Embed Size (px)
Citation preview

Review
10.1517/17425250802407463 © 2008 Informa UK Ltd ISSN 1742-5255 1245All rights reserved: reproduction in whole or in part not permitted
A critical overview of the infl uence of infl ammation and infection on P-glycoprotein expression and activity in the brain Derek J Roberts & Kerry B Goralski † † College of Pharmacy, Faculty of Health Professions, Dalhousie University, 5968 College Street, Halifax, N.S., B3H 3J5, Canada
Background : P-glycoprotein is a blood–brain barrier efflux transporter that limits drug accumulation in the brain. Objective : To review recent in vivo and in vitro investigations that examined the influence of inflammation, infection and related clinical neuroinflammatory disorders on P-glycoprotein expression and activity in the brain. Methods : Critical overview of English-language studies. Results/conclusions : Inflammation and infection produce dynamic changes in P-glycoprotein expression and activity in the blood–brain barrier. In vitro , blood–brain barrier P-glycoprotein activity is down-regulated after short-term exposure to inflammatory mediators whereas its activity is upregulated following more prolonged exposure. In vivo studies in both humans and animals have linked CNS inflammation, peripheral inflammation and related clinical neuroinflammatory disorders with altera-tions in the expression and activity of blood–brain barrier P-glycoprotein. The direction and degree of change in P-glycoprotein activity depends on the in vivo or in vitro model examined, the cell type examined (e.g., endothelial or glial), the inflammatory mediator utilized, the anatomic site in which the inflammatory response was first generated, the time points chosen for observation and the substrates analyzed. Alterations in P-glycoprotein activity affect drug activity in the CNS and seem clinically important.
Keywords: blood–brain barrier , cytokines , inflammation , infection , P-glycoprotein
Expert Opin. Drug Metab. Toxicol. (2008) 4(10):1245-1264
1. Introduction
1.1 Effect of neuroinfl ammation and -infection on P-glycoprotein activity and drug accumulation in the brain A host of clinical neuroinflammatory conditions, including bacterial meningo-encephalitis, acute traumatic brain injury, multiple sclerosis, Parkinson’s and Alzheimer’s diseases and HIV-related dementia are associated with altered blood–brain barrier permeability [1] . The first examples of this association were reports of enhanced accumulation of antibiotics in the cerebrospinal fluid (CSF) and/or brain of patients and experimental animals with CNS infections [2-4] . Although the presence of energy-dependent drug efflux transporters in the blood–brain barrier and choroid plexus (the anatomic location of the blood–CSF barrier) have been recognized for at least 50 years [5] , the connection among CNS inflammation, altered drug transporter activity and enhanced brain accumulation of drugs has only recently begun to be appreciated.
The genes ABCB1 ( MDR1 ) in humans and abcb1a ( mdr1a ) and abcb1b ( mdr1b ) in mice encode the ATP-binding cassette (ABC) drug efflux transporter proteins
1. Introduction
2. Regulation of brain
P-glycoprotein expression
and activity by infl ammation
and infection
3. Conclusion
4. Expert opinion
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Infl ammation, infection and brain-derived P-glycoprotein
1246 Expert Opin. Drug Metab. Toxicol. (2008) 4(10)
ABCB1, Abcb1a and Abcb1b, respectively, which are commonly referred to collectively as P-glycoprotein [6] . In the rodent blood–brain barrier, the Abcb1a isoform acts to prevent CNS drug accumulation by transporting a diverse array of commonly used therapeutic compounds out of the brain against a concentration gradient [7-10] . In humans, evidence supporting a reduction in blood–brain barrier P-glycoprotein activity during CNS infection dates back at least 40 years. Early clinical studies revealed that patients with meningitis had greater CSF concentrations of rifampicin and ethambutol when compared to control subjects [11,12] . However, such studies predated the discovery of P-glycoprotein in cerebral capillaries and the identification of rifampicin and ethambutol as P-glycoprotein substrates [8,13,14] . Therefore, the link between neuroinflammation and drug efflux transporter proteins such as P-glycoprotein probably went unrecognized.
Animal work completed over the past 5 or 6 years has since demonstrated that acute and chronic inflammatory and infectious processes generated within the CNS are capable of decreasing the expression and/or activity of blood–brain barrier P-glycoprotein. Decreased activity of this transporter promotes enhanced substrate drug accumulation in the CNS, which may alter pharmacologic and/or toxicologic response. Recent studies have also revealed that brain P-glycoprotein expression and activity is modified by inflammatory and infectious responses that originate in peripheral tissues, such as the peritoneum, rather than first within the CNS. Collectively, these studies highlight the presence of a newly recognized interaction between inflammation, infection and the expression and activity of brain-derived P-glycoprotein.
In this review, we discuss recent animal and human investigations that examined the influence of inflammation, infection and related clinical neuroinflammatory disorders on P-glycoprotein expression and activity in the brain. We also describe the molecular mechanisms by which these changes are thought to occur and the implications that altered P-glycoprotein activity has on CNS drug delivery, pharmacotherapy and toxicology. Finally, we propose goals for future research in this area.
1.2 Function of P-glycoprotein in the blood–brain and blood–CSF barriers Cerebral capillaries within the neural parenchyma constitute the anatomic ultrastructure of the blood–brain barrier. To a varying extent, these specialized neurovascular units allow passage of drugs, nutrients and hormones into the brain from the systemic circulation while simultaneously limiting passage of potentially harmful and toxic substances into the CNS. The physical component of the barrier is formed by a continuous polarized monolayer of non-fenestrated brain capillary endothelial cells with interposed high-resistance tight junctions (zonulae occludentes) [15] . This physical ultrastructure, combined with a lack of cerebral
endothelial cell pinocytotic activity, nonspecifically limits paracellular and transcellular permeability of macromolecules (especially high molecular mass, polar substances) and provides a stringent barrier against entry of hydrophilic xenobiotics into CSF and brain parenchyma [16] . To comple-ment these physical characteristics, brain capillary endothelial cells also express energy-dependent efflux transporter systems, which contribute to selective blood–brain barrier permeability by actively transporting drugs and other xenobiotics out of the CNS [16,17] .
The initial suggestion that P-glycoprotein contributed to this selective component of the blood–brain barrier began with its detection in human and rat brain capillaries ∼ 20 years ago, 13 years following its original discovery by Juliano and Ling (1976) in mutant Chinese hamster ovary cells ( Figure 1A , B ) [8,14,18] . This suggestion was then supported by the landmark animal investigation by Schinkel et al. (1994), which found that knockout mice devoid of the abcb1a gene could obtain a 20- and 100-fold higher accumulation of the P-glycoprotein substrates vin-blastine and ivermectin in the brain following peripheral administration as compared to wild-type animals, respectively [9] . Not surprisingly, abcb1a -deficient mice were also more susceptible to the toxic effects of these substrates, revealing the potential clinical importance of brain P-glycoprotein activity [9] . Subsequent studies utilizing abcb1a knockout mice or CF-1 mice with mutated, nonfunctional abcb1a genes have since confirmed that the absence of P-glycoprotein expression is associated with increased brain accumulation and/or toxicity of a multitude of commonly used therapeutic agents (e.g., anticonvulsants, antidepressants, antineoplastics, antiretrovirals, antipsychotics, calcineurin inhibitors, calcium channel blockers, glucocorticoids and opioids, among others) [10,19-26] . It is also clear that the localization of P-glycoprotein in brain capillaries and its associated protective function extends to other species. For example, King and co-workers (2001) found that downregulation of abcb1a with antisense technology reduces brain-to-blood transport of opioids in rats [27] . Collie dogs homozygous for the ABCB1-1 ∆ polymorphism (which produces a truncated and nonfunctional form of the transporter) are also more sensitive to the adverse CNS effects of loperamide and ivermectin, drugs that are normally excluded from the brain by P-glycoprotein [28-30] .
Less is known about the function of P-glycoprotein in the human brain. However, several recent clinical studies have suggested that humans with ABCB1 polymorphisms, or those receiving P-glycoprotein inhibitors, exhibit different pharmacological responses or even neurotoxicity in response to the administration of loperamide, chemotherapeutics, antidepressants and antiepileptics when compared with control subjects [23,31-33] . Taken together, all of the aforementioned studies highlight the conserved nature of P-glycoprotein among different species and its vital contribution to the protective function of the blood–brain barrier.
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Roberts & Goralski
Expert Opin. Drug Metab. Toxicol. (2008) 4(10) 1247
A.
B.
C.
Capillaryendothelium
Blood capillary
Brain extracellularfluid
Luminalmembrane
Basalmembrane
AstrocyteNeuron
Astrocyte
Tightjunction
PGP PGP Drug substrate
Choroidplexus
epithelium
Cerebrospinal fluidEpendyma
Luminalmembrane
Basalmembrane
Capillaryendothelium
Tightjunction
Neuron
Astrocytefoot process
Capillaryendothelial cell
Capillarylumen
Pericyte
Tight junction
Brainparenchyma
Basementmembrane
Blood capillaryFenestra
PGP
PGP ?
Figure 1 . Location and function of P-glycoprotein in the blood–brain and blood–CSF barriers. A. Cross section of the blood–brain barrier. The ultrastructure of the barrier consists of non-fenestrated cerebral capillary endothelial cells with high-resistance intercellular tight junctions. The barrier is contacted and reinforced circumferentially by astrocyte foot processes, which probably infl uence overall barrier function. P-glycoprotein is primarily expressed in cerebral capillary endothelial cells with lower expression in astrocytes. B. Longitudinal section through the blood–brain barrier. P-glycoprotein is localized to the luminal membrane of blood–brain barrier endothelial cells and transports substrate drugs from brain-to-blood, thereby preventing drug accumulation in the CNS. C. Longitudinal section of the blood–CSF barrier. P-glycoprotein is expressed on the subapical surface of the choroid plexus epithelium facing the CSF-containing spaces of the brain (e.g., lateral and third ventricles, among others) and may mediate transport of substrates into CSF. CSF: Cerebrospinal fl uid; PGP: P-glycoprotein.
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Infl ammation, infection and brain-derived P-glycoprotein
1248 Expert Opin. Drug Metab. Toxicol. (2008) 4(10)
Of note, a second interface between the systemic circulation and the brain is the blood–CSF barrier, formed by the specialized, convoluted epithelial cell layer of the choroid plexus, which invaginates into the lateral, third and fourth ventricles [34] . As the main function of the choroid plexus is secretion of CSF into the CSF-containing spaces of the brain, this barrier could represent an alternative site for xenobiotic entry into the CNS. The choroid plexus contains numerous uptake and efflux transporter systems as well as intercellular tight junctions akin to those in the blood–brain barrier [16] . P-glycoprotein in the choroid plexus is localized to the subapical surface facing the CSF compartment in which its function is largely unknown [35] . A study by Rao et al. (1999) suggested that choroid plexus P-glycoprotein might facilitate entry of drug and other xenobiotic substrates into CSF from blood and thus could oppose the action of this transporter in the blood–brain barrier ( Figure 1C ) [35] . However, this potential blood-to-CSF transport action of choroid plexus P-glycoprotein might also be viewed as a route of elimination from the CNS as drugs circulating in CSF might ultimately gain access to cerebral sinus blood through the arachnoid villi. Whichever the case, choroid plexus P-glycoprotein activity probably has only a minimal effect on overall CNS drug penetration as its expression in this location is substantially lower compared to that in the cerebral capillaries of the blood–brain barrier ( ∼ 0.5% based on a recent study) [36] .
