Embed Size (px)
Citation preview

安全管理・調査担当者必携
国内対応からグローバル対応へ
企画・編集一般財団法人 医薬品医療機器レギュラトリーサイエンス財団
PVの概要とノウハウ
安全管理・
調査担当者
一般財団法人 医薬品医療機器
レギュラトリーサイエンス財団
必携 PVの概要と
ノウハウ

序
開発から審査段階に重点が置かれていた医薬品の安全対策は,ICHにおけるE2Eガイドラインの合意を経て,2000年代初めからは,開発から審査,市販後に至るまでの医薬品のライフサイクル全般にわたるリスクマネジメント強化へと国際的に大きく舵が切られました。 わが国においても,欧米に遅れながらも RMPの考え方が導入され,徐々に定着しつつありますが,製薬企業のグローバル化に伴い,安全対策におけるわが国と欧米との規制の違いが随所で指摘されております。 本書は,1998年に初版が発行され,2008年の改訂を経て,医薬品の製造販売後安全管理や製造販売後調査担当の方々に幅広く活用されてきた「PMSの概要とノウハウ」を,その後の,RMP制度の本格的な導入や薬事法の改正等,医薬品の安全対策を巡る大きな環境の変化を取り入れるため,今回,全面的な内容の見直しを行ったものです。 特に,最新の知見や経験等をより幅広い方々に活用していただくため,2014年 5
月と 7月に開催した「2014年度 製造販売後安全管理・調査 基礎研修講座‒RMPの導入と改正 GVP/改正 GPSP等への対応‒」を基に,関係する講師の方々のご協力を得て,その内容をまとめたものです。執筆にご協力いただいた講師の方々には深くお礼申し上げます。 また,2014年 11月には医薬品医療機器等法が施行され,関連通知も多数発出されておりますので,これらについてもその内容を反映しております。 わが国をはじめ欧米各国の医薬品規制当局や製薬企業等は,医薬品等の安全対策に如何に科学性を導入するか,如何に定性的評価から定量的評価への転換を図るか,如何に効率的に実施するかに苦慮しております。本書が,このような内外の最新の動向を取り入れるための一助となることを強く期待しております。
理事長 土 井 脩
一般財団法人 医薬品医療機器レギュラトリーサイエンス財団
2015年 5月
B_i-x_PVの概要_前付-目次.indd iB_i-x_PVの概要_前付-目次.indd i 15.6.29 11:13:22 AM15.6.29 11:13:22 AM

1.2 製造販売後のファーマコビジランス 61
いて承認に係る用法・用量,効能・効果に従い,行う試験をいう(GPSP省令 第 2条第 4項)。
(d) その他の手順 このほか,製造販売後調査等に関する業務の「自己点検」,製造販売後調査等
業務に従事する者の「教育訓練」,製造販売後調査等業務の「委託」および製造販売後調査等業務に係る「記録の保存」に関する手順を定めなければならない。なお,製造販売後調査等業務の委託にあたっては GPSPにしたがって,製造販売後臨床試験業務の委託にあたっては GCPにしたがって,委託契約書を締結する。
(e) GPSP適合性調査 再審査あるいは再評価申請後に適合性調査が,申請者,受託者および製造販売
後臨床試験受託医療機関に対して行われる。 適合性評価は,「適合」,「不適合」の 2段階に分類される。不適合と判断され
た場合,当該調査などに基づき作成された資料の一部または全部について,再審査または再評価の対象から除外する措置がとられる。その結果を受けて,厚生労働省は承認の取り消し,または承認の一部変更を命ずることがある。詳細については「5.2 再審査適合性調査の実際」を参照されたい。
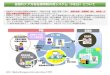
( f) RMPの導入と GVP・GPSP省令の改正 2013年 4月から導入された RMPは 2014年 10月に施行された改正 GVP・
GPSP省令により制度化された(図 1.2.9)。すなわち,GVP省令の第 2条に「医薬品リスク管理」について定義され,第 9条の 2に医薬品リスク管理計画書の策
図 1.2.9 RMPの実装と改正 GVP,GPSP省令(2014年 10月改正)
行政当局(厚生労働省,PMDA,都道府県等)
製造販売業者
医療関係者
総括製造販売責任者(総責)
品質保証責任者
製造所
安全管理責任者(安責)〈GVP〉〈GVP〉
副作用等症例報告使用上の注意改訂等+ RMP
★業許可更新時 GVP/GQP調査 【医薬品医療機器 等法 第 12条】
★再審査申請に 伴うGPSP適 合性調査
再審査・再評価申請安全性定期報告【医薬品医療機器等法 第 14条 4, 6】
GVP実施責任者(部門) GPSP実施責任者(部門)
○○本部長(製販責と同じまたは別の組織長)
〈GPSP〉〈GPSP〉〈GPSP〉製造販売後調査等管理責任者(製販責)
〈GQP〉〈GQP〉 〈GVP〉〈GQP〉
医薬品リスク管理計画
製造販売後調査等基本計画書(従来品)
連携
RMPに関する連携・報告
034-079_PVの概要_1章-2.indd 61034-079_PVの概要_1章-2.indd 61 15.6.29 11:14:36 AM15.6.29 11:14:36 AM

