Embed Size (px)
Citation preview

Ab Initio and DFT Investigation of theStructures and Properties ofChloromethyl and ChlorofluoromethylPeroxyl Radicals
S. EL-TAHERDepartment of Chemistry, Faculty of Science, Cairo University, Giza, Egypt
Received 11 May 2004; accepted 28 June 2004Published online 17 November 2004 in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/qua.20363
ABSTRACT: Ab initio and Density Functional Theory (DFT) calculations wereperformed to determine the equilibrium geometries, charge distributions, spin densitydistributions, dipole moments, electron affinities (EAs), and C–O bond dissociationenergies (BDEs) of CH2ClO2
• CHCl2O2•, CCl3O2
•, CF2ClO2•, CFCl2O2
•, and CHFClO2•
peroxyl radicals. The C–H BDEs of the parent methanes were calculated using the samelevels of theories. Both MP2(full) and B3LYP methods, using the 6-31G(d,p) basis set,were found to be capable of accurately predicting the geometries of peroxyl radicals.The B3LYP/6-31G(d,p) method was found to be comparable to high ab initio levels inpredicting C–O BDEs of studied peroxyl radicals and C–H BDEs of the parent alkanes.The progressive chlorine substitution of hydrogen atoms in methyl peroxyl radicalsresults in an increase (decrease) of the spin density on the terminal (inner) oxygen, adecrease in dipole moments, and an increase in electron affinities. The substitution offluorine by chlorine in the series CF3O2
• – CCl3O2• was found to lengthen (destabilize)
the C–O bonds. Both C–O BDEs and EAs of peroxyl radicals (RO2• ) were found to
correlate well with Taft �* substituent constants for the R groups. © 2004 WileyPeriodicals, Inc. Int J Quantum Chem 102: 178–188, 2005
Key words: ab initio; DFT; peroxyl radicals; bond dissociation energy (BDE);electron affinity
Introduction
P eroxyl radicals (RO2• ) have received much
attention because of the essential roles theyplay in atmospheric chemistry [1–4] as well as in
biological systems [5–9]. They are known to formvia reactions of alkyl radicals with oxygen [10]:
R• � O2 � RO2• (1)
The equilibrium described in (1) is of fundamentalimportance in radical chain oxidation in chemistryand biology [11]. The important role of peroxylradicals has stimulated a number of theoreticalstudies of their structures and reactivity [12–26].
Current address and Correspondence to: S. El-Taher, TeachersCollege in Dammam, P.O. Box 2375, Dammam 31451, SaudiArabia. E-mail: [email protected]
International Journal of Quantum Chemistry, Vol 102, 178–188 (2005)© 2004 Wiley Periodicals, Inc.

However, the studies concerning the structure andproperties of RO2
•, particularly the halogenatedones, are limited. Moreover, the substituent effectson the structure and properties of RO2, and on theenergetics of the C–OO bonds that entirely governthe kinetics of reaction (1) have not been studied ina systematic fashion.
Most recently [27], we presented a systematicinvestigation of the halogen substituent effects onthe geometries, charge distributions, spin densities,dipole moments, electron affinities, vibrational fre-quencies, and C–O bond dissociation energies(BDEs) of the fluoromethyl peroxyl radicals, usingab initio and Density Functional Theory (DFT) cal-culations. These properties are found to be impor-tant parameters in the understanding of the elec-tronic structure of peroxyl radicals as well as theirreactivity. In accord with previous investigations[18, 19, 22–24], the study reported that DFT pro-vides values for the studied parameters that arecomparable to high levels of ab initio calculations.Additionally, the B3LYP/6-31G(d,p) level wasfound to be a good choice for prediction of substitu-ent effects on the thermochemical properties of al-kyl and peroxyl radicals.
Here, we report ab initio and DFT results of theparameters: geometries, charge distributions, spindensities, dipole moments, electron affinities, andC–O BDEs of the chloromethyl and chlorofluoro-methyl peroxyl radicals, and C–H BDEs of the par-ent halomethanes. The study aims to: 1) explore theeffects of halogen substituents (Cl, F) on the studiedparameters, and 2) to justify the performance of theDFT, compared to ab initio methods, in predictingthe values of these parameters.
Methods of Calculation
All ab initio molecular orbital theory and densityfunctional theory calculations were performed usingGaussian 98W [28]. Molecular geometries were fullyoptimized at the HF/6-31G(d,p) and MP2(full)/6-31G(d,p) levels of theory [29]. MP2(full) indicates allelectrons were included in electron correlation calcu-lations. The unrestricted Hartree-Fock (UHF) wasused for all open-shell free radicals. The default opti-mization criteria were employed. Zero-point vibra-tional energies (ZPEs) were scaled by 0.8929 in thecalculation of thermochemical parameters. Single-point energy calculations were carried out at theMP4(SDTQ)/6-311�G(d,p)//MP2(full)/6-31G(d,p)level of theory [29].
Optimized geometries were also computed at theB3LYP/6-31G(d,p) level of DFT. The B3LYP [30]functional is a modification of the Becke’s three-parameter hybrid exchange functional (B3) [31] to-gether with the correlation functional of Lee, Yang,and Parr (LYP) [32]. This functional has been shownto provide reliable geometries, frequencies, andbond energies [33, 34]. ZPEs were scaled by 0.9806in the calculation of thermochemical parameters.Single-point energy calculations were performedat the B3LYP/6-311�G(2df,2p) level using theB3LYP/6-31G(d,p) geometries. To properly evalu-ate the electron affinities, the 6-31�G(d,p) basis setwas used for both the peroxyl anions and peroxylradicals.
