Embed Size (px)
Citation preview

A dentofacial deformity associated with incontinentia pigmenti: Report of a case Philip E. Milam, DDS,a Thomas J. Grifin, DDS,b and Robert D. Shapiro, DDS,c Harrison, Ark., and Chicago, Ill.
THE UNIVERSITY OF CHICAGO MEDICAL CENTER
I ncontinentia pigmenti (IP) is a genetic disease with an X-linked dominant mode of inheritance. This dis- tinctive multisystem disorder is characterized by its abnormalities of the skin pigmentation.le3 IP, also known as the Bloch-Sulzberger syndrome, has a va- riety of ectodermal and mesodermal aberrations including major dental anomalies.3 Described is a pa- tient with IP who developed not only the characteris- tic skin anomalies and dental defects of oligodontia and malformed dentition but also a severe dentofacial deformity.
CASEREPORT
A 16-year-old white girl was referred to the oral and maxillofacial surgery service for evaluation of her multiple dental disorders. The patient’s history revealed a diagnosis of IP. This syndrome manifested itself early after birth with multiple cutaneous vesicular lesions (Fig. 1). These pro- gressed through infancy and childhood by formation of verrucous and hyperpigmented lesions. They eventually be- came dispersed, resulting in subtle atrophic type of scarring of the skin. No other abnormalities were noted during childhood except for dental anomalies noted during routine dental examinations.
Family history revealed no known cases of IP in either parent. Siblings consisted of three daughters and one son. There was one miscarriage. The sex of the aborted fetus is unknown. Another daughter was noted to have developed crania1 synostosis that required surgery.
Physical examination disclosed subtle stellate scarring and retained hyperpigmentation on the patient’s back, thighs, hands, and face (Fig. 2). Clinical and radiographic
aFormerly Chief Resident, Division of Oral and Maxillofacial Sur- gery, The University of Texas Medical Branch, Galveston. Pres- ently in private practice in Harrison, Ark. bClinical Instructor, Section of Dentistry, The University of Chi- cago Medical Center.
CFormerly Assistant Professor and Chief, Division of Oral and Maxillofacial Surgery, The University of Texas Medical Branch, Galveston. Presently Associate Professor of Clinical Surgery and
Chief, Oral and Maxillofacial Surgery Service, The University of Chicago Medical Center. 7/12/12587
420
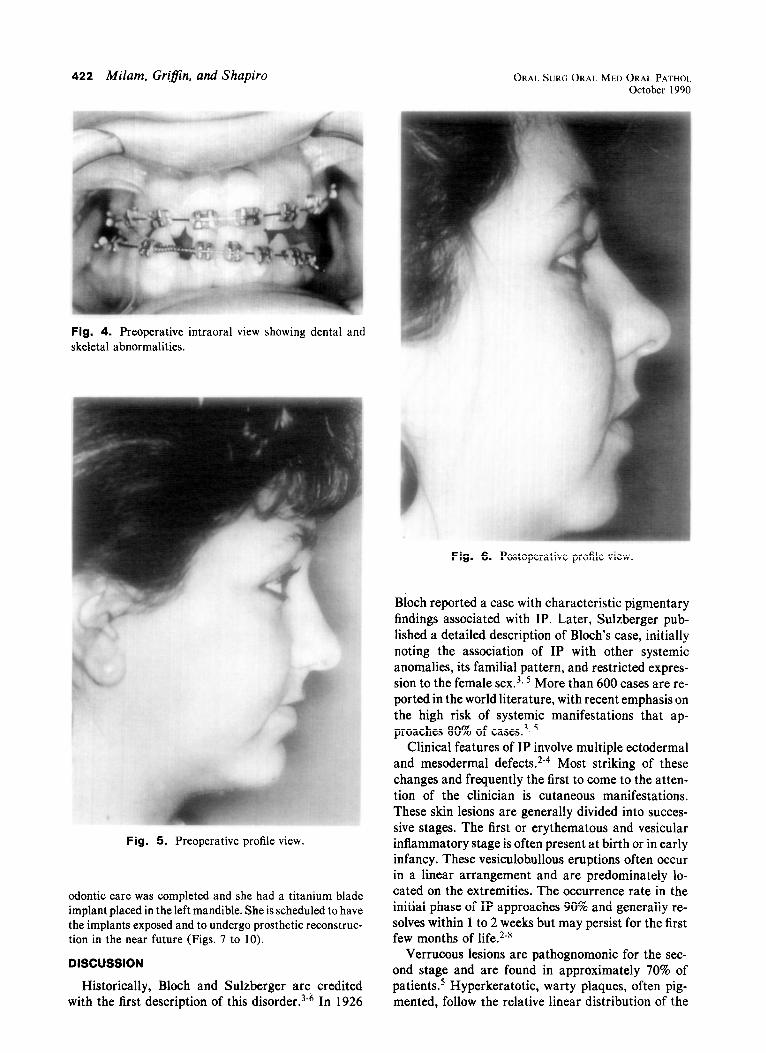
evaluation of the dentoskeletal anomalies revealed oli- godontia, retained primary dentition, malformed teeth, im- pacted right maxillary canine, maxillary and mandibular anteroposterior hypoplasia, maxillary vertical deficiency with cant, facial asymmetry, and left temporomandibular joint dysfunction (Figs. 3 to 6).