1.3 Anatomical and cellular localization of P-glycoprotein within the brain Some controversy exists regarding the localization of P-glycoprotein in the brain and in the cells of the blood–brain barrier secondary to variations in the fixation procedure and the specific antibody used for immunological localization of P-glycoprotein [36-41] . Most evidence supports that in normal brain tissue P-glycoprotein is primarily localized to the luminal membrane of cerebral capillary endothelial cells with lower levels of transporter expression in astrocytes ( Figure 1A ) [7,37,42] . Although neuronal P-glycoprotein has been detected, its expression in these cells is low and limited to brain regions affected by pathological conditions such as epilepsy and hypoxia/ischemia [37,43] . One recent investigation found that P-glycoprotein was also expressed in pericytes and on the abluminal membrane of blood–brain barrier endothelial cells and within numerous intracellular locations, including the nuclear envelope, cytoplasmic vesicles, Golgi complex and endoplasmic reticulum [40] , although these findings are presently controversial. P-glycoprotein expression within the apical membrane of cerebral capillary endothelial cells is consistent with its ability to limit intracerebral accumulation of xenobiotics; however, its detection in other brain cells suggests that P-glycoprotein may also be important for limiting cellular toxicity [40] . P-glycoprotein activity in astrocytes and pericytes may also act in parallel with that in the apical membrane of endothelial cells to further impede drug entry into the brain.
1.4 The innate CNS immune response The CNS is capable of mounting an immune response characterized by production of inflammatory mediators first in circumventricular organs (e.g., the area postrema, median eminence, pineal gland, posterior lobe of the pituitary, vascular organ of the lamina terminalis and subfornical organ) and other brain regions devoid of blood–brain barrier [44-46] . Such a response is initiated when centrally-located or circulating antigens permeate low-resistance, leaky cerebral capillaries and interact with toll-like receptors (TLRs) positioned on circumventricular organ-resident macro-phages and microglia [44-46] . TLRs recognize immunogenic elements of pathogens known as pathogen-associated molecular patterns. Distinct subtypes of TLRs recognize the principal pathogen-associated molecular patterns associated with Gram-positive and Gram-negative bacteria such as lipoteichoic acid and peptidoglycan for TLR2 and the bacterial endotoxin lipopolysaccharide (LPS) for TLR4 [46-48] . Once activated, some TLRs result in an intracellular signaling cascade that culminates in activation of the transcription factor NF- κ B [46,47] . Intracellular NF- κ B activation then leads to heightened transcription of chemokines, cytokines and other essential inflammatory mediators by CNS inflammatory cells [46,47] . These events result in the production of a localized inflam-matory response in the CNS that may subsequently generalize and involve the entire brain.
Numerous in vitro and in vivo investigations have examined the influence of such inflammatory processes on P-glycoprotein expression and activity in the whole brain, blood–brain barrier or in individual brain cells. Please see Tables 1 – 4 for a detailed and descriptive overview of the principal findings of these studies.
2. Regulation of brain P-glycoprotein expression and activity by infl ammation and infection
2.1 In vivo animal models of CNS infl ammation and infection Bacterial and fungal meningitis are associated with inflam-mation of the brain and leptomeninges and the release of pro-inflammatory cytokines such as TNF- α and IL-1 β [1] . Cryptococcal meningitis in particular is associated with severe leptomeningeal inflammation [49] . In a recent study of mice infected with Cryptococcus neoformans , Imbert et al. (2003) demonstrated a twofold increase in the C max and AUC of the P-glycoprotein substrate itraconazole in the brain of infected mice as compared to uninfected mice ( Table 1 ) [49,50] . In C. neoformans infected mice, brain itraconazole levels and antifungal efficacy (as measured by mouse survival) were further increased by the addition of the potent P-glycoprotein inhibitor GF120918 [49] . However, as GF120918 has previously also been identified as an inhibitor of the apical ABC-half transporter breast cancer resistance protein (BCRP), a partial involvement of this
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Roberts & Goralski
Expert Opin. Drug Metab. Toxicol. (2008) 4(10) 1249
Table 1 . Summary of in vivo studies examining the effect of CNS infl ammation or infection on PGP expression and activity in the rodent brain.
Study [author(s), year] Species Model of infl ammation or infection
Effect of treatment on brain abcb1a mRNA, PGP expression and pharmacokinetics of PGP substrates
Goralski et al. (2003) [54] Rat Escherichia coli O127:B8 LPS (25 µg ICV)
Brain abcb1a ⇓ at 6 h with ⇔ at 24 and 48 h Brain digoxin ⇑ at 6 and 24 h
Mouse E. coli O127:B8 LPS (2.5 µg ICV) Brain digoxin ⇑ at 24 h in mdr1a +/+ mice Brain digoxin ⇓ at 24 h in mdr1a -/- mice
Imbert et al. (2003) [49] Mouse CNS infection with Cryptococcus neoformans
⇑ C max and AUC of itraconazole in the brain ⇑ ⇑ C max and AUC of itraconazole in the brain of infected mice co-treated with GF120918
Spudich et al. (2006) [87] Mouse 30 and 90 min of MCAO PGP in cortex and striatum ⇑ between 3 and 24 h Brain:blood ratio of rifampicin and FK506 ⇔ at 6 h Brain:blood ratio of rifampicin and FK506 ⇑ at 6 h in MCOA mice co-treated with tariquidar
Lazarowski et al. (2007) [43] Rat CoCl 2 (2 – 400 mM) hypoxia/ischemia
PGP detected in endothelial cells, astrocytes and neurons 6 days after CoCl 2 injection Absence of PGP detection in brain of control rats
Morgan et al. (2007) [59] Mouse E. coli O127:B8 LPS (2.5 µg ICV) Brain abcb1a mRNA ⇓ at 4 h with ⇔ at 24 h in C3H/HeouJ mice Brain abcb1a mRNA ⇔ at 4 and 24 h in C3H/HeJ mice
Yu et al. (2007) [85] Rat 2 h of MCAO or neuronal injury induced by unilateral intra-strial injection of quinolinic acid
⇑ C max of cryptotanshinone in the brain ⇑ Brain/plasma AUC ratio of cryptotanshinone
Chen et al. (2007) [86] Rat ⇑ C max of brain tanshinone IIA ⇑ Brain/plasma AUC ratio of tanshinone IIA
⇑ : Increased compared to controls; ⇑ ⇑ : Large increase compared to controls; ⇓ : Decreased compared to controls; ⇔ : Unchanged compared to controls at the indicated time point. ICV: Intracerebroventricular; LPS: Lipopolysaccharide; MCAO: Middle cerebral artery occlusion; PGP: P-glycoprotein.
Table 2 . Summary of in vivo studies examining the effect of peripheral infl ammation on PGP expression and activity in the rodent brain.
Study [author(s), year] Species Model of infl ammation or infection
Effect of treatment on brain abcb1a mRNA, PGP expression and pharmacokinetics of PGP substrates
Zhao et al. (2002a) [63] Mouse Klebsiella pneumoniae LPS (10 mg/kg injected through tail vein)
Brain abcb1a ⇓ at 6 h with ⇔ at 12 and 24 h Brain and plasma doxorubicin ⇑ at 6 h with ⇔ at 12 h Brain/plasma doxorubicin ⇔ at 6 and 24 h
Zhao et al. (2002b) [64] Mouse Escherichia coli O157:H7 Shiga-like toxin II (0.2 µg injected into the tail vein)
Brain abcb1a ⇑ at 6, 12 and 24 h Brain doxorubicin and brain/plasma doxorubicin ⇔ at 6 h and ⇑ 24 h Brain distribution of doxorubicin further ⇑ by the PGP inhibitor ciclosporin A
Wang et al. (2005) [65] Rat E. coli O55:B5 endotoxin (5 mg/kg injected i.p.)
Brain abcb1a ⇓ at 4, 6 and 12 h with ⇔ at 24 h Brain PGP ⇓ between 6 and 12 h with ⇔ at 24 h 99m T-sestamibi accumulation in brain ⇑ at 24 h
Seelbach et al. (2007) [66] Rat λ -carrageenan peripheral infl ammatory hyperalgesia
Brain microvessel PGP ⇑ at 3 h Brain effl ux ( k out ) of 3 H-morphine ⇑ at 3 h Brain infl ux ( k in ) of 3 H-morphine was ⇔ at 3 h Net 3 H-morphine uptake into the brain was ⇓ Morphine analgesia in tail-fl ick latency test was ⇓
⇑ : Increased compared to controls; ⇓ : Decreased compared to controls; ⇔ : Unchanged compared to controls at the indicated time point. LPS: Lipopolysaccharide; PGP: P-glycoprotein.
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Infl ammation, infection and brain-derived P-glycoprotein
1250 Expert Opin. Drug Metab. Toxicol. (2008) 4(10)
Table 3 . Summary of in vitro studies examining the effect of infl ammatory mediators on PGP expression and activity in blood–brain barrier capillaries or endothelial cells.
Study [author(s), year] Preparation Treatment(s) Effect of treatment on abcb1a mRNA, PGP expression and PGP activity
Theron et al. (2003) [70] Immortalized rat brain capillary endothelial cell line, GPNT
0.1, 1 and 10 ng/ml of TNF- α
abcb1a ⇑ at 6 h and ⇔ at 24 h abcb1b ⇔ at 6 h and ⇑ at 24 h PGP ⇔ at 24, 48, 72 and 96 h ⇑ intracellular 3 H-vinblastine accumulation (maximal after 36 h)
Hartz et al. (2004) [67] Isolated rat brain capillaries 0.1 – 100 nM of ET-1 Rapid (0 – 90 min) and reversible ⇓ in steady state capillary lumen NBD-CYA accumulation ET B agonist, NO donor and PKC activator mimicked the effects of ET-1 Inhibition of ET B , NOS and PKC signaling blocked the effects of ET-1
Hartz et al. (2006) [58] Isolated rat brain capillaries 0.01 – 10 ng/ml of TNF- α
Rapid (0 – 150 min) and reversible ⇓ in steady-state capillary lumen NBD-CYA accumulation Inhibition of TNF- α , ET B , NOS and PKC signaling blocked the effects of TNF- α
0.01 – 10 ng/ml of Escherichia coli LPS
Rapid ⇓ in steady-state capillary lumen NBD-CYA accumulation Inhibition of TLR4, TNF- α , ET B , NOS and PKC signaling blocked the effects of LPS
Veszelka et al. (2006) [71] Isolated rat brain endothelial cells
0.01 – 10 µg/ml of Escherichia coli LPS (O55:B5)
⇓ cellular rhodamine-123 accumulation at 16 h (10 µg/ml dose of LPS)
Bauer et al. (2007) [61] Isolated rat brain capillaries 0.5 – 5 ng/ml of TNF- α 100 nM of ET-1
Capillary lumen NBD-CYA accumulation ⇓ between 1 and 3 h following treatment Capillary lumen NBD-CYA accumulation ⇑ 5 and 6 h following treatment PGP ⇑ at 6 h
Yu et al. (2007) [69] Rat brain endothelial cells (RBE4)
5 ng/ml TNF- α abcb1a ⇑ at 2, 6 and 12 h PGP ⇑ at 6, 12 and 24 h Cellular 3 H-vinblastine accumulation ⇓ at 6 h
Hembury et al. (2008) [74] Primary human endothelial and glia cell co-culture
10 – 100 nM of ET-1 PGP ⇔ at 24 h ⇓ in basal-apical transport of 3 H-digoxin at 24 h ⇑ ⇑ in apical-basal transport of 3 H-digoxin at 24 h
Hartz et al. (2008) [73] Isolated rat brain capillaries 5 – 100 µg/ml of DEP PGP and capillary lumen NBD-CYA accumulation ⇑ at 6 h following treatment Free radical scavengers and inhibition of NADPH oxidase, TNF- α , NOS and JNK blocked the effects of DEP
⇑ : Increased compared to controls; ⇑ ⇑ : Large increase compared to controls; ⇓ : Decreased compared to controls; ⇔ : Unchanged compared to controls at the indicated time point. DEP: Diesel exhaust particles; ET B : Endothelin-1 receptor B; ET-1: Endothelin-1; JNK: c-Jun N-terminal kinase; LPS: Lipopolysaccharide; NADPH: Nicotinamide adenine dinucleotide phosphate reduced; NBD-CYA: NBD-ciclosporin A [ N - ε (4-nitrobenzofurazan-7-yl)- D -Lys 8 ]-cyclopsorine A; NO: Nitric oxide; NOS: Nitric oxide synthase; PGP: P-glycoprotein; TLR4: Toll-like receptor 4.
transporter in the CNS uptake of itraconazole cannot be excluded [49] .
