Embed Size (px)
Citation preview

A Dual Role for Apolipoprotein E in NeuroinflammationAnti- and Pro-Inflammatory Activity
Ling Guo,1,2 Mary Jo LaDu,2,3 and Linda J. Van Eldik*,1,2
1Department of Cell and Molecular Biology, 2Drug Discovery Program,Northwestern University Feinberg School of Medicine, Chicago, IL 60611;
and 3Department of Medicine, Evanston NorthwesternHealthcare Research Institute, Evanston, IL 60201
Received May 29, 2003; Accepted February 4, 2004
Abstract
Chronically activated glia associated with amyloid plaques might contribute to neuronal dysfunction inAlzheimer’s disease (AD) through generation of neuroinflammatory molecules. Apolipoprotein E (apoE), alsofound associated with amyloid plaques, has been hypothesized to serve an anti-inflammatory role in the CNSthrough its ability to modulate β-amyloid (Aβ)-induced glial activation. To further characterize the effect of apoEon inflammation, we examined the ability of exogenously added human apoE3 and apoE4 to modulate neuro-inflammatory responses of cultured rat glia. Apolipoprotein E3 (apoE3) and apoE4 suppressed oligomericAβ-induced production of inducible nitric oxide synthase and cyclo-oxygenase-2, supporting an anti-inflammatory role for apoE. Exogenous apoE also inhibited Aβ-induced production of endogenous apoE.However, exogenous apoE in the absence of Aβ stimulated production of the pro-inflammatory cytokineinterleukin-1β in an isoform-dependent manner, with apoE4 inducing a significantly greater response than apoE3.These data support the idea that Aβ stimulation of glial apoE limits neuroinflammation but that overproductionof apoE by activated glia might exacerbate inflammation. In addition, the observation that apoE4 has more robustpro-inflammatory activity than apoE3 provides a mechanistic link between the APOE4 allele and AD, andsuggests potential apoE-based therapeutic strategies.
Index Entries: Alzheimer’s disease; apoE; glial activation; Aβ; cytokine.
Journal of Molecular NeuroscienceCopyright © 2004 Humana Press Inc.All rights of any nature whatsoever reserved.ISSN0895-8696/04/23:205–212/$25.00
Journal of Molecular Neuroscience 205 Volume 23, 2004
Introduction
A common feature of Alzheimer’s disease (AD)pathology is an abundance of activated glia associ-ated with amyloid plaques. Alarge number of neuro-inflammatory molecules are produced by theseactivated glia in the AD brain, including complementcomponents, acute phase proteins, pro-inflammatorycytokines and chemokines, and oxidative stress-related enzymes (Akiyama et al., 2000). Although theprecise relationship between the inflammatory
responses of activated glia and the onset or progres-sion of AD pathology remains unclear, there is com-pelling evidence that neuroinflammation is an earlyand critical event driving the disease process (Griffinet al., 1998; Van Eldik, 2001).
Neuroinflammation is attributable to dysregula-tion of glia (e.g., microglia and astrocytes), the mostabundant cell type in the brain. The normal role ofglia is to cooperate with the other major cell type,the neurons, to maintain homeostasis in the centralnervous system (CNS). However, in many diseases,
ORIGINAL ARTICLE
*Author to whom all correspondence and reprint requests should be addressed. E-mail: [email protected]

206 Guo et al.
Journal of Molecular Neuroscience Volume 23, 2004
including AD, there is an abnormally high or chronicactivation of glia, suggesting disruption of the bal-ance between pro- and anti-inflammatory processesin the brain. This overactive state of the glia causesdangerous levels of inflammatory molecules that canlead to neuron dysfunction and/or death. Neurondysfunction, in turn, can lead to further glial acti-vation, resulting in the propagation of a localized,detrimental cycle of neuroinflammation (Griffinet al., 1998). An increasing body of evidence basedon epidemiological data, feasibility studies inhumans, and investigations with preclinical animalmodels is consistent with uncontrolled neuro-inflammation and resultant neuron dysfunction asa potential contributor to disease progression in ADand other neurodegenerative disorders (Akiyamaet al., 2000). This suggests modulation of neuro-inflammation as a therapeutic approach to AD.
