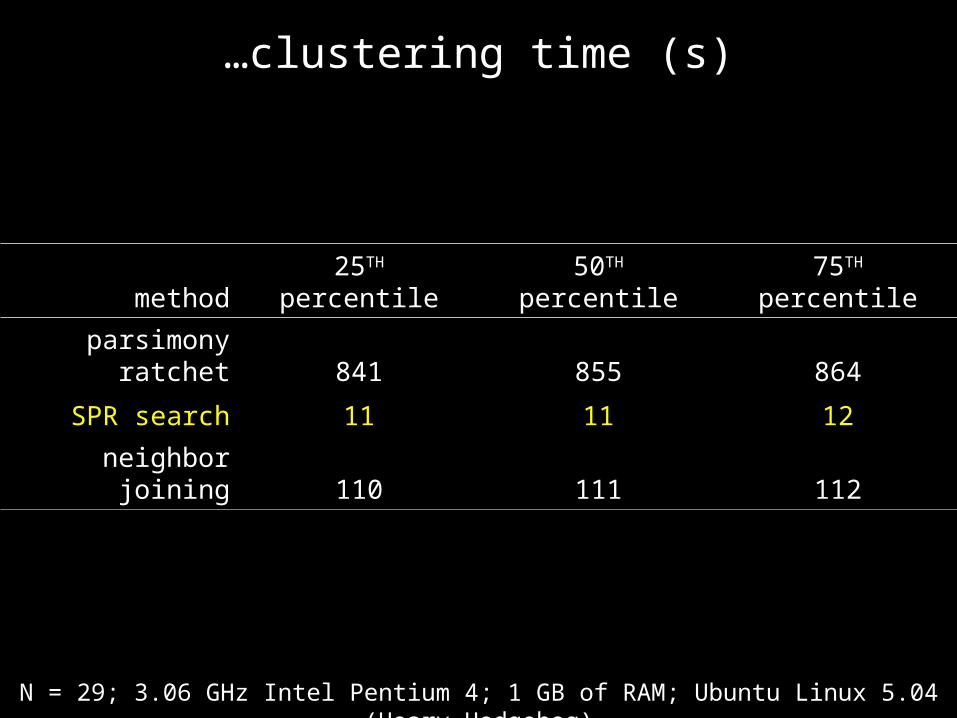
…clustering time (s)
method 25TH percentile 50TH percentile 75TH percentile
parsimony ratchet 841 855 864
SPR search 11 11 12
neighbor joining 110 111 112
N = 29; 3.06 GHz Intel Pentium 4; 1 GB of RAM; Ubuntu Linux 5.04 (Hoary Hedgehog)

similarity methods

similarity methods
BLASTn (version 2.2.10) BLAT (version 32)megaBLAST (version 2.2.10)
default parameters
best match(es) taken as ID