2.1 副作用・感染症報告,研究報告および外国措置報告 103
主治医からMRへ連絡され,その情報が安全管理部門に伝えられ,評価後,法規制に基づき PMDAへ報告されることになる。この医療関係者からの自発報告は,使用上の注意の改訂に結びつくケースが他の情報源に比べて多いことから有用な情報源と考えられる。
また,文献を通して情報を入手するケースや,医療関係者が直接,PMDAへ報告した症例を PMDAからフィードバックされるケース等がある。② 一般使用者からの自発報告
患者やその関係者から企業のお客様相談室等への問い合わせや連絡の中に安全性に関する情報が含まれているケースがある。このときに症例が validとなる以下の情報は必ず入手できるよう心がけておく必要がある。また,評価に必要な詳細情報についても,可能な限り入手するよう努める。
・ 患者識別情報(イニシャル,年齢,性別等) ・ 情報源(医師,薬剤師,消費者等) ・ 副作用・感染症名 ・ 被疑薬名③ 社内関連部門からの情報
品質保証部門に寄せられる品質クレーム情報や,広報部門に寄せられる情報の中に,安全性に関する情報が含まれている可能性があるため,これらの部門からももれなく安全性情報が入手できるようにルートを確立しておく。
④ 市販直後調査 販売開始後 6か月間,医薬品の適正使用を促し,その際に重篤な副作用の発生を
迅速に把握するために行う市販直後調査において入手した副作用も,自発報告として取り扱う。
表 2.1.15 副作用・感染症の情報源
自発的な情報源(unsolicited sources) ・ 医療関係者からの自発報告(市販直後調査からの報告も含む) ・ 一般使用者 ・ 文献 ・ 社内関連部門(品質保証部門,広報等)企業からの依頼に基づく非自発的な情報源(solicited sources) ・ 製造販売後調査,製造販売後臨床試験 ・ 臨床試験(治験) ・ 企業主導臨床研究その他 ・ 提携企業 ・ 規制当局
095-113_PVの概要_2章-1.indd 103095-113_PVの概要_2章-1.indd 103 15.6.29 11:15:32 AM15.6.29 11:15:32 AM

2.2 安全性情報の医学的評価 117
目となる。これらの項目に関する規定は 2.1節をご参照いただくことにして,本節ではとくに医学的評価の考え方が必要となる場合について簡単に説明する。
(2)個別症例評価 - 重篤性
重篤の定義は基本的に規制どおりであり,死亡・入院等の事実関係に基づく外形的評価であるが,そういった情報がない場合には,「その他の医学的に重要な状態」に該当するかどうかを企業として適切に判断しなくてはならない。この判断は,報告者により重篤とされていない有害事象についてのみ行うことで,報告要否決定のためには事足りるのであるが,理論上は全部の事象に対して行わなくてはならない。 「その他の医学的に重要な状態」か否かの評価方法としては, ① 個々の症例ごとに経過を考慮して判断 ② 事象名のみで判断という 2つの方向性がある。「常に重篤と取り扱う事象のリスト:Always Serious Term
List」を定め,そのリストに載っている事象なら重篤とする,という方式が米国系企業ではしばしばみられる方法である。一方,「医学的に重要な事象のリスト:Medically
Signifi cant List」を定め,そのリストに載っている事象については個々の症例経過を考慮して検討し,非重篤と考える根拠がない場合には重篤とする,という方式が,欧州系企業で多く用いられているようである。 リストに載せるべき事象としては,事象名自体が重症であることを示唆しているもの,例えば横紋筋融解症,劇症肝炎,中毒性表皮壊死融解症などが代表的である。 グローバル企業では,リストの記載事象名を英語にせざるをえないのだが,その場合は日本語で入手した事象名のMedDRA用語による英訳に注意が必要である。例えば「痙攣」はMedDRA では PT 10010904 Convulsion であり,通常リストに載っており重篤となる。しかし,日本語ではてんかんの大発作にみられるような代表的な強直性間代性痙攣でなくても,物をとろうとしたときに手がゆらゆらと震える,顔や指がピクピクと引きつる,何かの拍子に腕全体がピクンと動く,こむら返りのようにふくらはぎがつる,などいろいろな状態が「けいれん」と呼ばれる。これらは英語なら tremor,myoclonus,twitch,clampなどの多彩な表現になり,必ずしも全部が重篤ではないのだが,convulsion と訳せば重篤となってしまうようだ。
(3)個別症例評価 - 予測性
予測性の根拠は添付文書になるが,そこに記載されていても,副作用の性質・症状・特異性などが記載内容と一致しないものは「予測できない」とするのがルールである。しかし,これはなかなかに難しい医学的判断である。この点を考慮したていねいな予測性の評価は容易ではない。適切に運用されていることを正当に抗弁しうる企業が現在どの程度あるのかは不明である。
114-134_PVの概要_2章-2.indd 117114-134_PVの概要_2章-2.indd 117 15.6.29 11:15:55 AM15.6.29 11:15:55 AM

