Embed Size (px)
Citation preview

Absence of CCL2 and CCL3 Ameliorates Central NervousSystem Grey Matter But Not White Matter Demyelinationin the Presence of an Intact Blood–Brain Barrier
Katharina Janssen & Mira Rickert & Tim Clarner &
Cordian Beyer & Markus Kipp
Received: 13 November 2014 /Accepted: 22 January 2015 /Published online: 8 February 2015# Springer Science+Business Media New York 2015
Abstract A broad spectrum of diseases is characterized bymyelin abnormalities, oligodendrocyte pathology, and con-comitant glia activation, among multiple sclerosis (MS). Ourknowledge regarding the factors triggering gliosis and demy-elination is scanty. Chemokines are pivotal for microglia andastrocyte activation and orchestrate critical steps during theformation of central nervous system (CNS) demyelinating le-sions. Redundant functions of chemokines complicate, how-ever, the study of their functional relevance. We used thecuprizone model to study redundant functions of twochemokines, CCL2/MCP1 and CCL3/MIP1α, which are crit-ically involved in the pathological process of cuprizone-induced demyelination. First, we generated a mutant mousestrain lacking functional genes of both chemokines and dem-onstrated that double-mutant animals are viable, fertile, and donot present with gross abnormalities. Astrocytes and peritone-al macrophages, cultured form tissues of these animals didneither express CCL2 nor CCL3. Exposure to cuprizone re-sulted in increased CCL2 and CCL3 brain levels in wild-typebut not mutant animals. Cuprizone-induced demyelination,oligodendrocyte loss, and astrogliosis were significantly ame-liorated in the cortex but not corpus callosum of chemokine-deficient animals. In summary, we provide a novel powerfulmodel to study the redundant function of two importantchemokines. Our study reveals that chemokine function in theCNS redounds to region-specific pathophysiological events.
Keywords Chemokines . CNS . Cuprizone .
Multiple sclerosis
Introduction
Multiple sclerosis (MS) is an inflammatory disease of thecentral nervous system (CNS) characterized by myelin loss,oligodendrocyte death, inflammation, and various degrees ofaxonal damage [1]. For long,MSwas exclusively discussed asan autoimmune disorder with auto-reactive T cells directedagainst myelin components as the disease trigger [2]. Thisviewwas partially built on and supported by research obtainedin one of the most commonly used MS animal models, exper-imental autoimmune encephalomyelitis (EAE). EAE leads toT cell-driven inflammatory demyelination in the spinal cord,brain stem, and cerebellum due to immunization with myelinproteins or spinal cord homogenates [3–5]. Although MS andEAE have some features in common, like involvement of theadaptive immune response in CNS inflammation or clinicaldisease course, several differences exist between the modeland MS, which cannot be explained with an exclusive auto-immune view of the disease [6–9]. Biopsies and autopsies ofMS patients, for example, revealed that pathological changesalso occur in the absence of T cell-mediated inflammation anddemyelination [10–16] and show activation of microglial cells[11, 15, 17], increased expression of anti-apoptotic genes inoligodendrocytes, as well as apoptotic oligodendrocytes [10,18]. The cuprizone model is approved to selectively imagepathological aspects of MS, which are not primarily regulatedand controlled by the peripheral immune system [19, 20].Feeding of cuprizone induces demyelination of distinct CNSwhite and grey matter regions, among the white matter tractcorpus callosum or cortical grey matter [19]. In this model,oligodendrogliopathy results in significant oligodendrocyte
K. Janssen and M. Rickert contributed equally to this work as firstauthors.
K. Janssen :M. Rickert : T. Clarner :C. Beyer :M. Kipp (*)Institute of Neuroanatomy, Faculty of Medicine, RWTH AachenUniversity, Wendlingweg 2, 52074 Aachen, Germanye-mail: [email protected]
M. KippDepartment of Anatomy II, Ludwig-Maximilians-University ofMunich, Munich, Germany
Mol Neurobiol (2016) 53:1551–1564DOI 10.1007/s12035-015-9113-6

apoptosis with concomitant microglial activation,astrocytosis, and finally active demyelination [20–23] despitefunctional integrity of the blood–brain barrier (BBB) and ab-sence of profound peripheral leucocyte recruitment.
Recent focus has turned to the role of chemokines in CNSpathologies. Chemokines represent a group of chemotacticproteins implicated in leucocyte attraction and activation un-der inflammatory conditions [24–27]. Chemokines are smallproteins with a molecular weight ranging from 8 to 14 kDaand can be subdivided into four groups depending on thenumber and spacing of conserved cysteine residues in theiramino termini [28, 29]. Today, approximately 50 differentchemokines [30] and 20 corresponding receptors [31] havebeen described in humans with orthologous homologies inother mammalian species [25, 32]. Besides leucocyte attrac-tion/activation, another critical function of chemokines is theregulation of astrocyte and microglia function [33–39].Chemokines are upregulated in various CNS pathologies[40]. The expression and release of chemokines is inducedin demyelinating diseases and animal models, includingEAE [41] and the toxic cuprizone model [21]. The importanceof chemokines has also been demonstrated in other diseaseswith myelin abnormalities and/or oligodendrocyte pathology,including amyotrophic lateral sclerosis [42], Alzheimer’s dis-ease [43], or schizophrenia [44].
It has been recently shown that numerous chemokines areupregulated during cuprizone-induced demyelination and thatchemokines modulate cuprizone-induced pathology [41,45–48]. The absence of CCL3, also called macrophage-inflammatory protein-1α (MIP-1 α), delays demyelinationduring cuprizone treatment [41], however, this effect is justtransient and not profound. This points at other chemokinesand their receptors to be synergistically and/or supplementaryinvolved in cuprizone-induced pathology. RNase protectionassays support this view and show that the induction ofCCL2 expression is more distinct in CCL3−/− compared withwild-type animals. Due to a postulated redundant function ofchemokines, we created a mouse strain lacking both, CCL2and CCL3, and investigated, whether a combined deficiencyof both chemokines substantially impacts on cuprizone-induced demyelination and astrocyte/microglia activation.
Materials and Methods
Mice and Demyelination
C57BL/6 J mice were obtained from The JacksonLaboratory's stock (000664) at Charles River (Sulzfeld,Germany) and served as controls for CCL2/3 knockout (ko;CCL2/3−/−) mice. CCL2/3−/− mutants were generated by ex-tensive crossbreeding of CCL2 and CCL3 knockout mice.CCL2−/− and CCL3−/− single deficient animals were obtained
from The Jackson Laboratory (B6.129S4-Ccl2tm1Rol/J,B6.129P2-Ccl3tm1Unc/J). All animals were kept under stan-dard laboratory conditions according to the Federation ofEuropean Laboratory Animal Science Association's recom-mendations. The procedures were approved by the ReviewBoard for the Care of Animal Subjects of the district govern-ment (Nordrhein-Westfalen, Germany) and performed accord-ing to international guidelines on the use of laboratory mice.Demyelination was induced by feeding 10-week-old (19–21 g) female mice with ground standard rodent chow contain-ing 0.25 % cuprizone (bis-cyclohexanone oxaldihydrazone,Sigma-Aldrich Inc., Germany) for the indicated period as pub-lished previously [19, 23]. Control mice were fed groundstandard chow.
