Embed Size (px)
Citation preview

INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY, VOL. XV, 567-578 (1979)
Ab initio Molecular Fragment Calculations with Pseudopo t entials:
Hydrocarbon Calculations of Double-Zeta Quality
R. GASPAR, JR. Department of Biophysics, Medical University of Debrecen, H-4012 Debrecen, Hungary
R. GASPAR Institute of Theoretical Physics, Kossuth Lajos University, H-4010 Debrecen, Hungary
Abstracts
Pseudopotential theory is introduced into the ab initio FSGO molecular fragment method. A theoretical background for the pseudopotential fragment description and a method for large molecule formation is presented. Core-valence electron separation is achieved at both levels of the calculations with the resulting simplification of the molecular calculations. Using pseudopotentials of double-zeta quality a detailed description of pseudopotential molecular fragments CH4 (tetrahedral) and CH3 (planar) is presented. Applications of the pseudo-FSGO molecular fragment method to hydrocarbons are discussed. The results are compared to those of the original FSGO method and experiment.
La thiorie des pseudopotentiels est combinee avec la methode FSGO-ab-initio pour traiter des fragments moliculaires. On presente la theorie pour une description des fragments par un pseudopo- tentiel et une mtthode pour la construction d’une grande molecule. La separation coeur-valence a ete atteinte aux deux niveaux des calculs avec, par consequence, une simplification des calculs molecu- laires. A l’aide de pseudopotentiels de qualite double-zeta on a obtenu une description detaillee des fragments moleculaires CH, (tetrakdrique) et CH3 (plan). On discute des applications de la methode pseudo-FSGO a des hydrocarbures. Les resultats sont compares a ceux obtenus par la mkthode originale FSGO et des experiences.
Pseudopotentiale sind in die Molekularfragmentmethode von Typ ah-initio-FSGO eingefuhrt worden. Ein theoretischer Hintergrund der Pseudopotentialfragmentbeschreibung und eine Methode fur die Bildung grosser Molekule werden angegeben. Die Separation von Rumpf- und Valenzelek- tronen ist an beiden Stufen der Berechnungen erreicht worden, was die Molekulrechnungen verein- facht. Mit Pseudopotentialen von Doppelzetaqualitat wird eine detaillierte Beschreibung der Mole- kulfragmente CH4 (tetraedrisch) und CH3 (eben) erhalten. Anwendungen auf Kohlenwasserstoffen werden diskutiert. Die Ergebnisse werden mit entsprechenden Resultaten von Berechnungen mittels der ursprunglichen FSGO-Methode und mit Experimentaldaten verglichen.
Introduction
Molecular quantum mechanics has always been driven by necessity to develop calculational methods for the treatment of large molecular systems. The inspira- tion for this has been drawn mainly from other fields of science such as chemistry, biology, and pharmacology (see, e.g., Ref. 1). However, for a long time, the application of theoretical ideas of quantum mechanics to large molecular systems
0 1979 John Wiley & Sons, Inc. 0020-7608/79/0015-0567$01.00

568 GASPAR AND GASPAR
has been restricted by numerous technical difficulties. With the advent of large- scale digital computers and the development of semiempirical methods the size of the largest investigated molecules has rapidly increased. The semiempirical methods, however, provide a first-level approximation only, which is to a certain extent realistic and informative and which has found many applications in large chemical systems [2,31.
Recent developments in ab initio methods, with the introduction of new mathematical and computational ideas, also increased the size of the molecules that can be treated by these methods [4-61. Furthermore, “molecular fragment approach” has been proposed for ab initio treatment of large molecules on the basis of the floating-spherical-Gaussian-orbital (FSGO) method of Frost. The FSGO fragment method is especially suited for calculating the properties of large molecular systems [7-91.
Pseudopotential theory developed fairly early and has found its applications mainly in the quantum theory of atoms, ions, and solids [lo, 111. The pseudopo- tential method replaces the atomic cores by appropriate potentials in such a manner that only valence electrons remain to be treated explicitly during the atomic calculations. This idea has also been extended to molecular calculations [12]. Recently quite a few attempts have been made to incorporate pseudopoten- tials into the FSGO scheme [13]. Moreover, a new form of pseudopotential for general application in ab initio molecular calculations has been suggested. By the aid of this pseudopotential the pseudo-FsGo method has been established. It has been shown that the pseudo-FsGo method helps to reduce the computational requirement of the molecular calculations with the simultaneous improvement of their accuracy [14,15].