One potential approach to modulate neuroin-flammation is via regulation of β-amyloid (Aβ)(1–42)peptide induction of glial activation. β-Amyloid (Aβ)is a potent stimulator of glial-mediated neuro-inflammatory responses both in vitro and in vivo.Identification of endogenous CNS-derived modu-lators of Aβ-induced neuroinflammatory responsesmight yield insight into mechanisms by which thebrain maintains a normal state of inflammation andhow this process becomes dysregulated in AD. Onesuch potential endogenous mediator is apolipo-protein E (apoE). ApoE is a 34,000-molecular-weightprotein that, in the periphery, is involved in choles-terol and lipid transport and plasma lipoproteinmetabolism through the binding of apoE-contain-ing particles to a variety of low-density lipoprotein(LDL) receptor family members (for reviews, see Bef-fert et al., 1998; Mahley and Rall, 2000). In the CNS,apoE is synthesized and secreted primarily by astro-cytes and microglia, and has been postulated to beinvolved in a variety of functions, including lipidand cholesterol transport, neurite outgrowth andneuronal plasticity, and regulation of Aβ structure,clearance, or neurotoxicity (for references, see LaDuet al., 2001; Manelli et al., 2003). Human apoE existsas three isoforms, designated apoE2, apoE3, andapoE4, which are encoded from three allelic variantsof a single APOE gene located on chromosome 19.Inheritance of the APOE4 allele has been shown ina large number of studies to confer an increased rel-ative risk and an earlier age of onset of late-onsetAD (for references, see Bales et al., 2002; Manelli etal., 2003). This observation, that the apoE4 isoform
is associated with modified susceptibility to devel-opment of AD, has spurred an increased interestin the roles of apoE in the AD neuropathologicalcascade.
Much current research has centered on isoform-specific effects of apoE on neuronal function, espe-cially the ability of apoE3 to enhance neuronal repairand remodeling and protect against Aβ-inducedneurotoxicity (Mahley and Rall, 2000; Manelli et al.,2003). However, another mechanism by which apoEcould influence AD pathology and progression isthrough modulation of the neuroinflammatoryresponses of activated glia. We reported previouslythat Aβ selectively induced an increase in apoEproduction in activated glia (LaDu et al., 2000b), thatglia derived from apoE knockout mice showed morepronounced neuroinflammatory responses than gliafrom wild-type mice (LaDu et al., 2001), and thatexogenously added human apoE-containing parti-cles blocked Aβ-induced morphological activationof cultured glia (Hu et al., 1998). To extend theselatter findings, we tested the ability of exogenousapoE to modulate Aβ-induced neuroinflammatoryresponses. We report here that apoE can block selec-tive Aβ-induced neuroinflammatory responses.However, unexpectedly, apoE in the absence of Aβalso stimulated production of the pro-inflammatorycytokine interleukin-1β (IL-1β) in a concentration-and isoform-dependent manner, with apoE4 stimu-lating IL-1β at significantly lower concentrationsthan apoE3. These data suggest that apoE serves bothan anti-inflammatory and pro-inflammatory role inthe CNS, observations that have implications fordevelopment of apoE-based therapeutic approachesto AD.
Materials and MethodsMaterials
β-Amyloid (Aβ)(1–42) peptide was obtained fromAmerican Peptide (Sunnyvale, CA). The Aβ peptidewas pretreated with hexafluoroisopropanol andaged as a 100 µM stock under conditions that pro-mote formation of oligomeric Aβ (Dahlgren et al.,2002). β-Amyloid (Aβ)(1–42) peptide was used oncells at a final concentration of 10 µM. Lipopolysac-charide (LPS) from Salmonella typhimurium (Sigma,St. Louis, MO) was resuspended in sterile phosphate-buffered saline (PBS) at 100 µg/mL and was usedat a final concentration of 100 ng/mL. Apolipopro-tein E (apoE) particles were isolated from concen-

ApoE: Anti- and Pro-Inflammatory Activity 207
Journal of Molecular Neuroscience Volume 23, 2004
trated conditioned media of HEK-293 cells stablytransfected with human cDNA encoding apoE3 orapoE4 by size exclusion chromatography as describedpreviously (LaDu et al., 1994; Tokuda et al., 2000).
Glial Cultures and Cell TreatmentMixed glia (~95% astrocytes, 5% microglia) were
prepared from the cerebral cortex of neonatalSprague-Dawley rats as described previously (Huet al., 1996) and maintained in α-minimum essentialmedium (α-MEM, Invitrogen) supplemented with10% fetal bovine serum (HyClone), 2 mM glutamine,and antibiotics (100 U/mL penicillin, 100 µg/mLstreptomycin). For experiments, cells were trypsi-nized and seeded into 12-well tissue culture platesat a density of 1 × 105 cells per well. Cells were grownfor 24 h, washed twice with PBS to remove serum,and incubated in α-MEM containing N2 supple-ments (GIBCO/Life Technologies) for an additional24 h before treatment. Cells were then treatedwith either diluent or a glial activation stimulus(Aβ or LPS) in the presence or absence of apoE3or apoE4 for 12–36 h. When apoE was used, it wasadded to the cells 1 h before the activation stimulusor diluent.
