Embed Size (px)
Citation preview

R E S EARCH ART I C L E
PARK INSON ’S D I S EASE
http://sD
ownloaded from
a-Synuclein binds to TOM20 and inhibits mitochondrialprotein import in Parkinson’s diseaseRoberto Di Maio,1,2,3* Paul J. Barrett,1,2* Eric K. Hoffman,1,2 Caitlyn W. Barrett,1,2
Alevtina Zharikov,1,2,4 Anupom Borah,1,5 Xiaoping Hu,1,2 Jennifer McCoy,1,2 Charleen T. Chu,1,6
Edward A. Burton,1,2,4 Teresa G. Hastings,1,2 J. Timothy Greenamyre1,2,4†
a-Synuclein accumulation and mitochondrial dysfunction have both been strongly implicated in the patho-genesis of Parkinson’s disease (PD), and the two appear to be related. Mitochondrial dysfunction leads to accumu-lation and oligomerization of a-synuclein, and increased levels of a-synuclein cause mitochondrial impairment,but the basis for this bidirectional interaction remains obscure. We now report that certain posttranslationallymodified species of a-synuclein bind with high affinity to the TOM20 (translocase of the outer membrane 20)presequence receptor of the mitochondrial protein import machinery. This binding prevented the interaction ofTOM20 with its co-receptor, TOM22, and impaired mitochondrial protein import. Consequently, there were deficientmitochondrial respiration, enhanced production of reactive oxygen species, and loss of mitochondrial membranepotential. Examination of postmortem brain tissue from PD patients revealed an aberrant a-synuclein–TOM20 in-teraction in nigrostriatal dopaminergic neurons that was associated with loss of imported mitochondrial proteins,thereby confirming this pathogenic process in the human disease. Modest knockdown of endogenous a-synucleinwas sufficient to maintain mitochondrial protein import in an in vivo model of PD. Furthermore, in in vitro systems,overexpression of TOM20 or a mitochondrial targeting signal peptide had beneficial effects and preserved mito-chondrial protein import. This study characterizes a pathogenic mechanism in PD, identifies toxic species of wild-type a-synuclein, and reveals potential new therapeutic strategies for neuroprotection.
tm
by guest on May 25, 2018.sciencem
ag.org/
INTRODUCTION
Parkinson’s disease (PD) is a common neurodegenerative disorderthat results in motor impairment, cognitive and psychiatric symp-toms, and autonomic dysfunction (1). Both genetic and environ-mental factors have been implicated in PD pathogenesis, and itappears that mitochondrial defects and accumulation of the synap-tic protein a-synuclein are common to most forms of the disease(2). Moreover, there is evidence of a bidirectional interaction be-tween mitochondrial dysfunction and a-synuclein accretion andaggregation. Inhibition of mitochondrial complex I leads to accu-mulation and oligomerization of a-synuclein (3–5), and increasedlevels of a-synuclein cause mitochondrial impairment and produc-tion of reactive oxygen species (ROS) (6). The nature of the inter-action between a-synuclein and mitochondria remains obscure, asdoes the basis for the vulnerability of dopamine neurons of the ni-grostriatal tract. Furthermore, it is unclear whether unmodifiedmonomeric a-synuclein is responsible for these effects or whetherposttranslational modifications that have been implicated in patho-genesis, such as oligomerization, dopamine modification, phospho-rylation, or nitration, are important.
Although mitochondria contain their own genome, it encodes only13 proteins (7). Because mitochondria may contain up to 1500 distinctproteins (8), theymust import roughly 99%of these. Themitochondrialprotein import and sorting machinery is complex and highly regulated
1Pittsburgh Institute for Neurodegenerative Diseases, University of Pittsburgh, PittsburghPA 15213, USA. 2Department of Neurology, University of Pittsburgh, Pittsburgh, PA 15213USA. 3Ri.MED Foundation, Palermo, Italy. 4Geriatric Research Education and Clinical CenterVA Pittsburgh Healthcare System, Pittsburgh, PA 15240, USA. 5Department of Life Scienceand Bioinformatics, Assam University, Silchar 788011, India. 6Department of PathologyUniversity of Pittsburgh, Pittsburgh, PA 15213, USA.*These authors contributed equally to this work.†Corresponding author. Email: [email protected]
www
,,,
,
.Sc
(9, 10), and its components and mechanisms vary by the compartmentto which a protein is to be sorted. The best-characterized system is oneby which nuclear-encoded, matrix-targeted proteins that contain amitochondrial targeting signal (MTS; presequence) are recognizedby the translocase of the outer membrane (TOM) receptors and trans-located through the outer membrane to the translocase of the innermembrane (TIM) and into the matrix, where the MTS is cleaved toyield the mature protein (10). In this system, import of a presequence-containing preprotein starts with recognition of theMTS via interactionof its hydrophobic face with the TOM20 receptor (10). Subsequently,the hydrophilic side of the presequence is recognized by TOM22. Giventhe normally close proximity of TOM20 and TOM22, it is also possiblethat both receptors recognize theMTS simultaneously. However, recentcryo-EM (electron microscopy) studies suggest that, in addition to thecentral TOMcomplex core, there are “peripheral”TOM20 components,which are in a dynamic equilibrium with the preassembled TOMcomplex (11).
Althoughmonomeric a-synuclein is an intrinsically disordered pro-tein in solution, in association with anionic lipids in membranes, itforms an amphipathic helix (12) similar to known MTS motifs. In thiscontext, we hypothesized that, under some conditions, a-synucleinmight interact with TOM20 and interfere with import of mitochond-rially targeted proteins. As such, this may represent a new pathogenicmechanism and a potential target for therapeutic intervention in PD.
RESULTS
a-Synuclein interacts with TOM20 in vivoSystemic defects in mitochondrial complex I have been describedrepeatedly in PD, and systemic inhibition of complex I with rotenone
ienceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 1

R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
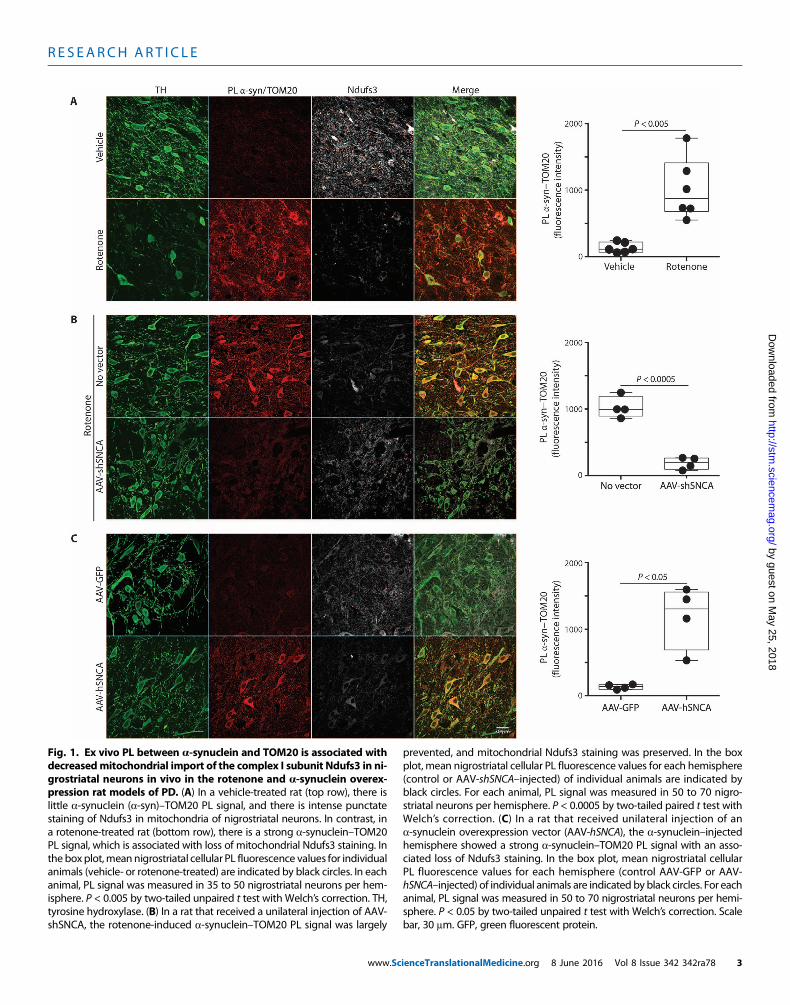
in rodents reproduces many features of PD, including Lewy pathologyand accumulation and oligomerization of a-synuclein in the substan-tia nigra (4, 5, 13). We have also found that in vivo rotenone treatmentincreases S129 phosphorylation of a-synuclein in nigrostriatal neu-rons (213% of control; P < 0.0001, two-tailed unpaired t test; fig. S1).Moreover, occupational exposure to rotenone is a risk factor for de-veloping the disease (14). Therefore, we used the rotenone model toassess potential interactions between a-synuclein and mitochondrialprotein import machinery. Using proximity ligation assays (PLAs)(15), we demonstrated a marked increase in the interaction betweena-synuclein and TOM20 in nigrostriatal dopamine neurons of ratstreated with rotenone relative to rats treated with vehicle (n = 6 pergroup; P < 0.001, two-tailed unpaired t test, or P < 0.005 with Welch’scorrection for unequal variances; Fig. 1A). There was no such inter-action between a-synuclein and TOM22 or TOM40 or a componentof the translocase of the inner membrane, TIM23 (fig. S2). As an initialassessment of whether this apparent interaction between a-synucleinand TOM20 resulted in loss of mitochondrially targeted protein, weexamined levels of the nuclear-encoded, mitochondrially targetedsubunit of complex I, NADH (reduced form of nicotinamide adeninedinucleotide)–ubiquinone oxidoreductase core subunit S3 (Ndufs3),and found a decrease in this imported protein after rotenone treatment.Furthermore, the remaining Ndufs3 immunoreactivity was more dif-fuse than punctate, as verified by a drop in the TOM20-Ndufs3 Pearsonindex from 0.77 ± 0.02 in control animals to 0.23 ± 0.02 in rotenone-treated rats (P < 0.0001, two-tailed unpaired t test; fig. S3).
a-Synuclein in nigrostriatal dopamine neurons increases withnormal aging, in PD, and in rats treated with rotenone (4, 16). Becausethe amount of oligomer formation and other posttranslational modifi-cations are dependent on the concentration of a-synuclein, we exam-ined the in vivo effects of reducing a-synuclein on its interaction withTOM20 in rotenone-treated rats. Three weeks before rotenone treat-ment, rats received a unilateral injection into substantia nigra of adeno-associated virus type 2 (AAV2) virus containing a short hairpin RNAspecifically targeting rata-synuclein (17).Quantitative confocal immuno-fluorescence showed a 30 to 40%knockdown of endogenousa-synucleinprotein in transduced dopamine neurons 3 weeks after injection (17).Using tissue from this previous study, we found that after rotenone treat-ment, in the untransduced hemisphere, there was a strong PL signal be-tween a-synuclein and TOM20, which was almost completely preventedby the modest reduction in a-synuclein in the AAV2-injected hemi-sphere (n = 4; P < 0.0001, two-tailed paired t test, or P < 0.0005 withWelch’s correction; Fig. 1B). Consistent with the decreased interactionof a-synuclein with TOM20, knockdown was associated with a pre-servation of punctate (mitochondrial) Ndufs3 staining, suggesting nor-mal protein import. Knockdown of endogenous a-synuclein providedbehavioral protection and preservation of nigrostriatal neurons, termi-nals, and dendrites (17), possibly indicating the importance of reducedmitochondrial protein import.