The present work makes an attempt to introduce the pseudopotential theory into the basic framework of the FSGO fragment method of Christoffersen, which has been specially designed for treating large molecules. In this way the ab initio nature of the molecular calculations will be preserved and the computational requirement of the resulting pseudo-FsGo fragment method is reduced to approximately that of an all-valence electrons SCF calculation.
Pseudopotential Molecular Fragments and Large Molecule Formation
Molecular fragments are small molecules that appear as entities in larger molecules, and they also can be used to build up large molecules by appropriately combining them. An adequate description of the electron distribution in molecu- lar fragments can be established in the framework of the floating-spherical- Gaussian-orbital method of Frost [7] . In the FSGO method normalized FSGOS are used as basis orbitals, and both the position and size of the FSGOS and the location of the nuclei are varied until the energy of the molecule is minimized [8,9]. A substantial fraction of the basis orbitals is used to accommodate the core electrons of the atoms only. These orbitals can be replaced by appropriate pseudopotentials within the general framework of the FSGO method in such a manner that only valence electrons remain to be treated explicitly in the molecular calculations

DOUBLE-ZETA QUALITY HYDROCARBON CALCULATIONS 569
[ 14,151. Molecular fragments in which the effect of the core orbitals is replaced by pseudopotentials will be called pseudopotential molecular fragments.
Using the pseudopotential approach the Nv valence electrons in a molecular fragment containing n atoms can be described by the valence pseudo-Hamil- tonian
N" NV 1 XPS= c ( - f ~ ( i ) + k = l i: vp(i)) + i > , c - rij (1)
i = l
where V? is the pseudopotential associated with the core of atom k. (All quantities in this report are given in atomic units except where otherwise stated.)
For a given atom we have
L V r = - - + C A I exp ( -a f r2 )Pf
r i
where Z is the net charge of the ion core consisting of the core electrons and the nucleus of the atom, A[ and aI are the pseudopotential parameters, r, 1, and P, have been defined in Ref. 15. A( and a[ are determined so that the two lowest eigensolutions of the atomic pseudo-Hamiltonian coincide with the valence orbital energies of the one-electron ion in question. The actual determination of the parameters has been made by a variational procedure using trial functions of double-zeta quality [14]. For the carbon atom the following pseudopotential parameters have been obtained: A. = 31.6899; a. = 8.0076; A l = -15.2716; a I = 20.6316. Ai and a1 with I r 2 has been set equal to zero because the corresponding term values of the ions and atom, respectively, are Coulombic to a good approximation.
The normalized FSGOS have the following form:
(3) Gz(r)=(2a p 1 1 exp{-p12(r-Rl)21
where p, is the orbital radius and R, is the vector determining the location of the center of the FSGOS. Both R, and pl of the molecular fragment are varied until the valence electronic energy Eel associated with the pseudo-Hamiltonian Xps is minimized. No change in the nuclear geometry of the fragment during the above minimization process is allowed. The total valence energy Eva, of a fragment can be determined as the sum of the valence electronic energy E,, and of the Coulombic repulsion energy between the atomic cores:
-1 -2 3 / 4
where z k and Zm are the charges of the atomic cores and Rkm is the distance between atoms k and m.
To be able to compare the results of the present investigation to those gained by other methods, as, for example, by the original FSGO fragment method, the total energy ET of the fragment and the molecules constructed from them has to be determined. For this purpose, EFo,,, the electronic energy of the core electrons

570 GASPAR AND GASPAR
of a carbon atom, is calculated according to the electronic energy of a He-like ion (EFore = -32.4061 hartree) [16]. The total energy of a fragment or molecule containing Nc carbon atoms is
ET = E v a l + N & L z ( 5 )
Table I contains a list of several molecular fragments of interest. Table I also contains the number of independent nonlinear parameters that have been varied
TABLE I. Molecular fragments of interest.
