Embed Size (px)
Citation preview

Indian Journal of Biochemistry & Biophysics Vol. 36, April 1999, pp. 101-106
A geometry optimization and molecular electrostatic potential mapping study of structure-activity relationship for some anti-Alzheimer agents
P S Kushwaha, M K Shukla and P C Mishra* Department of Physics, Banaras Hindu University, Varanasi 221 005 (India)
Received 25 September 1998; accepted 4 January 1999
Molecular geometries of some substituted (pyrroloamlno)pyridines which possess anti-Alzheimer activity were optimized and potential-derived CHelpG point charges were computed using ab initio SCF molecular orbital approach employing the 3-21 G basis set. AM I molecular orbital calculations were performed using these optimized geometries and thus optimized Hybridization Displacement Charges (HOC) combined with Lo wdin charges continuously di stributed in three dimension were obtained. Molecular electrostatic potential (MEP) maps of the molecules were obtained in two ways : (i) using the HOC-based model with the help of which MEP minima near the molecules were located, and (ii) using the CHelpG point charges, MEP values on the van der Waals surfaces of the molecules were computed . The MEP maps computed using both the methods have negative MEP regions near the pyridine nitrogen atom which appears to be the. main binding si te of the molecules with the appropriate receptor. Both electrostatic interaction and lipophilic association between these molecules and the receptor appear to contribute to biological activity.
Alzheimer's disease (AD) is related to age, occurring in middle or late life of humans and leads to dramatic personality changes including loss of cognition ' .2 . AD is a disease of the central nervous system and is caused by breakdown of the central cholinergic transmission. It can be treated with cholinomimetic agents which induce release of acetylcholine, inhibit hydrolysis of acetylcholine or act directly at cholinergic receptors3
,4. Thus AD can be controlled using muscarinic agonists which act directly at postsynaptic receptors5
. Several classes of molecules have been studied in this context4
-6
• Aminopyridines are known to induce increased release of actylchol ine7
. Davis et al. have synthesised several (pyrroloamino )pyridine analogs with lipophilic substituents and evaluated their activity against AD6.
Electrostatic interactions of drug molecules with receptors usually play important role with regard to their action. Such interactions are usually studied employing molecular electrostatic potential (MEP) or molecular elec.tric field (MEF) mapping usin~ point charge distributions e .g. Mulliken charges8
- ' . It is well known that these charge distributions are not accurate enough to yield reliable results as they do not even preserve molecular dipole moments obtained at the SCF level. However, when hybridization displacement charges (HOC) introduced recently by
*Corresponding author. E-mail : [email protected]
USl3 are combined with Lb wdin charges, molecu lar
dipole moments computed at the SCF level are fully preserved. A parametrically optimized approach to compute HDC has recently been developed l4
-17 and it
has been shown that one can obtain very reliable MEP patterns using these charges combined with Lbwdin charges obtained from AM I or MNDO semiempirical molecular orbital calculations. Wadsworth el a/.5
studied MEP maps of certain anti-Alzheimer agents which were derivatives of oxadiazole, triazol e and tetrazole and they found electrostatic interactions to be important for the binding of these molecules with the receptor. It is desirable to extend such a study to different types of anti-Alzheimer agents in order to examine the generality of importance of electrostatic interactions in this context. The molecules known as (pyrroloamino )pyridines synthesized and evaluated against AD by Davis el a/.6 appear to be suitable for such a study. We have investigated the structures and MEP maps of this class of molecules here .
Computational Method Ground state geometries of the molecu les were
optimized at the restricted Hartree-Fock selfconsistent field ab initio level of theory employing the 3-21 G basis set and the Window version of the Gaussian 94 program 18 running on a Pentium 200 MHz machine. Molecular e' ~trostatic potential (MEP)-derived charges were "btained using the

102 INDIAN J BIOCHEM. BIOPHYS, VOL. 36, APRlL 1999
CHelpG procedure l9. Optimized hybridization
displacement charges (HDC) were obtained using the ab initio optimized geometries and the AM I molecular orbital method20. MEP maps were obtained using the HDC model as explained below 13.17.