To validate our finding of an in vivo interaction between a-synucleinand TOM20, we used another model of PD that does not rely on aneurotoxin, namely, AAV2 viral-mediated overexpression of humana-synuclein (hSNCA) in nigrostriatal neurons. Quantitative immu-nofluorescence indicated a threefold overexpression of a-synuclein inthe transduced nigral neurons (P < 0.01) along with a marked increasein S129 phosphorylation (394% of control; P < 0.005, paired t test; fig.S1). In this model, which induces a-synuclein oligomerization (18)and causes delayed and progressive neurodegeneration (19), we also
www.Sc
found a strong PL signal between a-synuclein and TOM20 with aparallel loss of mitochondrial Ndufs3 in dopamine neurons in thevector-injected hemisphere, but not in the control hemisphere (n = 4;P < 0.005, two-tailed paired t test, or P < 0.05 with Welch’s correction;Fig. 1C).
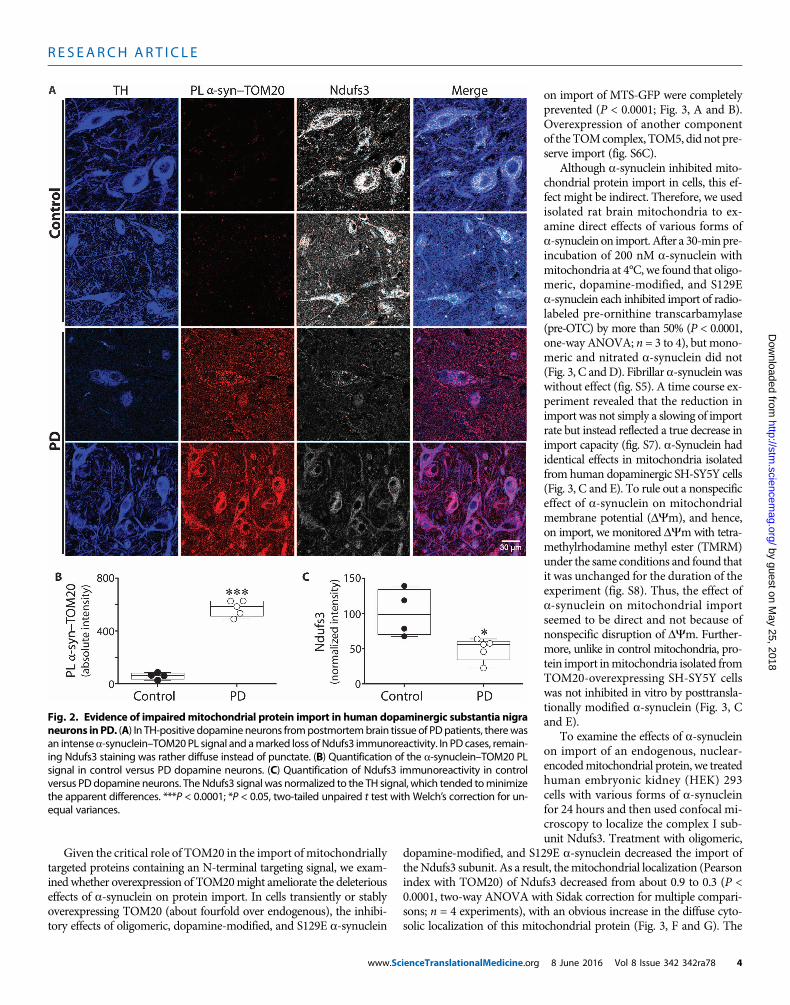
a-Synuclein interacts with TOM20 in dopaminergic neuronsin the PD brainOur results from rotenone-treated and hSNCA-overexpressing ratsdemonstrated an interaction between a-synuclein and TOM20, whichwas associated with reduced import and a decrease in mitochondriallylocalized Ndufs3. To determine whether this process was relevant toidiopathic humanPD,weperformedPLAs assays (a-synuclein–TOM20)and immunocytochemistry for the Ndufs3 subunit in blinded post-mortem substantia nigra sections from individuals with PD (n = 5) andfrom controls (n = 4). Compared to controls, nigrostriatal dopamineneurons from all PD cases had a strong PL signal (P < 0.0001), indicatingan interaction between a-synuclein and TOM20 (Fig. 2). Furthermore,relative to the controls, there was a prominent loss of mitochondrialNdufs3 staining (P < 0.02 or P < 0.05 with Welch’s correction), and thestaining that remained tended to be diffuse rather than punctate. Thissuggested that a-synuclein–induced impairment of mitochondrial pro-tein import may occur in the human disease.
a-Synuclein inhibits import of mitochondrial proteinsTo explore inmore detail the functional significance of thea-synuclein–TOM20 interaction, we turned to cellular and in vitro assays of mito-chondrial protein import in the presence or absence of various forms ofmonomeric, oligomeric, or otherwise posttranslationally modified re-combinant a-synuclein. For this purpose, we used (i) confocal imagingof dopaminergic SH-SY5Y cells expressing mitochondrially targetedGFP (MTS-GFP), (ii) direct assays of mitochondrial protein import inisolatedmitochondria, and (iii) confocalmeasurements ofmitochondriallocalization of endogenous, presequence-containing, nuclear-encoded,and imported proteins. a-Synuclein binds to lipidmembranes and caneasily cross the plasmamembrane, a property we took advantage of inin vitro studies (20). Control experiments using fluorescently labeleda-synuclein confirmed that each species of a-synuclein used in subse-quent experiments entered cells to an equivalent extent (fig. S4). Fur-thermore, when cells were treated with exogenous a-synuclein species(200 nM monomer equivalent), there was no detectable change in thetotal cellular content of a-synuclein, consistent with the data suggestingthat endogenous intraneuronal concentrations are in the range of 2 to5 mM (21), or even higher (22).
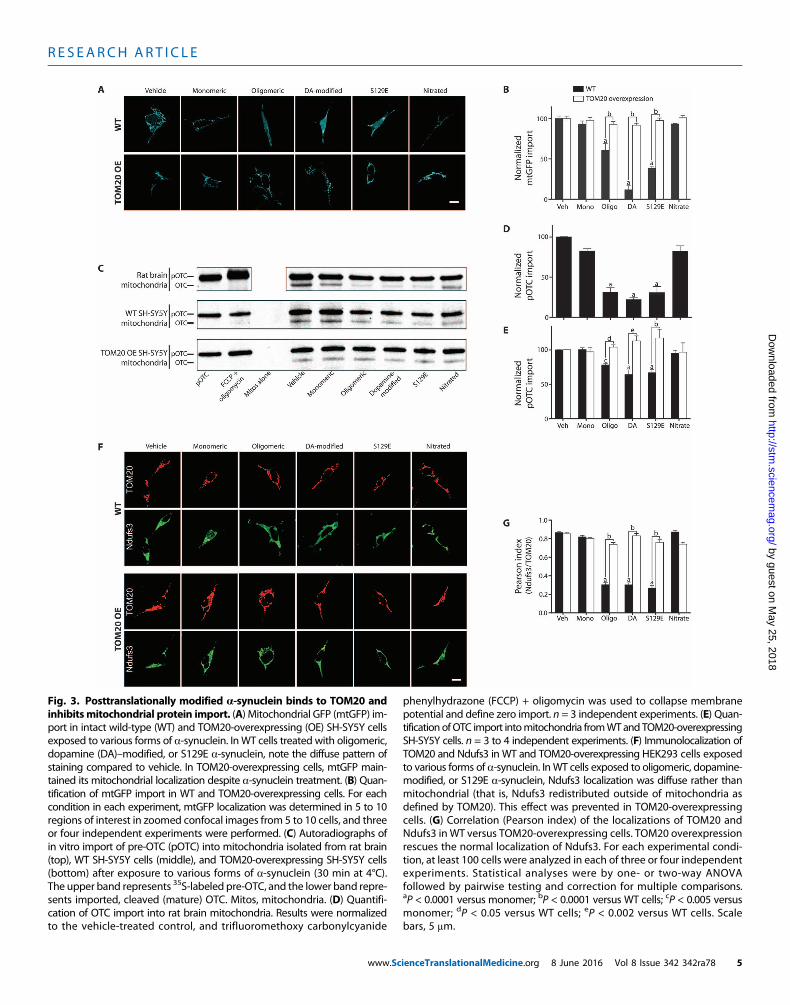
Treatment of SH-SY5Y cells with 200 nM monomeric a-synucleinfor 24 or 48 hours had no effect on import (mitochondrially localizedGFP), but the sameamount ofa-synuclein, in the formof small oligomers,potently inhibited import by about 50% [P < 0.0001, two-way analysis ofvariance (ANOVA); n = 3; Fig. 3, A and B]. Dopamine stably modifiesa-synuclein, and although the molecular nature of this modification iscontroversial, this formofa-synuclein has been implicated in pathogenesis(23, 24).When applied to SH-SY5Y cells, dopamine-modified a-synucleinalso potently inhibited import (P < 0.0001). Similarly, S129E a-synuclein,a phosphomimetic mutant, strongly inhibited MTS-GFP import (P <0.0001). In contrast, unlike the other posttranslational modifications, ni-trated a-synuclein behaved like the monomeric, unmodified protein anddid not inhibit import. Additionally, thioflavin T–positive fibrils ofa-synuclein had no effect on import (fig. S5, A to D).
ienceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 2
R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
Fig. 1. Ex vivo PL between a-synuclein and TOM20 is associated withdecreasedmitochondrial import of the complex I subunit Ndufs3 in ni-
prevented, and mitochondrial Ndufs3 staining was preserved. In the boxplot, mean nigrostriatal cellular PL fluorescence values for each hemisphere
grostriatal neurons in vivo in the rotenone and a-synuclein overex-pression rat models of PD. (A) In a vehicle-treated rat (top row), there islittle a-synuclein (a-syn)–TOM20 PL signal, and there is intense punctatestaining of Ndufs3 in mitochondria of nigrostriatal neurons. In contrast, ina rotenone-treated rat (bottom row), there is a strong a-synuclein–TOM20PL signal, which is associated with loss of mitochondrial Ndufs3 staining. Inthe box plot,meannigrostriatal cellular PL fluorescence values for individualanimals (vehicle- or rotenone-treated) are indicated by black circles. In eachanimal, PL signal was measured in 35 to 50 nigrostriatal neurons per hem-isphere. P < 0.005 by two-tailed unpaired t test with Welch’s correction. TH,tyrosine hydroxylase. (B) In a rat that received a unilateral injection of AAV-shSNCA, the rotenone-induced a-synuclein–TOM20 PL signal was largely
www.Sc
(control or AAV-shSNCA–injected) of individual animals are indicated byblack circles. For each animal, PL signal was measured in 50 to 70 nigro-striatal neurons per hemisphere. P < 0.0005 by two-tailed paired t test withWelch’s correction. (C) In a rat that received unilateral injection of ana-synuclein overexpression vector (AAV-hSNCA), the a-synuclein–injectedhemisphere showed a strong a-synuclein–TOM20 PL signal with an asso-ciated loss of Ndufs3 staining. In the box plot, mean nigrostriatal cellularPL fluorescence values for each hemisphere (control AAV-GFP or AAV-hSNCA–injected) of individual animals are indicated by black circles. For eachanimal, PL signal was measured in 50 to 70 nigrostriatal neurons per hemi-sphere. P < 0.05 by two-tailed unpaired t test with Welch’s correction. Scalebar, 30 mm. GFP, green fluorescent protein.
ienceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 3
R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
Given the critical role of TOM20 in the import ofmitochondriallytargeted proteins containing an N-terminal targeting signal, we exam-inedwhether overexpression of TOM20might ameliorate the deleteriouseffects of a-synuclein on protein import. In cells transiently or stablyoverexpressing TOM20 (about fourfold over endogenous), the inhibi-tory effects of oligomeric, dopamine-modified, and S129E a-synuclein
www.ScienceTranslationalMedicine.org
on import of MTS-GFP were completelyprevented (P < 0.0001; Fig. 3, A and B).Overexpression of another componentof theTOMcomplex, TOM5, did not pre-serve import (fig. S6C).