…similarity methods with nrITS 2 and matK
locus method precision accuracy to genus accuracy to species
BLAST 94% (81%) 100% (100%)! 67% (63%)
nrITS 2 BLAT 94% (82%) 99% (99%) 66% (62%)
megaBLAST 94% (80%) 95% (95%) 72% (68%)
BLAST 99% (67%) 100% (100%)! 84% (68%)
matK BLAT 99% (69%) 99% (99%) 82% (67%)
megaBLAST 99% (61%) 100% (99%) 84% (64%)
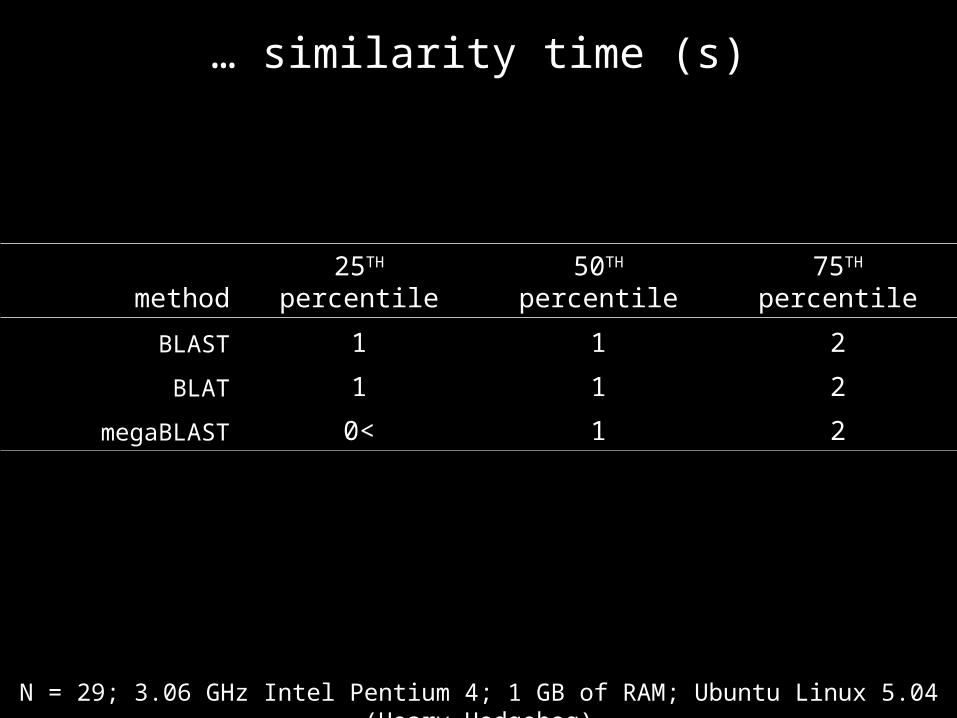
… similarity time (s)
method 25TH percentile 50TH percentile 75TH percentile
BLAST 1 1 2
BLAT 1 1 2
megaBLAST 0< 1 2
N = 29; 3.06 GHz Intel Pentium 4; 1 GB of RAM; Ubuntu Linux 5.04 (Hoary Hedgehog)

combination methods (cf. BOLD–ID)

combination methods (cf. BOLD–ID):
(1) get the top 100 BLAST hits
(2) align with MUSCLE
(a) 200 iteration ratchet holding 1 tree
(b) SPR holding 1 tree
(c) neighbor joining with Jukes–Cantor distances
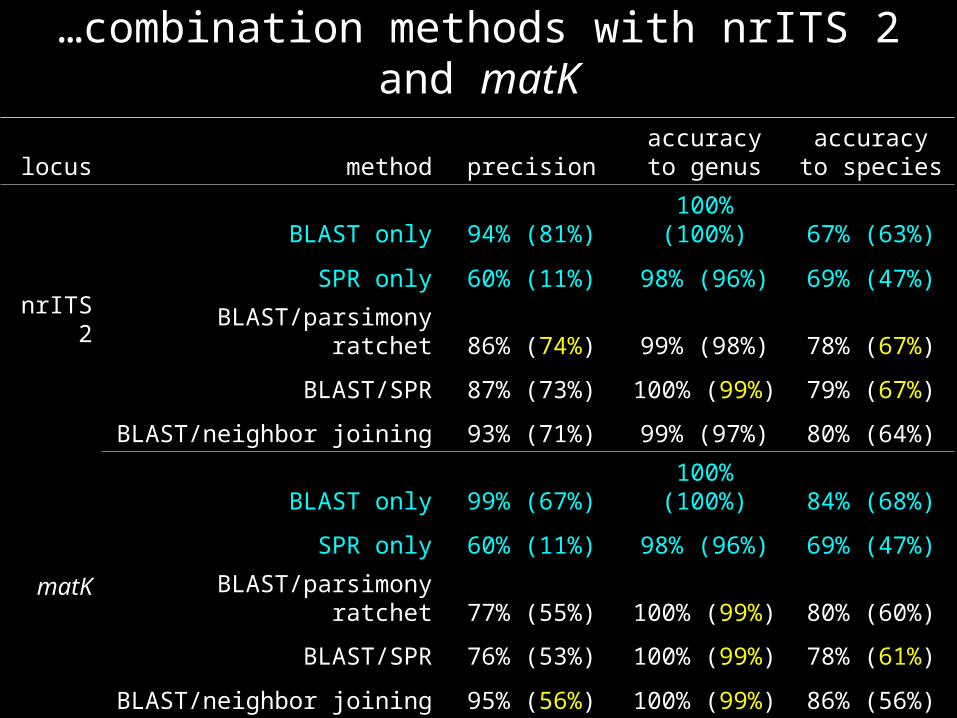
…combination methods with nrITS 2 and matK
locus method precisionaccuracy to
genusaccuracy to
species
nrITS 2
BLAST only 94% (81%) 100% (100%) 67% (63%)
SPR only 60% (11%) 98% (96%) 69% (47%)
BLAST/parsimony ratchet 86% (74%) 99% (98%) 78% (67%)
BLAST/SPR 87% (73%) 100% (99%) 79% (67%)
BLAST/neighbor joining 93% (71%) 99% (97%) 80% (64%)
matK
BLAST only 99% (67%) 100% (100%) 84% (68%)
SPR only 60% (11%) 98% (96%) 69% (47%)
BLAST/parsimony ratchet 77% (55%) 100% (99%) 80% (60%)
BLAST/SPR 76% (53%) 100% (99%) 78% (61%)
BLAST/neighbor joining 95% (56%) 100% (99%) 86% (56%)

