Embed Size (px)
Citation preview

Journal of Molecular Structure (Theochem), 205 (1990) 223-234 Elsevier Science Publishers B.V., Amsterdam - Printed in The Netherlands
223
AB INITIO GRADIENT CONFORMATIONAL ANALYSIS OF POLYAZOCYCLOHEXANES: 1,4-DIAZOCYCLOHEXANE, 1,3-DIAZOCYCLOHEXANE AND 1,3,6-TRIAZOCYCLOHEXANE
LUIS CARBALLEIRA, BERTA FERNAP;IDEZ, RICARDO A. MOSQUERA, MIGUEL A. RiOS, JESUS RODRIGUEZ OTERO and SAUL0 VAZQUEZ
Departamento de Quimica Fkica, Universidad de Santiago, Santiago de Compostela, E-15706, Galicia (Spain)
(Received 24 April 1989 j
ABSTRACT
The chair conformations of 1,4-diazocyclohexane, 1,3diazocyclohexane and 1,3,5-triazocyclo- hexane were determined by ab initio methods using the 4-21G basis set and complete geometrical optimization. The resuha are compared with available data from experimental and molecular mechanics studies.
INTRODUCTION
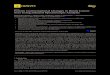
Although there is now a large body of information on the conformation of saturated six-member heterocycles with more than one heteroatom [ 11, there have hitherto been no ab initio studies of 1,4diaxocyclohexane (piperaxine, PIPZ), 1,3diaxocyclohexane (DACH) or 1,3,5-triazocyclohexane (TACH), whose numbering is illustrated in Fig. 1. As far as we are aware, the published work on these compounds consists of just two electron-diffraction studies of piperazine [ 2,3] together with molecular-mechanics [ 41 calculations for these molecules and various alkyl derivatives using the program MM2 (80) [ 5-91 or the modified version MMB-AE [lo]. With a view to enabling the results of these studies to be evaluated, we have now investigated the conformations of these molecules using Pulay’s ab initio method [ 121 and program [ 131 with the 4-21G basis set [ll] and complete optimization of the geometry. The ini- tial conformations used (three each for DACH and PIPZ and four for TACH) were chair forms with axial (A) or equatorial (E) amino hydrogen8 referred to in the name of the conformer in increasing order of nitrogen atom number (e.g. EEA denotes the TACH conformer with equatorial H on Nl and N3 and axial H on N5). Optimization was in all cases continued, with no constraints
0166-1280/90/$03.50 0 1990 EIsevier Science Publishers B.V.
224
PIP2
s- ‘2 =
?
C4 /=
1-c
ci
6 N
I
14
A 7 10
16 C2’
I
I 13
TACH
Fig. 1. 1,4-Diazocyclohexane (PIPZ), 1,3-diazocyclohexane (DACH) and 1,3,5Mazocyclohex- ane (TACH), with the numbering used in this paper.
other than those imposed by symmetry, until all the cartesian components of the forces on the atoms were less than 0.001 mdyn.
RESULTS AND DISCUSSION
1,4-Diazocyclohexan
The most stable of the three conformers studied was EE. The relative ener- gies of the three conformers are listed in Table 1 together with the values ob- tained in an MM2 (80) study [ 71 which also identified EE as the most stable conformer. Experimentally, the chair conformation of PIPZ has been deduced from electron-diffraction results [ 2,3], while Jones et al. [ 141, on the basis of dipole-moment measurements and the overtone of the N-H stretching vibra- tion in the infrared (IR) spectrum, inferred that the predominant conformer

225
TABLE 1
Absolute (E) and relative (dE) energies (in kcaI mol-‘) of the chair conformers of the polycy- clohexanes PIPZ, DACH and TACH (Fig. 1) as obtained by 4-21G//4-21G and molecular- mechanics calculations
Molecule Conformer 4-21G//4-21G MM2 (8O)a MM2-AEb AE AE
E All
PIPZ EE - 166660.02970 0.00 0.00 EA - 166659.60608 0.42 0.58 AA - 166658.84956 1.18 0.59
DACH EE - 166657.80472 5.45 0.00 4.9 EA - 166662.30128 0.95 0.75 1.1 AA - 166663.24985 0.00 1.05 0.0
TACH EEE - 176647.855 16.66 1.81 EEA - 176657.446 7.07 0.01 EAA - 176662.603 1.91 0.00 AAA - 176664.509 0.00 1.43 _
“Refs. 6-8. bRef. 10.
in the gaseous phase and inert solvents is EE and estimated the relative energy of EA as 0.4-0.5 kcal mol-l.
