Embed Size (px)
Citation preview


AAPS Advances in the Pharmaceutical Sciences Series
Volume 18
Series Editors Daan J. A. Crommelin Utrecht, Utrecht, The Netherlands
Robert A. LipperBack Cove Pharma, LLC, Waldoboro, Maine, USA

The AAPS Advances in the Pharmaceutical Sciences Series, published in partner-ship with the American Association of Pharmaceutical Scientists, is designed to de-liver well written volumes authored by opinion leaders and authorities from around the globe, addressing innovations in drug research and development, and best prac-tice for scientists and industry professionals in the pharma and biotech industries.
More information about this series at http://www.springer.com/series/8825

Feroz Jameel • Susan Hershenson Mansoor A. Khan • Sheryl Martin-MoeEditors
Quality by Design for Biopharmaceutical Drug Product Development
1 3

ISSN 2210-7371 ISSN 2210-738X (electronic)AAPS Advances in the Pharmaceutical Sciences SeriesISBN 978-1-4939-2315-1 ISBN 978-1-4939-2316-8 (eBook)DOI 10.1007/978-1-4939-2316-8
Library of Congress Control Number: 2015934451
Springer New York Heidelberg Dordrecht London© Springer Science+Business Media, LLC 2015This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed.The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use.The publisher, the authors and the editors are safe to assume that the advice and information in this book are believed to be true and accurate at the date of publication. Neither the publisher nor the authors or the editors give a warranty, express or implied, with respect to the material contained herein or for any errors or omissions that may have been made.
Printed on acid-free paper
Springer is part of Springer Science+Business Media (www.springer.com)
EditorsFeroz JameelParenteral Product and Process DevelopmentAmgen Inc.Thousand OaksCaliforniaUSA
Susan HershensonBill and Melinda Gates Foundation, Chemistry, Manufacturing and ControlsSeattleWashingtonUSA
Mansoor A. KhanDivision of Product Quality ResearchFood and Drug Administration, Center for Drug Evaluation and Research, Office of Testing and Research and Office of Pharmaceutical SciencesSilver SpringMarylandUSA
Sheryl Martin-MoeEnterprise Catalyst Group Inc.Palo AltoCaliforniaUSA

v
This work is dedicated to the memory of our friend and colleague Ronald Taticek as a tribute to the imagination that led him to undertake a project of this magnitude and made him a pioneer in this field. It is also a tribute to the dedication that inspired Ron to carry this effort so far to completion before he passed away on April 23, 2014. Ron will be greatly missed.

vii
Preface
Occasionally in one’s professional career you become aware that the hand of history is resting on your shoulder. So it was in July 2003, in Brussels, when the members of the International Conference on Harmonisation (ICH) Expert Working Groups (EWG) for quality agreed on a new vision and strategy for ICH. Summarized in the statement, “A harmonized pharmaceutical quality system applicable across the life cycle of the product emphasizing an integrated approach to quality risk manage-ment and science,” ICH agreed to progress three paradigm-changing guidelines. These were Q8 (pharmaceutical development), Q9 (quality risk management), and Q10 (pharmaceutical quality system). When I called to order the first Q8 EWG, we all thought that we might be able to take the existing European Note for Guidance on Development Pharmaceutics and convert it into an appropriate ICH format and that would be it: a simple task. It took us a little while to appreciate the futility of this approach, especially given the growing interest in the application of process analytical technology (PAT) and the growing appreciation that the goal of pharma-ceutical development is to design a quality product and its manufacturing process to deliver consistently the intended performance of the product. The only way to achieve that consistency would be by designing a product from the outset that would meet patients’ needs, acquiring comprehensive product and process understanding, and establishing a properly controlled manufacturing process. We needed to tell the world that quality cannot be tested into a product; it has to be designed into a product. But, of course, everyone already knew this, so there was nothing new here, but how could we help move the industry from its traditional 3-sigma processes toward 6-sigma? We needed to talk about Deming, Juran, kaizen, risk assessments, experimental designs, even the value of “failed” experiments. We needed to give the industry permission to share the fullness of their scientific knowledge without the fear of creating an ever-increasing list of regulatory questions that added little value but much time to the review and approval processes.
With these things in mind, the EWG drafted the ICH Q8 guideline. Recognizing that traditional development processes would still be needed, we referred to the new thinking as an “enhanced approach,” deliberately avoiding the moniker of “quality by design.” Even as Q8 went through its final revisions and adoption, it became clear that outside the confines of the EWG, neither the industry nor regulators had a clear understanding of the new paradigm. We were asked to use the addendum to

viii Preface
Q8 to define and exemplify “quality by design,” and we did our best, comparing traditional approaches with an enhanced quality-by-design approach. But even with this effort, and with subsequent Implementation Working Group efforts (which have included question and answer documents, points to consider), there is still mystery and confusion about what QbD really means for the pharmaceutical industry.
Fortunately, our journey has been helped by the foresight and commitment of a number of early adopters. Before the ink was dry on the first part of Q8, a team within the European Federation of Pharmaceutical Industries and Associations de-veloped a mock section P2 (Examplain), which demonstrated some of the key ele-ments of QbD including a quality target product profile, risk assessments, design of experiments, and design space. Two more comprehensive case studies, intended for discussion and teaching purposes, quickly followed. The first, ACE tablets, was aspirational in many respects and explored a number of innovative concepts that industry was contemplating. The second, A-Mab, discussed the application of QbD principles to a biotechnology product, stimulating much discussion between indus-try and regulators at the same time as the FDA was introducing its pilot programs. Other case studies such as the Sakura mock P2 from Japan and A-Vax (QbD for vaccines) and the several mock ANDA submissions have strengthened our under-standing and appreciation of both business and regulatory opportunities.
Many would regard QbD for chemical substances as straightforward: our under-standing of kinetics and thermodynamics enables rapid building on prior knowledge to provide scalable syntheses. On the other hand, drug product development still remains a complex blend of art and science which may be behind the often expe-rienced challenges of establishing well characterized, robust manufacturing pro-cesses that can be described by reliable models. For biologics, it could be argued that the opposite situation pertains. The drug substance is the process: the processes are often exquisitely designed and engineered with feed-forward and feedback con-trol strategies. While the quality is designed from the outset, the many degrees of freedom and the characterization challenges mean that full application of QbD prin-ciples is not easy. The list of critical quality attributes is generally extensive, our ability to directly connect them through analytical techniques back to the critical process parameters and forward to the patient is often not straightforward, and the realization of design spaces becomes challenging, especially when you consider the risks associated with movement with a design space. However, application of QbD principles to the final steps, the drug product, is much more straightforward.
Into one insightful volume is collected a wide range of discussions and prac-tical examples of the application of QbD to biological drug products. For those still uncertain about the business benefit, this is the area to start. Biological drug product manufacturing processes lend themselves to the enhanced approach. The risks, science and engineering are all much better understood than those in many other areas of our industry. The degrees of freedom are manageable. QbD prin-ciples facilitate developing an effective control strategy, arguably the most critical deliverable of a well planned and executed development program, including real-time release-testing opportunities.

