Embed Size (px)
Citation preview

PHARMACOCINETIQUEPHARMACOCINETIQUE
cible pharmacologique
administration
PKPK
Absorption:Résorption & Premier Passage
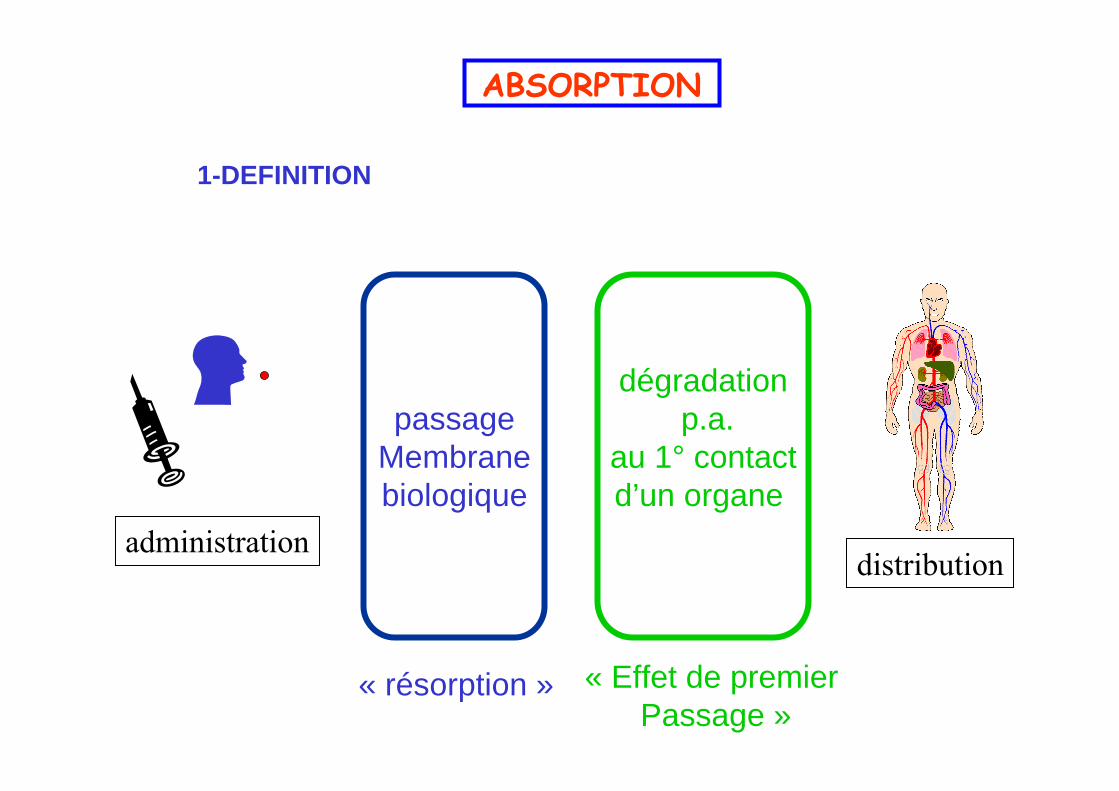
ABSORPTION
1-DEFINITION
distributionadministration
passageMembranebiologique
dégradationp.a.
au 1° contactd’un organe
« résorption » « Effet de premier Passage »

RESORPTION
1-DEFINITION
« Processus par lequel le médicament passe de sonLieu d ’application dans la circulation générale. »
Voies d ’administration concernées:
- ORALE (+++)
- INTRA-MUSCULAIRE
- SOUS-CUTANEE
- RECTALE + TRANS-MUQUEUSES
- TRANS-CUTANEE (+++)

Données générales.
Essentiellement par diffusion passive.
Loi de Fick: V = Kperm. x S x (Ce - Ci)
Conditionnée par:- pH du milieu.- pKa du principe actif.- Liposolubilité- Forme galénique. pH = pKa + log (F.I./F.N.I)
pH = pKa + log (F.N.I./F.I)
La résorption se fait:

Données générales.
Une bonne résorption nécessite:
1. Un p.a. soluble (hydrophile, ionisé).
2. Un p.a. pouvant passer une membrane biologiquelipidique (lipophile, non ionisé).

Données générales.
Forme galénique capitale:
Sirop, solution, ampoules: p.a. déjà dissout, résorption + rapide, atteinte de l’effet + rapide!
Formes sèches: variable!
Résorption peut être optimisée par formes galéniquesspécifiques (libération prolongée, différée, enrobages gastro-résistants…).
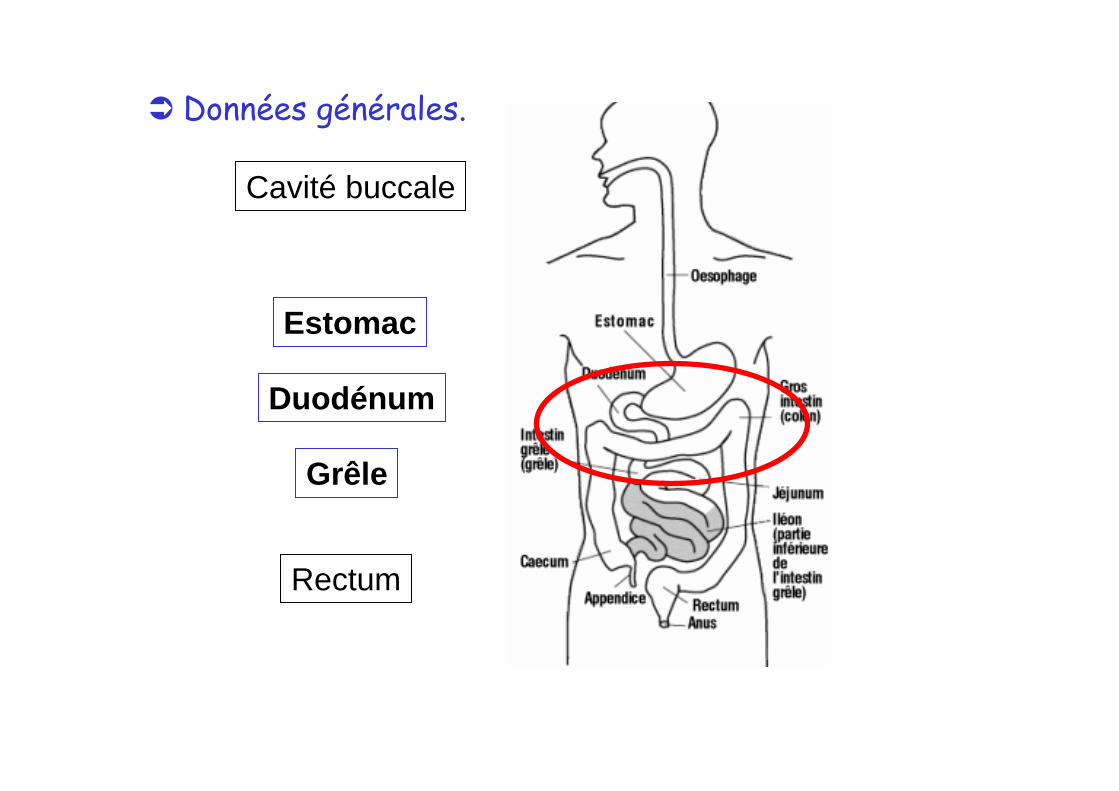
Données générales.
Estomac
Cavité buccale
Duodénum
Grêle
Rectum

2- RESORPTION GASTRO-INTESTINALE
Cavité buccale, œsophage.
a) RAPPELS PHYSIOLOGIQUES
- Pas de résorption (temps de latence insuffisant!).
- Exception: voies sub-linguales: si effet de premier passage hépatique important – permet obtention effet rapide (veines linguales puis jugulaire) mais de courte durée!Ex: dérivés nitrés, angine de poitrine.
- Développement de nouvelles formes galéniques (comprimés adhésifs): effet local recherché, pas d’exposition systémique!
- Risque d’exposition systémique si application locale fréquenteet lésions de la muqueuse buccale!
- Limites: compliance + non irritant!
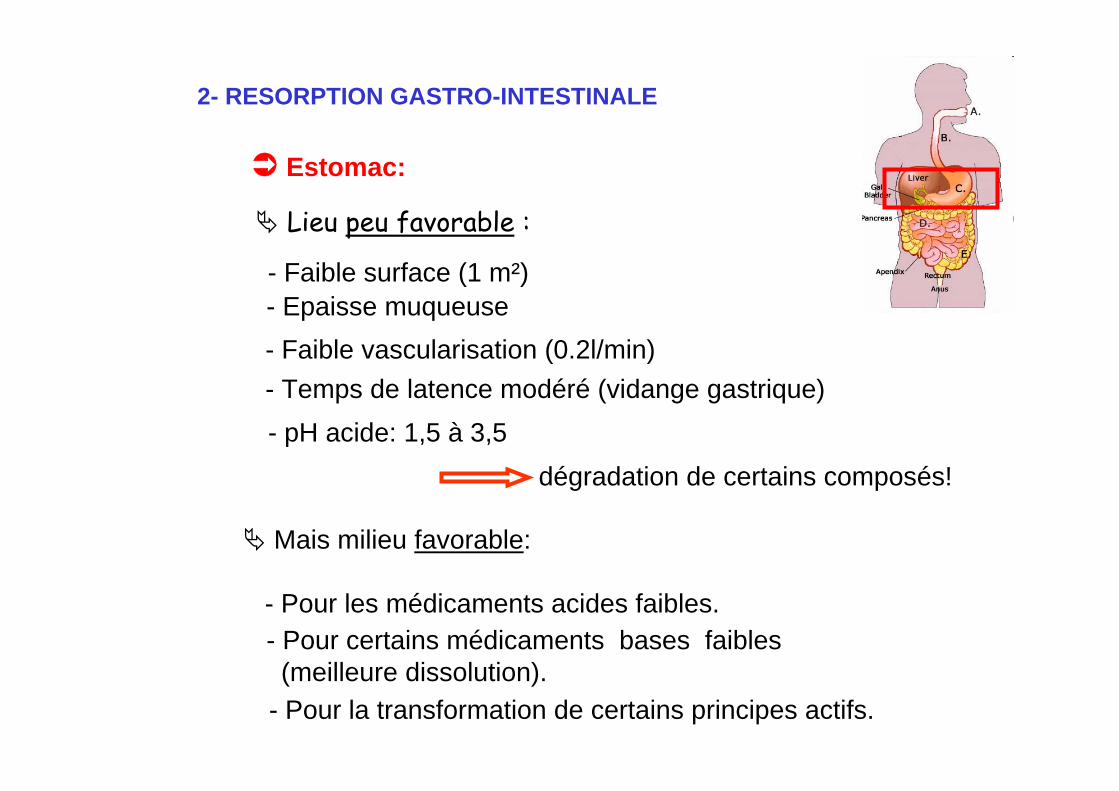
2- RESORPTION GASTRO-INTESTINALE
Mais milieu favorable:
- Pour les médicaments acides faibles.
Lieu peu favorable :
Estomac:
- Epaisse muqueuse- Faible vascularisation (0.2l/min)
- pH acide: 1,5 à 3,5
- Pour certains médicaments bases faibles(meilleure dissolution).
- Pour la transformation de certains principes actifs.
dégradation de certains composés!
- Faible surface (1 m²)
- Temps de latence modéré (vidange gastrique)
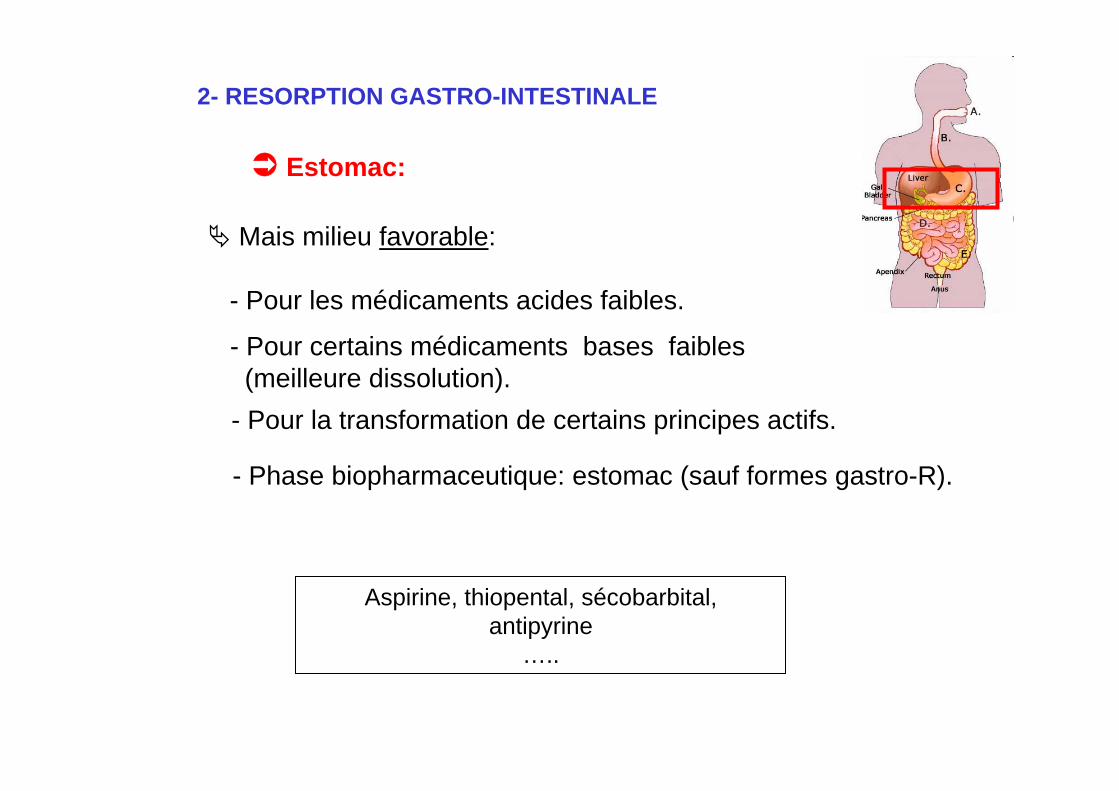
2- RESORPTION GASTRO-INTESTINALE
Mais milieu favorable:
- Pour les médicaments acides faibles.
- Pour certains médicaments bases faibles(meilleure dissolution).
- Pour la transformation de certains principes actifs.
Aspirine, thiopental, sécobarbital,antipyrine
…..
- Phase biopharmaceutique: estomac (sauf formes gastro-R).
Estomac:
2- RESORPTION GASTRO-INTESTINALE
Lieu plus favorable :
Duodénum/Jéjunum:
- Surface importante.
- pH moins acide (4-5).
- Présence de bile favorisant la dissolution des p.a.
- Concerne la majorité des formes per os!

