Embed Size (px)
Citation preview

21 August 1998
Ž .Chemical Physics Letters 293 1998 72–80
A systematic ab initio investigation of the open and ringstructures of ozone
Thomas Muller a, Sotiris S. Xantheas b, Holger Dachsel b, Robert J. Harrison b,¨Jaroslaw Nieplocha b, Ron Shepard c, Gary S. Kedziora c,1, Hans Lischka a,)
a Institut fur Theoretische Chemie und Strahlenchemie der UniÕersitat Wien, A-1090 Vienna, Austria¨ ¨b Pacific Northwest National Laboratory, Richland, WA 99352, USA
c Theoretical Chemistry Group, Chemistry DiÕision, Argonne National Laboratory, Argonne, IL 60439, USA
Received 11 May 1998
Abstract
˜ 1Ž Ž .The energy difference between the open and the ring isomer of ozone as well as the dissociation energy O X, A ™3 1Ž 3 y. Ž3 .. Ž .O X, S qO P have been determined at the CCSD T , MR-CISD and MR-AQCC levels of theory. Using correlation2 g
Ž .consistent basis sets up to quintuple-zeta quality, the estimated complete basis set limits for CCSD T and MR-AQCC liewithin 1 kcalrmol of the experimental value of 26.1"0.4 kcalrmol and place the ring isomer by 4.8 and 5.3 kcalrmol,respectively, above the dissociation limit. Zero-point vibrational corrections increase the latter two values by 1.4 kcalrmol.q 1998 Elsevier Science B.V. All rights reserved.
1. Introduction
Ozone plays a fundamental role in atmosphericchemistry. Its importance has lead to extensive theo-retical and experimental research efforts focused onboth its ground state and excited state properties.Extensive investigations of the potential energy sur-
Ž .faces PES of O have been performed by3w x w xBanichevich et al. 1 and by Xantheas et al. 2 .
Several previous studies have focused especially onŽ .the characterization of the global ‘open’ C and2v
˜ 1Ž . Ž .local ‘ring’ D minima on the ground state X, A3h 1Žw x.PES 3–6 . Although the ring isomer has not been
observed experimentally yet, interest in this species
) Corresponding author.1 Present address: Department of Chemistry, Northwestern Uni-
versity, Evanston, Il 60208, USA.
stems from its potential use as a high-energy fuelŽ w x.see, e.g., Refs. 7,8 . In order to assess the suitabil-ity of the ring isomer for such use, it is important to:Ž .1 accurately determine its energy separation withrespect to the global minimum and whether it liesabove or below the lowest dissociation limit O2Ž 3 y. Ž3 . Ž .X, S qO P ; and 2 investigate accessibleg
pathways for its formation, an effort involving thecharacterization of its excited states. The aim of thisstudy is to address the first question. Future studieswill concentrate on the characterization of the ex-cited states for this system.
It has been noted earlier from the analysis of theŽ w x.CI wavefunction see, e.g., Ref. 4 as well as
recently using the T diagnostic in the coupled-clus-1w xter expansion 5 that both the open and the ring
isomers do not represent true single-reference cases.Moreover, the importance of triple excitations in
0009-2614r98r$ - see front matter q 1998 Elsevier Science B.V. All rights reserved.Ž .PII: S0009-2614 98 00798-2

( )T. Muller et al.rChemical Physics Letters 293 1998 72–80¨ 73
coupled-cluster calculations has been stressed by Leew xand Scuseria 5,6 . The recent investigation by Wattsw xand Bartlett 9 even suggests non-negligible contri-
butions from quadruple and higher excitations. Inparticular, we note the strong dependence of theenergy separation D E between the open and theor
ring structure on the inclusion of triple excitations.D E amounts to y23.3 and y29.1 kcalrmol at theor
Ž . w xCCSD and CCSD T levels, respectively 5 .˜ 1Ž .The dissociation energy E of O X, A intodiss 3 1
Ž 3 y. Ž3 .O X, S qO P is even more difficult to com-2 gŽ .pute than D E . A FORS CASSCF calculationor
gave only 20% of the experimental E and even andiss
internally contracted multi-reference configurationŽinteraction with single and double excitations ic-
.MR-CISD using a cc-pVDZ basis produced only60% of the experimental value. Only the much larger
w xcc-pVQZ basis gave acceptable results 10 .Consideration of the multi-reference character of
the ozone molecule at the open, ring and dissociationgeometries is the main focus of our investigation.The most straightforward method to use would beMR-CI. However, truncated MR-CI calculations suf-fer from the well-known size-extensivity problemw x11 . This error can be reduced substantially by usingmethods such as MR averaged coupled-pair func-
Ž . w xtional MR-ACPF 12 or MR average quadraticŽ . w xcoupled cluster MR-AQCC 13 . Results from
MR-AQCC calculations are compared with thoseŽ .obtained from single-reference CCSD T calcula-
tions.Conceptually, the simplest reference space is the
Ž .complete active space CAS . However, with anincreasing number of electrons andror active or-bitals, its use soon leads to an extremely large com-putational effort. Therefore, another aspect of ourwork is to search for more economical referencespaces which give results comparable to those ob-tained from CAS references.