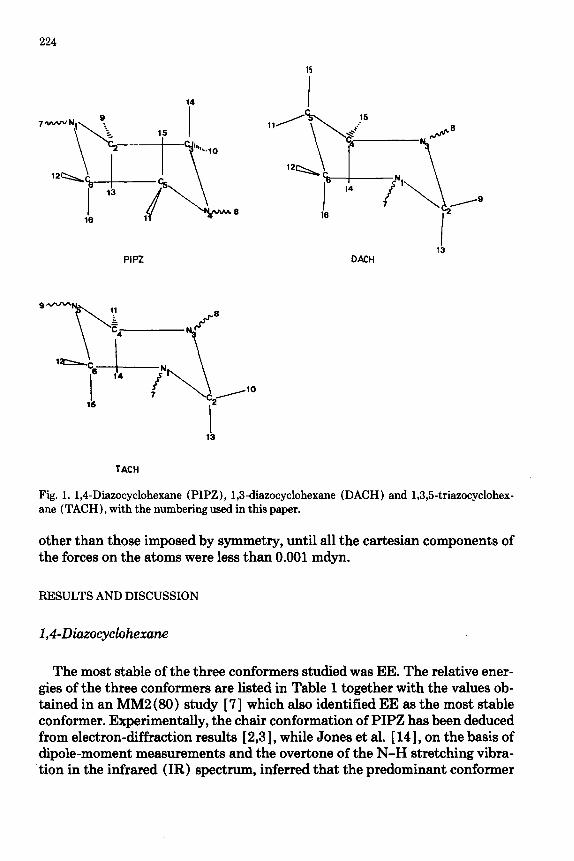
The geometries of the three PIPZ conformers (Table 2) exhibit the same anomeric effects as have been observed in other molecules with Lp-X-A-B units [ 15,161: when the A-B bond is antiperiplanar to the lone pair, the bond is stretched and the angle X-A-B widened. In the present case, this effect accounts for the following findings. (i) The C-C bond lengths are greater in AA than in EE (1.5406 versus 1.5312 A), and have an intermediate value (1.5371 A) in EA. (ii) For a given conformer, the longest C-H bonds are those lying antiperiplanar to a nitrogen lone pair. In EA, for example, the longest C- H bonds (1.0917 A) are C2-H13 and C6-H16, while in EE C-H bonds are significantly longer for axial than for equatorial H (1.0876 versus 1.0819 A). (iii) N-C-C angles are wider for axial than for equatorial N-H: 111.82” and 111.95” in AA and EA, respectively, as against 108.00’ and 108.07’ in EA and EE, respectively. (iv) N-C-H angles in which the hydrogen is anti to the ni- trogen lone pair are considerably wider than other N-C-H angles, reaching 112.39” in EE and 112.25” in EA. It may also be noted that for axial amino hydrogens, the N-H bond is longer (in EA, by 0.0026 A) and the C-N-H angle narrower (in EA, 111.73” as against 112.64” ) than for equatorial amino H.
The calculated ring-puckering coordinates [ 171 (Table 3) show the ring skeleton to be flattest in the conformer with no equatorial N-H bond, followed by EA, while the values of 8 and @ show all three conformers to be almost
226
TABLE 2
Completely optimized geometries of the chair conformers of PIPZ (Fig. 1) as obtained by 4-21G//4-21G calculations
Feature AA
Bond lengths (A) C2-Nl 1.4798 C3-C2 1.5406 N4-C3 1.4798 C5-N4 1.4797 C6-Nl 1.4797 C6-C5 1.5406 H7-Nl 1.0042 H8-N4 1.0042 H9-C2 1.0822 HlO-C3 1.0822 Hll-C5 1.0822 H12-C6 1.0822 H13-C2 1.0838 H14-C3 1.0838 H15-C5 1.0838 H16-C6 1.0838
Bond angles (“) C3-C2-Nl 111.82 N4-C3-C2 111.82 C5-N4-C3 112.74 C6-Nl-C2 112.74 C6-C5-N4 111.82 C5-C6-Nl 111.82 H7-Nl-C2 111.61 H7-Nl-C6 111.59 H8-N4-C3 111.61 H8-N4-C5 111.59 H9-C2-Nl 108.72 H9-C2-C3 111.16 HlO-C3-C2 111.16 HlO-C3-N4 108.72 Hll-C5-N4 108.73 Hll-C5-C6 H12-C6-Nl H12-C6-C5 H13-C2-Nl H13-C2-C3 H13-C2-H9 H14-C3-C2 H14-C3-N4 H14-C3-HlO
111.17 108.73 111.17 107.61 109.55 107.83 109.55 107.61 107.83
H15-C5-N4 107.56 H15-C5-C6 109.56 H15-C5-Hll 107.85 H16-C6-Nl 107.56 H16-C6-C5 109.56 H16-C6-H12 107.85
EA
1.4755 1.5371 1.4775 1.4775 1.4755 1.5371 1.0018 1.0044 1.0821 1.0818 1.0818 1.0821 1.0917 1.0806 1.0806 1.0917
108.00 108.07 111.95 108.07 112.18 112.72 113.33 112.72 111.95 108.07 108.00 108.07 112.64 112.46 112.64 112.46 111.73 112.46 111.73 112.46 108.77 108.72 110.30 110.13 110.89 110.13 108.84 108.72 108.84 108.72 110.89 110.13 108.77 108.72 110.30 110.13 112.25 112.39 109.31 108.55 108.21 108.97 108.62 108.55 107.75 112.39 108.68 108.97 107.75 112.39 108.62 108.55 108.68 108.97 112.25 112.39 109.31 108.55 108.21 108.97