ixPreface
Most of the leading pharma companies now consider QbD to be “business as usual” for the current development portfolio. An increasing number of publications attest to the business benefits that have accrued from QbD programs and filings. Experience is growing with successful regulatory submissions and approvals. For sure, both industry and agencies have been on a steep learning curve with the new paradigm, but in the USA, the small molecule pilot program followed by the bio-logics pilot program have provided valuable insight and learning. Similar initiatives have occurred elsewhere. The international agencies have mounted joint assessment and inspection programs—our new paradigm is here to stay, and the publication of this book could not be better timed. Now is the time to wholeheartedly grasp the op-portunities, to do the great science that surely motivates us all and comprehensively tell the story to the regulators. What are you afraid of? The patient is waiting.
John Berridge, Kent, UK([email protected])

xi
Contents
1 Challenges and Opportunities for Biotech Quality by Design ............... 1Cyrus Agarabi, Mansoor A. Khan and Rakhi B. Shah
2 Lessons Learned from Monoclonal Antibody Applications to the Office of Biotechnology Products Quality by Design Pilot Program ...................................................................................................... 17Barbara L. Rellahan, Steven Kozlowski and Patrick Swann
3 Definitions and Scope of Key Elements of QbD ...................................... 31Ron Taticek and Jun Liu
4 An Overview of Quality by Design for Drug Product ............................. 47Sheryl Martin-Moe and Carol Nast
5 Development of Drug Product Formulations: Molecular Design and Early Candidates Screening .................................................. 61Michael Siedler, Vineet Kumar, Ravi Chari, Sonal Saluja and Wolfgang Fraunhofer
6 Approaches for Early Developability Assessment of Proteins to Guide Quality by Design of Liquid Formulations .............................. 87Bernardo Perez-Ramírez, Nicholas Guziewicz, Robert Simler and Alavattam Sreedhara
7 Application of QbD Principles to Late-Stage Formulation Development for Biological Liquid Products ........................................... 115Alavattam Sreedhara, Rita L. Wong, Yvonne Lentz, Karin Schoenhammer and Christoph Stark
8 Application of QbD Principles for Lyophilized Formulation Development ............................................................................................... 137Ambarish Shah, Sajal M. Patel and Feroz Jameel

xii Contents
9 Drug Substance Frozen Storage and Thawing ........................................ 159Philippe Lam, Fredric J. Lim and Samir U. Sane
10 Quality by Design as Applied to Drug Substance Formulation Using Ultrafiltration and Diafiltration .............................. 191Joseph Edward Shultz, Herb Lutz and Suma Rao
11 A QbD Approach in the Development and Scale-Up of Mixing Processes ......................................................................................... 211Feroz Jameel and Sonja Wolfrum
12 Application of QbD Elements in the Development and Scale-Up of a Commercial Filtration Process .......................................... 237Feroz Jameel
13 Application of QbD Elements in the Development and Scale-up of Commercial Filling Process ................................................... 265Feroz Jameel, Cenk Undey, Paul M. Kovach and Jart Tanglertpaibul
14 Lyophilization Process Design and Development Using QbD Principles .................................................................................................... 303Sajal M. Patel, Feroz Jameel, Samir U. Sane and Madhav Kamat
15 Visible and Subvisible Protein Particle Inspection Within a QbD-Based Strategy .................................................................................. 331Erwin Freund and Shawn Cao
16 Quality by Design for Distribution of Environmentally Sensitive Pharmaceutical Products .......................................................... 353Paul Harber
17 Quality by Design for Primary Container Components ......................... 365Fran DeGrazio and Lionel Vedrine
18 Devices and Combination Products for Biopharmaceuticals ................ 403Rey T. Chern, Jeffrey C. Givand, Robin Hwang and Thomas J. Nikolai
19 Applicability of QbD for Vaccine Drug Product Development .............. 437Liuquan (Lucy) Chang, Jeffrey T. Blue, Joseph Schaller, Lakshmi Khandke and Bruce A. Green
20 Automation and High-Throughput Technologies in Biopharmaceutical Drug Product Development with QbD Approaches ................................................................................................. 475Vladimir Razinkov, Jerry Becker, Cenk Undey, Erwin Freund and Feroz Jameel

xiiiContents
21 Critical Quality Attributes, Specifications, and Control Strategy......... 511Timothy Schofield, David Robbins and Guillermo Miró-Quesada
22 Multivariate Analysis for Process Understanding, Monitoring, Control, and Optimization of Lyophilization Processes ... 537Theodora Kourti
23 Using Mathematical Modeling and Prior Knowledge for QbD in Freeze-Drying Processes .............................................................. 565Davide Fissore, Roberto Pisano and Antonello A. Barresi
24 Application of Multivariate Statistical Process Monitoring to Lyophilization Process ............................................................................... 595Fuat Doymaz
25 Application of PAT in Real-time Monitoring and Controlling of Lyophilization Process ........................................................................... 605Feroz Jameel, William J. Kessler and Stefan Schneid
26 Product Homogeneity Assessment During Validation of Biopharmaceutical Drug Product Manufacturing Processes ................ 649Fuat Doymaz, Frank Ye and Richard K. Burdick
27 Application of Quality by Design Principles to the Drug Product Technology Transfer Process ...................................................... 661Fredric J. Lim, Jagannathan Sundaram and Alavattam Sreedhara
28 Regulatory Considerations for Implementation of the QbD Paradigm for Biologics: Laying the Foundation for Product and Process Lifecycle Management.......................................................... 693Lynne Krummen
Index .................................................................................................................. 707