…combination time (s)
method 25TH percentile 50TH percentile 75TH percentile
BLAST only 1 1 2
SPR only 11 11 12
BLAST/parsimony ratchet 186 219 278
BLAST/SPR search 170 196 262
BLAST/neighbor joining 171 198 264
N = 29; 3.06 GHz Intel Pentium 4; 1 GB of RAM; Ubuntu Linux 5.04 (Hoary Hedgehog)

diagnostic methods
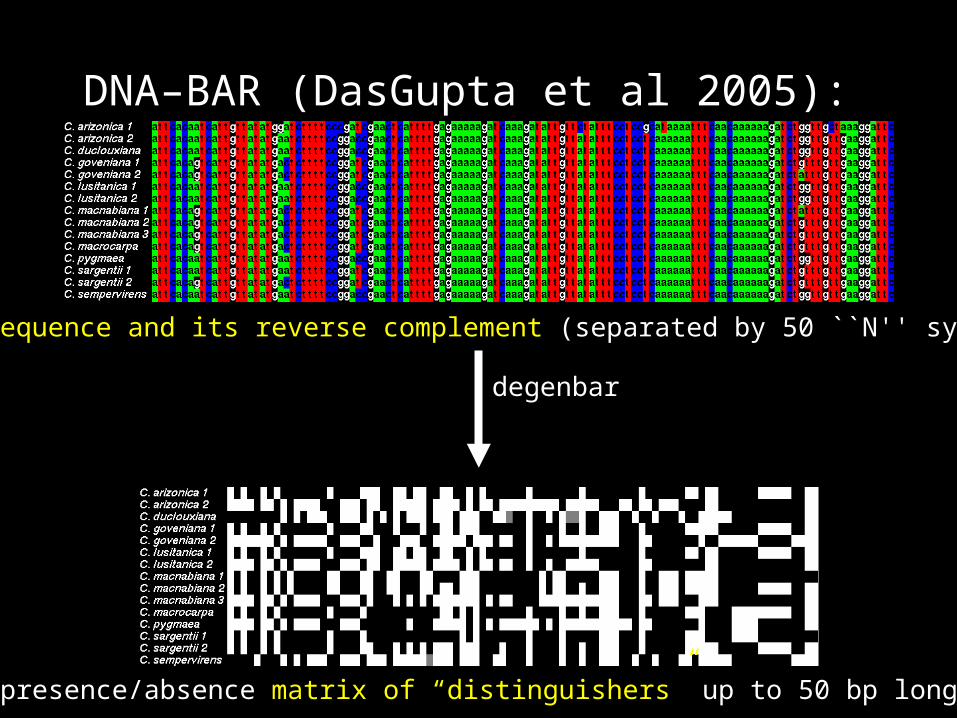
DNA–BAR (DasGupta et al 2005):
each sequence and its reverse complement (separated by 50 ``N'' symbols)
presence/absence matrix of “distinguishers” up to 50 bp long
degenbar
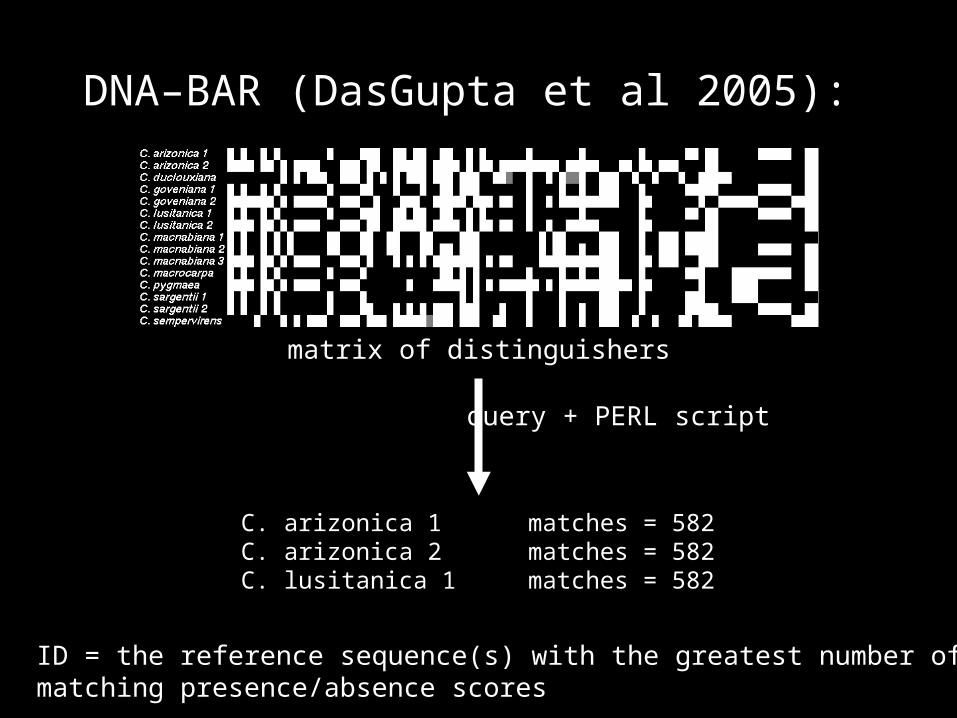
DNA–BAR (DasGupta et al 2005):
matrix of distinguishers
query + PERL script
ID = the reference sequence(s) with the greatest number of matching presence/absence scores
C. arizonica 1 matches = 582C. arizonica 2 matches = 582C. lusitanica 1 matches = 582
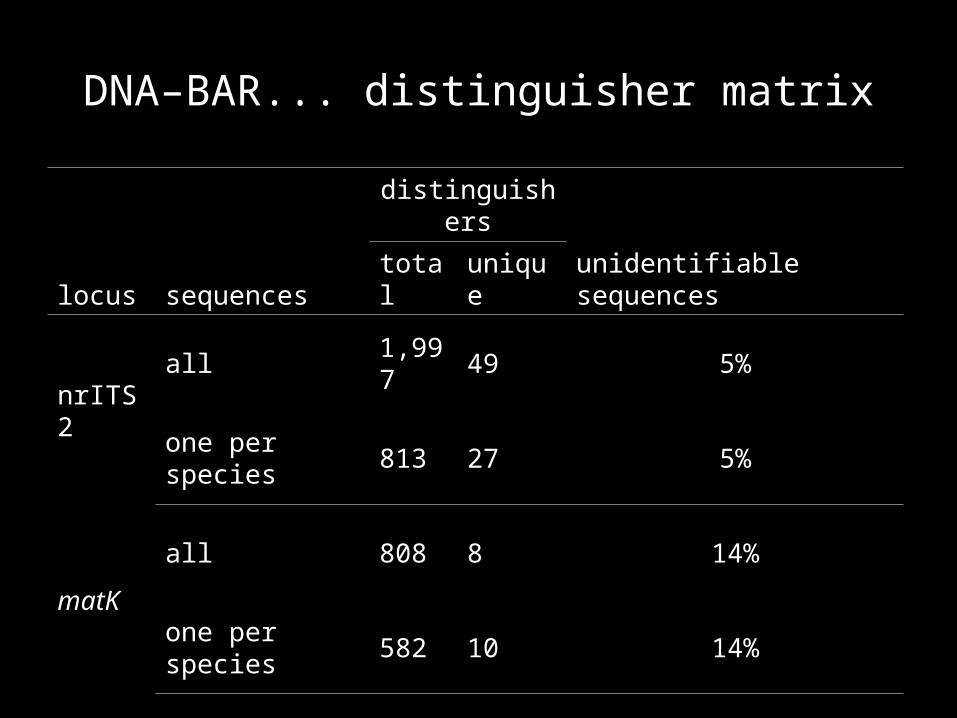
DNA–BAR... distinguisher matrix
locus sequences
distinguishers
unidentifiable sequencestotal unique
nrITS 2
all 1,997 49 5%
one per species 813 27 5%
matK
all 808 8 14%
one per species 582 10 14%
diagnostic methods: DOME ID
reference database (via PERL and MySQL): (1) all sequence strings of 10 nucleotides offset by 5 nucleotides were extracted from the reference sequences(2) each string was classified as diagnostic (unique to a particular species) or non–diagnostic(3) diagnostic strings were inserted into the diagnostic barcode