Results and Discussion
EQUILIBRIUM GEOMETRIES
It has been noted experimentally [12a] that theelectronic state of all peroxyl radicals of Cs symme-try is 2A�, i.e., the radicals are �-radicals. As hasbeen pointed out [12b], CH3O2
• has two Cs symmet-ric minima: with the �H–C–O–O torsion angle ofeither 0.0° (cis) or 180° (trans). The trans structure isfound to be more stable than the cis one by about0.4 kcal. On the other hand, for CHCl2O2
• whileMP2 predicts the trans structure to be more stableby 0.1 kcal than the cis one, B3LYP predicts the cisstructure to be more stable by 0.4 kcal. However,the trans structure is adopted as the global mini-mum for the studied peroxyl radicals, except for theCHFClO2
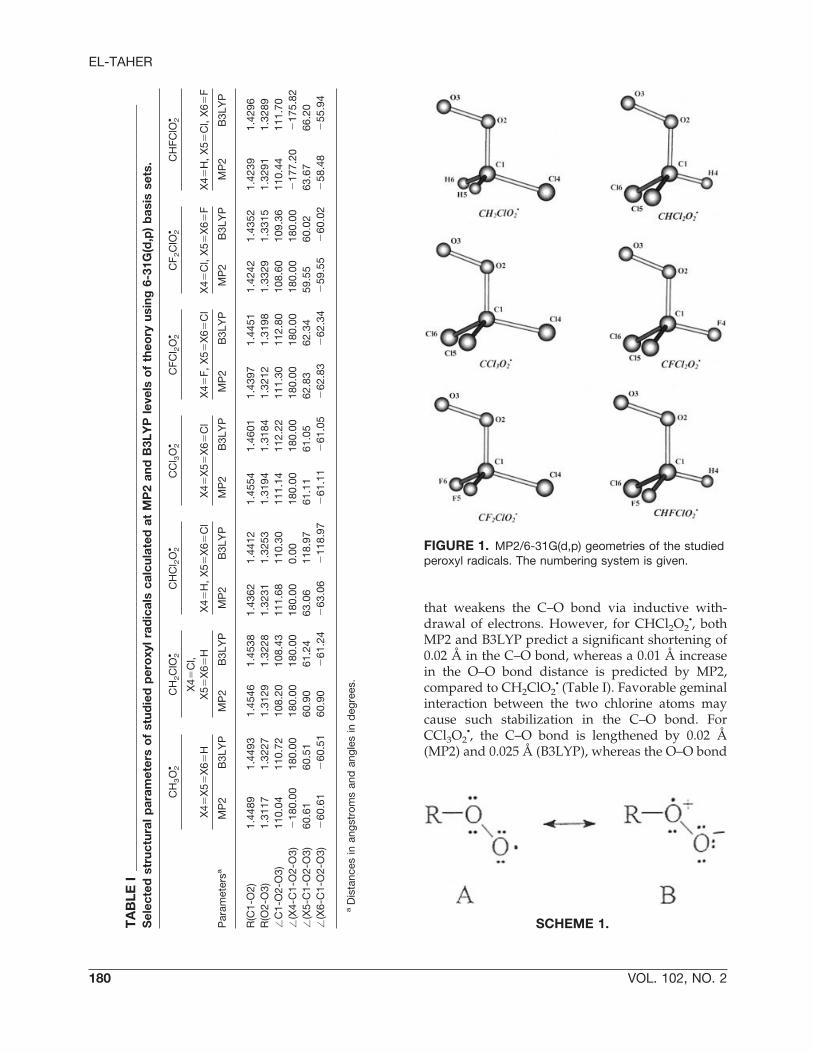
•, where its equilibrium structure has a C1symmetry. Table I shows the most important geo-metrical parameters of the studied peroxyl radicalscalculated at MP2(FULL) and B3LYP, using the6-31G(d,p) basis set. In addition, the MP2(FULL)/6-31G(d,p) structures and the numbering systemare depicted in Figure 1. Scheme 1 shows two majorresonance structures for peroxyl radicals: structureA, which has the unpaired electron localized on theterminal oxygen atom, and structure B, where theunpaired electron localized on the inner oxygenatom with a formal positive charge. Previous stud-ies [13, 18, 22] indicated that the resonance structureA is more important than the charged one B.
When going from CH3O2• to CH2ClO2
• the C–Obond lengthens by 0.006 Å (MP2) and 0.005 Å(B3LYP), and the O–O bond lengthens by 0.001 Å(MP2). This may be explained in terms of the factthat the chlorine atom acts as a powerful � acceptor
STRUCTURES OF PEROXYL RADICALS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 179
that weakens the C–O bond via inductive with-drawal of electrons. However, for CHCl2O2
•, bothMP2 and B3LYP predict a significant shortening of0.02 Å in the C–O bond, whereas a 0.01 Å increasein the O–O bond distance is predicted by MP2,compared to CH2ClO2
• (Table I). Favorable geminalinteraction between the two chlorine atoms maycause such stabilization in the C–O bond. ForCCl3O2
•, the C–O bond is lengthened by 0.02 Å(MP2) and 0.025 Å (B3LYP), whereas the O–O bond
TA
BLE
I__
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
__S
elec
ted
stru
ctur
alp
aram
eter
so
fst
udie
dp
ero
xylr
adic
als
calc
ulat
edat
MP
2an
dB
3LY
Ple
vels
of
theo
ryus
ing
6-31
G(d
,p)
bas
isse
ts.
Par
amet
ersa
CH
3O
2•C
H2C
lO2•
CH
Cl 2
O2•
CC
l 3O
2•C
FCl 2
O2•
CF 2
ClO
2•C
HFC
lO2•
X4�
X5�
X6�
HX
4�C
l,X
5�X
6�H
X4�
H,
X5�
X6�
Cl
X4�
X5�
X6�
Cl
X4�
F,X
5�X
6�C
lX
4�C
l,X
5�X
6�F
X4�
H,
X5�
Cl,
X6�
F
MP
2B
3LY
PM
P2
B3L
YP
MP
2B
3LY
PM
P2
B3L
YP
MP
2B
3LY
PM
P2
B3L
YP
MP
2B
3LY
P
R(C
1-O
2)1.
4489
1.44
931.
4546
1.45
381.
4362
1.44
121.
4554
1.46
011.
4397
1.44
511.
4242
1.43
521.
4239
1.42
96R
(O2-
O3)
1.31
171.
3227
1.31
291.
3228
1.32
311.
3253
1.31
941.
3184
1.32
121.
3198
1.33
291.
3315
1.32
911.
3289
�C
1-O
2-O
3)11
0.04
110.
7210
8.20
108.
4311
1.68
110.
3011
1.14
112.
2211
1.30
112.
8010
8.60
109.
3611
0.44
111.
70�
(X4-
C1-
O2-
O3)
�18
0.00
180.
0018
0.00
180.
0018
0.00
0.00
180.
0018
0.00
180.
0018
0.00
180.
0018
0.00
�17
7.20
�17
5.82
�(X
5-C
1-O
2-O
3)60
.61
60.5
160
.90
61.2
463
.06
118.
9761
.11
61.0
562
.83
62.3
459
.55
60.0
263
.67
66.2
0�
(X6-
C1-
O2-
O3)
�60
.61
�60
.51
60.9
0�
61.2
4�
63.0
6�
118.
97�
61.1
1�
61.0
5�
62.8
3�
62.3
4�
59.5
5�
60.0
2�
58.4
8�
55.9
4
aD
ista
nces
inan
gstr
oms
and
angl
esin
deg
rees
.
FIGURE 1. MP2/6-31G(d,p) geometries of the studiedperoxyl radicals. The numbering system is given.
SCHEME 1.