In addition to studying the correlation require-ments for the accurate calculation of E and D E ,diss or
we study the convergence with respect to the one-electron basis set. The systematic series of correla-
w Žtion-consistent basis sets cc-pVnZ sets n s.xD, T, Q, 5 developed by Dunning and co-workers
are used in order to determine the complete basis setŽ . Ž w xlimits CBS cf. Refs. 14–18 and references.therein of the energies and geometries calculated
using the various methods and reference selectionschemes.
2. Computational details
The calculations were carried out using uncon-Ž . w xtracted and internally contracted ic- MR-CISD 19 ,
w x Ž . w xMR-AQCC 13 and CCSD T 20,21 levels of the-Žory. Standard polarized valence cc-pVnZ, aug-cc-
. Ž .pVnZ and core-valence cc-pCVnZ basis sets de-Žveloped by Dunning et al. nsD, T, Q, 5; for refer-
w x.ences see Ref. 17 have been used. A CASSCFcalculation, used in this study as a reference calcula-tion, with 18 electrons in 12 valence orbitalsŽ Ž .. w xCAS 18r12 2 was performed first. The three 1soxygen orbitals were kept doubly occupied. Follow-
w xing the notation used in Ref. 2 the dominant orbitaloccupations and the list of active orbitals as well as
Žthe doubly-occupied orbitals are as follows C2v.symmetry adapted basis sets were used throughout :
open structure:Ž .2Ž .2Ž .2Ž .2Ž .2Ž .2Ž .2Ž .21a 1b 2a 3a 4a 2b 5a 3b1 1 1 1 1 1 1 1Ž .2Ž .2Ž .2Ž .2Ž .0Ž .0Ž .06a 4b 1b 1a 2b 7a 5b ;1 1 2 2 2 1 1
ring structure:Ž .2Ž .2Ž .2Ž .2Ž .2Ž .2Ž .2Ž .21a 1b 2a 3a 4a 2b 5a 3b1 1 1 1 1 1 1 1Ž .2Ž .2Ž .2Ž .2Ž .0Ž .0Ž .06a 1b 1a 2b 4b 7a 5b .1 2 2 2 1 1 1
These configurations constitute ;83% and ;
87%, respectively, of the weight of the CASSCFwavefunctions.
Based on these CASSCF calculations, a series ofreference configuration sets of increasing complexity
Ž .starting from a single-reference SR to a full-va-Ž .lence space CAS 18r12 reference were constructed
for the use in subsequent uncontracted MR-CISDand MR-AQCC calculations. The three oxygen 1sorbitals were kept frozen throughout. The variousreference configuration spaces are characterized bythe number of inactive and active orbitals, the totalnumber of reference configurations and configura-
Ž .tions, respectively see Table 1 . The following ref-erence configuration sets have been used.
Ø The single-reference set SR containing only thedominant configuration as given above.
Ž .Ø A minimal reference space denoted MIN-REFadditionally includes the next important configura-
Ž .2 Ž .2tion, i.e. the double excitation 1a ™ 2b for2 2
the open structure and the pair of degenerate double

( )T. Muller et al.rChemical Physics Letters 293 1998 72–80¨74
Ž .2 Ž .2 Ž .2 Ž .2excitations 6a ™ 4b and 3b ™ 4b for1 1 1 1
the ring structure.Ø SEL-REF denotes a threshold selected set with
a threshold criterion of 0.05 for the absolute value ofthe CI or AQCC CSF expansion coefficients. Theselection is carried out iteratively. Based on an SRcalculation an initial reference space is determined.The corresponding MR calculation yields an im-proved set of expansion coefficients to which theselection criterion is applied. This procedure is re-peated until the reference remains unchanged. Closeto the equilibrium geometries the reference space isinsensitive to geometry relaxation. Therefore the ref-erence space is kept fixed during the optimization ofeach structure.
Ø A reference configuration selection schemeŽbased on 5 primary configurations the combined set
of MIN-REF references for the open and ring struc-.ture from which all reference configurations are
generated by single and double excitations within theset of active orbitals. The set PRIM1-REF containsthe 10 active orbitals which are occupied in theprimary references only, i.e. the orbitals 7a and 5b1 1
are excluded from the original list of 12 orbitals.PRIM2-REF contains 12 internal orbitals, but re-
Ž .stricts the three 2s-type orbitals 3a , 4a , 2b to be1 1 1
inactive. PRIM3-REF is the same as PRIM2-REFbut lifts the restriction on the 2s-type orbitals.PRIM2-REF and PRIM3-REF are approximations to
Ž . Ž .CAS 12r9 and CAS 18r12 , respectively, reducing
Žthe total number of configurations by up to 80% see.Table 1 . The two most important configurations
were selected as primary references for the calcula-tion of the dissociated system.
Ž .Ø A CAS 12r9 reference space with 12 elec-trons in the valence space of 9 oxygen p orbitals.
Ž .Ø A full valence space CAS 18r12 with 18electrons in the valence space of 12 orbitals.