50 . of independent n o n l i n e a r Molecular fragment h 0 . O f FSGO’S parameters
P s e u d o p o t e n t i a l FSGO f r a p e n t I s e u o o p o t e n t i a l ?‘SGC fragmer.t fragment method method fragn.ent method method
CH4 ( t e t r a h e d r a l ) 4 5 2 3
CHJ ( p l a n a r )
6H ( t e t r a h e d r a l )
IUH ( p l a n a r )
hH2 ( p l a n a r )
NH+ ( p l a n a r )
NH; ( t e t r a h e d r a l )
Ii20 ( b e n t )
H20 (sP’)
OH ( s p 2 )
H;O ( p l a n a r )
3
3
3
5a
5a Sa
5a
4
4
4
5a
5a
5a
6a
5
6a
ba
6a
5
5
6a
6a
6a
4
5 4
6
4
3
5
6
6
4
a These fragments use the v orbital description defined in Ref. 7. For v orbital FSGO positions 10.1 bohr was taken above and below the plane of the fragment.
for each fragment according to the above-described procedure and the number of FSGOS that proved to be sufficient to describe the electronic structure of the fragment in the pseudopotential approach. The selection and chemical appli- cability of the above set of fragments need no consideration in the present article, because the usefulness of the listed fragments as building blocks of large molecules has already been proved [17,18].
Table I1 presents a detailed characterization of the fragments CH4 (tetra- hedral) and CH3 (planar) bearing special importance in hydrocarbon investiga- tions. A modified version of the FSGO program of Frost has been developed, and the resulting pseudo-FsGo procedure has been used to gain the nonlinear frag- ment parameter and the energy values listed in the table [14, 151. During the calculation of the fragment parameters and energies no variation in the nuclear geometry of the fragments was allowed.
For large molecule formation from the pseudopotential molecular fragments a method similar to that of the pseudo-FsGo procedure can be used. The VY

DOUBLE-ZETA QUALITY HYDROCARBON CALCULATIONS 571
TABLE 11. Optimized parameters of the CH4 (tetrahedral) and CH3 (planar) fragments.”
- CH4 ( t e t r a h e d r a l ) : CH d i s t a n c e = 2.0598 bohr
Gaussian parameters
Dis tance from S carbon atom Csrbi ta l t y p e
CH 1-6735 1.1897
T o t a l energy = -39.5719
CH3 ( p l a n a r ) : CH d i s t a n c e = 1.7856b Gaussian parameters
O r b i t a l t y p e 3 Dis tance from
carbon atom
CH 1.5027 1.0838 1.9529 zo.1
T o t a l energy = -38.9695 -
a All distances and energies in the table are given in a.u. bThis distance was chosen according to the formula originally
proposed by Christoffersen et al. in Ref. 7.
pseudopotentials are introduced into the FSGO procedure by restricting the treatment to the valence electrons only and replacing the effect of the core with pseudopotentials. Furthermore, the atomic orbitals of the pseudo-FsGo pro- cedure are replaced by the fragment FSGOS. The first step of large molecule formation requires the removal of those hydrogen atoms from the fragments, which served only to polarize the environment for the determination of the nonlinear parameters in the molecular fragments. However, the FSGOS are retained in these regions and new chemical bonds are formed by taking the linear combination of pairs of these fragment FSGOS.
Hydrocarbon Investigations
Following the rules outlined in the previous section, ethane, ethylene, and propane molecules have been constructed from CH4 (tetrahedral) and CH3 (planar) pseudopotential molecular fragments. The application of the pseudopo- tential fragment method to the calculation of the above molecules bears special importance not only because they are widely investigated hydrocarbons but they also serve as useful models for larger molecules in the present series of investiga- tions. The choice of the particular pseudopotential molecular fragments and the correctness of their description according to the pseudopotential approach can be supported only by results gained on larger molecules assembled from them.

572 GASPAR AND GASPAR
Geometrical as well as energy predictions of the method should be examined. As precise experimentally determined geometries are usually available only for smaller molecules, we feel justified in replacing larger molecules by these smaller hydrocarbons in the course of the present investigation.