Tissue Preparation
Preparation of tissues was performed as previously described[49–51]. For histological and immunohistochemical studies,mice were transcardially perfused with 2 % paraformaldehydein phosphate-buffered saline (PBS; pH 7.2). After overnightpost-fixation in the same fixative, brains were dissected, em-bedded in paraffin, and then coronary sectioned into 5 μmsections from approximately the levels 215 to 525 (study forCCL2/3−/− mice characterization) or the levels 255 to 295(study for impact of CCL2/3-deficiency on cuprizone-induced demyelination) according to the mouse brain atlasof Sidman et al. For gene expression analyses, mice weretranscardially perfused with ice-cold PBS, brains subsequent-ly removed, and the entire corpus callosum (CC) separatedfrom the cortex (Cx) as described previously [52, 53].Tissues were immediately snap-frozen in liquid nitrogen andkept at −80 °C until further use.
Histochemistry and Immunohistochemistry
For immunohistochemistry (IHC), sections were placed onsilane-coated slides, de-paraffinized, rehydrated, heat-unmasked if necessary, blocked with PBS containing 1 %normal horse or goat serum, and incubated overnight at 4 °Cwith the primary antibodies diluted in blocking solution.Primary antibodies and dilutions used in the study are givenin Table 1. After washing and blocking of endogenous perox-idase with 0.3 % hydrogen peroxide (in PBS) for 30 min,sections were incubated with appropriate biotinylated second-ary antibodies for 1 h at room temperature, followed byperoxidase-coupled avidin-biotin-complex (ABC kit, VectorLaboratories). The di-amino-benzidine-reaction (DAB;DAKO Deutschland GmbH, Germany) was used to visualizeperoxidase-avidin-biotin complexes. Sections were counter-stained with standard hematoxylin to visualize cell nuclei ifappropriate. Secondary antibodies and dilutions used are giv-en in Table 2. Stained sections were analyzed using a Nikon
1552 Mol Neurobiol (2016) 53:1551–1564

ECLIPSE 80i microscope. To estimate myelination, stainingintensity in anti-PLP-stained sections was quantified usingImageJ after automatic setting of a threshold. Due to the abun-dance of microglia cells and astrocytes in the midline of thecorpus callosum after cuprizone-induced demyelination, thesame strategy was followed to estimate differences betweenastrogliosis/microgliosis in wild-type vs. chemokine deficientanimals. In contrast, extent of astrogliosis/microgliosis in thecortical grey matter was estimated by manual counting ofcells. For cell quantification, two consecutive sections permice were evaluated, and values of both sections were aver-aged. Adenomatous polyposis coli (APC)+, ionized calcium-binding adaptor molecule 1 (IBA1)+, and glial fibrillary acidicprotein (GFAP)+ cells were only counted when a cell nucleuswas clearly visible. Cell numbers are given in cells per squaremillimeter. Two blinded examiners performed manual cellquantification twice and results were averaged. LFB/PASstains were performed following established protocols [49]and evaluated as described elsewhere from our group [54].Hypertrophy of microglia cells was analyzed by quantifyingthe mean area covered per microglia cell in IBA-1-stainedsections using the area measurement tool integrated in theNikon NIS Elements software (version 3.22.14, Nikon,Germany).
Cell Culture and Treatment
All cells were kept at 37 °C and 5 % CO2 in a humidifiedincubator.
Primary mouse astrocyte cultures were prepared from thebrains of 1 to 2-day-old wild-type or CCL2/3−/− mice as pub-lished previously [55]. Briefly, meninges were carefully re-moved from cerebral hemispheres and the tissue mechanicallydisrupted using a pipette. Single-cell suspensions were trans-ferred to culture dishes (3 brains/10 cm dish) and kept inDulbecco's modified Eagle's medium (DMEM; Gibco, LifeTechnologies, Germany) supplemented with 10 % heat-
inactivated fetal calf serum (FCS; PAA Laboratories GmbH,Germany), 50 U/ml penicillin, and 50 μg/ml streptomycin(P/S; Gibco, Life Technologies, Germany) until 80–90% con-fluence was reached. The medium was replaced every otherday. Before starting the experiment, cells were sub-cultured1:10 in fresh 10 cm dishes to minimize microglial contamina-tion and grown till 80–90 % confluence. For treatment, cellswere seeded into six-well plates, grown to 80 % confluenceand treated with 1–10μg/ml lipopolysaccharide (LPS; L4391,Sigma-Aldrich, Germany) or vehicle for 24 h.
Isolation of mouse peritoneal macrophageswas performedfrom adult wild-type and CCL2/3−/− mice. Animals werekilled and the abdominal skin carefully removed leaving theperitoneum intact; 5 ml of PBS containing 3 % FCS wasinjected with a 25-gauge needle into the peritoneal cavity.After gently massaging the abdominal walls for a fewminutes,the liquidwas removed with a 25-gauge needle and placed in afalcon tube on ice. The peritoneal cavity was opened, and theremaining liquid carefully collected with a syringe withoutaspirating blood. The number of isolated macrophages wasapproximately 106 cells per mouse. The suspension was cen-trifuged for 5 min at 400×g, washed once with PBS, and re-suspended in 10 ml RPMI (Gibco, Life Technologies,Germany) containing 10 % FCS and P/S; 2×105 cells weresubsequently seeded per six-well plate and left in the incuba-tor. To remove dead cells and cell debris, the medium waschanged after 2 h. After overnight incubation, cells were treat-ed with 1 μg/ml LPS for 2 h in RPMI containing 10 % FCSand P/S. After treatment, the supernatant was removed andstored at −80 °C until further use.
Real-time Reverse Transcriptase-Polymerase Chain Reaction
Gene expression levels were investigated using the real-timereverse transcriptase-polymerase chain reaction (rtRT-PCR)technology (BioRad, Germany), SensiMix SYBR Green(Bioline, Germany), and a standardized protocol as described
Table 1 Primary antibodies used for IHC/ICC
Antibody Host Directed against Dilution AGR Supplier cat. No.
GFAP Rabbit Glial fibrilary acidic protein (astrocytes) 1:5000 Tris/EDTA Encore, RPCA-GFAP
IBA1 Rabbit Ionized calcium binding adaptor molecule-1 (microglia) 1:3000 Tris/EDTA Wako, 019-19741
APC/CC1 Mouse Adenomatous polyposis coli (mature oligodendrocytes) 1:250 Tris/EDTA Calbiochem OP80
OLIG2 Rabbit Oligodendrocyte transcription factor 2 (oligodendrocytes) 1:2000 Tris/EDTA Millipore, AB9610
PLP Mouse Proteolipid protein (myelin) 1:1500 None AbD Serotec, MCA839G
Table 2 Secondary antibodiesfor IHC/ICC Antibody Host Directed against Dilution Supplier cat. No.
Biotinylated anti-mouse IgG Horse Mouse IgGs 1:50 Vector Lab. BA-2000
Biotinylated anti-rabbit IgG Goat Rabbit IgGs 1:50 Vector Lab. BA-1000
Mol Neurobiol (2016) 53:1551–1564 1553

previously [50, 55, 56]. Primer sequences and respective an-nealing temperatures are given in Table 3. The expressionlevel of the ribosomal RNA 18 s (18 s) was used as a referencefor mouse samples. Constant expression of the reference geneunder all experimental conditions was routinely tested.Furthermore, melting-curve analyses and gel-electrophoresisof the products was performed to assess specificity of theamplification (not shown).
Genotyping of CCL2/3−/− Mice
Genotyping was performed using standard PCRwith genomicDNA isolated from a small piece of mouse tail. For DNAisolation, the Nucleospin tissue Kit (Macherey-Nagel,Germany) was used according to the manufacturer's recom-mendations for mouse tails. Briefly, the PCR reaction wasconducted using BioXAct short DNA polymerase (Bioline,Germany; BIO-21065), PCR optimization Kit buffers G, C,or I and 2 μl of genomic DNA. The primer sequences, productsizes, and the buffer used for PCR reaction are given inTable 4. The genotyping strategy is illustrated in Fig. 1.