3.1 医薬品リスク管理計画の現状と対策 195
らの判断と方針で創意工夫して J‒RMPを策定する。逆にこの機会を利用して,製薬企業は J‒RMPの公開情報を通じて医療関係者や薬剤師の協力を得ることができる。ベネフィットがリスクを上回ることを継続的かつ定期的に確認する「育薬」の活動は,患者にとってもメリットがある。このように医療関係者と患者をまき込んでともに「育薬」活動を行うことができれば,製薬企業は製品のライフサイクルの延長とポテンシャルの最大化も実現できる。
3.1.9 RMP重要なリスクへの対策
RMPで把握された重要なリスク(「安全性検討事項」)に対応するために,どのような考え方で対策を立てるべきかそのコンセプトを表 3.1.2に示した。RMPの中で追加の措置が必要になった場合は,以下のような対策が求められる。
(1)「重要な特定されたリスク」への対応
「重要な特定されたリスク」は,すでに医薬品との関連性がわかっているので,リスク最小化計画に基づく追加の安全対策が必要になる場合がある。患者にとって,製品のベネフィットが該当する重要なリスクを上回るようにするために創意工夫して,添付文書の改訂だけに頼らない「追加のリスク最小化策」を講じる必要がある。
(2)「重要な潜在的リスク」の対応
「重要な潜在的リスク」の対応は,関連性が疑われるが十分に薬剤との因果関係が確認されていない「グレー」な重要なリスクの状態から,そのリスクが「白」か「黒」かの結論を出すために「追加の安全性監視計画」を実施する場合がある。従前の使用成績調査 3,000例で課題が解決するのかを検討する必要がある。適切なツールや方法をリス
表 3.1.2 RMP重要なリスクへの対応方針
安全性検討事項
安全性監視計画 リスク最小化計画
通常 追加 通常 追加
重要な特定されたリスク ●●
重要な潜在的リスク ●●
重要な不足情報 ○ ○
有効性 ●●
179-198_PVの概要_3章-1.indd 195179-198_PVの概要_3章-1.indd 195 15.6.29 11:17:36 AM15.6.29 11:17:36 AM

364 6 ファーマコビジランスの国際動向と契約
タベースである EudraVigilanceを使って EMAにより監視されている。 個別症例レベルの活動で日本と大きく違うのは報告される情報の範囲であり,
表 6.2.1で示すように,EUでは以下にあげた使用時の重篤副作用および非重篤副作用を,それぞれ 15暦日および 90暦日で報告することが要求されている。
・ 承認範囲内での使用(表 6.2.1で示す Special situationを含む) ・ 過量投与,off‒label使用,誤用,濫用,医療過誤など,承認範囲外での使用 ・ 職業的曝露 また,個別症例や定期報告に該当せず,これまで当局報告が曖昧だった文献や基
礎・臨床試験から得られる情報を,Emerging Safety Issue(新規安全性情報)と位置付け,当局報告を必須とするとともに,シグナル検出での活用が求められている。これは日本における研究報告や海外措置報告に該当する。
② 集積情報レベル 上記に沿って収集された個別症例の安全性情報が集積されたデータベースや,
Emerging Safety Issueなどを使って,ベネフィット‒リスクバランスが評価されるが,その機会は以下の 3つに分類される。
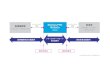
図 6.2.1 市販後の医薬品安全性監視システムの日本と EUの比較
RMP(最新化される)
0年 5年 10年
安全性定期報告・PBRER
製造販売後調査(調査・試験)
市販直後調査(6か月)副作用・感染症報告/研究報告/外国措置報告
添付文書,患者向医薬品ガイド
RMP(最新化される)
PBRER ACO*1
PASS/PAES
PBRERRenewal Renewal
PBRERACO
PASS/PAES
Additional monitoring
副作用報告(Special situation report,Emerging Safety Issue含む)
SmPC*2,PIL*3
措置報報措
ng Safet
Renewal
tnal situa
Renewal
ata
日本
承認
EU
承認
再審査
申請
再審査
*1 Addendum to clinical overview, *2 Summary of Product Characteristics, *3 Patient Information Leaflet
注)EUと日本では,市販後の医薬品安全性監視に供せられる,ツール(RMP,添付文書,定期報告など),活動(副作用報告,臨床研究など)や,市販後の情報に基づき再度評価されるシステムなど似ている点が多い。
362-379_PVの概要_6章-2.indd 364362-379_PVの概要_6章-2.indd 364 15.6.29 11:23:18 AM15.6.29 11:23:18 AM