EL-TAHER
180 VOL. 102, NO. 2
is shortened by 0.005 Å, compared to that inCH2ClO2
•. With the three bulky Cl atoms on thecarbon, steric interactions involving the terminaloxygen atom might also contribute to the destabi-lization of the C–O bond, together with the unfa-vorable geminal interactions between the three Clatoms. Consistent with this explanation is the in-crease in �C–O–O when going from CH2ClO2
•
(108.43°) to CCl3O2• (112.22°), which is due to the
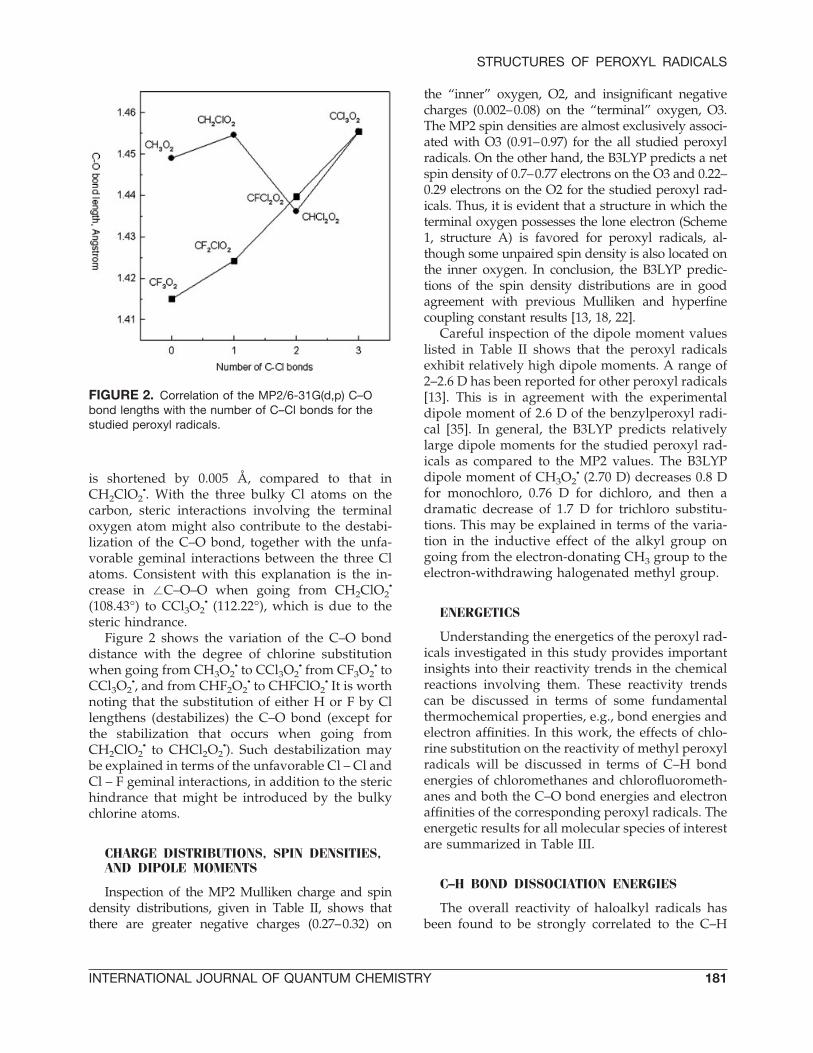
steric hindrance.Figure 2 shows the variation of the C–O bond
distance with the degree of chlorine substitutionwhen going from CH3O2
• to CCl3O2• from CF3O2
• toCCl3O2
•, and from CHF2O2• to CHFClO2
• It is worthnoting that the substitution of either H or F by Cllengthens (destabilizes) the C–O bond (except forthe stabilization that occurs when going fromCH2ClO2
• to CHCl2O2•). Such destabilization may
be explained in terms of the unfavorable Cl – Cl andCl – F geminal interactions, in addition to the sterichindrance that might be introduced by the bulkychlorine atoms.
CHARGE DISTRIBUTIONS, SPIN DENSITIES,AND DIPOLE MOMENTS
Inspection of the MP2 Mulliken charge and spindensity distributions, given in Table II, shows thatthere are greater negative charges (0.27–0.32) on
the “inner” oxygen, O2, and insignificant negativecharges (0.002–0.08) on the “terminal” oxygen, O3.The MP2 spin densities are almost exclusively associ-ated with O3 (0.91–0.97) for the all studied peroxylradicals. On the other hand, the B3LYP predicts a netspin density of 0.7–0.77 electrons on the O3 and 0.22–0.29 electrons on the O2 for the studied peroxyl rad-icals. Thus, it is evident that a structure in which theterminal oxygen possesses the lone electron (Scheme1, structure A) is favored for peroxyl radicals, al-though some unpaired spin density is also located onthe inner oxygen. In conclusion, the B3LYP predic-tions of the spin density distributions are in goodagreement with previous Mulliken and hyperfinecoupling constant results [13, 18, 22].
Careful inspection of the dipole moment valueslisted in Table II shows that the peroxyl radicalsexhibit relatively high dipole moments. A range of2–2.6 D has been reported for other peroxyl radicals[13]. This is in agreement with the experimentaldipole moment of 2.6 D of the benzylperoxyl radi-cal [35]. In general, the B3LYP predicts relativelylarge dipole moments for the studied peroxyl rad-icals as compared to the MP2 values. The B3LYPdipole moment of CH3O2
• (2.70 D) decreases 0.8 Dfor monochloro, 0.76 D for dichloro, and then adramatic decrease of 1.7 D for trichloro substitu-tions. This may be explained in terms of the varia-tion in the inductive effect of the alkyl group ongoing from the electron-donating CH3 group to theelectron-withdrawing halogenated methyl group.
ENERGETICS
Understanding the energetics of the peroxyl rad-icals investigated in this study provides importantinsights into their reactivity trends in the chemicalreactions involving them. These reactivity trendscan be discussed in terms of some fundamentalthermochemical properties, e.g., bond energies andelectron affinities. In this work, the effects of chlo-rine substitution on the reactivity of methyl peroxylradicals will be discussed in terms of C–H bondenergies of chloromethanes and chlorofluorometh-anes and both the C–O bond energies and electronaffinities of the corresponding peroxyl radicals. Theenergetic results for all molecular species of interestare summarized in Table III.
C–H BOND DISSOCIATION ENERGIES
The overall reactivity of haloalkyl radicals hasbeen found to be strongly correlated to the C–H
FIGURE 2. Correlation of the MP2/6-31G(d,p) C–Obond lengths with the number of C–Cl bonds for thestudied peroxyl radicals.
STRUCTURES OF PEROXYL RADICALS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 181
bond dissociation energies (BDEs) of the parenthaloalkanes [36]. Because the reactivity of the per-oxyl radicals is strongly dependent on the stabilityof their alkyl radicals, we will discuss such stabilityin terms of the C–H BDEs of the parent halometh-anes. Although the C–H BDEs of halomethaneshave been previously estimated at high ab initiolevels of theory [36], the present estimation willenable us to judge the DFT performance with re-spect to high ab initio methods in predicting theBDEs and the substituent effects trends.
The direct calculation of C–H BDEs from thereaction RH3 R• � H• is known to be in error dueto insufficient treatment of electron correlation andincompleteness of the basis sets. An indirect way toaccurately estimate the C–H BDEs is to useisodesmic reactions in which the same number andtype of bonds are there on each side. Thus, a goodbalance is introduced in the estimation of the cor-relation energy between reactants and products.The reactions CHX3 � CH3 3 CX3 � CH4, whose
enthalpy changes, �H298, between products and re-actants is BDE (H-CX3) – BDE (H-CH3), where X �H, F, and Cl are selected. Using the experimentalvalue of BDE (H-CH3) � 104.9 kcal mol�1 [37], theBDE (H-CX3) of the studied halomethanes are esti-mated at various levels of theory and given in TableIV, together with the available experimental data[37].
The MP2/6-31G(d,p) C–H BDEs of CH3Cl andCH2Cl2 are overestimated by only 0.9 and 1.3 kcal/mol, respectively, whereas those of CHCl3 andCHFCl2 are underestimated by 1.7 and 1.9 kcal/mol, compared with the experimental results. It isnoted that the MP4(SDTQ)/6-311�G(d,p) level per-forms worse in estimating the C–H BDEs. On theother hand, the B3LYP/6-31G(d,p) underestimatesthe C–H BDEs of CH4, CH3Cl, CH2Cl2, CHCl3, andCHFCl2 by 0.3, 0.4, 0.9, 3.7, and 6.0 kcal/mol, re-spectively. It is worth noting that the use of a rela-tively large basis set, 6-311�G(2df,2p), improvesthe B3LYP BDEs, in contrast to that of fluorometh-
TABLE II ______________________________________________________________________________________________Charge distributions, spin densities, and dipole moments of studied peroxyl radicals.