Although a-synuclein inhibited mito-chondrial protein import in cells, this ef-fect might be indirect. Therefore, we usedisolated rat brain mitochondria to ex-amine direct effects of various forms ofa-synuclein on import. After a 30-minpre-incubation of 200 nM a-synuclein withmitochondria at 4°C, we found that oligo-meric, dopamine-modified, and S129Ea-synuclein each inhibited import of radio-labeled pre-ornithine transcarbamylase(pre-OTC) by more than 50% (P < 0.0001,one-way ANOVA; n = 3 to 4), but mono-meric and nitrated a-synuclein did not(Fig. 3, C andD). Fibrillar a-synuclein waswithout effect (fig. S5). A time course ex-periment revealed that the reduction inimport was not simply a slowing of importrate but instead reflected a true decrease inimport capacity (fig. S7). a-Synuclein hadidentical effects in mitochondria isolatedfrom human dopaminergic SH-SY5Y cells(Fig. 3, C and E). To rule out a nonspecificeffect of a-synuclein on mitochondrialmembrane potential (DYm), and hence,on import, we monitored DYmwith tetra-methylrhodamine methyl ester (TMRM)under the same conditions and found thatit was unchanged for the duration of theexperiment (fig. S8). Thus, the effect ofa-synuclein on mitochondrial importseemed to be direct and not because ofnonspecific disruption of DYm. Further-more, unlike in control mitochondria, pro-tein import inmitochondria isolated fromTOM20-overexpressing SH-SY5Y cellswas not inhibited in vitro by posttransla-tionally modified a-synuclein (Fig. 3, Cand E).
To examine the effects of a-synucleinon import of an endogenous, nuclear-encodedmitochondrial protein, we treatedhuman embryonic kidney (HEK) 293cells with various forms of a-synucleinfor 24 hours and then used confocal mi-croscopy to localize the complex I sub-unit Ndufs3. Treatment with oligomeric,
dopamine-modified, and S129E a-synuclein decreased the import ofthe Ndufs3 subunit. As a result, themitochondrial localization (Pearsonindex with TOM20) of Ndufs3 decreased from about 0.9 to 0.3 (P <0.0001, two-way ANOVA with Sidak correction for multiple compari-sons; n = 4 experiments), with an obvious increase in the diffuse cyto-solic localization of this mitochondrial protein (Fig. 3, F and G). The
Fig. 2. Evidence of impairedmitochondrial protein import in human dopaminergic substantia nigra
neurons inPD. (A) In TH-positive dopamineneurons frompostmortembrain tissue of PDpatients, therewasan intensea-synuclein–TOM20PL signal and amarked loss of Ndufs3 immunoreactivity. In PD cases, remain-ing Ndufs3 staining was rather diffuse instead of punctate. (B) Quantification of the a-synuclein–TOM20 PLsignal in control versus PD dopamine neurons. (C) Quantification of Ndufs3 immunoreactivity in controlversus PD dopamine neurons. The Ndufs3 signal was normalized to the TH signal, which tended tominimizethe apparent differences. ***P < 0.0001; *P < 0.05, two-tailed unpaired t test with Welch’s correction for un-equal variances.8 June 2016 Vol 8 Issue 342 342ra78 4
R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
Fig. 3. Posttranslationally modified a-synuclein binds to TOM20 andinhibitsmitochondrial protein import. (A) Mitochondrial GFP (mtGFP) im-
phenylhydrazone (FCCP) + oligomycin was used to collapse membranepotential and define zero import. n = 3 independent experiments. (E) Quan-
port in intact wild-type (WT) and TOM20-overexpressing (OE) SH-SY5Y cellsexposed to various forms of a-synuclein. InWT cells treated with oligomeric,dopamine (DA)–modified, or S129E a-synuclein, note the diffuse pattern ofstaining compared to vehicle. In TOM20-overexpressing cells, mtGFP main-tained its mitochondrial localization despite a-synuclein treatment. (B) Quan-tification of mtGFP import in WT and TOM20-overexpressing cells. For eachcondition in each experiment, mtGFP localization was determined in 5 to 10regions of interest in zoomed confocal images from 5 to 10 cells, and threeor four independent experiments were performed. (C) Autoradiographs ofin vitro import of pre-OTC (pOTC) into mitochondria isolated from rat brain(top), WT SH-SY5Y cells (middle), and TOM20-overexpressing SH-SY5Y cells(bottom) after exposure to various forms of a-synuclein (30 min at 4°C).The upper band represents 35S-labeled pre-OTC, and the lower band repre-sents imported, cleaved (mature) OTC. Mitos, mitochondria. (D) Quantifi-cation of OTC import into rat brain mitochondria. Results were normalizedto the vehicle-treated control, and trifluoromethoxy carbonylcyanide
www.Sc
tification ofOTC import intomitochondria fromWTand TOM20-overexpressingSH-SY5Y cells. n = 3 to 4 independent experiments. (F) Immunolocalization ofTOM20 and Ndufs3 in WT and TOM20-overexpressing HEK293 cells exposedto various forms of a-synuclein. InWT cells exposed to oligomeric, dopamine-modified, or S129E a-synuclein, Ndufs3 localization was diffuse rather thanmitochondrial (that is, Ndufs3 redistributed outside of mitochondria asdefined by TOM20). This effect was prevented in TOM20-overexpressingcells. (G) Correlation (Pearson index) of the localizations of TOM20 andNdufs3 in WT versus TOM20-overexpressing cells. TOM20 overexpressionrescues the normal localization of Ndufs3. For each experimental condi-tion, at least 100 cells were analyzed in each of three or four independentexperiments. Statistical analyses were by one- or two-way ANOVAfollowed by pairwise testing and correction for multiple comparisons.aP < 0.0001 versus monomer; bP < 0.0001 versus WT cells; cP < 0.005 versusmonomer; dP < 0.05 versus WT cells; eP < 0.002 versus WT cells. Scalebars, 5 mm.
ienceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 5
R E S EARCH ART I C L E
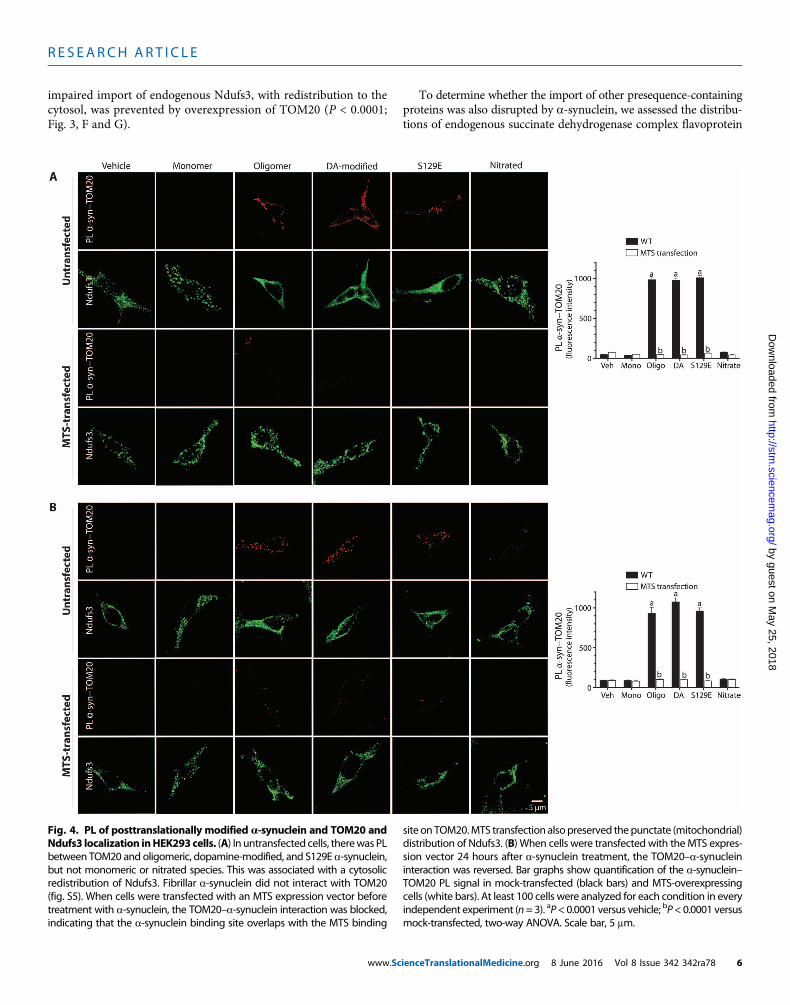
impaired import of endogenous Ndufs3, with redistribution to thecytosol, was prevented by overexpression of TOM20 (P < 0.0001;Fig. 3, F and G).
www.Sc
To determine whether the import of other presequence-containingproteins was also disrupted by a-synuclein, we assessed the distribu-tions of endogenous succinate dehydrogenase complex flavoprotein
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
Fig. 4. PL of posttranslationally modified a-synuclein and TOM20 and site onTOM20.MTS transfection alsopreserved thepunctate (mitochondrial)
Ndufs3 localization inHEK293cells. (A) In untransfected cells, therewas PLbetween TOM20 and oligomeric, dopamine-modified, and S129E a-synuclein,but not monomeric or nitrated species. This was associated with a cytosolicredistribution of Ndufs3. Fibrillar a-synuclein did not interact with TOM20(fig. S5). When cells were transfected with an MTS expression vector beforetreatment with a-synuclein, the TOM20–a-synuclein interaction was blocked,indicating that the a-synuclein binding site overlaps with the MTS bindingdistribution of Ndufs3. (B) When cells were transfected with theMTS expres-sion vector 24 hours after a-synuclein treatment, the TOM20–a-synucleininteraction was reversed. Bar graphs show quantification of the a-synuclein–TOM20 PL signal in mock-transfected (black bars) and MTS-overexpressingcells (white bars). At least 100 cells were analyzed for each condition in everyindependent experiment (n=3). aP<0.0001 versus vehicle; bP<0.0001 versusmock-transfected, two-way ANOVA. Scale bar, 5 mm.
ienceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 6
R E S EARCH ART I C L E
by guest on May 25, 201
http://stm.sciencem
ag.org/D
ownloaded from
subunit A (SDHA; a subunit of complex II), cytochrome c oxidasesubunit 4 (COX4; a subunit of complex IV), and mitochondrial heatshock protein 70 (mtHSP70; a matrix chaperone). As with Ndufs3,there was a cytosolic redistribution of each of these endogenous pro-teins after treatment of cells with oligomeric, dopamine-modified, andS129Ea-synuclein, but not aftermonomeric or nitrated species (fig. S9).Additionally, wemonitored the expression and distribution of two pro-teins encoded by the mitochondrial genome (which do not require im-port). Mitochondrially encoded NADH–ubiquinone oxidoreductasecore subunit 1 (ND1; a subunit of complex I) and mitochondrially en-coded COX1 (mtCO1; a subunit of complex IV) were unaffected bya-synuclein during the time course of this experiment (fig. S9).