Western Blot AnalysisWestern blots of cell lysates were done as described
previously (Petrova et al., 1999). The primary anti-bodies used were goat anti-COX-2 (1�250 dilution,Santa Cruz), goat anti-rat IL-1β (1�1000 dilution,R&D Systems), monoclonal anti-GFAP (glial fibril-lary acidic protein) (1�108 dilution, Sigma), mono-clonal anti-iNOS (inducible nitric oxide synthase)(1�1000 dilution, Transduction Laboratories), andrabbit anti-apoE prepared and used as described pre-viously (LaDu et al., 2000b). Secondary goat anti-mouse (1�2000 dilution), goat anti-rabbit (1�5000dilution), and rabbit anti-goat (1:5000 dilution)antibodies conjugated to horseradish peroxidase(Jackson ImmunoResearch) were used.
Data AnalysisIntensity of the protein bands detected by Western
blots was determined by using ImageQuant imageanalysis software. The difference of means amonggroups of data was analyzed with ANOVA, followedby the posttest Neuman-Keul multiple comparisontest. Statistical significance was established at a levelof p < 0.05.
Results
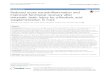
Previously, we reported that addition of exoge-nous apoE3- or apoE4-containing particles to rat glialcultures leads to an attenuation of Aβ-induced mor-phological activation, with no apoE isoform selec-tivity observed (Hu et al., 1998). To extend those initialfindings and also to determine whether exogenousapoE alone affects glial-mediated inflammation, weexamined additional glial activation responses. Ratglial cultures were stimulated with Aβ(1–42) (10 µM)in the presence or absence of exogenously addednative apoE3 or apoE4 (30 µg/mL; 0.9 µM), and thelevels of three standard neuroinflammatory pro-teins (iNOS, COX-2, and IL-1β) were measured byWestern blot analysis of cell lysates. As shown inFig. 1, apoE had selective effects on Aβ-inducedneuroinflammatory responses. ApoE3 and apoE4significantly inhibited Aβ-induced production ofiNOS (Fig. 1A) and COX-2 (Fig. 1B), and no apoE iso-form selectivity was observed. In contrast, neitherapoE3 nor apoE4 blocked Aβ-induced IL-1β pro-duction (Fig. 1C). The levels of the astrocytic protein,GFAP, which do not change upon activation of astro-cytes in culture (Hu et al., 1998), were used as a load-ing control (Fig. 1D).
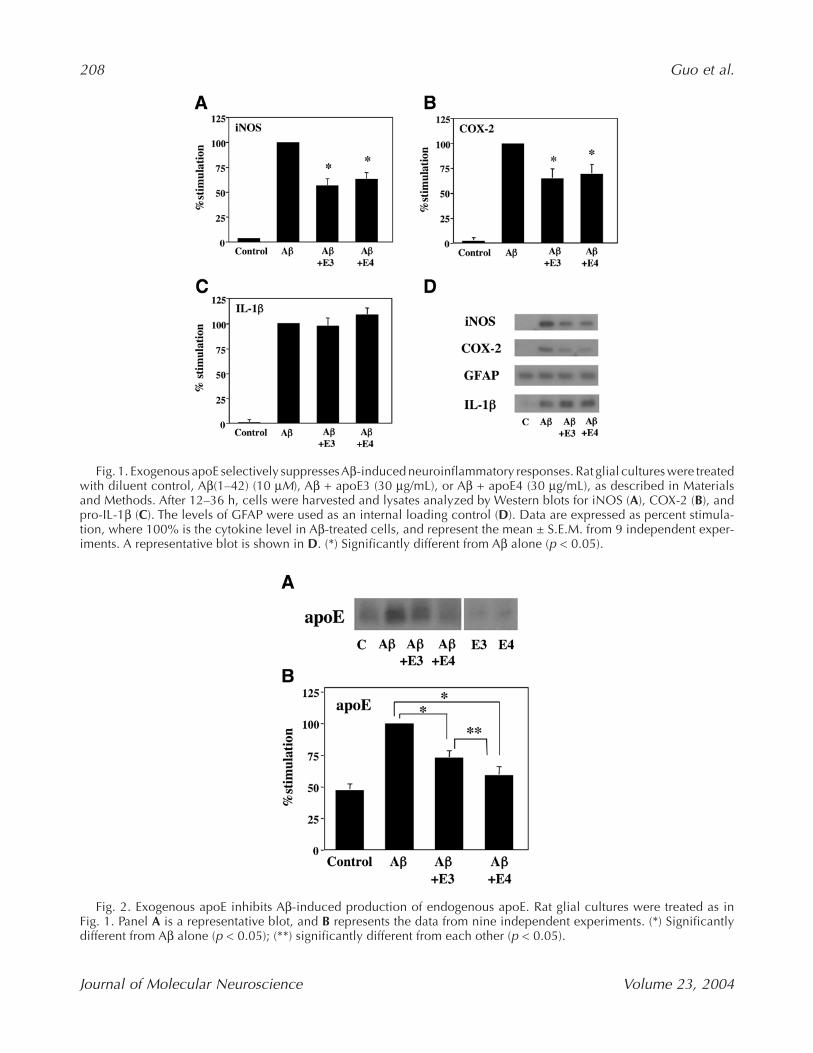
Previously, we found that Aβ(1–42) could inducea robust increase in apoE levels in glial cultures (LaDuet al., 2000b). Therefore, we examined if exogenousapoE3 and apoE4 would have an effect on the amountof endogenous apoE in response to Aβ stimulation.Interestingly, we found that treatment of glia with30 µg/mL apoE3 or apoE4 suppressed Aβ-inducedendogenous apoE levels (Fig. 2). In addition, therewas an apoE isoform selectivity, in that apoE4 wasmore effective than apoE3 in blocking Aβ-inducedendogenous apoE (Fig. 2).