Ž .Internally-contracted ic- MR-SDCI calculationshave been carried out with a full-valence CASŽ . Ž .18r12 reference space exclusively. The CCSD Tcalculations are based on the RHF reference configu-rations as given above for the two minima. Uncon-tracted MR-SDCI and MR-AQCC geometry opti-mizations have been limited to the smaller referencespaces SR, MIN-REF and SEL-REF. The calculation
Ž .of the dissociation energy E is based on: 1 thediss
supermolecule approach with a geometry optimizedŽ3 y . Ž3O molecule S state and an oxygen atom P2 g
˚. Ž .state at a distance of 100 A; and 2 on separatecalculations for the oxygen molecule and oxygenatom, respectively, with appropriately split referenceconfiguration spaces. The differences in E ob-diss
tained from these two approaches illustrate the size-extensivity error. The CBS limit E was determined`
by a fit of the total energies E of a series ofn
cc-pVnZ results to the exponential form E sE qn `
BeyC n.The uncontracted MR-CISD and MR-AQCC cal-
culations have been carried out using the COLUMBUS
Table 1Characterization of the various configuration selection schemes in the CI calculations for the open and ring structures of ozone
aReference set Internal orbitals Number of references Number of configurations in MRCIcinactive active cc-pVTZ cc-pVQZ cc-pV5Z
SR 9 0 1 64 242 536d d dcMIN-REF 7 2r3 2r3 184 708 1570d d dcŽ .SEL-REF MRCI 5r4 7r8 7r14 125 4644 10317d d dcŽ .SEL-REF MR-AQCC 4 8 10r14 1205 4644 10317
PRIM1-REF 0 10 20 1127 4383 9768PRIM2-REF 3 9 169 11126 43506 97156PRIM3-REF 0 12 437 24547 97171 215873
Ž .CAS 12r9 3 9 666 38790 152744 341929Ž .CAS 18r12 0 12 4067 129053 514747 1157095
a In thousands.b h-functions omitted.c Pairs of values separated by slashes refer to the numbers of orbitalsrconfigurations for the open and ring structures, respectively.dRing structure.
( )T. Muller et al.rChemical Physics Letters 293 1998 72–80¨ 75
w xprogram package 22–24 . CI expansions up to 515million configurations for single-point calculations
Žare obtained with the larger reference spaces cf..Table 1 . For these cases, the recently developed
w xparallel CI program pciudg 25 of the COLUMBUS
suite has been used. The pciudg module is a mas-sively parallel implementation of the standard ciudg
w xCI program using the global array tools 26 . Thepciudg calculations were performed on a CRAY T3Eusing up to 512 processors with a parallel efficiencyof more than 99%.
The geometry optimizations for the uncontractedCI and AQCC calculations have been performed