Enzyme-Linked Immunosorbent Assay
CCL2 and CCL3mouse enzyme-linked immunosorbent assay(ELISA; Peprotech, Germany) experiments were carried outaccording to the manufacturer's protocol. Color developmentof the ABTS substrate (Sigma-Aldrich, Germany) was moni-tored with a Tecan infinite 200-plate reader at 405 nm with awavelength correction at 650 nm. Chemokine release wasnormalized to total protein content of cell lysate as determinedwith the Pierce BCA protein Assay (Thermo Fisher Scientific,Germany).
Statistical Analysis
Statistics were performed using absolute data. Intergroup dif-ferences were tested by one-way ANOVA followed byTukey's post hoc test using GraphPad Prism 5 (GraphPadSoftware Inc.) if not stated otherwise. All data are given asarithmetic means±SEM. p values are indicated as *p≤0.05;**p≤0.01; ***p≤0.001. Two separate animal cohorts wereperformed with N=5 per experimental group, each.
Results
CCL2/3−/− Mice Are Viable, Fertile, and Show No GrossPhysical or Behavioural Abnormality
To study redundant functions of CCL2 and CCL3 in thecuprizonemodel, we generated CCL2/CCL3 double knockoutanimals by extensive breeding. Both genes are located onchromosome 11 with a distance of 1.24 cM, meaning thatthe genes have a recombination frequency of around 1.24 %.We crossbred the two commercially available single knockoutstrains from Jackson laboratory. The F1 generation of hetero-zygous CCL2−/+CCL3−/+ mice was further mated to obtainhomozygous double knockout mice.
After extensive breading cycles, we finally obtained ani-mals lacking the genes for both chemokines, CCL2 and CCL3as verified by genotyping. The obtained homozygousCCL2/3−/− mice are viable and fertile. The genotyping strate-gy is illustrated in Fig. 1a, b. To detect either wild-type ormutantCcl2, one single PCRwith a common antisense primer(Ccl2-P1 in Fig. 1a), and two distinct wild-type (Ccl2-P2) vs.mutant (Ccl2-P3) sense primers have been conducted. To de-tect either wild-type or mutant Ccl3, two separate PCRs wereperformed. One primer pair (Ccl3-P1 and Ccl3-P2) detects thewild-type locus, whereas the other primer pair (Ccl3-P3 andCcl3-P4) detects the mutant locus. Sense primers for bothcases (i.e.,Ccl2 orCcl3 genotyping) were located at the regionof the initially introduced neomycin cassette [57, 58]. In astandard PCR approach, homozygous wild-type mice displayone band of 287 bp for theCcl2 gene and one of 150 bp for theCcl3 gene, whereas knockout mice show a 179-bp band forthe disrupted Ccl2 and a 600-bp band for the disrupted Ccl3gene (compare Fig. 1b). In contrast, heterozygous CCL2−/+
mice show a 287-bp band for the Ccl2 wild-type gene and a179-bp band for the Ccl2 knockout gene as they posse bothgene variants, one on each chromosome 11. This also applies
Table 3 List of primers used for rtRT-PCR and PCR
Name Forward sequence 5′-3′ Reverse sequence 5′-3′
Ccl2 mouse tta aaa acc tgg atc gga acc aa gca tta gct tca gat tta cgg gt
Ccl3 mouse ttc tct gta cca tga cac tct gc cgt gga atc ttc cgg ctg tag
18 s mouse cgg cta cca cat cca agg aa gct gga att acc gcg gct
Table 4 List of primers and buffers used for genotyping
Name Sequence 5′-3′ Size (bp) Buffer
Ccl2 Mut s s GCC AGA GGC CAC TTGTGTAG
179 G
Ccl2 WT s s TGA CAG TCC CCA GAGTCA CA
287
Ccl2 common as as TCATTG GGATCATCTTGC TG
Ccl3 WT s s ATA CAA GCA GCA GCGAGTACC
150 C
Ccl3 WT as as ATG GCG CTG AGA AGACTT G
Ccl3 Mut s s TAA AGC GCATGC TCCAGA CT
600 I
Ccl3 Mut as as AGA GTC CCT CGATGTGGC TA
1554 Mol Neurobiol (2016) 53:1551–1564

for CCL3−/+ heterozygous mice which show two bands with150 and 600 bp for the Ccl3 wild-type and Ccl3 knockoutgene, respectively.
To further verify double-gene deletion on the level of ge-nomic DNA, we sequenced the PCR products of the
genotyping PCR for the knockout sequence and compared itwith the wild-type sequence of Ccl2 and Ccl3 as published atENSEMBL (Ccl2, ENSMUSG00000035385 and Ccl3,ENSMUSG00000000982). Figure 1c shows a part of the se-quencing result at the transition site of the neomycin cassette
Ccl3
Ccl2
179 bp
287 bp
150 bp
600 bp
product size
genotyping products used for sequencing
wild type sequence
mutant sequence neomycin cassette
common primer
common primer
mutant primer
wild type primer
X X
neomycin cassette
wild type primer as
mutant primer as
mutant primer s
wild type primer s
X X
wild type sequence
mutant sequence
mo
use
ch
rom
oso
me
11
wild type sequence
wild type sequence
mutant sequence
mutant sequence
homologous site
homologous site
Ccl3-/-Ccl2-/-
A B
C
Ccl2-P1
Ccl2-P1
Ccl2-P2
Ccl2-P3
Ccl3-P1Ccl3-P2
Ccl3-P3
Ccl3-P4
Fig. 1 Genotyping strategy and DNA sequencing of CCL2/3−/− mice. aGenotyping strategy for wild-type (wt) and CCL2/3-deficient (CCL2/3−/−)mice is shown. Note that the sense primers (Ccl2-P3 and Ccl3-P4)detecting the knockout is placed in the neomycin cassette, which wasintroduced into the gene of interest when generating the original singleknockout mouse. b Gel electrophoresis of PCR-based genotyping ofCCL2/3−/− mice and expected product size. c Breeding scheme and genesequence obtained by sequencing the genotyping PCR products. Both
genes are located on chromosome 11 with a distance of 1.24 cM(recombination frequency=1.24 %). Note that the mutant sequence ispartially homologous to the wild-type sequence representing the sites ofhomologous recombination during initial generation of the singleknockout mice. The non-homologous part of the mutant sequence likelyrepresents a part of the neomycin cassette and did not give a specific resultin a BLAST mouse genome search
Mol Neurobiol (2016) 53:1551–1564 1555
and the homologous recombination site of the mutant se-quences paired with the wild-type sequence of Ccl2 (uppersequence) or Ccl3 (lower sequence). We found that the se-quenced DNA is partly homologous to the Ccl2 or Ccl3 generepresenting the homologous recombination sites present in
the vector which was used to introduce the disrupted sequenceinto the genome of the single knockout mice [57, 58]. Theadjacent non-homologous sequence did not give a specificresult in a BLAST search and very likely represents a part ofthe neomycin cassette present in the disrupted gene.
