Embed Size (px)
Citation preview

Mutation Research, 286 (1993) 189-197 189 © 1993 Elsevier Science Publishers B.V. All rights reserved 0027-5107/93/$06.00
MUT 05218
A proflavin-induced frameshift hotspot in the thymidylate synthase gene of bacteriophage T4
Michael D. Brown *, Lynn S. Ripley a and Dwight H. Hall School of Biology, Georgia Institute of Technology, Atlanta, GA 30332-0230, USA and a Department of Microbiology and Molecular Genetics, University of Medicine and Dentistry of New Jersey Medical School, Newark, NJ 07103, USA
(Received 6 July 1992) (Revision recived 24 August 1992)
(Accepted 11 September 1992)
Keywords: Hotspot, mutagenic; Proflavin; Topoisomerase II; Bacteriophage T4; Thymidylate synthase gene
Summary
Twenty-one independent thymidylate synthase deficient ( td) mutants were isolated after proflavin mutagenesis of T4Do phage. A strikingly high proportion of these mutations (17 of 21; 80%) mapped in a small 122 nucleotide (nt) region which spans the 5' splice site of this intron-containing gene. This region comprises only 14% of the total td exon sequence. RNA sequence analysis of these mutants identified a series of frameshift insertion/deletion mutations and indicated a hotspot for proflavin-in- duced mutations in the 3' end of exon I of the td gene. The mutant sequences at the hotspot site fully support a previously proposed mutagenic mechanism for proflavin-induced mutations in which frameshifts are produced as a consequence of exonuclease or DNA polymerase activity at the 3' ends of nicks in the DNA produced by perturbation of the T4-encoded type II topoisomerase activity by the acridine. Sixteen of the seventeen DNA mutations in the hotspot region can be explained by the model as a consequence of enzymatic processing of nicks at two phosphodiester bonds staggered by 4 base pairs (bp) and located on opposite strands of the DNA. Thus, these mutants exhibit precisely the symmetry expected of topoisomerase-mediated mutagenesis. The DNA sequences of the td hotspot mutants, when considered with the sequences of proflavin-induced mutants in the T4 r l I B and lysozyme genes, confirm the view that proflavin-induced mutations in diverse bacteriophage T4 DNA sequences are all produced by the topoisomerase-dependent mechanisms and do not support the view that classical misalignments in DNA repeats are hotspots for proflavin-induced mutagenesis in T4.
Correspondence: Dr. Dwight H. Hall, School of Biology, Georgia Institute of Technology, Atlanta, GA 30332-0230, USA. Tel. 404-894-3702; Fax 404-853-0048.
* Present address: Department of Genetics and Molecular Medicine, Emory University School of Medicine, Atlanta, GA 30322, USA.
The general mutagenicity of proflavin (3,6-di- aminoacridine) was initially observed in bacterio- phage T2 (DeMars, 1953). Subsequently, a variety of acridines have been shown to induce frameshift mutations in bacteriophage T4 as well as in T2 (Orgel and Brenner, 1961; Lerman, 1964). Early models to explain acridine-induced frameshifts

190
attempted to link what was known of the speci- ficity of frameshift mutation (Streisinger et al., 1966) to the binding of the acridine mutagens to the DNA (Lerman, 1961) which was character- ized by "weak-binding" to the exterior of the DNA helix and by "strong-binding" through in- tercalation between DNA base pairs. A model (Brenner et al., 1961) in which intercalation be- tween adjacent bases in the DNA template led to miscopying errors by the polymerase was gener- ally unsupported by several in vivo experiments (Drake, 1970). However, in vitro assays later demonstrated that the presence of acridines dur- ing DNA polymerization can promote errors (Shearman and Loeb, 1983; Conrad and Topal, 1986; Revich and Ripley, 1990). Another early model (Lerman, 1963), proposed that acridines induce unequal crossing over by promoting mis- takes in homologous pairing. Moreover, acridine treatments in T4 lead to an excess of low molecu- lar weight intracellular DNA (Altman and Ler- man, 1970), a situation that might well favor enhanced recombination. However, at present no strong evidence exists for an obligate role for recombination in the mutational process induced by proflavin in the bacteriophage T4 system. Proflavin-induced T4 frameshifts, studied by Strigini (1965), were not found to be preferen- tially associated with recombinational heterozy- gotes. In bacteriophage lambda, the association of acridine-induced frameshifts with monotonic base pair runs is consistent with models for mis- aligned pairing in these sequences (Skopek and Hutchinson, 1984), but no studies have identified the molecular events responsible. Indeed, in E. coli lacI sequences, strong sequence-specific ef- fects suggest that the presence of a run of identi- cal base-pairs is not sufficient to define an acri- dine-induced frameshift hotspot (Gordon et al., 1991).