w xwith the analytic gradient program 27,28 of COLUM-BUS using the AO integral and AO derivative integral
w xpackages of DALTON 29 . The internally contractedŽ .ic-MR-CISD and the CCSD T calculations were
w xcarried out using the MOLPRO 96 30 suite of pro-grams.
3. Results and discussion
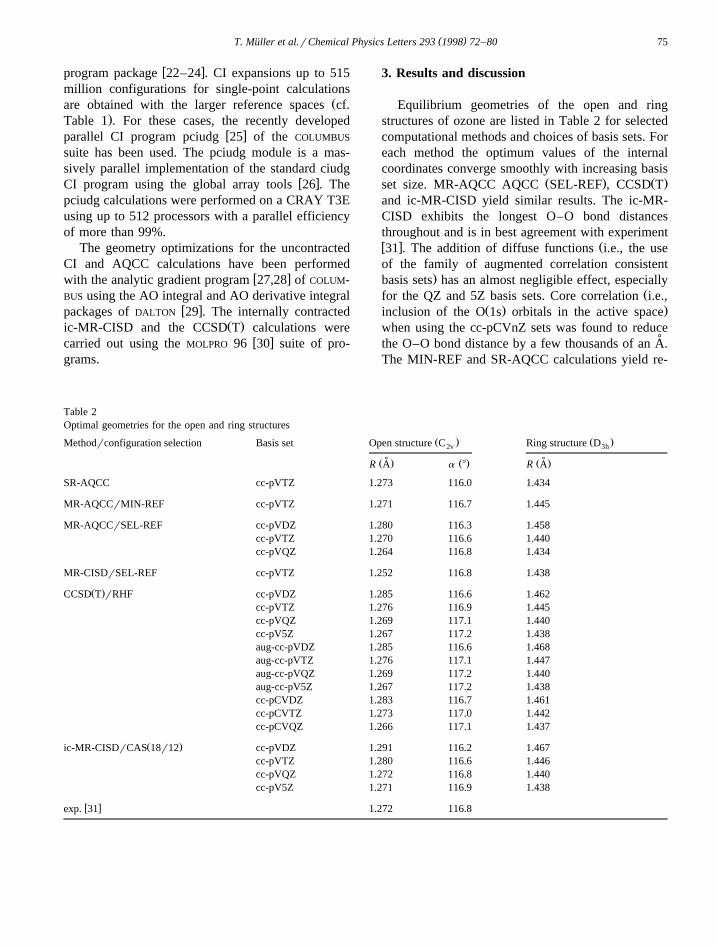
Equilibrium geometries of the open and ringstructures of ozone are listed in Table 2 for selectedcomputational methods and choices of basis sets. Foreach method the optimum values of the internalcoordinates converge smoothly with increasing basis
Ž . Ž .set size. MR-AQCC AQCC SEL-REF , CCSD Tand ic-MR-CISD yield similar results. The ic-MR-CISD exhibits the longest O–O bond distancesthroughout and is in best agreement with experimentw x Ž31 . The addition of diffuse functions i.e., the useof the family of augmented correlation consistent
.basis sets has an almost negligible effect, especiallyŽfor the QZ and 5Z basis sets. Core correlation i.e.,
Ž . .inclusion of the O 1s orbitals in the active spacewhen using the cc-pCVnZ sets was found to reduce
˚the O–O bond distance by a few thousands of an A.The MIN-REF and SR-AQCC calculations yield re-
Table 2Optimal geometries for the open and ring structures
Ž . Ž .Methodrconfiguration selection Basis set Open structure C Ring structure D2v 3h
˚ ˚Ž . Ž . Ž .R A a 8 R A
SR-AQCC cc-pVTZ 1.273 116.0 1.434
MR-AQCCrMIN-REF cc-pVTZ 1.271 116.7 1.445
MR-AQCCrSEL-REF cc-pVDZ 1.280 116.3 1.458cc-pVTZ 1.270 116.6 1.440cc-pVQZ 1.264 116.8 1.434
MR-CISDrSEL-REF cc-pVTZ 1.252 116.8 1.438
Ž .CCSD T rRHF cc-pVDZ 1.285 116.6 1.462cc-pVTZ 1.276 116.9 1.445cc-pVQZ 1.269 117.1 1.440cc-pV5Z 1.267 117.2 1.438aug-cc-pVDZ 1.285 116.6 1.468aug-cc-pVTZ 1.276 117.1 1.447aug-cc-pVQZ 1.269 117.2 1.440aug-cc-pV5Z 1.267 117.2 1.438cc-pCVDZ 1.283 116.7 1.461cc-pCVTZ 1.273 117.0 1.442cc-pCVQZ 1.266 117.1 1.437
Ž .ic-MR-CISDrCAS 18r12 cc-pVDZ 1.291 116.2 1.467cc-pVTZ 1.280 116.6 1.446cc-pVQZ 1.272 116.8 1.440cc-pV5Z 1.271 116.9 1.438
w xexp. 31 1.272 116.8
( )T. Muller et al.rChemical Physics Letters 293 1998 72–80¨76
sults of quite acceptable quality. The largest devia-tions are found for MR-CISD calculations with thesmaller reference spaces which produced O–O bond
˚distances that are too low by as much as 0.03 A. Atypical example consists of the SEL-REF MR-CISDcalculation. Accurate results are obtained only withthe much larger reference spaces used in the inter-nally contracted CI calculations.