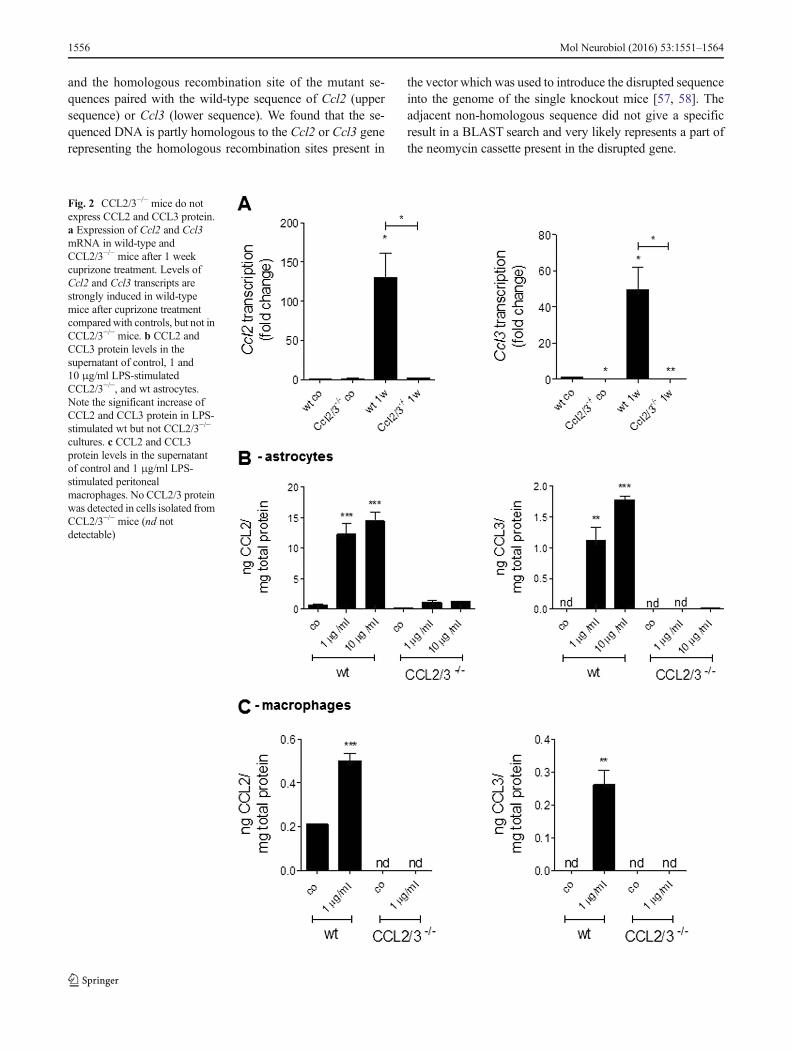
Fig. 2 CCL2/3−/− mice do notexpress CCL2 and CCL3 protein.a Expression of Ccl2 and Ccl3mRNA in wild-type andCCL2/3−/− mice after 1 weekcuprizone treatment. Levels ofCcl2 and Ccl3 transcripts arestrongly induced in wild-typemice after cuprizone treatmentcomparedwith controls, but not inCCL2/3−/− mice. b CCL2 andCCL3 protein levels in thesupernatant of control, 1 and10 μg/ml LPS-stimulatedCCL2/3−/−, and wt astrocytes.Note the significant increase ofCCL2 and CCL3 protein in LPS-stimulated wt but not CCL2/3−/−
cultures. c CCL2 and CCL3protein levels in the supernatantof control and 1 μg/ml LPS-stimulated peritonealmacrophages. No CCL2/3 proteinwas detected in cells isolated fromCCL2/3−/− mice (nd notdetectable)
1556 Mol Neurobiol (2016) 53:1551–1564

Fig. 3 CCL2/3−/− mice show normal brain morphology and histology.Immunohistochemical and histochemical characterization of untreatedCCL2/3−/− mice compared with wild-type controls. GFAP (a), IBA1(b), OLIG2 (c), and PLP (d); immunehistochemistry of CCL2/3−/− miceand wild-type mice at the level 305 according to the mouse brain atlas ofSidman et al. e Quantification and a representative picture of microglia
area in the respective genotypes within the somatosensory cortex.Regions of interest are indicated in the hematoxylin stained overviewpicture (f; corpus callosum (CC) and layer 4 of the somatosensorycortex (Cx)). No gross abnormality was observed in CCL2/3−/− micecompared with the wild-type controls. Scale bars, 50/25 μm
Mol Neurobiol (2016) 53:1551–1564 1557

Ccl2 and Ccl3 Expression Is Disrupted in CCL2/3−/− Mice
In a next step, we verified the knockout of Ccl2 and Ccl3 onthe messenger RNA (mRNA) and protein level. To studymRNA levels of Ccl2 and Ccl3 in double knockout mice,we fed wild-type and CCL2/3−/− mice with 0.25 % cuprizonefor 8 days. As shown in Fig. 2a, in cuprizone-exposed wild-type but not CCL2/3−/−mice, there was a strong expression ofCcl2 and Ccl3 transcripts. To study CCL2 and CCL3 deletionon the protein level, we performed ELISA experiments withprimary astrocytes and peritoneal macrophages isolated fromrespective mutants or wild-type animals. Astrocytes and mac-rophages were treated with LPS as indicated in the figurelegend, and CCL2 and CCL3 protein levels were quantifiedin the supernatant of these cell cultures. As shown in Fig. 2b, c,both cell types released significant amounts of CCL2 andCCL3 after LPS stimulation. LPS-induced chemokine releasewas not observed in chemokine-deficient animals. Taken to-gether, these results clearly illustrate that both chemokines,CCL2 and CCL3, are not expressed in double-deficientanimals.
CCL2/3−/− Mice Display Normal Brain Morphology
To address the morphology and histology of the brains ofCCL2/3−/− mice, coronary sections of 6-week-old wild-typeand knockout mice were investigated using immunohisto-chemistry (Fig. 3a–d). Brains were sectioned from the levels215–525 according to the mouse brain atlas of Sidman et al.Figure 3 shows representative pictures of the level 305 fromwild-type and CCL2/3−/− mice. The main regions of interestwere the medial corpus callosum (CC) and the layer 4 of thesomatosensory cortex (Cx) as indicated in the hematoxylineosin-stained overview. No gross abnormalities were detectedon the cellular level regarding GFAP+ astrocytes (Fig. 3a),IBA1+ microglia (Fig. 3b), OLIG2+ oligodendrocytes(Fig. 3c), or PLP+ myelin fibers (Fig. 3d). To substantiatethese observations, the number of astrocytes, microglia, andoligodendrocytes was systematically evaluated within differ-ent parts of the mouse brain, namely the medial and lateral partof the CC, the telencephalic Cx, the gyrus dentatus of thehippocampus formation, and the cerebellar white matter. Nosignificant difference in cell numbers was found betweenwild-type and CCL2/3− /− mice (data not shown).Furthermore, we quantified the area covered per IBA1+ mi-croglia cell, as another indicator of microglia reactivity. Asshown in Fig. 3e, no significant difference was observed forthis cell parameter. Finally, we investigated the morphology ofthe liver, spleen, and kidney using hematoxylin eosin stain butdid not observe any gross abnormalities compared with wild-typemice (not shown). Taken together, CCL2/3−/−mice revealnormal brain morphology and histology and no abnormalitiesin the three major organ systems.
CCL2/3−/− Mice Display Less Severe Demyelination,Astrogliosis, and Oligodendrocyte Loss in the CortexAfter Cuprizone-Induced Demyelination
To test the function of CCL2 and CCL3 in vivo, CCL2/3−/−
mice were treated with 0.25 % cuprizone for 5 weeks and theextent of demyelination compared with cuprizone-treatedwild-type C57BL/6 mice. As shown in Fig. 4a, severe demy-elination was evident in the midline of the corpus callosum inboth genotypes. Extent of white matter demyelination wascomparable in both mice strains. In PLP-stained sections,myelination values were 10.6±4.5 % (mean±standard error)in wild-type animals and 12.4±3.3 % in CCL2/3−/− animals.Comparably, myelination evaluated in LFB/PAS-stained sec-tions did not show any significant difference between bothgenotypes (3.2±0.8 % in wild-type vs. 5.8±1.3 % inCCL2/3−/−). In line with these findings, activation of astro-cytes and microglia was similar in CCL2/3−/− and wild-typemice. Thus, the absence of CCL2 and CCL3 obviously doesnot ameliorate cuprizone-induced demyelination in the stud-ied white matter tracts.