EE
1.4757 1.5312 1.4757 1.4757 1.4757 1.5312 1.0021 1.0021 1.0819 1.0819 1.0819 1.0819 1.0876 1.0876 1.0876 1.0876
Feature AA
Torsional angles (“) C3-C2-Nl-C6 - 52.99 C3-C2-Nl-H7 73.54 H9-C2-Nl-C6 - 176.11 H9-C2-Nl-H7 - 49.58 H13-C2-Nl-C6 67.35 H13-C2-Nl-H7 - 166.12 N4-C3-C2-Nl 52.50 N4-C3-C2-H9 174.22 HlO-C3-C2-Nl 174.22 HlO-C3-C2-H9 - 64.06 N4-C3-C2-H13 - 66.71 HlO-C3-C2-H13 55.01 H14-C3-C2-Nl - 66.71 H14-C3-C2-H9 55.01 H14-C3-C2-H13 174.08 C5-N4-C3-C2 - 52.99 H8-N4-C3-C2 73.54 C5-N4-C3-HlO - 176.11 H8-N4-C3-HlO - 49.58 C5-N4-C3-H14 67.35 H8-N4-C3-H14 - 166.12 C6-C5-N4-C3 52.99 C6-C5-N4-H8 - 73.55 Hll-C5-N4-C3 176.12 Hll-C5-N4-H8 49.58 H15-C5-N4-C3 - 67.34 H15-C5-N4-H8 166.12 C5-C6-Nl-C2 52.99 C5-C6-Nl-H7 -73.55 H12-C6-Nl-C2 176.12 H12-C6-Nl-H7 49.58 H16-C6-Nl-C2 -67.34 H16-C6-Nl-H7 166.12 Nl-C6-C5-N4 -52.50 Nl-C6-C5-Hll - 174.23 H12-C6-C5-N4 - 174.23 H12-C6-C5-Hll 64.03 Nl-C6-C5-H15 66.66 H12-C6-C5-H15 -55.08 H16-C6-C5-N4 66.66 H16-C6-C5-Hll - 55.08 N16-C6-C5-H15 - 174.19
EA EE
-59.88 170.78
- 179.59 51.06 60.69
- 68.65 55.11
173.86 176.88
-64.37 - 67.28
54.49 -63.75
54.99 173.86
-53.73 72.62
- 176.67 -50.32
65.64 - 168.00
53.73 - 72.62 176.67 50.32
- 65.64 168.00 59.88
- 170.78 179.59
-51.06 - 60.69
68.65 -55.11
- 176.88 - 173.86
64.37 63.75
- 54.99 67.28
- 54.49 - 173.86
60.63 - 170.95 - 179.85 -51.43 -59.13
69.29 - 57.73
- 176.36 - 176.36
65.01 64.41
-54.21 64.41
-54.21 - 173.44
60.63 - 170.95 - 179.85 -51.43 -59.13
69.29 - 60.63 170.95 179.85 51.43 59.13
-69.29 - 60.63 170.95 179.85 51.43 59.13
-69.29 57.73
176.36 176.36
-65.01 -64.41
54.21 -64.41
54.21 173.44

227
TABLE 3
Values of the puckering coordinates* Q, 0 and # and the N environment planarity parame&’ h as obtained by 4-2lG//4-21G and MM2(80) [8] calculations
PIPZ DACH TACH
AA EA EE AA EA EE AAA EAA EEA EEE
4-21G Q 0.521 0.566 0.604 0.527 0.566 0.584 0.500 0.543 0.561 0.561 e 0.0 4.3 0.0 3.1 3.4 1.8 0.0 4.9 1.1 0.0 $ 91.2 0.0 120.0 120.0 22.4 120.0 60.3 0.0 120.0 0.0 hN1 0.369 0.347 0.354 0.370 0.345 0.344 0.374 0.345 0.336 0.343 hN2 0.369 0.371 0.354 0.370 0.397 0.344 0.374 0.397 0.336 0.343 hN3 _ _ _ _ _ _ 0.314 0.397 0.420 0.343
MM2 Q 0.566 0.565 0.564 0.559 0.561 0.558 0.547 0.553 0.553 0.547 0 0.0 0.4 -0.1 0.4 1.2 0.2 0.0 1.0 0.6 0.0 $ 64.8 2.2 175.2 60.2 46.5 57.9 68.0 118.1 2.3 84.0 hN1 0.414 0.408 0.409 0.409 0.414 0.406 0.408 0.407 0.407 0.410 hN2 0.414 0.414 0.409 0.409 0.414 0.406 0.415 0.407 0.415 0.410 hN3 _ _ _ _ _ _ 0.408 0.419 0.415 0.410
“Ref. 17. bin A.
perfect chairs. The environment of the N atoms is flattest (i.e. the distance h between N and the plane defined by its three substituents least) when N-H is equatorial.