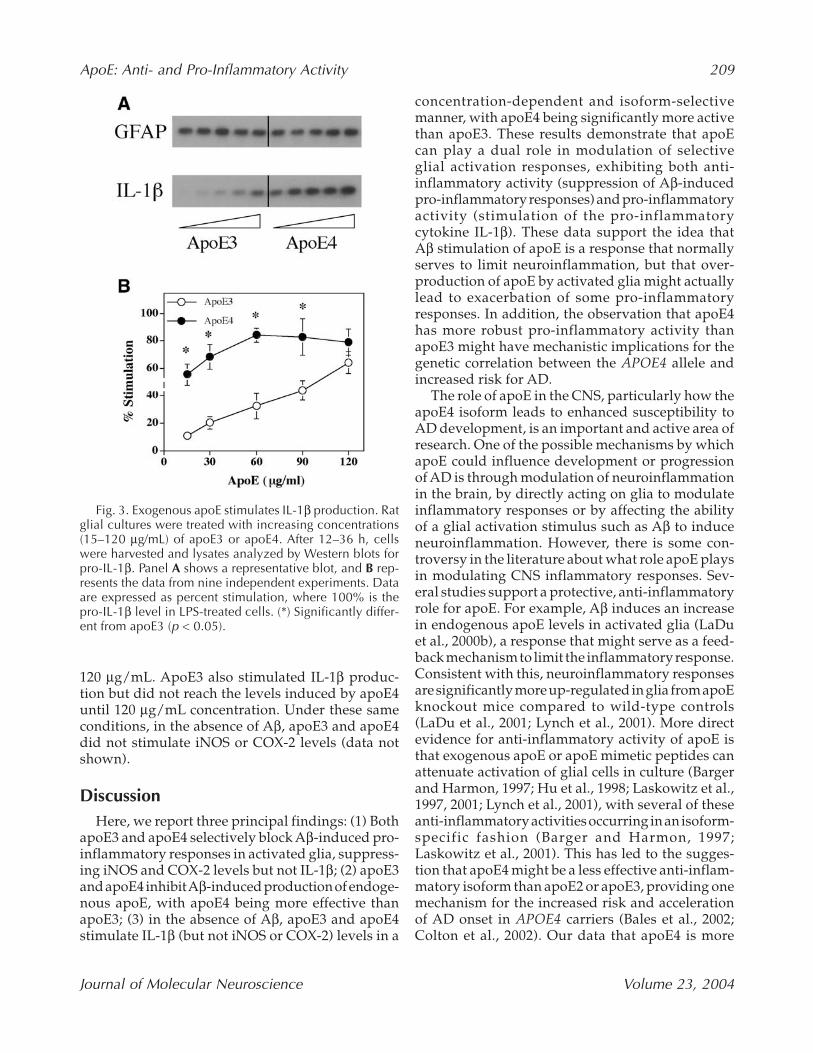
The observation that apoE could suppress Aβ-induced iNOS and COX-2 production, but notAβ-induced IL-1β levels (Fig. 1), prompted us toexamine the effects of apoE3 and apoE4 on IL-1β inthe absence of Aβ. We treated cells with increasingconcentrations (15 to 120 µg/mL) of exogenousapoE3 and apoE4, and measured the levels of IL-1βproduced. ApoE3 and apoE4 stimulated IL-1β pro-duction in a concentration-dependent manner, andapoE4 was significantly more active than apoE3 instimulating IL-1β (Fig. 3). Even at the lowest con-centration of apoE4 tested (15 µg/mL), there was arobust stimulation of IL-1β levels that reacheda plateau at 60 µg/mL and was sustained up to
208 Guo et al.
Journal of Molecular Neuroscience Volume 23, 2004
Fig. 1. Exogenous apoE selectively suppresses Aβ-induced neuroinflammatory responses. Rat glial cultures were treatedwith diluent control, Aβ(1–42) (10 µM), Aβ + apoE3 (30 µg/mL), or Aβ + apoE4 (30 µg/mL), as described in Materialsand Methods. After 12–36 h, cells were harvested and lysates analyzed by Western blots for iNOS (A), COX-2 (B), andpro-IL-1β (C). The levels of GFAP were used as an internal loading control (D). Data are expressed as percent stimula-tion, where 100% is the cytokine level in Aβ-treated cells, and represent the mean ± S.E.M. from 9 independent exper-iments. A representative blot is shown in D. (*) Significantly different from Aβ alone (p < 0.05).