xv
Contributors
Cyrus Agarabi Division of Product Quality Research, Office of Testing and Research and Office of Pharmaceutical Sciences, Center for Drug Evaluation and Research, Food and Drug Administration, Silver Spring, MD, USA
Antonello A. Barresi Dipartimento di Scienza Applicata e Tecnologia, Politecnico di Torino, Torino, corso Duca degli Abruzzi 24, Italy
Jerry Becker Drug Product Development, Amgen Inc., Seattle, WA, USA
Jeffrey T. Blue Vaccine Drug Product Development, Merck, West Point, PA, USA
Richard K. Burdick Amgen Inc. One Amgen Center Drive, Longmont, CO, USA
Shawn Cao Process and Product Development, Amgen Inc., Thousand Oaks, CA, USA
Liuquan (Lucy) Chang Biopharm Development, Vaccine Research & Early Development, Pfizer Inc., Teva Biopharmaceuticals, Rockville, MD, USA
Ravi Chari Preformulation, Bioresearch Center, AbbVie, Worcester, MA, USA
Rey T. Chern Merck Manufacturing Division, Pharmaceutical Packaging Tech-nology & Development, West Point, PA, USA
Fran DeGrazio Global R & D, Strategic Program Management and Technical Customer Support, West Pharmaceutical Services, Exton, PA, USA
Fuat Doymaz Global Quality Engineering, Amgen Inc., Thousand Oaks, CA, USA
Davide Fissore Dipartimento di Scienza Applicata e Tecnologia, Politecnico di Torino, Torino, corso Duca degli Abruzzi 24, Italy
Wolfgang Fraunhofer Combination Products-Biologics, Drug Product Develop-ment, AbbVie, Chicago, IL, USA
Erwin Freund Parenteral Product and Process Development, Amgen, Inc., Thousand Oaks, CA, USA

xvi Contributors
Jeffrey C. Givand Device Development, Merck Research Laboratories, West Point, PA, USA
Bruce A. Green Vaccine Research and Early Development, Pfizer, Pearl River, NY, USA
Nicholas Guziewicz Drug Product Process Technology, Amgen Inc., Thousand Oaks, CA, USA
Paul Harber Modality Solutions, LLC, Indianapolis, IN, USA
Robin Hwang ICP Consulting Corp., Thousand Oaks, CA, USA
Feroz Jameel Parenteral Product and Process Development, Amgen Inc., Thousand Oaks, CA, USA
Drug Product Engineering, Amgen, Inc., Thousand Oaks, CA, USA
Madhav Kamat Bristol-Myers Squibb, New Brunswick, NJ, USA
William J. Kessler Physical Sciences Inc., Andover, MA, USA
Mansoor A. Khan Division of Product Quality Research, Office of Testing and Research and Office of Pharmaceutical Sciences, Center for Drug Evaluation and Research, Food and Drug Administration, Silver Spring, MD, USA
Lakshmi Khandke Vaccine Research and Early Development, Pfizer, Pearl River, NY, USA
Theodora Kourti Global Manufacturing & Supply, GSK, London, UK
Paul M. Kovach Drug Product Commercialization Technology Center, Manu- facturing Science and Technology, Eli Lilly and Company, Indianapolis, IN, USA
Steven Kozlowski Office of Biotechnology Products, Office of Pharmaceutical Sciences, Center for Drug Evaluation and Research, Food and Drug Administration, Bethesda, MD, USA
Lynne Krummen Regulatory Affairs Department, Genentech, South San Francisco, CA, USA
Vineet Kumar Pharmaceuticals, Johnson and Johnson, Malvern, PA, USA
Philippe Lam Pharmaceutical Processing and Technology Development, Genentech, San Francisco, CA, USA
Yvonne Lentz Global Manufacturing Sciences and Technology Biologics, Genentech, San Francisco, CA, USA
Fredric J. Lim Pharmaceutical Processing and Technology Development, Gene- ntech, South San Francisco, CA, USA
Jun Liu Pharma Technical Operations, Development, Genentech Late Stage Pharmaceutical Development, San Francisco, CA, USA

xviiContributors
Herb Lutz Biomanufacturing Sciences Network, EMD Millipore Corp., Darmstadt, Germany
Sheryl Martin-Moe Enterprise Catalyst Group Inc., Palo Alto, CA, USA
Guillermo Miró-Quesada Quantitative Sciences, MedImmune, Gaithersburg, MD, USA
Carol Nast Enterprise Catalyst Group Inc., Palo Alto, CA, USA
Thomas J. Nikolai Biologics Processing Development, Hospira, Lake Forest, IL, USA
Sajal M. Patel Formulation Sciences, Biopharmaceutical Development, MedIm-mune, Gaithersburg, MD, USA
Bernardo Perez-Ramírez BioFormulations Development, Global Biotherapeutics, Sanofi Corporation, Framingham, MA, USA
Lynn Phelan Vaccine Research and Early Development, Pfizer, Pearl River, NY, USA
Roberto Pisano Dipartimento di Scienza Applicata e Tecnologia, Politecnico di Torino, Torino, corso Duca degli Abruzzi 24, Italy
Suma Rao Process and Product Development, Amgen Inc, Thousand Oaks, CA, USA
Vladimir Razinkov Drug Product Development, Amgen Inc., Seattle, WA, USA
Barbara L. Rellahan Division of Monoclonal Antibodies, Office of Biotechnology Products, Office of Pharmaceutical Sciences, Center for Drug Evaluation and Research, Food and Drug Administration, Bethesda, MD, USA
Amgen Inc., Rockville, MD, USA
David Robbins Purification Process Sciences, MedImmune, Gaithersburg, MD, USA
Sonal Saluja Bioresearch Center, AbbVie, Worcester, MA, USA
Samir U. Sane Pharmaceutical Processing and Technology Development, Genentech, San Francisco, CA, USA
Samir U. Sane Pharmaceutical Development, Genentech, San Francisco, CA, USA
Joseph Schaller Sterile/Liquids Commercialization, Merck, West Point, PA, USA
Stefan Schneid Syntacoll GmbH, Saal, Germany
Karin Schoenhammer Technical Research and Development, Biologics, Novartis Pharma AG, Basel, Switzerland
Timothy Schofield Regulatory Sciences & Strategy, Analytical Biotechnology, MedImmune, Gaithersburg, MD, USA