Intestin grêle (iléum):
- surface, longueur, élevées : 200 m2, 4-5 m
- Forte vascularisation (1 l/min) , villosités, capillaires lymphatiques
- pH 5 à 8 : favorise la forme non ionisée.
Lieu très favorable :
- Transporteurs actifs
- Bile, surfactant: accroît la solubilisation des p.a.
- Concerne la majorité des formes per os!
Absorption favorisée, également, par de nombreusessécrétions:
- Pancréatiques (enzymes protéolytiques).
- Biliaires: les sels biliaires indispensables pourl ’absorption des molécules liposolubles.
- Intestinales: nombreuses enzymes présentes dans la lumière intestinale et /ou la membrane: disaccharisades, dipeptidases, entérokinase, cytochromes …)
BIOTRANSFORMATIONS
Intestin grêle:
« effet de premier passage intestinal »

Surface et longueur plus faibles.
Colon:
- Les composés à dissolution lente.
Intéresse essentiellement:
- Les formes galéniques à délitement progressif.
ABSORPTION PLUS LENTE!
Faible surface d ’échange (pas de villosités).

Surface et longueur plus faible. Captage par:
Voie rectale
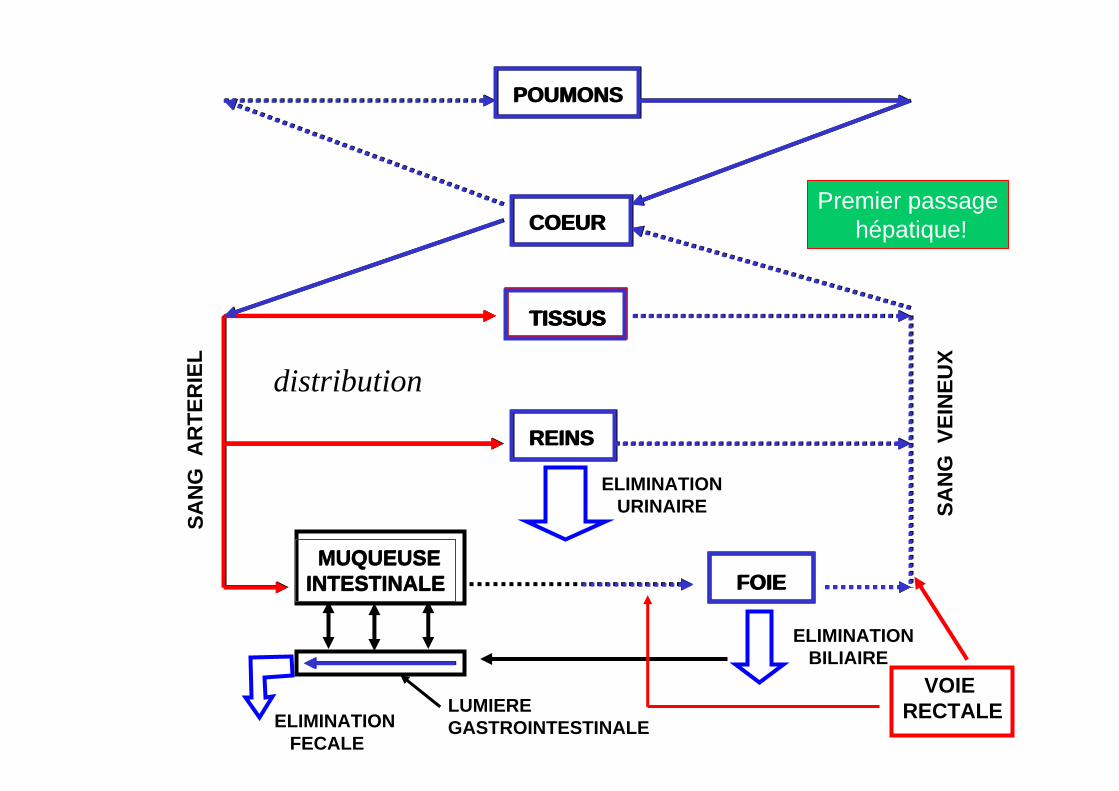
- Veines hémorroïdaires inférieures + moyennes (puis: foie!).
- Atteinte partielle du tronc porte.
- Résorption plus rapide que voie orale!
Limitation partielle et aléatoire de l’effet de premier passage hépatique!
- Veines hémorroïdaires supérieures (évite le foie!).

3- INFLUENCE DES CARACTÉRISTIQUES PHYSICO-CHIMIQUES DU MÉDICAMENT.
- Pour les médicaments ‘’acides ’’ faibles:
La majorité des médicaments sont :
- RÔLE DU pKa
donc ionisables en fonction du pH!
Evaluation des proportions en forme ionisée et non ionisée par les équations de Henderson-Hasselbach:
- Pour les médicaments ‘’ bases ’’ faibles:
Des ‘’acides faibles ’’Des ‘’bases faibles ’’
IONISEE]NON[FORMEIONISEE][FORMElogpKapH +=
IONISEE][FORMEIONISEE]NON[FORMElogpKapH +=
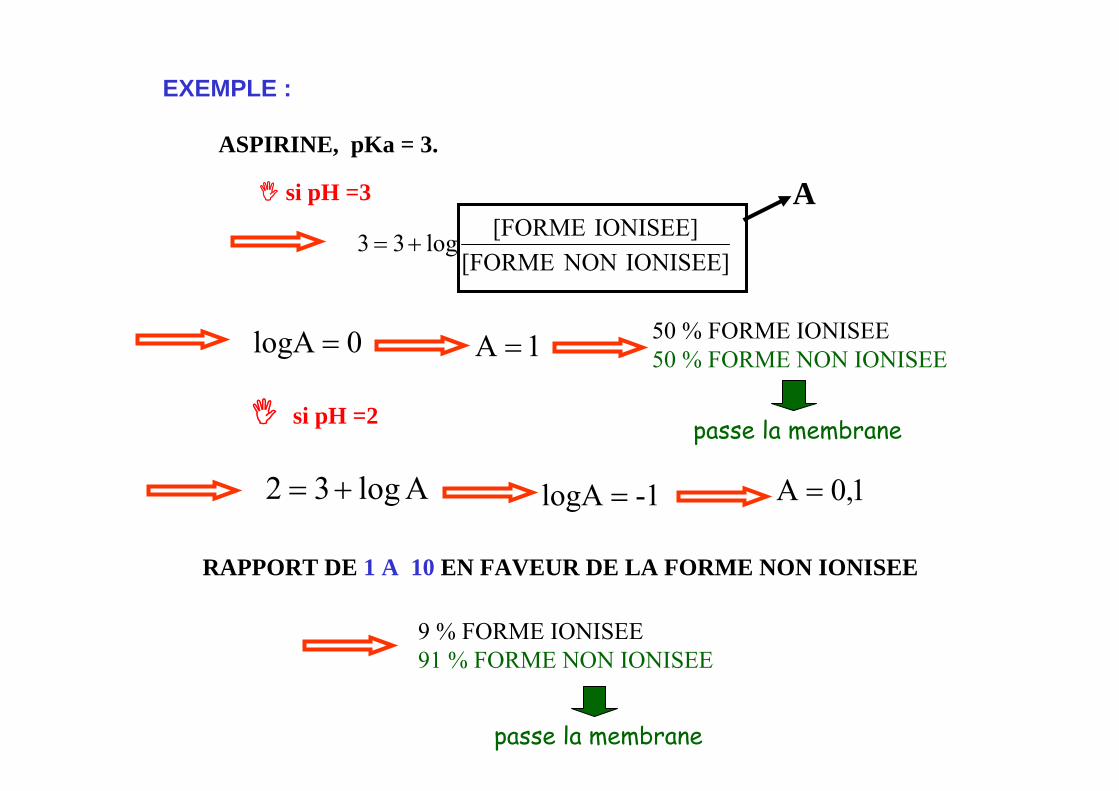
EXEMPLE :
si pH =3
ASPIRINE, pKa = 3.
IONISEE]NON[FORMEIONISEE][FORMElog33 +=
A
0logA = 1A = 50 % FORME IONISEE50 % FORME NON IONISEE
si pH =2
Alog32 += 1-logA = 1,0A =
9 % FORME IONISEE91 % FORME NON IONISEE
RAPPORT DE 1 A 10 EN FAVEUR DE LA FORME NON IONISEE
passe la membrane
passe la membrane

POUR LES MEDICAMENTS ‘’ACIDES FAIBLES’’
Si pKa < 2,5 et si 2,5 < pH < 8 F.N.I. faible (peu de passage!)
Si pKa > 7,5 quel que soit le pH, F.N.I. prédomine (passage!)
Si 2,5 < pKa < 7,5 variation importante de la F.I. avec le pH!
POUR LES MEDICAMENTS ‘ ’BASES FAIBLES’’
Si pKa < 5 insensible aux variations de pH
Si 5 < pKa < 11 variation importante de la F.N.I avec le pH!
3- INFLUENCE DES CARACTÉRISTIQUES PHYSICO-CHIMIQUES DU MÉDICAMENT.

Caractérise la liposolubilité d ’une substance
- RÔLE DU COEFFICIENT DE PARTAGE (Liposolubilité)
Solvant 1 (apolaire) : huile, benzène, heptane...
Plus le ks est élevé, plus le médicament est liposoluble.
Solvant 2 (polaire) : phase aqueuse….
2SOLVANTDANS][1SOLVANTDANS][Ks =
Plus le ks est élevé, plus le p.a. passe une membrane lipidique!
Log P: coef de partage octanol/H2O (= Log Kow (octanol/water)).

Log P (= log Kow) : référence en R&D pharma!
• Passage B.H.E: 2,0 • Résorption orale: 1,8 • Résorption sub-linguale: 5,5 • Résorption percutanée: 2,6
Etudes statistiques permettent d’évaluer la capacité d’un actif à passer +/- les membranes à partir de la détermination du Log P:
Permet un premier crible in silico!
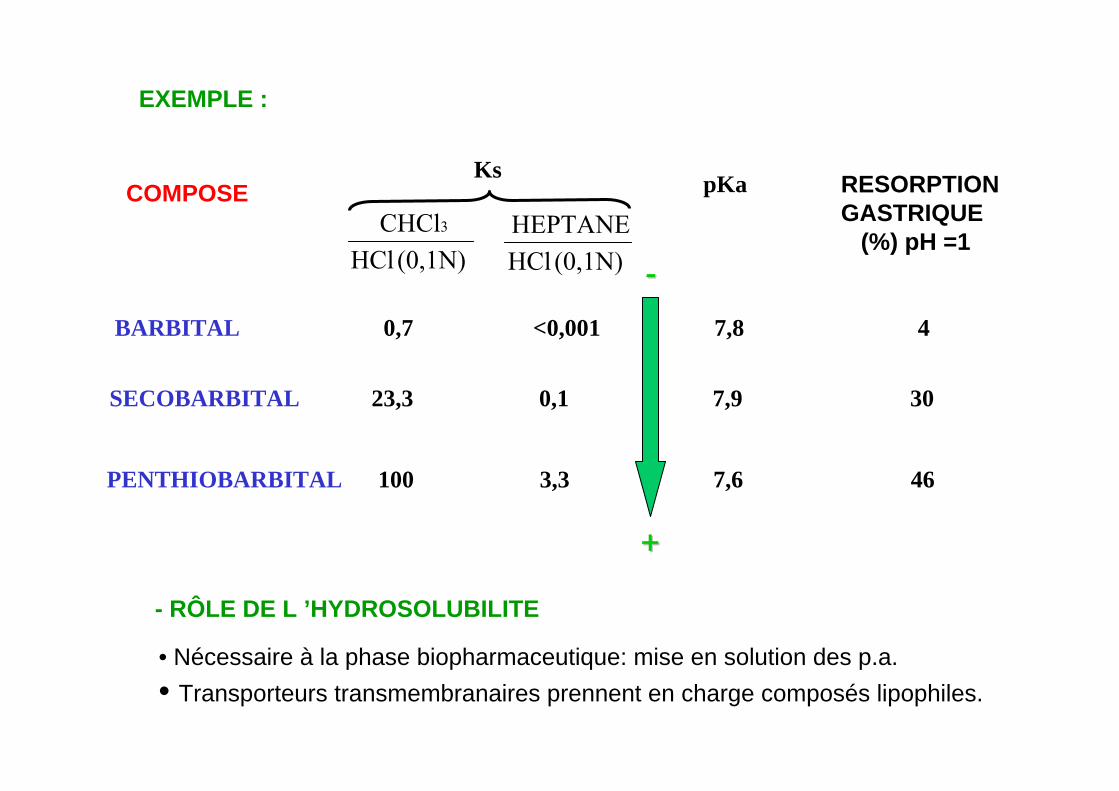
EXEMPLE :
(0,1N)HClCHCl3
(0,1N)HClHEPTANE
COMPOSEKs pKa RESORPTION
GASTRIQUE(%) pH =1
- RÔLE DE L ’HYDROSOLUBILITE
BARBITAL 0,7 <0,001 7,8 4
PENTHIOBARBITAL 100 3,3 7,6 46
• Nécessaire à la phase biopharmaceutique: mise en solution des p.a.• Transporteurs transmembranaires prennent en charge composés lipophiles.
++
--
SECOBARBITAL 23,3 0,1 7,9 30
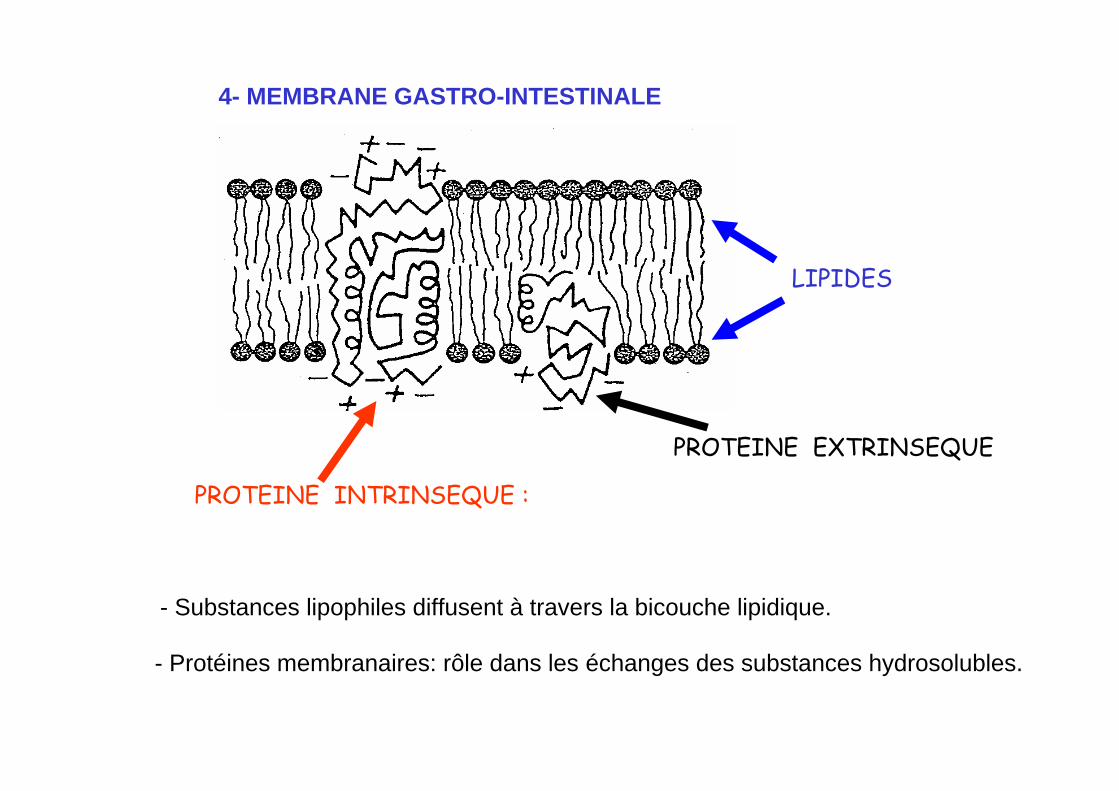
4- MEMBRANE GASTRO-INTESTINALE
- Protéines membranaires: rôle dans les échanges des substances hydrosolubles.
PROTEINE EXTRINSEQUE
PROTEINE INTRINSEQUE :
LIPIDES
- Substances lipophiles diffusent à travers la bicouche lipidique.