Radical
Charge distribution Spin density Dipole moment
MP2/6-31G(d,p)
B3LYP/6-31G(d,p)
MP2/6-31G(d,p)
B3LYP/6-31G(d,p)
MP2/6-31G(d,p)
B3LYP/6-31G(d,p)
C1 �0.022 �0.077 �0.022 �0.024CH3OO• O2 �0.318 �0.166 0.097 0.289 2.54 2.70
O3 �0.077 �0.170 0.913 0.703
C1 0.015 �0.051 �0.019 �0.021CH2ClO2
• O2 �0.300 �0.144 0.091 0.287 2.14 1.90O3 �0.059 �0.149 0.920 0.709
C1 0.010 �0.058 �0.013 �0.011CHCl2O2
• O2 �0.289 �0.134 0.053 0.229 1.67 1.94O3 �0.012 �0.097 0.954 0.755
C1 �0.014 �0.079 �0.013 �0.011CCl3O2
• O2 �0.271 �0.118 0.056 0.237 0.84 1.03O3 �0.002 �0.087 0.952 0.751
C1 0.478 0.310 �0.012 �0.009CFCl2O2
• O2 �0.290 �0.140 0.055 0.234 0.88 0.86O3 �0.007 �0.093 0.951 0.753
C1 0.935 0.658 �0.007 �0.005CF2ClO2
• O2 �0.324 �0.172 0.040 0.226 0.70 0.97O3 �0.008 �0.101 0.970 0.772
C1 0.486 0.327 �0.010 �0.007CHFClO2
• O2 �0.315 �0.161 0.045 0.224 1.80 2.05O3 �0.013 �0.103 0.962 0.764
EL-TAHER
182 VOL. 102, NO. 2

TA
BLE
III__
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
_T
ota
lele
ctro
nic
ener
gie
s,ze
ro-p
oin
ten
erg
ies
(ZP
E),
and
ther
mal
corr
ecti
ons
(TC
,29
8.15
K)a
Sp
ecie
s
6-31
G(d
,p)
6-31
1�G
(d,p
)//6
-31G
(d,p
)B
3LY
P/
6-31
G(d
,p)
B3L
YP
/6-
311�
G(2
df,2
p)
ZP
ETC
HF
MP
2M
P2
MP
4(S
DTQ
)H
FB
3LY
PH
FB
3LY
P
CH
3C
l�
499.
0977
9�
499.
3932
9�
499.
4282
5�
499.
4654
4�
500.
1125
5�
500.
1582
322
.63
23.3
41.
911.
90C
H2C
l•�
498.
4645
3�
498.
7285
9�
498.
7641
8�
498.
7995
0�
499.
4413
8�
499.
4890
213
.74
14.1
02.
052.
37C
H2C
lO2•
�64
8.09
982
�64
8.71
796
�64
8.82
976
�64
8.88
888
�64
9.80
797
�64
9.91
145
20.9
521
.06
2.87
2.90
(�64
8.10
679)
(�64
8.73
383)
(�64
8.82
974)
(�64
8.88
891)
(�64
9.81
923)
(�64
9.91
146)
20.9
621
.03
2.86
2.90
CH
2C
lO2�
�64
8.12
627
�64
8.80
104
�64
8.89
328
�64
8.94
949
�64
9.92
308
�65
0.01
235
19.8
820
.15
3.02
2.93
CH
2C
l 2�
957.
9881
2�
958.
4167
1�
958.
4784
6�
958.
5269
9�
959.
6989
0�
959.
7786
017
.82
18.1
32.
252.
26C
HC
l 2•
�95
7.35
968
�95
7.75
836
�95
7.81
956
�95
7.86
650
�95
9.03
558
�95
9.11
652
9.52
9.86
2.29
2.30
CH
Cl 2
O2•
�11
06.9
8946
�11
07.7
4332
�11
07.8
8122
�11
07.9
5194
�11
09.3
9393
�11
09.5
2940
15.5
215
.14
2.66
2.90
(�11
06.9
9659
)(�
1107
.761
55)
(�11
07.8
8120
)(�
1107
.951
98)
(�11
09.4
0423
)(�
1109
.529
39)
15.5
015
.24
3.28
3.36
CH
Cl 2
O2�
�11
07.0
3140
�11
07.8
4459
�11
07.9
6151
�11
08.0
2955
�11
09.4
9560
�11
09.6
1924
14.4
614
.34
3.52
3.66
CH
Cl 3
�14
16.8
7109
�14
17.4
3744
�14
17.5
2261
�14
17.5
8097
�14
19.2
8035
�14
19.3
9448
12.2
612
.22
2.80
2.83
CC
l 3•�
1416
.248
16�
1416
.784
30�
1416
.871
97�
1416
.930
52�
1418
.623
46�
1418
.738
364.
454.
452.
832.
81C
Cl 3
O2•
�15
65.8
6849
�15
66.7
6305
�15
66.9
2737
�15
67.0
0991
�15
68.9
7120
�15
69.1
4086
9.25
8.69
3.97
4.09
(�15
65.8
7419
)(�
1566
.784
96)
(�15
66.9
2737
)(�
1567
.009
96)
(�15
68.9
8230
)(�
1569
.140
87)
9.22
8.63
3.98
4.10
CC
l 3O
2��
1565
.924
08�
1566
.880
24�
1567
.020
01�
1567
.099
88�
1569
.086
61�
1569
.243
328.
247.
994.
234.
35
CH
FCl 2
�10
56.8
3006
�10
57.4
2641
�10
57.5
5636
�10
57.6
0765
�10
58.9
2414
�10
59.0
4807
13.3
213
.30
2.59
2.62
CFC
l 2•
�10
56.2
0092
�10
56.7
6746
�10
56.8
9588
�10
56.9
4585
�10
58.2
6140
�10
58.3
8505
5.43
5.43
2.59
2.58
CFC
l 2O
2•
�12
05.8
3473
�12
06.7
5791
�12
06.9
6013
�12
07.0
3411
�12
08.6
2177
�12
08.7
9931
10.4
19.
953.
713.
81(�
1205
.847
65)
(�12
06.7
8703
)(�
1206
.958
81)
(�12
07.0
3176
)(�
1208
.640
59)
(�12
08.7
9926
)10
.36
9.83
3.72
3.82
CFC
l 2O
2��
1205
.897
95�
1206
.883
41�
1207
.053
27�
1207
.124
33�
1208
.743
49�
1208
.900
169.
389.
163.
934.
03
CH
F 2C
l�
696.
7970
6�
697.
4230
5�
697.
5917
1�
697.
6340
7�
698.
5734
2�
698.
7053
414
.47
14.4
62.
392.
41C
F 2C
l•�
696.
1618
2�
696.
7584
9�
696.
9248
3�
696.
9652
2�
697.
9045
3�
698.
0354
26.
486.
482.
382.