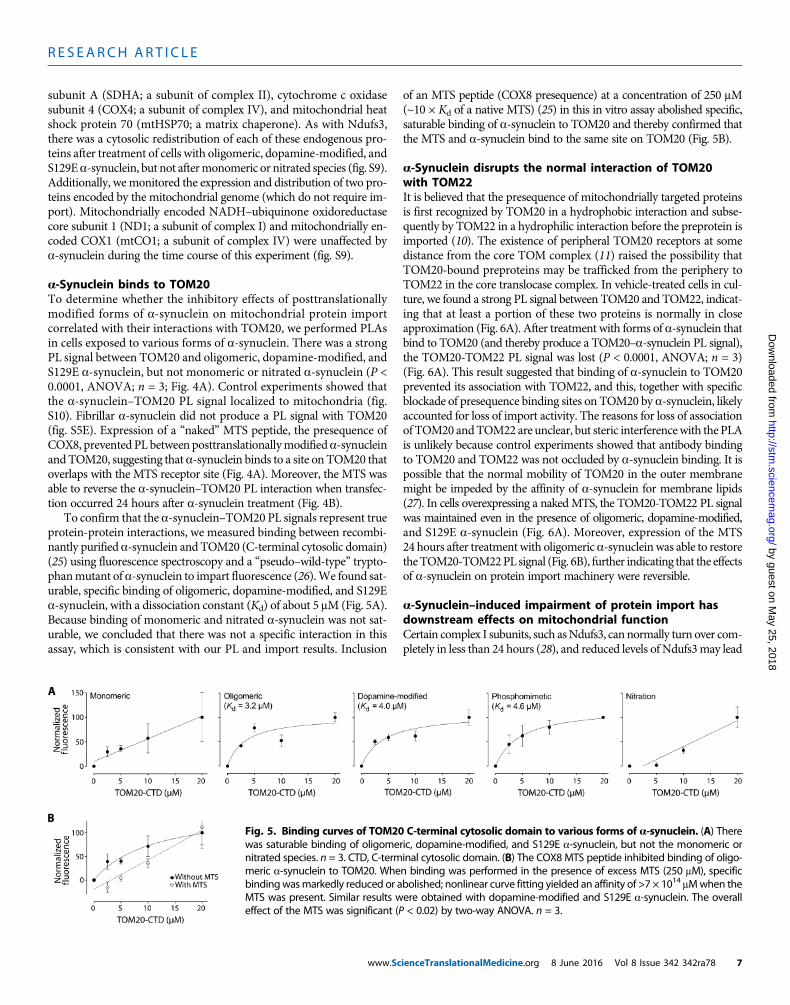
a-Synuclein binds to TOM20To determine whether the inhibitory effects of posttranslationallymodified forms of a-synuclein on mitochondrial protein importcorrelated with their interactions with TOM20, we performed PLAsin cells exposed to various forms of a-synuclein. There was a strongPL signal between TOM20 and oligomeric, dopamine-modified, andS129E a-synuclein, but not monomeric or nitrated a-synuclein (P <0.0001, ANOVA; n = 3; Fig. 4A). Control experiments showed thatthe a-synuclein–TOM20 PL signal localized to mitochondria (fig.S10). Fibrillar a-synuclein did not produce a PL signal with TOM20(fig. S5E). Expression of a “naked” MTS peptide, the presequence ofCOX8, prevented PL between posttranslationallymodifieda-synucleinandTOM20, suggesting thata-synuclein binds to a site on TOM20 thatoverlaps with the MTS receptor site (Fig. 4A). Moreover, the MTS wasable to reverse the a-synuclein–TOM20 PL interaction when transfec-tion occurred 24 hours after a-synuclein treatment (Fig. 4B).
To confirm that the a-synuclein–TOM20 PL signals represent trueprotein-protein interactions, we measured binding between recombi-nantly purified a-synuclein and TOM20 (C-terminal cytosolic domain)(25) using fluorescence spectroscopy and a “pseudo–wild-type” trypto-phanmutant of a-synuclein to impart fluorescence (26).We found sat-urable, specific binding of oligomeric, dopamine-modified, and S129Ea-synuclein, with a dissociation constant (Kd) of about 5 mM (Fig. 5A).Because binding of monomeric and nitrated a-synuclein was not sat-urable, we concluded that there was not a specific interaction in thisassay, which is consistent with our PL and import results. Inclusion
www.Sc
of an MTS peptide (COX8 presequence) at a concentration of 250 mM(~10 × Kd of a native MTS) (25) in this in vitro assay abolished specific,saturable binding of a-synuclein to TOM20 and thereby confirmed thatthe MTS and a-synuclein bind to the same site on TOM20 (Fig. 5B).
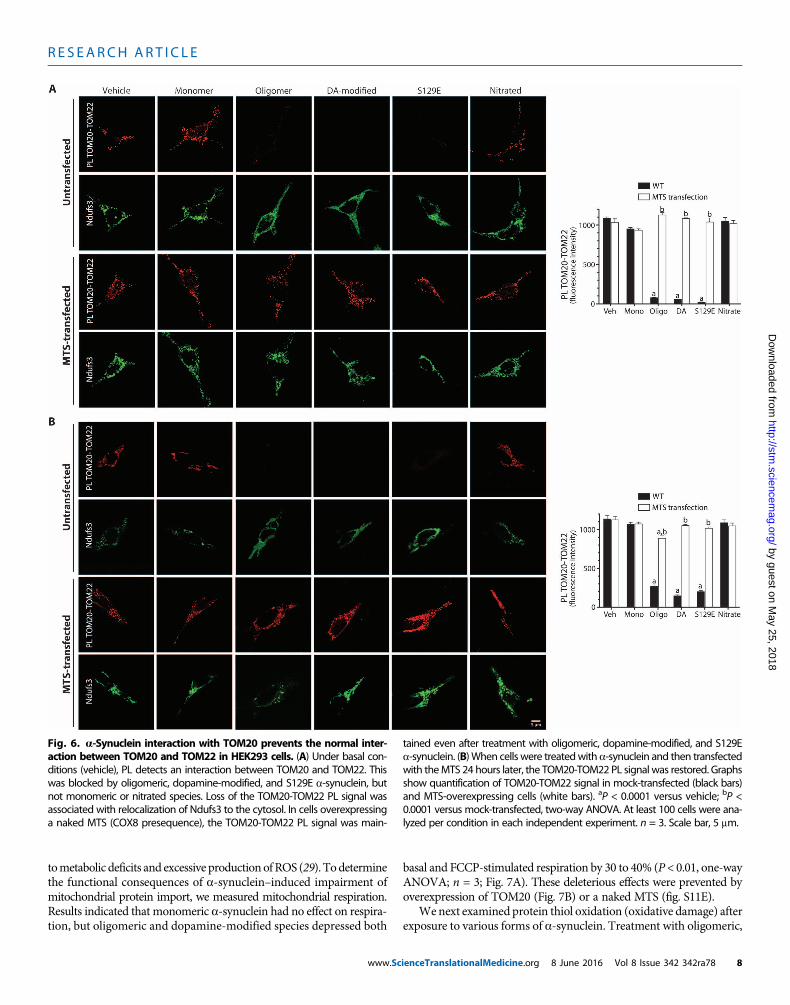
a-Synuclein disrupts the normal interaction of TOM20with TOM22It is believed that the presequence of mitochondrially targeted proteinsis first recognized by TOM20 in a hydrophobic interaction and subse-quently by TOM22 in a hydrophilic interaction before the preprotein isimported (10). The existence of peripheral TOM20 receptors at somedistance from the core TOM complex (11) raised the possibility thatTOM20-bound preproteins may be trafficked from the periphery toTOM22 in the core translocase complex. In vehicle-treated cells in cul-ture, we found a strong PL signal between TOM20 and TOM22, indicat-ing that at least a portion of these two proteins is normally in closeapproximation (Fig. 6A). After treatment with forms of a-synuclein thatbind to TOM20 (and thereby produce a TOM20–a-synuclein PL signal),the TOM20-TOM22 PL signal was lost (P < 0.0001, ANOVA; n = 3)(Fig. 6A). This result suggested that binding of a-synuclein to TOM20prevented its association with TOM22, and this, together with specificblockade of presequence binding sites on TOM20 by a-synuclein, likelyaccounted for loss of import activity. The reasons for loss of associationof TOM20 andTOM22 are unclear, but steric interferencewith the PLAis unlikely because control experiments showed that antibody bindingto TOM20 and TOM22 was not occluded by a-synuclein binding. It ispossible that the normal mobility of TOM20 in the outer membranemight be impeded by the affinity of a-synuclein for membrane lipids(27). In cells overexpressing a nakedMTS, the TOM20-TOM22 PL signalwas maintained even in the presence of oligomeric, dopamine-modified,and S129E a-synuclein (Fig. 6A). Moreover, expression of the MTS24 hours after treatmentwith oligomerica-synucleinwas able to restoretheTOM20-TOM22PL signal (Fig. 6B), further indicating that the effectsof a-synuclein on protein import machinery were reversible.
a-Synuclein–induced impairment of protein import hasdownstream effects on mitochondrial functionCertain complex I subunits, such asNdufs3, cannormally turn over com-pletely in less than 24 hours (28), and reduced levels of Ndufs3may lead
8
Fig. 5. Binding curves of TOM20 C-terminal cytosolic domain to various forms of a-synuclein. (A) Therewas saturable binding of oligomeric, dopamine-modified, and S129E a-synuclein, but not the monomeric ornitrated species. n = 3. CTD, C-terminal cytosolic domain. (B) The COX8 MTS peptide inhibited binding of oligo-meric a-synuclein to TOM20. When binding was performed in the presence of excess MTS (250 mM), specificbindingwasmarkedly reduced or abolished; nonlinear curve fitting yielded an affinity of >7 × 1014 mMwhen theMTS was present. Similar results were obtained with dopamine-modified and S129E a-synuclein. The overalleffect of the MTS was significant (P < 0.02) by two-way ANOVA. n = 3.
ienceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 7
R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
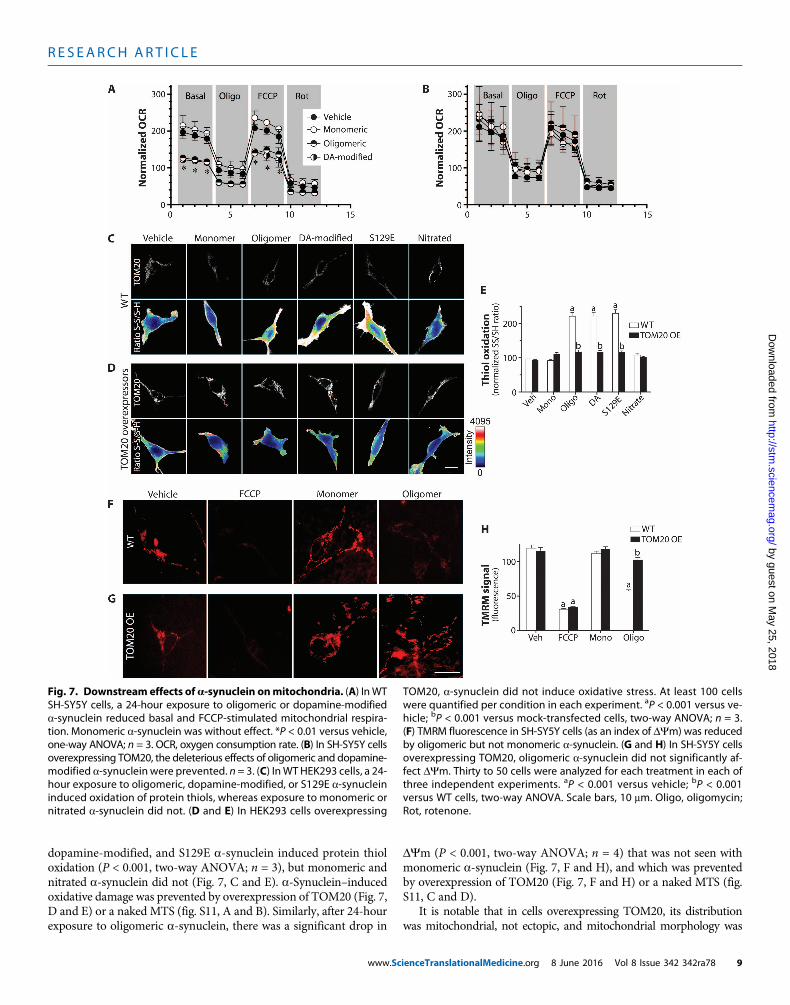
tometabolic deficits and excessive productionofROS (29). Todeterminethe functional consequences of a-synuclein–induced impairment ofmitochondrial protein import, we measured mitochondrial respiration.Results indicated that monomeric a-synuclein had no effect on respira-tion, but oligomeric and dopamine-modified species depressed both
www.Sc
basal and FCCP-stimulated respiration by 30 to 40% (P < 0.01, one-wayANOVA; n = 3; Fig. 7A). These deleterious effects were prevented byoverexpression of TOM20 (Fig. 7B) or a naked MTS (fig. S11E).