To correct these problems the following treatment plan was devised and performed: (1) presurgical orthodontic treatment to level and align the maxillary and mandibular arches; (2) Le Fort I maxillary osteotomy with interposi- tional bone grafting to reposition the maxilla inferiorly 5 mm (right) and 3 mm (left) along with a 4 mm anterior ad- vancement; (3) bilateral mandibular sagittal split ramus osteotomies to advance and rotate the mandible approxi- mately 4 mm and achieve a normal midline, overbite, and overjet relationship; (4) genioplasty with interpositional bone grafting to increase chin height and width; (5) rigid fixation of maxilla and mandible with bone plates and screws.
The maxillary surgery was performed through an incision in the vestibule from first molar to first molar. Reflection of the mucoperiosteum was carried superiorly until the in- fraorbital nerve and foramen were identified. After the maxilla had been measured and marked appropriately, a modified Le Fort I step osteotomy was created from the piriform rim to the posterior maxilla with the vertical step in the zygomatic buttress of the maxilla to facilitate place- ment of bone plates. Sectioning of the lateral nasal wall, the nasal septum, and the pterygomaxillary fissure was per- formed in the routine fashion and the maxilla “downfrac- tured.” Sharp areas of the lateral nasal wall were removed, and the descending palatine arteries were freed and pre- served. With the maxilla mobilized, an interim splint was placed to facilitate the downward and forward moves. At the same time, corticocancellous bone was harvested from the inner table of the left hip. Properly fashioned wedges were placed in the osteotomy area to maintain the previ- ously determined amount of maxillary lengthening. The maxilla was then stabilized by the application of Wiirzburg titanium bone plates that had been contoured to be passive when the screws were tightened. Corticocancellous bone was then wedged between the posterior aspect of the max- illa and the pterygoid plates. The entire osteotomy was then packed with corticocancellous chips and the incision closed.
Attention was then directed to the mandible, where an

Volume 70 Number 4
Dentofacial deformity associated with incontinentia pigmenti 42 I
Fig. 1. Vesicular lesion phase manifested early in in- fancy.
incision approximately 2.5 cm in length was made along the external oblique ridge. The mucoperiosteum was dissected to expose the lingual aspect of the mandibular ramus to the lingula. A horizontal osteotomy was perfomred on the lin- gual aspect of the ramus with a reciprocating saw approx- imately 20 mm in length. The osteotomy was continued an- teriorly down the superior aspect of the mandible to the buccal aspect of the first molar area. At that point a verti- cal bone cut was made through the buccal cortex to the in- ferior border and through the border to the medial aspect of the mandible. With a spatula osteotome the bone cuts were completed and the mandibular sagittal split accom- plished. The contralateral side was completed in a like fashion. The permanent occlusal splint was then placed and the planned occlusion obtained. With the teeth in the splint and the condyles seated, the mandibular segments were rigidly fixed by the insertion of Wiirzburg titanium screws placed transcutaneously.