38C
F 2C
lO2•
�84
5.81
141
�84
6.75
936
�84
6.99
693
�84
7.06
171
�84
8.27
694
�84
8.46
001
11.6
011
.19
3.45
3.54
(�84
5.82
686)
(�84
6.79
178)
(�84
6.99
677)
(�84
7.06
167)
(�84
8.30
101)
(�84
8.55
431)
11.5
110
.99
3.46
3.55
CF 2
ClO
2��
845.
8734
0�
846.
8849
9�
847.
0861
9�
847.
1477
4�
848.
3977
6�
848.
5543
110
.48
10.1
73.
663.
69
CH
2FC
l�
597.
9393
0�
598.
3989
9�
598.
5031
5�
598.
5430
0�
599.
3347
0�
599.
4260
518
.87
19.1
62.
112.
12C
HFC
l•�
597.
3076
2�
597.
7375
0�
597.
8407
0�
597.
8788
0�
598.
6682
6�
598.
7591
610
.63
10.9
62.
102.
09C
HFC
lO2•
�74
6.95
313
�74
7.73
450
�74
7.91
113
�74
7.97
327
�74
9.03
868
�74
9.18
332
16.6
516
.44
3.06
3.13
(�74
6.96
539)
(�74
7.76
051)
(�74
7.91
104)
(�74
7.97
326)
(�74
9.05
794)
(�74
9.18
333)
16.5
916
.31
3.07
3.14
CH
FClO
2��
746.
9972
3�
747.
8419
4�
747.
9890
3�
748.
0481
7�
749.
1463
7�
749.
2702
515
.47
15.4
03.
333.
47
H•
�0.
4982
3�
0.49
823
�0.
4998
1�
0.49
981
�0.
5002
7�
0.50
216
0.00
0.00
0.89
0.89
O2
�14
9.61
791
�14
9.95
432
�15
0.02
934
�15
0.04
860
�15
0.32
004
�15
0.37
832
1.80
2.33
1.49
1.48
aTo
tale
lect
roni
cen
ergi
esar
egi
ven
inha
rtre
es.
Zer
o-p
oint
ener
gies
and
ther
mal
corr
ectio
ns(g
iven
inkc
al.m
ol�
1)a
reca
lcul
ated
atth
eH
Fan
dB
3LY
Ple
vels
ofth
eory
usin
g6-
31G
(d,p
)b
asis
set
with
0.89
29an
d0.
9806
scal
ing
ofth
evi
bra
tiona
lfre
que
ncie
s,re
spec
tivel
y.
STRUCTURES OF PEROXYL RADICALS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 183

anes [27]. This is due to the fact that chlorine atomsmay be much better described by a relatively largeand more diffuse basis set, compared to the rela-tively small fluorine atoms. Inspection of the data inTable IV shows that the successive substitution ofchlorine for hydrogen atoms in CH4 results in adecrease in the C–H BDE, accompanied by a corre-sponding lengthening of the C–H bond length,whereas the substitution of chlorine atoms inCHCl3 by fluorine atoms (on going from CHCl3 toCHF3) results in an increase in the C–H BDE, ac-companied by a corresponding shortening in theC–H bond length (Fig. 3). This may be explained interms of the fact that the geminal interaction be-tween two or even three fluorine atoms stronglystabilizes the structures of fluoromethanes, whereasthe fluorine–chlorine or chlorine–chlorine interac-tion results in a relatively weak stabilization. Ingeneral, the B3LYP C–H BDEs are in much betteragreement with experiment than previously ob-tained results at high ab initio levels (Table IV). Thissuggests that the B3LYP method with a suitablebasis set may be a good alternative to the muchmore computationally expensive high ab initiomethods in predicting the C–H BDEs for relativelylarge alkanes.
C–O BOND DISSOCIATION ENERGIES
The substituent effects on the reactivity of thestudied peroxyl radicals will be discussed in terms
of their C–O BDEs that correspond to the negative�H298 of reaction (1). To our knowledge, only theC–O BDEs of CH2ClO2, CHCl2O2
•, and CCl3O2•
have been experimentally determined [25, 38]. Atheoretical determination of C–O BDE of CCl3O2
• atthe B3LYP/6-31G(d) level has been reported [24].Neither experimental nor theoretical determination
TABLE IV _____________________________________________________________________________________________Calculated C–H bond dissociation energies (BDEs).
Levels of calculations CH4 CH3Cl CH2Cl2 CHCl3 CHFCl2 CHF2Cl CH2FCl
MP2/6-31G(d,p) 101.1 100.4 96.9 94.1 98.8 101.1 98.9MP2/6-311�G(d,p)//MP2/6-31G(d,p) 99.2 101.2 98.1 93.4 100.6 103.4 100.4MP4(SDTQ)/6-311�G(d,p)//MP2/6-31G(d,p) 100.6 100.7 97.7 91.9 100.0 103.2 100.1B3LYP/6-31G(d,p) 105.2 99.1 94.7 92.1 94.7 98.5 96.7B3LYP/6-311�G(2df,2p) 102.7 99.2 95.2 92.9 96.1 100.4 98.2
MP2/cc-pVtza 110.6 105.9 101.1 100.0CCSD(T)/6-311��G(3df,3pd)a 103.8 96.6G2MP2 105.8b
G2 105.8c
expt 104.9 � 0.1b 99.5 � 0.6d 95.6 � 0.6d 95.8 � 1e 100.7 � 2f
a Ref. 36.b Ref. 37(a).c Ref. 38(a).d Ref. 37(b).e Ref. 37(c).f Ref. 37(d).
FIGURE 3. Correlation of the B3LYP/6-31G(d,p) C–HBDEs with the C–H bond lengths for the parent meth-anes.
EL-TAHER
184 VOL. 102, NO. 2
of the C–O BDEs of CHFCl2O2•, CHF2ClO2
•, andCH2FClO2
• has been previously reported.Displayed in Table V are the indirectly estimated
values of C–O BDEs of the studied RO2•, using the
isodesmic reactions: CX3O2 � CH3 3 CX3 �CH3O2
•, whose enthalpy changes, �H298, betweenproducts and reactants are given by BDE (CX3-O2)– BDE (CH3-O2), where X � H, F, and Cl. Theindirect method is adopted to minimize errorsstemming from incomplete basis sets and limitedtreatment of electron correlation. Using the experi-mental value of BDE(CH3-O2) � 32.7 kcal/mol [25],the BDE(CX3-O2) of the studied peroxyl radicals areestimated. It is worth noting that the indirectlyestimated C–O BDEs values are significantly im-proved over the C–O BDEs values that are directlyestimated from Eq. 1 (given in parentheses). TheC–O BDEs of CH2ClO2, CHCl2O2
•, and CCl3O2• are
overestimated by only 1.4 (2.2), 3.3 (4.4), and 5.8(7.0) kcal/mol at the MP2/6-31G(d,p) (MP4(SDTQ)/6-311�G(d,p)), respectively. The B3LYP/6-31G(d,p)level underestimates the C–O BDEs of CH2ClO2 andCHCl2O2
• by only 1.1 and 2.0 kcal/mol, whereas itoverestimates that of CCl3O2
• by 2.0 kcal/mol. Thisconfirms the importance of electron correlation inestimating C–O BDEs for chloromethyl peroxylradicals. In other words, the errors due to electroncorrelation are greatly minimized when the C–OBDEs are indirectly estimated, resulting in valuesthat are in good agreement with experiment. It canbe inferred from the results in Table V that theB3LYP/6-31G(d,p) level underestimates the C–O
BDEs of CH3O2•, CH2ClO2•, CHCl2O2
•, and CCl3O2•
by only 1.7, 1.1, 2.0, and 2.0 kcal/mol, whereasthe B3LYP/6-311�G(2df,2p) level underestimatesthem by 3.2, 1.1, 2.5, and 2.7 kcal/mol, respectively,with respect to the experimental values. Thus, theuse of the larger 6-311�G(2df,2p) basis set does notmake any improvement on the 6-31G(d,p); instead,it gives worse results. However, these findings areimproved over what has been previously reported[39] that the B3LYP/6-31G(d,p) level systematicallypredicts lower BDEs values by 3-6 kcal/mol forbonds involving oxygen, e.g., C–O, O–H, and O–O.The reason is certainly due to the use of the indirectmethod.