Sequencing of much larger numbers of proflavin-induced mutations have recently pro- vided critical information necessary to explain the novel mutagenic molecular events involved in acridine-induced mutation in bacteriophage T4. Initial observations of the unusual symmetry of the DNA sequence changes found in an acridine-induced mutational hotspot in the r l IB gene implicated mutagenic activity within the
hotspot at two separate phosphodiester bonds on opposite DNA strands staggered by four bp (Rip- ley and Clark, 1986). Because nicking of opposite DNA strands four bp apart is characteristic of prokaryotic type II topoisomerases such as E. coli DNA gyrase (Morrison and Cozzarelli, 1979), the role of the T4-encoded type II topoisomerase in the mutagenic process was examined (Ripley et al., 1988). These studies demonstrated a require- ment for active T4-encoded topoisomerase for in vivo mutagenesis and revealed a sequence-depen- dent correlation between acridine-induced DNA cleavage measured in vitro and the specificity of frameshift mutagenesis in ~'ivo. This correlation suggested that topoisomerase-mediated DNA cleavage was a required step whose sequence-de- pendence directed the specificity of the acridine- induced duplications which are produced by the templated addition of nucleotides by DNA poly- merase at the 3'-OH end produced in the cleav- age reaction, followed by joining to the original 5' end (Ripley et al., 1988). Similarly, the less fre- quent acridine-induced deletions could be ac- counted for by the action of a 3' exonuclease at the same 3'-OH end and rejoining of the short- ened strand to the original 5' end. A prediction of this model for acridine-induced, topoiso- merase-mediated frameshift mutagenesis is that DNA sequence changes that produce altered cut- ting by the topoisomerase will alter mutagenesis. This prediction has been confirmed as nucleotide changes in the T4 r l IB gene that altered specific sites of topoisomerase cutting in vitro also al- tered the specific sites of proflavin mutagenesis (Masurekar et al., 1991).
In this study, we extend our examination of proflavin-induced mutational specificity to the td gene of bacteriophage T4. We demonstrate that the most frequent proflavin-induced mutations in the entire td gene of bacteriophage T4 are frameshifts which occur at a hotspot site whose characteristics implicate the same topoisomer- ase-mediated frameshift mutagenic mechanism.
Materials and methods
Media Broth medium contained 10 g Bacto-tryptone,
5 g NaCI and distilled-deionized water (d2H20)

to one liter. Broth agar plates included 12 g Bacto agar per liter. Broth top agar included 7 g Bacto agar per liter. Luria Broth Medium (LB) contained 10 g NaC1, 10 g Bacto-tryptone, 5 g yeast extract, 1 g glucose and dEH20 to one liter. The pH of the medium was adjusted to 7.4 with 1 N NaOH prior to sterilization. LB agar plates contained 12 g Bacto agar. LB top agar contained 7 g Bacto agar. M9 medium contained 1 g NH4C1, 3 g KH2PO 4, 5.8 g NaEHPO 4, 0.5 g NaC1, 1 mM MgESO4, 10 ml of 50% dextrose and d2H20 to 1 liter. GPTG medium contained 5.8 g NaC1, 3.7 g KC1, 0.11 g CaCI2, 0.1 g MgCl 2 .6H20 1.1 g NH4C1, 12.1 g Trizma (Tris) base (pH 7.4), 0.14 g NaESO4, 0.10 g B-glycerophosphoric acid, 10 ml of 50% glucose (w/v) solution, 10 ml vitamin-free casein hydrolysate (Nutritional Biochemical Com- pany), and dEH20 to one liter. GPTG agar plates included 12 g Bacto agar and 20 /xg/ml L-tryp- tophan. Diluting fluid (DF) contained 5 g NaCl, 1 g Bacto-tryptone and dEH20 to one liter.
Bacterial strains, phage and plasmids E. coli B or BB cells were used for phage
crosses and the preparation of phage lysates. Genetic selection and screening for T4 td and dihydrofolate reductase (frd) mutations were performed by plating presumptive mutants on E. coli GM201 or E. coli OK305 cells. E. coli GM201 (thyAdeoB) is a low thymine-requiring derivative of E. coli B (Lomax and Greenberg, 1968). E. coli OK305 is a uracil-requiring E. coli B mutant that has little cytidine deaminase activity and no detectable deoxycytidine deaminase activity (Hall and Tessman, 1966; Karlstr6m, 1968).
E. coli lambda lysogen RRI (Ac+), a recA + derivative of HB101, was used as host cell for a collection of plasmids carrying T4 td gene inserts that were used in marker rescue experiments (Belfort et al., 1986; Hall et al., 1987). These td subclones were constructed in Dr. M. Belfort's laboratory by subcloning restriction endonuclease fragments from a 2.85 kb T4 EcoRI fragment containing the entire td gene into the plasmid vector pKC30 (Hall et al., 1987).