We next examined protein thiol oxidation (oxidative damage) afterexposure to various forms of a-synuclein. Treatment with oligomeric,
Fig. 6. a-Synuclein interaction with TOM20 prevents the normal inter-action between TOM20 and TOM22 in HEK293 cells. (A) Under basal con-
tained even after treatment with oligomeric, dopamine-modified, and S129Ea-synuclein. (B)When cells were treatedwith a-synuclein and then transfected
ditions (vehicle), PL detects an interaction between TOM20 and TOM22. Thiswas blocked by oligomeric, dopamine-modified, and S129E a-synuclein, butnot monomeric or nitrated species. Loss of the TOM20-TOM22 PL signal wasassociated with relocalization of Ndufs3 to the cytosol. In cells overexpressinga naked MTS (COX8 presequence), the TOM20-TOM22 PL signal was main-
with theMTS 24 hours later, the TOM20-TOM22 PL signal was restored. Graphsshow quantification of TOM20-TOM22 signal in mock-transfected (black bars)and MTS-overexpressing cells (white bars). aP < 0.0001 versus vehicle; bP <0.0001 versus mock-transfected, two-way ANOVA. At least 100 cells were ana-lyzed per condition in each independent experiment. n = 3. Scale bar, 5 mm.
ienceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 8
R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
dopamine-modified, and S129E a-synuclein induced protein thioloxidation (P < 0.001, two-way ANOVA; n = 3), but monomeric andnitrated a-synuclein did not (Fig. 7, C and E). a-Synuclein–inducedoxidative damage was prevented by overexpression of TOM20 (Fig. 7,D and E) or a naked MTS (fig. S11, A and B). Similarly, after 24-hourexposure to oligomeric a-synuclein, there was a significant drop in
www.Sc
DYm (P < 0.001, two-way ANOVA; n = 4) that was not seen withmonomeric a-synuclein (Fig. 7, F and H), and which was preventedby overexpression of TOM20 (Fig. 7, F and H) or a naked MTS (fig.S11, C and D).
It is notable that in cells overexpressing TOM20, its distributionwas mitochondrial, not ectopic, and mitochondrial morphology was
Fig. 7. Downstream effects ofa-synuclein onmitochondria. (A) InWTSH-SY5Y cells, a 24-hour exposure to oligomeric or dopamine-modified
TOM20, a-synuclein did not induce oxidative stress. At least 100 cellswere quantified per condition in each experiment. aP < 0.001 versus ve-
a-synuclein reduced basal and FCCP-stimulated mitochondrial respira-tion. Monomeric a-synuclein was without effect. *P < 0.01 versus vehicle,one-way ANOVA; n = 3. OCR, oxygen consumption rate. (B) In SH-SY5Y cellsoverexpressing TOM20, the deleterious effects of oligomeric and dopamine-modified a-synuclein were prevented. n = 3. (C) InWT HEK293 cells, a 24-hour exposure to oligomeric, dopamine-modified, or S129E a-synucleininduced oxidation of protein thiols, whereas exposure to monomeric ornitrated a-synuclein did not. (D and E) In HEK293 cells overexpressing
hicle; bP < 0.001 versus mock-transfected cells, two-way ANOVA; n = 3.(F) TMRM fluorescence in SH-SY5Y cells (as an index of DYm) was reducedby oligomeric but not monomeric a-synuclein. (G and H) In SH-SY5Y cellsoverexpressing TOM20, oligomeric a-synuclein did not significantly af-fect DYm. Thirty to 50 cells were analyzed for each treatment in each ofthree independent experiments. aP < 0.001 versus vehicle; bP < 0.001versus WT cells, two-way ANOVA. Scale bars, 10 mm. Oligo, oligomycin;Rot, rotenone.
ienceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 9

R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
unchanged (Fig. 3F). Moreover, although such cells were protected againsta-synuclein–induced import defects and resultant respiratory defects,oxidative stress, and depolarization, the mitochondrial a-synuclein–TOM20 PL signal remained intact (Fig. 3F). The mechanism by whichoverexpression of TOM20 was protective remains unclear, but it ap-pears to enhance the efficiency of import by increasing the initial rateof mitochondrial protein import. Additionally, the protective effectwas saturable because higher concentrations of toxic a-synuclein species(1.7 or 3.4 mM versus 200 nM) could overcome the beneficial effects ofenhanced TOM20 expression (fig. S12).
Toxic species of a-synuclein may be trimers and tetramersEvidence presented here suggests that oligomeric, dopamine-modified,and S129E a-synuclein species impair protein import, whereas mono-meric, nitrated, and fibrillar formsdonot. To look for possible structuralexplanations for these differences, we used circular dichroism spectros-copy to examine the a-synuclein species used in biological experiments(fig. S13, A and B). However, this did not reveal clear differences be-tween the toxic and nontoxic species; all were predominantly in arandom coil conformation with a similar small component of a-helixconformation. As expected, only the fibril preparation had a significantamount of b-sheet structure. In contrast, there were some apparent dif-ferences when the species were separated by SDS–polyacrylamide gelelectrophoresis (SDS-PAGE) (fig. S13C). As intended, the nontoxicmonomer preparation was predominantly monomeric, with a smallcomponent of dimer. The nitrated species was composed mostly ofmonomer and dimer and high–molecular weight material, with littletrimer or tetramer. On the other hand, each of the toxic species—oligomeric, dopamine-modified, and S129E a-synuclein—had relative-ly large amounts of trimer and tetramer, which are about 25 to 35% ofthe total (P < 0.05 to 0.001 versusmonomer or nitrated species, ANOVA;fig. S13D). Under conditions of our preparation, the fibril sample was acontinuous “smear,” and individual bands could not be resolved. Fromthese experiments, we tentatively concluded that a trimeric and/ortetrameric structure may be important for mitochondrial toxicity ofa-synuclein. It is also important to note that each species of a-synucleinwas used in our experiments at a concentration of 200 nM (monomerequivalent), so the actual concentrationof oligomerwas substantially lower.
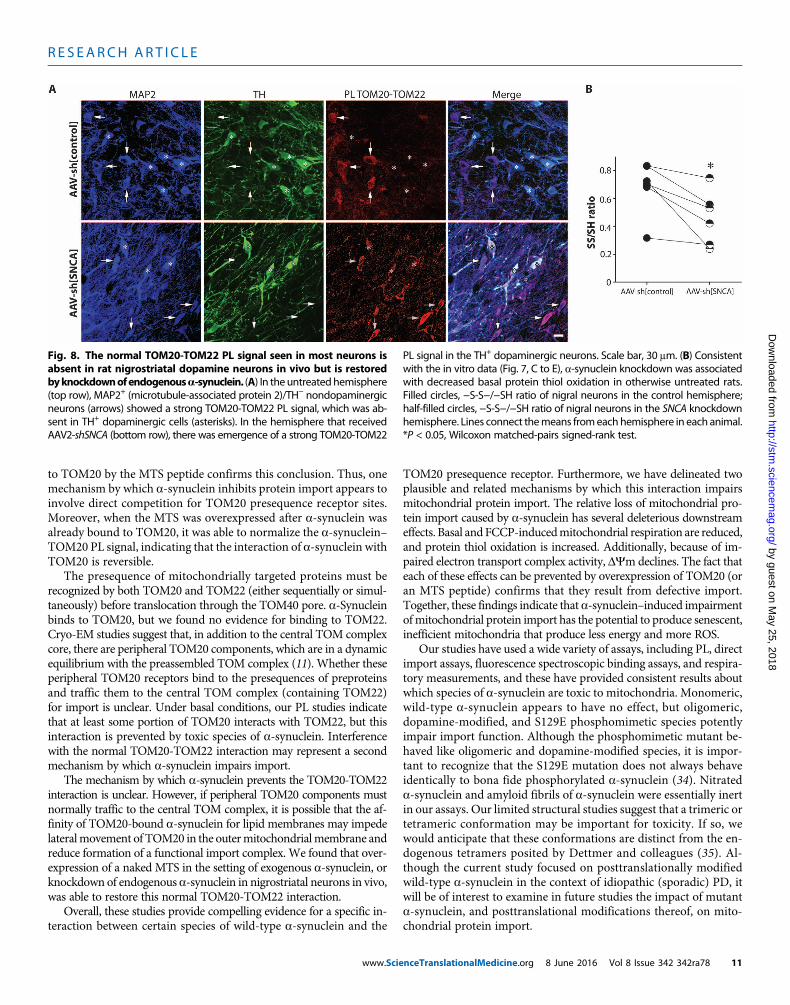
The effects of endogenous a-synuclein on protein importmachinery may explain the selective vulnerability ofdopamine neuronsIn cultured cells, there was a strong PL signal between TOM20-TOM22,which could be disrupted by posttranslationally modified a-synuclein.Similar TOM20-TOM22 interactions were seen in neurons in rat brainsections (Fig. 8A). However, there was a conspicuous absence of theTOM20-TOM22 PL signal in nigrostriatal dopamine neurons in con-trol animals. In the midbrain, these neurons have, by far, the highestexpression of SNCAmRNA(17), raising the possibility that endogenousa-synuclein (perhaps combined with dopamine modification) may besufficient to impair import in these neurons at baseline. Consistent withthis hypothesis, knockdown of endogenous a-synuclein in control rats“restored” a TOM20-TOM22 PL signal in nigrostriatal neurons relativeto the untransduced hemisphere (Fig. 8A). Furthermore, we have re-ported previously that, under basal conditions, nigrostriatal neuronsexist in a more oxidized state than cortical or other dopaminergicneurons (30). The results in Fig. 7 suggest that a-synuclein may con-tribute to this process. In a separate cohort of animals, we found that
www.Scie
AAV-shRNA–mediated knockdown of endogenous a-synuclein de-creased basal levels of protein thiol oxidation in nigrostriatal neuronsrelative to the contralateral hemisphere, which received AAV-shControlvector (P < 0.05; Fig. 8B). Thus, neuron-specific differences in theamount of endogenous a-synuclein (or its posttranslational state)may confer differences in mitochondrial protein import efficiencyand, possibly, selective vulnerability.