The ethane molecule is assembled from two pseudopotential CH4 (tetra- hedral) fragments. The two hydrogen atoms along the carbon-carbon bond to be formed are removed from the fragments, but the Gaussians belonging to them are retained in their original positions with respect to the carbon atoms. The two fragments are then placed at a distance until the separation of the two carbon atoms corresponds to the experimentally determined carbon-carbon distance of ethane (Rc-c = 2.90 bohr) [7]. The linear combination of the two Gaussians along the C-C bond is formed and the FsGo-pseudopotential procedure is performed. Now the relative position of the CH3 groups in ethane can be varied in order to describe the barrier to rotation in the molecule. Table I11 contains a
TABLE 111. Ethane barrier to rotation.”
AnKle of rotationb T o t a l energy (deg.) ( h a r t r e e )
0.0
0.5 1.5 2 -0
2.5 3 .0 3.5
4.5 5.0
15.0 30 .o 45.0
60 .0
4 .0
-78.087654 -78 -083653 -78.083642
-78,063653 -78.083620 -78 -083605 -7a.083588 -78.083567
-78 .oa35m -78.087544
-78 .O82484 -78.079645 -78.076785 -7a.075595
a During the calculation of the barrier to rotation in ethane experimental distances and tetrahedral angles were used.
bAngle of rotation is measured from the staggered conformer.
detailed description of the results. The angle of rotation 0 is measured from the staggered conformer and the total energy ET of the molecule is determined for selected values of 0. The height of the rotational barrier is found to be 5.06 kcal/mol compared to the experimentally observed 2.98 kcal/mol. Furthermore, the shape of the rotational barrier presented in Table I11 is analyzed according to the formula suggested to describe the precise shape of the rotational barrier for

DOUBLE-ZETA QUALITY HYDROCARBON CALCULATIONS 573
comparison with experimentally determined data [ 191:
E ( 0 ) = $ V,( 1 - cos 3 0 )
Figure 1 displays the deviation between the points of the theoretical curve predicted by the above equation and those of the curve gained by the pseudopo- tential fragment method. Figure 2 shows the variation of total energy of ethane
8 (Degrees )
Figure 1. Relative percentage deviation of the potential barrier hindering free rotation in ethane as predicted by the pseudopotential fragment method and by the E(0) = iV , ( l - cos 30) equation for selected values of the internal torsional angle 0.
V, has the value of 5.06 kcal/mol.
t -78.083
t
Figure 2 . Total energy of ethane as a function of the C-C distance.

574 GASPAR AND GASPAR
with Rc-c, the separation of the carbon atoms. The minimum of the curve predicts an equilibrium C-C distance of 2.80 bohr in ethane compared to the experimentally observed value of 2.90 bohr [20].
The capability of the present method to reproduce an unsaturated carbon- carbon double bond may be investigated on the ethylene molecule. Experimental internuclear distances and idealized bond angles (120") are used [21]. FSGOS of both CH4 (tetrahedral) and CH3 (planar) type fragments are applied to charac- terize the electronic structure of the molecule. CH4 (tetrahedral) type FSGOS are placed between nuclei C and H to construct CH bond in ethylene, for this bond corresponds to a normal CH bond. The carbon-carbon double bond of the molecule is formed by using T type and CH type orbitals taken from the CH3 (planar) fragment. Linear combinations of the FSGOS are then formed both from the T type and the CH type orbitals for the carbon-carbon bond, respectively, and the total energy of the molecule is determined. The results of such calculations for different C=C distances are displayed in Figure 3. The equilibrium C=C distance of 2.57 bohr is deduced from the above curve compared to the experimentally observed 2.47 bohr [21].
The propane molecule is assembled within the framework of the molecular fragment approach from CH, (tetrahedral) fragment FSGOS only. Selecting experimentally determined internuclear distances and tetrahedral bond angles, investigations regarding conformational properties of propane can be performed
t -77.1651 1 I I 1
2.55 2.60 2.65 2.70 R ( B o h r s )
Figure 3 . Total energy of ethylene as a function of the C=C distance.

DOUBLE-ZETA QUALITY HYDROCARBON CALCULATIONS 575
[22]. Varying the position of the terminal methyl groups relative to the central CH2 group three main conformations of the molecule can be established. Table IV presents calculated total energy values belonging to different conformers of propane. The table also contains the barriers to rotation separating the three main conformers on the energy scale. The present results can be compared only to
TABLE IV. Conformational studies of propane molecule.”