Bacteriophage T4Do, a standard T4 phage type (Hall and Tessman, 1966), was used for proflavin mutagenesis. The T4Do genotype does not influence the td or frd assays.
191
Restrictive plating conditions for td and frd mu- tants
T4 mutants defective in thymidylate synthase production (td mutants) or dihydrofolate reduc- tase (frd mutants) were screened for by assaying phage for the "white halo" phenotype (Hall, Tessman and Karlstr6m, 1967). Phage were plated with 2 x 108 E. coli OK305 cells on GPTG + cytidine (CR) plates (GPTG plates with 20 mg/1 CR added to the agar before plate pouring) sup- plemented with 4/zl of CR (10 mg/ml) added to the top agar. Plates were incubated at 37°C for 10-12 h. Under these conditions, T4 td and frd mutants show a strong "white halo" around the plaque.
T4 td mutants were identified by small plaque size on agar plates where thymidine was limiting (Lomax and Greenberg, 1968). Phage were plated with 2 x 108 E. coli GM201 cells on GPTG plates supplemented with thymidine (TdR) by adding 4 tzg of TdR (2/xl of a 2 mg/ml stock solution) to the top agar prior to plating. The plates were incubated at 37°C for 10-12 h. T4 td mutants form very small plaques under this plating condi- tion and are easily distinguished from td + phage including frd mutants. Alternatively, approxi- mately 30 /~l of phage suspension were spotted on the hardened top agar and incubated as above. This spot test allowed a number of phage to be screened for a td phenotype simultaneously.
To discern which phage producing a white halo phenotype were frd mutants, 2-4 x 108 E. coli OK305 cells, 14 /zg trimethoprim, 800 /.~g pyrimethamine, and 4/zg CR were added to 2.5 ml GPTG top agar and poured onto a GPTG + CR agar plate. Approximately 30 ~l of phage dilution were spotted onto the agar plate with a capillary tube. Plates were incubated at 37°C for 10-14 h or until a visible cell lawn formed. T4 frd mutants form no plaques under these conditions, while frd +, including td mutants, form plaques (Johnson and Hall, 1973).
Proflavin mutagenesis Proflavin mutagenesis of phage T4 was as de-
scribed by Brenner, Benzer and Barnett (1958) with minor modification. To 10 ml of E. coli B cells (approximately 2 × 109 cells), L-tryptophan was added to a final concentration of 20/zg/ml,

192
proflavin to a final concentration of 4 Izg/ml and 2 x 108 T4Do phage. The mutagenesis lysate was aerated during a 10 minute 37°C incubation at which time chloramphenicol (CP) was added to a final concentration of 20 ~ g / m l . After a one hour 37°C incubation, the lysate was diluted 100- fold in M9 media and one drop of the dilution was added to each of 42 tubes containing one ml of M9 medium. Following a one hour 37°C incu- bation, the one ml lysates were chloroformed and 0.2 ml plated with E. coli 0K305 cells on G P T G + CR agar plates. From these plates, only one "white halo" (wh) plaque was picked from each of tubes 1-40 to obtain independent mutants. From tubes 1, 2 and 5, three wh plaques were picked and designated by the mutagenesis tube number and "A", "B" or "C" respectively. An overall frequency of 0.7% wh plaques was ob- served after proflavin mutagenesis. The fre- quency of spontaneous (non-induced) wh plaque formation obtained by plating T4Do on E. coli OK305 cells was less than 0.02%. A total of 29 independent proflavin-induced wh mutants are discussed in this report.
T4 phage crosses Phage crosses were performed with the two T4
phage being crossed at a titer of 2 × 109 P F U / m l , usually by dilution of overnight liquid lysates which contain approximately 5 x 109 P F U / m l (Hall, Tessman and Karlstr6m, 1967). One drop of each phage from a 1 ml glass pipet was added to a large test tube followed by one drop of broth liquid medium and one drop of fresh E. coli B or BB cells at 2 -5 x 108 cells/ml. The mixture was incubated for 90 min at 37°C in a shaking water bath followed by the addition of 3 drops of chlo- roform and 9.9 ml of diluting fluid. Dilutions of the cross were further diluted and spotted or plated under the appropriate conditions depend- ing on the phage being crossed. Two types of control crosses were performed with each set of crosses: (1) phage to be experimentally crossed were crossed to themselves (negative control), and (2) known td marker phage were crossed with each other (positive control).
Marker rescue The recombination-based marker rescue tech-
nique described by Mattson et al. (1977) and used
by Hall et al. (1987) was used to map putative td mutations. Briefly, marker rescue [ysates for td marker rescue were made by infecting a series of E. co# cells containing plasmid td subclones with a td mutant phage. The td gene restriction frag- ments used in plasmid subclone construction are shown in Fig. 2. Marker rescue lysates were di- luted 100-fold and spotted under plating condi- tions which restrict the growth of td mutants. The appearance of wild-type (td +) phage in the spots after 37°C overnight incubation was indica- tive of positive marker rescue recombination. By analyzing which of the library of td subclones a particular td mutant phage showed positive marker rescue with, td mutations were mapped to a particular restriction fragment which defined the td subclone inserts. If a mutation maps near an end of a particular cloned restriction frag- ment, a lower number of td + recombinants is observed due to the decreased amount of se- quence homology.