Demyelination in the cuprizone model is evident not onlyin the white matter but also in distinct grey matter regions [59,60]. To evaluate whether chemokine deficiencymight result inmyelin preservation in less severe inflamed regions such asthe cerebral cortex, we analyzed the extent of myelin losswithin the somato-sensory cortex region. As demonstrated inFig. 5a, severe cortical demyelination was evident after5 weeks of cuprizone exposure. Interestingly, myelin losswas less prominent in CCL2/3−/− compared with wild-typemice. In wild-type mice, myelination index declined from100 to 20.5 %, whereas the myelination index dropped from100 to 70.4% (p<0.001) in CCL2/3−/−mice. In line with thesefindings, loss of OLIG2+ oligodendrocytes was reduced inCCL2/3−/−mice (Fig. 1b). Comparably, the number of mature,myelinating oligodendrocytes, as estimated by anti-APC im-munohistochemistry, was higher in CCL2/3−/− compared withwild-type mice (data not shown). We furthermore analyzed theresponse of cortical astrocytes and microglia. As demonstratedin Fig. 4c, d, astrocytosis but not microgliosis was less strongin the cortex of CCL2/3−/− compared with wild-type mice.
�Fig. 4 Ccl2/3−/− mice show equal demyelination and gliosis in thecorpus callosum after cuprizone treatment compared with wt mice. aEvaluation of PLP staining intensity in wild-type and CCL2/3−/− miceafter 5 weeks cuprizone treatment in the corpus callosum. Scale bars,100/50 μm. CCL2/3−/− and wild-type mice show similar demyelination.Representative pictures are shown on the right side. Furthermore, LFB/PAS-stained pictures are shown in the lower row. b Evaluation of GFAP+
cells in the corpus callosum of the control and cuprizone-treatedCCL2/3−/− and wild-type mice. c Analysis of IBA1+ cells in the corpuscallosum of wild-type and CCL2/3−/− mice treated 5 weeks withcuprizone and representative pictures of IBA1 immunohistochemistry.Cuprizone treatment induces comparable astrogliosis and microgliosisin both wild-type and CCL2/3−/− mice. Scale bar, 100 μm
1558 Mol Neurobiol (2016) 53:1551–1564

Mol Neurobiol (2016) 53:1551–1564 1559

1560 Mol Neurobiol (2016) 53:1551–1564

Discussion
Actively demyelinating MS lesions are separated into fourpatterns, comprising the vast majority of analyzed biopsy ma-terial. It was suggested that pattern III lesions occur whenoligodendrocytes are subjected to environmental factors (vi-ral, chemical, or vascular dysfunction) that produce metabolicstress [61]. These pattern III MS lesions might involve a pro-cess called “dying back gliopathy,” as the distal elements ofoligodendrocyte processes show the earliest signs of degener-ation [62]. The cuprizone model reflects various aspects ofpattern III lesion development. Feeding mice the copper che-lator cuprizone is believed to inhibit mitochondrial functionand cause CNS demyelination. More than 25 years ago,dying-back gliopathy in cuprizone-induced demyelinationwas described [63]. Our group was recently able to verifythese findings in very early stages of cuprizone-inducedoligodendrogliopathy [64]. At least during the early stages,cuprizone-induced oligodendrogliopathy is clearly mediatedthrough classical apoptotic signaling cascades [21, 65].Notably, cuprizone-induced demyelination lesions have addi-tional similarities with pattern III lesions of MS, includingindistinct lesion borders and abundant accumulation oflesion-located microglia but only a sparse hemic leukocyteresponse [66]. To conclude, distinct but important aspects ofMS lesion development and progression are mimicked by thecuprizone model, and a better understanding of cuprizone-induced oligodendrocyte loss, inflammation, and demyelin-ation will help to get insight into what is going on in theMS-diseased brain.
We and others have shown that chemokines are upregulat-ed in the cuprizone-induced model of demyelination in whichthe BBB remains intact and T cells are not involved in thedisease process [19]. Induced chemokines includeCCL5/RANTES, CCL4/MIP-1β, CCL3/MIP-1α, CXCL10/IP-10, or CCL2/MCP-1. An induction of these chemokinescan be detected as early as 1 week after the start of treatment
well before demyelination is observed [21, 41, 48, 67]. Thus,it is likely that chemokines orchestrate microgliosis andastrogliosis in the cuprizone model.
In the current study, we focused on the two chemokinesCCL2 and CCL3. CCL2 is a potent chemo-attractant that canbe produced by various cell types including astrocytes, mi-croglia, endothelial cells, and macrophages [68] and can inter-act with different target cells among monocytes, activated Tcells, natural killer (NK) cells, or microglia [69]. CCL2 medi-ates its effects by binding to and activating its main chemokinereceptor CCR2, a seven-transmembrane-spanning protein thatis functionally linked to downstream signaling pathwaysthrough heterotrimeric G proteins [70]. CCL2-deficient miceare markedly resistant to the induction of EAE and show asignificant reduction in macrophage recruitment into the CNS[71]. Furthermore, CCR2 knockout mice display an amelio-rated disease course compared with their wild-type littermates[72–74]. Importantly, CCR2 disruption did not result in 100%protection in EAE but disease course was characterized by adelayed onset and lower maximum disease score [73]. In thiscontext, it is important to notice that chemokines often act inconcert with other chemokines and cytokines to affect tissueinfiltration and inflammation [75]. The functional relevance ofCCL3 during EAE disease induction and progression is con-troversially discussed. On one hand, CCL3-deficient mice arefully susceptible to MOG-induced EAE [76], whereas on theother hand, anti-CCL3 antibody treatment inhibited the devel-opment of acute EAE [77]. In line with the finding that CCL3neutralization improves autoimmune-triggered inflammation,it has been shown that this chemokine plays a pivotal role inthe development of recurrent anterior uveitis [78]. To con-clude, both chemokines are potent regulators duringlymphocyte-driven inflammatory events and might be thera-peutic targets in the future to ameliorate the deleterious effectsof peripheral immune cells in the MS diseased CNS.