A mandibular genioplasty was then performed through the conventional vestibular incision. The bone cuts were made with a reciprocating saw. The inferior fragment was sectioned vertically and widened 4 mm, and an interposi- tional bone graft was placed. The segments were then sta- bilized with 24-gauge stainless steel wire.
The patient had an uneventful postoperative course and
Fig. 2. Retained hyperpigmentation present on patient’s back.
Fig. 3. Preoperative radiograph revealing partial an- odontia with malformed, retained, and impacted dentition.
was discharged on the third day after surgery. Maxillo- mandibular fixation was released before discharge, with night elastics continued for 1 month. Oral physical therapy was continually emphasized with regular manipulation by the surgeon. Seven months postoperatively the patient has excellent oral opening without temporomandibular joint pain and “popping.” Six months postoperatively her orth-
422 Milam, Grifin, and Shapiro ORAL SURC ORAL MED ORAL PATHOL October 1990
Fig. 4. Preoperative intraoral view showing dental and skeletal abnormalities.
Fig. 6. Postoperative profile view.
Fig. 5. Preoperative profile view.
odontic care was completed and she had a titanium blade implant placed in the left mandible. She is scheduled to have the implants exposed and to undergo prosthetic reconstruc- tion in the near future (Figs. 7 to 10).
DISCUSSION
Historically, Bloch and Sulzberger are credited with the first description of this disorder.3-6 In 1926
Bloch reported a case with characteristic pigmentary findings associated with IP. Later, Sulzberger pub- lished a detailed description of Bloch’s case, initially noting the association of IP with other systemic anomalies, its familial pattern, and restricted expres- sion to the female sex.‘, 5 More than 600 cases are re- ported in the world literature, with recent emphasis on the high risk of systemic manifestations that ap- proaches 80% of cases.3T s
Clinical features of IP involve multiple ectodermal and mesodermal defects.2-4 Most striking of these changes and frequently the first to come to the atten- tion of the clinician is cutaneous manifestations. These skin lesions are generally divided into succes- sive stages. The first or erythematous and vesicular inflammatory stage is often present at birth or in early infancy. These vesiculobullous eruptions often occur in a linear arrangement and are predominately lo- cated on the extremities. The occurrence rate in the initial phase of IP approaches 90% and generally re- solves within 1 to 2 weeks but may persist for the first few months of life.2-8
Verrucous lesions are pathognomonic for the sec- ond stage and are found in approximately 70% of patients5 Hyperkeratotic, warty plaques, often pig- mented, follow the relative linear distribution of the

Volume 70 Number 4
Dentofacial deformity associated with incontinentia pigmenti 423
Fig. 7. Intraoral view revealing improved occlusal rela- tion.
, I
‘\ I
Fig. 8. Preoperative and postoperative cephalometric tracing overlay.
vesicular lesions. These lesions commonly persist for 6 to 12 months.3~5*8~9
The pigmented stage is the third and most distinc- tive phase of IP. It is characterized by the presence of linear and whorled hyperpigmentation of the skin and generally does not correspond to the involved areas of the previous two stages. 3* 5, lo, 1 1 This pigmentation is attributed to descent of pigment to the dermal layer where melanophages reveal increased amounts of melanin.** 4 Occurrence rate approaches 98% to 100% of affected persons. Occasionally patients show the pigmented stage at birth, a fact that prompts investi- gators to propose in utero occurrence of the first two stages. Hyperpigmentation may persist indefinitely, although it usually tends to fade in late childhood or adolescence.
Fading of pigmented lesions and atrophic scar de-
Fig. 9. Illustration of position of bony cuts and various types of fixation used and their locations.