RNA preparation from T4-infected cells T4 infected E. coli BB cells were prepared
according to the method of Belfort et al. (1985). E. coli BB cells from a fresh overnight culture grown in LB medium were diluted 100-fold into LB liquid medium (0.3 ml overnight culture into 30 ml LB) and grown at 30°C in a shaking water bath until cell concentration reached 2 × 108 cells/ml. Cell concentration was determined by microscopic analysis using a Petroff-Hauser cell counter. Cells to be infected were then trans- ferred to a 50 ml Oak Ridge centrifuge tube and centrifuged at 10,000 rpm for 10 min at 4°C. The cell pellet was resuspended in 0.5 ml LB followed by the addition of the infecting phage and place- ment of the tubes on ice for 5 min. Ceils were infected with a phage multiplicity of infection of 7. The cel l /phage mixture was added to 30 ml of pre-warmed (30°C) LB and incubated at 30°C for 7 min in a shaking water bath. Following infec- tion, 1.2 ml of chloramphenicol (CP) at 2.5 m g /m l were added and the cells were held at 30°C for 1 min. The infected cells were cooled rapidly in an ice-water bath until 4°C and then transferred to chilled centrifuge tubes and centrifuged at 10,000 rpm for 10 min at 4°C. The cell pellet was resus- pended in 1 ml of 10 mM Tris (pH 7.5)-CP (100
~g/ml) solution without vortexing. The infected cells were transferred to a 1.5 ml Eppendorf microfuge tube and centrifuged at 4°C for 2 min. The cell pellet was resuspended with vortexing in 0.5 ml of Tris-CP solution. Following a 2 min centrifugation, the cell pellets were quick-frozen in a CO2-EtOH bath and either used immediately or stored at -80°C for up to a month.
RNA extraction and sequencing RNA isolation from T4-infected E. coli BB
cells was performed as described by Belfort et al. (1985). Total RNA extraction was initiated after a seven minute infection to maximize the amount of td mRNA isolated. RNA preparations were stored at -80°C or used directly for sequencing.
Dideoxy chain-terminating RNA sequence analysis was performed as previously described (Belfort et al., 1985). 20 pmoles of gamma-32p - dATP 5'-end labelled (as described by Wallace et
193
al., 1981) synthetic oligonucleotide were used in the annealing reaction. Sequencing reactions were loaded on to an 8% polyacrylamide-7 M urea sequencing gel, dried, and visualized by autora- diography.
Results
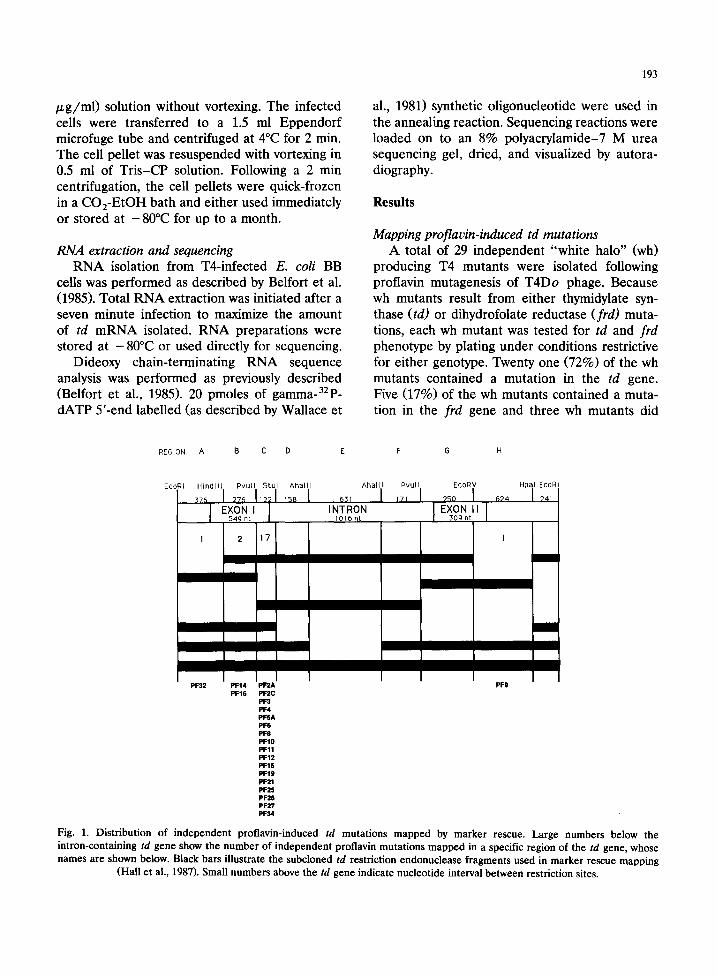
Mapping proflavin-induced td mutations A total of 29 independent "white halo" (wh)
producing T4 mutants were isolated following proflavin mutagenesis of T4Do phage. Because wh mutants result from either thymidylate syn- thase (td) or dihydrofolate reductase (frd) muta- tions, each wh mutant was tested for td and frd phenotype by plating under conditions restrictive for either genotype. Twenty one (72%) of the wh mutants contained a mutation in the td gene. Five (17%) of the wh mutants contained a muta- tion in the frd gene and three wh mutants did
REGION: A B C D E F G H
EcoRI H i n d l l l Pvuf l S tu l A h a l l l A h a l l l Pvu l l EcoRV Hp~ EcoRI
5 4 9 n t 1016 n t 3 0 9 n t
1 2 17 I
m
/
PF32 PF14 PF2A PF9 PF16 PF2C
pF3 PF4 PFSA PF6 PF8 PF10 PF11 PF12 PF15 PF19 PF21 PF25 PF26 PF27 PF34