The main geometrical elements of EE determined in this study (once trans- formed to rg and r, values by application of empirical corrections [ 181) are compared with those obtained by MM2 [ 71 and the rB and F, values deduced from gas-phase electron-diffraction results [ 31 in Table 4. The differences be- tween the 4-21G//4-21G results and the experimental conclusions are, on the whole, within the limits of experimental error, and the discrepancies with re- spect to the MM2 figures are also no more than might be expected [ 191. Spe- cifically, MM2 (80) fails to detect either the anomeric effects or the N-H lengthening predicted by the ab initio calculations [6] [both the N-H bonds in EA are 1.017 A long according to MM2 (80) 1, and that the values of the ring- planarity parameter Q calculated by MM2 are virtually the same for all three conformers, such small differences as are predicted placing the conformers in the reverse order to that obtained by the ab initio method (Table 3). The MM2 findings nevertheless agree with the present results in predicting only minimal deviation of the ring from the perfect chair conformation, and as regards the effect of N-H orientation on the planarity of the environment of the N atoms (though all the h values are significantly larger for MM2 (80) than for the 4-21G calculations).

229
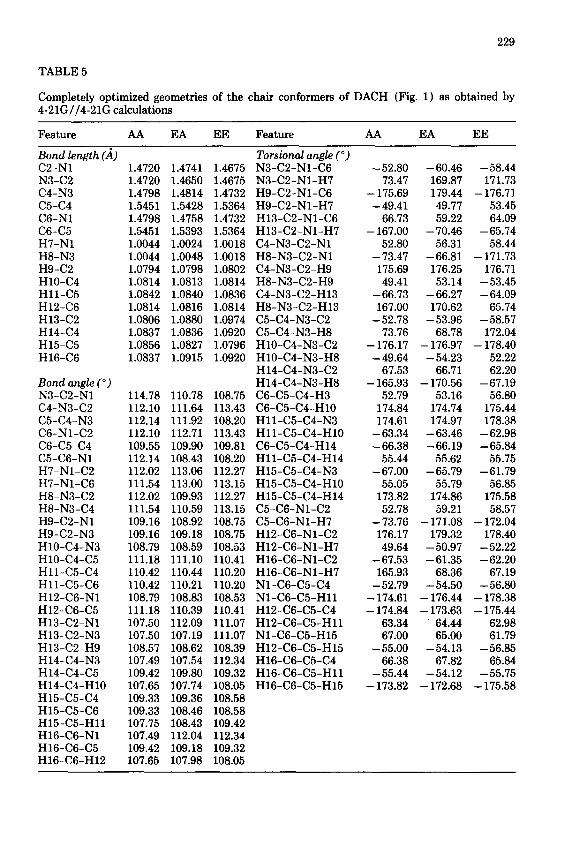
TABLE 5
Completely optimized geometries of the chair conformers of DACH (Fig. 1) as obtained by 4-21G//4-21G calculations
Feature AA EA EE Feature AA EA EE
Bond length (A) C2-Nl - N3-C2 C4-N3 c5-c4 C6-Nl C6-C5 H7-Nl H8-N3 H9-C2 HlO-C4 Hll-C5 H12-C6 H13-C2 H14-C4 H15-C5 H16-C6
1.4720 1.4720 1.4798 1.5451 1.4798 1.5451 1.0044 1.0044 1.0794 1.0814 1.0842 1.0814 1.0806 1.0837 1.0856 1.0837
Torsional angle Co) 1.4741 1.4675 1.4650 1.4675 1.4814 1.4732 1.5428 1.5364 1.4758 1.4732 1.5393 1.5364 1.0024 1.0018 1.0048 1.0018 1.0798 1.0802 1.0813 1.0814 1.0840 1.0836 1.0816 1.0814 1.0880 1.0974 1.0836 1.0920 1.0827 1.0796 1.0915 1.0920
Bond angle (“) N3-C2-Nl C4-N3-C2 C5-C4-N3 C6-Nl-C2 C6-C5-C4 C5-C6-Nl H7-Nl-C2 H7-Nl-C6 H8-N3-C2 H8-N3-C4 H9-C2-Nl H9-C2-N3 HlO-C4-N3 HlO-C4-C5 Hll-C5-C4 Hll-C5-C6 H12-C6-Nl H12-C6-C5 H13-C2-Nl H13-C2-N3 H13-C2-H9 H14-C4-N3 H14-C4-C5 H14-C4-HlO H15-C5-C4 H15-C5-C6 H15-C5-Hll H16-C6-Nl H16-C6-C5 H16-C6-H12