5- LES MECANISMES DE LA RESORPTION.
processus :
A- DIFFUSION PASSIVE.
- sens du gradient de concentration.
conduit à un état d ’équilibre entre les deux milieuxde part et d ’autre de la membrane.
- non saturable
- non spécifique
- ne présente pas de phénomènes d ’inhibition!
- pas de dépense d ’énergie!

La vitesse nécessaire pour atteindre l’état d ’équilibre obéit à la Loi de Fick :
S = surface de la membrane
D = coefficient de diffusion à l ’intérieur de la membrane
a = épaisseur de la membrane
(Ce-ci) = différence de [ ] de part et d ’autre de la membrane
Ks = coefficient de partage
aCi)(CeKsSDV −×××
=
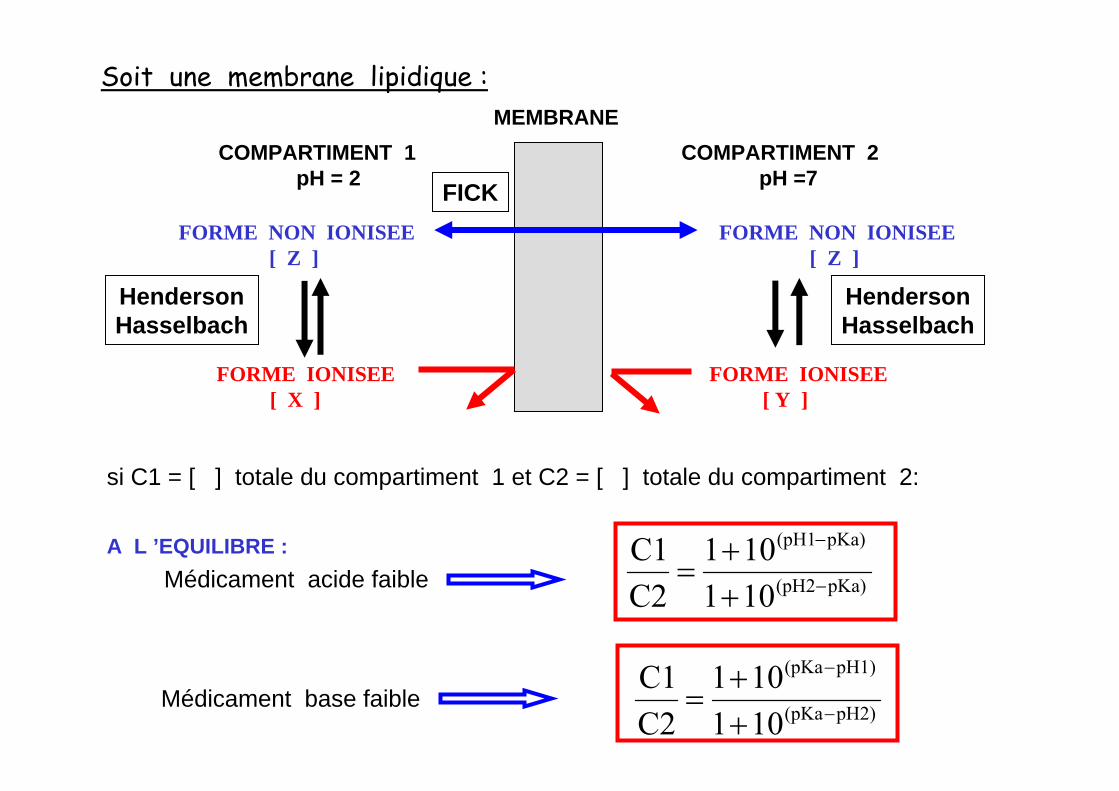
MEMBRANECOMPARTIMENT 1
pH = 2COMPARTIMENT 2
pH =7
FORME NON IONISEE[ Z ]
FORME NON IONISEE[ Z ]
FORME IONISEE[ X ]
FORME IONISEE[ Y ]
Soit une membrane lipidique :
si C1 = [ ] totale du compartiment 1 et C2 = [ ] totale du compartiment 2:
A L ’EQUILIBRE :
pKa)(pH2
pKa)(pH1
101101
C2C1
−
−
++
=Médicament acide faible
pH2)(pKa
pH1)(pKa
101101
C2C1
−
−
++
=Médicament base faible
FICK
HendersonHasselbach
HendersonHasselbach

MEMBRANECOMPARTIMENT 1
(MILIEU GASTRIQUE)pH = 1
COMPARTIMENT 2(PLASMA)
pH =7FORME NON IONISEE
[ 1 ]FORME NON IONISEE
[ 1 ]
FORME IONISEE[ 0,01 ]
FORME IONISEE[ 10000 ]
EXEMPLE 1 : Médicament à pKa = 3
C1 = [ ] TOTALE DU COMPARTIMENT 1 = 1,01
C2 = [ ] TOTALE DU COMPARTIMENT 2 = 10001
A L ’EQUILIBRE :
1000101,1
101101
C2C1
)3(7
)3(1
=++
= −
−
>99% résorbable!Rapide
Indépendant tempsde latence
>99% résorbable!Rapide
Indépendant tempsde latence
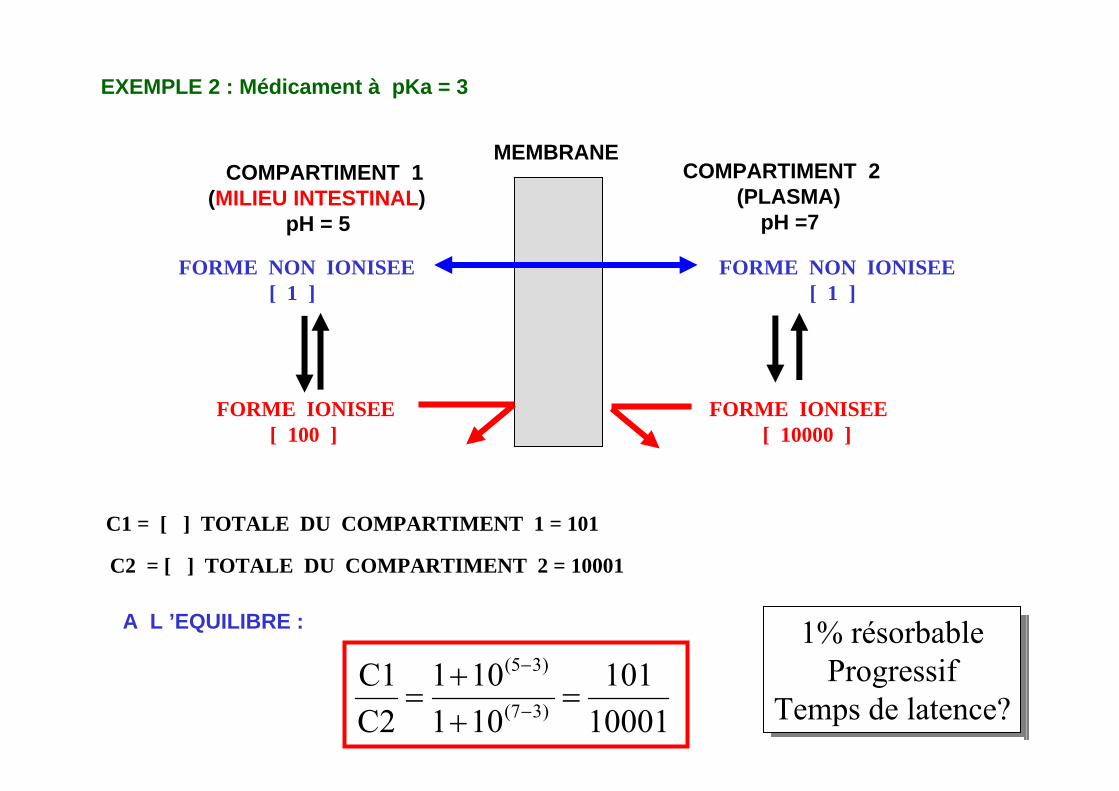
MEMBRANECOMPARTIMENT 1
(MILIEU INTESTINAL)pH = 5
COMPARTIMENT 2(PLASMA)
pH =7
FORME NON IONISEE[ 1 ]
FORME NON IONISEE[ 1 ]
FORME IONISEE[ 100 ]
FORME IONISEE[ 10000 ]
EXEMPLE 2 : Médicament à pKa = 3
C1 = [ ] TOTALE DU COMPARTIMENT 1 = 101
C2 = [ ] TOTALE DU COMPARTIMENT 2 = 10001
A L ’EQUILIBRE :
10001101
101101
C2C1
)3(7
)3(5
=++
= −
−1% résorbable
ProgressifTemps de latence?
1% résorbableProgressif
Temps de latence?
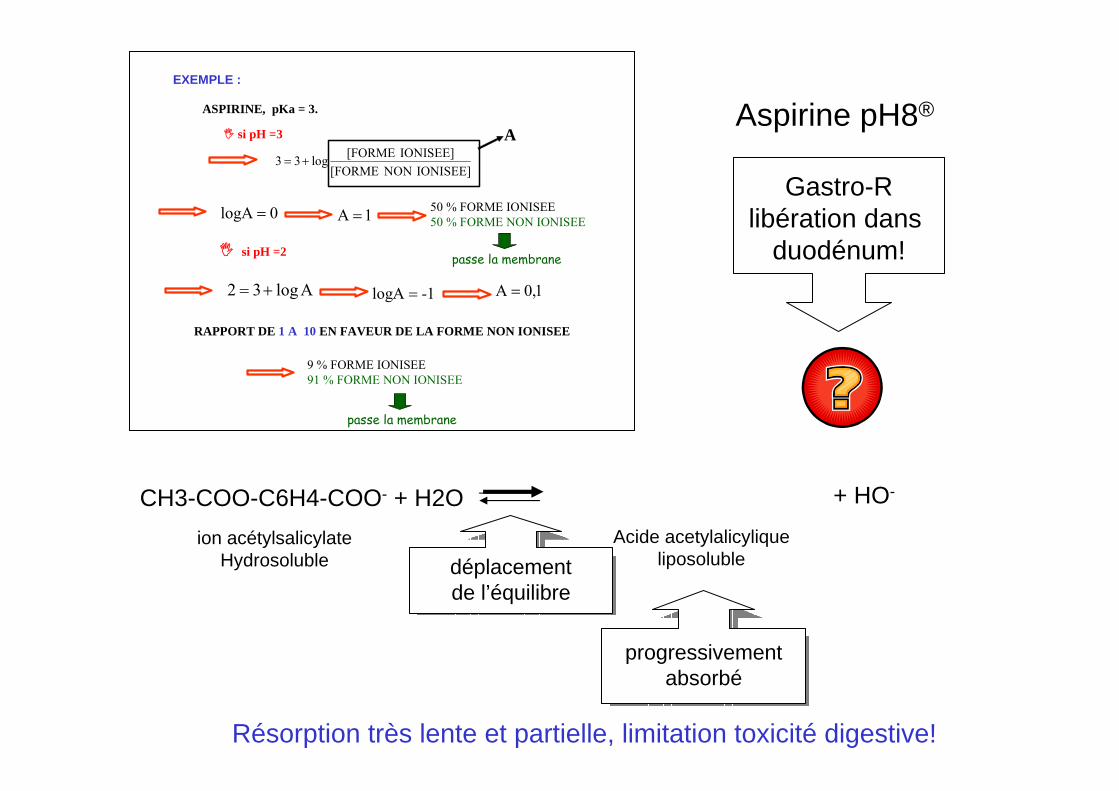
EXEMPLE :
si pH =3
ASPIRINE, pKa = 3.
IONISEE]NON[FORMEIONISEE][FORMElog33 +=
A
0logA = 1A = 50 % FORME IONISEE50 % FORME NON IONISEE
si pH =2
Alog32 += 1-logA = 1,0A =
9 % FORME IONISEE91 % FORME NON IONISEE
RAPPORT DE 1 A 10 EN FAVEUR DE LA FORME NON IONISEE
passe la membrane
passe la membrane
Aspirine pH8®
Gastro-Rlibération dans
duodénum!
CH3-COO-C6H4-COO- + H2O CH3-COO-C6H4-COOH + HO-
ion acétylsalicylateHydrosoluble
Acide acetylalicyliqueliposoluble
progressivementabsorbé
progressivementabsorbé
déplacementde l’équilibre
déplacementde l’équilibre
Résorption très lente et partielle, limitation toxicité digestive!