Fig. 1. Distribution of independent proflavin-induced td mutations mapped by marker rescue. Large numbers below the intron-containing td gene show the number of independent proflavin mutations mapped in a specific region of the td gene, whose names are shown below. Black bars illustrate the subcloned td restriction endonuclease fragments used in marker rescue mapping
(Hall et al., 1987). Small numbers above the td gene indicate nucleotide interval between restriction sites.

194
A 1 ) DNA FOLYMERASE AND RELIGATION - DUPLICATION
I OH 2) 3' EXONUCLEASE AND RELIGATION - DELETION !
B 4 5 0 4 S 4
446 459 ' 5'- A A T A T A T G G ~ C A T T ~ T A A A T A T A T G G C A T T A C C G C C T T G T C • I1! i
TTATA ~"TACC GTAAT -5'
I J HO I 1 ) DNA POLYMERASE AND RELIGATION - DUPLICATION I I • DUPLICATION
Z ) 3' EXONUCLEASE AND REUGATION. DELETION [ ] DELETION
Fig. 2. Proflavin-induced, topoisomerase-mediated frameshift hotspot in the phage T4 td gene. (a) Model showing topoisomerase nicking at phosphodiester bonds 450-451 and 454-455 (opposite D N A strands) of td exon I sequence produces 3 ' -OH termini. Fur ther processing by D N A polymerase or 3 '-exonuclease and religation to the original 5 ' -PO 4 termini can generate template-di- rected duplications or deletions, respectively. Bold bases show four base pair stagger characteristic of acridine-induced, topoiso- merase-mediated frameshift mutan t sequences. (b) Clustering of the 16 proflavin-induced hotspot mutat ions around phosphodi- ester bonds 450-451 and 454-455 of td exon I. The above model therefore accounts for all mutant sequences at the td gene
hotspot.
not show either a strong frd or td mutant pheno- type.
The 21 td mutations were mapped in the in- tron-containing td gene by marker rescue using a series of six plasmid td subclones containing over- lapping restriction endonuclease fragments and encompassing the entire td gene. All 21 muta- tions mapped in just four regions of the td gene (Fig. 1). A very high proportion (17 of 21) of the mutations mapped in the 122 nt region C of the td gene defined by a PuulI-StuI restriction frag- ment. This restriction fragment encompasses the
3' end of exon I and the first 10 nucleotides of the intron and represents only 7% of the entire td gene and only 14% of the polypeptide coding region (exon I and exon II). The four remaining td mutations are dispersed in three different seg- ments of the gene.
The distribution of the mutations mapping in region C was measured by crossing the mutants to each other. Pair-wise crosses between each of 12 mutants and the remaining 11 produced no wild type recombinants except in the case of PF19. In fact, PF19 showed substantial recombi-
TABLE 1
S E Q U E N C E CHANGES OF P R O F L A V I N (PF)- INDUCED td M U T A T I O N S
PF-induced td mutants Sequence changes a Mutat ion
448 PF2C, PF3, PF4, PF6, PF8, PF10, T A T A T G G --* T A T A A T G G A duplication
PF12, PF15, PF25
444 PF2A, PF5A, PF21, PF26 T A A A T A T A T G G C ~ T A A A T A T A T A T G G C AT duplication
450 PF27, PF34 T A T G G C A T ~ T A T G G G C A T G duplication
452 PFI 1 T G G C A T T A C C G C C T T G T C ~ T G G G T C 12 base pair deletion
475 PF19 T F C T A T C --* T TCATC T deletion
a Number above first nucleotide in sequence corresponds to td gene exon I nucleotide. All mutagenic events, except for PF19, could occur between td exon I bases 449-450 or 453-454. Underl ine shows range of nucleotides where dupl icat ions/delet ions occurred.