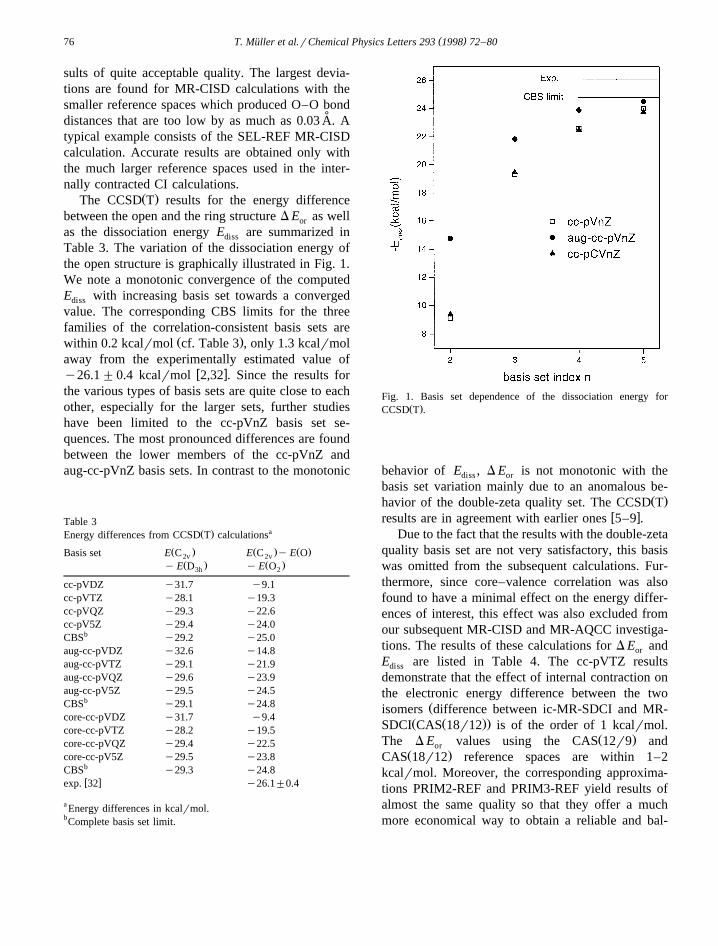
Ž .The CCSD T results for the energy differencebetween the open and the ring structure D E as wellor
as the dissociation energy E are summarized indiss
Table 3. The variation of the dissociation energy ofthe open structure is graphically illustrated in Fig. 1.We note a monotonic convergence of the computedE with increasing basis set towards a convergeddiss
value. The corresponding CBS limits for the threefamilies of the correlation-consistent basis sets are
Ž .within 0.2 kcalrmol cf. Table 3 , only 1.3 kcalrmolaway from the experimentally estimated value of
w xy26.1"0.4 kcalrmol 2,32 . Since the results forthe various types of basis sets are quite close to eachother, especially for the larger sets, further studieshave been limited to the cc-pVnZ basis set se-quences. The most pronounced differences are foundbetween the lower members of the cc-pVnZ andaug-cc-pVnZ basis sets. In contrast to the monotonic
Table 3Ž . aEnergy differences from CCSD T calculations
Ž . Ž . Ž .Basis set E C E C y E O2v 2vŽ . Ž .y E D y E O3h 2
cc-pVDZ y31.7 y9.1cc-pVTZ y28.1 y19.3cc-pVQZ y29.3 y22.6cc-pV5Z y29.4 y24.0
bCBS y29.2 y25.0aug-cc-pVDZ y32.6 y14.8aug-cc-pVTZ y29.1 y21.9aug-cc-pVQZ y29.6 y23.9aug-cc-pV5Z y29.5 y24.5
bCBS y29.1 y24.8core-cc-pVDZ y31.7 y9.4core-cc-pVTZ y28.2 y19.5core-cc-pVQZ y29.4 y22.5core-cc-pV5Z y29.5 y23.8
bCBS y29.3 y24.8w xexp. 32 y26.1"0.4
a Energy differences in kcalrmol.bComplete basis set limit.
Fig. 1. Basis set dependence of the dissociation energy forŽ .CCSD T .
behavior of E , D E is not monotonic with thediss or
basis set variation mainly due to an anomalous be-Ž .havior of the double-zeta quality set. The CCSD T
w xresults are in agreement with earlier ones 5–9 .Due to the fact that the results with the double-zeta
quality basis set are not very satisfactory, this basiswas omitted from the subsequent calculations. Fur-thermore, since core–valence correlation was alsofound to have a minimal effect on the energy differ-ences of interest, this effect was also excluded fromour subsequent MR-CISD and MR-AQCC investiga-tions. The results of these calculations for D E andor
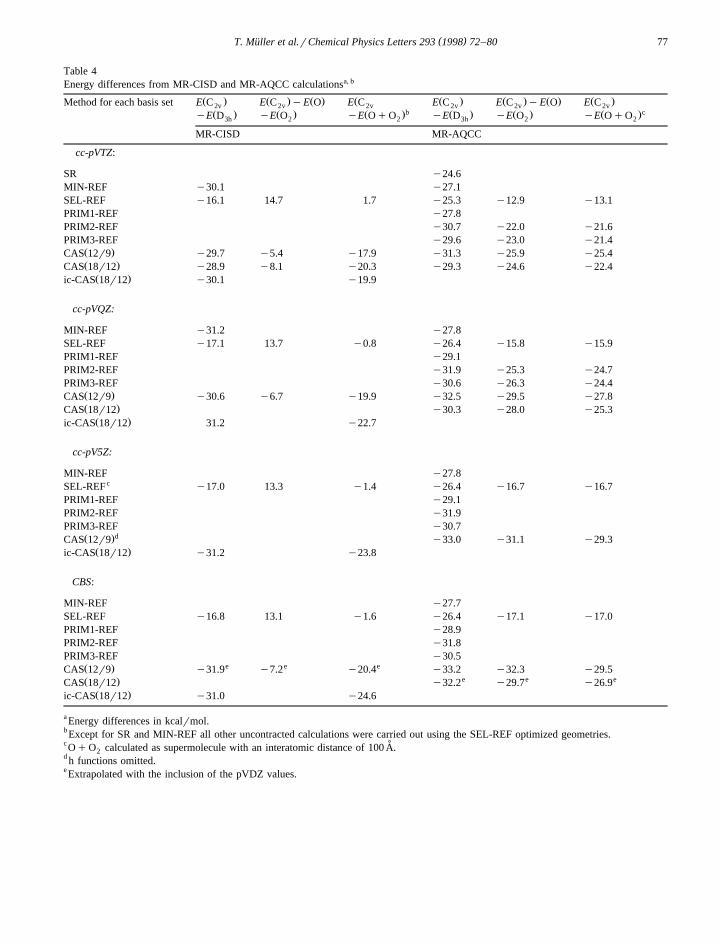
E are listed in Table 4. The cc-pVTZ resultsdiss
demonstrate that the effect of internal contraction onthe electronic energy difference between the two
Žisomers difference between ic-MR-SDCI and MR-Ž Ž ..SDCI CAS 18r12 is of the order of 1 kcalrmol.