Careful inspection of the data presented in TableV shows that the progressive substitution of chlo-rine for hydrogen atoms in CH3O2
• results in adecrease in the C–O BDEs. This is predicted by alllevels of calculations, and it is consistent with thetrend noted for the substituent effects on the C–HBDEs in chloromethanes (Table IV). The decrease inthe C–H BDE when the hydrogen substitution bychlorine increases is in agreement with the decreasein energy barriers of the hydrogen abstraction re-actions of chloromethanes [40]. This trend is accom-panied by an increase in the stability of the result-ant alkyl radicals and, hence, a consequent decreasein the stability of the corresponding peroxyl radi-cals. The weakening of the C–O bond with thesubstitution of chlorine for hydrogen atoms inCH3O2
• has been experimentally observed [38b,c].On the other hand, the substitution of chlorine at-
TABLE V ______________________________________________________________________________________________Calculated C–O bond dissociation energies (BDEs).
Levels of calculations CH3–O2• CH2Cl–O2
• CHCl2–O2• CCl3–O2
• CFCl2–O2• CF2Cl–O2
• CHFCl–O2•
MP2/6-31G(d,p) 19.7 30.3 (17.2) 29.2 (15.5) 25.7 (12.7) 32.9 (19.9) 36.1 (26.3) 36.1 (23.1)MP2/6-311�G(d,p)//MP2/
6-31G(d,p) 19.5 31.3 (18.0) 30.5 (16.6) 27.0 (13.7) 32.3 (19.1) 37.2 (23.9) 35.3 (22.1)MP4(SDTQ)/6-311�G(d,p)//
MP2/6-31G(d,p) 22.5 31.1 (20.9) 30.3 (19.4) 26.9 (16.7) 32.3 (22.1) 37.4 (27.2) 35.3 (25.1)B3LYP/6-31G(d,p) 30.5 27.8 (25.5) 23.3 (21.0) 17.9 (15.7) 25.6 (23.4) 33.0 (30.8) 31.1 (28.9)B3LYP/6-311�G(2df,2p) 29.0 27.8 (24.0) 22.8 (19.0) 17.2 (13.5) 24.4 (20.6) 30.8 (27.0) 29.8 (26.1)
expt 32.2 � 1.5a 28.9 � 2.6b 25.3 � 1.4b 19.9 � 1.0c
32.7 � 0.9d 29.3 � 2.5d 25.9 � 2.0d 22.0 � 1.5d
a Ref. 38(a).b Ref. 38(b).c Ref. 38(c).d Ref. 25.All values (in kcal/mol) are calculated for 298 K and 1 atm.Values in parentheses are the directly estimated C–O BDEs.
STRUCTURES OF PEROXYL RADICALS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 185
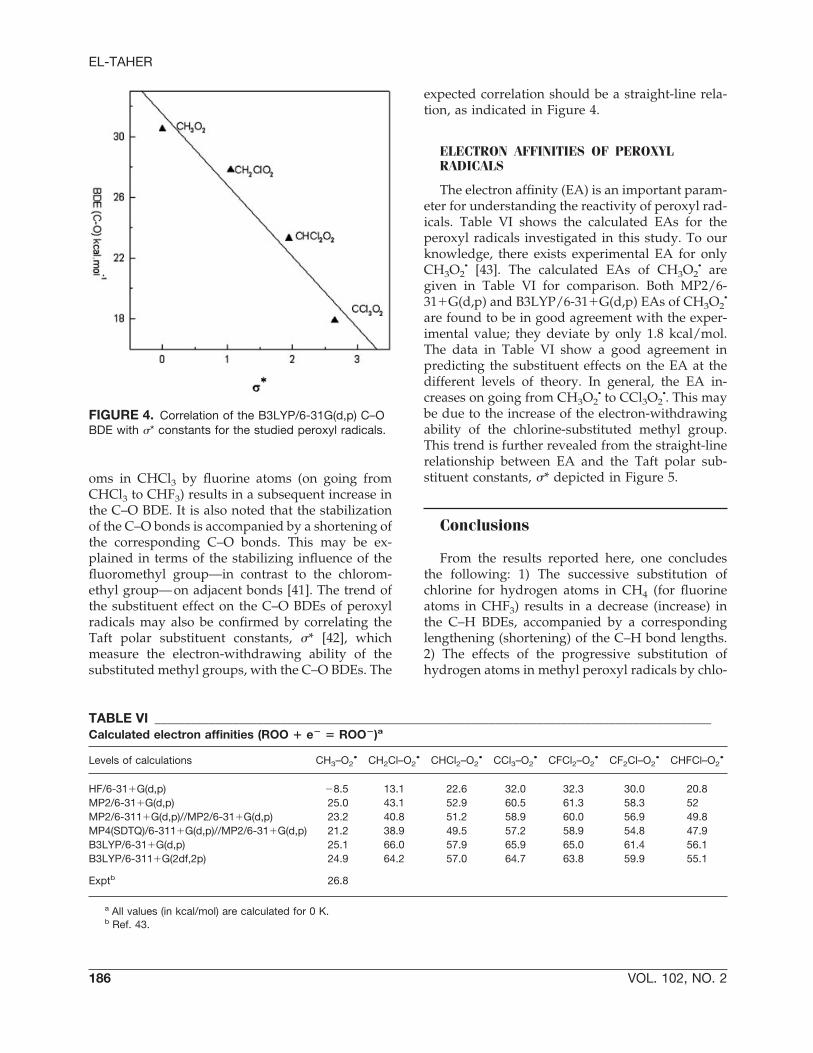
oms in CHCl3 by fluorine atoms (on going fromCHCl3 to CHF3) results in a subsequent increase inthe C–O BDE. It is also noted that the stabilizationof the C–O bonds is accompanied by a shortening ofthe corresponding C–O bonds. This may be ex-plained in terms of the stabilizing influence of thefluoromethyl group—in contrast to the chlorom-ethyl group—on adjacent bonds [41]. The trend ofthe substituent effect on the C–O BDEs of peroxylradicals may also be confirmed by correlating theTaft polar substituent constants, �* [42], whichmeasure the electron-withdrawing ability of thesubstituted methyl groups, with the C–O BDEs. The
expected correlation should be a straight-line rela-tion, as indicated in Figure 4.