In the context of cuprizone-induced pathology, where mi-croglia activation is an outstanding histopathological hallmark(compare Figs. 4 and 5), it is important to notice that bothchemokines regulate microglia responses. CCL2 and CCL3induce migration [79–81], proliferation [82], and activation[83, 84] of microglia. The functional relevance of CCL2 andCCL3 during cuprizone-induced demyelination is not well-addressed in the literature. The absence of CCL3 delays de-myelination during cuprizone treatment [41]. CCL2 expres-sion was compensatory induced in the brain of CCL3-deficient animals. Unexpectedly, CCL2/3−/−mice in our studywere fully susceptible to cuprizone-induced white matter de-myelination. The extent of corpus callosum demyelinationwas comparable in both mice strains. Thus, double-deficientanimals display the same vulnerability against a 5-weekcuprizone intoxication challenge as compared with theCCL3 single-knockout strain [41]. In contrast to the study ofMcMahon et al., we extended our analysis to the adjacent
�Fig. 5 CCL2/3−/−mice show less severe demyelination, oligodendrocyteloss, and astrogliosis after cuprizone treatment in the cortex comparedwith wt animals. a Evaluation of PLP staining intensity in wild-typeand CCL2/3−/− mice after 5 weeks cuprizone intoxication. CCL2/3−/−
mice show considerably less demyelination in the cortex region. Theright panel shows representative pictures of PLP-stained sections ofcontrol and 5 weeks cuprizone-treated wild-type and CCL2/3−/− mice.Note that CCL2/3−/− mice show more OLIG2+ cells in the cortex areacompared with wild-type mice. c Evaluation of GFAP+ cells in the cortexof cuprizone-treated CCL2/3−/− and wild-type mice and representativepictures of GFAP immunohistochemistry. Note that CCL2/3−/− micedisplay less pronounced astrogliosis after 5 weeks of cuprizonetreatment in the cortex compared with wild-type mice. d Analysis ofIBA1+ cells in the cortex of control and cuprizone-treated wild-type andCCL2 /3 − / − mice and r ep r e s en t a t i v e p i c t u r e s o f IBA1immunohistochemistry. Cuprizone treatment induces similarmicrogliosis in both wild-type and CCL2/3−/− mice. Quantification wasperformed in layer 4 of the somatosensory cortex. Scale bar, 50 μm
Mol Neurobiol (2016) 53:1551–1564 1561

cortical grey matter. The reason for that was that myelin lossand the concomitant activation of microglia is less severe inthe cortex compared with the white matter corpus callosum[59]. Due to the lower magnitude of chemokine-triggered lo-cal inflammation, we speculated that under these circum-stances the absence of distinct chemokines might result in apreservation of inflammation and in consequence myelin loss.Our findings show that cuprizone-induced myelin loss is sig-nificantly lower in CCL2/3−/− compared with wild-type ani-mals. Astrocytosis but not microgliosis was less distinct in theknockout mice, raising the possibility that microglia are theprincipal source of CCL2 and CCL3 in this model, whereasastrocytes and oligodendrocytes are the respective target cells.In line with this assumption, it has been shown that astrocytesexpress in an activated state the respective chemokine recep-tors such as CCR1 [85], CCR2 [86], and CCR5 [87].
In summary, our results suggest that CCL2 and CCL3 inconcert orchestrate cuprizone-induced demyelination and as-trocyte activation in a region-specific manner with particularefficacy in the grey matter. Future studies have to address thecell-type specificity of these effects, i.e., unravel the underly-ing cell–cell interactions, and apply CCL2-deficient mice inEAE pathology.
This study, however, harbors several limitations. First, wedid not systematically address the effect of either CCL2 orCCL3 deficiency on cuprizone-induced demyelination. Dataobtained from a limited number of animals indicate that thecombined deletion of both chemokines is required to effec-tively prevent cortical myelin loss (data not shown). Second,we have not yet addressed which cell types express thechemokines, andwhether this cellular expression changes dur-ing lesion progression. Last but not least it will be essential tostudy the impact of combined CCL2/3 deficiency on myelinpathology in other MS-related animal models such as EAE inthe future. Nevertheless, this is the first report describing thatCCL2/3−/− animals are viable, fertile, and do not show anygross abnormalities, and that the combined deficiency of thesechemokines results in a partial protection of the grey matterfrom experimentally induced demyelination. These animalsare now available for future mechanistic studies which aimto address redundant chemokine function.
Acknowledgments We would like to thank Helga Helten and PetraIbold for the excellent technical assistance. This study was supportedby financial support from ProMyelo-SFZ (MK) and by a START grant(TC) from the Faculty of Medicine (RWTH Aachen University).
References
1. van der Valk P, De Groot CJ (2000) Staging of multiple sclerosis(MS) lesions: pathology of the time frame of MS. NeuropatholAppl Neurobiol 26:2–10
2. Lassmann H (2005) Multiple sclerosis pathology: evolution of path-ogenetic concepts. Brain Pathol 15:217–222
3. Kabat EA, Wolf A, Bezer AE (1946) Rapid production of acutedisseminated encephalomyelitis in rhesus monkeys by injection ofbrain tissue with adjuvants. Science 104:362–363. doi:10.1126/science.104.2703.362
4. Lorentzen JC et al (1995) Protracted, relapsing and demyelinatingexperimental autoimmune encephalomyelitis in DA rats immunizedwith syngeneic spinal cord and incomplete Freund's adjuvant. JNeuroimmunol 63:193–205
5. van der Star BJ, Vogel DY, Kipp M, Puentes F, Baker D, Amor S(2012) In vitro and in vivo models of multiple sclerosis. CNS NeurolDisord Drug Targets 11:570–588
6. Corthals AP (2011) Multiple sclerosis is not a disease of the immunesystem. Q Rev Biol 86:287–321
7. Nakahara J, Aiso S, Suzuki N (2010) Autoimmune versusoligodendrogliopathy: the pathogenesis of multiple sclerosis. ArchImmunol Ther Exp (Warsz) 58:325–333. doi:10.1007/s00005-010-0094-x
8. Nakahara J, Maeda M, Aiso S, Suzuki N (2012) Current concepts inmultiple sclerosis: autoimmunity versus oligodendrogliopathy. ClinRev Allergy Immunol 42:26–34. doi:10.1007/s12016-011-8287-6
9. Stys PK (2013) Pathoetiology of multiple sclerosis: are we barkingup the wrong tree? F1000Prime Rep 5:20. doi:10.12703/P5-20
10. Barnett MH, Prineas JW (2004) Relapsing and remitting multiplesclerosis: pathology of the newly forming lesion. Ann Neurol 55:458–468. doi:10.1002/ana.20016
11. Gay FW, Drye TJ, Dick GW, Esiri MM (1997) The application ofmultifactorial cluster analysis in the staging of plaques in early mul-tiple sclerosis. Identification and characterization of the primary de-myelinating lesion. Brain J Neurol 120(Pt 8):1461–1483
12. Lassmann H (2003) Hypoxia-like tissue injury as a component ofmultiple sclerosis lesions. J Neurol Sci 206:187–191
13. Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ (2007) Lesiongenesis in a subset of patients with multiple sclerosis: a role for innateimmunity? Brain 130:2800–2815. doi:10.1093/brain/awm236