Fig. 2. Exogenous apoE inhibits Aβ-induced production of endogenous apoE. Rat glial cultures were treated as inFig. 1. Panel A is a representative blot, and B represents the data from nine independent experiments. (*) Significantlydifferent from Aβ alone (p < 0.05); (**) significantly different from each other (p < 0.05).
ApoE: Anti- and Pro-Inflammatory Activity 209
Journal of Molecular Neuroscience Volume 23, 2004
120 µg/mL. ApoE3 also stimulated IL-1β produc-tion but did not reach the levels induced by apoE4until 120 µg/mL concentration. Under these sameconditions, in the absence of Aβ, apoE3 and apoE4did not stimulate iNOS or COX-2 levels (data notshown).
DiscussionHere, we report three principal findings: (1) Both
apoE3 and apoE4 selectively block Aβ-induced pro-inflammatory responses in activated glia, suppress-ing iNOS and COX-2 levels but not IL-1β; (2) apoE3and apoE4 inhibit Aβ-induced production of endoge-nous apoE, with apoE4 being more effective thanapoE3; (3) in the absence of Aβ, apoE3 and apoE4stimulate IL-1β (but not iNOS or COX-2) levels in a
concentration-dependent and isoform-selectivemanner, with apoE4 being significantly more activethan apoE3. These results demonstrate that apoEcan play a dual role in modulation of selectiveglial activation responses, exhibiting both anti-inflammatory activity (suppression of Aβ-inducedpro-inflammatory responses) and pro-inflammatoryactivity (stimulation of the pro-inflammatorycytokine IL-1β). These data support the idea thatAβ stimulation of apoE is a response that normallyserves to limit neuroinflammation, but that over-production of apoE by activated glia might actuallylead to exacerbation of some pro-inflammatoryresponses. In addition, the observation that apoE4has more robust pro-inflammatory activity thanapoE3 might have mechanistic implications for thegenetic correlation between the APOE4 allele andincreased risk for AD.
The role of apoE in the CNS, particularly how theapoE4 isoform leads to enhanced susceptibility toAD development, is an important and active area ofresearch. One of the possible mechanisms by whichapoE could influence development or progressionof AD is through modulation of neuroinflammationin the brain, by directly acting on glia to modulateinflammatory responses or by affecting the abilityof a glial activation stimulus such as Aβ to induceneuroinflammation. However, there is some con-troversy in the literature about what role apoE playsin modulating CNS inflammatory responses. Sev-eral studies support a protective, anti-inflammatoryrole for apoE. For example, Aβ induces an increasein endogenous apoE levels in activated glia (LaDuet al., 2000b), a response that might serve as a feed-back mechanism to limit the inflammatory response.Consistent with this, neuroinflammatory responsesare significantly more up-regulated in glia from apoEknockout mice compared to wild-type controls(LaDu et al., 2001; Lynch et al., 2001). More directevidence for anti-inflammatory activity of apoE isthat exogenous apoE or apoE mimetic peptides canattenuate activation of glial cells in culture (Bargerand Harmon, 1997; Hu et al., 1998; Laskowitz et al.,1997, 2001; Lynch et al., 2001), with several of theseanti-inflammatory activities occurring in an isoform-specific fashion (Barger and Harmon, 1997;Laskowitz et al., 2001). This has led to the sugges-tion that apoE4 might be a less effective anti-inflam-matory isoform than apoE2 or apoE3, providing onemechanism for the increased risk and accelerationof AD onset in APOE4 carriers (Bales et al., 2002;Colton et al., 2002). Our data that apoE4 is more
Fig. 3. Exogenous apoE stimulates IL-1β production. Ratglial cultures were treated with increasing concentrations(15–120 µg/mL) of apoE3 or apoE4. After 12–36 h, cellswere harvested and lysates analyzed by Western blots forpro-IL-1β. Panel A shows a representative blot, and B rep-resents the data from nine independent experiments. Dataare expressed as percent stimulation, where 100% is thepro-IL-1β level in LPS-treated cells. (*) Significantly differ-ent from apoE3 (p < 0.05).