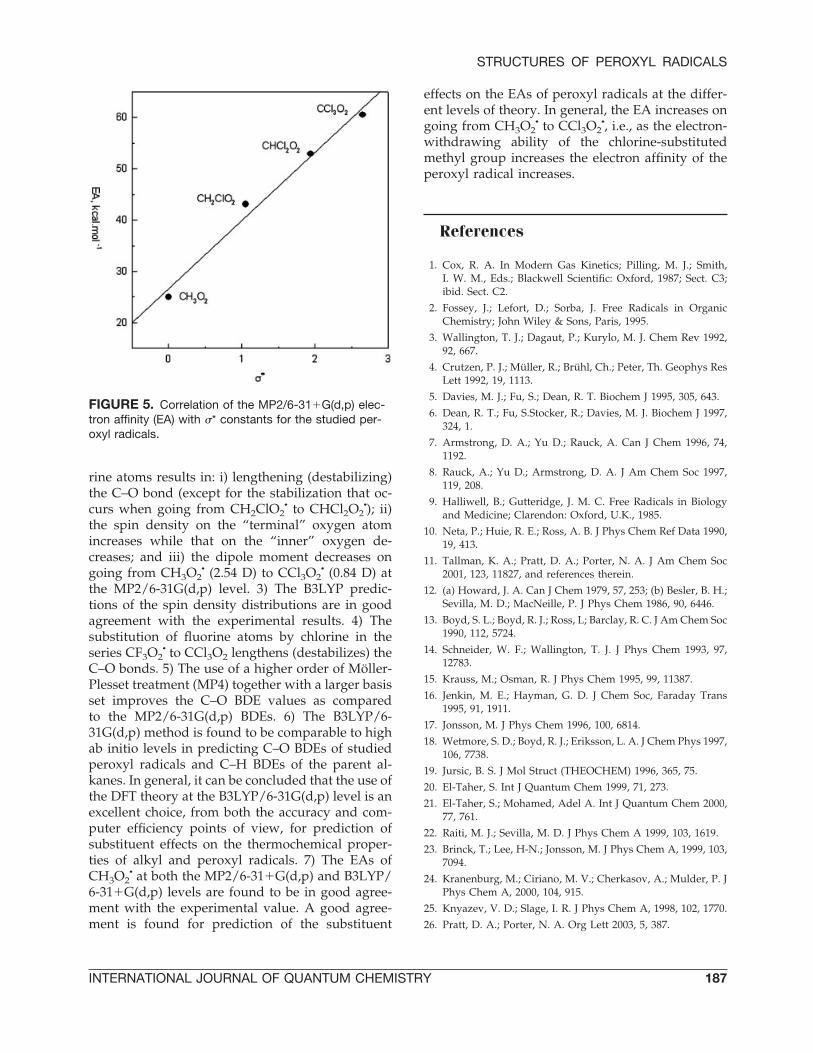
ELECTRON AFFINITIES OF PEROXYLRADICALS
The electron affinity (EA) is an important param-eter for understanding the reactivity of peroxyl rad-icals. Table VI shows the calculated EAs for theperoxyl radicals investigated in this study. To ourknowledge, there exists experimental EA for onlyCH3O2
• [43]. The calculated EAs of CH3O2• are
given in Table VI for comparison. Both MP2/6-31�G(d,p) and B3LYP/6-31�G(d,p) EAs of CH3O2
•
are found to be in good agreement with the exper-imental value; they deviate by only 1.8 kcal/mol.The data in Table VI show a good agreement inpredicting the substituent effects on the EA at thedifferent levels of theory. In general, the EA in-creases on going from CH3O2
• to CCl3O2•. This may
be due to the increase of the electron-withdrawingability of the chlorine-substituted methyl group.This trend is further revealed from the straight-linerelationship between EA and the Taft polar sub-stituent constants, �* depicted in Figure 5.
Conclusions
From the results reported here, one concludesthe following: 1) The successive substitution ofchlorine for hydrogen atoms in CH4 (for fluorineatoms in CHF3) results in a decrease (increase) inthe C–H BDEs, accompanied by a correspondinglengthening (shortening) of the C–H bond lengths.2) The effects of the progressive substitution ofhydrogen atoms in methyl peroxyl radicals by chlo-
FIGURE 4. Correlation of the B3LYP/6-31G(d,p) C–OBDE with �* constants for the studied peroxyl radicals.
TABLE VI _____________________________________________________________________________________________Calculated electron affinities (ROO � e� � ROO�)a
Levels of calculations CH3–O2• CH2Cl–O2
• CHCl2–O2• CCl3–O2
• CFCl2–O2• CF2Cl–O2
• CHFCl–O2•
HF/6-31�G(d,p) �8.5 13.1 22.6 32.0 32.3 30.0 20.8MP2/6-31�G(d,p) 25.0 43.1 52.9 60.5 61.3 58.3 52MP2/6-311�G(d,p)//MP2/6-31�G(d,p) 23.2 40.8 51.2 58.9 60.0 56.9 49.8MP4(SDTQ)/6-311�G(d,p)//MP2/6-31�G(d,p) 21.2 38.9 49.5 57.2 58.9 54.8 47.9B3LYP/6-31�G(d,p) 25.1 66.0 57.9 65.9 65.0 61.4 56.1B3LYP/6-311�G(2df,2p) 24.9 64.2 57.0 64.7 63.8 59.9 55.1
Exptb 26.8
a All values (in kcal/mol) are calculated for 0 K.b Ref. 43.
EL-TAHER
186 VOL. 102, NO. 2
rine atoms results in: i) lengthening (destabilizing)the C–O bond (except for the stabilization that oc-curs when going from CH2ClO2
• to CHCl2O2•); ii)
the spin density on the “terminal” oxygen atomincreases while that on the “inner” oxygen de-creases; and iii) the dipole moment decreases ongoing from CH3O2
• (2.54 D) to CCl3O2• (0.84 D) at
the MP2/6-31G(d,p) level. 3) The B3LYP predic-tions of the spin density distributions are in goodagreement with the experimental results. 4) Thesubstitution of fluorine atoms by chlorine in theseries CF3O2
• to CCl3O2 lengthens (destabilizes) theC–O bonds. 5) The use of a higher order of Moller-Plesset treatment (MP4) together with a larger basisset improves the C–O BDE values as comparedto the MP2/6-31G(d,p) BDEs. 6) The B3LYP/6-31G(d,p) method is found to be comparable to highab initio levels in predicting C–O BDEs of studiedperoxyl radicals and C–H BDEs of the parent al-kanes. In general, it can be concluded that the use ofthe DFT theory at the B3LYP/6-31G(d,p) level is anexcellent choice, from both the accuracy and com-puter efficiency points of view, for prediction ofsubstituent effects on the thermochemical proper-ties of alkyl and peroxyl radicals. 7) The EAs ofCH3O2
• at both the MP2/6-31�G(d,p) and B3LYP/6-31�G(d,p) levels are found to be in good agree-ment with the experimental value. A good agree-ment is found for prediction of the substituent
effects on the EAs of peroxyl radicals at the differ-ent levels of theory. In general, the EA increases ongoing from CH3O2
• to CCl3O2•, i.e., as the electron-
withdrawing ability of the chlorine-substitutedmethyl group increases the electron affinity of theperoxyl radical increases.
