Embed Size (px)
Citation preview

• We plan to extend the amount of information present in the database;
• Include MD input and output files and setups to guarantee reproducibility;
• Use of replica-exchange MD to improve sampling for “difficult” cases;
• Improvement of key parameters, e.g. dihedral angles, by comparison with QM
dynamics in presence of implicit/explicit solvents, using biased MD techniques;
• Setup of parameters at different pHs (different atomic charges);
• Use of different solvents (e.g. hydrophobic solvents, organic “broths”) to improve
conformational sampling while (possibly) mimicking interactions with functional
groups in biomolecules such as proteins, nucleic acids and membranes.
We present an on-line database of all-atom force-field parameters and molecular properties of compounds with antimicrobial activity (mostly antibiotics and some beta-lactamase inhibitors). For each compound, we provide the General Amber Force Field parameters for the most likely conformer at physiological pH, together with an analysis of properties of interest as extracted from µs-long molecular dynamics simulations in water and counter ions (0.1M KCl). The properties include number and population of structural clusters, molecular flexibility, hydrophobic and hydrophilic molecular surfaces, the statistics of intra- and inter-molecular H-bonds, as well as structural and dynamical properties of solvent molecules within first and second solvation shells. In addition, the database contains several key molecular parameters, such as energy of the frontier molecular orbitals, vibrational properties, rotational constants, atomic partial charges and electric dipole moment, computed by Density Functional Theory. This work is part of a wider project aiming to build-up a database containing structural, physico-chemical and dynamical properties of medicinal compounds using different force-field parameters with increasing level of complexity and reliability. The database is accessible at http://www.dsf.unica.it/translocation/db/
AcknowledgmentsThe research leading to these results was conducted as part of the Translocation consortium (www.translocation.eu) and has received support from the Innovative Medicines Initiatives Joint Undertaking under Grant Agreement n115525, resources which are composed of financial contribution from the European Union 7th framework programme (FP7/2007-2013) and EFPIA companies in kind contribution.
References ● Wang J, Wolf RM, Caldwell JW, Kollman PA, & Case DA (2004) Development and testing of
a general amber force field. Journal of Computational Chemistry 25, 1157-1174.● Marvin 6.2.0, 2013, ChemAxon (www.chemaxon.com).● Frisch MJ, et al. (2009) Gaussian 09, Revision A.1 (Gaussian, Inc., Wallingford CT).● Case DA, et al. (2014) AMBER 14 (University of California, San Francisco).● Pyrkov TV et al. (2009) PLATINUM: A web tool for analysis of hydrophobic/hydrophilic
organization of biomolecular complexes, Bioinformatics 25,1201-1202.● Bonomi et al. (2009) PLUMED: A portable plugin for free-energy calculations with
molecular dynamics, Comput. Phys. Comm. 180, 1961-1972.
A database of force-field parameters, dynamics and properties of antimicrobial compoundsG. Malloci, A. V. Vargiu, G. Serra, A. Bosin, P. Ruggerone, and M. Ceccarelli
Physics Department - University of Cagliari, Citt. Univ. Monserrato (CA) - ITALY [email protected] - [email protected]
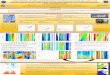
Abstract Protocol
Homepage
General properties
MD setup files MD properties
Isoelectric point
PDB file (QM structure)
Cluster analysis
Intra- inter-molecular HB statistics
Asphericity Shape anisotropy
Principal moments of gyration tensor
Perspectives
●Water structure/dynamics●Root mean square fluctuation
●Minimal projection area
A= S1 − 1
2(S2 + S3) κ 2 = 1− 3
S1S2 + S1S3 + S2S3
S1 + S2 + S3( ) 2
Si
AMBER .prep and .frcmod
files
Compound Summary
Polar/Hydrophobic surfaces
QM properties
Electric dipole
Molecular orbital data
Vibrational analysis andIR absorption spectrum


![Database Practices- DB 12c Oracle Banking Payments...Database Practices- DB 12c Oracle Banking Payments Release 14.1.0.0.0 [May] 2018 Table of Contents 1. DATABASE INITIALIZATION PARAMETERS](https://img.pdfslide.net/doc/110x75/60a0e711e33eea186a5019ff/database-practices-db-12c-oracle-banking-payments-database-practices-db-12c.jpg)