210 Guo et al.
Journal of Molecular Neuroscience Volume 23, 2004
effective than apoE3 in blocking endogenous apoEproduction in activated glia support this interpre-tation. Finally, apoE could potentially serve an anti-inflammatory role through its ability to influence Aβpolymerization or clearance and thereby reduce theconcentration of this potent pro-inflammatory glialstimulus (Bales et al., 2002). All of the evidence abovepoints to an anti-inflammatory, protective role forapoE in modulation of neuroinflammation.
However, there are also a number of studies sug-gesting that apoE might be pro-inflammatory andactually contribute to AD pathogenesis. One of themore compelling observations comes from analysisof apoE knockout mice crossed with transgenic miceoverexpressing a human mutant amyloid precursorprotein (APP) gene (Bales et al., 1999; Holtzmanet al., 2000). The absence of apoE resulted in asignificant delay of both amyloid deposition andastrocyte and microglial activation. In addition,neuroinflammation was found to accelerate amyloiddeposition in APP transgenic mice, a process thatrequired apoE expression (Qiao et al., 2001). Otherstudies have also supported a pro-inflammatory,detrimental role for apoE. For example, activation ofglial cells has been postulated to enhance neurode-generation through the generation of neurotoxic apoEor apoE fragments (Clay et al., 1995; Tolar et al., 1997,1999; Moulder et al., 1999). Our findings here, thatapoE can induce production of IL-1β in the absenceof Aβ, support a pro-inflammatory potential for apoEand demonstrate that apoE4 has much more robustpro-inflammatory activity than apoE3 does.
Based on the literature overall, there does notappear to be a general consensus about the rela-tionship between glial inflammation and apoE, andwhether apoE should be thought of as an anti- orpro-inflammatory mediator in AD. We propose thatapoE has the potential to be either beneficial or detri-mental, and that proper control of apoE levels andthe interactions of apoE with Aβ are important formaintaining the appropriate balance between anti-and pro-inflammatory activities. The observationthat AD brain exhibits chronic neuroinflammationsuggests that the homeostatic mechanisms that nor-mally hold glial inflammation in check have beendisrupted or overwhelmed (Janciauskiene et al.,2002; Wyss-Coray and Mucke, 2002). It is straight-forward to envision several potential ways in whichdysregulation of apoE production or Aβ–apoE inter-actions could contribute to chronic neuroinflamma-tion. For example, overproduction of apoE inactivated glia might initially function as a natural
homeostatic modulator of Aβ inflammatory andneurotoxic activity. However, if apoE levels are nottightly regulated, there is the potential for thishomeostatic mechanism to break down, particularlywith apoE4. ApoE4 might reach levels that can stim-ulate IL-1β, accelerating the neuroinflammatorycycle (Griffin et al., 1998; Mrak and Griffin, 2001).Unregulated apoE production could also affectneuronal function if neurotoxic forms of apoE areproduced. In addition, disruption of Aβ–apoE inter-actions could affect the ability of apoE to modulateAβ aggregation, metabolism, internalization, orclearance (Hyman et al., 2000; LaDu et al., 2000a).Finally, a dysregulation of apoE receptors or recep-tor signaling mechanisms could have pleiotropiceffects on both glial and neuronal responses (Got-thardt et al., 2000; LaDu et al., 2001). These consid-erations emphasize the critical importance of futureresearch to define the mechanisms of regulation ofapoE production and its isoform-dependent inter-actions with Aβ.
In summary, apoE might act at multiple points inthe neuroinflammation/neuropathology cascadethat leads to AD, and isoform-specific activities ofapoE3 versus apoE4 at different points of the cas-cade might underlie the association of the apoE4 iso-form with enhanced susceptibility to AD. Thesefunctions of apoE relevant to AD are diagrammati-cally summarized in Fig. 4. In terms of its relation-ship to neuroinflammation, apoE4 might be both aless effective anti-inflammatory agent and a morepotent pro-inflammatory agent than apoE3. Theseobservations have implications for development ofapoE-based therapeutics for AD. For example, devel-opment of inhibitors of apoE production or activitymight be a useful strategy for individuals who carrythe APOE4 allele. In particular, small moleculeinhibitors that could specifically suppress the pro-inflammatory activity of apoE4 without affecting itsanti-inflammatory activity would be extremelyattractive. For individuals who do not carry anAPOE4 allele, development of an apoE agonist ormimetic to boost the neuroprotective effects of theapoE3 or E2 protein might also be a feasible approach.What is critically needed in the field as a foundationfor future development of any apoE-based thera-peutic approach to AD is more research on the mol-ecular mechanisms by which apoE modulates theneuroinflammatory/neuropathology cascade. Thisresearch must include elucidation of the glial sig-naling pathways induced by apoE4 vs apoE3, delin-eation of the molecular mechanisms by which apoE

ApoE: Anti- and Pro-Inflammatory Activity 211
Journal of Molecular Neuroscience Volume 23, 2004
modulates Aβ-induced glial and neuronal responses,and exploration of points of intervention in the path-ways to identify new drug discovery targets for pre-venting or treating AD.