Ž .The D E values using the CAS 12r9 andorŽ .CAS 18r12 reference spaces are within 1–2
kcalrmol. Moreover, the corresponding approxima-tions PRIM2-REF and PRIM3-REF yield results ofalmost the same quality so that they offer a muchmore economical way to obtain a reliable and bal-
( )T. Muller et al.rChemical Physics Letters 293 1998 72–80¨ 77
Table 4Energy differences from MR-CISD and MR-AQCC calculationsa, b
Ž . Ž . Ž . Ž Ž . Ž . Ž . Ž .Method for each basis set E C E C yE O E C E C E C yE O E C2v 2v 2v 2v 2v 2vb cŽ . Ž . Ž . Ž . Ž . Ž .yE D yE O yE OqO yE D yE O yE OqO3h 2 2 3h 2 2
MR-CISD MR-AQCC
cc-pVTZ:
SR y24.6MIN-REF y30.1 y27.1SEL-REF y16.1 14.7 1.7 y25.3 y12.9 y13.1PRIM1-REF y27.8PRIM2-REF y30.7 y22.0 y21.6PRIM3-REF y29.6 y23.0 y21.4
Ž .CAS 12r9 y29.7 y5.4 y17.9 y31.3 y25.9 y25.4Ž .CAS 18r12 y28.9 y8.1 y20.3 y29.3 y24.6 y22.4
Ž .ic-CAS 18r12 y30.1 y19.9
cc-pVQZ:
MIN-REF y31.2 y27.8SEL-REF y17.1 13.7 y0.8 y26.4 y15.8 y15.9PRIM1-REF y29.1PRIM2-REF y31.9 y25.3 y24.7PRIM3-REF y30.6 y26.3 y24.4
Ž .CAS 12r9 y30.6 y6.7 y19.9 y32.5 y29.5 y27.8Ž .CAS 18r12 y30.3 y28.0 y25.3
Ž .ic-CAS 18r12 31.2 y22.7
cc-pV5Z:
MIN-REF y27.8cSEL-REF y17.0 13.3 y1.4 y26.4 y16.7 y16.7
PRIM1-REF y29.1PRIM2-REF y31.9PRIM3-REF y30.7
dŽ .CAS 12r9 y33.0 y31.1 y29.3Ž .ic-CAS 18r12 y31.2 y23.8
CBS:
MIN-REF y27.7SEL-REF y16.8 13.1 y1.6 y26.4 y17.1 y17.0PRIM1-REF y28.9PRIM2-REF y31.8PRIM3-REF y30.5
e e eŽ .CAS 12r9 y31.9 y7.2 y20.4 y33.2 y32.3 y29.5e e eŽ .CAS 18r12 y32.2 y29.7 y26.9
Ž .ic-CAS 18r12 y31.0 y24.6
a Energy differences in kcalrmol.b Except for SR and MIN-REF all other uncontracted calculations were carried out using the SEL-REF optimized geometries.c ˚OqO calculated as supermolecule with an interatomic distance of 100 A.2d h functions omitted.eExtrapolated with the inclusion of the pVDZ values.

( )T. Muller et al.rChemical Physics Letters 293 1998 72–80¨78
anced description of both structures. The PRIM1-REFresults are generally too low by ;1–2 kcalrmol.The SR-, MIN-REF- and SEL-REF-MR-AQCC re-sults differ from the corresponding CAS referencespace result by several kcalrmol. The SEL-REFMR-CISD results are poor. The AQCC method im-proves the quality of the SEL-REF results substan-tially but cannot overcome the inherent bias of thisselection scheme. The MIN-REF CI results are sur-prisingly good in spite of the relatively small CIexpansion. This behavior is certainly fortuitous since,for example, the size-extensivity corrections givenby AQCC deteriorate the results. Moreover, it shouldbe noted that in contrast to the CAS and PRIM-REFschemes neither the MIN-REF nor the SEL-REFschemes provide a simultaneously balanced descrip-tion of the open and ring structure. In particular,PRIM1-REF is superior to the SEL-REF schemewhile costing roughly the same. However, the disso-ciation energy cannot be computed by the PRIM1-REF procedure since there is no unambiguous wayto construct a compatible set of primary referencesfor the dissociated system.
Further inspection of Table 4 reveals that keepingthe 2s orbitals inactive favors the open structure,which may be expected because the dominant con-figuration of the ring structure is of srs) type incontrast to the prp) type in the open structure.Reducing the number of available srs) excitationsby deleting the 2s orbitals from the active orbitals
Ž .should thus slightly favor the open structure.An accurate and affordable calculation of E isdiss
more difficult than that for D E . MR-CISD failsor
badly in terms of size-extensivity as can be seenfrom a comparison of the second and third column ofthe MR-CISD section of Table 4. This is true even
Ž .for the CAS 18r12 reference space. The super-molecule approach works much better and leads to
Ž .results which agree quite well with the CCSD Tvalues. It is evident that MR-AQCC performs wellwith respect to size-extensivity as may be seen bycomparing columns two and three of the MR-AQCCsection of Table 4. For small reference configurationspaces they differ by ;0.1 kcalrmol, for the largerreference spaces size-extensivity corrections tend toovershoot by ;2 kcalrmol. Employing the SEL-REF scheme introduces a considerable bias and isthus not well suited for the calculation of the dissoci-
ation energy. However, even in this unfavorable caseMR-AQCC largely improves the otherwise com-pletely unacceptable small reference space MR-CISDresults. Restricting the CAS to the oxygen p-orbitalsŽ Ž ..CAS 12r9 gives rise to a systematic error of ;3kcalrmol. The primary reference schemes appear tobe useful approximations but are certainly less bal-anced than for the D E case.or
The CBS limits of the most accurate methods,Ž .namely CCSD T , ic-MR-SDCI, MR-SDCI
Ž Ž .. Ž Ž ..CAS 18r12 and MR-AQCC CAS 18r12 sup-port a value of D E between y32.2 and y29.1or
kcalrmol. The analogous CBS limit for the dissocia-tion energy ranges from y26.9 to y24.6 kcalrmol,which is in good agreement with the experimentalvalue of y26.1"0.4 kcalrmol. Therefore, our bestestimates places the ring isomer 4.8, 5.3 and 6.4
Ž .kcalrmol, for CCSD T , MR-AQCC and ic-MRSDCI, respectively, above the dissociation limit.