195
nation with all 16 of the other region C proflavin-induced mutants. Because the failure to detect wild type recombinants in this system indi- cates that the mutations are probably less than 12 bp apart, these recombination results suggest that the proflavin-induced mutations, with the excep- tion of PF19, are very tightly clustered in td region C. Crosses between PF19 and PFl l yielded fewer recombinants than other crosses, suggest- ing that PFl l was distinct from most of the mutants in the cluster.
Phage crosses between the td region C proflavin-induced mutants and previously se- quenced td mutants isolated by nitrous acid or hydroxylamine mutagenesis (Brown et al., 1992) were used to more precisely localize the position of the clustered mutations. Subsets of the mu- tants were crossed to tdN57 (a C to T change at nt 559), tdH6 (a G to A change at nt 548), tdN43 (an amber mutation at nt 514), tdN54 (an amber mutation at nt 518), and tdH12 (a G to A change at nt 467). Extensive recombination was found for all the markers except tdH12. Reduced recombi- nation was detected in crosses between tdH12 and PF2A, PF2C, PF3, PF4, PF6, PF8, PF15 and PF25. A further reduction in recombination was detected in crosses between tdH12 and PF19, PF27 and PF34 and no recombinants were de- tected with PFl l . These results confirmed the somewhat unusual nature of PF19 and PFl l ob- served when the proflavin-induced mutants were crossed to each other and suggested that the majority of mutants lie within exon I, closer to nt 467 (tdH12) than to the other downstream mark- ers tested.
Sequences of proflavin-induced td mutations The sequence change in each of the 17 clus-
tered proflavin-induced td mutants was deter- mined by dideoxy sequencing of phage RNA. The sequencing results (Table 1) revealed that the td phenotypes were likely due to exon I frameshifts, as expected from the proflavin treatment. All of the mutations, with the exception of PF19, are located physically close to each other. Thus, the mutant sequences are fully consistent with the mapping data. All added bases are duplications and duplications sharply outnumber deletions. The hotspot for proflavin-induced mutations is
located 96-100 bp upstream from the 3' end of exon I (td exon I bp 450-454). The PF19 muta- tion is located approximately 24 bp downstream from the hotspot region. Both the deletion size (12 bp) and the position of PF l l are consistent with the slightly reduced recombination frequen- cies observed with this marker in crosses with PF19.
Discussion
A collection of 29 white halo (wh) producing phage induced by proflavin mutagenesis was screened to determine if the white halo pheno- type was due to a td or frd mutation. A high number (21 of 29, or 72%) of the white halo mutants proved to be the result of a td mutation. Marker rescue mapping of the mutations re- vealed that most of these (17 of 21) were con- tained in region C of the td gene defined by a 122 nucleotide PvulI-StuI restriction fragment en- compassing the 3' end of td exon I (Fig. 1). Within this fragment, phage crosses indicated that all but one of the mutations were tightly clustered and were quite close to the previously-sequenced tdH12 mutation at nt 467.
RNA sequence analysis verified the mapping and phage cross results and unambiguously iden- tified a proflavin mutagenic hotspot at nucleotide positions 450-451 and 454-455 in the td exon I sequence. Only PF19 was located outside the hotspot, 24 bases downstream at nucleotide 477. As expected from the frameshift mutagen proflavin, all the sequence alterations within the hotspot are duplications or deletions. The most frequent (9 of 16) mutation within the hotspot was a duplication of an A at position 451. Four mutants were AT duplications beginning at this same site. The three remaining mutations at the hotspot lie 4 bp away: the duplications of G at 454 and the deletion of 12 bp from 455 to 466. Extensive nitrous acid and hydroxylamine muta- genesis of the td gene has not identified a similar mutagenic hotspot (Brown et al., 1992).
Only 6 of the 16 hotspot mutant sequences could potentially be accounted for by classical slipped pairing or DNA misalignment models. The four AT duplication mutations at 451-452 could be accounted for by misaligned pairing

196
within the initial A T A T A T (AT) 3 sequence, leading to (AT)4; and the two G duplication mutations at 454 could be the consequence of slipped pairing in the initial GG sequence leading to GGG. The slipped pairing model cannot ex- plain the most frequent mutations, the duplica- tion of A at 451, nor the 12 bp deletion.