114.78 110.78 108.75 112.10 111.64 113.43 112.14 111.92 108.20 112.10 112.71 113.43 109.55 109.90 109.81 112.14 108.43 108.20 112.02 113.06 112.27 111.54 113.00 113.15 112.02 109.93 112.27 111.54 110.59 113.15 109.16 108.92 108.75 109.16 109.18 108.75 108.79 108.59 108.53 111.18 111.10 110.41 110.42 110.44 110.20 110.42 110.21 110.20 108.79 108.83 108.53 111.18 110.39 110.41 107.50 112.09 111.07 107.50 107.19 111.07 108.57 108.62 108.39 107.49 107.54 112.34 109.42 109.80 109.32 107.65 107.74 108.05 109.33 109.36 108.58 109.33 108.46 108.58 107.75 108.43 109.42 107.49 112.04 112.34 109.42 109.18 109.32 107.65 107.98 108.05
N3-C2-Nl-C6 N3-C2-Nl-H7 H9-C2-Nl-C6 H9-C2-Nl-H7 H13-C2-Nl-C6 H13-C2-Nl-H7 C4-N3-C2-Nl H8-N3-C2-Nl C4-N3-C2-H9 H8-N3-C2-H9 C4-N3-C2-H13 HS-N3-C2-H13 C5-C4-N3-C2 C5-C4-N3-H8 HlO-C4-N3-C2 HlO-C4-N3-H8 H14-C4-N3-C2 H14-C4-N3-H8 C6-C5-C4-H3 C6-C5-C4-HlO Hll-C5-C4-N3 Hll-C5-C4-HlO C6-C5-C4-H14 Hll-C5-C4-H14 H15-C5-C4-N3 H15-C5-C4-HlO H15-C5-C4-H14 C5-C6-Nl-C2 C5-C6-Nl-H7 H12-C6-Nl-C2 H12-C6-Nl-H7 H16-C6-Nl-C2 H16-C6-Nl-H7 Nl-C6-C5-C4 Nl-C6-C5-Hll H12-C6-C5-C4 H12-C6-C5-Hll Nl-C6-C5-H15 H12-C6-C5-H15 H16-C6-C5-C4 H16-C6-C5-Hll H16-C6-C5-H15
- 52.80 73.47
- 175.69 -49.41
66.73 - 167.00
52.80 - 73.47 175.69 49.41
-66.73 167.00
-52.78 73.76
- 176.17 - 49.64
67.53 - 165.93
52.79 174.84 174.61
- 63.34 - 66.38
55.44 - 67.00
55.05 173.82 52.78
- 73.76 176.17 49.64
-67.53 165.93
-52.79 - 174.61 - 174.84
63.34 67.00
- 55.00 66.38
- 55.44 - 173.82
- 60.46 - 58.44 169.87 171.73 179.44 - 176.71 49.77 53.45 59.22 64.09
- 70.46 - 65.74 56.31 58.44
-66.81 - 171.73 176.25 176.71 53.14 - 53.45
-66.27 - 64.09 170.62 65.74
- 53.96 - 58.57 68.78 172.04
- 176.97 - 178.40 - 54.23 52.22
66.71 62.20 - 170.56 -67.19
53.16 56.80 174.74 175.44 174.97 178.38
- 63.46 -62.98 -66.19 - 65.84
55.62 55.75 -65.79 -61.79
55.79 56.85 174.86 175.58 59.21 58.57
- 171.08 - 172.04 179.32 178.40
- 50.97 -52.22 -61.35 -62.20
68.36 67.19 - 54.50 - 56.80
- 176.44 - 178.38 - 173.63 - 175.44
64.44 62.98 65.00 61.79
-54.13 - 56.85 67.82 65.84
-54.12 -55.75 - 172.68 - 175.58

230
vironments of the nitrogen atoms are slightly flatter with equatorial than with axial N-H bonds (Table 3 ) .
The order of stability among the conformers predicted using MM2 (80) is the opposite to that predicted using 4-21G//4-21G, but there is no such dis- crepancy when the molecular-mechanics program used is MMB-AE [lo], which takes anomeric effects into account (Table 1). As expected, MM2 (80) also fails to reproduce the geometric anomeric effects described above [ 6,9], and conclusions concerning ring planarity likewise differ from those of the ab initio study (Table 3). With regard to the N atom environments, the same holds as for PIPZ.