The hybridization dipole moment along the xdirection arising due to the mixing of 2s and 2px atomic orbitals of an atom can be written as
llx = OxQI . (1)
where QI represents the (2s,2px) density matrix element for the atom under consideration and Dx is the displacement of the electronic charge QI along the x-direction from the atom under consideration. Similar expressions can be written for the components lly and llz of hybridization dipole moment. Now, we define a distance R from the atom under consideration as
R = (Ox2 + 0/ + Oz2)112 .. . (2)
The total hybridization dipole moment
llh= (11/ + 11/' + 11/ )112 ... (3)
can be thought to arise from a charge, called hybridization displacement charge (HDC), placedl at a distance R from the atom under consideration. The magnitude of HDC is now obtained as
Q = llJR. = [(QI2 + Q/ + Q/ )/3 ]112 ... (4)
The equation (J), considering all the threr
components llx, lly and llz of llh can also be written as
lli = (KOi) (Q/K) . . . (5)
= D;' Q/ (i= x,y,z and j =.1 ,~,3 respectively)
where K is a constant (parameter). By varying K, the distance D;' and charge Q/ can be changed. Further, the charge Q can be distributed spherically symmetrically and continuously according to the form of square of the valence Slaters type of atomic orbital (STO) of the atom under consideration. The exponents (l;;) of the STO's for HDC and K defined above for different atoms have been adjusted so as to reproduce ab initio MEP features of molecules as closely as possible (Table 1). Further details of the method of calculations are available in the literature 13.17.
Results and Discussion The general molecular structure of (Pyrrolo
amino)pyridines is presented in Fig. L Here X represents the substituent attached to the C2 or C3 position of the pyrrole ring. The different substituents
Table I - Optimized values of the parameter K and Slater exponents (I;) for hybridization displacement charge calculations for different non-hydrogen atoms under the AMI method
Atom
C 1'1 (NH,NH2)
1'1 (Pyridine) N (HCN,NNO) o (CO) o (NNO,N02)
o (OH) F CI S (SH) S (Ring) S (CS)
K
0.3 0.1
0.85 0.45 0.8 0.8
0.25 0.7
0.83 0.12 0. 12 0.86
1;'