xviii Contributors
Ambarish Shah Formulation Sciences, Biopharmaceutical Development, MedIm-mune, Gaithersburg, MD, USA
Rakhi B. Shah Division of Product Quality Research, Office of Testing and Research and Office of Pharmaceutical Sciences, Center for Drug Evaluation and Research, Food and Drug Administration, Silver Spring, MD, USA
Joseph Edward Shultz Biologics Process Research and Development, Novartis Pharma AG, Basel, Switzerland
Michael Siedler NBE Formulation Sciences & Process Development, AbbVie, Ludwigshafen, Germany
Robert Simler Formulation and Process Development, Biogen Idec., Cambridge, MA, USA
Alavattam Sreedhara Late Stage Pharmaceutical Development, Genentech, San Francisco, CA, USA
Christoph Stark Technical Research and Development, Biologics, Novartis Pharma AG, Basel, Switzerland
Jagannathan Sundaram Global Biologics Manufacturing Science and Technology, Genentech, San Francisco, CA, USA
Patrick Swann Division of Monoclonal Antibodies, Office of Biotechnology Products, Office of Pharmaceutical Sciences, Center for Drug Evaluation and Research, Food and Drug Administration, Bethesda, MD, USA
Biogen Idec, Cambridge, MA, USA
Jart Tanglertpaibul Drug Product Commercialization Technology Center, Manufacturing Science and Technology, Eli Lilly and Company, Indianapolis, IN, USA
Ron Taticek Pharma Technical Operations, Biologics, Genentech Vacaville Operations, South San Francisco, CA, USA
Cenk Undey Process Development, Amgen Inc, Thousand Oaks, CA, USA
Lionel Vedrine Device Development, Genentech, San Francisco, CA, USA
Sonja Wolfrum Institute of Particle Technology, University of Erlangen-Nuremberg, Erlangen, Germany
Rita L. Wong Global Manufacturing Sciences and Technology Biologics, Genentech, South San Francisco, CA, USA
Frank Ye Amgen Inc. One Amgen Center Drive, Thousand Oaks, CA, USA

xix
About the Editors
Dr. Feroz Jameel is a Principal Scientist for Parenteral Product & Process Development at Amgen Inc., in Thousand Oaks, CA, where he is involved in the development, optimization, scale-up and transfer to manufacturing of biopharmaceutical products. Feroz received his Master’s degree in Pharmaceutics from the University of Delhi and his Ph.D in Pharmaceutics from the University of Connecticut. His publications include a co-edited book, several book chapters and more than 40 peer-reviewed manuscripts and presentations. He has held multiple leadership positions at the American Association of Pharmaceutical Scientists (AAPS) including chair of the freezing and drying technologies group and lead of the industrial consortium for application of QbD to lyophilization. He is a recipient of two patents in lyophilization formulation and lyophiliztion process development. Dr. Jameel has received several awards including the AAPS and Parental Drug Association’s Fred Simon Award for the best paper published in the Journal of Pharmaceutical Science and Technology. He has also chaired several symposia on the development of biological products.
Dr. Susan A. Hershenson is the Deputy Director of Chemistry, Manufacturing and Controls at the Bill and Melinda Gates Foundation, where her major responsibilities are to support the CMC development and drug delivery needs for therapeutic projects funded by the Foundation. She has many years of experience in drug development, including biologics, small molecules, combination products and drug delivery systems. Prior to the Gates Foundation, she served as President of Pharmaceutical Transformations LLC, a consulting service for the pharmaceutical, biotechnology, drug delivery and related industries. Her clients included a wide range of biotechnology, pharmaceutical and drug delivery companies, start-ups, venture capitals, university labs and non-profit organizations. Before starting her own practice, she served in a number of positions in the biopharmaceutical industry, including Vice President of Pharmaceutical and Device Development at Genentech and Vice President of Pharmaceutics at Amgen. During her career, Dr. Hershenson has made significant contributions to the development and commercialization of numerous therapeutic products including BETASERON®, Stemgen®, Kepivance®, Aranesp®, Neulasta®, Sensipar®, Nplate®, Vectibix®, Prolia®, XGEVA® and

xx
Nutropin AQ NuSpin® and has supported the development of numerous clinical candidates. She received her Ph.D. in Biochemistry from Yale University and held a postdoctoral fellowship in the laboratory of Dr. Robert Stroud at the University of California, San Francisco. Dr. Hershenson publishes and teaches actively and serves on multiple scientific advisory boards.
Dr. Mansoor A. Khan is the Director of Product Quality Research and a Senior Biomedical Research Scientist at the Center for Drug Evaluation and Research at the Food and Drug Administration (FDA), where he overseas research teams on biotech products, chemistry and stability, drug delivery systems and bioavailability/bioequivalence. Prior to joining the FDA, he was a Professor of Pharmaceutics and Director of the graduate program in the School of Pharmacy at Texas Tech University Health Sciences Center. He is a registered pharmacist and earned his Ph.D. in Industrial Pharmacy from the St. John’s University School of Pharmacy. He has published more than 255 peer-reviewed manuscripts, four texts, 25 book chapters, 200 poster presentations, and more than 175 invited presentations worldwide. He has held several leadership positions at the American Association of Pharmaceutical Scientists (AAPS) including elected chair of pharmaceutics and drug delivery (PDD) and the founding chair of formulations design and development (FDD). He serves on the editorial board of Pharmaceutical Technology, the International Journal of Pharmaceutics, AAPS Pharmsci Tech, and Drug Delivery and Translational Research. He has received the AAPS Research Achievement Award in Formulations Design and Development and is an AAPS Fellow
Dr. Sheryl Martin-Moe is an Executive Vice President at Enterprise Catalyst Group in Palo Alto, CA, where she consults for biotechnology, pharmaceutical and related companies and serves on scientific advisory boards. She has managed the development of more than 90 diverse drugs and combination products. She received her Ph.D. in Cell Biology from the University of Vermont and was a postdoctoral fellow in Biochemistry at the University of California, Berkeley. She worked at Sterling Drug in its research division and held management positions in various Development and Operation areas at Centocor, Bayer, and Genentech and was most recently Global Head of Pharmaceutical Development at Novartis Biologics in Basel, Germany. Her publications include two patents, two book chapters and 21 publications and invited presentations, including a publication that won the Parenteral Drug Association’s Fred Simon Award. Dr. Martin-Moe was a member of the CMC Biotech Working Group and co-authored its A-Mab case study for QbD.
About the Editors