processus :
B- DIFFUSION FACILITE.
- Sens du gradient de concentration
Vitesse souvent > à celle de la diffusion passive
- Par transporteur (protéines membranaires intrinsèques).- Saturation et inhibition possibles!
- Pas de dépense d ’énergie.
MEMBRANECOMPARTIMENT 1 COMPARTIMENT 2
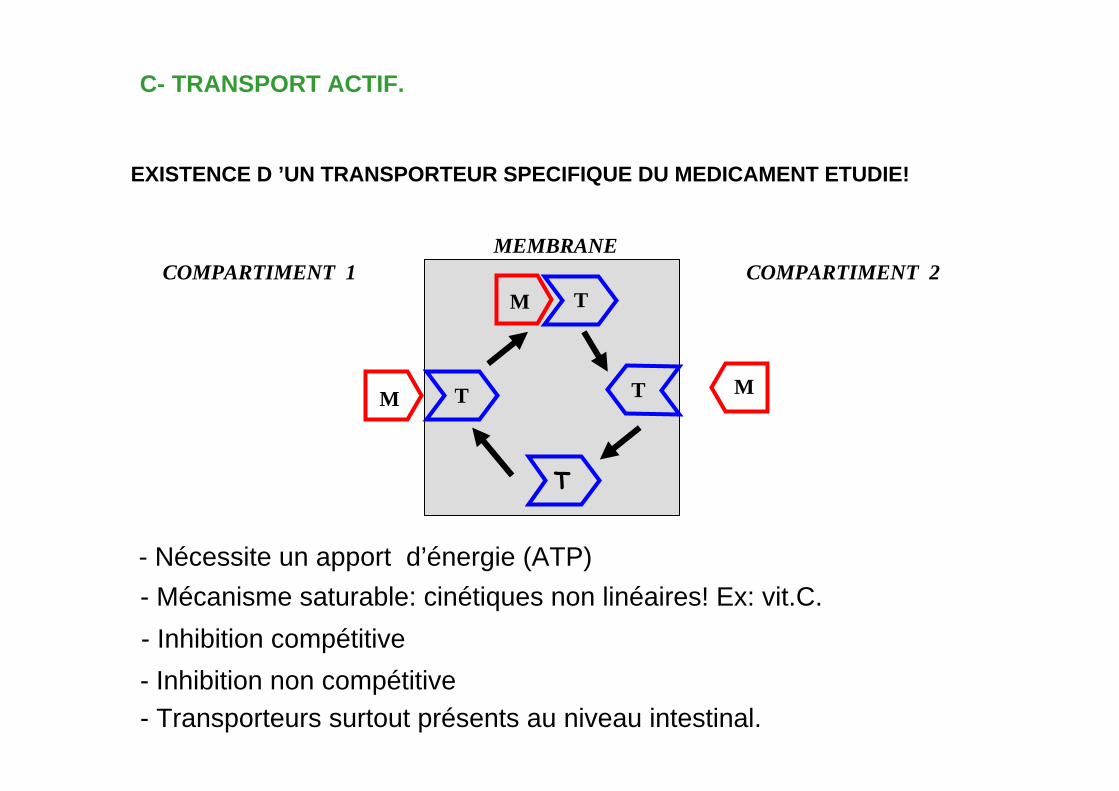
C- TRANSPORT ACTIF.
EXISTENCE D ’UN TRANSPORTEUR SPECIFIQUE DU MEDICAMENT ETUDIE!
- Nécessite un apport d’énergie (ATP)
M
M T
T
T
T
- Mécanisme saturable: cinétiques non linéaires! Ex: vit.C.- Inhibition compétitive - Inhibition non compétitive
M
- Transporteurs surtout présents au niveau intestinal.

6- FACTEURS MODIFIANT LA RESORPTION DIGESTIVE.
- DEGRADATION CHIMIQUE (hydrolyse acide, estomac)
- METABOLISATION (effet de 1° passage intestinal)
- COMPLEXATION
- ALIMENTATION
- INTERACTIONS MEDICAMENTEUSES (variations pH, complexation)
- ETATS PHYSIO-PATHOLOGIQUES
- VIDANGE GASTRIQUE
- DEBITS SANGUINS
- FORTES VARIABILITES INTER- ET INTRA-INDIVIDUELLE!!
± 30%+/-30%
± 30%+/-30%
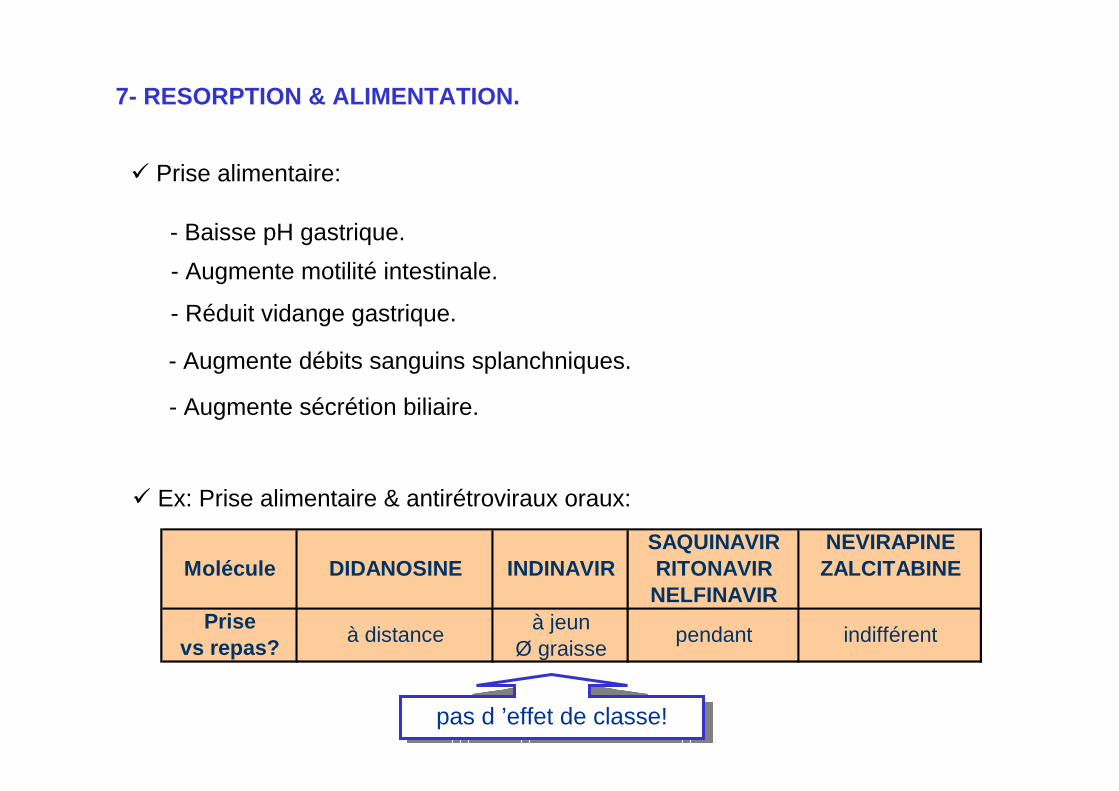
7- RESORPTION & ALIMENTATION.
Prise alimentaire:
- Baisse pH gastrique. - Augmente motilité intestinale.
- Augmente débits sanguins splanchniques.
- Augmente sécrétion biliaire.
SAQUINAVIR NEVIRAPINEMolécule DIDANOSINE INDINAVIR RITONAVIR ZALCITABINE
NELFINAVIRPrise à jeun
vs repas? Ø graisseà distance pendant indifférent
Ex: Prise alimentaire & antirétroviraux oraux:
- Réduit vidange gastrique.
pas d ’effet de classe!pas d ’effet de classe!
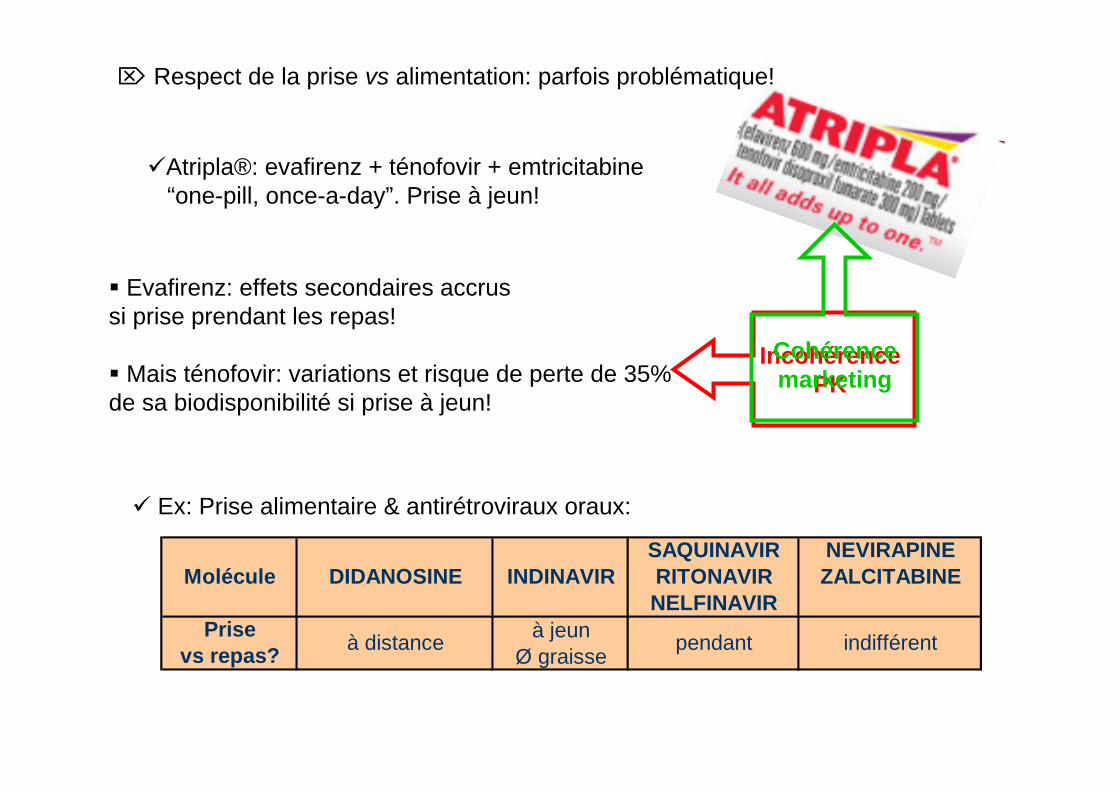
SAQUINAVIR NEVIRAPINEMolécule DIDANOSINE INDINAVIR RITONAVIR ZALCITABINE
NELFINAVIRPrise à jeun
vs repas? Ø graisseà distance pendant indifférent
Ex: Prise alimentaire & antirétroviraux oraux:
Atripla®: evafirenz + ténofovir + emtricitabine“one-pill, once-a-day”. Prise à jeun!
Evafirenz: effets secondaires accrussi prise prendant les repas!
Mais ténofovir: variations et risque de perte de 35% de sa biodisponibilité si prise à jeun!
IncohérencePK
Cohérencemarketing
⌦ Respect de la prise vs alimentation: parfois problématique!

- Luméfantrine: résorption OK uniquement si prise alimentaire!
- Prise pendant les repas (ou: 1.5 g lipides)
- Accès palustre: syndrome anorexique + nausées/vomissements!
- Perte de 90% de la biodisponibilité initiale luméfantrine si prise à jeun!
Echappement thérapeutique, risque de résistances?Echappement thérapeutique, risque de résistances?
- Equivaut à monothérapie arteméther!
Ex: Riamet® (arteméther + luméfantrine): traitementdes accès palustres non-compliqués.
Nécessité PK de prise pendant repas!
⌦ Respect de la prise vs alimentation: parfois problématique!
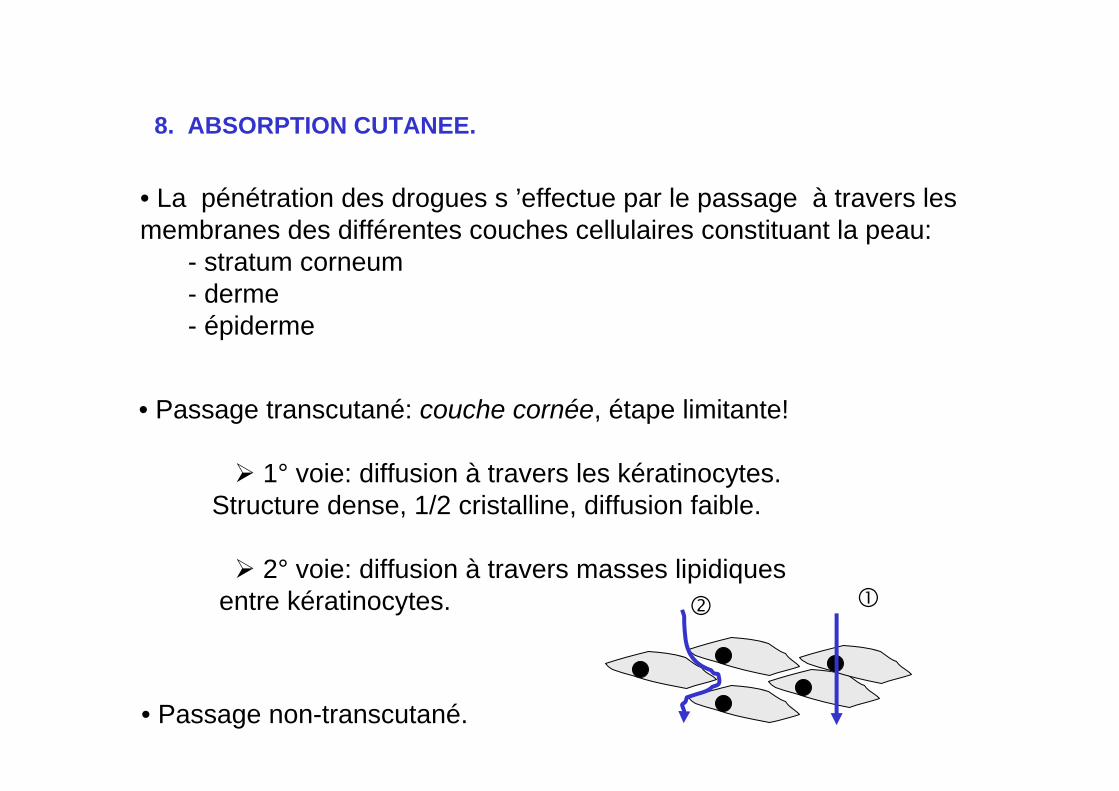
8. ABSORPTION CUTANEE.
• La pénétration des drogues s ’effectue par le passage à travers les membranes des différentes couches cellulaires constituant la peau:
- stratum corneum- derme- épiderme
• Passage transcutané: couche cornée, étape limitante!
1° voie: diffusion à travers les kératinocytes. Structure dense, 1/2 cristalline, diffusion faible.
2° voie: diffusion à travers masses lipidiques entre kératinocytes.
• Passage non-transcutané.