1.625 2.360 1.480 1.600 1.580 1.100 2.470 1.550 1.750 1.250 1.180 1.280
" Zeta for HDC under the AM I method.
I;b
1.625 1.950 1.950 1.950 2.275 2.275 2.275 2.600 2.033 1.817 1.817 1.817
b Atomic zeta values obtained from the Slater's rules were used for the charges located on the atomic sites.
Fig. I - The general molecular structure of (pyrroloamino) pyridines and ab initio optimized bond lengths (A) and bond angles (deg.) of pyrroloaminopyridine. [R and X are specified in Table 2].
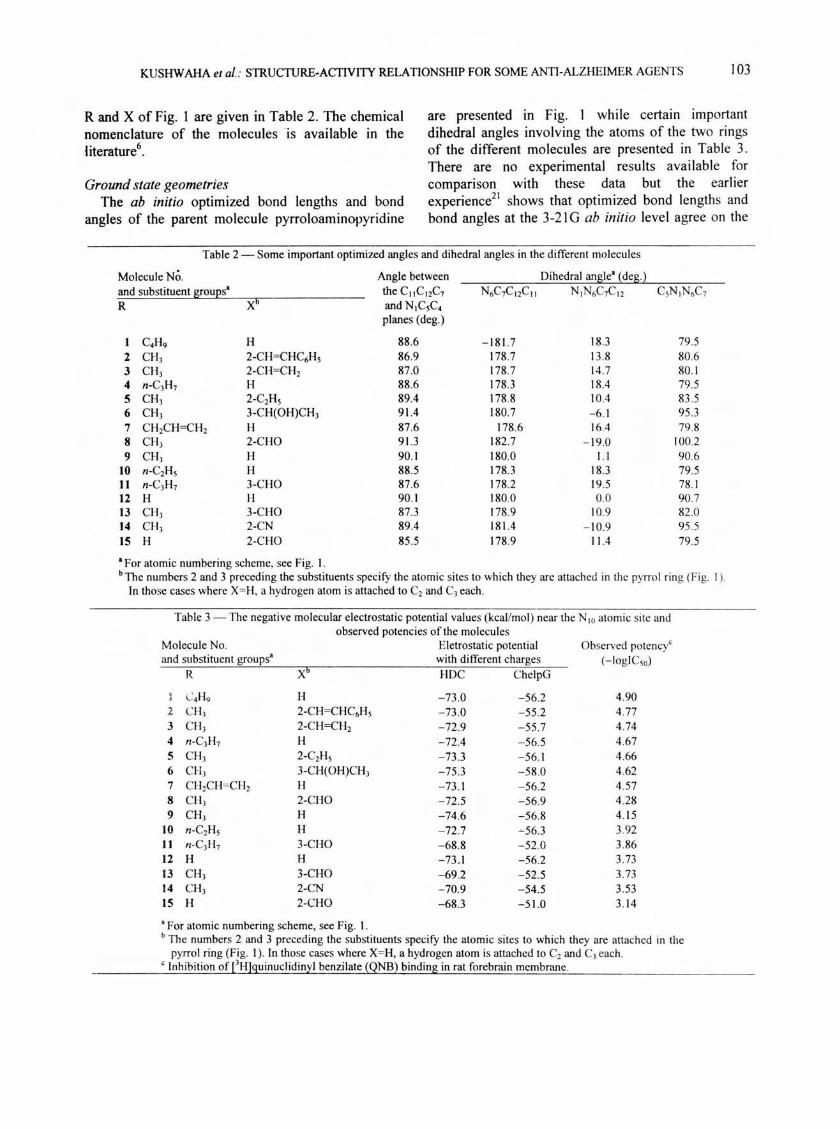
KUSHWAHA et at.: STRUCTURE-ACfIVITY RELATIONSHIP FOR SOME ANTI-ALZHEIMER AGENTS 103
R and X of Fig. 1 are given in Table 2. The chemical nomenclature of the molecules 'is available in the literature6
.
Ground state geometries
are presented in Fig. I while certain important dihedral angles involving the atoms of the two rings of the different molecules are presented in Table 3. There are no experimental results available for comparison with these data but the earlier experience21 shows that optimized bond lengths and bond angles at the 3-21 G ab initio level agree on the
The ab initio optimized bond lengths and bond angles of the parent molecule pyrroloaminopyridine
Table 2 - Some important optimized angles and dihedral angles in the different molecules
Molecule No. Angle between Dihedral angle- (deg.)
and substituent S!:ouQs' the C"C12C7 N6C7C'2C , , N,N6C7C'2 CsN,N6C7
R X6 and N,CSC4
planes (deg.)
C4H9 H 88.6 -181.7 18.3 79.5 2 CH3 2-CH=CHC6HS 86.9 178.7 13.8 80.6 3 CH3 2-CH=CH2 87.0 178.7 14.7 80.1 4 n-C3H7 H 88.6 178.3 18.4 79 .5 5 CH3 2-C2HS 89.4 178.8 10.4 83 .5 6 CH3 3-CH(OH)CH3 91.4 180.7 - 6.1 95 .3 7 CH2CH=CH2 H 87.6 178.6 16.4 79.8 8 CH3 2-CHO 91.3 182.7 - 19.0 100.2 9 CH3 H 90.1 180.0 1.1 90.6
10 n-C2Hs H 88.5 178.3 18.3 79.5 11 n-C3H7 3-CHO 87.6 178.2 19.5 78 .1 12 H H 90.1 180.0 0.0 90.7 13 CH 3 3-CHO 87.3 178.9 10.9 82 .0 14 CH3 2-CN 89.4 181.4 - 10.9 95 .5 15 H 2-CHO 85.5 178.9 11.4 79.5
• For atomic numbering scheme, see Fig. I . bThe numbers 2 and 3 preceding the substituents specify the atomic sites to which they are attached in the pyrrol ring (Fig. I ).
In those cases where X=H, a hydrogen atom is attached to C2 and C3 each.