DISCUSSION
Our results tie together two central pathogenic mechanisms in PD:a-synuclein accumulation and mitochondrial impairment. We havefound that certain species of a-synuclein bind specifically to TOM20,prevent its interaction with the co-receptor TOM22, and inhibit mito-chondrial protein import. This leads to impairment of mitochondrialfunction, with reduced respiration and excessive ROS production.Our data suggest that this process is operative in human PD and maycontribute to pathogenesis. These findings help to explain the deleteri-ous effects of a-synuclein on mitochondria, and they suggest newtherapeutic strategies.
There is a single published report showing that a-synuclein is a sub-strate for the import machinery and is imported into themitochondrialmatrix, where it binds to and inhibits complex I (31). To the best of ourknowledge, this has not been replicated, and our results suggest thatthere is no specific interaction between monomeric a-synuclein andthe TOM machinery. Another report suggests that TOM40 levels arereduced when a-synuclein is overexpressed, but import was notmeasured, and no mechanism was elucidated (32). Finally, it has beenreported that efficient import prevents PTEN-induced putative kinase1 (PINK1) from accumulating on the mitochondrial surface, where itcan recruit parkin and promote autophagic mitochondrial clearance(33). We hypothesize that by inhibiting protein import, a-synucleinmay also alter PINK1-parkin signaling, and this is under investigation.
Here, we used complementary techniques (PL, multiple assays ofmitochondrial protein import, and fluorescence spectroscopy) to pro-vide convergent evidence that certain posttranslationally modified formsof a-synuclein (soluble oligomers, dopamine-modified, and S129E) bindto TOM20 and impair mitochondrial import of endogenous or exoge-nous presequence-containing nuclear-encoded proteins. Our bindingassays indicate that these species of a-synuclein bind to TOM20 withaffinities of about 5 mM. By contrast, the affinity of a normal TOM20substrate, the aldehyde dehydrogenase presequence, is lower—about20 mM(25). The intraneuronal concentration of a-synuclein has been es-timated to be 2 to 5 mM (21), and in our experiments, a-synuclein spe-cies were applied to cells or isolated mitochondria at a concentrationequivalent to 200 nMmonomeric protein. Control experiments confirmedthat all forms of a-synuclein used here entered cells to an equivalent ex-tent and did not alter total a-synuclein in treated cells. Thus, we concludethat local conditions that predispose to relatively minor oligomer forma-tion, dopamine modification, or S129 phosphorylation (or some com-bination thereof) are likely to have pronounced effects on proteinimport into mitochondria.
Whereas binding assays provided strong evidence that a-synucleininteracts specifically with TOM20, the fact that overexpression of anaked MTS (COX8 presequence) blocked the a-synuclein–TOM20 PLsignal indicates that the a-synuclein binding site overlaps with the MTSrecognition site on TOM20. Direct inhibition of a-synuclein binding
nceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 10
R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
to TOM20 by the MTS peptide confirms this conclusion. Thus, onemechanism by which a-synuclein inhibits protein import appears toinvolve direct competition for TOM20 presequence receptor sites.Moreover, when the MTS was overexpressed after a-synuclein wasalready bound to TOM20, it was able to normalize the a-synuclein–TOM20 PL signal, indicating that the interaction of a-synuclein withTOM20 is reversible.
The presequence of mitochondrially targeted proteins must berecognized by both TOM20 and TOM22 (either sequentially or simul-taneously) before translocation through the TOM40 pore. a-Synucleinbinds to TOM20, but we found no evidence for binding to TOM22.Cryo-EM studies suggest that, in addition to the central TOM complexcore, there are peripheral TOM20 components, which are in a dynamicequilibrium with the preassembled TOM complex (11). Whether theseperipheral TOM20 receptors bind to the presequences of preproteinsand traffic them to the central TOM complex (containing TOM22)for import is unclear. Under basal conditions, our PL studies indicatethat at least some portion of TOM20 interacts with TOM22, but thisinteraction is prevented by toxic species of a-synuclein. Interferencewith the normal TOM20-TOM22 interaction may represent a secondmechanism by which a-synuclein impairs import.
The mechanism by which a-synuclein prevents the TOM20-TOM22interaction is unclear. However, if peripheral TOM20 components mustnormally traffic to the central TOM complex, it is possible that the af-finity of TOM20-bound a-synuclein for lipid membranes may impedelateralmovement of TOM20 in the outermitochondrialmembrane andreduce formation of a functional import complex. We found that over-expression of a naked MTS in the setting of exogenous a-synuclein, orknockdown of endogenous a-synuclein in nigrostriatal neurons in vivo,was able to restore this normal TOM20-TOM22 interaction.
Overall, these studies provide compelling evidence for a specific in-teraction between certain species of wild-type a-synuclein and the
www.Scie
TOM20 presequence receptor. Furthermore, we have delineated twoplausible and related mechanisms by which this interaction impairsmitochondrial protein import. The relative loss of mitochondrial pro-tein import caused by a-synuclein has several deleterious downstreameffects. Basal and FCCP-inducedmitochondrial respiration are reduced,and protein thiol oxidation is increased. Additionally, because of im-paired electron transport complex activity, DYmdeclines. The fact thateach of these effects can be prevented by overexpression of TOM20 (oran MTS peptide) confirms that they result from defective import.Together, these findings indicate that a-synuclein–induced impairmentofmitochondrial protein import has the potential to produce senescent,inefficient mitochondria that produce less energy and more ROS.
Our studies have used a wide variety of assays, including PL, directimport assays, fluorescence spectroscopic binding assays, and respira-tory measurements, and these have provided consistent results aboutwhich species of a-synuclein are toxic to mitochondria. Monomeric,wild-type a-synuclein appears to have no effect, but oligomeric,dopamine-modified, and S129E phosphomimetic species potentlyimpair import function. Although the phosphomimetic mutant be-haved like oligomeric and dopamine-modified species, it is impor-tant to recognize that the S129E mutation does not always behaveidentically to bona fide phosphorylated a-synuclein (34). Nitrateda-synuclein and amyloid fibrils of a-synuclein were essentially inertin our assays. Our limited structural studies suggest that a trimeric ortetrameric conformation may be important for toxicity. If so, wewould anticipate that these conformations are distinct from the en-dogenous tetramers posited by Dettmer and colleagues (35). Al-though the current study focused on posttranslationally modifiedwild-type a-synuclein in the context of idiopathic (sporadic) PD, itwill be of interest to examine in future studies the impact of mutanta-synuclein, and posttranslational modifications thereof, on mito-chondrial protein import.
Fig. 8. The normal TOM20-TOM22 PL signal seen in most neurons isabsent in rat nigrostriatal dopamine neurons in vivo but is restored
PL signal in the TH+ dopaminergic neurons. Scale bar, 30 mm. (B) Consistentwith the in vitro data (Fig. 7, C to E), a-synuclein knockdown was associated
byknockdownofendogenousa-synuclein. (A) In the untreatedhemisphere(top row), MAP2+ (microtubule-associated protein 2)/TH− nondopaminergicneurons (arrows) showed a strong TOM20-TOM22 PL signal, which was ab-sent in TH+ dopaminergic cells (asterisks). In the hemisphere that receivedAAV2-shSNCA (bottom row), there was emergence of a strong TOM20-TOM22
with decreased basal protein thiol oxidation in otherwise untreated rats.Filled circles, −S-S−/−SH ratio of nigral neurons in the control hemisphere;half-filled circles, −S-S−/−SH ratio of nigral neurons in the SNCA knockdownhemisphere. Lines connect themeans fromeach hemisphere in each animal.*P < 0.05, Wilcoxon matched-pairs signed-rank test.
nceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 11

R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
To determine the relevance of our findings to the human disease, weexamined postmortem brain specimens from controls and individualswho died with PD. Just as in rats treated with rotenone or injected withan AAV overexpression vector, we found that nigrostriatal dopamineneurons from PD cases showed a marked increase in the a-synuclein–TOM20 PL signal compared to controls. Similarly, this a-synuclein–TOM20 PL signal was associated with a relative loss and cytosolicredistribution of the endogenous, imported complex I subunit, Ndufs3.Notably, another recent study reported a large loss of a presequence-containing imported protein (Ndufb8) relative to amitochondrially en-coded protein (COX1) in nigral neurons in PD (36). This discovery ofan apparent protein import defect in humanPDadds to the list of findingsthat have been predicted by the rotenone rat model of PD (13, 37, 38).
The results of this study may also shed light on the basis of theselective vulnerability of nigrostriatal neurons to degeneration in PD.Atbaseline, these cells express more SNCAmRNA than surroundingmid-brain neurons, and they exist in a higher state of oxidation (17, 30). Wefound that these neurons do not show the normal TOM20-TOM22 in-teraction seen in other neurons (and presumably needed for efficientprotein import); however, when endogenous a-synuclein was knockeddown in vivo, the TOM20-TOM22 PL signal emerged. Furthermore,the knockdown of endogenous a-synuclein decreased the oxidationstate of these neurons. Thus, under basal conditions, it appears that en-dogenous a-synuclein levels may be sufficient to affect protein importfunction; this situation is likely to be exacerbated in aging asa-synucleinaccumulates in these cells (16). Moreover, elevated a-synuclein increasedthe redistribution of dopamine from vesicles to the cytosol (39) where itmay modify a-synuclein. Dopamine-modified a-synuclein is particu-larly potent at inhibiting mitochondrial protein import.
Although the results of our studies are unanticipated and providenew insights into pathogenesis, there are limitations and unknownsas well. For example, thus far, we have not identified the structural char-acteristics that define the toxic species of a-synuclein, although a tri-meric or tetrameric structure may be important. We also found thatnitrated and fibrillar species of a-synuclein were essentially inert inour system; however, this does not exclude the possibility that theymight exert toxicity by other mechanisms. Moreover, our study fo-cused on wild-type a-synuclein, so we do not know yet how pathogenica-synuclein mutations affect mitochondrial protein import. Addition-ally, many of our experiments used cell lines rather than primary neu-rons for technical reasons; however, key aspects of our findings wereverified ex vivo using rat or human brain tissue. Finally, although weshowed that mitochondrial protein import impairment caused cellulartoxicity in forms of reduced respiration, oxidative damage, and mito-chondrial depolarization, the relative extent towhich this specificmech-anism contributes to the overall neurotoxicity of a-synuclein remains tobe determined.
Despite these caveats, the results presented here have several im-portant therapeutic implications. First, together with the recent reportby Zharikov et al. (17), they suggest that even the modest reduction ofendogenous a-synucleinmay have beneficial effects onmitochondrialfunction and nigral redox state—and may be neuroprotective in PD.Given that multiple posttranslational modifications render a-synucleintoxic to mitochondria, it appears that strategies aimed at a general re-duction of a-synuclein, rather than targeting specific modifications oraggregation states, may be most efficacious.
Additionally, we found that overexpression of TOM20 preventeda-synuclein–induced impairment ofmitochondrial protein import, as
www.Scie
well as its downstream consequences, such as respiratory defects, ROSproduction, and loss of DYm. The mechanism by which a moderate(two- to threefold) increase in TOM20 protein levels is protective is notyet clear, but preliminary studies indicate that TOM20-overexpressingmitochondria have a faster initial rate of protein import. This result isconsistent with previous work showing that TOM20 overexpressioncan increase import (40). Additionally, however, we found that mito-chondria isolated fromTOM20-overexpressing cells and assayed in vitrowere resistant to the inhibitory effects of the otherwise toxic species ofa-synuclein. In this context, it will be of interest to determine whetheroverexpression of TOM20 in vivo is protective in models of PD.