Conformer Energy crimonent
Staggered-staggered Staggered-eclipsed Eclipsed-eclipsed
hinetic 14.3479 14.3679 14,3866
blectronic Coulomhic r e p u l s i o n 58 2906 56.3108 58.3407
Point core-electron attraction -144.:,732 -144,6270 -1U.7074
Point core -po in t core revulsion 43.4229 a9.9469 49.9851
Fseudo core-electron repulsion 2.5969 7.5979 2 - 5991 Total energ7 -116.6331 -116.6256 -116.6143
Barriers to rotation
E(sta~perPd-st3fifered) - E(stasgered-eclipsea) = 4.68 kcal/mole E(staf~ered-stapFered) - E(eclipsed-ecliDqed) = 11.78 kcal/mole
a During the calculations experimental distances and tetrahedral angles were used.
similar ones gained by other calculational methods, as, for example, by the original FSGO fragment method, for no experimental data exist for these barrier heights in propane. The lower barrier to rotation can also be favorably compared to that of the methyl groups in ethane. Changing the C-C-C bond angle in propane the total energy of the molecule is determined and displayed in Figure 4. From the minimum of the curve the equilibrium bond angle of 112.2’ can be deduced in good agreement with the experimentally determined angle of 112.4” W I .
Discussion
A characteristic feature of the present investigation is the introduction of the pseudopotential theory within the general framework of the ab inifio FSGO fragment method. A theoretical background to the pseudopotential molecular fragment description and large molecule formation has been established. Pseudopotentials have been included in the description of the individual molecu- lar fragments and also in the large molecule “assembling” FSGO procedure. Replacing the atomic cores by pseudopotentials simplifies the description of the molecular fragments. This simplification results in the reduction of the number of

576
- 111 0.J aJ L
L ... 2 -116.63395-
F
- x
C W
0
0 +
-116.63400- - -
GASPAR AND GASPAR
c -116.63390
i
-116.63405 Lo ' 112 ' 0 I 1130 ' C - C - C Angle ( Degrees)
'
Figure 4. Total energy of the staggered-staggered conformer of propane as a function of the C-C-C angle.
FSGOS required to characterize the individual molecular fragmmts, as can be seen in Table I. This is an important technical advantage of the present method, for the molecular fragments are further involved in a FSGO type of calculational pro- cedure during their assembly into larger molecules. The time consumption of this latter procedure strongly depends on the number of FSGOS involved in the description of the large molecule under consideration. The saving of computation time, compared to the requirements of the original FSGO fragment method, further increases if atoms from rows of the periodic table with higher atomic number are contained by the investigated molecules. The simplifications intro- duced in the description of the molecular fragments result not only in calculational but conceptual advantages too. The core-valence electron separation achieved by the introduction of the pseudopotential theory enables one to use well-known chemical ideas directly in order to select the appropriate molecular fragments and construct large molecules from them.
Furthermore, Table V demonstrates that geometrical and energy predictions for hydrocarbons gained by the pseudopotential fragment method are more accurate than those gained by the original FSGO fragment method [7]. It must be emphasized at this point that highly restricted basis sets have been employed in the molecular calculations, for the hydrocarbons have been assembled from the molecular fragments characterized in Table 11. The accuracy of the total energy determinations by the pseudopotential fragment method is most striking if one studies the appropriate figures in Table V.