ŽZero-point energy corrections reduce in absolute. w xvalue the energy gap D E by 0.4 kcalrmol 5 andor
w xthe dissociation energy by 1.8 kcalrmol 2 and thusincrease the relative position of the ring structurewith respect to the dissociation limit by 1.4 kcalrmolw x2,5 . As expected, the PRIM-REF results also com-pare favorably with the more elaborate calculations.
4. Conclusions
A variety of computational methods have beenused to investigate size-extensivity and multi-refer-ence effects on the relative stability of the open andring structure of ozone and the position of the ringisomer with respect to the lowest dissociation limit.Complete basis set limits were estimated by usingthe sequences of correlation consistent basis sets.Our calculations indicate that the energy differencebetween the open and ring isomers of O , D E , is in3 or
the range between y32.2 and y29.1 kcalrmol.Ž .CCSD T and MR-AQCC calculations with a basis
set of at least quadruple-zeta quality lie within 1kcalrmol of the experimental dissociation energy of26.1"0.4 kcalrmol, therefore suggesting that thering isomer of O lies above the lowest dissociation3
Ž .limit. Although CCSD T is considered to be mostaccurate for true single-reference cases, we obtainreliable results also for O , which certainly does not3

( )T. Muller et al.rChemical Physics Letters 293 1998 72–80¨ 79
Ž .belong to this category. The CCSD T results requireonly a fraction of the cost of the more expensive MRschemes. However, in critical cases it is necessary toverify the validity of the single-reference CC results
Žnot only through internal criteria such as the T1.diagnostic but also through comparative calculations
with true multi-reference methods. The characteriza-tion of energy surfaces requires multi-referencemethods anyway.
In order to reduce the well-known size-extensivityerror of the MR-CISD method to an acceptableamount, large CAS reference spaces had to be em-ployed. Requiring such large reference spaces makesaccurate CI calculations expensive. Even internallycontracted MR-SDCI schemes become unmanage-able for somewhat larger systems. MR-AQCC com-bines the virtues of the CC and MR-CI methods. Thesize-extensivity problem is largely reduced, and it isapplicable to cases of increasing multi-referencecharacter. MR-AQCC can provide good results withmuch less effort than MR-CI. However, the choiceof the reference space is crucial. A threshold drivenselection scheme for the reference configurationsgave discouraging results, whereas a scheme basedon a set of primary references from which additionalreferences were generated by single and double exci-tations into the valence orbital space was successful.
Acknowledgements
This work was performed under the auspices ofthe Austrian ‘‘Fonds zur Forderung der wis-¨senschaftlichen Forschung’’, project No. P10681-CHE, the High Performance Computing and Com-munication Program of the Office of Scientific Com-puting and the Division of Chemical Sciences, Officeof Basic Energy Sciences, U.S. Department of En-ergy, under contract DE-AC06-76RLO 1830 withBattelle Memorial Laboratory, which operates thePacific Northwest National Laboratory; and the Of-fice of Basic Energy Sciences, Division of ChemicalSciences, U.S. Department of Energy, under contractW-31-109-ENG-38 with the University of Chicago,which operates the Argonne National Laboratory.This research used resources of the National EnergyResearch Scientific Computing Center, which is sup-ported by the Office of Energy Research of the U.S.
Department of Energy under Contract No. DE-AC03-76SF00098. We are grateful for computer timeat the DEC Alpha Server of the Computer Center ofthe University of Vienna.
References
w x1 A. Banichevich, S.D. Peyerimhoff, F. Grein, Chem. Phys.Ž .178 1993 155.
w x2 S.S. Xantheas, G.J. Atchity, St.T. Elbert, K. Ruedenberg, J.Ž .Chem. Phys. 94 1991 8054.
w x3 S. Shih, R.J. Buenker, S.D. Peyerimhoff, Chem. Phys. Lett.Ž .28 1974 463.
w x4 P.J. Hay, T.H. Dunning Jr., W.A. Goddard III, J. Chem.Ž .Phys. 62 1975 3912.