• Passage du derme.
Dissolution des molécules dans l ’eau cytosolique. Diffusion aisée, sauf lorsque les p.a sont:
- Très hydrophobes (dissolution OK?)- Très hydrophiles (passage membranaire OK?).
• Atteinte des capillaires.
Diffusion médicamenteuse dans la substance basale comprise entre le collagène (derme) et les fibres d ’élastine (capillaires).
• Passage par les follicules pileux.
Possibilité de dissolution dans le sébum capillaires.
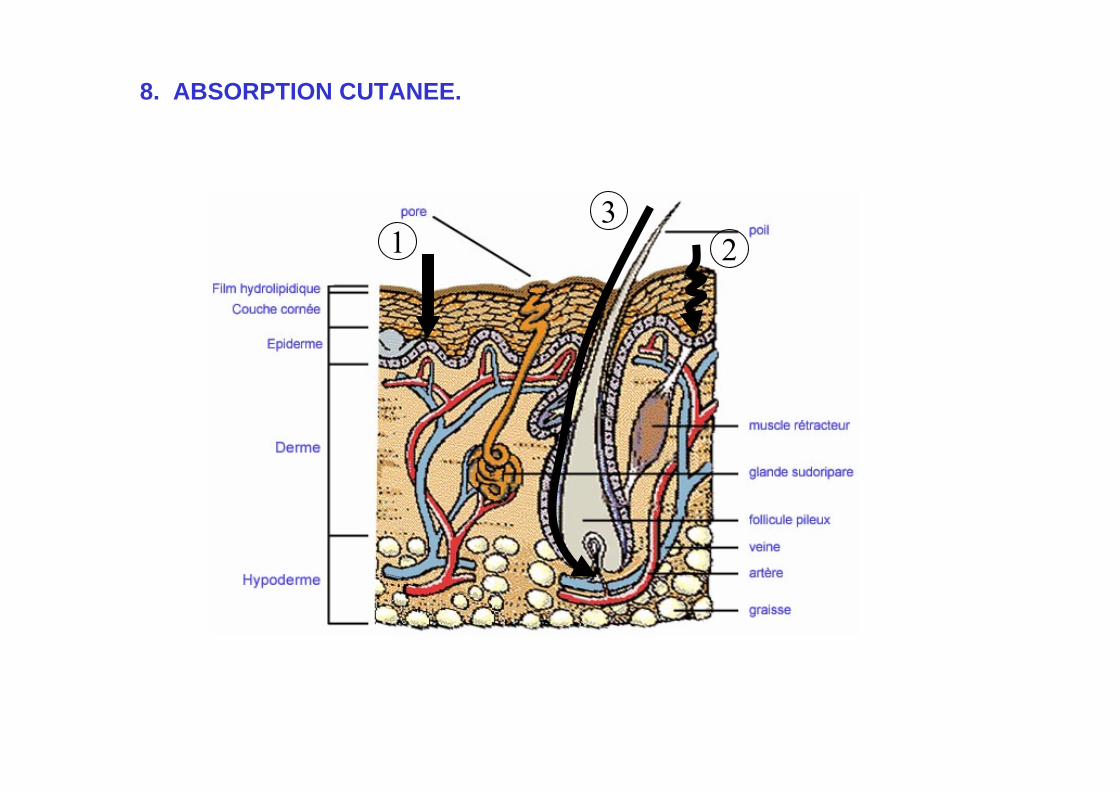
8. ABSORPTION CUTANEE.
1 23

- Patient:
- Etat physiopathologique: hydratation et abrasion de la peau, existence de lésions/brûlures, syndromes inflammatoires...
PÉNÉTRATION PÉNÉTRATION ∆∆ !!
• Facteurs de variations:
- Surface d ’application.
- Température du tissu cutané.
- Tissu adipeux, hydratation.
- Localisation.
- Age.
- Origine ethnique: variations dans les vitesses de résorption!

• Facteurs de variations:
- Principe actif: taille de la molécule, lipophilie.
Petites Molécules diffusent mieux: coefficient de diffusion inversement proportionnel à la racine carrée de la masse molaire (quand transport passif).
Molécule Lipophiles passent membranes biologiques.Coefficient de partage Huile/Eau (Ks) doit être élevé.
- Forme galénique: couple véhicule/principe actif.
Mise à disposition du p.a meilleure si couple hétéro (ex: véhicule lipophile/ p.a. Hydrophile).
Principe Actif non ionisé passe mieux les membranes.
Système occlusif: pénétration.

• Peu de molécules pénètrent la peau et ont un effet systémique à partir d ’une application topique (nécessité de développer des systèmes transdermiques: patchs pour palier biodisponibilité faible à nulle). Agents favorisant pénétration: couches profondes, pas systémique!
En résumé:
• Absorption proportionnelle à la surface d ’exposition.
• Véhicule lipophile, massage: pénétration et risque de passage dans la circulation générale.
• Risque d ’exposition systémique non souhaitée si lésions étendues!

9. AUTRES RESORPTIONS
• Autres sites de résorption:
- Intra-musculaire.
- Sous-cutanée.
- Intranasale.
- Ophtalmique.
- Vaginale.
Vitesse de résorption dépend de vascularisation au site d ’injection, volume injecté, liposolubilité, osmolarité...
Vitesse et quantité résorbée très variable: forme galénique, pH, états physiopathologiques…Effet local recherché le plus souvent, mais risque de passage systémique! Ex: β-bloquants ophtalmiques, diffusion systémique fréquente!
- Pulmonaire.Dépend de la taille des particules, débit sanguins, états physiopathologiques: conditionne atteinte desalvéoles!

10. QUE FAIRE SI MAUVAISE RESORPTION ORALE?
• Objectif: faire une forme orale simple!
• Si faible résorption: formulation galénique plus complexe pour:
- Protéger le p.a si dégradation enzymatique/ hydrolyse.
- Contrôler la phase biopharmaceutique: libération du p.a à un endroit précis du tractus digestif.
- Formes à libération contrôlée.
- Si pas de solution galénique: solution pharmacochimique: pro-drugs?
• Si aucune solution: faire du parentéral?
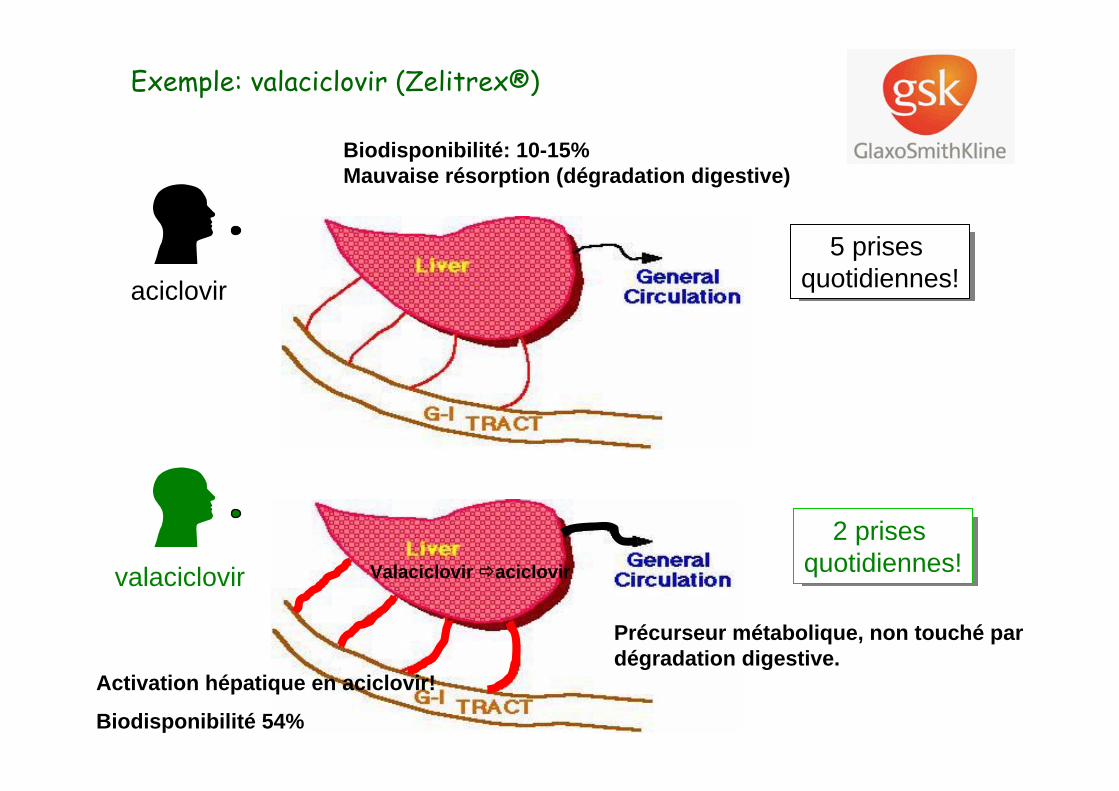
valaciclovir
Exemple: valaciclovir (Zelitrex®)
aciclovir5 prises
quotidiennes!5 prises
quotidiennes!
Biodisponibilité: 10-15%Mauvaise résorption (dégradation digestive)
2 prises quotidiennes!
2 prises quotidiennes!
Précurseur métabolique, non touché pardégradation digestive.
Valaciclovir aciclovir
Activation hépatique en aciclovir!
Biodisponibilité 54%
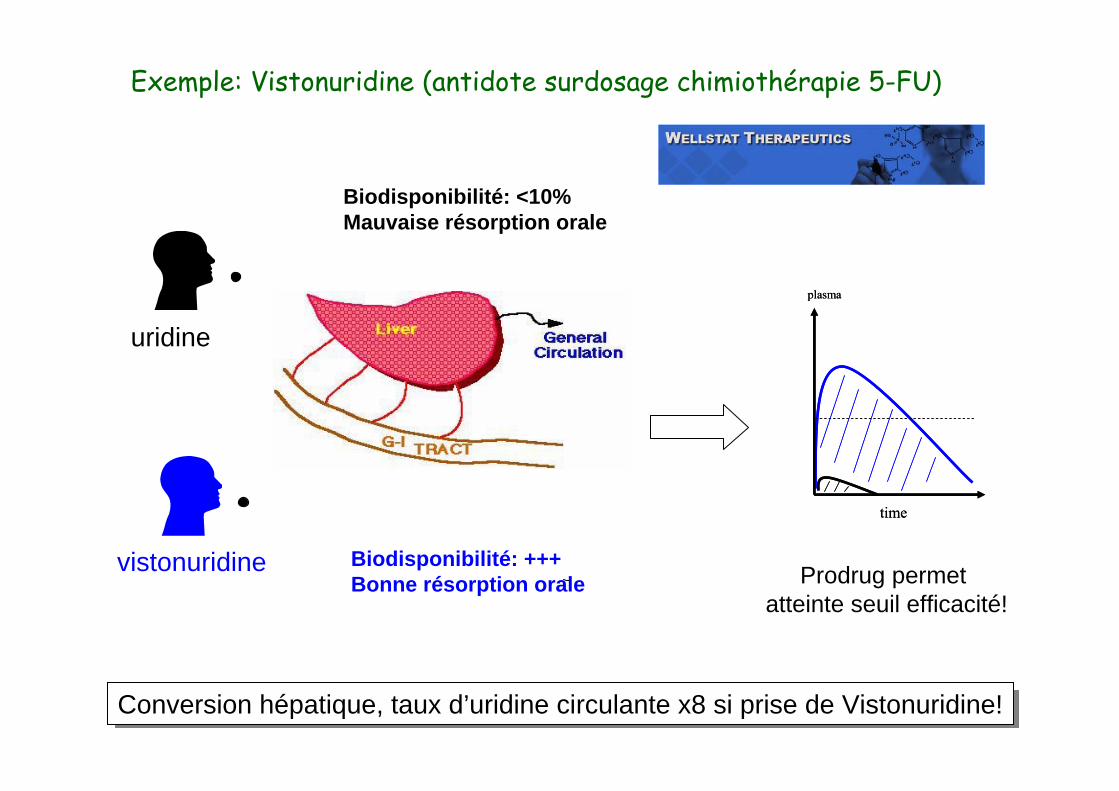
Exemple: Vistonuridine (antidote surdosage chimiothérapie 5-FU)
uridine
Biodisponibilité: <10%Mauvaise résorption orale
vistonuridine Biodisponibilité: +++Bonne résorption orale
time
plasma
time
plasma
Prodrug permet atteinte seuil efficacité!
Conversion hépatique, taux d’uridine circulante x8 si prise de Vistonuridine!Conversion hépatique, taux d’uridine circulante x8 si prise de Vistonuridine!

EFFET DE PREMIER PASSAGEEFFET DE PREMIER PASSAGE
1-DEFINITION
« perte de médicament par métabolisme avant son arrivée dans la circulation générale, des son premier contact avec l ’organe responsable de la biotransformation »

Notion de biodisponibilité:
« Quantité de médicament atteignant la circulation générale après administration. »
Conjonction de 2 phénomènes:
- Résorption (f).
- Effets de 1° passage (f ’).
F = f x f ’
10070
30
30%
f = 0.7 f ’ = 0.42
F = 0.7 x 0.42 = 30%

Notion de biodisponibilité:
« Quantité de médicament atteignant la circulation générale après administration. »
Conjonction de 2 phénomènes:
- Résorption.
- Effets de 1° passage.
• Essentiellement: premier passage hépatique
• Existe effet de premier passage digestif!
• Existe premier passage pulmonaire!
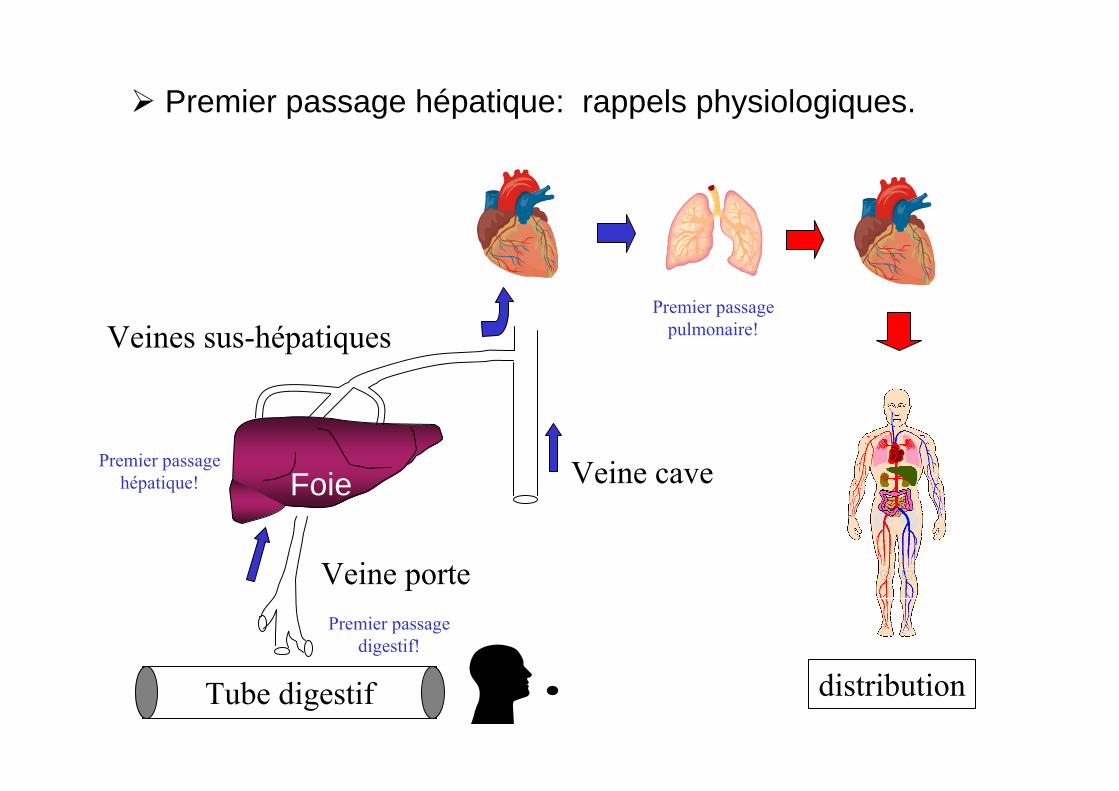
Premier passage hépatique: rappels physiologiques.
distribution
Premier passagepulmonaire!
Veine porte
Veine caveFoie
Tube digestif
Veines sus-hépatiques
Premier passagedigestif!
Premier passagehépatique!