database
GCGTTGATGG GTTGGGCGTT CATACGTTGG GTCACCATAC CCTTTGTTTG AGGGACCTTT CTGAGCATCG GTGCACTGAG TTCTCGATGC GGCGTTTCTC TAGCTGGCGT AGGTCTAGCT GGCTGAGGTC GCTTGCATCG CCCTAGCTTG AATGTGCGCA GATGCAATGT TAGCCGGCGT CTGTCTAGCC GCCTTGCCCC ATGCCCCCTG ATCGTGGTGC CCCTGCAAGT AGTGTGCGCA TAGACGACGT CTGTCTAGAC GACTTGCCCC CTTGCGGATC CGGCCTGACT ACCCCCGGCC CGTGAACCCC CTGCCTGACT CCCCCCTGCC TGGGCCGTCA CGCGATGGGC ATACGCGCGA GCCCTTTGAG TGCGGTGGGA CAAGTGAGGA TCGGGCAAGT TAAAATCGTC CAAACCCGTC GTGCATGTGC CGTGCGTGCA CTTCCCACGA CCGTCCCGCA GCATTTGCGG CTCGGGGAGC AAGACCCGTC GCGGCAAGAC GTGCGTGCGT TGCAGAGGGG TTCTCACGAA AGGTTCTCCC GTGCCAGGTT TGCGTCCCGC TTGTTTGCGT TTTCATTGTT GGCGGCATGA TCCCCTGCCC CTTGCTTTTT GGCGGCTTGC CGGCGGGCGG CGGCACGGCG CTTTACGGCA AGACTCCGCG GATCGAGACT CAAGTGATCG GGTGTCAAGT GGTGGCCCCC GGCTCATCAT TGAAACGTGC CCCAAGACGG CGTGCCCCAA AGGACCGGGA TGGGGGTGGG CCGCGTGGGG GACCTCCATT AAACCGACCT AAAGAAAAGA TCCAAGAAAA GCCTGTTTTC GGTCAGCCTG CATGCGTGCG TCAAGGATCC CGGTTTCAAG CGACGCGGTT GTGCTCGGAA GGGATGTGCT CTACGGTCGA GTCGCCTACG ATAGTCTTCA CGGCGATAGT TGTTTTCATG GATGGTGTTT GTCCCTATCA ATTAAAATAC CGATCCGAGT GCGGGTGAGA TCCCCCCCAA AGGATGACGA GCAAAAGGAT ACATGATTCG AATACAACTC CGCAAGCGGC GGCGTGGAAT TCAGCGTTGG ACGGGTCAGC GATAGTCCGT GATCCGATAG GCATTGGGGG GATATTTGAT TAGCCCAAAA