The intracerebroventricular (ICV) injection of LPS, a glycolipid endotoxin derived from the outer membrane of Gram-negative bacteria, is commonly used as an alternative to rodent models of infection as LPS is a principal stimulus of CNS inflammation in humans induced by Gram-negative pathogens [51] . In rodents, ICV administration of LPS
produces a robust inflammatory response in the brain characterized by activation of microglia and astrocytes with the consequent production of the pro-inflammatory cytokines TNF- α , IL-6 and IL-1 β , among others [52-55] . In agreement with the findings of Imbert and co-workers (2003), a previous study by our research group demonstrated a rapid downregulation of abcb1a mRNA to 50% of control levels 6 h after injection of Escherichia coli LPS
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Roberts & Goralski
Expert Opin. Drug Metab. Toxicol. (2008) 4(10) 1251
(serotype O127:B8) into the rat CNS ( Table 1 ). This downregulation correlated with a 1.6-fold increase in the accumulation of the P-glycoprotein substrate 3 H-digoxin in the brain. However, 24 h following treatment with LPS, brain abcb1a mRNA returned to baseline levels although 3 H-digoxin accumulation in the brain remained ∼ 1.4-fold higher compared to control. After 48 h, brain abcb1a mRNA and 3 H-digoxin were similar in LPS- versus saline-treated controls. To confirm that increased digoxin accumulation in the brain subsequent to LPS injection was P-glycoprotein-dependent, we repeated the 3 H-digoxin distribution study in abcb1a- expressing ( abcb1a +/+ ) versus abcb1a -deficient ( abcb1a -/- ) mice. Treatment of abcb1a +/+ mice with ICV LPS for 24 h was linked with a twofold increase in brain digoxin concentra-tion when compared to saline controls [54] . In comparison, saline-treated abcb1a -/- mice displayed a 10-fold higher basal accumulation of 3 H-digoxin in the brain compared to wild-type mice. Thus, blood–brain barrier P-glycoprotein activity appeared to be only partially inhibited or downregulated following an acute inflammatory response in the brain of wild-type mice. Surprisingly, abcb1a -/- mice treated with ICV LPS displayed a 30% reduction in brain digoxin levels when compared with saline injected abcb1a -/- mice [54] . This
effect was postulated to involve organic anion transport-ing polypeptide 2 (oatp2) downregulation, a bidirectional blood–brain barrier transporter that mediates high affinity digoxin transport [54,56,57] .
Recent work by Hartz et al. (2006) (described in detail later in this review) has indicated that activation of TLR4 signaling is a major pathway involved in regulating P-glycoprotein expression and activity in isolated rat brain capillaries following treatment with LPS [58] . When evaluated in vivo , ICV administration of E. coli LPS (serotype O127:B8) to TLR4-expressing mice (the C3H/HeouJ strain) produced a 50% reduction in brain abcb1a expression after 4 h [55,59] . However, 24 h following LPS administration, brain abcb1a expression returned to baseline ( Table 1 ). In comparison, ICV administration of E. coli LPS to C3H/HeJ mice with a mutated nonfunctional TLR4 did not result in a downregulation of abcb1a expression in brain [55,59,60] . Thus, activation of TLR4 is required for reduction of brain abcb1a expression during LPS-induced CNS inflammation in vivo . Interestingly, following ICV administration of LPS, there is an induction of TNF- α and IL-6 expression in the brain of TLR4-expressing mice but not in that of TLR4-deficient mice. Thus, in agreement with the in vitro
Table 4 . Summary of studies examining the effect of HIV infection on PGP expression and activity in post-mortem human brain, mouse brain and astrocytes.
Study [author(s), year] Species Model of infl ammation or infection
Effect of treatment on brain abcb1a , PGP expression, and pharmacokinetics of PGP substrates
Langford et al. (2004) [75] Human Postmortem brain from HIV - , HIVE - and HIVE + subjects
PGP ⇓ in brain capillaries of HIVE - and HIVE +
PGP ⇑ ⇑ in astrocytes and microglia of HIVE + compared HIVE - and HIV -
Ronaldson and Bendayan (2006) [62]
Primary rat astrocytes 1 nM HIV-1 96zm651 gp120 protein abcb1a and PGP ⇓ at 6, 12 and 24 h ⇑ cellular accumulation of 3 H-digoxin and 3 H-saquinavir Inhibition of IL-6, but not TNF- α and IL-1 β , blocked the effects of HIV gp120
0.5 or 10.0 ng/ml TNF- α PGP ⇑ at 6, 12 and 24 h
0.4 or 10 ng/ml IL-1 β PGP ⇑ at 6, 12 and 24 h
0.3 or 10 ng/ml IL-6 PGP ⇓ ⇓ ⇓ at 6, 12 and 24 h
Hayashi et al. (2005) [79] Primary mouse brain endothelial cells and astrocytes
100 – 400 ng/ml HIV-TAT 1-72 abcb1a ⇑ in endothelial cells with ⇔ in astrocytes at 6 h PGP ⇑ in endothelial cells between 2 and 6 h ⇓ endothelial cell accumulation of rhodamine-123 at 12 h
Mouse Intra-hippocampal injection of TAT 1-72
PGP ⇑ localized to brain endothelial cells at 24 h
⇑ : Increased compared to controls; ⇓ : Decreased compared to controls; ⇓ ⇓ ⇓ : Very large decrease compared to controls; ⇔ : Unchanged compared to controls or HIV - subjects at the indicated time point. HIVE - : HIV infected without encephalitis; HIVE + : HIV infected with encephalitis; HIV TAT: HIV transactivator; PGP: P-glycoprotein.
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Infl ammation, infection and brain-derived P-glycoprotein
1252 Expert Opin. Drug Metab. Toxicol. (2008) 4(10)
studies presented later in this review, inflammatory cytokine-mediated pathways are likely to modulate brain abcb1a mRNA expression and P-glycoprotein activity in vivo [58,61,62] .
2.2 In vivo animal models of systemic infl ammation and infection It is important to recognize that the blood–brain barrier divides the systemic circulation from the interstitial fluid of the CNS. The brain parenchyma is, therefore, exposed to cytokines and related inflammatory mediators from both sides of the barrier. Thus, inflammatory and infectious responses generated outside of the CNS (e.g., in blood or peripheral tissues such as the peritoneum) should be capable of altering blood–brain barrier P-glycoprotein activity as long as they produce an adequate concentration of circulating inflammogens at the level of cerebral capillary endothelial cells.
Indeed, there is already a body of in vivo evidence describing modulation of P-glycoprotein activity at the blood–brain barrier by inflammatory responses generated first in the periphery, rather than in the CNS ( Table 2 ). Zhao et al. (2002a) found that whole brain P-glycoprotein decreased by 30% 6 h following injection of Klebsiella pneumoniae endotoxin into the tail vein of mice, with no effect at 12 and 24 h post-injection. In comparison, the plasma and brain concentrations of the P-glycoprotein substrate doxorubicin were marginally (20 – 30%) higher in Klebsiella -treated versus control mice 6 h after initiation of the inflammatory insult [63] . The lack of effect of a 6 or 24 h treatment of K. pneumoniae endotoxin on the brain/plasma ratio of doxorubicin led the authors to conclude that this endotoxin does not affect the blood–brain barrier transport of doxorubicin [63] . Somewhat different results were obtained in a subsequent study by the same group that examined the CNS distribution of doxorubicin in mice 6 and 24 h following tail vein injection of Shiga-like toxin II from the E. coli strain O157:H7 [64] . Twenty-four hours following injection of the toxin, treated mice displayed a threefold increase in the brain doxorubicin level and the brain:plasma ratio of doxorubicin as compared to control mice [64] . The effect of Shiga-like toxin II on brain doxorubicin distribution was partially reversed with the TNF- α -inhibitor pentoxy-fylline, indicating that TNF- α contributed to the modulation of blood–brain barrier transport in the whole animal [64] . The P-glycoprotein inhibitor ciclosporine A further enhanced the brain levels of doxorubicin in Shiga-like toxin II-treated mice, suggesting that inflammatory responses potentiate the effect of P-glycoprotein inhibitors. However, enhanced brain accumulation of doxorubicin did not correlate with whole brain P-glycoprotein levels, which were upregulated 1.6- to 1.7-fold that of control levels between 6 and 24 h after treatment with Shiga-like toxin II. Thus, P-glycoprotein activity may be inhibited despite higher immunodetectable levels of this transporter. In a third study, Wang et al. (2005) examined the effect of systemic inflammation induced by
intraperitoneal injection of E.coli O55:B5 LPS on blood–brain barrier P-glycoprotein expression and activity. Similar to CNS inflammation induced by ICV administration of E. coli LPS [54] , these investigators found that intraperitoneal injection of this endotoxin strain reversibly reduced brain abcb1a mRNA by 60, 50 and 45% at 4, 6 and 12 h after treatment, respectively, with full recovery of mRNA expression by 24 h [65] . In addition, a similar time-dependent loss of immunodetectable P-glycoprotein was observed in membrane fractions from rat brain following injection of LPS. In comparison to the study by Zhao et al. (2002b), however, no increase in brain abcb1a mRNA or P-glycoprotein levels were observed over the 24-h treatment period. In the same study, biodistribution and imaging studies utilizing the radiopharmaceutical 99 mTc-sestamibi (a substrate of both P-glycoprotein and multidrug resistance transporters) revealed that P-glycoprotein downregulation was accompanied by heightened 99 mTc–sestamibi distribution into brain 24 h after LPS treatment. In a study by Seelbach et al. (2007) that utilized a different methodology, morphine pharmaco-kinetics and brain P-glycoprotein expression were examined after inducing localized peripheral inflammatory hyperalgesia by injection of the rat hind paw with 3% λ -carrageenen. Three hours post-injection, λ -carrageenen-treated rats displayed a peripheral blood neutrophilia and a 40% increase in P-glycoprotein expression in isolated brain capillaries [66] . Consistent with the increase in P-glycoprotein expression, the rate of morphine flux from brain-to-blood increased whereas the rate of blood-to-brain flux of morphine remained constant during in situ brain perfusion [66] . Overall, peripheral inflammatory hyperalgesia resulted in a net decrease (30%) in the brain accumulation of 3 H-morphine and resultant analgesic efficacy in λ -carrageenen-treated rats compared to non-inflamed controls [66] . Given the conflicting nature of the aforementioned study results, the link between peripheral inflammation and brain P-glycoprotein expression and activity seems to be highly dependent on the specific inflammatory mediator examined or used [63-66] .