1,3,5-Triazocyclohne
According to the present calculations, the most stable conformer is AAA (Table 1 ), although there are no experimental data confirming this. MM2 (80) calculations [8] point to EAA and EEA as the most stable conformers, but agree with the present work in identifying EEE as the least stable.
The N-C distances exhibit the same trend as in DACH, being longer when the other N bound to the C bears an axial H; in the case of EAA, for example, the distance between C and N atoms bearing axial H is 1.4758 A when the other N bound to the carbon atom also bears an axial H and 1.4687 A when it bears an equatorial H. The pattern of the C-H bond lengths (Table 6) can likewise be explained in terms of anomeric interaction between the equatorial lone pair of a nitrogen with axial H and the corresponding C-N bonds.
As in PIPZ and DACH, N-H bonds are longer when axial than when equa- torial, 1.0046 A in AAA as against 1.0012 A in EEE or, intramolecularly, 1.0048 as against 1.0026 A in EAA and 1.0054 as against 1.0018 A in EEA. The lengths of both N-H,, and N-H, bonds decreases as their number increases, the for- mer from 1.0054 A in EEA to 1.0046 A in AAA and the latter from 1.0026 A in EAA to 1.0012 A in EEE.
The pattern exhibited by the N-C-N and H-C-N bond angles (Table 6) is explicable in terms of anomeric effects. The H-C-N angles are wider when the hydrogen on the N atom is axial than when it is equatorial, 109.52’ in AAA as against 108.88” in EEE. The N-C-N angles increase with the number of axial H on their N atoms: in EAA, N3-C4-N5 is 3.6’ wider than Nl-C2-N3; in EEA Nl-C6-N5 is 1.6” wider than Nl-C2-N3; and the N-C-N angles of AAA are 5.9’ wider than those of EEE.
The C-N-C and C-N-H angles are both narrower when the H on the nitro- gen atom is axial than when it is equatorial. For C-N-C the difference is 0.560 ’ in EAA, 2.230” in EEA, and 1.96” on comparing AAA with EEE; while for C- N-H it is 2.50” in EAA, 3.81’ in EEA, and 0.95’ on comparing AAA with EEE.
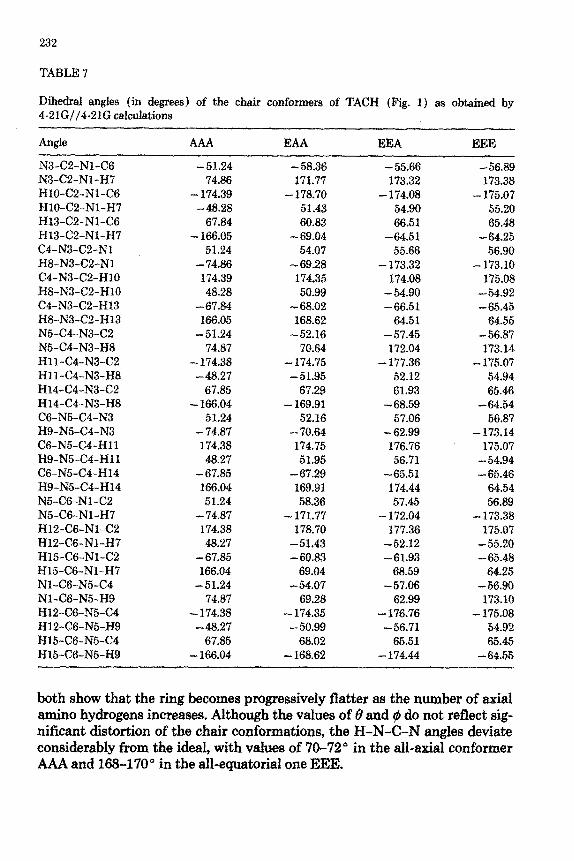
The dihedral angles (Table 7) and the puckering amplitudes Q (Table 3)

231
TABLE 6
Bond lengths and bond angles of the chair conformers of TACH (Fig. 1) as obtained by 4-21G//4-21G calculations
Feature AAA EAA EEA EEE
Bond length (A) C2-Nl N3-C2 C4-N3 N5-C4 C6-Nl C6-N5 H7-Nl H&N3 H9-N5 H10-C2 Hll-C4 H12-C6 H13-C2 H14-C4 H15-C6
Bond angle (“) N3-C2-Nl C4-N3-C2 N5-C4-N3 C6-Nl-C2 C6-N5-C4 N5-C6-Nl H7-Nl-C2 H7-Nl-C6 H8-N3-C2 H8-N3-C4 H9-N5-C4 H9-N5-C6 HlO-CZ-Nl HlO-C2-N3 Hll-C4-N3 Hll-C4-N5 H12-C6-Nl H12-C6-N5 H13-C2-Nl H13-C2-N3 H13-C2-HlO H14-C4-N3 H14-C4-N5 H14-C4-Hll H15-C6-Nl H15-C6-N5 H15-C6-H12
1.4757 1.4771 1.4705 1.4668 1.4757 1.4687 1.4705 1.4663 1.4757 1.4758 1.4728 1.4664 1.4757 1.4758 1.4682 1.4664 1.4757 1.4771 1.4728 1.4668 1.4757 1.4687 1.4682 1.4663 1.0046 1.0026 1.0018 1.0013 1.0046 1.0048 1.0018 1.0012 1.0046 1.0048 1.0054 1.0012 1.0789 1.0793 1.0798 1.0794 1.0789 1.0787 1.0791 1.0794 1.0789 1.0793 1.0791 1.0794 1.0805 1.0879 1.0969 1.0981 1.0805 1.0804 1.0882 1.0981 1.0805 1.0879 1.0882 1.0981