1
Chapter 1Challenges and Opportunities for Biotech Quality by Design
Cyrus Agarabi, Mansoor A. Khan and Rakhi B. Shah
© Springer Science+Business Media, LLC 2015F. Jameel et al. (eds.), Quality by Design for Biopharmaceutical Drug Product Development, AAPS Advances in the Pharmaceutical Sciences Series 18, DOI 10.1007/978-1-4939-2316-8_1
R. B. Shah () · C. Agarabi · M. A. KhanDivision of Product Quality Research, Office of Testing and Research and Office of Pharmaceutical Sciences, Center for Drug Evaluation and Research, Food and Drug Administration, 10903 New Hampshire Ave, Silver Spring, MD 20993, USAe-mail: [email protected]
1.1 Introduction
The goal of biotechnological product development is to design and establish a for-mulation composition and robust manufacturing process to consistently and reliably meet all the quality standards intended for its therapeutic purpose. Traditionally, products are released onto the market only after successful ‘end product testing’, however, with the introduction of ‘Quality by Design’ (QbD) for pharmaceuticals (ICH Q8 2009), quality standards need to be built into the product by design and cannot be met merely at the end-product-testing stage. A scientific knowledge base along with appropriate quality risk management principles (ICH Q9 2005, ICH Q10 2008) and enhanced process and product understanding through process analytical technology (PAT) principles (PAT guidance 2004) can offer advantages for biotech product manufacturing over a traditional approach (Table 1.1).
Bioprocessing is generally divided into two stages; the upstream operations for the generation of the active biological ingredient referred to as the drug substance, and the fill-finish activities that are required to generate a finished drug product. For the scope of this chapter, the terms, biotech molecules and proteins, are limited to monoclonal antibodies or therapeutic proteins.
These active biological ingredients are more complex than small molecules, as their biological activity requires a unique 3-D structural conformation. Addition-ally, proteins are prone to degradation throughout bioprocessing; examples include
The findings and conclusions in this chapter have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.

2 C. Agarabi et al.
deamidation, oxidation, hydrolysis, aggregation, and denaturation, which can result in activity loss and/or immunogenicity. Often, proteins undergo a post-translational modification in the upstream drug substance processing during biosynthesis. The site of the post-translational modification can vary and potentially produce a protein with more than one form, for example, various glycosylated forms of a monoclonal antibody. Such structural heterogeneity is sometimes inevitable and is challenging to address throughout drug substance and drug product manufacturing.
Due to the complex physicochemical and stability issues, the majority of biotech products are administered via parenteral routes with intravenous and subcutaneous being the most common routes of administration. Biotech-finished drug products can be broadly classified as liquids and lyophilized powders for reconstitution prior to injection. Relative to small molecules, the fill-finish manufacturing steps for bio-tech drug products do not involve complex multi-step processes, with lyophilization a notable exception. Due to the complex nature of the molecules, there are signifi-cant challenges in consistently manufacturing high-quality biotech drug products. Yielding consistent product quality with minimum or no failed batches is a goal for any biopharmaceutical scientists. Rejected or failed batches not only results in loss of revenue but can also bring about negative criticism from the stakeholders and users. Therefore, QbD principles are based upon the idea that quality cannot be tested in the products but should be built-in by design. QbD can offer advantages for complex protein products even as the science and technology to support sev-eral elements of QbD are still evolving. Application of QbD to biotech products is not trivial and some of the challenges presented include (i) structural complexity
Table 1.1 Salient features of pharmaceutical development under traditional and QbD paradigm (ICH Q8 2009)Aspects Traditional QbDPharmaceutical development Empirical; univariate
experimentsSystematic; multivariate experiments
Manufacturing process Fixed; validation on three ini-tial full-scale batches; focus on reproducibility
Adjustable within design space; continuous verifica-tion; focus on control strategy and robustness
Process control In-process testing for go/no-go; offline analysis with slow response
PAT utilized for feedback and feed forward, real time
Product specification Primary means of quality control; based on batch data
Part of the overall quality control strategy; based on desired product performance
Control strategy Mainly by intermediate and end product testing
Risk-based; controls shifted upstream; real-time release
Lifecycle management Reactive to problems and OOS; post-approval changes needed
Continuous improvement enabled within design space

31 Challenges and Opportunities for Biotech Quality by Design
of the biotech drug substance, (ii) a lack of understanding of interactions between drug substance with formulation excipients, (iii) assigning clinically relevant specifications to a biotech product, and (iv) constructing a multidimensional design space for a biotech product at various scales.
Despite so many challenges, it is possible for biotech industries and regulato-ry agencies to mutually benefit by adopting QbD principles (Rathore and Winkle 2009; Rathore 2009; Shah et al 2010). In 2008, the FDA announced a notice of a pilot program in Federal Register regarding voluntary submissions of applications under the QbD paradigm for biotech drugs following the successful voluntary pro-gram for small molecules QbD (FDA notice, 2008).
1.2 QbD Implementation in Biomanufacturing
QbD implementation is a multi-step approach and is well defined in ICH guidance documents (ICH Q8 2009, ICH Q9 2005, ICH Q10 2008). It is schematically rep-resented in Fig. 1.1, as an iterative risk assessment process in which the quality tar-get product profile is initially predetermined. QbD principles can be very helpful to understand critical quality attributes (CQA), process parameters, and impact of variations in formulation or process on CQAs. Through risk management and sta-tistical approaches a design space can be constructed and followed for a bioprocess manufacturing. The overall approach detailed in the section below for the biotech drug substance and products which are mainly categorized as liquid or lyophilized formulations.
Process
Design Space
Product Performance
PAT
Product quality/ performance achieved by ef-fective and efficient manu-facturing processes
Product specifications based on mecha-nistic understanding of how formulation and process variables impact biotech product performance
Continuous “real time” assurance of quality
Fig. 1.1 Quality roadmap for a bioprocess