2.3 In vitro and cellular models of infl ammation and infection Several recent in vitro studies have attempted to describe the pathways responsible for temporal changes in the expression and function of blood–brain barrier P-glycoprotein following exposure to E. coli LPS and other pivotal inflammatory mediators such as TNF- α and endothelin-1 (ET-1) ( Table 3 ) [46,58,61,67-71] . The investigations by Bauer et al. (2007) and Hartz et al. (2004 and 2006) deserve primary mention as they account for much of the existing knowledge on this topic [58,61,67] . Each of these studies utilized a novel model of assessing P-glycoprotein function that utilizes confocal microscopy and quantita tive image analysis to measure alterations in the steady-state accumulation of a fluorescent P-glycoprotein substrate ( N - ε -(4-nitrobenzofurazan-7-yl)- D -Lys 8 -ciclosporine A,
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Roberts & Goralski
Expert Opin. Drug Metab. Toxicol. (2008) 4(10) 1253
NBD-CSA) in isolated rat cerebral capillary lumens in the presence or absence of inflammatory mediators [58,61,67] . The first study by Hartz et al. (2004) found that capillary lumen NBD-CSA (P-glycoprotein activity) was rapidly and reversibly reduced in a dose-dependent manner by escalating doses of ET-1. The inhibitory effect of ET-1 was similar in magnitude to PSC833, a potent P-glycoprotein inhibitor, indicating essentially complete inhibition of transporter activity. This inhibition was maximal between 15 and 60 min postexposure and quickly reverted back to baseline in ∼ 60 min of ET-1 removal [67] . Subsequent studies by this group revealed a similar dose-dependent, transient reduction in brain capillary P-glycoprotein activity after treatment with TNF- α or E. coli LPS [58] . ET-1, TNF- α or LPS did not alter intercellular tight junction permeability when measured by a kinetic dye assay, supporting that the observed changes in NBD-CSA transport were due to altered P-glycoprotein function and not a frank physical disruption of the barrier [58,67] . Interestingly, such brief exposures of intact rat brain capillaries to ET-1, TNF- α and LPS were not associated with decreases in P-glycoprotein expression and, therefore, must have reflected transporter inhibition and/or inactivation.
In comparison, prolonged exposure of intact cerebral capillaries to low concentrations of ET-1 or TNF- α results in a markedly different effect on P-glycoprotein activity. The previously mentioned inhibition of P-glycoprotein activity after short-term inflammatory mediator exposure is followed by a return to baseline function 3 – 4 h after cytokine exposure and then a subsequent induction of P-glycoprotein activity above control levels at 5 – 6 h [61] . The in vitro activity of P-glycoprotein in response to a continuous exposure to ET-1 and TNF- α , therefore, exhibits a time-dependent, biphasic response [61] . In agreement with the above studies, Yu and co-workers demonstrated an induction of abcb1a mRNA and P-glycoprotein expression with reduced cellular accumulation of 3 H-vinblastine 6 h after treatment of rat brain endothelial cells with TNF- α [69] . They also revealed that the inductive effect of TNF- α on abcb1a mRNA and P-glycoprotein was maintained for at least 24 h [69] . In contrast, another study reported increased cellular accumulation of 3 H-vinblastine without a concomitant change in P-glycoprotein levels following treatment of a rat brain endothelial cell line (GPNT) with TNF- α for 6 – 96 h [70] .
The previously mentioned studies by Bauer et al. (2007) and Hartz et al. (2004 and 2006) have also provided a highly detailed and thorough description of the biochemical pathways responsible for biphasic changes in brain capillary P-glycoprotein activity ( Figure 2 ) [58,61,67] . Endothelial cells possess receptors for TNF- α , IL-1, ET-1 and LPS, many of which are present on both the luminal and abluminal surfaces of blood–brain barrier endothelial cells [58,61,67] . Following acute exposure of rat brain capillaries to LPS, TLR4 is activated and leads to TNF- α release, which then binds to TNF-R1, leading to pro-ET1 release ( Figure 2A ).
Pro-ET1 is then converted to active ET-1 by endothelin-converting enzyme. In the ensuing steps, ET-1 binding to the ET B receptor activates nitric oxide synthase (NOS), which produces nitric oxide (NO) and stimulates PKC activity [58] . Putative mechanisms hypothesized to be responsible for inhibition of P-glycoprotein activity by PKC include direct phosphorylation of P-glycoprotein or a protein capable of modulating P-glycoprotein activity [58] . PKC may also alter trafficking of P-glycoprotein between some intracellular compartments and the plasma membrane [58,72] .
The aforementioned pathway is responsible for the majority (80%) of reduction in P-glycoprotein activity after treatment with LPS whereas a second signaling pathway, which acts directly through NOS, contributes to a smaller degree (20%) [58] . With longer-term exposure to TNF- α , such as might occur in chronic inflammatory diseases, an alternate sequence of events culminates in induction of P-glycoprotein activity ( Figure 2B ). As before, TNF- α binds to TNF-R1 on the basal membrane, which leads to release of active ET-1. ET-1 subsequently binds to both the ET A (Endothelin-1 receptor A) and ET B (Endothelin-1 receptor B) receptors, which are colocalized on the basal membrane with TNF-R1. NOS is then activated leading to NO production and subsequently PKC activation. PKC activation leads to translocation of the nuclear transcription factor, NF- κ B, from the cytoplasm to the nucleus, ultimately resulting in increased P-glycoprotein expression and activity [61] . Of interest, a more recent study by this group also identified that short-term exposure of rat brain capillaries to diesel exhaust particles triggers oxidative stress and results in an inflammatory cascade, which ultimately upregulates P-glycoprotein expression and activity, albeit by a some-what different mechanism than that described in the proceeding paragraphs [73] .
In contrast to the relative wealth of in vitro studies using animal cell culture models, only one investigation presently exists that describes the effect of ET-1 on P-glycoprotein function in a human blood–brain barrier model [74] . This study reported no detectable change in P-glycoprotein expression as determined by FACS analysis 24 h following treatment with ET-1. However, basal-to-apical transport of 3 H-digoxin across a cell monolayer was reduced by 35%, suggesting a slight reduction in P-glycoprotein activity [74] . Further, drug uptake into human cerebral capillary endo-thelial cells seemed to be enhanced as apical-to-basal transport of digoxin increased fourfold after 24 h of treat ment with ET-1 [74] . Although these findings need to be confirmed in future studies, this investigation represents an important step toward translating the findings previously described exclusively in rodents to man.
2.4 HIV infection and HIV encephalopathy HIV-associated dementia (also referred to as AIDS dementia complex and HIV encephalopathy) is a clinical syndrome of HIV infection characterized by various cognitive, behavioral
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.
Infl ammation, infection and brain-derived P-glycoprotein
1254 Expert Opin. Drug Metab. Toxicol. (2008) 4(10)
A.
B.
PKC
ECE
Pro-ET-1
ET-1
NO
TNFR1
NOS
ETB
Pro-ET-1 releaseand conversion
TranscriptionTranslation
(+)
ETA
Long-termregulation
Capillary lumen
Brainextracellular fluid
LPS
TNF-α release
Pro-ET-1ET-1
ETBTLR4 TNFR1
(-)
NOS
Pro-ET-1 releaseand conversion
Short-termregulation
Capillary lumen
Brainextracellular
fluid
NO
PKC
ECE
PGP
PGP
TACE
NF-κB
NF-κB
TNF-α
TNF-α
Figure 2 . Signaling pathways involved in regulation of P-glycoprotein activity in the blood–brain barrier as adapted from Hartz et al. (2006), Bauer et al. (2007) and Hartz et al. (2004) [58,61,67] . A. Downregulation of P-glycoprotein activity after short-term exposure to Escherichia coli LPS, TNF- α , or ET-1. B. Upregulation of P-glycoprotein expression and activity following prolonged exposure to TNF- α and ET-1. The (+) and (-) symbols denote inhibition and activation, respectively. ECE: Endothelin-1 converting enzyme; ET A : Endothelin-1 receptor A; ET B : Endothelin-1 receptor B; ET-1: Endothelin-1; LPS: Lipopolysaccharide; NO: Nitric oxide; NOS: Nitric oxide synthase; PGP: P-glycoprotein; TACE: TNF- α converting enzyme; TLR4: Toll-like receptor 4; TNFR1: TNF- α receptor 1.
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Roberts & Goralski
Expert Opin. Drug Metab. Toxicol. (2008) 4(10) 1255
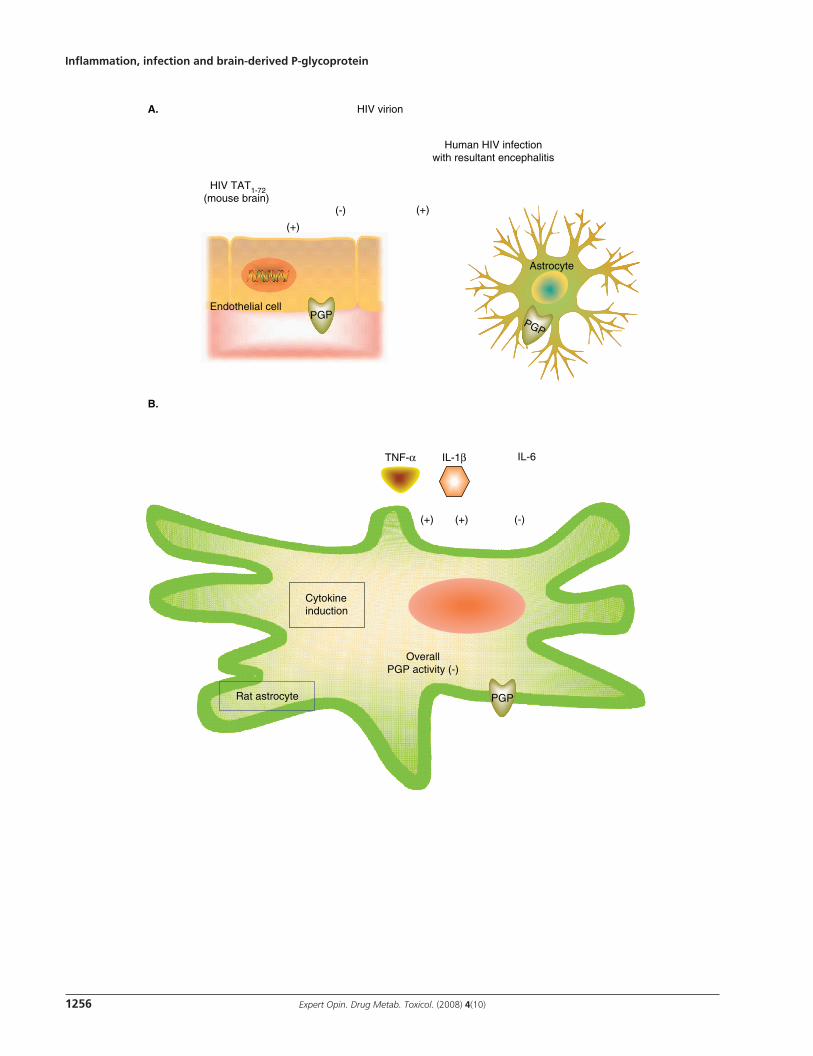
and motor deficits [75,76] . Patients with HIV-associated dementia may develop encephalitis with subsequent damage to cerebral microvasculature, formation of microglia nodules and generation of multinucleated giant cells with pro-inflammatory cytokine (TNF- α , IL-6 and IL-1 β ) production and release within the CNS [77] . Because a number of HIV protease inhibitors are restricted from the CNS by P-glycoprotein [22,78] , some have hypothesized that increased P-glycoprotein expression decreases penetration of anti-HIV therapy into the CNS and enhances neuropathology in HIV-infected individuals [75] . Langford et al. (2004) examined P-glycoprotein levels in postmortem human brain from HIV negative controls (HIV - ), HIV positive patients without encephalitis (HIVE - ) and HIV positive patients with encephalitis (HIVE + ) [75] . This group observed differential effects of HIVE + on P-glycoprotein expression in glia and endothelial cells. In HIV negative controls, P-glycoprotein expression was localized to brain capillary endothelial cells [75] . In comparison, HIVE - patients displayed less intense P-glycoprotein immunoreactivity in the capillaries of the neocortex with no immunoreactivity in subarachnoid vessels [75] . The level of neocortical microvessel P-glycoprotein was further reduced in HIVE + patients [75] . In contrast, P-glycoprotein levels were increased in astrocytes and microglia of HIVE + compared to seronegative controls and HIVE - patients ( Figure 3A ) [75] . Indeed, a direct correlation (r = 0.523) was found between HIV viral burden and P-glycoprotein expression in astrocytes and microglia [75] .