Premier passage hépatique: rappels physiologiques.
On peut limiter l’effet de premier passage par le choix de la voie d’administration!
On peut limiter l’effet de premier passage par le choix de la voie d’administration!
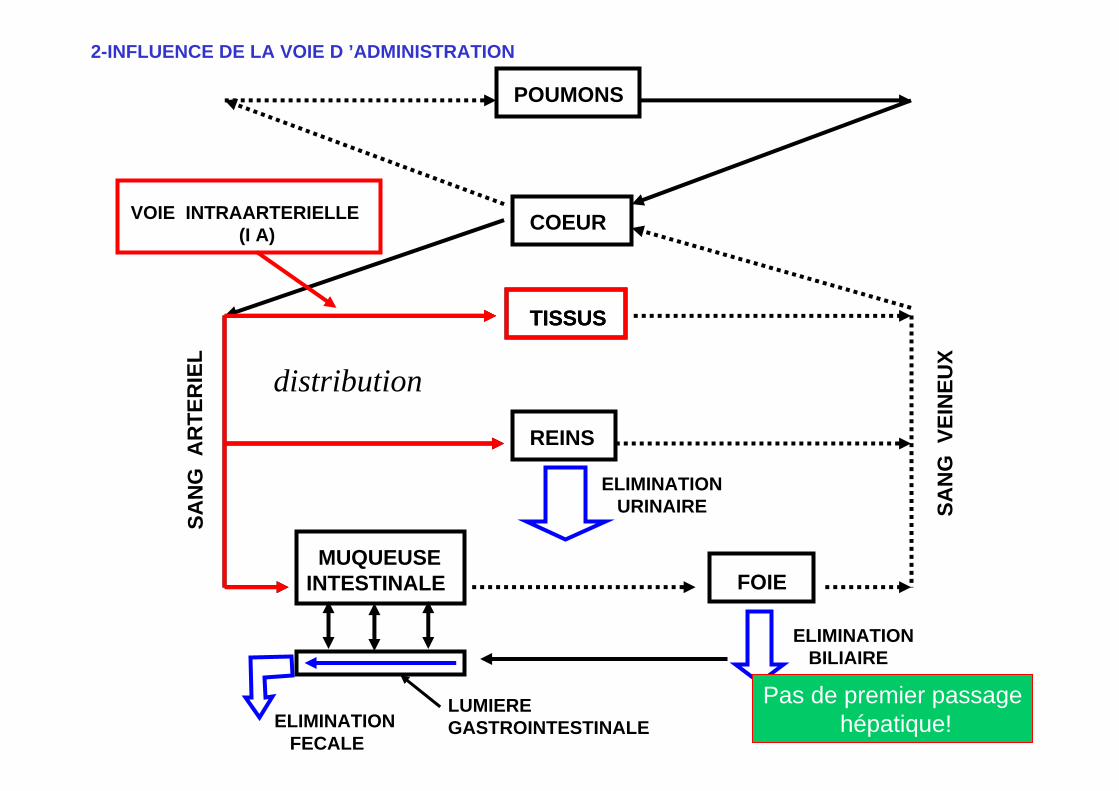
POUMONS
COEUR
TISSUS
REINS
FOIEMUQUEUSE
INTESTINALE
ELIMINATIONURINAIRE
ELIMINATIONBILIAIRE
ELIMINATIONFECALE
LUMIEREGASTROINTESTINALE
VOIE INTRAARTERIELLE(I A)
TISSUS
SAN
G A
RTE
RIE
L
SAN
G V
EIN
EUX
2-INFLUENCE DE LA VOIE D ’ADMINISTRATION
Pas de premier passagehépatique!
distribution
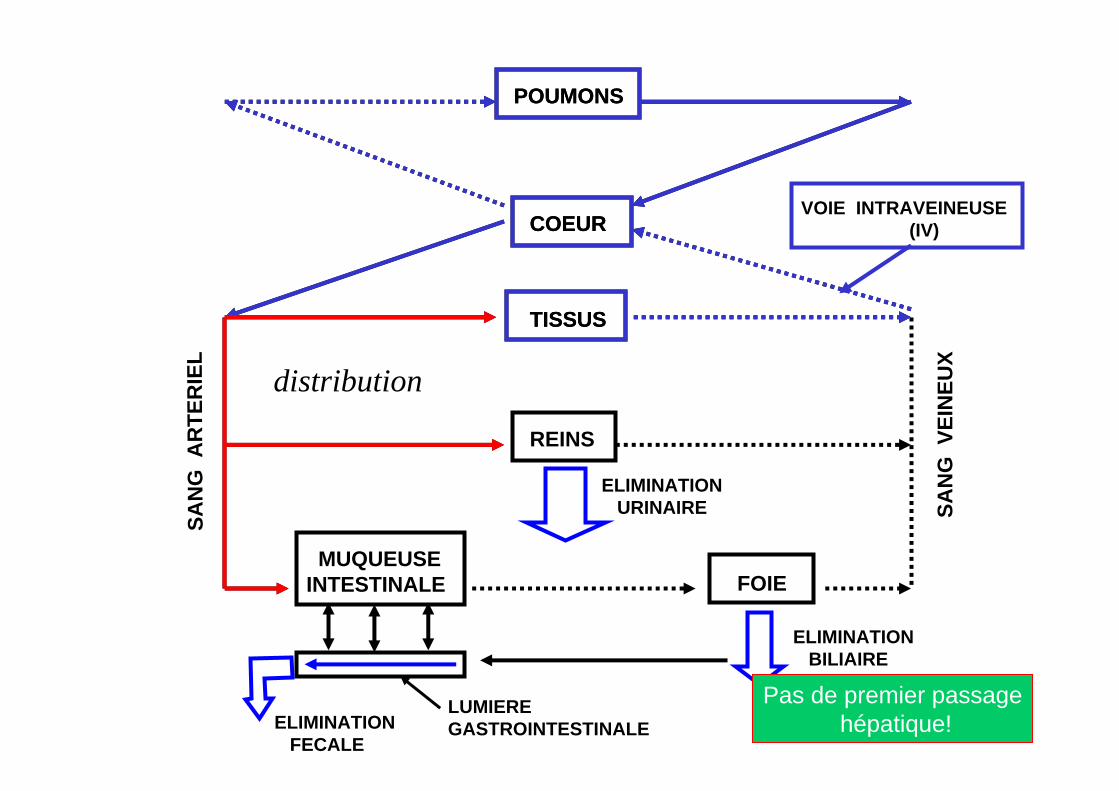
POUMONS
COEUR
TISSUS
REINS
FOIEMUQUEUSE
INTESTINALE
ELIMINATIONURINAIRE
ELIMINATIONBILIAIRE
ELIMINATIONFECALE
LUMIEREGASTROINTESTINALE
VOIE INTRAVEINEUSE(IV)
POUMONS
COEUR
TISSUS
SAN
G V
EIN
EUX
SAN
G A
RTE
RIE
L
Pas de premier passagehépatique!
distribution
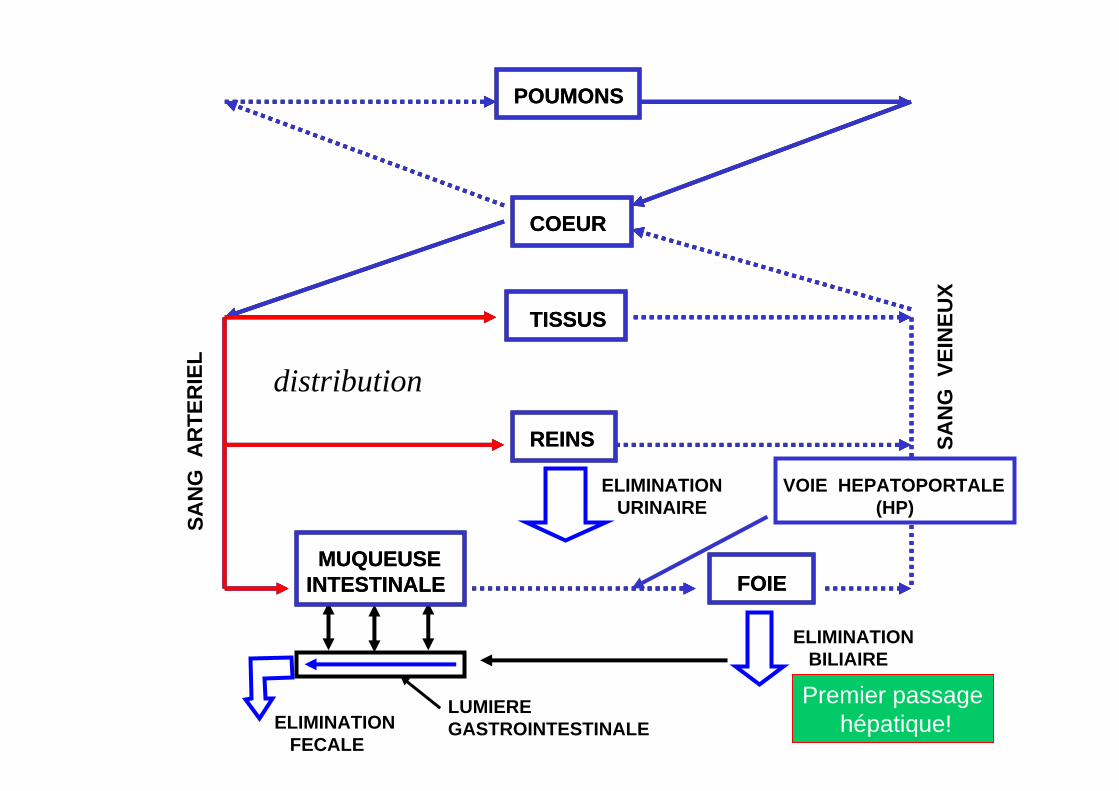
SAN
G V
EIN
EUX
POUMONS
COEUR
TISSUS
REINS
FOIEMUQUEUSE
INTESTINALE
ELIMINATIONURINAIRE
ELIMINATIONBILIAIRE
ELIMINATIONFECALE
LUMIEREGASTROINTESTINALE
FOIE
POUMONS
COEUR
TISSUS
REINS
MUQUEUSEINTESTINALE
VOIE HEPATOPORTALE(HP)
SAN
G A
RTE
RIE
L
Premier passagehépatique!
distribution
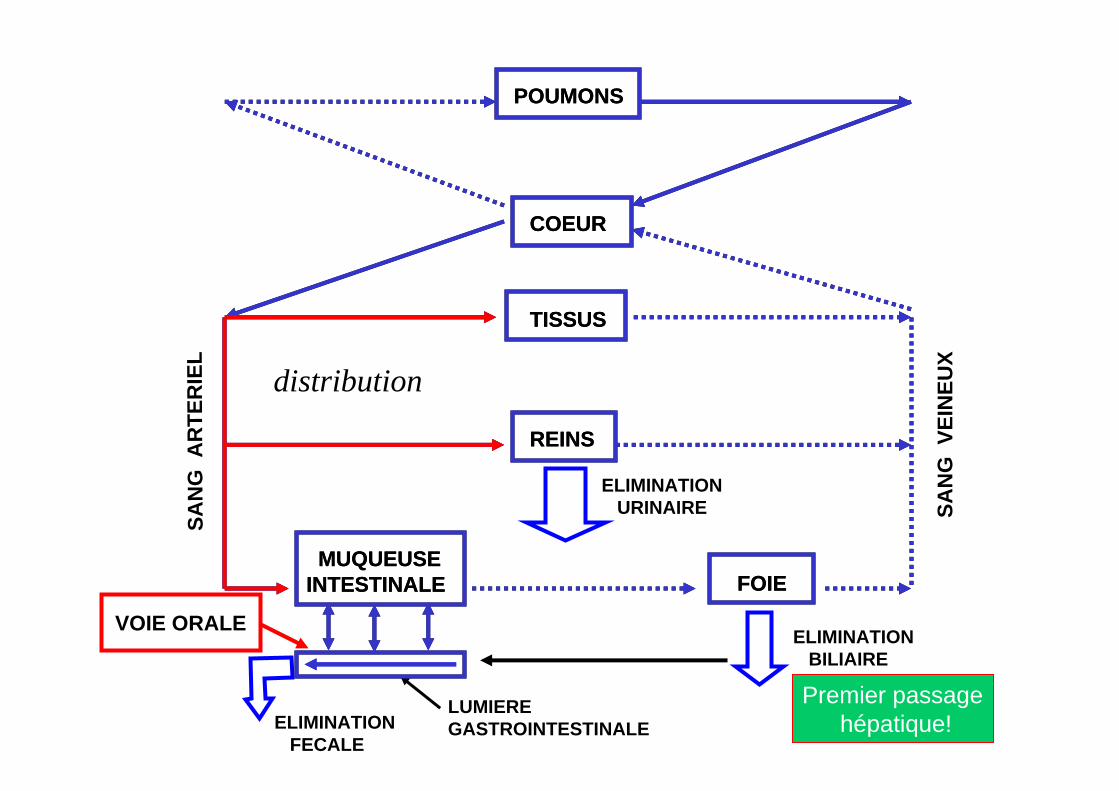
POUMONS
COEUR
TISSUS
REINS
FOIEMUQUEUSE
INTESTINALE
ELIMINATIONURINAIRE
ELIMINATIONBILIAIRE
ELIMINATIONFECALE
LUMIEREGASTROINTESTINALE
POUMONS
COEUR
TISSUS
REINS
FOIEMUQUEUSE
INTESTINALE
VOIE ORALE
SAN
G V
EIN
EUX
SAN
G A
RTE
RIE
L
Premier passagehépatique!
distribution
POUMONS
COEUR
TISSUS
REINS
FOIEMUQUEUSE
INTESTINALE
ELIMINATIONURINAIRE
ELIMINATIONBILIAIRE
ELIMINATIONFECALE
LUMIEREGASTROINTESTINALE
TISSUS
SAN
G A
RTE
RIE
L
SAN
G V
EIN
EUX
POUMONS
COEUR
TISSUS
REINS
FOIEMUQUEUSE
INTESTINALE
VOIE RECTALE
Premier passagehépatique!
distribution
3- CONSEQUENCES CLINIQUES.
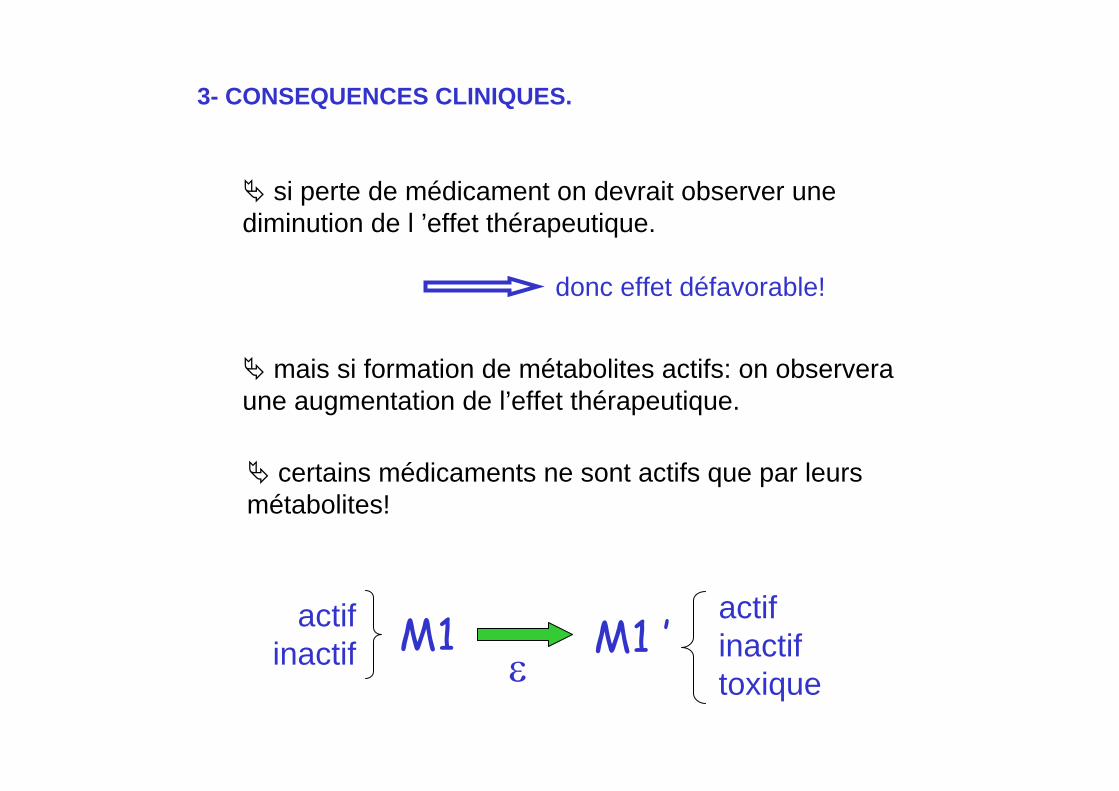
certains médicaments ne sont actifs que par leurs métabolites!
si perte de médicament on devrait observer une diminution de l ’effet thérapeutique.
mais si formation de métabolites actifs: on observeraune augmentation de l’effet thérapeutique.
donc effet défavorable!
M1 M1 ’ε
actifinactiftoxique
actifinactif