4 C. Agarabi et al.
1.2.1 Drug Substance Manufacturing
Figure 1.2 is an example of the unit operations, which comprise drug substance manufacturing. The process begins with the thawing of cells from a working cell bank, the growth and expansion of the cells through different scales into a com-mercial scale bioreactor. Once the cell culture process is complete, the material is removed from the reactor and concentrated via centrifugation. The concentrate is clarified and purified, usually via chromatographic methods, of unwanted host cell proteins and other impurities to yield a pure protein. The analytics listed in Fig. 1.2 are examples of techniques which may be used in various unit operations throughout the drug substance manufacturing process. The media composition, pH, dissolved oxygen (DO), amino acid (AA) analysis, optical density (OD), viable cell density (VCD), and off gas analysis may be commonly utilized in-line, on-line, or off-line to monitor parameters in the bioreactor during cell culture. The chromatographic methods, such as size exclusion (SEC), cation exchange (CEX), anion exchange (AEX) with pulsed amperometric detector (PAD), are generally studied off-line. While all of the unit operations offer opportunities to explore QbD principles, the cell culture of materials in stirred tank bioreactors is an area of particular interest. Due to the high costs and complexities of utilizing living systems to generate active biologic materials and the potential for irreversible damage which may travel down-stream to the final drug product, there is a great demand for the enhanced process understanding which a QbD approach can establish.
1.2.2 Liquid Formulations
Many biologics are formulated as liquid formulations at the end of the downstream purification process. Liquid manufacturing involves mixing the drug substance with other excipients including pH modifiers, tonicity agents, stabilizers, surfac-tants, chelators, etc. followed by filtration, fill/finish operations. Inspection at the end of line has been done in automated mode by using automated machines for the clarity of the solution (Knapp and Abramson 1990). However, understanding the stability during shelf life in various buffer systems, pH, ionic strength, stabilizers, and preservatives is an important quality attribute. Additionally, there is a grow-ing trend towards more complex delivery systems for liquid formulations, which include prefilled syringes. A prefilled syringe is a single-dose unit of a biologic to which a needle is fixed. Disposable syringes are used for this purpose in which the liquid drug product is filled so that exact dose of the drug is available for the patient without the need of a pre-injection step, i.e. withdrawal from the vial. This eliminates waste due to vial overfilling, it is easier to handle and more convenient for the patients. However, interaction of the drug product with the syringe material poses a technological challenge for such delivery systems (Soikes 2011). Thus in a systematic QbD development approach, the compatibility of the syringe material, stability, and safety should become an integral part of the novel delivery systems for biotech products.
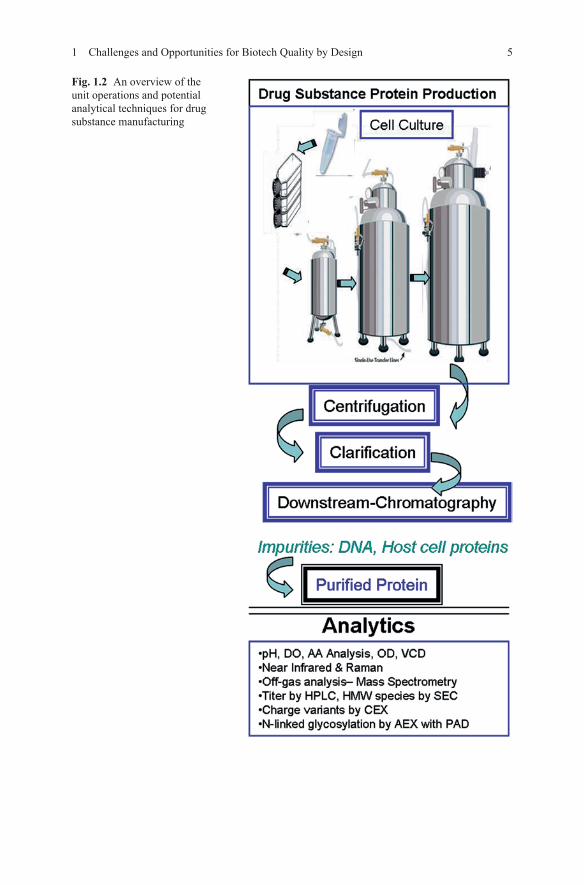
51 Challenges and Opportunities for Biotech Quality by Design
Fig. 1.2 An overview of the unit operations and potential analytical techniques for drug substance manufacturing

6 C. Agarabi et al.
1.2.3 Lyophilized Formulations
Lyophilization or freeze drying is a low temperature drying to convert solutions of heat-labile materials into solids. For a biotech drug substance, this process is employed if the molecule is unstable in liquid form to increase the shelf-life of the product. Lyophilization is a multi-step process which includes a (i) freezing, (ii) primary drying, and (iii) secondary drying. In this case, phase transformations oc-cur in this process and therefore present unique challenges during manufacturing to maintain stability of the molecule. The following section describes how a QbD approach can be useful for a lyophilization process and some of the challenges encountered.
1.3 Challenges and/or Opportunities of QbD for Biotech Products
1.3.1 Quality Target Product Profile (QTPP) and Critical Quality Attributes (CQAs) for Biotech Formulations
A quality target product profile (QTPP) is defined as ‘a prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy of the drug product’ (ICH Q8 2009). QTPP should consider route of administration, dosage form and strength, delivery system, container closure system, attributes affecting pharma-cokinetic characteristics, and drug product quality criteria. Commonly identified QTPP for a biotech liquid product includes clarity, subvisible particulates, pH, concentration, bioactivity, sterility, stability, color, odor, etc. For a lyophilized product, QTPP include pharmaceutical elegance in terms of cake appearance, reconstitution time, moisture content in addition to applicable requirements of liquid dosage forms.
Once the QTPP has been identified, the next step is identifying the relevant CQAs. A CQA is defined as ‘a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or dis-tribution to ensure the desired product quality’ (ICH Q8 2009). CQAs for drug substance, excipients, intermediates (in-process materials), and drug products can be determined based on either prior experience or from experimental designs. The key consideration is an assessment of the extent to which individual CQA variation can have an impact on the overall quality of the drug product. Examples of poten-tial drug substance CQAs include host cell protein (HCP) content, glycan forma-tion/heterogeneity, aggregates, potency, titer, etc. For a liquid drug product, this might include clarity, stability, sterility, etc. Additionally, in the case of pre-filled syringes, compatibility between container closure (i.e. syringe material) and drug substance need to be studied thoroughly. CQAs usually are thought to be a subset

71 Challenges and Opportunities for Biotech Quality by Design
of QTPP. For a lyophilized product, it may include reconstitution time in addition to other applicable attributes listed for a liquid product. Thus, there may be multiple CQAs for a product, and it can become very complex while applying risk assess-ment principles or other QbD tools.
1.3.2 Setting Specification for a Biotech Drug Product
Traditionally specifications for a biotech drug product were set on the basis of end product testing rather than by design. Under the science and risk-based approaches, QbD may help set the specifications based on user requirements rather than those obtained in the current state of manufacturing. A ‘specification’ is defined as a list of tests, reference to analytical procedures, and appropriate acceptance criteria that are numerical limits, ranges or other criteria (ICH Q6A 1999). For a biotech drug product, conformance to specifications would mean that when tested according to the listed analytical procedure, a liquid or lyophilized product will meet the listed acceptance criteria.
For a liquid or lyophilized biotech product, aggregation of drug substance may be performed as an in-process test, or may be performed as a release test, depend-ing on its relevance to product performance. Justification of specification should include either linkage to performance, stability, safety, efficacy, etc. Thus QbD can help set clinically relevant or meaningful specifications for a biotech drug product.
1.3.3 Quality Risk Management (QRM) for Biotech Formulations
Quality risk management (QRM) helps to identify and control potential quality is-sues from early stages of development to marketing and large-scale manufacturing for biopharmaceutical drug products. It gives a higher level of product quality as-surance. It increases cost saving and efficiency for industry as well as regulators. For sponsors, it facilitates innovation, increases manufacturing efficiency, reduces cost and product rejections, minimizes or eliminates potential compliance actions, enhances opportunities for first cycle approval, and streamlines post-approval changes and regulatory processes. It also offers opportunities for continuous im-provement. For regulators, it helps in more focused inspections and reduces burden of post-approval supplements.
The most daunting risk factors are those that occur sparsely and have a cata-strophic impact on the product and can only be detected after it is too late to correct the problem. Microbial or viral contamination of a bioreactor can bring a manu-facturer to a grinding halt if not handled correctly. During the risk assessment, it is important to understand the causes of contamination, examples include: improper cleaning/sterilization of equipment, improper aseptic techniques by operators, and