14. Prineas JW et al (2001) Immunopathology of secondary-progressivemultiple sclerosis. Ann Neurol 50:646–657
15. Sanders V, Conrad AJ, Tourtellotte WW (1993) On classification ofpost-mortem multiple sclerosis plaques for neuroscientists. JNeuroimmunol 46:207–216
16. van der Valk P, Amor S (2009) Preactive lesions in multiple sclerosis.Cu r r Op in Neu ro l 22 : 207–213 . do i : 10 . 1097 /WCO.0b013e32832b4c76
17. Li H, Newcombe J, Groome NP, Cuzner ML (1993) Characterizationand distribution of phagocytic macrophages in multiple sclerosisplaques. Neuropathol Appl Neurobiol 19:214–223
18. Kuhlmann T, Lucchinetti C, Zettl UK, Bitsch A, Lassmann H, BruckW (1999) Bcl-2-expressing oligodendrocytes in multiple sclerosislesions. Glia 28:34–39. doi:10.1002/(SICI)1098-1136(199910)28:1<34::AID-GLIA4>3.0.CO;2-8
19. KippM, Clarner T, Dang J, Copray S, Beyer C (2009) The cuprizoneanimal model: new insights into an old story. Acta Neuropathol 118:723–736. doi:10.1007/s00401-009-0591-3
20. Skripuletz T, Gudi V, Hackstette D, Stangel M (2011) De- andremyelination in the CNS white and grey matter induced bycuprizone: the old, the new, and the unexpected. Histol Histopathol26:1585–1597
21. Buschmann JP, Berger K, Awad H, Clarner T, Beyer C, Kipp M(2012) Inflammatory response and chemokine expression in thewhite matter corpus callosum and gray matter cortex region duringcuprizone-induced demyelination. J Mol Neurosci MN 48:66–76.doi:10.1007/s12031-012-9773-x
22. Gudi V et al (2009) Regional differences between grey and whitematter in cuprizone induced demyelination. Brain Res 1283:127–138. doi:10.1016/j.brainres.2009.06.005
1562 Mol Neurobiol (2016) 53:1551–1564

23. Kipp M et al (2011) The hippocampal fimbria of cuprizone-treatedanimals as a structure for studying neuroprotection in multiple scle-rosis. Inflamm Res 60:723–726. doi:10.1007/s00011-011-0339-0
24. Baggiolini M (1998) Chemokines and leukocyte traffic. Nature 392:565–568. doi:10.1038/33340
25. Charo IF, Ransohoff RM (2006) The many roles of chemokines andchemokine receptors in inflammation. N Engl J Med 354:610–621.doi:10.1056/NEJMra052723
26. Luster AD (1998) Chemokines—chemotactic cytokines that mediateinflammation. N Engl J Med 338:436–445. doi:10.1056/NEJM199802123380706
27. Ubogu EE, Cossoy MB, Ransohoff RM (2006) The expression andfunction of chemokines involved in CNS inflammation. TrendsPharmacol Sci 27:48–55. doi:10.1016/j.tips.2005.11.002
28. Fernandez EJ, Lolis E (2002) Structure, function, and inhibition ofchemokines. Ann Rev Pharmacol Toxicol 42:469–499. doi:10.1146/annurev.pharmtox.42.091901.11583842/1/469
29. Rossi D, Zlotnik A (2000) The biology of chemokines and theirreceptors. Annu Rev Immunol 18:217–242. doi:10.1146/annurev.immunol.18.1.217
30. Laing KJ, Secombes CJ (2004) Chemokines. Dev Comp Immunol28:443–460. doi:10.1016/j.dci.2003.09.006
31. Murphy PM (2002) International Union of Pharmacology. XXX.Update on chemokine receptor nomenclature. Pharmacol Rev 54:227–229
32. Mackay CR (2001) Chemokines: immunology's high impact factors.Nat Immunol 2:95–101. doi:10.1038/84298
33. Andjelkovic AV, Song L, Dzenko KA, Cong H, Pachter JS (2002)Functional expression of CCR2 by human fetal astrocytes. J NeurosciRes 70:219–231. doi:10.1002/jnr.10372
34. Biber K, Dijkstra I, Trebst C, De Groot CJ, Ransohoff RM, BoddekeHW (2002) Functional expression of CXCR3 in cultured mouse andhuman astrocytes and microglia. Neuroscience 112:487–497
35. Grizenkova J, Akhtar S, Brandner S, Collinge J, Lloyd SE (2014)Microglial Cx3cr1 knockout reduces prion disease incubation time inmice. BMC Neurosci 15:44. doi:10.1186/1471-2202-15-44
36. Heesen M et al (1996) Mouse astrocytes respond to the chemokinesMCP-1 and KC, but reverse transcriptase-polymerase chain reactiondoes not detect mRNA for the KC or new MCP-1 receptor. JNeurosc i Res 45 :382–391 . do i :10 .1002 / (SICI )1097-4547(19960815)45:4<382::AID-JNR7>3.0.CO;2-5
37. McMillin M et al (2014) Neuronal CCL2 is upregulated during he-patic encephalopathy and contributes tomicroglia activation and neu-rological decline. J Neuroinflammation 11:121. doi:10.1186/1742-2094-11-121
38. Odemis V, Moepps B, Gierschik P, Engele J (2002) Interleukin-6 andcAMP induce stromal cell-derived factor-1 chemotaxis in astrogliaby up-regulating CXCR4 cell surface expression. Implications forBrain Inflammation. J Biol Chem 277:39801–39808. doi:10.1074/jbc.M200472200
39. Tanabe S et al (1997) Functional expression of the CXC-chemokinereceptor-4/fusin on mouse microglial cells and astrocytes. J Immunol159:905–911
40. Glabinski AR, Ransohoff RM (1999) Chemokines and chemokinereceptors in CNS pathology. J Neurovirol 5:3–12
41. McMahon EJ, CookDN, Suzuki K,Matsushima GK (2001) Absenceof macrophage-inflammatory protein-1alpha delays central nervoussystem demyelination in the presence of an intact blood-brain barrier.J Immunol 167:2964–2971
42. Henkel JS et al (2004) Presence of dendritic cells, MCP-1, and acti-vated microglia/macrophages in amyotrophic lateral sclerosis spinalcord tissue. Ann Neurol 55:221–235. doi:10.1002/ana.10805
43. Nash KR et al (2013) Fractalkine overexpression suppresses tau pa-thology in a mouse model of tauopathy. Neurobiol Aging 34:1540–1548. doi:10.1016/j.neurobiolaging.2012.12.011
44. Asevedo E et al (2013) Impact of peripheral levels of chemokines,BDNF and oxidative markers on cognition in individuals withschizophrenia. J Psychiatr Res 47:1376–1382. doi:10.1016/j.jpsychires.2013.05.032
45. Krauthausen M, Saxe S, Zimmermann J, Emrich M, Heneka MT,Muller M (2014) CXCR3 modulates glial accumulation and activa-tion in cuprizone-induced demyelination of the central nervous sys-tem. J Neuroinflammation 11:109. doi:10.1186/1742-2094-11-109
46. Patel JR, McCandless EE, Dorsey D, Klein RS (2010) CXCR4 pro-motes differentiation of oligodendrocyte progenitors andremyelination. Proc Natl Acad Sci U S A 107:11062–11067. doi:10.1073/pnas.1006301107
47. Patel JR et al (2012) Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation anddifferentiation within the adult CNS. Acta Neuropathol 124:847–860. doi:10.1007/s00401-012-1034-0
48. Skripuletz T et al (2013) Astrocytes regulate myelin clearancethrough recruitment of microglia during cuprizone-induced demye-lination. Brain J Neurol 136:147–167. doi:10.1093/brain/aws262
49. Acs P et al (2009) 17beta-estradiol and progesterone preventcuprizone provoked demyelination of corpus callosum in male mice.Glia 57:807–814. doi:10.1002/glia.20806
50. Groebe A, Clarner T, Baumgartner W, Dang J, Beyer C, Kipp M(2009) Cuprizone treatment induces distinct demyelination,astrocytosis, and microglia cell invasion or proliferation in the mousecerebellum. Cerebellum 8:163–174. doi:10.1007/s12311-009-0099-3
51. Kipp M et al (2008) Brain-region-specific astroglial responsesin vitro after LPS exposure. J Mol Neurosci MN 35:235–243. doi:10.1007/s12031-008-9057-7
52. Clarner T, Parabucki A, Beyer C, Kipp M (2011) Corticosteroidsimpair remyelination in the corpus callosum of cuprizone-treatedmice. J Neuroendocrinol 23:601–611. doi:10.1111/j.1365-2826.2011.02140.x