3-CONSEQUENCES CLINIQUES
Métabolisme des xénobiotiques: importance des cytochromes P450.
M1 M1 ’ε inactif ?actif?
Quatre isoenzymes (Cyp 1A2, Cyp 2C9, Cyp 2D6, Cyp 3A4) impliquésdans 90% des spécialités!
Risque d’interactions médicamenteuses!
Polymorphisme génétique!
Variabilités inter- et intra-individuelles

3-CONSEQUENCES CLINIQUES
Interactions médicamenteuses possibles!
M1 M1 ’ε inactifactif
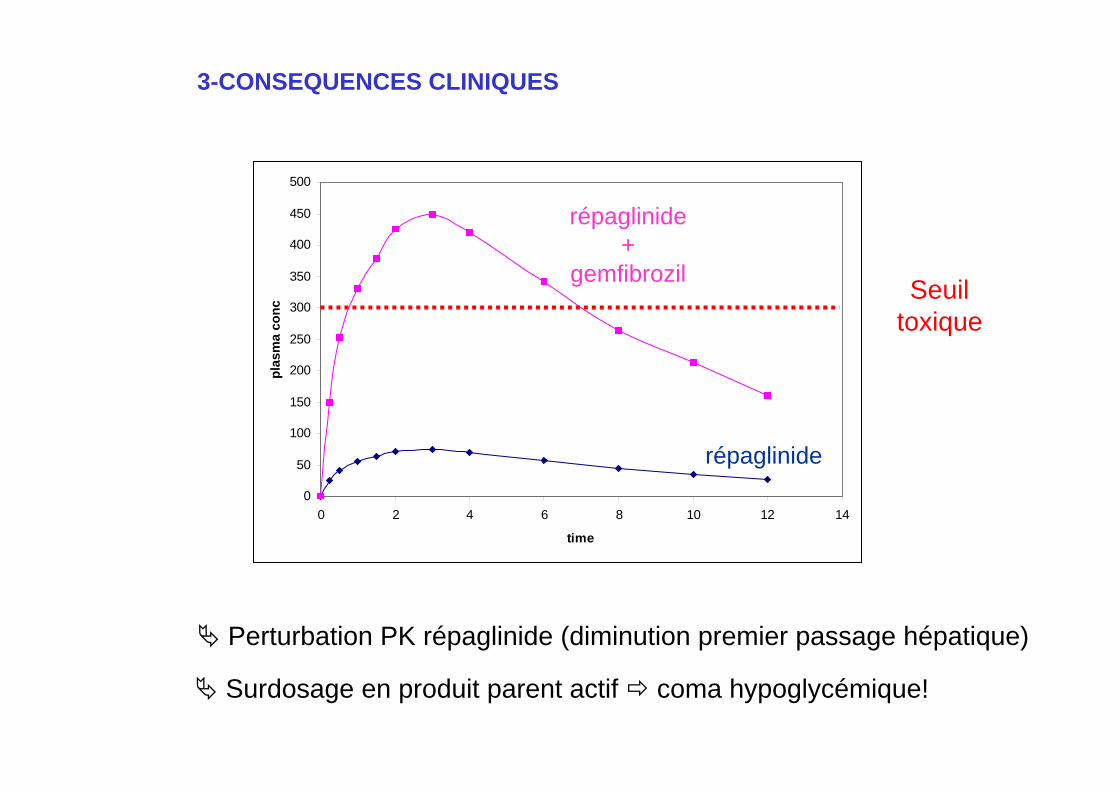
Ex: répaglinide (Novornorm®): hypoglycémiant.
- métabolites inactifs!- métabolisé par le Cyp2C8 hépatique.- 100% d ’élimination sous forme de métabolites.
Répaglinide + gemfibrozil (Lipur®): inhibition du Cyp2C8!
+ + +
- t1/2: 1.3 3.7 h- AUC x8 perturbation paramètres PK!
3-CONSEQUENCES CLINIQUES
Perturbation PK répaglinide (diminution premier passage hépatique)
Surdosage en produit parent actif coma hypoglycémique!
0
50
100
150
200
250
300
350
400
450
500
0 2 4 6 8 10 12 14
time
plas
ma
conc
répaglinide
répaglinide+
gemfibrozil Seuiltoxique
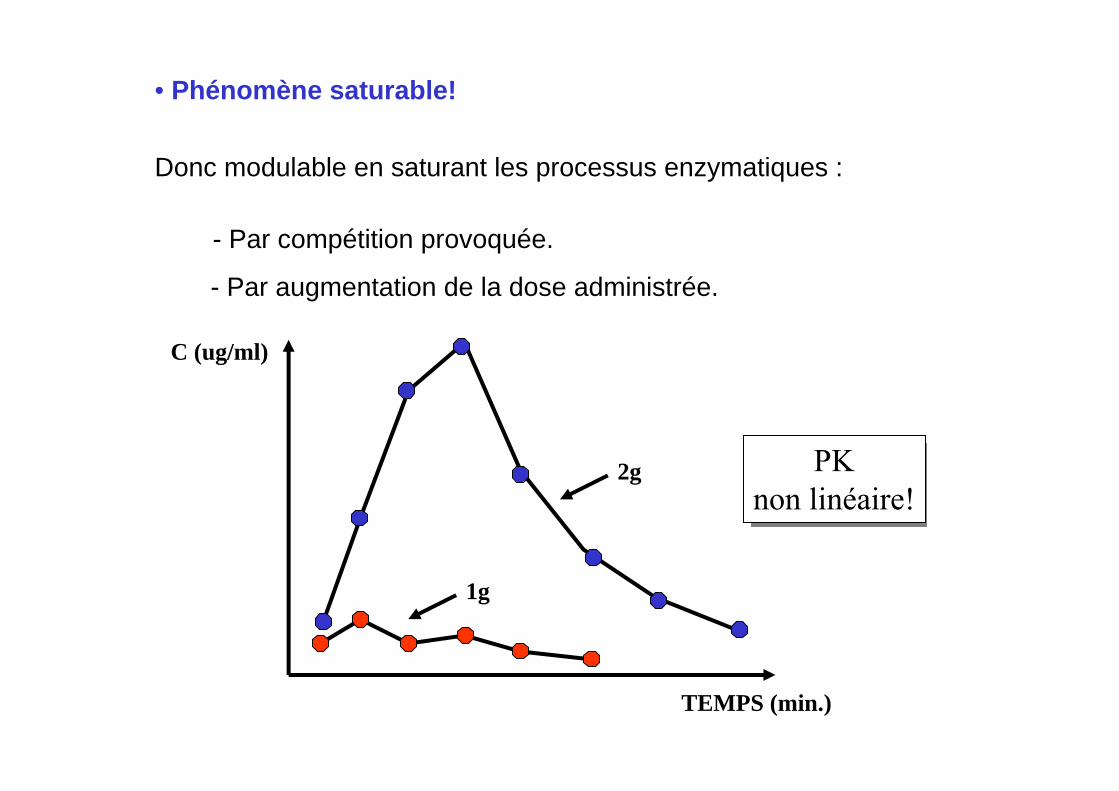
• Phénomène saturable!
Donc modulable en saturant les processus enzymatiques :
- Par compétition provoquée.
- Par augmentation de la dose administrée.
1g
2g
C (ug/ml)
TEMPS (min.)
PKnon linéaire!
PKnon linéaire!

• Modulation effet 1° passage: « Effet Booster »
Principe: associer deux molécules substrats des mêmes équipementsenzymatiques lors du premier passage hépatique.
Une des deux molécules sert de « leurre biochimique ».
La seconde molécule est moins touchée par le métabolisme hépatique!
Ex: Association ritonavir + lopinavir(Kaletra®): « effet booster ».


4- NATURE DE L ’EFFET DE PREMIER PASSAGE
Exemples :
- Nortriptyline (Motival®, ..)- Prostaglandines.
A- AU NIVEAU PULMONAIRE
⌦ Biotransformations limitées :
Oxydation, réduction, désalkylation, hydrolyse, Sulfoconjugaison.