8 C. Agarabi et al.
contamination during sampling. By identifying potential failure modes during de-velopment, proper safeguards and risk mitigation techniques can be established.
For a liquid drug product, challenges to develop a stable solution formulation include multiple degradation pathways and chemical modifications to the molecule. Therefore, assessing the stability risk is of great importance in the overall QRM process for biotech liquid products. Moreover, formulation excipients, processing factors, as well as container-closure systems play an important role in the stability of the molecule. Initial risk assessment can be done by various methods, one of which is by using Ishikawa or Fishbone diagram.
For a lyophilized drug product, risk assessment could be performed for all the identified CQAs, which include but are not limited to cake appearance, reconstitution time, moisture content, stability, potency, etc. An example for a risk assessment is provided in Fig. 1.3. Once the initial risk assessment is performed, all the factors can be identified that could impact the CQAs. However, in order to manage and/control those that pose a significant risk, risk analysis at times is often performed. This process assigns a score depending upon the severity (impact) and occurrence (frequency) as well as ease of detectability in case of an excursion. Risk priority number (RPN) scores are calculated as shown below.
It is often challenging to assign a rating for severity, occurrence, and detectability, especially if there is little institutional knowledge regarding the product or pro-cess. A consistent scale should be used each time to prevent bias or variable results (Table 1.2). Often times, low scoring can be justified based on experience, litera-ture, or expert knowledge. However, since this exercise is done without any prior experiments, it is often times semi-quantitative. Once the experimental values are
Risk priority number Severity Occurrence Detectability.= × ×
Lyophilized mAb stability
Plant
Raw materials Manufacturing
Analytical
Drug substanceBuffers
Stabilizers
Cell-culture
Freeze-drying
Filling
Sampling
Instrument RH
Temp
Location
Operator
Method
Size
conformation
Duration
Temperature
downstreampH
composition
Impurities
Fig. 1.3 Fishbone (Ishikawa) diagram for CQA of stability for a biotech-lyophilized drug product

91 Challenges and Opportunities for Biotech Quality by Design
available, it can be treated as more quantitative and is a very useful tool to focus on high-risk factors. Risk review and reassignment of risk numbers is an iterative pro-cess and the model is continuously improved and updated with additional knowl-edge gained about the impact of the risk factors on the CQAs.
1.3.4 Biotech Formulation Design Space
After QRM, once the risk factors are identified and prioritized, a statistical approach may be useful to perform experiments in a multidimensional manner. This is espe-cially important for the design space determination. Design Space is defined as ‘the multidimensional combination and interaction of input variables (e.g. material at-tributes) and process parameters that have been demonstrated to provide assurance of ’quality’ (ICH Q8 2009). Design space is proposed by an applicant and is subject to approval by the regulatory agency. The development of a bioprocess design space requires a careful analysis of process data obtained during development and routine manufacturing along with data from the statistical design of experiments to develop appropriate process models and predict process performance.
Design of experiments (DoE) has been found to be useful for understanding the impact of input process and/or formulation factors on the CQAs. This has been done for small molecules in a two-tiered way (Zidan et al. 2007; Shah and Khan 2005; Shah et al. 2004; Nazzal et al. 2002). In this approach, a screening DoE is first used to identify the main effects of input factors on the dependent variable and those with the highest impact can be studied further using optimization or higher resolu-tion DoEs. In the response surface DoEs, three or four factors can be studied in a multidimensional way that can provide interaction of the input factors in addition to the main effects. It is critical to decipher the impacts of main and interaction effects when creating a design space.
Due to the staggering costs of experimentation at the manufacturing scale, it is essential to have lab scale systems, which accurately reflect the commercial process. The implications for robust low-volume parallel apparatuses with high-throughput
Table 1.2 Risk priority numbera score based on ratings of severity, occurrence and detectability (ICH Q9 2005)Ratings Severity Occurrence Detectability1 Minimal or no impact Not likely High2 Minor impact Low likelihood Moderate3 Moderate impact; workarounds possible Likely Medium4 Unacceptable impact; workarounds
possibleHighly likely Low
5 Unacceptable impact; no alternatives exist
Near certainly None
a RPN score (severity × occurrence × detectability) will vary from 1 to 125

10 C. Agarabi et al.
capabilities, including cost-effective disposable systems for supporting DOE and QbD principles have been proposed (Rao et al. 2009). Micro-reactors, stirred tank assemblies below 100 mL have been studied for rapid screening and have shown promise with regard to scalability. Although, significant hurdles remain due to small volumes with regard to emulating equivalent control of gassing resulting in excess foaming, and uneven gas distribution (Chen et al. 2009).
At the bench top scale, commercially available cell culture reactors are avail-able with working volumes ranging from approximately 100 mL to 14 L. Through mathematical modelling of the oxygen mass transfer rates via the calculation of the volumetric mass transfer coefficient ( KLa) values, bench top-scale bioreactors have been modelled against commercial scale reactors to guide process scale up (Garcia-Ochoa and Gomez 2003). A case study of 2 L bioreactors was experimen-tally evaluated against 15 and 110 L reactors and modelled for 2000 L vessel opera-tion (Zhang and Mostafa 2009). The authors successfully used a fractional factorial experimental design for clone selection and process optimization for temperature, pH, and pH shift from parallel 2 L reactors, which was ultimately transferred to the commercial scale.
There are various other approaches to determine design space which includes a first-principle approach where combination of experimental data and mechanistic knowledge of chemistry, physics, and engineering is used to model and predict the performance. Scale-up correlations are also important when the experiments are conducted at a laboratory scale. Verification experiments at a larger scale may be performed for a stronger correlation of the design space at various scales.
1.3.5 Bioprocess Parameters and Control
Process analytical technology (PAT) is gaining momentum after the guidance docu-ment was published by the FDA in September 2004 (PAT guidance 2004). A fun-damental element of PAT is to increase process understanding and control, and is useful to verify product quality prior to release. A control strategy is defined as ‘A planned set of controls, derived from current product and process understanding that assures process performance and product quality’ (PAT guidance 2004). For a biotech drug product, PAT offers the possibility to increase the product quality, consistency, and reduce product risk through increased process knowledge and un-derstanding with the optimized process control through the use of various tools. PAT has a potential to offer ‘real-time release’ (RTR) in lieu of end product test-ing. In RTR, material attributes are measured and controlled along with process parameters. Material attributes can be assessed using direct and/or indirect process analytical methods. However, for bioprocesses, it is challenging to obtain RTR due to the complexity of the products. Additional research is required for bioprocessing to help monitor and control the manufacturing process.
During the cell culture process, each cell line requires a unique blend of nutri-ents, buffers, and other additives to produce a highly active protein with desirable critical quality attributes with a cost effective yield. There are a wide variety of