Table 3 - The negative molecular electrostatic potential values (kcal/mol) near the N ' 0 atomic site and observed potencies of the molecules
Molecule No. Eletrostatic potential Observed potency< and substituent groups' with different charges ( -logICso)
R X6 HDC CtielpG
L4H9 H - 73.0 - 56.2 4.90 .2 CH3 2-CH=CHC6HS -73.0 -55.2 4.77 3 CH3 2-CH=CH2 -72.9 - 55.7 4.74 4 n-C3H7 H -72.4 - 56.5 4.67 5 CH3 2-C2HS - 73 .3 - 56.1 4.66 6 CI-l) 3-CH(OH)CH3 - 75 .3 - 58.0 4.62 7 CH2CH=CH2 H -73. 1 - 56.2 4.57 8 CH) 2-CHO - 72 .5 -56.9 4.28 9 CH3 H -74.6 -56.8 4.15
10 n-C2Hs H - 72.7 - 56.3 3.92 II n-C3H7 3-CHO - 68 .8 - 52.0 3.86 12 H H -73 .1 -56.2 3. 73 13 CH3 3-CHO -69.2 -52.5 3.73 14 CH3 2-CN -70.9 - 54.5 3.53 15 H 2-CHO -68.3 -51.0 3. 14
• For atomic numbering scheme, see Fig. I . b The numbers 2 and 3 preceding the substituenls specify the atomic sites to which they are attached in the
pyrrol ring (Fig. 1). In those cases where X=H, a hydrogen atom is attached to C2 and C) each. e Inhibition of [3H]quinuclidinyl benzilate (QNB) binding in rat forebrain membrane.

104 INDlAN J BIOCHEM BIOPHYS, VOL. 36, APRIL 1999
average with experiment within about 0.0 I A and I deg. respectively, and the same is expected to be true in the present case also. The pyridine and pyrro le rings of the molecules were found separately to be planar but the two ring planes were found to be very much off from each other . The dihedral angle between the planes CIICI2C7 and N IC5C4 representing the planes of the pyridine and pyrrol e rings respective ly, in the parent molecule 12, was found to be 90 . 1 deg. (Tab le 2). Thus the two (pyrrole and pyridine) rings are almost perpendicular to each other in the parent molecule 12. For the other molecules which are derivatives of 12, the corresponding dihedral ang les were found to lie between 85.5 to 91.4 deg. T hus the different substitutions (Tab)e 2) on Iy have mild effects on the relative ori entations of the two rings.
When the alkyl chain length at R changes from methy l (mo lecule 9) to ethyl, propy l or butyl (molecules 10, 4 or 1 respectively), X remaining the same (H), the dihedra l ang le N I 6C7CI2 increases from about 1. 1 deg. to about 18 deg. in a ll the ca ses. As Table 2 shows, the overall shapes of the molecules 12 and 9 are sim ilar. Thus the substitution of a C H; group at R fo r a H atom does not change shape of the molecu le noticeably . The sums of the optimized bond angles N IN6R, C7N6R and N IN6C7 in the molecules 12 and 9 were found to be 360 deg. whi le the corresponding sums changed to around 355 deg. in the molecules 10, 4 and 1 where the chain at R is longer than the methyl group. This shows that the nitrogen atom N6 acquires a pyramidal character when the substituent R is larger than a methyl group.
A comparison of the opti mi zed va lues of the dihedral angle NIN6C7CI2 in the molecules 5, 8 and 14 where R=CH) and · X are different Crsubstituents (Table 2), reveals that substitutions at the Cr position in the pyrrole ring also cause significant change in the ori entation of pyrrole ring with respect to that o f the pyridine ring. A comparison of the values of the same dihedral angle in the molecules 6 and 13 in both of which R=CH) and X are two different substituents shows that relative orientations of the two rings are sens itive to substitution at the C)-position also. Since the Crsubstituents would not have significant stearic interactions with the substituents represented by R, the sensitivity of the relative orientations of the two rings to substitutions at the C3-position shows that the relative orientations of the rings are controlled by
electronic factors also besides the possi ble stearic interactions which would be particularly significant between certain Crsubstituents and R (Fig. I).