In contrast, an alternative frameshift model previously proposed to explain proflavin and m- AMSA induced mutation in the r l I B gene of T4 can explain all 16 of the hotspot mutants and account for their positions relative to one another (Ripley et al., 1988). The A and AT duplications at 451 and 452 are precisely 4 bp upstream from the G duplication and 12 bp deletion. The dupli- cations are just like those shown previously to coincide with the sites where acridines have been directly demonstrated to induce cutting of the DNA by the T4-encoded type II topoisomerase. The enzyme produces cuts at sites that are 4 bp apart on opposite DNA strands, and thus ac- counts for the symmetrical arrangement of mu- tant sequences around the broken phosphodi- ester bonds (Fig. 2a). Interestingly, there are no obvious similarities between the topoisomerase I1 target D N A sequences at the hotspots in the T4 r l I B , lysozyme and td genes.
All of the 16 hotspot td sequences are consis- tent with a single frameshifting mechanism (Fig. 2b). Combined consideration of DNA sequences of the td gene and site-specific mutation frequen- cies of proflavin-induced frameshifts strongly sup- ports the view that proflavin-induced frameshifts are not well explained by slipped pairing, even though some of the mutations in the hotspot coincided with DNA sequences in which slipped- pairing might have occurred. There are much more impressive repeats in other portions of the gene. For example, after proflavin treatment, only 1 mutant was located in the first 160 bp of exon I. However, this region contains one 5-bp long A : T run and two 4-bp long A : T runs. In the next 276 bp of the exon, only two mutants were found, yet this region contains one 6-bp long A : T run, one 5-bp long A : T run and six 4-bp long A : T runs. Indeed, 6-bp long runs of A : T pairs are such excellent targets for spontaneous mutation in T4 (Pribnow et al., 1981; Streisinger and Owen, 1985) that it would be expected that this site might well
be the site of "spontaneous" background mutants that might occur in the sample of mutants se- lected after proflavin treatment. The failure of coincidence between proflavin-induced mutations and monotonic A : T bp runs in T4 supports and extends studies in a proflavin mutation hotspot in the T4 r l I B gene (Masurekar et al., 1991). To- gether, the results firmly argue that despite occa- sional coincidence of A : T runs within the hotspot sites, the repeated nature of the bases in a run is not a crucial factor in defining mutagenic sites.
Acknowledgments
The authors would like to thank Dr. Marlene Belfort for oligonucleotides and helpful advice. This work was supported by National Institutes of Health grants GM16306 and GM36714 to D.H.H and by American Cancer Society grant CN-50E to L.S.R.
References
Altman, S. and L.S. Lerman (1970) Effects of 9-aminoacridine on bacteriophage T4 deoxyribonucleic acid synthesis, J. Mol. Biol., 50, 263-277.
Belfort, M., J. Pedersen-Lane, K. Ehrenman, F. Chu, G.F. Maley, F. Maley, D.S. McPheeters and L. Gold (1986) RNA splicing and in vivo expression of the intron-contain- ing td gene of bacteriophage T4, Gene, 41, 93-102.
Belfort, M., J. Pedersen-Lane, D. West, K. Ehrenman, G. Maley, F. Chu and F. Maley (1985) Processing of the intron-containing thymidylate synthase (td) gene of phage T4 is at the RNA level, Cell, 41,375-382.
Brenner, S., L. Barnett, F.H.C. Crick and A. Orgel (1961) The theory of mutagenesis, J. Mol. Biol., 3, 121-124.
Brenner, S., S. Benzer and L. Barnett (1958) Distribution of proflavin-induced mutations in the genetic fine structure, Nature, 182, 983-985.
Brown, M.D., C.M. Povinelli and D.H. Hall (1992) Distribu- tion of nitrous acid and hydroxylamine-induced mutations in the intron-containing thymidylate synthase (td) gene of bacteriophage T4, Genetics, submitted.
Conrad, M. and M.D. Topal (1986) Induction of deletion and insertion mutations by 9-aminoacridine. An in vitro model, J. Biol. Chem., 261, 16226-16232.
DeMars, R.I. (1953) Chemical mutagenesis in bacteriophage T2, Nature, 72, 964.
Drake, J.W. (1970) The molecular basis of mutation. Holden- Day, San Francisco, CA. pg 123-145.
Gordon, A.J.E., J.A. Halliday, M.J. Horsfall and B.W. Glick- man (1991) Spontaneous and 9-aminoacridine-induced frameshift mutagenesis: second-site frameshift mutation within the N-terminal region of the lacI gene of Es- cherichia coli. Mol. Gen. Genet., 227, 160-164.

Hall, D.H., I. Tessman and O. Karlstrrm (1967) Linkage of T4 genes controlling a series of steps in pyrimidine biosyn- thesis, Virology, 31, 442-448.
Hall, D.H., C.M. Povinelli, K. Ehrenman, J. Pedersen-Lane, F. Chu and M. Belfort (19~7) Two domains for splicing in the intron of the phage T4 thymidylate synthase (td) gene established by nondirected mutagenesis, Cell, 48, 63-71.
Hall, D.H. and I. Tessman (1966) T4 mutants unable to induce deoxycytidylate deaminase activity, Virology, 29, 339-345.