Results from this human investigation have subsequently been followed by an in vitro analysis of the effect of HIV-1 gp 120 and TAT (transactivator) proteins on P-glycoprotein expression and function in astrocytes and endothelial cells, respectively. Contrary to the Langford study, both abcb1a mRNA and P-glycoprotein levels decreased substantially 6 – 24 h following treatment of cells with the HIV viral envelope protein, HIV-1 96ZM651 gp120 [62] . Such P-glycoprotein downregulation was also linked with a 1.5- to 1.8-fold higher cellular uptake of 3 H-digoxin and 14 C-saquinavir, respectively ( Figure 3B ) [62] . The investigators postulated that inhibition of P-glycoprotein expression and activity could have been secondary to the release of the inflammatory cytokines TNF- α , IL-1 β and IL-6 from astrocytes after treatment with HIV-1 96ZM651 gp120 [62] . Further analysis revealed that P-glycoprotein expression in astrocytes was increased 1.6- to 2.9-fold by IL-1 β and TNF- α , respectively, whereas IL-6 decreased transporter expression by 90% [62] . IL-6 (and not TNF- α or IL-1 β ) neutralizing antibodies reversed the inhibitory effect of gp120 on P-glycoprotein expression [62] , suggesting that IL-6 was the principle cytokine responsible for P-glycoprotein downregulation following gp120 treatment. In contrast, Hayashi et al. (2005) noted an upregulation of P-glycoprotein transcription and translation in primary cultures of mouse brain microvascular endothelial cells following treatment with HIV-Tat 1-72 protein ( Figure 3A ) [79] . Tat protein injection was also linked with enhanced
P-glycoprotein expression and increased rhodamine-123 efflux from cerebral capillary endothelial cells [79] . In support of these in vitro findings, upregulation of blood–brain barrier P-glycoprotein expression was confirmed in vivo by intra-hippocampal injection of Tat protein in mice [79] .
2.5 Stroke and neuronal injury Ischemic and hemorrhagic stroke have been linked with the production of a multitude of inflammatory mediators (e.g., NO, IL-1 and IL-6) and increased expression of transcription factors such as hypoxia inducible factor-1 α , some of which are capable of modulating P-glycoprotein activity [61,80-84] . Yu and co-workers (2007) demonstrated a 2.5-fold increase in the brain/plasma AUC 0 – 24 h of crypto-tanshinone (a natural antioxidant and P-glycoprotein substrate) in rats subjected to 2 h of middle cerebral artery occlusion as compared to control rats ( Table 1 ) [85] . In a similar study, Chen et al. (2007) found a 2.3-fold increase in the brain/plasma AUC 0 – 24 h of the related P-glycoprotein substrate tanshinone IIA following middle cerebral artery occlusion as compared to control rats [86] .
In the study by Yu et al. (2007), neurotoxicity induced by unilateral intra-striatal injection of quinolinic acid was also investigated. Quinolinic acid injection increased the C max and brain/plasma AUC 0 – 24 h levels of cryptotanshinone, albeit to a lesser degree than did middle cerebral artery occlusion. Quinolinic acid-induced neuronal injury also produced similar effects on the CNS pharmacokinetics of tanshinone IIA [86] . However, no attempt was made in either study to correlate alterations in CNS drug distribution following stroke or neuronal injury with reduced P-glycoprotein mRNA or protein expression. In comparison, Spudich et al. (2006) demonstrated increased expression of P-glycoprotein in capillary endothelial cells within the striatum and cortex of the ischemic compared to the non-ischemic cerebral hemisphere in the mouse ( Table 1 ) [87] . The overall induction of P-glycoprotein expression was more pronounced following a 90 min versus a 30 min occlusion period, suggesting an injury dose-response [87] . An increase in P-glycoprotein expression was not noted in astrocytes or neurons [87] . Although increased expression of P-glycoprotein following cerebral ischemia would logically result in reduced penetration of substrate drugs into the CNS, no change in the accumula-tion of FK506 or rifampicin was observed in the ischemic versus the non-ischemic hemisphere when examined 6 h after 30 min of middle cerebral artery occlusion. The inter-pretative value of this pharmacokinetic data, however, is limited by the use of a single time point (6 h) for analysis. Furthermore, data are not reported for the 90 min cerebral arterial occlusion period, during which there were pro-nounced changes in P-glycoprotein expression. In this study, the P-glycoprotein inhibitor tariquidar enhanced the brain uptake of the neuroprotectant agents FK506 and rifampicin to a greater degree in the ischemic versus the non-ischemic hemisphere [87] . P-glycoprotein inhibition or deactivation
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.
Infl ammation, infection and brain-derived P-glycoprotein
1256 Expert Opin. Drug Metab. Toxicol. (2008) 4(10)
A.
B.
Cytokineinduction
TNF-α IL-1β IL-6
(-)(+)(+)
OverallPGP activity (-)
Rat astrocyte
Human HIV infectionwith resultant encephalitis
Astrocyte
Endothelial cell
(+)(-)
HIV TAT1-72(mouse brain)
(+)
HIV virion
PGP
PGPPGP
protein synthesis
PGP, mRNA, or
HIV gp120
Figure 3 . Effect of HIV infection on P-glycoprotein activity in blood–brain barrier endothelial cells and astrocytes. A. Post-mortem analysis of brain samples from HIV infected individuals with encephalitis (HIVE + ) revealed that P-glycoprotein expression was increased in astrocytes and decreased in cerebral capillary endothelial cells compared to brains from patients with HIV infection without encephalitis (HIVE - ) and uninfected individuals [75] . The HIV protein TAT 1-72 induced P-glycoprotein expression and transport activity in mouse brain endothelial cells [79] . B. In primary cultures of rat astrocytes, the HIV protein gp120 decreased abcb1a mRNA and P-glycoprotein expression and activity whereas TNF- α and IL-1 β produced a small elevation in P-glycoprotein levels [62] . IL-6 reduced P-glycoprotein levels in astrocytes by 90% [62] . During gp120 treatment, the effect of IL-6 on P-glycoprotein downregulation predominated, resulting in a net loss of transporter levels and activity in astrocytes [62] . The (+) and (-) symbols denote inhibition and activation, respectively. HIV TAT: HIV transactivator; PGP: P-glycoprotein.
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Roberts & Goralski
Expert Opin. Drug Metab. Toxicol. (2008) 4(10) 1257
also enhanced the neuroprotective efficacy of FK506 and rifampicin [87] . This result is not unlike the previous demonstration of enhanced brain uptake and efficacy of itraconazole in C. neoformans infected rats co-treated with a P-glycoprotein inhibitor [49] . Taken together, these studies support the hypothesis that P-glycoprotein inhibition may facilitate drug efficacy by increasing the brain distribution of therapeutically active agents that are normally excluded from the CNS by this transporter. A fourth study examined the effect of CoCl 2 -induced cerebral ischemia on P-glycoprotein expression in the rat brain [43] . P-glycoprotein expression was detectable in capillary endothelial cells, astrocyte foot processes and neurons of the injured brain area of CoCl 2 -treated rats [43] . It was suggested that P-glycoprotein expression in hypoxic neurons and astrocytes could potentially impair drug access to brain parenchyma [43] . Surprisingly, however, P-glycoprotein was not detected in the brains of saline-treated control rats, which could have been a result of para formaldehyde fixation, a technique known to mask epitopes on P-glycoprotein [37] . Nonetheless, the lack of basal P-glycoprotein expression makes it difficult to quantitate the extent of induction produced by CoCl 2 -induced hypoxia. After comparing the results of the studies by Spudich et al. (2006) and Lazarowski et al. (2007), it seems that the overall effects on neuronal and astrocyte P-glycoprotein expression are dependent on the model of hypoxia/ischemia utilized.
2.6 Parkinson’s and Alzheimer’s diseases and epilepsy A characteristic pathologic feature of Alzheimer’s disease is β -amyloid angiopathy, which is believed to result from accumulation of amyloid- β (A β ) in the walls of arteries and arterioles following its secretion by neurons into the interstitium of the brain parenchyma [88,89] . Deposition of extracellular fibrillar A β protein in the CNS is associated with microglia activation and pro-inflammatory cytokine and acute phase reactant production [90] . Production of these mediators is believed to lead to a chronic neuroinflammatory response closely associated with β -amyloid plaques [90] . Several animal and cellular investigations have indicated that A β elimination from the brain is partially mediated through active transport by P-glycoprotein at the level of the blood–brain barrier [88,89,91,92] . An interesting translational autopsy study suggested that a similar relationship may exist between P-glycoprotein and A β accumulation in human cerebral vasculature [89] . Vogelgesang et al. (2004) analyzed P-glycoprotein expression and A β deposition in the cerebral vessels of 243 non-demented, elderly adults and found that vessels with low transporter expression had A β deposition in their walls whereas vessels with high P-glycoprotein expression had no A β accumulation in their walls. Low P-glycoprotein expression may, therefore, present a novel risk factor for Alzheimer’s disease or, alternatively, may simply be reduced with progression of the disease [88] .
Relatively less is known about the impact of P-glycoprotein on the development of Parkinson’s disease. However, several
studies have linked the development of this neurodegenerative disease with environmental factors such as exposure to pesticides or insecticides, living in rural areas and drinking well water, among others [93,94] . Other investigations have suggested that P-glycoprotein polymorphisms predispose to the damaging effects of environmental toxins such as pesticides and have a significant association with Parkinson’s disease [93-95] . In support of this view, an in vivo human study found that the uptake of the P-glycoprotein substrate [ 11 C]-verapamil in the midbrain of patients with Parkinson’s disease was significantly higher than that of control patients, suggesting a reduction in P-glycoprotein activity in patients with Parkinson’s disease versus control patients [96] . As neuroinflammation is a universal finding in Parkinson’s disease, with increased numbers of inflammatory cells and pro-inflammatory cytokines, including TNF- α and IL-1 β , being found in the brain of affected individuals [97] , it is possible that the associated CNS inflammatory response could alter blood–brain barrier P-glycoprotein activity.
There are a number of investigations linking refractory epilepsy with increased blood–brain barrier efflux transporter expression and activity (please refer to one of the following excellent articles for an overview of the extensive literature on this topic [31,98,99] ). In a recently published rodent study, van Vliet et al. (2007) reported P-glycoprotein overexpression in the ventral hippocampus and parahippocampal cortex of chronic epileptic rats as compared to control rats [100] . This localized limbic P-glycoprotein overexpression was thought to occur in the blood–brain barrier and was linked with reduced brain/plasma levels of the widely used antiepileptic drug phenytoin. It has been postulated that P-glycoprotein over-expression in the epileptic brain could stem from constitutive transporter overexpression owing to genetic polymorphisms, seizure-induced overexpression and/or induction by antiepileptic drug therapies [31] . However, recent studies have implicated inflammatory pathways as a causative link between epilepsy and P-glycoprotein induction [101] . The neuroinflammatory reaction that commonly occurs following epileptic seizures involves excessive glutamate release, activation of the NMDA glutamate receptor, and subsequently enhanced COX-2 expression [101] . Bauer et al. (2008) found that exposing isolated cerebral capillaries to escalating doses of glutamate resulted in increased COX-2 expression and a dose-dependent increase in P-glycoprotein expression and transport activity. Unilateral intracerebral microinjections of glutamate into the hippocampus of rats also increased local P-glycoprotein expression in the cerebral microvessels of the neighboring hippocampal hilus as compared to vehicle-treated control rats whereas transporter expression in more distant brain regions was unaffected. Using a pilocarpine status epilepticus model, Bauer et al. (2008) further demonstrated seizure-induced increases in cerebral capillary P-glycoprotein expression, could be attenuated by administration of the non-selective COX-1/COX-2 inhibitor indomethacin [101] . Importantly, this study is the first investigation to identify
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Infl ammation, infection and brain-derived P-glycoprotein
1258 Expert Opin. Drug Metab. Toxicol. (2008) 4(10)
a feasible inflammatory target for the prevention of drug-resistant epilepsy.