Molecular electrostatic potential (MEP) maps
The HOC charges combined with Lb wdin charges computed us ing the optimized molecular geometries and the AM I method2o
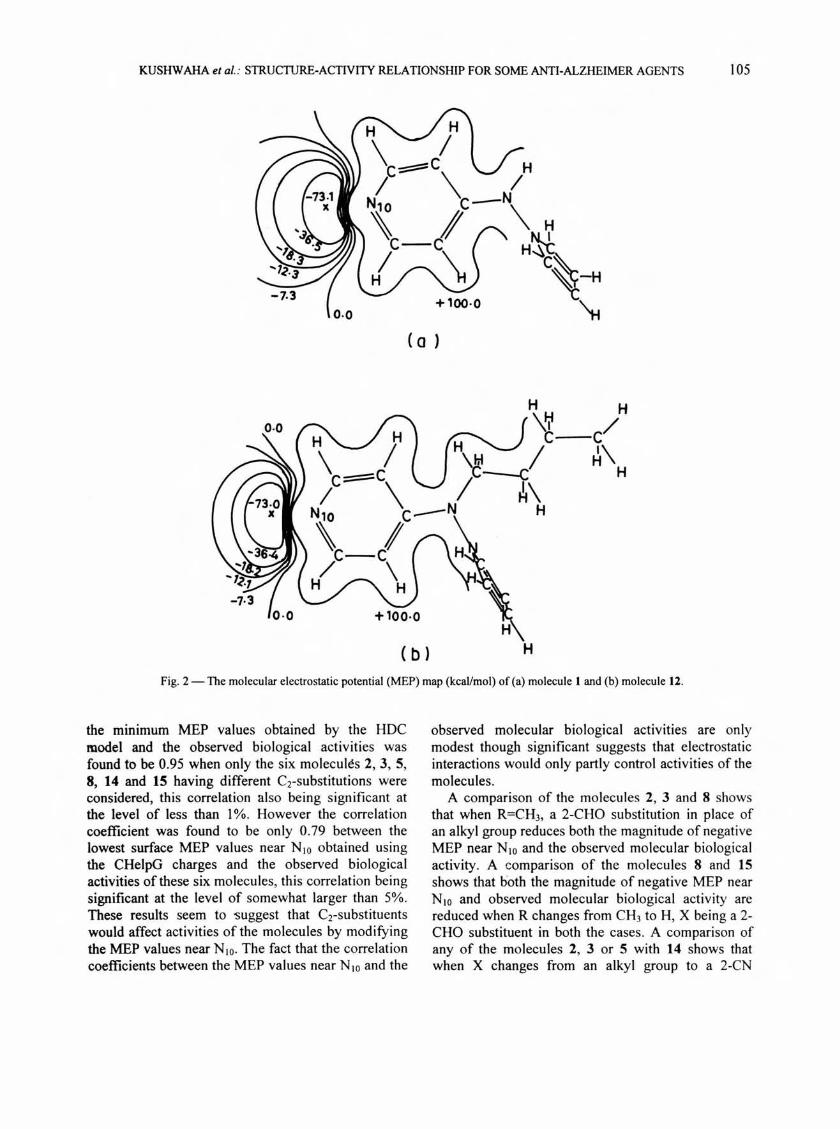
, d~stributed continuously in three dimension, were used to compute isopotential contour MEP maps. The MEP map of each molecule computed in thi s way shows MEP minimum near the pyridine nitrogen atom (N 10) (Fig. I ). The minimum MEP values near N IO in th e different cases are presented in Table 3 along with the observed bio logica l act iv iti es of the molecules. The MEP va lues near the same si te on the van der Waals surfaces of the mo lecules obtained us ing the ab initio C HelpG po int charges are also presented in Table 3. The computed contour MEP maps fo r two molecules i.e . molecules 1 and 12 obtained us ing the HOC model , as an example, are presented in Fig. 2. There are two types of interactions between biomolecules or drugs and the ir receptors wh ich usua lly playa crucial ro le 111 biolog ical activity22.23 : ( i) Electrostatic interaction and (ii) Hydrophobic or lipophilic aSSOCiation . The e lectric polarization energy is approximately p;~~ortional to th~ e lectros~atic 1I1teractlon energy--· . . Hydrogen bonding II1teractlons
. I fl· ) 4 '5 F I are promll1ent y 0 e ect rostatlc type- .- . urt ler, out of the two types of interact ion energies given above, the former is usually of greater importance but the role of the la tter canilOt be neglected. While electrostatic interactions are quantifiable in terms of the correspond i ng M EP va I ues, there is no potentia l fUllction that can be used to describe hydrophobic or !ipophilic association . Study of linear correlation coefficients between M EP values and biological activities of molecules forms the com mon approach to eva luate the importance of electrostatic interacti ons.