Finally, contrary to our expectations, overexpression of a nakedMTShad beneficial effects. We had anticipated that expression of the MTSwould be similar to a-synuclein, competing for TOM20 presequencebinding sites and inhibiting import.However,whereas theMTSblockedthe a-synuclein–TOM20 PL signal as expected, it also preserved themitochondrial import of the endogenous Ndufs3 subunit. In addition,MTS overexpression was able to prevent or even reverse the loss of theTOM20-TOM22 interaction induced by a-synuclein. As a consequence,overexpression of the naked MTS prevented downstream toxic effectsof a-synuclein, such as reduced respiration, increased ROS production,and mitochondrial depolarization. On this basis, examination of anMTS peptide as a neuroprotective strategy is warranted.
In conclusion, we have defined a mechanism by which a-synucleinimpairs a criticalmitochondrial function, protein import, and have shownthat this likely occurs in human PD.Our findings suggest new therapeuticstrategies that should be tested in future studies.
MATERIALS AND METHODS
Study designThis study was designed to determine the potential role of variousspecies of wild-type a-synuclein on mitochondrial protein importand downstream mitochondrial function. For this purpose, we usedseveral in vivo manipulations, including rotenone-treated rats and ratswith AAV2-mediated knockdown or overexpression of a-synuclein inthe substantia nigra. To confirm relevance to the human disease, weexamined measures of mitochondrial protein import and binding ofa-synuclein to TOM20 in postmortem human brain tissue. Additionalmechanistic experiments were conducted in intact cells (HEK293 andSH-SY5Y) and isolated mitochondria from brains and cultured cells.Most experiments used recombinant a-synuclein in the followingforms: monomeric, oligomeric, dopamine-modified, S129E phospho-mimetic mutant, nitrated, and fibrillar. Mitochondrial protein importwas assessed by (i) direct import assays in isolated mitochondria, (ii)assessment of “mtGFP” uptake in transfected cells, and (iii) localizationof endogenous presequence (MTS)–containing proteins in cells or brainsections. The binding of a-synuclein species to TOM20was assessed byPL in intact cells or in brain sections and in vitro by fluorescence spec-troscopy using recombinant proteins. In response to different species ofa-synuclein, mitochondrial functions were assessed in intact cells bymeasurement of respiration (Seahorse), DYm (TMRM fluorescence),and ROS production (thiol staining). Finally, to test potential protectivetherapeutic strategies, cells were transfected to overexpress TOM20 orthe COX8 presequence (MTS).
a-Synuclein expression and purification. hSNCA complementaryDNA (cDNA) (wild type or S129E) was transfected into BL21-DE3
nceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 12

R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
Escherichia coli, and a-synuclein was purified as described by Vollesand Lansbury (41).
a-Synuclein modificationFor oligomers, monomeric a-synuclein was diluted to a concentrationof 5mg/ml and shaken at ~300 to 500 rpm at 37°C for 3 days. Treatmentfor dopamine modification was carried out as described by Martinez-Vicente et al. (23). For nitration, monomeric a-synuclein was diluted toa concentration of 5 mg/ml, and 50 ml of 1% tetranitromethane wasadded per 500 ml of a-synuclein solution. The solution was vortexedtwice for 10min. For S129 phosphomimetic, S129Emutant SNCA cDNAwas transfected into E. coli as described above. All samples (monomers,oligomers, dopamine-modified, nitrated, and S129E) received the fol-lowing treatment after modification: dialysis in the dark in 4 liters ofphosphate-buffered saline with gentle stirring overnight (10-kDmolec-ular weight cutoff). After dialysis, all samples were spun at 14,000g for5 min to pellet any fibrils that formed. The supernatant contained sol-uble oligomers and monomers as confirmed by SDS-PAGE, and thepellet contained fibrils. Samples were prepared fresh weekly.
Proximity ligationassay. PLAwasperformed in4%paraformaldehyde–fixed tissue or cells. Samples were incubated with specific primary anti-bodies to the proteins to be detected. Secondary antibodies conjugatedwith oligonucleotides were added to the reaction and incubated. Liga-tion solution, consisting of two oligonucleotides and ligase, was added.In this assay, the oligonucleotides hybridize to the two PLA probes andjoin to a closed loop if they are in close proximity. Amplification solu-tion, consisting of nucleotides and fluorescently labeled oligonucleo-tides, was added together with polymerase. The oligonucleotide armof one of the PLAprobes acts as a primer for “rolling-circle amplification”(RCA) using the ligated circle as a template, and this generates a conca-temeric product. Fluorescently labeled oligonucleotides hybridize to theRCA product. The PL signal was visible as a distinct fluorescent spot andwas analyzed by confocal microscopy (Duolink, Sigma-Aldrich). Controlexperiments included routine immunofluorescence staining of proteinsof interest under identical experimental conditions.
Fluorescencemeasurements.Quantitative fluorescencemeasurementsweremadewith anOlympus upright three-laser scanning confocalmicro-scope, taking care to ensure that images contained no saturated pixels. Forquantitative comparisons, all imaging parameters (for example, laserpower, exposure, and pinhole) were held constant across specimens.
Statistical analysesEach result presented here was derived from three to six independent ex-periments. For simple comparisons of two experimental conditions, two-tailed unpaired t tests were used. Where variances were not equal, Welch’scorrection was used.When virus was injected into one hemisphere ofthe brain and theotherhemispherewasused as a control, two-tailedpairedt tests or Wilcoxon matched-pairs signed-rank tests were used. For com-parisons of multiple experimental conditions, one- or two-way ANOVAwas used, and if significant overall, post hoc corrections (Bonferroni orSidak) for multiple pairwise comparisons were made. P values less than0.05 were considered significant. All bar graphs show means ± SEM.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/8/342/342ra78/DC1Materials and MethodsFig. S1. Amounts of a-synuclein and S129-phosphorylated a-synuclein in vivo.
www.Scie
Fig. S2. Positive and negative PL interactions with a-synuclein in substantia nigra pars compacta.Fig. S3. Rotenone induces a loss of mitochondrial localization of the nuclear-encoded,imported protein Ndufs3.Fig. S4. All species of a-synuclein used in this study enter cells to an equivalent extent, andwhen added at 200 nM, they do not change intracellular concentrations of a-synuclein or itslocalization.Fig. S5. Fibrillar a-synuclein does not affect mitochondrial protein import.Fig. S6. Overexpression of TOM20 and TOM5.Fig. S7. Time course of isolated brain mitochondrial protein import in the absence or presenceof monomeric and oligomeric a-synuclein.Fig. S8. Lack of mitochondrial depolarization by a-synuclein during import assays.Fig. S9. Effects of a-synuclein on import and localization of other mitochondrial proteins.Fig. S10. The a-synuclein–TOM20 PL signal colocalizes with mitochondria.Fig. S11. Downstream effects of a-synuclein on mitochondria are blocked by MTS overexpression.Fig. S12. The protective effects of TOM20 overexpression on mitochondrial protein import canbe overcome by increased concentrations of a-synuclein.Fig. S13. Structural analysis of the a-synuclein species used in this study.
REFERENCES AND NOTES
1. L. V. Kalia, A. E. Lang, Parkinson disease in 2015: Evolving basic, pathological and clinicalconcepts in PD. Nat. Rev. Neurol. 12, 65–66 (2016).
2. M. Zaltieri, F. Longhena, M. Pizzi, C. Missale, P. Spano, A. Bellucci, Mitochondrial dysfunc-tion and a-synuclein Synaptic pathology in Parkinson’s disease: Who’s on first? ParkinsonsDis. 2015, 108029 (2015).
3. R. Betarbet, T. B. Sherer, G. MacKenzie, M. Garcia-Osuna, A. V. Panov, J. T. Greenamyre,Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci.3, 1301–1306 (2000).
4. R. Betarbet, R. M. Canet-Aviles, T. B. Sherer, P. G. Mastroberardino, C. McLendon, J.-H. Kim,S. Lund, H.-M. Na, G. Taylor, N. F. Bence, R. Kopito, B. B. Seo, T. Yagi, A. Yagi, G. Klinefelter,M. R. Cookson, J. T. Greenamyre, Intersecting pathways to neurodegeneration in Parkinson’sdisease: Effects of the pesticide rotenone on DJ-1, a-synuclein, and the ubiquitin–proteasomesystem. Neurobiol. Dis. 22, 404–420 (2006).
5. J. R. Cannon, V. Tapias, H. M. Na, A. S. Honick, R. E. Drolet, J. T. Greenamyre, A highlyreproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 34, 279–290 (2009).
6. L. J. Hsu, Y. Sagara, A. Arroyo, E. Rockenstein, A. Sisk, M. Mallory, J. Wong, T. Takenouchi,M. Hashimoto, E. Masliah, a-synuclein promotes mitochondrial deficit and oxidativestress. Am. J. Pathol. 157, 401–410 (2000).
7. E. A. Schon, S. DiMauro, M. Hirano, Human mitochondrial DNA: Roles of inherited andsomatic mutations. Nat. Rev. Genet. 13, 878–890 (2012).
8. M. Elstner, C. Andreoli, U. Ahting, I. Tetko, T. Klopstock, T. Meitinger, H. Prokisch, MitoP2: Anintegrative tool for the analysis of the mitochondrial proteome. Mol. Biotechnol. 40, 306–315(2008).
9. C. M. Koehler, Protein translocation pathways of the mitochondrion. FEBS Lett. 476, 27–31(2000).
10. C. Schulz, A. Schendzielorz, P. Rehling, Unlocking the presequence import pathway. TrendsCell Biol. 25, 265–275 (2015).
11. K. Model, C. Meisinger, W. Kühlbrandt, Cryo-electron microscopy structure of a yeastmitochondrial preprotein translocase. J. Mol. Biol. 383, 1049–1057 (2008).
12. W. S. Davidson, A. Jonas, D. F. Clayton, J. M. George, Stabilization of a-synuclein secondarystructure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449 (1998).
13. J. T. Greenamyre, J. R. Cannon, R. Drolet, P.-G. Mastroberardino, Lessons from the rotenonemodel of Parkinson’s disease. Trends Pharmacol. Sci. 31, 141–142 (2010).
14. C. M. Tanner, F. Kamel, G. W. Ross, J. A. Hoppin, S. M. Goldman, M. Korell, C. Marras,G. S. Bhudhikanok, M. Kasten, A. R. Chade, K. Comyns, M. B. Richards, C. Meng, B. Priestley,H. H. Fernandez, F. Cambi, D. M. Umbach, A. Blair, D. P. Sandler, J. W. Langston, Rotenone,paraquat and Parkinson’s disease. Environ. Health Perspect. 119, 866–872 (2011).
15. B. Koos, L. Andersson, C.-M. Clausson, K. Grannas, A. Klaesson, G. Cane, O. Söderberg, Analysisof protein interactions in situ by proximity ligation assays. Curr. Top. Microbiol. Immunol. 377,111–126 (2014).
16. Y. Chu, J. H. Kordower, Age-associated increases of a-synuclein in monkeys and humansare associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease?Neurobiol. Dis. 25, 134–149 (2007).
17. A. D. Zharikov, J. R. Cannon, V. Tapias, Q. Bai, M. P. Horowitz, V. Shah, A. El Ayadi, T. G. Hastings,J. T. Greenamyre, E. A. Burton, shRNA targeting a-synuclein prevents neurodegeneration in aParkinson’s disease model. J. Clin. Invest. 125, 2721–2735 (2015).