3. Conclusion
The studies described herein demonstrated that P-glycoprotein expression and activity in the brain and blood–brain barrier is modulated by numerous inflammatory and infectious stimuli generated within the brain and peripheral tissues. These stimuli include bacterial, fungal and viral infections; neuronal injury produced by hypoxia/ischemia, neurotoxins and epilepsy; and neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases. Studies in isolated rat brain capillaries have been particularly illuminating with respect to identifying the molecular signaling pathways that modulate P-glycoprotein expression following exposure to E. coli LPS and cytokines. Indeed, the identification of such signaling pathways may ultimately lead to the development of small molecule therapeutics capable of altering the therapeutic efficacy and/or toxicity of CNS-acting drugs.
4. Expert opinion
The findings of the investigations summarized herein collectively support that altered blood–brain barrier P-glycoprotein activity during inflammation and infection is a general phenomenon that may have wide-ranging effects on the CNS accumulation, pharmacological activity and neurotoxicity of drugs. It is important to note that the direction and degree of change in P-glycoprotein activity depends on the in vivo or in vitro model used, the cell type examined (i.e., endothelial or glial), the inflammatory mediator utilized, the anatomic site in which the inflammatory response was first generated, the time points chosen for observation and the P-glycoprotein substrates analyzed. Because of the potential for inter-species differences, future studies should describe the effect of inflammatory and infectious processes on drug transport systems within the human blood–brain barrier. Further, animal pharmacokinetic studies of inflammation and infection should incorporate several time point measure-ments for P-glycoprotein expression and activity given the time-dependent effect observed in several studies. Despite the inter-study variations described above and earlier in this review, there are a number of common themes that have been identified, which are summarized in the following model ( Figure 4 ). In an otherwise healthy subject, P-glycoprotein prevents intracerebral accumulation of drugs by actively transporting substrate agents in a retrograde fashion across the blood–brain barrier (left hand panel of Figure 4A ). However, in the acute phase of a CNS inflammatory or infectious reaction, there is an initial loss of P-glycoprotein expression and/or activity, thus leading to increased accumula-tion of P-glycoprotein substrates in the brain (middle panel of Figure 4A ). As the acute inflammatory episode resolves, P-glycoprotein expression and activity returns to baseline
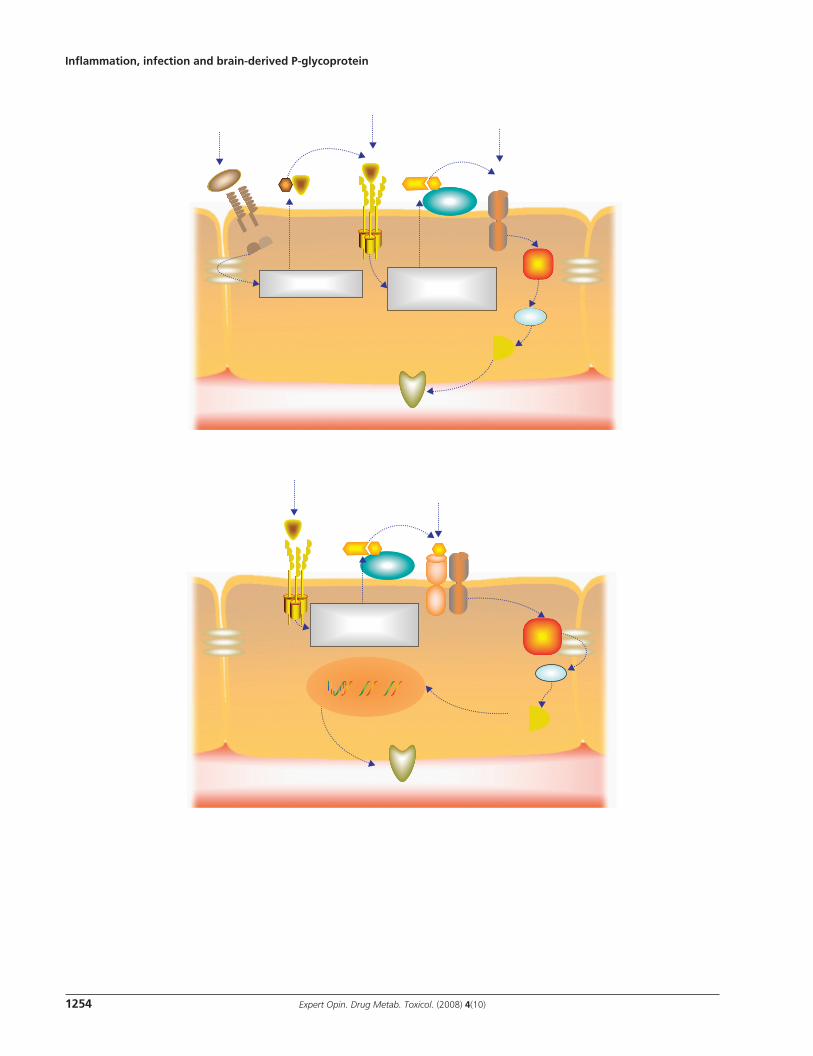
(right hand panel of Figure 4A ). If the inflammatory stimulus persists following the acute phase, then the persistent, chronic neuroinflammatory reaction promotes heightened expression of P-glycoprotein in the blood–brain barrier (right hand panel of Figure 4A ). This heightened P-glycoprotein activity enhances blood–brain barrier function and greatly limits CNS drug penetration. Whether this model applies to humans with neuroinflammatory illnesses can only be answered with translational clinical studies. However, we believe this model provides a modern general description of the interaction between inflammation, infection and P-glycoprotein activity while simultaneously affording a novel explanation for historical reports of enhanced CSF levels of P-glycoprotein substrate antibiotics during CNS infections [2] .
It is essential to recognize that alterations in P-glycoprotein activity during inflammatory or infectious conditions may have either positive or negative consequences, depending on the drug in question and the therapeutic goals for the patient. For example, in meningitis and encephalitis, the site of drug action is within the leptomeninges and neural parenchyma, respectively. Therefore, a reduction in blood–brain barrier P-glycoprotein activity during acute inflammation could result in higher drug levels within infected meninges and/or brain with the potential for enhanced therapeutic efficacy. A transient reduction in blood–brain barrier P-glycoprotein activity could also allow for improved delivery of neuro-protectant agents in diseases such as ischemic or hemorrhagic stroke and improve the possibility of pharmaco logic neuro-protection in humans. In contrast, chronic diseases such as HIV infection and epilepsy result in an upregulation of blood–brain barrier P-glycoprotein and may reduce CNS drug levels. Reduced CNS drug levels could then result in drug resistance and even pharmacotherapeutic failure.
A second finding worth reflecting on is the heightened effect of P-glycoprotein inhibitors during CNS inflammation and/or infection ( Figure 4B ) [49,69,87] . As discussed previously, competitive drug interactions and the pharmacological effects of P-glycoprotein substrate drugs may be more pronounced during episodes of CNS inflammation as the basal activity of P-glycoprotein is already compromised. This synergistic effect may be either beneficial or detrimental. Animal models of CNS infection and ischemia have clearly demonstrated that P-glycoprotein inhibitors enhance the CNS concentration and efficacy of itraconazole, rifampicin and FK506 above that of the disease process alone, suggesting a beneficial effect of this host-immune, drug–drug interaction [49,87] . Conversely, near-maximal inhibition of P-glycoprotein activity during CNS inflammation could contribute to neurotoxicity. A novel clinical example of such a situation is implied from data derived from critically ill patients with acute inflam-matory brain injury (e.g., closed head trauma, intracerebral or intraventricular hemorrhage, or subarachnoid hemorrhage). These patients receive upwards of 30 drugs concomitantly, including P-glycoprotein substrates (e.g., dexamethasone, morphine and ranitidine) and inhibitors (e.g., amiodarone
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Roberts & Goralski
Expert Opin. Drug Metab. Toxicol. (2008) 4(10) 1259
and diltiazem) as part of their routine care. We believe that such polypharmacy during an acute neuroinflammatory reaction places these patients at high risk for drug interactions involving the P-glycoprotein transport system. Indeed, our research group has already published preliminary data to support this [59] .
In this review, we focused solely on P-glycoprotein as it is presently the best-characterized energy-dependent drug efflux transporter in the brain and blood–brain barrier. However,
the influence of inflammation and infection on the accumulation of drugs within the CNS may also depend on alterations in the activity of other drug efflux and uptake transporters. Other transporters of importance include BCRP, multi-drug resistance transporters, organic anion transporting polypeptide, organic cation transporters and organic anion transporters [102] . Although a similar relationship between CNS inflammation, infection and changes in the activity of these other drug efflux and uptake transporters (e.g., BCRP
(-)
Acute inflammation or infection +competitive inhibition of PGP Greatly reduced PGP activity Greatly enhanced drug accumulation in the CNS
A.
Time
(-)
(-)
(+)
e.g.,LPS
(-)
e.g., TNF-αor ET-1
Capillary
Endothelial cell
PGP Inhibitor
B.
Administration of PGP Inhibitor(e.g., PSC833) inhibits drug efflux
across the blood–brain barrier
PGP substratePGPPGP
Healthy Normal PGP activity Low drug accumulation in the CNS
i. Acute inflammationor infection Reduced PGP activity Enhanced drug accumulation in the CNS
ii. Chronic inflammationor resolution phase Recovery or enhanced PGP activity Low drug accumulation in the CNS
iii.
Figure 4 . Proposed hypothesis for the effect of CNS infl ammation on blood–brain barrier P-glycoprotein activity and the accumulation of P-glycoprotein substrates in the brain. A. P-glycoprotein activity and expression in blood–brain barrier endothelial cells unexposed to infl ammation (i.), during an acute infl ammatory response (ii.) and after resolution of the infl ammatory response or during chronic infl ammation or infection (iii.). B. The effect of P-glycoprotein inhibitors are potentiated during a CNS infl ammatory response, resulting in an even greater accumulation of transporter substrates in the brain. The (+) and (-) symbols denote inhibition and activation, respectively. ET-1: Endothelin-1; LPS: Lipopolysaccharide; PGP: P-glycoprotein.
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Infl ammation, infection and brain-derived P-glycoprotein
1260 Expert Opin. Drug Metab. Toxicol. (2008) 4(10)
and MRP1, 2, and 4) has been suggested by a limited, but expanding, number of studies [54,61,73,103,104] , no firm conclusions can presently be made. However, to formulate a model that will provide a complete understanding of blood–brain barrier pharmacokinetics it is essential for future studies to address the impact of inflammation and infection on the function of these other, at present less well characterized, blood–brain barrier drug transporters.
The recognition of an association between inflammation and P-glycoprotein activity has been accompanied by numerous animal and human investigations linking acute and chronic neuroinflammatory conditions such as acute brain injury, epilepsy, HIV dementia, peripheral neuropathy, Alzheimer’s and Parkinson’s diseases with altered P-glycoprotein activity ( Figure 5 ) [37,59,75,89,93] . Taken together, the findings of these studies suggest that alterations in blood–brain barrier P-glycoprotein activity during neuroinflammation may have a broader clinical importance than now appreciated. The potential for clinical importance may be further broadened if diseases associated with peripheral inflammation such as inflammatory arthritis and related autoimmune conditions, obesity, Type 2 diabetes, surgery and trauma are also capable of modulating the activity of brain-derived
P-glycoprotein. In the ensuing years, there is an immediate need for well-designed clinical pharmacokinetic studies to address the relationship between such diseases and the activity of drug transporters such as P-glycoprotein within the blood–brain barrier. Such studies are likely to be facilitated by positron emission tomography imaging techniques, which provide a non-invasive mechanism to quantitatively assess P-glycoprotein activity in the human blood–brain barrier [24,105] . With this information in hand, it will then be possible to appropriately inform and caution the medical community about the potential positive and negative impact of diseases associated with central and peripheral inflammation on overall blood–brain barrier function.
Acknowledgements
The work was supported by a Nova Scotia Health Research Foundation operating grant.
Declaration of interest
D Roberts and K Goralski have no conflicts of interest to declare.