The linear correlation coefficient between the MEP minima obtained using the HDC model and the observed biological activi ties of the mo lecu les was found to be 0.69 when all the molecules were considered . The corresponding val ue of the linear correlation coefficient was found to be 0.67 between the surface MEP values near N 10 and the observed biological activities of the molecules. BJth the above correlation coefficients are s ign i ficant at the level of less than I % according to the t-test26 and hence they are quite reliable. The correlation coefficient between
KUSHWAHA et al.: STRUCTURE-ACTIVITY RELATIONSHIP FOR SOME ANTI-ALZHEIMER AGENTS 105
(0 )
( b ) H
Fig. 2 - The molecular electrostatic potential (MEP) map (kcaVmol) of (a) molecule I and (b) molecule 12.
the mlntmum MEP values obtained by the HOC model and the observed biological activities was found to be 0.95 when only the six molecules 2, 3, 5, 8, 14 and 15 having different Crsubstitutions were considered, this correlation also being significant at the level of less than 1 %. However the correlation coefficient was found to be only 0.79 between the lowest surface MEP values near N 10 obtained using the CHelpG charges and the observed biological activities of these six molecules, this correlation being significant at the level of somewhat larger than 5%. These results seem to 'Suggest that C2-substituents would affect activities of the molecules by modifying the MEP values near N 10. The fact that the correlation coefficients between the MEP values near N 10 and the
observed molecular biological actIvItIes are only modest though significant suggests that electrostatic interactions would only partly control activities of the molecules.
A comparison of the molecules 2, 3 and 8 shows that when R=CH), a 2-CHO substitution in place of an alkyl group reduces both the magnitude of negative MEP near N 10 and the observed molecular biological activity. A comparison of the molecules 8 and 15 shows that Doth the magnitude of negative MEP near N 10 and observed molecular biological activity are reduced when R changes from CH) to H, X being a 2-CHO substituent in both the cases. A comparison of any of the molecules 2, 3 or 5 with 14 shows that when X changes from an alkyl group to a 2-CN

106 INDIAN J BIOCHEM BIOPHYS, VOL. 36, APRIL 1999
substituent, both the magnitude of negative MEP near NIO and observed molecular biological activity are reduced substantially. Thus on the whole, when R is an alkyl group and the 2-substituent is also an alkyl group, its derivative, a hydrogen atom or a partly conjugated moiety but not a CHO or CN group, the magnitude of negative MEP near N 10 and observed molecular biological activity are both high . These results strongly suggest that the N 10 site of the molecules would be involved in electrostatic interaction e.g. hydrogen bonding with the appropriate receptor. In the earlier studl also., MEP minima near ring nitrogen atoms beside certain oxygen atoms were considered to be important in controlling activities of the molecules .
A consideration of biological activities of the molecules and nature and size of the substituents R and X suggests that there may be a lipophilic pocket on the receptor such that a lipophilic group R may fit or associate with the same, thereby enhancing molecular activity appreciably. It appears that a C4H9
group at R may have the most appropriate size among those of the different substituents (R) considered here to fit with the lipophilic pocket of the receptor. Thus on the whole, biological activities of the molecules appear to be partly controlled by electrostatic rnteraction and partly by lipophilic assoc iation between these molecules and the receptor.
Conclusion The present study leads us to the conclusion that
biological activities of the molecules under study are controlled partly by both of electrostatic interaction and lipophilic association between each of the molecules and the corresponding receptor. The s ite of the molecules which would be involved in hydrogen bonding with the receptor would be the pyridine nitrogen atom while the lipophilic association would involve the appropriate substituents (R) at the nitrogen atom which bridges the two rings, the planes of which are nearly perpendicular to each other.
Acknowledgement The authors are thankful to CSIR (New Delhi) and
UGC (New Delhi) for financial support. PSK thanks the Banaras Hindu University for a research fellowship.
References I Yan S D, Che[l X, Fu J, Chen M, Zhu H, Roher A, Slattery T,
Zhao L, Nagashima M, Morser J, Migheli A, Naworth P, Stern D & Schmidt A M (1996), Nature, London 382, 685-691.