Flg. 10. Six-month postoperative panoramic radiograph showing location of titanium blade implant (mandibular left).
velopment marks the final stage of the cutaneous le- sions. This reticulated scarring phase has been re- ported in less than one third of cases.3y 5, * 1
Extracutaneous anomalies involve the dentition, hair, eyes, central nervous system, and skeleton. These defects are seen in more than 50% of persons with IP. Major dental anomalies occurring in both deciduous and permanent dentition are noted in two thirds of patients with this disorder. Defects include partial or complete anodontia, peg or conical defor- mities of the teeth, enamel disorders, and delayed eruption. Forty-three percent of patients have oli- godontia, 18% display delayed eruption, and 33% manifest malformation of dentition.3-5, 12-14
Ophthalmic disorders are present in 25% to 35% of patients and are often unilateral. Abnormalities most commonly seen are strabismus and retinal detach- ment. Other ocular defects include nystagmus, ptosis,

424 Milam, GrijJin, and Shapiro
cataracts, blue sclera, pigmentation of the iris, ciliary body atrophy, vascular anomalies, myopia, and chori- oretinitis. Many ocular changes can be progressive and eventually lead to blindness.5, ‘. “3 I5
Central nervous system involvement occurs in up to one third patients with IP. The most commonly reported central nervous system disorder is seizures, which occur in approximately 13% of patients with IP. Mental ability of the patients ranges from superior intelligence to severe retardation.3, 5, ‘3 *, 16, ”
Alopecia occurs in more than one third of affected patients. This anomaly is generally considered to be a result of tissue scarring and generally appears on the vertex of the scalp. Agenesis of the eyebrows and eye- lashes has also been reported.3* ‘3 I8
Nail dystrophies are rare and include anomalies consisting of subungual keratotic tumors with under- lying lytic bone deformities, nail thinning, or total nail absence 5, 11, 19.20
A number of other structural anomalies may be present. These include dwarfism and small stature, club foot, spina bifida, cleft lip and palate, hemiatro- phy, chondrodystrophy, skull and ear deformities, congenital dislocation of the hip, hemivertebrae, ex- tra ribs, and syndactyly.3-5
Results of laboratory studies are generally within normal limits although a marked eosinophilia and leukocytosis may occur in a large percentage of patients. Eosinophilia reaching 50% and leukocytosis of 50,000/mm3 may be present at birth and generally peak at 1 to 10 weeks of age. The abnormal labora- tory levels appear to correlate with the inflammatory phase of the skin lesions.5q 1 I, 19, 2o
Several mechanisms have been proposed to explain the pathogenesis of IP, however, these theories remain unproven. Genetic family studies have described the disorder as consistent with an X-linked dominant pattern of inheritance with presumed lethality in hemizygous males. Recent studies have assigned the IP gene locus specifically to band Xp 11 and speculate the disorder may be a submicroscopic deletion. How- ever, further investigation is needed to resolve the is- sue as to the nature of the IP gene, its mode of action in producing the IP phenotype in females, and prena- tal lethality in males. Genetic counseling is advisable for patients with IP since the expected risk of offspring is one affected female, one normal female, one normal male, and one aborted male.‘, 5, I83 2o
The fact that this patient had partial anodontia and maxillomandibular hypoplasia created technical problems in the performance of orthognathic surgery. The ability to rigidly fix the maxilla and mandible made a stable result more likely in the presence of in- complete dentition. It should also be noted that the cancellous portion of the mandible was extremely
ORAL SURG ORAL MED ORAL PATHOL October 1990
limited and the sagittal split was largely through cor- tical bone. This finding also presented difficulty in the placement of the titanium blade implant.
SUMMARY
A case of IP in a 16-year-old girl has been presented. This patient manifested classic ectodermal and mesodermal anomalies. We present this case to illustrate a rare etiologic factor in the development of dentofacial deformities that can be treated in the conventional manner.
REFERENCES
1.
2.
3.
4.
5.
6.