53. Kipp M et al (2011) Brain lipid binding protein (FABP7) as modu-lator of astrocyte function. Physiol Res 60(Suppl 1):S49–S60
54. Slowik A, Schmidt T, Beyer C, Amor S, Clarner T, Kipp M (2015)The sphingosine 1-phosphate receptor agonist FTY720 is neuropro-tective after cuprizone-induced CNS demyelination. Br J Pharmacol172:80–92. doi:10.1111/bph.12938
55. Braun A, Dang J, Johann S, Beyer C, Kipp M (2009) Selectiveregulation of growth factor expression in cultured cortical astrocytesby neuro-pathological toxins. Neurochem Int 55:610–618. doi:10.1016/j.neuint.2009.06.004
56. Kipp M et al (2011) BLBP-expression in astrocytes during experi-mental demyelination and in human multiple sclerosis lesions. BrainBehav Immun 25:1554–1568. doi:10.1016/j.bbi.2011.05.003
57. Cook DN et al (1995) Requirement of MIP-1 alpha for an inflamma-tory response to viral infection. Science 269:1583–1585
58. Lu B et al (1998) Abnormalities in monocyte recruitment and cyto-kine expression in monocyte chemoattractant protein 1-deficientmice. J Exp Med 187:601–608
59. Clarner T et al (2012) Myelin debris regulates inflammatory re-sponses in an experimental demyelination animal model and multiplesclerosis lesions. Glia 60:1468–1480. doi:10.1002/glia.22367
60. Skripuletz T et al (2008) Cortical demyelination is prominent in themurine cuprizone model and is strain-dependent. Am J Pathol 172:1053–1061. doi:10.2353/ajpath.2008.070850
61. Liu L et al (2010) CXCR2-positive neutrophils are essential forcuprizone-induced demyelination: relevance to multiple sclerosis.Nat Neurosci 13:319–326. doi:10.1038/nn.2491
62. Lassmann H, Bruck W, Lucchinetti CF (2007) The immunopatholo-gy of multiple sclerosis: an overview. Brain Pathol 17:210–218. doi:10.1111/j.1750-3639.2007.00064.x
63. Ludwin SK, Johnson ES (1981) Evidence for a "dying-back"gliopathy in demyelinating disease. Ann Neurol 9:301–305. doi:10.1002/ana.410090316
Mol Neurobiol (2016) 53:1551–1564 1563

64. Krauspe BM et al (2014) Short-Term Cuprizone Feeding Verifies N-Acetylaspartate Quantification as a Marker of Neurodegeneration. JMol Neurosci MN. doi:10.1007/s12031-014-0412-6
65. Hesse A et al (2010) In toxic demyelination oligodendroglial celldeath occurs early and is FAS independent. Neurobiol Dis 37:362–369. doi:10.1016/j.nbd.2009.10.016
66. Remington LT, Babcock AA, Zehntner SP, Owens T (2007)Microglial recruitment, activation, and proliferation in response toprimary demyelination. Am J Pathol 170:1713–1724. doi:10.2353/ajpath.2007.060783
67. Biancotti JC, Kumar S, de Vellis J (2008) Activation of inflammatoryresponse by a combination of growth factors in cuprizone-induceddemyelinated brain leads to myelin repair. Neurochem Res 33:2615–2628. doi:10.1007/s11064-008-9792-8
68. Yao Y, Tsirka SE (2014) Monocyte chemoattractant protein-1 and theblood-brain barrier. Cell Mol Life Sci CMLS 71:683–697. doi:10.1007/s00018-013-1459-1
69. Szczucinski A, Losy J (2007) Chemokines and chemokine receptorsin multiple sclerosis. Potential targets for new therapies. Acta NeurolScand 115:137–146. doi:10.1111/j.1600-0404.2006.00749.x
70. Charo IF (1999) CCR2: from cloning to the creation of knockoutmice. Chem Immunol 72:30–41
71. Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM (2001)Absence of monocyte chemoattractant protein 1 in mice leads todecreased local macrophage recruitment and antigen-specific T help-er cell type 1 immune response in experimental autoimmune enceph-alomyelitis. J Exp Med 193:713–726
72. Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ (2000) CC chemo-kine receptor 2 is critical for induction of experimental autoimmuneencephalomyelitis. J Exp Med 192:899–905
73. Gaupp S, Pitt D, Kuziel WA, Cannella B, Raine CS (2003)Experimental autoimmune encephalomyelitis (EAE) in CCR2(−/−)mice: susceptibility in multiple strains. Am J Pathol 162:139–150.doi:10.1016/s0002-9440(10)63805-9
74. Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD (2000)Resistance to experimental autoimmune encephalomyelitis in micelacking the CC chemokine receptor (CCR)2. J Exp Med 192:1075–1080
75. Collins PD, Marleau S, Griffiths-Johnson DA, Jose PJ, Williams TJ(1995) Cooperation between interleukin-5 and the chemokineeotaxin to induce eosinophil accumulation in vivo. J Exp Med 182:1169–1174
76. Tran EH, Kuziel WA, Owens T (2000) Induction of experimentalautoimmune encephalomyelitis in C57BL/6 mice deficient in either
the chemokinemacrophage inflammatory protein-1alpha or its CCR5receptor. Eur J Immunol 30:1410–1415. doi:10.1002/(sici)1521-4141(200005)30:5<1410::aid-immu1410>3.0.co;2-l
77. Karpus WJ, Kennedy KJ (1997) MIP-1alpha and MCP-1 differen-tially regulate acute and relapsing autoimmune encephalomyelitis aswell as Th1/Th2 lymphocyte differentiation. J Leukoc Biol 62:681–687
78. ManczakM, Jiang S, Orzechowska B, Adamus G (2002) Crucial roleof CCL3/MIP-1alpha in the recurrence of autoimmune anterior uve-itis induced with myelin basic protein in Lewis rats. J Autoimmun 18:259–270
79. Cross AK,WoodroofeMN (1999) Chemokines inducemigration andchanges in actin polymerization in adult rat brain microglia and ahuman fetal microglial cell line in vitro. J Neurosci Res 55:17–23
80. Cudaback E, Yang Y, Montine TJ, Keene CD (2014) APOEgenotype-dependent modulation of astrocyte chemokine CCL3 pro-duction. Glia. doi:10.1002/glia.22732
81. El-Hage N et al (2006) HIV-1 Tat and opiate-induced changes inastrocytes promote chemotaxis of microglia through the expressionof MCP-1 and alternative chemokines. Glia 53:132–146. doi:10.1002/glia.20262
82. Hinojosa AE, Garcia-Bueno B, Leza JC, Madrigal JL (2011)CCL2/MCP-1 modulation of microglial activation and proliferation.J Neuroinflammation 8:77. doi:10.1186/1742-2094-8-77
83. Wang HK et al (2008) Free radical production in CA1 neurons in-duces MIP-1alpha expression, microglia recruitment, and delayedneuronal death after transient forebrain ischemia. J Neurosci Off JSoc Neurosci 28:1721–1727. doi:10.1523/jneurosci.4973-07.2008
84. Yang G et al (2011) Neuronal MCP-1 mediates microglia recruitmentand neurodegeneration induced by the mild impairment of oxidativemetabolism. Brain Pathol 21:279–297. doi:10.1111/j.1750-3639.2010.00445.x
85. Han Y, Wang J, Zhou Z, Ransohoff RM (2000) TGFbeta1 selectivelyup-regulates CCR1 expression in primary murine astrocytes. Glia 30:1–10
86. Quinones MP et al (2008) Role of astrocytes and chemokine systemsin acute TNFalpha induced demyelinating syndrome: CCR2-dependent signals promote astrocyte activation and survival via NF-kappaB and Akt. Mol Cell Neurosci 37:96–109. doi:10.1016/j.mcn.2007.08.017
87. Lee YK et al (2009) CCR5 deficiency induces astrocyte activation,Abeta deposit and impaired memory function. Neurobiol Learn Mem92:356–363. doi:10.1016/j.nlm.2009.04.003
1564 Mol Neurobiol (2016) 53:1551–1564







![[inserm-00630697, v1] The chemokine CCL2 protects against ... · The chemokine CCL2 protects against methylmercury neurotoxicity. David Godefroy, Romain-Daniel Gosselin, Akira Yasutake,](https://img.pdfslide.net/doc/110x75/5f071b327e708231d41b5617/inserm-00630697-v1-the-chemokine-ccl2-protects-against-the-chemokine-ccl2.jpg)





