It must be noted, however, that it is always the total valence energy that is calculated by the pseudopotential fragment method, and the percentage devia- tions of the experimental and calculated total valence energy values are much less than those of the experimental and the calculated total energy values determined by the FSGO fragment method. The magnitude of total valence energy is much
DOUBLE-ZETA QUALITY HYDROCARBON CALCULATIONS 577
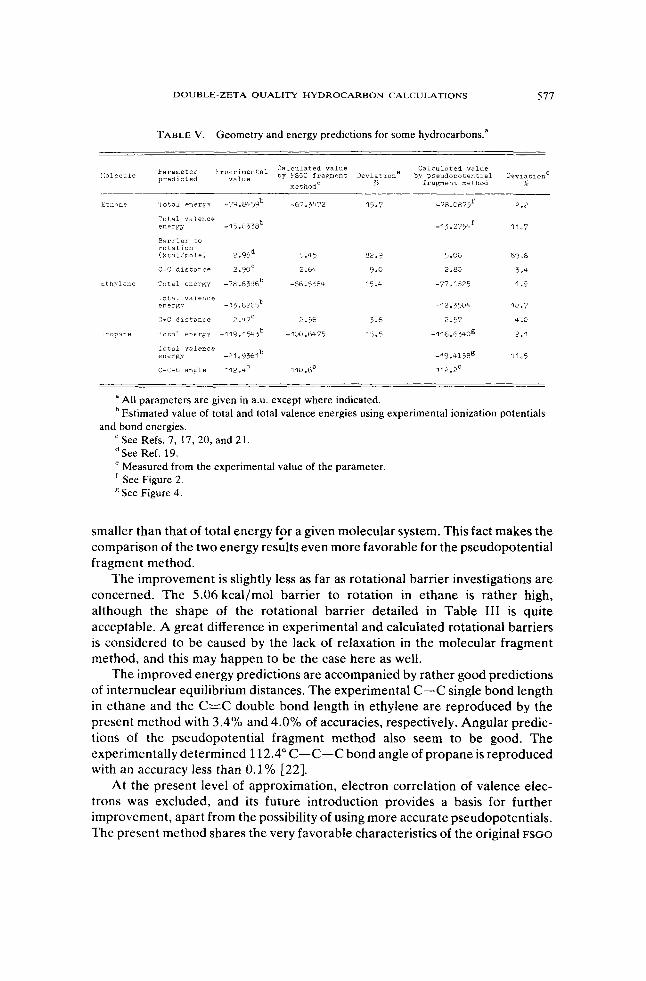
TABLE V. Geometry and energy predictions for some hydrocarbons."
Calcul?ted value
% fragnent method :.olecule ~ a r a n e t e r :-xperimental by ?SiO f r a m e n t Deviatione bv pseudoootential Deviatlone predicted value
nethod'
I ' o t a l valence enerpy -15.07385 -1 5.2754' 11.7
srrrr1er to rotation (kcal/mole) 7.96* 5.45 62*9 5.06 69.8
C-C d i s t a z c e 2.90' 2.w 9.0 2.80 3.4
r t h , v l e n e I o t a 1 e n t r p j -i8.63:16b -66.5584 15.4 -77.1675 1.9
' o t a i valer.ce energv -1 J.b265b -12.5504 16.7
:=c d i s t a n c e 2.47' 2.56 1.6 2.57 4 .0
I r 0 p ' ~ n e 1'07;51 entr&Y -11U.1Y13b -100.6475 15.5 -1 16.6 540' 2.1
. o t a l valence enerFy -21 .9561b
c-c-6 mRle 112.*0 110.60
-19.4158' 11.5
112.20
a All parameters are given in a.u. except where indicated. Estimated value of total and total valence energies using experimental ionization potentials
See Refs. 7, 17,20, and 21.
Measured from the experimental value of the parameter See Figure 2. See Figure 4.
and bond energies.
dSee Ref. 19.
smaller than that of total energy f2r a given molecular system. This fact makes the comparison of the two energy results even more favorable for the pseudopotential fragment method.
The improvement is slightly less as far as rotational barrier investigations are concerned. The 5.06 kcal/mol barrier to rotation in ethane is rather high, although the shape of the rotational barrier detailed in Table I11 is quite acceptable. A great difference in experimental and calculated rotational barriers is considered to be caused by the lack of relaxation in the molecular fragment method, and this may happen to be the case here as well.
The improved energy predictions are accompanied by rather good predictions of internuclear equilibrium distances. The experimental C-C single bond length in ethane and the C=C double bond length in ethylene are reproduced by the present method with 3.4% and 4.0% of accuracies, respectively. Angular predic- tions of the pseudopotential fragment method also seem to be good. The experimentally determined 112.4" C-C-C bond angle of propane is reproduced with an accuracy less than 0.1% [22].