2 Schnabel J (1993), New scientist 138, 22-26. 3 Garvey D S, Wasicak J T, Chung J Y -L, Shue Y -K, Carrera
G M, May P D, McKil\ney M M, Anderson D, Cadman E, Vella-Rountree L, Nadzan A M & Williams M, (1992), J Med Chem,35, 1550-1557.
4 Klein J T, Davi s L, Olsen G E, Wong G S, Huger F P, Smith
C P, Petko W W, Comfeldt M, Wilker J C, Blitzer R D. Landau E, Haroutunian V, Martin L L & Eflland R C ( 1996). J Med Chem, 39, 570-581.
5 Wadsworth H J, Jenkins S M, Orlek B S, Cassidy F, C lark M S G, Brown F, Riley G J, Graves D, Hawkin s J & Naylor C B (1992) , J Med Chern, 35, 1280-1290.
6 Davis L, Ol sen G E, Klein J T, Kapples K J. Huger F P. Smi th C P, Petko W W, Comfeldt M & Emand R C (1996), J Med Chern, 39, 582-587.
7 TheslelT S (1980), Ne uroscience 5, 1413-1419. 8 Scrocco E & Tomasi J (1978), Adv Quantum Chem, II. 11 5. 9 Po litzer P & Murray J S (1991) in Reviews of Computational
Chemistry (K B Lipkowitz and D B Boyd, eds) , Vol 2, Chap 7,VCH, New York.
10 Shukla M K & Mi shra P C (1995), J Mol Strllct (Theochem). 340, 159-167.
II Mi shra P C & Kumar A (1995), in Moiecli lar Similarity /I (Topics in Current Chemistry) (K D Sen, ed) Vol. 174. pp 27. Springer-Verlag, Heidelberg.
12 Mishra P C & Kumar A (1996), in Moieclilar Electrostatic Potentials : Concepts and Applications (J S Murray & K D Sen, ed), pp 257, Elsevier, Amsterdam.
13 Kumar A, Mohan C G & Mi shra P C ( 1995), Int J Quant Chern , 55, 53-60.
14 Mohan C G, Kumar A & Mishra P C (1996), Int J QlIant Chem, 60, 699-708 .
15 Mohan C G, Kumar A & Mi shra P C ( 1997), Int J QlIant Chem, 62, 67-76.
16 Santhosh C & Mishra P C ( 1996), Indian J Biochem Biophys. 33,458-464
17 Mohan C G & Mishra P C ( 1998), In! J Quant Chem. 66. 149-156.
18 Fri sch M J, Trucks G W, Schl egel H B, Gill P M W, Johnson B G, Robb M A, Cheeseman J R. Keith T, Peterson G A. Montgomery J A. Raghavachari K, AI-Laham M A, Zakrzewski V G, Ortz J V, Foresman J B, C ioslowski J, Stefanov B B, Nanayakkara A. Chall acombe M, Peng C Y, Ayala P Y, Chen W, Wong M W. Andres J L, Repl ogle E S. Gomperts R, Martin R L, Fox D J, Defrees D J, Baker J, Stewart J J P, Head-Gordon M, Gonza lez C & Pople J A, (1995) , Gaussian 94, Revision B I (Gaussian Inc, Pittsburgh.
PAl · 19 I3rencman C M & Wiberg K B (1990). J Comp Chelll. II ,
361. 20 Dewar M J S, Zoebisch E G, Hea ly E F & Stewart J J P
(1985) , J Am Chern Soc, 107, 3902-3909. 2: Shukla M K & Mi shra P C (1 998), Chem Phys , 230, 187-200 22 Naray-Szabo G & Ferenczy G G ( 1995), Chem Rev. 95 . 829-
847 . 23 Naray-Szabo G (1989), J Mol Graph , 7. 76. 24 Kumar A. Mohan C G & Mishra P C (1996), J Mol Struct
(rheochem), 361 , 135-144. 25 Nair A C & Mi shra P C (1996) J Mol Strllct (Theochem) . 364.
209-217 . 26 Downie. Heath N M (1970) Basic Statistical Methods R W 3rd
Edition (Harper & Row) pp 230, New York.





