18. H. Dimant, S. K. Kalia, L. V. Kalia, L. N. Zhu, L. Kibuuka, D. Ebrahimi-Fakhari, N. R. McFarland,Z. Fan, B. T. Hyman, P. J. McLean, Direct detection of alpha synuclein oligomers in vivo.Acta Neuropathol. Commun. 1, 6 (2013).
nceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 13

R E S EARCH ART I C L E
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from
19. M. Decressac, B. Mattsson, M. Lundblad, P. Weikop, A. Björklund, Progressive neuro-degenerative and behavioural changes induced by AAV-mediated overexpression ofa-synuclein in midbrain dopamine neurons. Neurobiol. Dis. 45, 939–953 (2012).
20. K. J. Ahn, S. R. Paik, K. C. Chung, J. Kim, Amino acid sequence motifs and mechanisticfeatures of the membrane translocation of a-synuclein. J. Neurochem. 97, 265–279 (2006).
21. C. H. Westphal, S. S. Chandra, Monomeric synucleins generate membrane curvature. J. Biol.Chem. 288, 1829–1840 (2013).
22. F.-X. Theillet, A. Binolfi, B. Bekei, A. Martorana, H. M. Rose, M. Stuiver, S. Verzini, D. Lorenz,M. van Rossum, D. Goldfarb, P. Selenko, Structural disorder of monomeric a-synuclein persistsin mammalian cells. Nature 530, 45–50 (2016).
23. M. Martinez-Vicente, Z. Talloczy, S. Kaushik, A. C. Massey, J. Mazzulli, E. V. Mosharov, R. Hodara,R. Fredenburg, D.-C. Wu, A. Follenzi, W. Dauer, S. Przedborski, H. Ischiropoulos, P. T. Lansbury,D. Sulzer, A. M. Cuervo, Dopamine-modified a-synuclein blocks chaperone-mediated autophagy.J. Clin. Invest. 118, 777–788 (2008).
24. K. A. Conway, J.-C. Rochet, R. M. Bieganski, P. T. Lansbury Jr., Kinetic stabilization of thea-synuclein protofibril by a dopamine-a-synuclein adduct. Science 294, 1346–1349 (2001).
25. Y. Abe, T. Shodai, T. Muto, K. Mihara, H. Torii, S.-i. Nishikawa, T. Endo, D. Kohda, Structuralbasis of presequence recognition by the mitochondrial protein import receptor Tom20.Cell 100, 551–560 (2000).
26. J. C. Lee, R. Langen, P. A. Hummel, H. B. Gray, J. R. Winkler, a-synuclein structures fromfluorescence energy-transfer kinetics: Implications for the role of the protein in Parkinson’sdisease. Proc. Natl. Acad. Sci. U.S.A. 101, 16466–16471 (2004).
27. M. Ramakrishnan, P. H. Jensen, D. Marsh, Association of a-synuclein and mutants with lipidmembranes: Spin-label ESR and polarized IR. Biochemistry 45, 3386–3395 (2006).
28. C. E. J. Dieteren, W. J. H. Koopman, H. G. Swarts, J. G. P. Peters, P. Maczuga, J. J. van Gemst,R. Masereeuw, J. A. M. Smeitink, L. G. J. Nijtmans, P. H. G. M. Willems, Subunit-specificincorporation efficiency and kinetics in mitochondrial complex I homeostasis. J. Biol. Chem.287, 41851–41860 (2012).
29. S. Suhane, H. Kanzaki, V. Arumugaswami, R. Murali, V. K. Ramanujan, Mitochondrial NDUFS3regulates the ROS-mediated onset of metabolic switch in transformed cells. Biol. Open 2,295–305 (2013).
30. M. P. Horowitz, C. Milanese, R. Di Maio, X. Hu, L. M. Montero, L. H. Sanders, V. Tapias, S. Sepe,W. A. van Cappellen, E. A. Burton, J. T. Greenamyre, P. G. Mastroberardino, Single-cell redoximaging demonstrates a distinctive response of dopaminergic neurons to oxidative insults.Antioxid. Redox Signal. 15, 855–871 (2011).
31. L. Devi, V. Raghavendran, B. M. Prabhu, N. G. Avadhani, H. K. Anandatheerthavarada,Mitochondrial import and accumulation of a-synuclein impair complex I in human do-paminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100(2008).
32. A. Bender, P. Desplats, B. Spencer, E. Rockenstein, A. Adame, M. Elstner, C. Laub, S. Mueller,A. O. Koob, M. Mante, E. Pham, T. Klopstock, E. Masliah, TOM40 mediates mitochondrialdysfunction induced by a-synuclein accumulation in Parkinson’s disease. PLOS One 8,e62277 (2013).
33. G. Bertolin, R. Ferrando-Miguel, M. Jacoupy, S. Traver, K. Grenier, A. W. Greene, A. Dauphin,F. Waharte, A. Bayot, J. Salamero, A. Lombès, A.-L. Bulteau, E. A. Fon, A. Brice, O. Corti, TheTOMM machinery is a molecular switch in PINK1 and PARK2/PARKIN-dependent mito-chondrial clearance. Autophagy 9, 1801–1817 (2013).
34. K. E. Paleologou, A. W. Schmid, C. C. Rospigliosi, H.-Y. Kim, G. R. Lamberto, R. A. Fredenburg,P. T. Lansbury Jr., C. O. Fernandez, D. Eliezer, M. Zweckstetter, H. A. Lashuel, Phosphorylationat Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of a-synuclein.J. Biol. Chem. 283, 16895–16905 (2008).
www.Scie
35. U. Dettmer, A. J. Newman, F. Soldner, E. S. Luth, N. C. Kim, V. E. von Saucken, J. B. Sanderson,R. Jaenisch, T. Bartels, D. Selkoe, Corrigendum: Parkinson-causing a-synuclein missensemutations shift native tetramers to monomers as a mechanism for disease initiation.Nat. Commun. 6, 8008 (2015).
36. A. Grünewald, N. Z. Lax, M. C. Rocha, A. K. Reeve, P. D. Hepplewhite, K. A. Rygiel, R. W. Taylor,D. M. Turnbull, Quantitative quadruple-label immunofluorescence of mitochondrial and cy-toplasmic proteins in single neurons from human midbrain tissue. J. Neurosci. Methods 232,143–149 (2014).
37. P. G. Mastroberardino, E. K. Hoffman, M. P. Horowitz, R. Betarbet, G. Taylor, D. Cheng,H. M. Na, C.-A. Gutekunst, M. Gearing, J. Q. Trojanowski, M. Anderson, C. T. Chu, J. Peng,J. T. Greenamyre, A novel transferrin/TfR2-mediated mitochondrial iron transport system isdisrupted in Parkinson’s disease. Neurobiol. Dis. 34, 417–431 (2009).
38. L. H. Sanders, J. McCoy, X. Hu, P. G. Mastroberardino, B. C. Dickinson, C. J. Chang, C. T. Chu,B. Van Houten, J. T. Greenamyre, Mitochondrial DNA damage: Molecular marker of vulner-able nigral neurons in Parkinson’s disease. Neurobiol. Dis. 70, 214–223 (2014).
39. E. V. Mosharov, R. G. W. Staal, J. Bové, D. Prou, A. Hananiya, D. Markov, N. Poulsen, K. E. Larsen,C. M. H. Moore, M. D. Troyer, R. H. Edwards, S. Przedborski, D. Sulzer, a-synuclein over-expression increases cytosolic catecholamine concentration. J. Neurosci. 26, 9304–9311(2006).
40. M. Yano, M. Kanazawa, K. Terada, C. Namchai, M. Yamaizumi, B. Hanson, N. Hoogenraad,M. Mori, Visualization of mitochondrial protein import in cultured mammalian cells withgreen fluorescent protein and effects of overexpression of the human import receptorTom20. J. Biol. Chem. 272, 8459–8465 (1997).
41. M. J. Volles, P. T. Lansbury Jr., Relationships between the sequence of a-synuclein and itsmembrane affinity, fibrillization propensity, and yeast toxicity. J. Mol. Biol. 366, 1510–1522(2007).
Acknowledgments: We thank H. Yano for the pGEM-3Zf(+)-pOTC plasmid. Funding: Thiswork was supported by research grants from the DSF Charitable Foundation, the Ri.MED Foun-dation, the Consolidated Anti-Aging Foundation, NIH (NS095387, NS059806, ES022644,ES020718, ES020327, NS065789, AG026389, and P50AG005133), the U.S. Department of VeteransAffairs (1I01BX000548), the Blechman Foundation, the American Parkinson Disease Association, andthe Department of Biotechnology, Government of India. Author contributions: R.D.M. and P.J.B.designed, performed, and analyzed the PL and protein import experiments and edited the manu-script; E.K.H. was responsible for molecular biology and created and validated cell lines; C.W.B. alsodid someof theprotein import experiments; J.M. did someof the PLexperiments; A.Z. andA.B. did invivo gene transfer experiments; X.H. was responsible for cell culture experiments; C.T.C. supervisedhuman neuropathological studies; E.A.B. and T.G.H. designed and analyzed the experiments andedited the manuscript; and J.T.G. supervised the project, designed and analyzed the experiments,and wrote the paper. Competing interests: The authors declare that they have no competing in-terests. J.T.G. is a paid consultant for Biogen and FORMA Therapeutics.
Submitted 23 February 2016Accepted 17 May 2016Published 8 June 201610.1126/scitranslmed.aaf3634
Citation: R. Di Maio, P. J. Barrett, E. K. Hoffman, C. W. Barrett, A. Zharikov, A. Borah, X. Hu,J. McCoy, C. T. Chu, E. A. Burton, T. G. Hastings, J. T. Greenamyre, a-Synuclein binds toTOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 8,342ra78 (2016).
nceTranslationalMedicine.org 8 June 2016 Vol 8 Issue 342 342ra78 14

disease-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson'sα
Jennifer McCoy, Charleen T. Chu, Edward A. Burton, Teresa G. Hastings and J. Timothy GreenamyreRoberto Di Maio, Paul J. Barrett, Eric K. Hoffman, Caitlyn W. Barrett, Alevtina Zharikov, Anupom Borah, Xiaoping Hu,
DOI: 10.1126/scitranslmed.aaf3634, 342ra78342ra78.8Sci Transl Med
consequences.species. This study also highlights potential ways to prevent this deleterious interaction and its downstreamsenescence of mitochondria, which show reduced respiration and increased production of reactive oxygen receptor TOM20. This results in impaired import of proteins required for mitochondrial function and leads todopamine-modified forms, but not the monomeric or fibrillar forms, bind with high affinity to the mitochondrial
-synuclein, such as oligomeric andαDi Maio and colleagues report that specific forms of wild-type Now,Parkinson's disease and appear to intersect, but how the two are related to each other has remained elusive.
-Synuclein accumulation and mitochondrial dysfunction are central to the pathogenesis of most forms ofα-Synuclein disrupts the mitochondrial protein import businessα
ARTICLE TOOLS http://stm.sciencemag.org/content/8/342/342ra78
MATERIALSSUPPLEMENTARY http://stm.sciencemag.org/content/suppl/2016/06/06/8.342.342ra78.DC1
CONTENTRELATED
http://science.sciencemag.org/content/sci/360/6386/267.fullhttp://science.sciencemag.org/content/sci/358/6369/1440.fullhttp://science.sciencemag.org/content/sci/353/6307/1498.full
REFERENCES
http://stm.sciencemag.org/content/8/342/342ra78#BIBLThis article cites 41 articles, 10 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.Science Translational Medicinetitle licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. TheScience, 1200 New York Avenue NW, Washington, DC 20005. 2017 © The Authors, some rights reserved; exclusive
(ISSN 1946-6242) is published by the American Association for the Advancement ofScience Translational Medicine
by guest on May 25, 2018
http://stm.sciencem
ag.org/D
ownloaded from





