References
1. Cox, R. A. In Modern Gas Kinetics; Pilling, M. J.; Smith,I. W. M., Eds.; Blackwell Scientific: Oxford, 1987; Sect. C3;ibid. Sect. C2.
2. Fossey, J.; Lefort, D.; Sorba, J. Free Radicals in OrganicChemistry; John Wiley & Sons, Paris, 1995.
3. Wallington, T. J.; Dagaut, P.; Kurylo, M. J. Chem Rev 1992,92, 667.
4. Crutzen, P. J.; Muller, R.; Bruhl, Ch.; Peter, Th. Geophys ResLett 1992, 19, 1113.
5. Davies, M. J.; Fu, S.; Dean, R. T. Biochem J 1995, 305, 643.
6. Dean, R. T.; Fu, S.Stocker, R.; Davies, M. J. Biochem J 1997,324, 1.
7. Armstrong, D. A.; Yu D.; Rauck, A. Can J Chem 1996, 74,1192.
8. Rauck, A.; Yu D.; Armstrong, D. A. J Am Chem Soc 1997,119, 208.
9. Halliwell, B.; Gutteridge, J. M. C. Free Radicals in Biologyand Medicine; Clarendon: Oxford, U.K., 1985.
10. Neta, P.; Huie, R. E.; Ross, A. B. J Phys Chem Ref Data 1990,19, 413.
11. Tallman, K. A.; Pratt, D. A.; Porter, N. A. J Am Chem Soc2001, 123, 11827, and references therein.
12. (a) Howard, J. A. Can J Chem 1979, 57, 253; (b) Besler, B. H.;Sevilla, M. D.; MacNeille, P. J Phys Chem 1986, 90, 6446.
13. Boyd, S. L.; Boyd, R. J.; Ross, L; Barclay, R. C. J Am Chem Soc1990, 112, 5724.
14. Schneider, W. F.; Wallington, T. J. J Phys Chem 1993, 97,12783.
15. Krauss, M.; Osman, R. J Phys Chem 1995, 99, 11387.
16. Jenkin, M. E.; Hayman, G. D. J Chem Soc, Faraday Trans1995, 91, 1911.
17. Jonsson, M. J Phys Chem 1996, 100, 6814.
18. Wetmore, S. D.; Boyd, R. J.; Eriksson, L. A. J Chem Phys 1997,106, 7738.
19. Jursic, B. S. J Mol Struct (THEOCHEM) 1996, 365, 75.
20. El-Taher, S. Int J Quantum Chem 1999, 71, 273.
21. El-Taher, S.; Mohamed, Adel A. Int J Quantum Chem 2000,77, 761.
22. Raiti, M. J.; Sevilla, M. D. J Phys Chem A 1999, 103, 1619.
23. Brinck, T.; Lee, H-N.; Jonsson, M. J Phys Chem A, 1999, 103,7094.
24. Kranenburg, M.; Ciriano, M. V.; Cherkasov, A.; Mulder, P. JPhys Chem A, 2000, 104, 915.
25. Knyazev, V. D.; Slage, I. R. J Phys Chem A, 1998, 102, 1770.
26. Pratt, D. A.; Porter, N. A. Org Lett 2003, 5, 387.
FIGURE 5. Correlation of the MP2/6-31�G(d,p) elec-tron affinity (EA) with �* constants for the studied per-oxyl radicals.
STRUCTURES OF PEROXYL RADICALS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 187

27. El-Taher, S.; El-Azhary, Adel A. Int J Quantum Chem 2004,98, 502.
28. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgom-ery, J. A.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Mil-lam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas,O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci,B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Peters-son, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.;Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski,J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz,P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.;Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.;Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B. G.;Chen, W.; Wong, M. W.; Andres, J. L.; Head-Gordon, M.;Replogle, E. S.; Pople, J. A. Gaussian 98W (Revision 5.1),Gaussian, Pittsburgh PA, 1998.
29. Hehre, W. J.; Radom, L.; Schleyer, P. v. R.; Pople, J. A. AbInitio Molecular Orbital Theory; John Wiley & Sons: NewYork, 1986.
30. Stephens, P. J.; Devlin, F. J.; Chablovski, C. F.; Frisch, M. J. JPhys Chem 1994, 98, 11623.
31. Becke, A. D. J Chem Phys 1993, 98, 1372.
32. Lee, C.; Yang, W.; Parr, R. G. Phys Rev B 1988, 37, 785.
33. Martin, J. M. L.; El-Yazal, J.; Francois, J-P. Mol Phys 1995, 86,1437.
34. Bauschlicher, C. W., Jr.; Partridge, H. Chem Phys Lett 1995,240, 533.
35. Fessenden, R. W.; Hitachi, A.; Nagarajan, V. J Phys Chem1984, 88, 107.
36. McGivern, W. S.; Derecskei-Kovacs, A.; North, S. W.; Fran-cisco, J. S. J Phys Chem A 2000, 104, 436.
37. (a) Berkowitz, J.; Ellison, G. B.; Gutman, D. J Phys Chem1994, 98, 2744; (b) Seetula, J. A. J Chem Soc Faraday Trans1996, 92 3069; (c) Tschuikow-Roux, E.; Paddison, S. IntJ Chem Kinet 1987, 19, 15; (d) Miyokawa, K.; Tschuikow-Roux, E. J Phys Chem 1992, 96, 7328.
38. (a) Benson, S. W. J Phys Chem 1996, 100, 13544; (b) Russel,J. J.; Seetula, J. A.; Gutman, D.; Melius, C. F.; Senkan, S. M.Symp (Int) Combust Proc 1990, 23, 163; (c) Russel, J. J.;Seetula, J. A.; Gutman, D.; Danis, F.; Caralp, F.; Lightfoot,P. D.; Lesclaux, R.; Melius, C. F.; Senkan, S. M. J Phys Chem1990, 94, 3277.
39. Van Scheppingen, W. B.; Dorrestijn, E.; Arends, I. W. C. E.;Mulder, P.; Korth, H.-G. J Phys Chem A 1997, 101, 5404.
40. Rayez, M-T; Rayez, J-C. J Phys Chem 1994, 98, 11342.41. McMillen, D. F.; Golden, D. M. Annu Rev Phys Chem 1982,
33, 493.42. Taft, Jr., R. W. J Chem Phys 1957, 26, 93.43. Blanksby, S. J.; Ramond, T. M.; Davico, G. E.; Nimlos, M. R.;
Kato, S.; Bierbaum, V. M.; Lineberger, W. C.; Ellison, G. B.;Okumura, M. J Am Chem Soc 2001, 123, 9585.
EL-TAHER
188 VOL. 102, NO. 2

![TRIS[2-CHLORO-1-(CHLOROMETHYL)ETHYL] PHOSPHATE …echa.europa.eu/documents/10162/6434698/orats...The report provides the environmental risk assessment of the substance tris[2-chloro-1-(chloromethyl)ethyl]](https://img.pdfslide.net/doc/110x75/5e3c26c0347c775bf42ed59b/tris2-chloro-1-chloromethylethyl-phosphate-echa-the-report-provides-the.jpg)



