At the present level of approximation, electron correlation of valence elec- trons was excluded, and its future introduction provides a basis for further improvement, apart from the possibility of using more accurate pseudopotentials. The present method shares the very favorable characteristics of the original FSGO

578 GASPAR AND GASPAR
fragment method, namely, the accuracy of the calculations increases as the molecules become more complicated. This advantage of the method arises from the fact that during the formation of each new chemical bond between the molecular fragments an additional orbital is added to the basis set. This results in an increasing relative percentage of virtual orbitals with the increasing size of the molecule [ 171.
We regard the present method as a potential challenger mainly of semiem- pirical procedures, although it must be noted that this method has no semiem- pirical character at the molecular level. The core-valence electron separation achieved by the introduction of the pseudopotentials simplifies a great deal the description of the molecular fragments and large molecules.
Bibliography
[l] B. Pullman, in Molecular Orbital Studies in Chemical Pharmacology, L. B. Kier, Ed. (Springer-
[2] J. A. Pople and D. L. Beveridge, Approximate Molecular Orbital Theory (McGraw-Hill, New
[3] R. Daudel and C. Sandorfy, Semiempirical Wave-Mechanical Calculations on Polyatomic
[4] E. Clementi, J. Mehl, and W. Niessen, J. Chem. Phys. 54, 508 (1971). [5] J. J. Kaufman and W. S. Koski, Int. J. Quantum Chem., Quantum Biol. Symp. 2, 35 (1975). [6] J. D. Dill, P. v. R. Schleyer, J. S. Binkley, R. Seeger, J. A. Pople, and E. Haselbach, J. Am. Chem.
[7] R. E. Christoffersen, D. W. Genson, and G. M. Maggiora, J. Chem. Phys. 54, 239 (1971). [8] A. A. Frost, J. Chem. Phys. 47, 3707 (1967); 47, 3714 (1967); J. Am. Chem. SOC. 89, 3064
[9] A. A. Frost, J. Am. Chem. SOC. 90, 1965 (1968); J. Phys. Chem. 72, 1289 (1968).
Verlag, New York, 1969), p. 1.
York, 1970).
Molecules (Yale U. P., London, 1971).
SOC. 98, 5428 (1976).
(1967).
[ lo] E. Schwarz, Theor. Chim. Acta 11, 307 (1968); J. N. Bardsley, Case Studies At. Phys. 4, 299
[Il l See, e.g., W. A. Harrison, Pseudopolentials in the Theory ofMetals (Benjamin, New York, 1966). [12] W. A. Bingel, R.-J. Koch, and W. Kutzelnigg, Acta Phys. Acad. Sci. Hungar. 27, 323 (1969); R.
[13] S. Topiol, A. A. Frost, M. A. Ratner, J. W. Moskowitz, and C. P. Melins, Theor. Chim. Acta 45,
[I41 R. Gaspar and R. Gaspar, Jr., Acta Phys. Acad. Sci. Hungar. (to be published). [15] R. Gaspar and R. Gaspar, Jr., Int. J. Quantum Chem. 15, 559 (1979). [ 161 P. Gombas, Theorie und Losungsmethoden des Mehrteilchenproblems der Wellenmechanik
[17] R. E. Christoffersen, Adv. Quantum Chem. 6 , 333 (1972). 1181 T. Oie, G. M. Maggiora, and R. E. Christoffersen, Int. J. Quantum, Quantum Chem. Symp. 3,
[19] S. Weiss and G. Leroi, J . Chem. Phys. 48, 962 (1968). [20] W. J . Lafferty and E. K. Plyer, J. Chem. Phys. 37, 2688 (1962). [21] J. W. Moskowitz and M. C. Harrison, J. Chem. Phys. 42, 1726 (1965). [22] D. R. Lide, Jr., J . Chem. Phys. 33, 1514 (1960).
(1974).
Gaspar and I. Tarnhssy-Lentei, Acta Phys. Acad. Sci. Hungar. 40, 283 (1976).
177 (1977).
(Birkhaser, Basel, 1950).
119 (1976).
Received March 1, 1978 Revised September 12, 1978 Accepted for publication October 18, 1978





