114.08 110.26 108.29 108.20 111.22 111.11 112.87 113.24 114.08 113.91 109.93 108.18 111.22 111.67 112.87 113.18 111.22 111.11 110.64 113.24 114.08 110.26 109.93 108.20 111.97 113.51 112.92 112.86 111.97 113.51 113.93 112.86 111.97 110.04 112.92 112.95 111.97 111.01 113.93 112.96 111.97 111.01 109.11 112.96 111.97 110.04 109.11 112.95 109.52 109.32 108.98 108.87 109.52 109.43 108.98 108.87 109.52 109.31 109.04 108.89 109.52 109.31 109.39 108.89 109.52 109.32 109.04 108.87 109.52 109.43 109.39 108.87 107.50 111.98 111.05 111.16 107.50 107.13 111.05 111.19 108.56 108.66 108.45 108.49 107.50 107.74 112.50 111.17 107.50 107.74 107.19 111.17 108.57 108.70 .108.73 108.49 107.50 111.98 112.50 111.16 107.50 107.13 107.19 111.19 108.57 108.66 108.73 108.49
232
TABLE 7
Dihedral angles (in degrees) of the chair nonfocal of TACH (Fig. 1) as obtained by 4-21G//4-21G calculations
Angle AAA EAA EEE
N3-C2-Nl-C6 N3-CZ-Nf-H7 H~O-C2-Nl-C6 HlO-C2-Nl-H7 H13-C2-Nl-C6 H13-CZ-Nl-H7 C4-N3-CZ-Nl H8-N3-CZ-Nl C4-N3-C2-HlO H8-N3-C2-HlO C4-N3-CZ-H13 H8-N3-C2-H13 N5-C4-N3-C2 N5-C4-N3-HS Hll-C4-N3-C2 Hll-C4-N3-H8 Hl4-C4-N3-C2 Hl4-C4.-N3-H8 C6-N5-C4-N3 H9-N5-C4-N3 C6-N5-C4-Hll H9-N&C4-Hll C6-N5-C4-H14 H9-N5-C4-H14 N5-C6-Nl-C2 N5-C6-Nl-H7 H12-C6-Nl-C2 Hl2-~6-Nl-H7 H15-C6-Nt-C2 H15-C6-Nl-H7 Nl-C6-N&-C4 Nl-C6-N5-H9 H12-C6-N&C4 H12-C6-N&H9 HWC6-N&-C4 Hl5-C6-N6-H9
-51.24 -58.36 74.86 171.77
- 174.39 - 178.70 -48.28 51.43
67.84 60.83 - 166.05 - 69.04
51.24 54.07 - 74.86 - 69.28 174.39 174.35 48.28 50.99
-67.64 - 66.02 166.05 168.62
-51.24 -52.16 74.87 70.64
- 174.38 - 174.75 -48.27 -51.95
67.85 67.29 - 166.04 - 169.91
51.24 52.16 - 74.87 - 70.64 174.38 174.75 48.27 51.95
- 67.65 - 67.29 166.04 169.91 51.24 58.36
- 74.87 - 171.77 174.38 178.10 48.27 -51.43
-67.85 -60.83 166.04 69.04
- 51.24 - 54.07 74.67 69.28
- 174.38 - 174.35 -48.27 - 50.99
67.85 68.02 - 166.04 - 168.62
- 55.66 - 56.89 173.32 173.38
- 174.08 - 175.07 54.90 55.20 66.51 65.48
-64.51 - 64.25 55.66 56.90
- 173.32 - 173.10 174.08 175.08
- 54.90 -54.92 -66.51 - 65.45
64.51 64.55 - 57.45 - 56.87 172.04 173.14
- 177.36 - 175.07 52.12 54.94 61.93 65.46
- 68.59 -64.54 57.06 56.87
-62.99 - 173.14 176.76 175.07 56.71 - 54.94
-65.51 - 65.46 174.44 64.54 57.45 56.89
- 172.04 - 173.38 177.36 175.07
-52.12 - 55.20 -61.93 - 65.48
68.59 64.25 - 57.06 - 56.90
62.99 173.10 - 176.76 - 175.08
-56.71 54.92 65.51 65.45
- s74.44 - 64.55
both show that the ring becomes progressively flatter as the number of axial amino hydrogens increases. Although the values of 8 and Q1 do not reflect sig- nificant distortion of the chair c~nfo~ations, the H-N-C-N angles deviate considerably from the ideal, with values of 70-72” in the all-axial conformer AAA and 168-170” in the all-~ua~rial one EEE.