Johnson, J.R. and D.H. Hall (1973) Isolation and characteri- zation of mutants of bacteriophage T4 resistant to folate analogs, Virology, 53, 413-426.
Karlstr6m, O. (1968) Mutants of Escherichia coli defective in ribonucleotide and deoxyribonucleotide catabolism, J. Bacteriol., 95, 1069-1077.
Lerman, L.S. (1961) Structural considerations in the interac- tion of DNA and acridines, J. Mol. Biol., 3, 18-30.
Lerman, L.S. (1963) The structure of the DNA-acridine com- plex, Proc. Natl. Acad. Sci. USA, 49, 94-102.
Lerman, L.S. (1964) Acridine mutagens and DNA structure, J. Cell. Comp. Physiol., 64, Suppl.1, 1-18.
Lomax, M.S. and G.R. Greenberg (1968) Characteristics of the deo operon: role in thymidine utilization and sensitiv- ity to deoxyribonucleotides, J. Bacteriol., 96, 501-514.
Masurekar, M., K.N. Kreuzer and L.S. Ripley (1991) The specificity of topoisomerase-mediated DNA cleavage de- fines acridine-induced frameshift specificity within a hotspot in bacteriophage T4, Genetics, 127, 453-462.
Mattson, T., G. Van Houwe, A. Bolle, G. Selzer and R. Epstein (1977) Genetic identification of cloned fragments of bacteriophage T4 DNA and complementation by some clones containing early T4 genes, Mol. Gen. Genet., 154, 319-326.
Morrison, A. and N.R. Cozzarelli (1979) Site-specific cleavage of DNA by E. coli DNA gyrase, Cell, 17, 175-184.
Orgel, A. and S. Brenner (1961) Mutagenesis of bacterio- phage T4 by acridines, J. Mol. Biol., 3, 762-768.
197
Pribnow, D., D.C. Sigurdson, L. Gold, B.S. Singer, C. Napoli, J. Brosius, T.J. Dull, and H.F. Noller (1981) rII cistrons of bacteriophage T4. DNA sequence around the inter- cistronic divide and positions of genetic landmarks, J. Mol. Biol., 149, 337-376.
Revich, G.G. and L.S. Ripley (1990) Effects of proflavin and photoactivated proflavin on the template function of sin- gle-stranded DNA, J. Mol. Biol., 211, 63-74.
Ripley, L.S. and A. Clark (1986) Frameshift mutations pro- duced by proflavin in bacteriophage T4, Proc. Natl. Acad. Sci. USA, 83, 6954-6958.
Ripley, L.S., J.S. Dubbins, J.G. deBoer, D.M. DeMarini, A.M. Bogerd and K.N. Kreuzer (1988) Hotspot sites for acri- dine-induced frameshift mutations in bacteriophage T4 correspond to sites of action of the T4 type II topoiso- merase, J. Mol. Biol., 200, 665-680.
Shearman, C.W., and L.A. Loeb (1983) On the fidelity of DNA replication. Specificity of nucleotide substitution by intercalating agents, J. Biol. Chem., 258, 4485-4491.
Skopek, T.R., and F. Hutchinson (1984) Frameshift mutagen- esis of lambda prophage by 9-aminoacridine, proflavin and ICR-191, Mol. Gen. Genet., 195, 418-423.
Streisinger, G., Y. Okada, J. Emrich, J. Newton, A. Tsugita, E. Terzaghi and M. Inouye (1966) Frameshift mutations and the genetic code, Cold Spring Harbor Symp. Quant. Biol., 31, 77-84.
Streisinger, G., and J.E. Owen (1985) Mechanisms of sponta- neous and induced frameshift mutation in bacteriophage T4, Genetics, 109, 633-659.
Strigini, P. (1965) On the mechanism of spontaneous rever- sion and genetic recombination in bacteriophage T4, Ge- netics, 52, 759-776.
Wallace, R.B., M.J. Johnson, T. Hirose, T. Miyake, E.H. Kawashima and K. Itakura (1981) The use of synthetic oligonucleotides as hybridization probes. II. Hybridization of oligonucleotides of mixed sequence to rabbit B-globin DNA, Nucl. Acids Res., 9, 879-894.




![BACTERIOPHAGE-RESISTANT AND BACTERIOPHAGE-SENSITIVE ...halsmith/phagemutantsubmitted_2.pdf · BACTERIOPHAGE-RESISTANT AND BACTERIOPHAGE-SENSITIVE BACTERIA IN A CHEMOSTAT ... [22],](https://img.pdfslide.net/doc/110x75/5b3839687f8b9a5a518d2ce1/bacteriophage-resistant-and-bacteriophage-sensitive-halsmithphagemutantsubmitted2pdf.jpg)
