Neurodegenerative diseases Alzheimer’s disease? Parkinson’s disease? Peripheral neuropathy?
Stroke
CNS infection HIV Bacterial or fungal meningitis
Traumatic braininjury?
Drug efficacyand toxicity
Acute peripheral inflammation and infection Bacteremia Localized tissue injury outside of the CNS Surgery?
Chronic systemic inflammation Obesity and Type 2 diabetes? Arthritis or related rheumatologic or autoimmune disorders
PGP
Figure 5 . Summary of the putative effects of infl ammation and infection on P-glycoprotein expression and activity in the human brain. A number of animal studies support that P-glycoprotein expression and activity in the CNS is modulated by infl ammatory and infectious responses generated in both the brain and in peripheral tissues such as the peritoneum. Studies reporting altered P-glycoprotein expression and/or activity or enhanced CSF drug levels during meningitis, HIV infection and epilepsy suggest this link may also exist in humans. Further investigations are needed to delineate exactly how these conditions affect the activity of this blood–brain barrier transporter in humans and whether these alterations impact overall CNS drug effi cacy and toxicity. PGP: P-glycoprotein.
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Roberts & Goralski
Expert Opin. Drug Metab. Toxicol. (2008) 4(10) 1261
Bibliography Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers. 1. De Vries HE, Kuiper J, De Boer AG,
et al. The blood-brain barrier in neuroinfl ammatory diseases. Pharmacol Rev 1997 ; 49 (2): 143 -55
2. Barling RW, Selkon JB. The penetration of antibiotics into cerebrospinal fl uid and brain tissue. J Antimicrob Chemother 1978 ; 4 (3): 203 -27
3. Dacey RG, Sande MA. Effect of probenecid on cerebrospinal fl uid concentrations of penicillin and cephalosporin derivatives. Antimicrob Agents Chemother 1974 ; 6 (4): 437 -41
4. Kearney BP, Aweeka FT. The penetration of anti-infectives into the central nervous system. Neurol Clin 1999 ; 17 (4): 883 -900
5. Pappenheimer JR, Heisey SR, Jordan EF. Active transport of Diodrast and phenolsulfonphthalein from cerebrospinal fl uid to blood. Am J Physiol 1961 ; 200 : 1 -10
6. Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Ann Rev Biochem 1993 ; 62 : 385 -427
7. Beaulieu E, Demeule M, Ghitescu L, et al. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem J 1997 ; 326 (Pt 2): 539 -44
8. Cordon-Cardo C, O’Brien JP, Casals D, et al. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci USA 1989 ; 86 (2): 695 -8
• The fi rst report of P-glycoprotein expression in human brain capillaries.
9. Schinkel AH, Smit JJ, Van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a defi ciency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994 ; 77 (4): 491 -502
•• The fi rst report of enhanced CNS drug accumulation in the mdr1a knockout mouse.
10. Schinkel AH, Wagenaar E, Van Deemter L, et al. Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest 1995 ; 96 (4): 1698 -705
11. Place VA, Pyle MM, De La Huerga J. Ethambutol in tuberculous meningitis. Am Rev Respir Dis 1969 ; 99 (5): 783 -5
12. Sippel JE, Mikhail IA, Girgis NI, et al. Rifampin concentrations in cerebrospinal fl uid of patients with tuberculous meningitis. Am Rev Respir Dis 1974 ; 109 (5): 579 -80
13. Hartkoorn RC, Chandler B, Owen A, et al. Differential drug susceptibility of intracellular and extracellular tuberculosis, and the impact of P-glycoprotein. Tuberculosis Edinb 2007 ; 87 (3): 248 -55
14. Thiebaut F, Tsuruo T, Hamada H, et al. Immunohistochemical localization in normal tissues of different epitopes in the multidrug transport protein P170: evidence for localization in brain capillaries and crossreactivity of one antibody with a muscle protein. J Histochem Cytochem 1989 ; 372 : 159 -64
15. Oldendorf WH. Blood-brain barrier permeabilty to drugs. Ann Rev Pharmacol 1974 ; 14 : 239 -48
16. Graff CL, Pollack GM. Drug transport at the blood-brain barrier and the choroid plexus. Curr Drug Metab 2004 ; 5 (1): 95 -108
• A very detailed overview of drug transporters in the CNS.
17. Kusuhara H, Sugiyama Y. Active effl ux across the blood-brain barrier: role of the solute carrier family. NeuroRx 2005 ; 2 (1): 73 -85
18. Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 1976 ; 455 (1): 152 -62
•• This seminal study reports the discovery of P-glycoprotein in cancer cells.
19. Thuerauf N, Fromm MF. The role of the transporter P-glycoprotein for disposition and effects of centrally acting drugs and for the pathogenesis of CNS diseases. Eur Arch Psychiatry Clin Neurosci 2006 ; 256 (5): 281 -6
20. Lankas GR, Cartwright ME, Umbenhauer D. P-glycoprotein defi ciency in a subpopulation of CF-1 mice enhances avermectin-induced neurotoxicity. Toxicol Appl Pharmacol 1997 ; 143 (2): 357 -65
21. Umbenhauer DR, Lankas GR, Pippert TR, et al. Identifi cation of a P-glycoprotein-defi cient subpopulation in the CF-1 mouse strain using a restriction fragment length polymorphism. Toxicol Appl Pharmacol 1997 ; 146 (1): 88 -94
22. Choo EF, Leake B, Wandel C, et al. Pharmacological inhibition of
P-glycoprotein transport enhances the distribution of HIV-1 protease inhibitors into brain and testes. Drug Metab Dis 2000 ; 28 (6): 655 -60
23. Uhr M, Tontsch A, Namendorf C, et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron 2008 ; 57 (2): 203 -9
24. Luurtsema G, Molthoff CF, Windhorst AD, et al. (R)- and (S)-[11C]verapamil as PET-tracers for measuring P-glycoprotein function: in vitro and in vivo evaluation. Nulc Med Biol 2003 ; 30 (7): 747 -51
25. Schinkel AH, Wagenaar E, Mol CA, et al. P-glycoprotein in the blood-brain barrier of mice infl uences the brain penetration and pharmacological activity of many drugs. J Clin Invest 1996 ; 97 (11): 2517 -24
26. Van Asperen J, Schinkel AH, Beijnen JH, et al. Altered pharmacokinetics of vinblastine in Mdr1a P-glycoprotein-defi cient Mice. J Natl Cancer Inst 1996 ; 88 (14): 994 -9
27. King M, Su W, Chang A, et al. Transport of opioids from the brain to the periphery by P-glycoprotein: peripheral actions of central drugs. Nat Neurosci 2001 ; 4 (3): 268 -74
28. Mealey KL, Greene S, Bagley R, et al. P-glycoprotein contributes to the blood-brain, but not blood-cerebrospinal fl uid, barrier in a spontaneous canine p-glycoprotein knockout model. Drug Metab Dispos 2008 ; 36 (6): 1073 -9
29. Mealey KL, Bentjen SA, Gay JM, et al. Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics 2001 ; 11 (8): 727 -33
30. Sartor LL, Bentjen SA, Trepanier L, et al. Loperamide toxicity in a collie with the MDR1 mutation associated with ivermectin sensitivity. J Vet Intern Med 2004 ; 18 (1): 117 -8
31. Loscher W. Drug transporters in the epileptic brain. Epilepsia 2007 ; 48 (Suppl 1): 8 -13
32. Greenberg ML, Fisher PG, Freeman C, et al. Etoposide, vincristine, and cyclosporin A with standard-dose radiation therapy in newly diagnosed diffuse intrinsic brainstem gliomas: a pediatric oncology group phase I study. Pediatr Blood Cancer 2005 ; 45 (5): 644 -8
33. Sadeque AJ, Wandel C, He H, et al. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin Pharmacol Ther 2000 ; 68 (3): 231 -7
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Infl ammation, infection and brain-derived P-glycoprotein
1262 Expert Opin. Drug Metab. Toxicol. (2008) 4(10)
34. Segal MB. The choroid plexuses and the barriers between the blood and the cerebrospinal fl uid. Cell Mol Neurobiol 2000 ; 20 (2): 183 -96
35. Rao VV, Dahlheimer JL, Bardgett ME, et al. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fl uid drug-permeability barrier. Proc Natl Acad Sci USA 1999 ; 96 (7): 3900 -5
36. Gazzin S, Strazielle N, Schmitt C, et al. Differential expression of the multidrug resistance-related proteins ABCb1 and ABCc1 between blood-brain interfaces. J Comp Neurol 2008 ; 510 (5): 497 -507
•• This report provides a relative quantifi cation of P-glycoprotein in brain capillaries versus the choroid plexus.
37. Volk H, Potschka H, Loscher W. Immunohistochemical localization of P-glycoprotein in rat brain and detection of its increased expression by seizures are sensitive to fi xation and staining variables. J Histochem Cytochem 2005 ; 53 (4): 517 -31
• This study compares various immunohistochemical techniques for P-glycoprotein detection in the rat brain.
38. Pardridge WM, Golden PL, Kang YS, et al. Brain microvascular and astrocyte localization of P-glycoprotein. J Neurochem 1997 ; 68 (3): 1278 -85
39. Schinkel AH. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv Drug Deliv Rev 1999 ; 36 (2-3): 179 -94
40. Bendayan R, Ronaldson PT, Gingras D, et al. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J Histochem Cytochem 2006 ; 54 (10): 1159 -67
41. Schlachetzki F, Pardridge WM. P-glycoprotein and caveolin-1alpha in endothelium and astrocytes of primate brain. Neuroreport 2003 ; 14 (16): 2041 -6
42. Virgintino D, Robertson D, Errede M, et al. Expression of P-glycoprotein in human cerebral cortex microvessels. J Histochem Cytochem 2002 ; 50 (12): 1671 -6
43. Lazarowski A, Caltana L, Merelli A, et al. Neuronal mdr-1 gene expression after experimental focal hypoxia: a new obstacle for neuroprotection? J Neurol Sci 2007 ; 258 (1-2): 84 -92
44. Lafl amme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating
gram-negative bacterial cell wall components. FASEB J 2001 ; 151 : 155 -63
45. Nadeau S, Rivest S. Endotoxemia prevents the cerebral infl ammatory wave induced by intraparenchymal lipopolysaccharide injection: role of glucocorticoids and CD14. J Immunol 2002 ; 169 (6): 3370 -81
46. Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev 2002 ; 3 (3): 216 -27
• This article provides a very good overview of the CNS infl ammatory response.
47. Beutler B, Hoebe K, Du X, et al. How we detect microbes and respond to them: the Toll-like receptors and their transducers. J Leukoc Biol 2003 ; 74 (4): 479 -85
48. Chow JC, Young DW, Golenbock DT, et al. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem 1999 ; 274 (16): 10689 -92
49. Imbert F, Jardin M, Fernandez C, et al. Effect of effl ux inhibition on brain uptake of itraconazole in mice infected with Cryptococcus neoformans. Drug Metab Dispos 2003 ; 31 (3): 319 -25
50. Miyama T, Takanaga H, Matsuo H, et al. P-glycoprotein-mediated transport of itraconazole across the blood-brain barrier. Antimicrob Agents Chemother 1998 ; 42 (7): 1738 -44
51. Scheld WM, Koedel U, Nathan B, et al. Pathophysiology of bacterial meningitis: mechanism(s) of neuronal injury. J Infect Dis 2002 ; 186 (Suppl 2): S225 -33
52. Renton KW, Nicholson TE. Hepatic and central nervous system cytochrome P450 are down-regulated during lipopolysaccharide-evoked localized infl ammation in brain. J Pharmacol Exp Ther 2000 ; 294 (2): 524 -30
53. Nicholson TE, Renton KW. Role of cytokines in the lipopolysaccharide-evoked depression of cytochrome P450 in the brain and liver. Biochem Pharmacol 2001 ; 62 (12): 1709 -17
54. Goralski KB, Hartmann G, Piquette-Miller M, et al. Downregulation of mdr1a expression in the brain and liver during CNS infl ammation alters the in vivo disposition of digoxin. Br J Pharmacol 2003 ; 139 (1): 35 -48
• The fi rst description that LPS-induced CNS infl ammation modifi es brain P-glycoprotein expression and function.