TCGCCTAGCC GCCCTTCGGC CATGCGCCCT CTACTCTTTC AACGTCTACT CACGCGAGAG CGCGTCACGC CGCGTATCTT AGCGTGCATC GGGGGAGCGT GCTACGGGGG CGAGGCGTCC GGAACCGAGG TTTCACGGGT GCCGATCCGG AATGCGCCGA GTACTCGCGA TGGCAAGGAT GCCGGTACCG CAACGGCCGG AAGCGGGCAG GCAGCAAGCG CGAGACGATG GACGACGAGA AGACCCGGGA CGAGCCTTCA CGGATGAGAA TTGCGCGGAT CTCCATAGGT TTCCCCCAAG AATCGTTCCC CGCCTCGATG CCGAGCCTCG TTCAAGAATC GTGAATTCAA AAAATTCACG TCGTCCGCCG GCGACCCAGC GAAGCGCGAC ACGGGTGCCG CGTGTAATGT AACGACGTGT AGTAAAGGTC GCTCAAGTAA GACGTGCTCA TGCTGGACGT TAGATGGCTG GGCGGTATGT CCGATGCGAT ATCCCCCGAT TCCTGTCCTC GAGACTCCAA ACCGGCGTTG CAAAGACCGG ACTGAAATGA AGGGCTCGGC ATATCGTCGG CAGGAATCCC AATTGCAGGA CCAACGATGA ACATCCCAAC TGTCAACATC CCTCTCCCGT GGTTGGACGG TTGATGGTTG GGGGATTGAT AATCTAGTTG AGGGGAATCT CTCTTTCCAA CGCCTCTCTT CTGTGCGCCT TCGACCTGTG CTTTCTCGAC CGCTACTTTC AGCGCCGCTA ATCTCAGCGC TGGGTATCTC CTCGTTGGGT TCGCGCTCGT GTGTGTCGCG CTTGACGTCC AAAGCCTCGT CTTCGAAAGC CCGATGCGCT TCTCGCCGAT CCCTGTCTCG GTTGGAGGGT TGATCGTTGG TTGATTGATC GGTGATTGAT TCGTGGGTGA TCTTCTCGTG GCTATTCTTC GACGGGCTAT TAGCTGACGG CTGGATAGCT CAGCACTGGA GGCTTCAGCA TCGCGGGCTT GTGATTGCTG CCGCCGTGAT CTGCCCCGCC CTTCTCTGCC CCTGACTTCT CGTTGCCTGA GCTGCCGTTG TGCTGGCTGC TCCAGTGCTG GGCTATCCAG CCGTGGGCTA GCGCCCCGTG CTGTTGCGCC CGAGGCTGTT CTTTACGCCT GCGCCCTTTA GAAAGGGCTT GATCGGAAAG TGTTGCATGT GGTCCTGTTG TTGTCGGTCC CATGGTTGTC