w x Ž .5 T.J. Lee, Chem. Phys. Lett. 169 1990 529.w x Ž .6 T.J. Lee, G.E. Scuseria, J. Chem. Phys. 93 1990 489.w x Ž .7 E.T. Seidl, H.F. Schaefer III, J. Chem. Phys. 88 1988 7043.w x8 R.A. Copeland, Ch.G. Bressler, M.J. Dyer, in: P.G. Carrick,
Ž .N.T. Williams Eds. , Proceedings of the High Energy Den-Ž .sity Matter HEDM Contractor’s Conference, Boulder, CO,
March 1997, p.96.w x Ž .9 J.D. Watts, R.J. Bartlett, J. Chem. Phys. 108 1998 2511.
w x Ž .10 G.J. Atchity, K. Ruedenberg, Theor. Chem. Acc. 96 1997176.
w x Ž .11 R.J. Bartlett, G.D. Purvis, Int. J. Quantum Chem. 14 1978561.
w x Ž .12 R.J. Gdanitz, R. Ahlrichs, Chem. Phys. Lett. 143 1988 413.w x Ž .13 P.G. Szalay, R.J. Bartlett, Chem. Phys. Lett. 214 1993 481.w x Ž .14 S.S. Xantheas, T.H. Dunning Jr., J. Phys. Chem. 97 1993
18.w x Ž .15 D. Feller, J. Chem. Phys. 96 1992 6104.w x Ž .16 S.S. Xantheas, J. Chem. Phys. 104 1996 8821.w x17 K.A. Peterson, A.K. Wilson, D.E. Woon, Th.H. Dunning Jr.,
Ž .Theor. Chem. Acc. 97 1997 251.w x Ž . Ž .18 J.M.L. Martin, J. Mol. Struct. Theochem 398-399 1997
135.w x Ž .19 H.-J. Werner, P.J. Knowles, J. Chem. Phys. 89 1988 5803.w x20 K. Raghavachari, G.W. Trucks, J.A. Pople, M. Head-Gordon,
Ž .Chem. Phys. Lett. 157 1989 479.w x21 R.J. Bartlett, J.D. Watts, S. Kucharski, J. Noga, Chem. Phys.
Ž .Lett. 165 1990 513.w x22 COLUMBUS, an ab initio electronic structure program, Release
Ž .5.3 1997 , written by: H. Lischka, R. Shepard, I. Shavitt,F.B. Brown, R.M. Pitzer, R. Ahlrichs, H.-J. Bohm, A.H.H.¨Chang, D.C. Comeau, H. Dachsel, M. Dallos, R. Gdanitz, C.Erhard, M. Ernzerhof, P. Hochtl, S. Irle, G. Kedziora, T.¨Kovar, Th. Muller, V. Parasuk, M. Pepper, P. Scharf, H.¨Schiffer, M. Schindler, M. Schuler, P.G. Szalay, J.-G. Zhao.¨
w x23 H. Lischka, R. Shepard, F. Brown, I. Shavitt, Int. J. QuantumŽ .Chem. S15 1981 91.
w x24 R. Shepard, I. Shavitt, R.M. Pitzer, D.C. Comeau, M. Pep-per, H. Lischka, P.G. Szalay, R. Ahlrichs, F.B. Brown, J.G.
Ž .Zhao, Int. J. Quantum Chem. S22 1988 149.w x25 H. Dachsel, H. Lischka, R. Shepard, J. Nieplocha, R.J.
Ž .Harrison, J. Comput. Chem. 18 1997 430.

( )T. Muller et al.rChemical Physics Letters 293 1998 72–80¨80
w x26 J. Nieplocha, R.J. Harrison, R.J. Littlefield, J. Supercomput.Ž .10 1996 169.
w x27 R. Shepard, H. Lischka, P.G. Szalay, T. Kovar, M. Ernzer-Ž .hof, J. Chem. Phys. 96 1992 2085.
w x28 G. Kedziora, R. Shepard, H. Lischka, Th. Muller, unpub-¨lished work.
w x29 DALTON, an ab initio electronic structure program, ReleaseŽ .1.0 1997 , written by T. Helgaker, H.J.Aa. Jensen, P.
˚Jørgensen, J. Olsen, K. Ruud, H. Agren, T. Andersen, K.L.Bak, V. Bakken, O. Christiansen, P. Dahle, E.K. Dalskov, T.Enevoldsen, H. Heiberg, H. Hettema, D. Jonsson, S. Kir-
pekar, R. Kobayashi, H. Koch, K.V. Mikkelsen, P. Norman,M.J. Packer, T. Saue, P.R. Taylor, O. Vahtras.
w x30 MOLPRO 96 is a package of ab initio programs written byH.-J. Werner, P.J. Knowles, with contributions from J.Almlof, R.D. Amos, M.J.O. Deegan, S.T. Elbert, C. Hampel,¨W. Meyer, K. Peterson, R. Pitzer, A.J. Stone, P.R. Taylor, R.Lindh, M.E. Mura, T. Thorsteinsson.
w x Ž .31 T. Tanaka, Y. Morino, J. Mol. Spectrosc. 33 1970 538.w x32 D.W. Arnold, C. Xu, E.H. Kim, D.M. Neumark, J. Chem.
Ž .Phys. 101 1994 912.





