233
MM2 failed to detect the anomeric effects described above, predicting max- imum inter-conformer variations of 0.4’ and 0.2’ for N-C-N and C-N-C an- gles, respectively [ 71, as against the 5.9’ and 2.6” calculated in this study. Also, the inter-conformer differences in the ring puckering amplitude Q found by MM2 (80) were minimal and ordered differently from those found by 4-21G. The two methods are nevertheless not too dissimilar as regards the trend (al- though not the value) of the planarity h of the N atom environment, which is flatter when N-H is equatorial.
CONCLUSIONS
MM2 (80) fails to order conformers by energy correctly when anomeric units are present. MMB-AE [lo] succeeds in doing so in the case of DACH, but no MMB-AE results are available for PIPZ or TACH. With regard to the struc- tural trends within the series of compounds studied, it may be concluded that: (i) increasing the number of nitrogen atoms flattens the chair conformation but not the environments of the nitrogen atoms; (ii) the planarity of both the ring itself and the N environments is virtually the same for both diazocyclo- hexanes; and (iii) increasing the number of axial N-H bonds flattens the ring and decreases the planarity of the N environments.
ACKNOWLEDGEMENTS
We thank Prof. B. Fuchs for providing us with a copy of the manuscript of ref. 10. This work was financially supported by the Xunta de Galicia and by Spanish Ministry of Education and Science F.P.I. grants awarded to B.F., R.A.M., J.R. and S.A.V.
REFERENCES
2 3 4
5
6 7 8 9
10 11 12
T.A. Crabb and A.R. Katritzky, Adv. Heterocycl. Chem., 36 (1984) 1. M. Davis and 0. Hassel, Acta Chem. Stand., 17 (1963) 1181. A. Yokozeki and K. Kuchitsu, Bull. Chem. Sot. Jpn., 44 (1971) 2352. U. Burkert and N.L. Allinger, Molecular Mechanics, American Chemical Society, Washing- ton, DC, 1982. (a) N.L. Allinger, J. Am. Chem. Soc.,99 (1977) 8127. (b) N.L. Allinger and Y. Yuh, QCPE, 12 (1980) 395. L. Carballeira, R.A. Mosquera and M.A. Rfos, J. Mol. Struct., 176 (1988) 89. L. Carballeira, R.A. Mosquera, M.A. Rios and C.A. Tovar, J. Mol. Struct., 193 (1989) 263. L. Carbaheira, R.A. Mosquera and M.A. Rfos, J. Mol. Struct., in press. R.A. Mosquera, Ph.D. Thesis, University of Santiago de Compostela, Spain, 1988. P. Aped, L. Schleifer, B. Fuchs and S. Wolfe, J. Comput. Chem., 10 (1989) 265. P. Pulay, G. Fogarasi, F. Pang and J.E. Boggs, J. Am. Chem. Sot., 101 (1979) 2550. P. Pulay, Mol. Phys., 17 (1969) 197.

234
13 P. Pulay, Theor. Chim. Acta, 50 (1979) 299. 14 R.A.Y. Jones, A.R. Katritzky, A.C. Richards, S. Saba, A.J. Sparrow and D.L. Trepanier,
Chem. Commun., (1972) 673. 15 C. Romers, C. Altona, H.R. Buys and E. Havinga, Topics Stereochem., 4 (1969) 39. 16 J.O. Williams, J.N. Scarsdale, L. Schafer and H.J. Geise, J. Mol. Struct. (Theochem), 76
(1981) 11. 17 D. Cremer and J.A. Pople, J. Am. Chem. Sot., 97 (1975) 1354. 18 L. Schafer, J. Mol. Struct., 100 (1983) 51. 19 K. Siam, O.V. Dorofeeva, V.S. Mastryukov, J.D. Ewbank, N.L. Allinger and L. Schafer, J.
Mol. Struct. (Theochem), 164 (1988) 93. 20 A.R. Katritzky, M. Moreno-Maiias, A.C. Richards, A.J. Sparrow and D.L. Trepanier, J. Chem.
Sot., Perkin Trans. 2, (1973) 325. 21 H. Booth and R.U. Lemieux, Can. J. Chem., 49 (1971) 777.