diagnostic methods: DOME ID
reference database (via PERL and MySQL): (1) all sequence strings of 10 nucleotides offset by 5 nucleotides were extracted from the reference sequences(2) each string was classified as diagnostic (unique to a particular species) or non–diagnostic(3) diagnostic strings were inserted into the diagnostic barcode
database
diagnostic barcode database
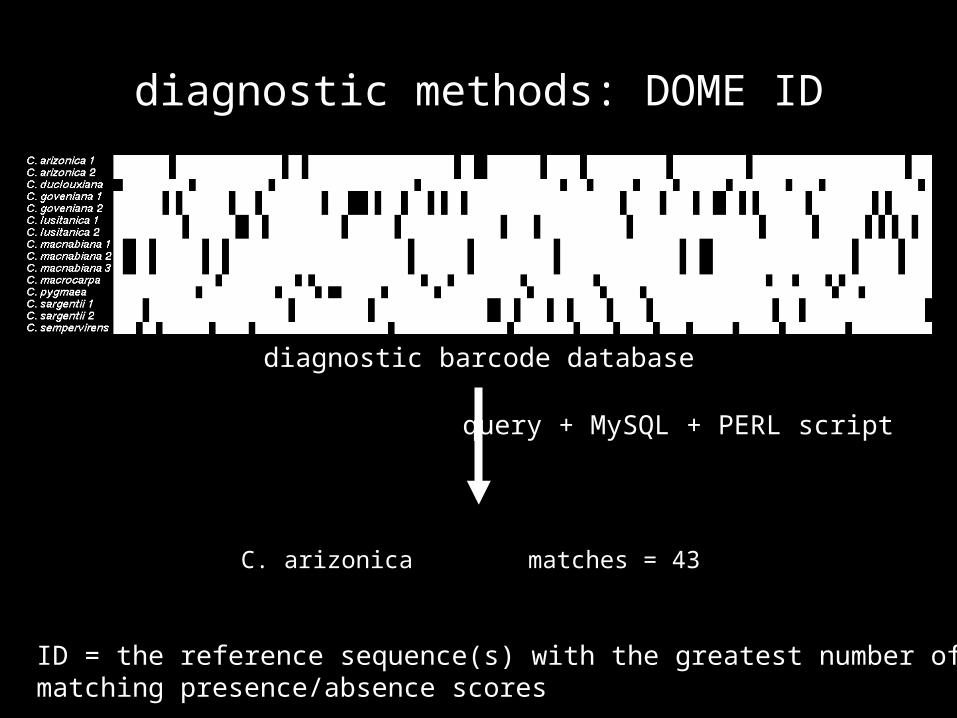
diagnostic methods: DOME ID
query + MySQL + PERL script
ID = the reference sequence(s) with the greatest number of matching presence/absence scores
C. arizonica matches = 43
diagnostic barcode database