AcknowledgmentsThese studies were supported in part by NIH
grants AG13939 (L. V. E.) and AG21184 (L. V. E. andM. J. L.). We thank Jeffrey Craft for assistance withthe statistical analyses.
ReferencesAkiyama H., Barger S., Barnu, S., Bradt B., Bauer J., Cole
G. M., et al. (2000) Inflammation and Alzheimer’s dis-ease. Neurobiol. Aging 21, 383–421.
Bales K. R., Dodart J. C., DeMattos R. B., Holtzman D. M.,and Paul S.M. (2002) Apolipoprotein E, amyloid, andAlzheimer disease. Mol. Interventions 2, 363–375.
Bales K. R., Verina T., Cummins D. J., Du Y., Dodel R. C.,Saura J., et al. (1999) Apolipoprotein E is essential foramyloid deposition in the APPV717F transgenic mousemodel of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA96, 15233–15238.
Barger S. W. and Harmon A. D. (1997) Microglial activa-tion by Alzheimer amyloid precursor protein and mod-ulation by apolipoprotein E. Nature 388, 878–881.
Beffert U., Danik M., Krzywkowski P., Ramassamy C.,Berrada F., and Poirier J. (1998) The neurobiology ofapolipoproteins and their receptors in the CNS andAlzheimer’s disease. Brain Res. Rev. 27, 119–142.
Clay M. A., Ananthraramaiah G. M., Mistry M. J.,Balasubramaniam A., and Harmony J. A. K. (1995)Localization of a domain in apolipoprotein E with bothcytostatic and cytotoxic activity. Biochemistry 34,11142–11151.
Colton C. A., Brown C. M., Czapiga M., and Vitek M. P.(2002) Apolipoprotein E allele specific regulation ofnitric oxide production. Ann. N. Y. Acad. Sci. 962,212–225.
Dahlgren K. N., Manelli A. M., Stine W. B., Baker L. K.,Krafft G. A. ,and LaDu M. J. (2002) Oligomeric and fib-rillar species of amyloid-β peptides differentially affectneuronal viability. J. Biol. Chem. 277, 32046–32053.
Gotthardt M., Trommsdorff M., Nevitt M. F., Shelton J.,Richardson J. A., Stockinger W., et al. (2000) Inter-actions of the low density lipoprotein receptor genefamily with cytosolic adaptor and scaffold proteinssuggest diverse biological functions in cellular com-munication and signal transduction. J. Biol. Chem. 275,25616–25624.
Griffin W. S. T., Sheng J. G., Royston M. C., GentlemanS. M., McKenzie J. E., Graham D. I., et al. (1998)Glial-neuronal interactions in Alzheimer’s disease: thepotential role of a “cytokine cycle” in disease progres-sion. Brain Pathol. 8, 65–72.
Fig. 4. Model depicting the potential beneficial and detrimental effects of apoE on Aβ-induced glial and neuronalresponses. β-Amyloid (Aβ)(1–42)induces an increase in apoE and pro-inflammatory molecules (e.g., iNOS, COX-2,IL-1β) in microglia/astrocytes in an apoE receptor-dependent manner. The increased levels of apoE, particularly apoE3,might have a neuroprotective role to stimulate neurite outgrowth and suppress Aβ-induced neurotoxicity, also via anapoE receptor-dependent process. Apolipoprotein E3 (apoE3) and apoE4 might also act in a beneficial manner as ahomeostatic modulator to limit the extent of the Aβ-induced glial inflammatory responses. However, dysregulation ofapoE production might have detrimental consequences. Overproduction of apoE, particularly apoE4, might result instimulation of pro-inflammatory molecules (e.g., IL-1β) and acceleration of the neuroinflammatory cycle.

212 Guo et al.
Journal of Molecular Neuroscience Volume 23, 2004
Holtzman D. M., Bales K. R., Tenkova T., Fagan A. M.,Parsadanian M., Sartorius L. J., et al. (2000) Apolipo-protein E isoform-dependent amyloid deposition andneuritic degeneration in a mouse model of Alzheimer’sdisease. Proc. Natl. Acad. Sci. USA 97, 2892–2897.
Hu J., Castets F., Guevara J. L., and Van Eldik L. J. (1996)S100β stimulates inducible nitric oxide synthase activ-ity and mRNA levels in rat cortical astrocytes. J. Biol.Chem. 271, 2543–2547.
Hu J., LaDu M. J., and Van Eldik L. J. (1998) ApolipoproteinE attenuates β-amyloid-induced astrocyte activation.J. Neurochem. 71, 1626–1634.