55. Goralski KB, Abdulla D, Sinal CJ, et al. Toll-like receptor-4 regulation of hepatic Cyp3a11 metabolism in a mouse model of LPS-induced CNS infl ammation. Am J Physiol 2005 ; 289 (3): G434 -43
56. Gao B, Stieger B, Noe B, et al. Localization of the organic anion transporting polypeptide 2 (Oatp2) in capillary endothelium and choroid plexus epithelium of rat brain. J Histochem Cytochem 1999 ; 47 (10): 1255 -64
57. Noe B, Hagenbuch B, Stieger B, et al. Isolation of a multispecifi c organic anion and cardiac glycoside transporter from rat brain. Proc Natl Acad Sci USA 1997 ; 94 (19): 10346 -50
58. Hartz AM, Bauer B, Fricker G, et al. Rapid modulation of P-glycoprotein-mediated transport at the blood-brain barrier by tumor necrosis factor-alpha and lipopolysaccharide. Mol Pharmacol 2006 ; 69 (2): 462 -70
•• An excellent investigation that describes the rapid and reversible downregulation of P-glycoprotein activity by TNF-α and LPS.
59. Morgan ET, Goralski KB, Piquette-Miller M, et al. Regulation of drug-metabolizing enzymes and transporters in infection, infl ammation, and cancer. Drug Metab Dispos 2008 ; 36 (2): 205 -16
60. Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (New York, NY) 1998 ; 282 (5396): 2085 -8
61. Bauer B, Hartz AM, Miller DS. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol Pharmacol 2007 ; 71 (3): 667 -75
•• This study describes the biphasic regulation of blood-brain barrier P-glycoprotein by ET-1 and TNF-α.
62. Ronaldson PT, Bendayan R. HIV-1 viral envelope glycoprotein gp120 triggers an infl ammatory response in cultured rat astrocytes and regulates the functional expression of P-glycoprotein. Mol Pharmacol 2006 ; 70 (3): 1087 -98
63. Zhao YL, Du J, Kanazawa H, et al. Effect of endotoxin on doxorubicin transport across blood-brain barrier and P-glycoprotein function in mice. Eur J Pharmacol 2002 ; 445 (1-2): 115 -23
64. Zhao YL, Du J, Kanazawa H, et al. Shiga-like toxin II modifi es brain distribution of a P-glycoprotein substrate, doxorubicin,
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Roberts & Goralski
Expert Opin. Drug Metab. Toxicol. (2008) 4(10) 1263
and P-glycoprotein expression in mice. Brain Res 2002 ; 956 (2): 246 -53
65. Wang JH, Scollard DA, Teng S, et al. Detection of P-glycoprotein activity in endotoxemic rats by 99mTc-sestamibi imaging. J Nucl Med 2005 ; 46 (9): 1537 -45
66. Seelbach MJ, Brooks TA, Egleton RD, et al. Peripheral infl ammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. J Neurochem 2007 ; 102 (5): 1677 -90
67. Hartz AM, Bauer B, Fricker G, et al. Rapid regulation of P-glycoprotein at the blood-brain barrier by endothelin-1. Mol Pharmacol 2004 ; 66 (3): 387 -94
•• The fi rst study to describe the rapid regulation of brain capillary P-glycoprotein activity by ET-1.
68. Nie XJ, Olsson Y. Endothelin peptides in brain diseases. Rev Neurosci 1996 ; 7 (3): 177 -86
69. Yu C, Kastin AJ, Tu H, et al. TNF activates P-glycoprotein in cerebral microvascular endothelial cells. Cell Physiol Biochem 2007 ; 20 (6): 853 -8
70. Theron D, Barraud De Lagerie S, Tardivel S, et al. Infl uence of tumor necrosis factor-alpha on the expression and function of P-glycoprotein in an immortalised rat brain capillary endothelial cell line, GPNT. Biochem Pharmacol 2003 ; 66 (4): 579 -87
71. Veszelka S, Pasztoi M, Farkas AE, et al. Pentosan polysulfate protects brain endothelial cells against bacterial lipopolysaccharide-induced damages. Neurochem Int 2007 ; 50 (1): 219 -28
72. Miller DS, Bauer B, Hartz AM. Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol Rev 2008 ; 60 (2): 196 -209
73. Hartz AM, Bauer B, Block ML, et al. Diesel exhaust particles induce oxidative stress, proinfl ammatory signaling, and P-glycoprotein up-regulation at the blood-brain barrier. FASEB J 2008 ; 22 (8): 2723 -33
74. Hembury A, Mabondzo A. Endothelin-1 reduces P-glycoprotein transport activity in an in vitro model of human adult blood-brain barrier. Cell Mol Neurobiol 2008 . [Epub ahead of print]
75. Langford D, Grigorian A, Hurford R, et al. Altered P-glycoprotein expression in AIDS
patients with HIV encephalitis. J Neuropathol Exp Neurol 2004 ; 63 (10): 1038 -47
76. Nath A, Schiess N, Venkatesan A, et al. Evolution of HIV dementia with HIV infection. Int Rev Psychiatry (Abingdon, England) 2008 ; 20 (1): 25 -31
77. Ghafouri M, Amini S, Khalili K, et al. HIV-1 associated dementia: symptoms and causes. Retrovirology 2006 ; 3 : 28
78. Kim RB, Fromm MF, Wandel C, et al. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest 1998 ; 101 (2): 289 -94
79. Hayashi K, Pu H, Tian J, et al. HIV-Tat protein induces P-glycoprotein expression in brain microvascular endothelial cells. J Neurochem 2005 ; 93 (5): 1231 -41
80. Sharp FR, Bergeron M, Bernaudin M. Hypoxia-inducible factor in brain. Adv Exp Med Biol 2001 ; 502 : 273 -91
81. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 2007 ; 87 (1): 315 -424
82. Rodriguez-Yanez M, Castillo J. Role of infl ammatory markers in brain ischemia. Curr Opin Neurol 2008 ; 21 (3): 353 -7
83. Comerford KM, Wallace TJ, Karhausen J, et al. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res 2002 ; 62 (12): 3387 -94
84. Sukhai M, Piquette-Miller M. Regulation of the multidrug resistance genes by stress signals. J Pharm Pharm Sci 2000 ; 3 (2): 268 -80
85. Yu XY, Lin SG, Chen X, et al. Transport of cryptotanshinone, a major active triterpenoid in Salvia miltiorrhiza Bunge widely used in the treatment of stroke and Alzheimer’s disease, across the blood-brain barrier. Curr Drug Metab 2007 ; 8 (4): 365 -78
86. Chen X, Zhou ZW, Xue CC, et al. Role of P-glycoprotein in restricting the brain penetration of tanshinone IIA, a major active constituent from the root of Salvia miltiorrhiza Bunge, across the blood-brain barrier. Xenobiotica 2007 ; 37 (6): 635 -78
87. Spudich A, Kilic E, Xing H, et al. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat Neurosci 2006 ; 9 (4): 487 -8
88. Cirrito JR, Deane R, Fagan AM, et al. P-glycoprotein defi ciency at the
blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 2005 ; 115 (11): 3285 -90
89. Vogelgesang S, Warzok RW, Cascorbi I, et al. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 2004 ; 1 (2): 121 -5
• This clinical study describes an inverse relationship between P-glycoprotein levels and amyloid beta deposition in brain capillaries.
90. Eikelenboom P, Bate C, Van Gool WA, et al. Neuroinfl ammation in Alzheimer’s disease and prion disease. Glia 2002 ; 40 (2): 232 -9
91. Kuhnke D, Jedlitschky G, Grube M, et al. MDR1-P-glycoprotein (ABCB1) mediates transport of Alzheimer’s amyloid-beta peptides – implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain pathol (Zurich, Switzerland) 2007 ; 17 (4): 347 -53
92. Lam FC, Liu R, Lu P, et al. Beta-amyloid effl ux mediated by p-glycoprotein. J Neurochem 2001 ; 76 (4): 1121 -8
93. Drozdzik M, Bialecka M, Mysliwiec K, et al. Polymorphism in the P-glycoprotein drug transporter MDR1 gene: a possible link between environmental and genetic factors in Parkinson’s disease. Pharmacogenetics 2003 ; 13 (5): 259 -63
94. Furuno T, Landi MT, Ceroni M, et al. Expression polymorphism of the blood-brain barrier component P-glycoprotein (MDR1) in relation to Parkinson’s disease. Pharmacogenetics 2002 ; 12 (7): 529 -34
95. Lee CG, Tang K, Cheung YB, et al. MDR1, the blood-brain barrier transporter, is associated with Parkinson’s disease in ethnic Chinese. J Med Genet 2004 ; 41 (5): e60
96. Kortekaas R, Leenders KL, Van Oostrom JC, et al. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol 2005 ; 57 (2): 176 -9
97. Whitton PS. Infl ammation as a causative factor in the aetiology of Parkinson’s disease. Br J Pharmacol 2007 ; 150 (8): 963 -76
98. Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug effl ux transporters. Nat Rev 2005 ; 6 (8): 591 -602
• An excellent overview of the role of multidrug resistance transporters in drug-resistant brain diseases.
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.

Infl ammation, infection and brain-derived P-glycoprotein
1264 Expert Opin. Drug Metab. Toxicol. (2008) 4(10)
99. Lazarowski A, Czornyj L, Lubienieki F, et al. ABC transporters during epilepsy and mechanisms underlying multidrug resistance in refractory epilepsy. Epilepsia 2007 ; 48 (Suppl 5): 140 -9
100. Van Vliet EA, Van Schaik R, Edelbroek PM, et al. Region-specifi c overexpression of P-glycoprotein at the blood-brain barrier affects brain uptake of phenytoin in epileptic rats. J Pharmacol Exp Ther 2007 ; 322 (1): 141 -7
101. Bauer B, Hartz AM, Pekcec A, et al. Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Mol Pharmacol 2008 ; 73 (5): 1444 -53
102. De Vries NA, Zhao J, Kroon E, et al. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res 2007 ; 13 (21): 6440 -9
103. Balayssac D, Cayre A, Authier N, et al. Involvement of the multidrug resistance transporters in cisplatin-induced neuropathy in rats. Comparison with the chronic constriction injury model and monoarthritic rats. Eur J Pharmacol 2006 ; 544 (1-3): 49 -57
104. Hayashi K, Pu H, Andras IE, et al. HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood-brain barrier. J Cereb Blood Flow Metab 2006 ; 26 (8): 1052 -65
105. Ikoma Y, Takano A, Ito H, et al. Quantitative analysis of 11C-verapamil transfer at the human blood-brain barrier for evaluation of P-glycoprotein function. J Nucl Med 2006 ; 47 (9): 1531 -7
• This study describes a non-invasive PET scanning method to quantitatively asses human blood-brain barrier P-glycoprotein function.
Affi liation Derek J Roberts 1 BSc (Pharm) & Kerry B Goralski † 2 PhD † Author for correspondence 1 Doctor of Medicine (MD) Candidate, Faculty of Medicine Dalhousie University, 5849 University Avenue, Halifax, N.S., B3H 4H7, Canada †2 Assistant Professor, College of Pharmacy, Faculty of Health Professions and, Department of Pharmacology, Faculty of Medicine,5968 College Street, Halifax, N.S., B3H 3J5, Canada Tel: +1 902 494 2052 ; Fax: +1 902 494 1396 ; E-mail: [email protected]
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f A
uckl
and
on 1
1/03
/14
For
pers
onal
use
onl
y.





