diagnostic methods: ATIM
presence/absence matrix of all possible of 10 bp combinations [1,048,576 motifs]
PERL script

diagnostic methods: ATIM
1,048,576 character presence/absence matrix
TNT (parsimony ratchet)
reference tree (strict consensus)

diagnostic methods: ATIM
query+
1,048,576 character presence/absence matrix+
reference tree (positive constraint)
TNT (TBR hold 20)
identification scored using “Least Inclusive Clade”

…diagnostic methods with nrITS 2 and matK
locus method precision accuracy to genus accuracy to species
nrITS 2
DNA–BAR 98% (89%)! 86% (86%) 65% (62%)
DOME ID 80% (80%) 86% (84%) 67% (66%)
ATIM 100% (83%) 99% (98%) 83% (71%)!
matK
DNA–BAR 100% (79%)! 96% (96%) 73% (62%)
DOME ID 60% (60%) 53% (53%) 50% (50%)
ATIM 100 (67%) 98% (97%) 87% (53%)
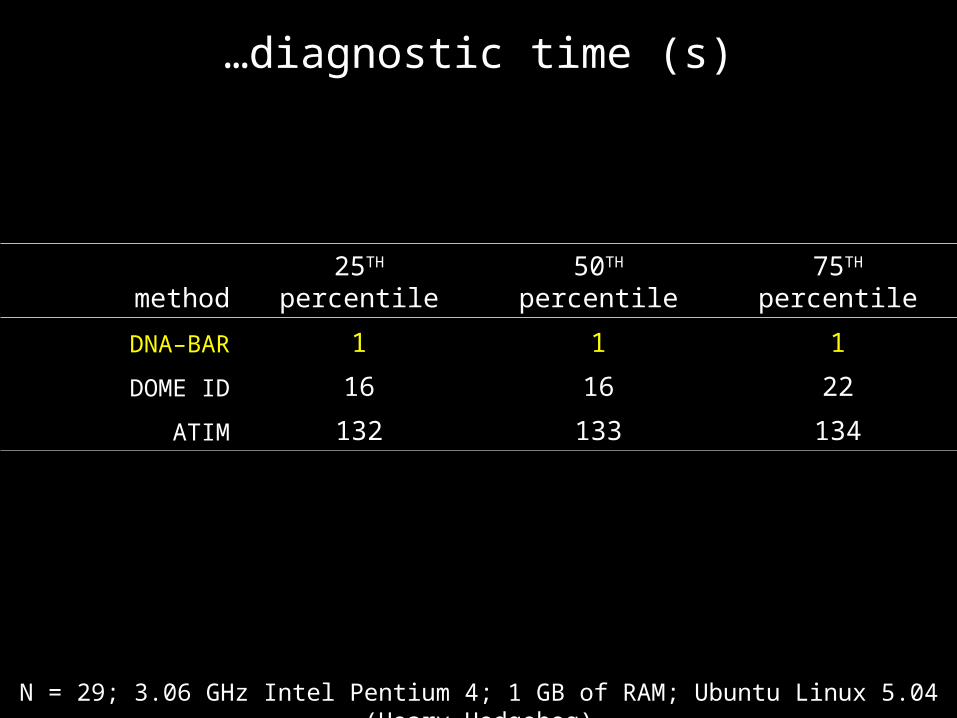
…diagnostic time (s)
method 25TH percentile 50TH percentile 75TH percentile
DNA–BAR 1 1 1
DOME ID 16 16 22
ATIM 132 133 134
N = 29; 3.06 GHz Intel Pentium 4; 1 GB of RAM; Ubuntu Linux 5.04 (Hoary Hedgehog)