Hyman B. T., Strickland D., and Rebeck G. W. (2000) Roleof the low-density lipoprotein receptor-related proteinin β-amyloid metabolism and Alzheimer disease. Arch.Neurol. 57, 646–650.
Janciauskiene S., Sun Y. X., and Wright H. T. (2002) Inter-actions of Aβ with endogenous anti-inflammatoryagents: a basis for chronic neuroinflammation inAlzheimer’s disease. Neurobiol. Dis. 10, 187–200.
LaDu M. J., Falduto M. T., Manelli A. M., Reardon C. A.,Getz G. S., and Frail D. E. (1994) isoform-specific bind-ing of apolipoprotein E to β-amyloid. J. Biol. Chem. 269,23403–23406.
LaDu M.J., Reardon C., Van Eldik L. J., Fagan A. M., BuG., Holtzman D., and Getz G. S. (2000a) Lipoproteinsin the central nervous system. Ann. N. Y. Acad. Sci. 903,167–175.
LaDu M. J., Shah J. A., Reardon C. A., Getz G. S., Bu G.,Hu J., et al. (2000b) Apolipoprotein E receptors medi-ate the effects of β-amyloid on astrocyte cultures. J. Biol.Chem. 275, 33974–33980.
LaDu M. J., Shah J. A., Reardon C. A., Getz G. S., Bu G.,Hu J., et al. (2001) Apolipoprotein E and apolipoproteinE receptors modulate Aβ-induced glial neuroinflam-matory responses. Neurochem. Int. 39, 427–434.
Laskowitz D. T., Goel S., Bennett E. R., and MatthewW. D. (1997) Apolipoprotein E suppresses glial cellsecretion of TNFα. J. Neuroimmunol. 76, 70–74.
Laskowitz D. T., Thekdi A. D., Thekdi S. D., Han S. K. D.,Myers J. K., Pizzo S. V., and Bennett E. R. (2001)Downregulation of microglial activation by apolipo-protein E and apoE-mimetic peptides. Exp. Neurol.167, 74–85.
Lynch J. R., Morgan D., Mance J., Matthew W. D., andLaskowitz D. T. (2001) Apolipoprotein E modulates
glial activation and the endogenous central nervoussystem inflammatory response. J. Neuroimmunol. 114,107–113.
Mahley R. W. and Rall S. C., Jr. (2000) Apolipoprotein E:far more than a lipid transport protein. Annu. Rev.Genomics Hum. Genet. 1, 507–537.
Manelli A. M., Stine W. B., Jr., Van Eldik L. J., and LaDuM. J. (2004) ApoE and Aβ1-42 interactions: effects ofisoform and conformation on structure and function.J. Mol. Neurosci.23, 231–242.
Moulder K. L., Narita M., Chang L. K., Bu G., and JohnsonE. M., Jr. (1999) Analysis of a novel mechanism of neu-ronal toxicity produced by an apolipoprotein E-derivedpeptide. J. Neurochem. 72, 1069–1080.
Mrak R. E. and Griffin W. S. T. (2001) Interleukin-1,neuroinflammation, and Alzheimer’s disease. Neurobiol.Aging 22, 903–908.
Petrova T. V., Akama K. T., and Van Eldik L. J. (1999)Cyclopentanone prostaglandins suppress activation ofmicroglia: down-regulation of inducible nitric oxidesynthase by 15-deoxy-∆12, 14-prostaglandin J2. Proc.Natl. Acad. Sci. USA 96, 4668–4673.
Qiao X., Cummins D. J., and Paul S. M. (2001)Neuroinflammation-induced acceleration of amyloiddeposition in the APPV717F transgenic mouse. Eur.J. Neurosci. 14, 474–482.
Tokuda T., Calero M., Matsubara E., Vidal R., Kumar A.,Permanne B., et al. (2000) Lipidation of apolipoproteinE influences its isoform-specific interaction withAlzheimer ’s amyloid β peptides. Biochem. J. 348,359–365.
Tolar M., Keller J. N., Chan S., Mattson M. P., MarquesM. A., and Crutcher K. A. (1999) Truncated apolipo-protein E (ApoE) causes increased intracellular calciumand may mediate ApoE neurotoxicity. J. Neurosci. 19,7100–7110.
Tolar M., Marques M. A., Harmony J. A., and CrutcherK. A. (1997) Neurotoxicity of the 22 kDa thrombin-cleavage fragment of apolipoprotein E and relatedsynthetic peptides is receptor-mediated. J. Neurosci. 17,5678–5686.
Van Eldik L. J. (2001) Glia and Alzheimer’s disease.Neurochem. Int. 39, 329–331.
Wyss-Coray T. and Mucke L. (2002) Inflammation inneurodegenerative disease—a double-edged sword.Neuron 35, 419–432.