111 Challenges and Opportunities for Biotech Quality by Design
suppliers, which manufacture chemically defined media and offer an opportuni-ty for evaluation during the development phase to study the effects of initial and in process feeding. At the commercial scale, where tens of thousands of litres are required, it is often more cost-effective to formulate the media in-house with the required nutrients and supplements and to devise a tailored feeding strategy for the individual cell line application. Stand alone or autosampler interfaced nutrient analyzers utilize electrochemical sensors and commonly measure glucose, gluta-mine, glutamate, lactate, ammonium, sodium, potassium, ionized calcium, partial pressure of oxygen (PO2), partial pressure of carbon dioxide (PCO2), pH, and use freezing point depression to measure osmolality.
Examples of on-line HPLC of amino acids for cell culture feeding strategies have been seen for over 15 years (Kurokawa et al. 1994) and can play a pivotal role in the quantitation of nutrient consumption during processing. Advances in HPLC capabilities for automated derivitization of amino acids through autosampler pro-gramming alleviates the need for tedious off-line manual sample preparation (Frank and Powers 2007) and supports automated feedback loop strategies. Quantitation of nutrient consumption patterns are essential components to develop additional capabilities, which require model building and chemometrics and do not directly measure concentration. A prominent example is near-infrared (NIR) spectroscopy, which has been studied by a number of authors as an in-line tool to monitor the cell culture process within the bioreactor. Quantitation of prevalent components, such as glucose, glutamine, and lactate, is commonly studied, and evaluated against commercially available nutrient analyzers. More advanced models have evaluated biomass (Tosi et al. 2003), protein titer (Mattes et al. 2007), and the application of multiplexing NIR probes for DoE studies (Roychoudhury et al. 2007). A challenge associated with NIR is its sensitivity to water, resulting in a large absorbance of water peaks at 1400 and 1900–2100 nm, which may limit its utility for cell culture medium, which are aqueous in nature. Peak overlap of nutrients has also been iden-tified as a complication of NIR, and may additionally limit the applications of the technique (Arnold et al. 2002).
Raman spectroscopy, which is not affected by water, has emerged as a potential spectroscopic alternative to NIR for cell culture monitoring, and has been demon-strated to be a capable in-line tool for real-time monitoring of nutrient consump-tion. Multiplexed raman systems to quantitate common media components, such as glucose, lactate, glutamate, and ammonium, as well as osmolarity and viable cell density (VCD) have been studied (Moretto et al. 2011). The applications of this technique are limited and require further investigation.
Process parameters in the case of liquid manufacturing include time, speed, and temperature of mixing, hold time, excipient quality. The process parameters for filtration unit operation include filtration pressure, flow rate, and the attributes that needs to be controlled include aggregate level or filtration type (Sharma et al. 2008). Pressure gauge and/or flow meters have been used to control the filtration unit op-eration (Sharma et al. 2008). For a filling unit operation, process parameters include fill weight/volume, fill temperature, vial size, filling speed, etc. A balance or even Nuclear magnetic resonance (NMR) for more accurate monitoring has been cited

12 C. Agarabi et al.
in the literature (Aldridge 2007). Some of these process control techniques are eas-ily adapted to various scales, for example, automated inspection, pressure gauge, weight measurements, etc. However, sophisticated techniques such as NMR have limitations for use at a commercial scale due to the time and specialized expertise required for the measurements.
For a lyophilized product, optimum freeze-drying process relies on the knowl-edge of critical properties of the formulation and process parameters and application of this information to process design. The critical formulation properties include the collapse temperature of the formulation, the stability of the drug, and the proper-ties of the excipients used (Tang and Pikal 2004). Above the glass temperature, the freeze-dried cake will be physically unstable, which may result in crystallization of one or more of the compounds in the formulation, or shrinkage of the cake. The sta-bility issue is particularly important for biotech drugs, as some of the biotech drugs may be sensitive to the stresses imposed by freeze drying and may be degraded or decomposed during the process. Also, interplay between the choice of proper excipients and the process have an important impact on quality and stability of the product. The glass transition temperature can be influenced by the moisture content and the choice of excipients.
There is a need for online and in-line measurement sensors which can act as an ‘eye’ of the process to monitor the real-time phenomena occurring during process (Shah et al. 2011). These techniques are crucial during the liquid or lyophilized formulations manufacturing process. Various sensors which are being considered to monitor the process of freeze-drying are described in the literature (Tang et al. 2006a, b, c; Roy and Pikal 1989; Schwegman et al. 2007; Read et al. 2010). Mano-metric Temperature Measurments (MTM) is a procedure to measure the product temperature at the sublimation interface during primary drying by quickly isolat-ing the freeze-drying chamber from the condenser for a short time and by subse-quent analysis of the pressure rise during this period (Tang et al. 2006a, b, c). The method is relatively easy to use and yields product dry-layer resistance in real time. Figure 1.4 shows a freeze drying process profile for a study of model monoclonal antibody formulations. The MTM was useful in detecting the end point of primary drying with three thermocouples placed in vials and using pirani gauge as well as capacitance manometer readings. Although, MTM is generally limited to lab-scale freeze-drying measurements, it has a potential to be applied for larger scales as well.
While PAT is still an evolving area, it is believed that over time it may be seen as an initiative that helped foster a period of innovation, efficiency, and expansion for the biopharmaceutical industry.
1.4 Summary
QbD methodologies with the identification and justification of target product pro-files, and product and process understanding to obtain preset specifications, with appropriate control strategies can ensure a state of control. This provides a potential





