1.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
Cannizzaro LA, Hecht F. Gene for incontinentia pigmenti maps to band Xpll with an (X:10) (pl l:g22) translocation. Clin Genet 1987;32:66-9. Foster SC, Album MM. Incontinentia pigmenti: Bloch- Sulzberger, Block-Seimens disease. J Oral Surg 1970;29:837- 45. Kegel MF. Dominant disorders with miltiple organ involve- ment, Dermatol Clin 1987;5:205-18. Baddour HM, Steed DL, Tilson HBL. Incontinentia pigmen- ti: report of a case. J Oral Surg 1981;38:157-9. Cohen BA: Incontinentia pigmenti. Neurol Clin 1987;5:361- 77. Jelinek JE. Sulzberger on incontinentia pigmenti. Int J Der- matol 1977;16:365-8. Francois J. Incontinenia pigmenti (B&h-Sulzberger syn- drome) and retinal changes. Br J Ophthalmol 1984;68:19-25. O’Brien JE, Feingold M. Incontinentia pigmenti, a longitudi- nal study. Am J Dis Child 1985;139:711-12. Morgan JD. Incontinentia pigmenti (Bloch-Sulzberger syn- drome). Am J Dis Child 1971;122:294-300. Carnev RG. Carnev RG Jr. Incontinentia uinmenti. Arch Dermatol 1970;102:i57-62.
. -
Simmons DA, Kegel MF, Scher RK, Hines YC. Subungual tumors in incontinentia pigmenti. Arch Dermatol 1986; 122:1431-4. Alper JC. The genodermatoses and their significance in pedi- atric dermatology. Dermatol Clin 1986;4:45-54. Bjellerup M. Incontinentia pigmenti with dental anomalies: a three generation study. Acta Derm Venereol (Stockh) 1982; 62~262-4. Burges MC. Incontinentia pigmenti. Six cases of Bloch- Sulzberger syndrome. Br Dent J 1982;152:195-6. Rosenfeld SI, Smith ME. Ocular findings in incontinentia pig- menti. Ophthalmology 1985;92:543-6. Auraham E, Hare1 S, Jurgenson U, et al. Computed tomo- graphic demonstration of brain changes in incontinentia pig- menti. Am J Dis Child 1985;139:372-4. Gureuital AW, Farrell W, Horock S, et al. Incontinentia pig- menti, a systemic genodermatosis with striking cutaneous findings. Clin Pediatr (Phila)1973;12:396-401. Wiklund DA, Weston WL. Incontinentia pigmenti: a four generation study. Arch Dermatol 1980;116:701-3. Hartman DL. Incontinentia pigmenti associated with subun- gual tumors. Arch Dermatol 1966;94:632-5.
20. Moscaro JM. Palou J, Vives P. Painful subungual keratotic tumors in incontinentia pigmenti. J Am Acad Dermatol 1985;13:913-8.
Reprint requests to: Dr. Robert D. Shapiro Section of Oral and Maxillofacial Surgery The University of Chicago Medical Center Room E212, Box 418 - Chicago, IL 60637















![Tnfa Signaling Through Tnfr2 Protects Skin Against ...eprints.whiterose.ac.uk/81541/1/Tnfa signaling through tnfr2 protects... · genodermatosis incontinentia pigmenti (IP) [17]](https://img.pdfslide.net/doc/110x75/5f3bedf6651a4c137761035c/tnfa-signaling-through-tnfr2-protects-skin-against-signaling-through-tnfr2-protects.jpg)




![First IKBKG Gene Mutation Study in Serbian Incontinentia ... · Incontinentia pigmenti (IP; Bloch-Sulzberg-er syndrome; MIM 308300) is a rare X-linked dominant genodermatosis [5]](https://img.pdfslide.net/doc/110x75/5f3bedf5651a4c1377610355/first-ikbkg-gene-mutation-study-in-serbian-incontinentia-incontinentia-pigmenti.jpg)