DAWG I “training” dataset
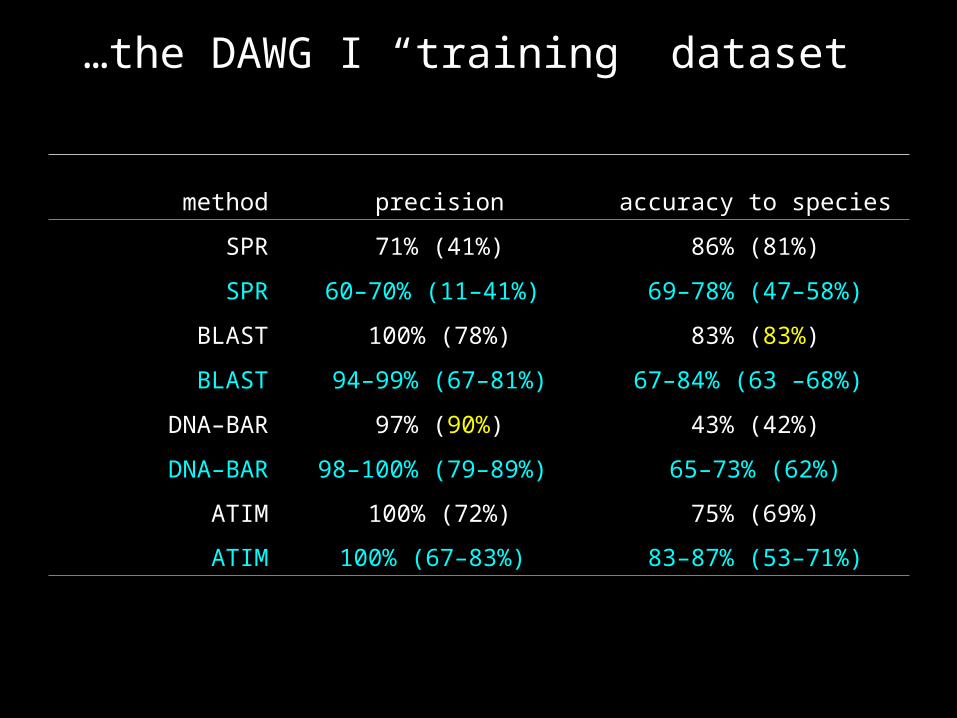
…the DAWG I “training” dataset
method precision accuracy to species
SPR 71% (41%) 86% (81%)
SPR 60–70% (11–41%) 69–78% (47–58%)
BLAST 100% (78%) 83% (83%)
BLAST 94–99% (67–81%) 67–84% (63 –68%)
DNA–BAR 97% (90%) 43% (42%)
DNA–BAR 98–100% (79–89%) 65–73% (62%)
ATIM 100% (72%) 75% (69%)
ATIM 100% (67–83%) 83–87% (53–71%)

conclusions:
all methods are relatively precise => expect accuracy to approximate precision
observed accuracy of species level identification is lower=> failure of the algorithms to correspond to species delimitations (shared haplotypes or haplotypes of a species are more similar to those of different species)=> for accurate identification, the reference database must contain virtually all haplotypes
none of the methods performed particularly well=> computer time
=> BLAST (BLAT and megaBLAST too)=> DNA–BAR

acknowledgments
brilliant insights &tc:
K. Cameron
C. Chaboo
T. Dikow
C. Martin
R. Meier
M. Mundry
money:
Cullman Program for Molecular Systematic Studies
DIMACS/NSF





