Exemples :
- Aspirine- certaines benzodiazépines (flurazépam)- Morphine- Isoprenaline (Isuprel®)- Pénicillines- …
B- AU NIVEAU INTESTINAL
⌦ Biotransformations abondantes :
Oxydation, réduction (notamment des dérivés nitrés), désalkylation, hydrolyse, glucuro- et sulfo-conjugaison.
Action de la flore et de la muqueuse intestinale.
Equipement enzymatique voisin de celui du foie; Mais activité plus faible!
C- AU NIVEAU HEPATIQUE
Métabolisation intense et abondante
On peut observer une excrétion biliaire de certains métabolites conjugués de taille moléculaire importante.
Très nombreux systèmes enzymatiques.
⌦ Biotransformations de tout type
PHENOMENE DE RECIRCULATION
CYCLE ENTERO-HEPATIQUE
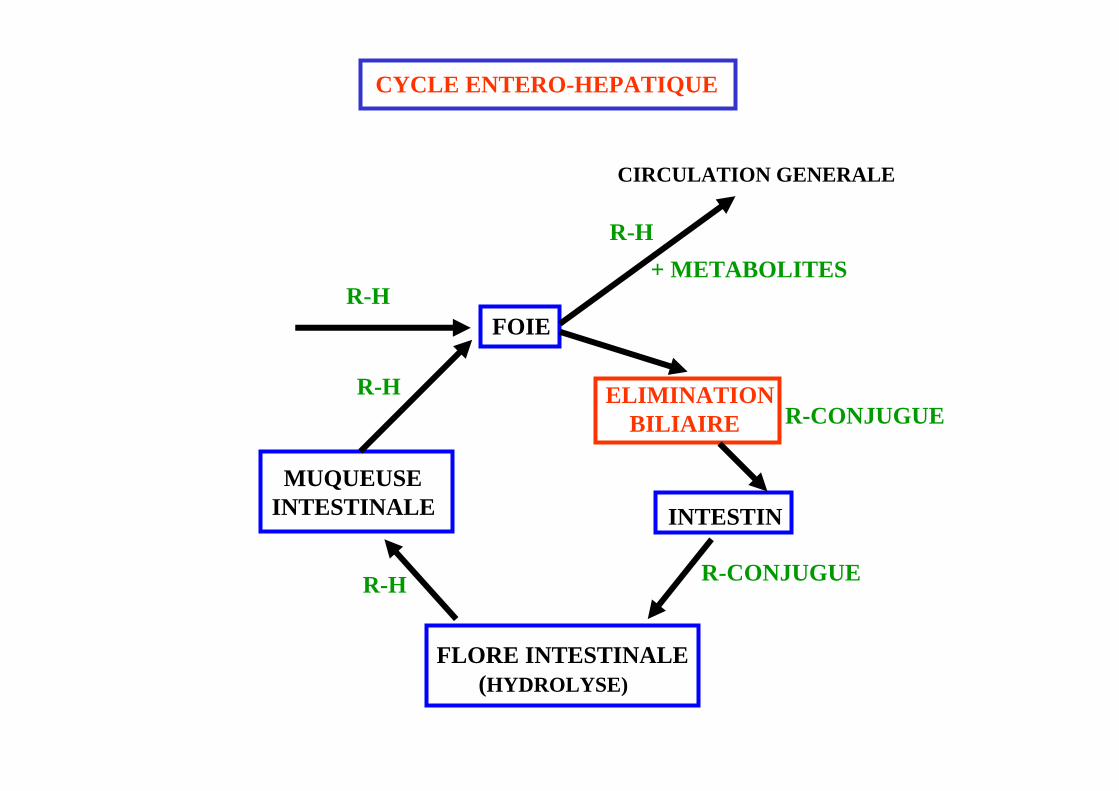
CYCLE ENTERO-HEPATIQUE
INTESTIN
FLORE INTESTINALE(HYDROLYSE)
MUQUEUSEINTESTINALE
FOIER-H
R-H
R-H
R-CONJUGUE
R-CONJUGUEELIMINATION
BILIAIRE
R-H+ METABOLITES
CIRCULATION GENERALE
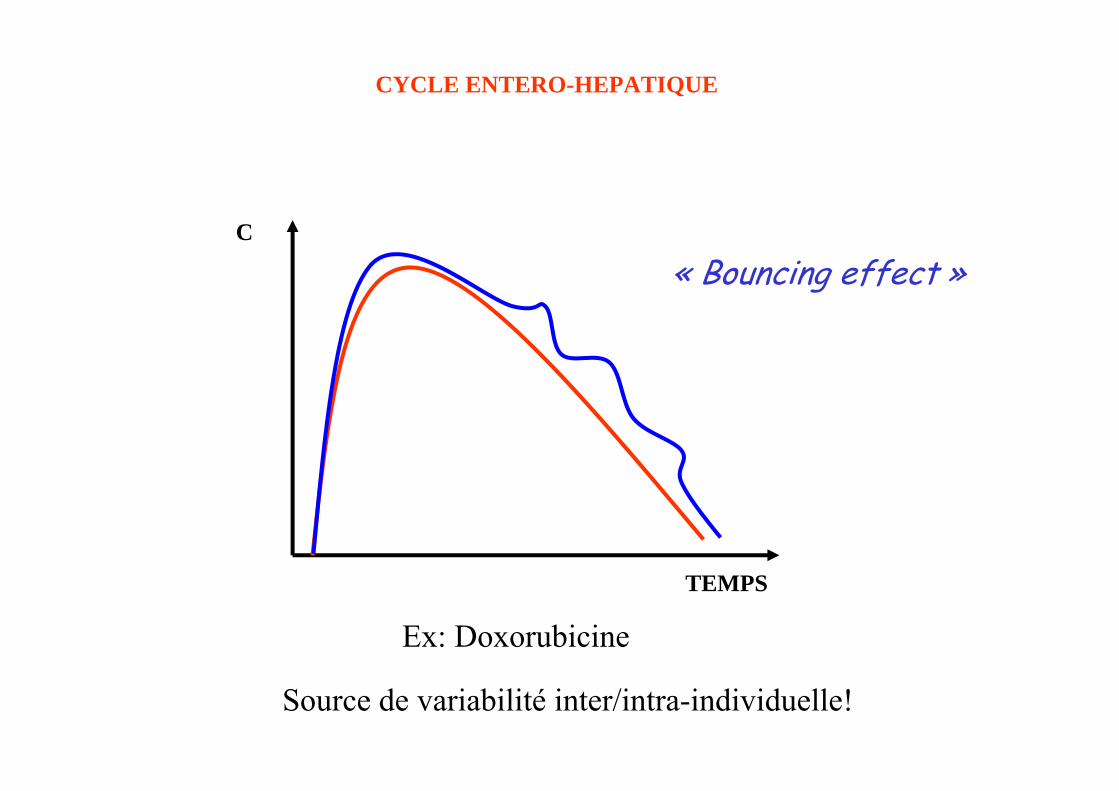
C
TEMPS
« Bouncing effect »
CYCLE ENTERO-HEPATIQUE
Ex: Doxorubicine
Source de variabilité inter/intra-individuelle!

5- EVALUATION DE L ’EFFET DE PREMIER PASSAGE.
⌦ En réalité ce phénomène correspond à une somme d’effets de premier passage :
⌦Conduit à une diminution des taux circulants d’un médicament.
⌦ Donc diminution de la surface sous la courbe (ssc) des concentrations plasmatiques ou sanguines.
PIH EEEE ++=

A) EVALUATION CHEZ L ’ANIMAL
Exemple : calcul de l ’effet de 1er passage hépatique
HP : passage par le foie et les poumons
fH: fraction qui échappe au métabolisme hépatique
HH f1E −=IV
HPH
SSCSSCf =
IV : passage par les poumons

B) CHEZ L ’HOMME
IV : VOIE DE REFERENCE!
- néglige la possibilité d ’une résorption partielle.
- fait abstraction d ’un éventuel effet de 1er passagepulmonaire.
IV
ORALE
SSCSSC1E −=

EXEMPLES :
1- Pour un médicament fp = 0,93 et fh = 0,81: déterminer la part de l ’élimination hépatique et pulmonaire.
2- Le métabolisme d’un médicament après administrationI.V. Représente 24 % de la dose. Si Ep = 0,14 quelle est la part du métabolisme hépatique ?
METABOLISME PULMONAIRE : 14 % DE LA DOSE
METABOLISME HEPATIQUE : 24 - 14 = 10 % DE LA DOSE
d’où
(7%)0,070,931Ep =−=
(19%)0,190,811EH =−=

6- FACTEURS MODIFIANT L ’EFFET DE 1er PASSAGE
Tous les facteurs qui peuvent modifier le bagage enzymatique:
- au niveau pulmonaire
- au niveau intestinal
- au niveau hépatique
⌦ Pathologies⌦ Interactions médicamenteuses⌦ Alimentation (modification débits splanchniques)⌦ Facteurs génétiques (polymorphismes)⌦ Age, grossesse…..

6- FACTEURS MODIFIANT L ’EFFET DE 1er PASSAGE
⌦ Pathologies: anastomose porto-cave (foie cirrhotique)
Veine porte
Veine cave

6- FACTEURS MODIFIANT L ’EFFET DE 1er PASSAGE
⌦ Polymorphisme génétique
Tamoxifène(inactif)
N-demethyl-tamoxifène(actif)
cyp2D6
• Hormonothérapie - cancers du sein: Tamoxifène (Novaldex®)
• Polymorphisme génétique: >10% population mutée sur Cyp2D6!
• Altération effet premier passage, défaut d’activation!
Echappement
Thérapeutique

6- FACTEURS MODIFIANT L ’EFFET DE 1er PASSAGE
⌦ Interactions médicamenteuses.
Tamoxifène(inactif)
N-demethyl-tamoxifène(actif)
cyp2D6
• Hormonothérapie - cancers du sein: Tamoxifène (Novaldex®)
• Altération de l ’effet de premier passage hépatique, défaut d’activation!
• Effets II: bouffées de chaleur.
• Prescription de venlafaxine (Effexor®)
Echappement
Thérapeutique
….. inhibiteur du Cyp2D6!

1- BIODISPONIBILITE ABSOLUE.
A- DEFINITION
= fraction ou % de médicament qui, après administration,orale atteint la circulation générale.
= quantité relative, de médicament, absorbée à partir d ’une forme orale et vitesse à laquelle se produit cephénomène.
BIODISPONIBILITE

B- INTERET DES ETUDES DE BIODISPONIBILITE ?
- nouveau médicament.
- nouveau dosage de la forme utilisée.
- nouvelle voie d’administration.
- nouvelle forme galénique.
OBLIGATION REGLEMENTAIRE!OBLIGATION REGLEMENTAIRE!OBLIGATION REGLEMENTAIRE!
- génériques & biosimilaires.
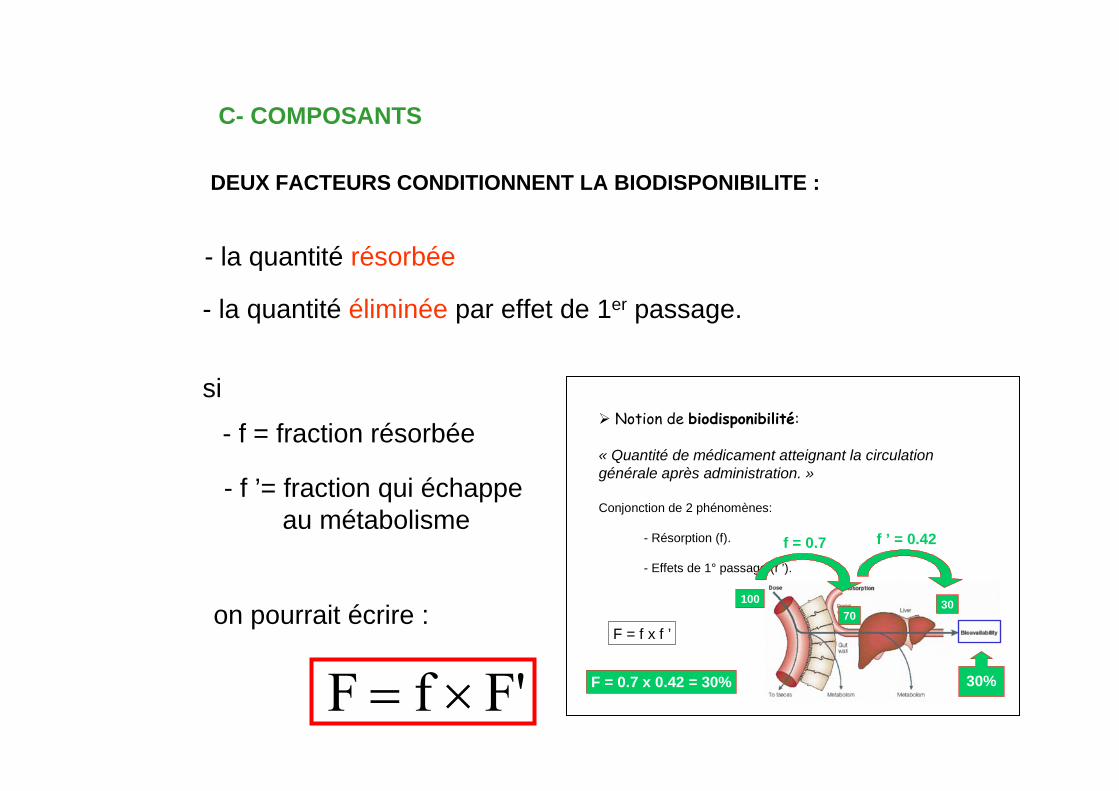
C- COMPOSANTS
DEUX FACTEURS CONDITIONNENT LA BIODISPONIBILITE :
- la quantité résorbée
- la quantité éliminée par effet de 1er passage.
si
- f = fraction résorbée
- f ’= fraction qui échappe au métabolisme
on pourrait écrire :
F'fF ×=
Notion de biodisponibilité:
« Quantité de médicament atteignant la circulation générale après administration. »
Conjonction de 2 phénomènes:
- Résorption (f).
- Effets de 1° passage (f ’).
F = f x f ’
10070
30
30%
f = 0.7 f ’ = 0.42
F = 0.7 x 0.42 = 30%

D- CALCUL
- A PARTIR DU PLASMA OU DU SANG:
Ae = quantité totale excrétée
- A PARTIR DES URINES
si doses Orale et IV différentes :
ivouiaSSCoraleSSCF =
oraleDoseivouiaDose
ivouiaSSCoraleSSCF ×=
oraleDoseivouiaDose
ivouiaAeoraleAeF ×=

E- LIMITES D’UTILISATION
- problème lié au métabolisme
si le médicament donne des métabolites actifs labiodisponibilité calculée à partir du produit parentsous-estime l ’effet thérapeutique.
donc:
obligation de bien connaître l ’activité des métaboliteset de les quantifier!
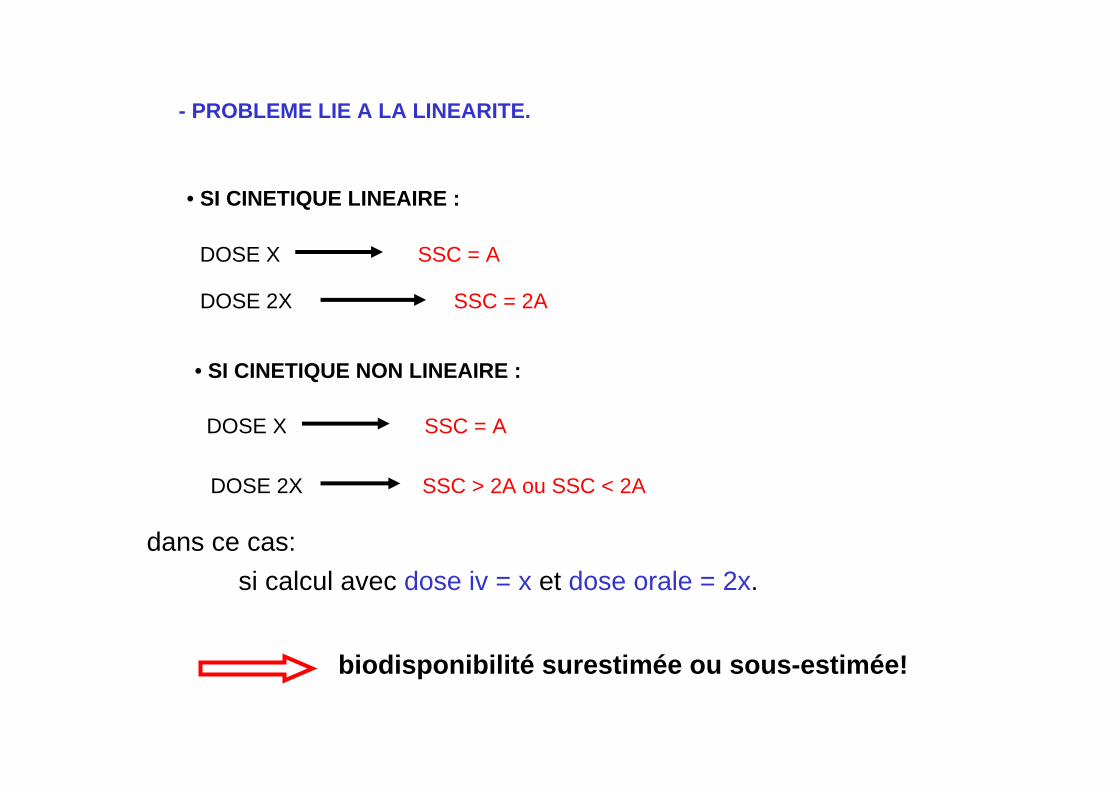
- PROBLEME LIE A LA LINEARITE.
• SI CINETIQUE LINEAIRE :
si calcul avec dose iv = x et dose orale = 2x.
DOSE X SSC = A
DOSE 2X SSC = 2A
• SI CINETIQUE NON LINEAIRE :
DOSE X SSC = A
DOSE 2X SSC > 2A ou SSC < 2A
dans ce cas:
biodisponibilité surestimée ou sous-estimée!
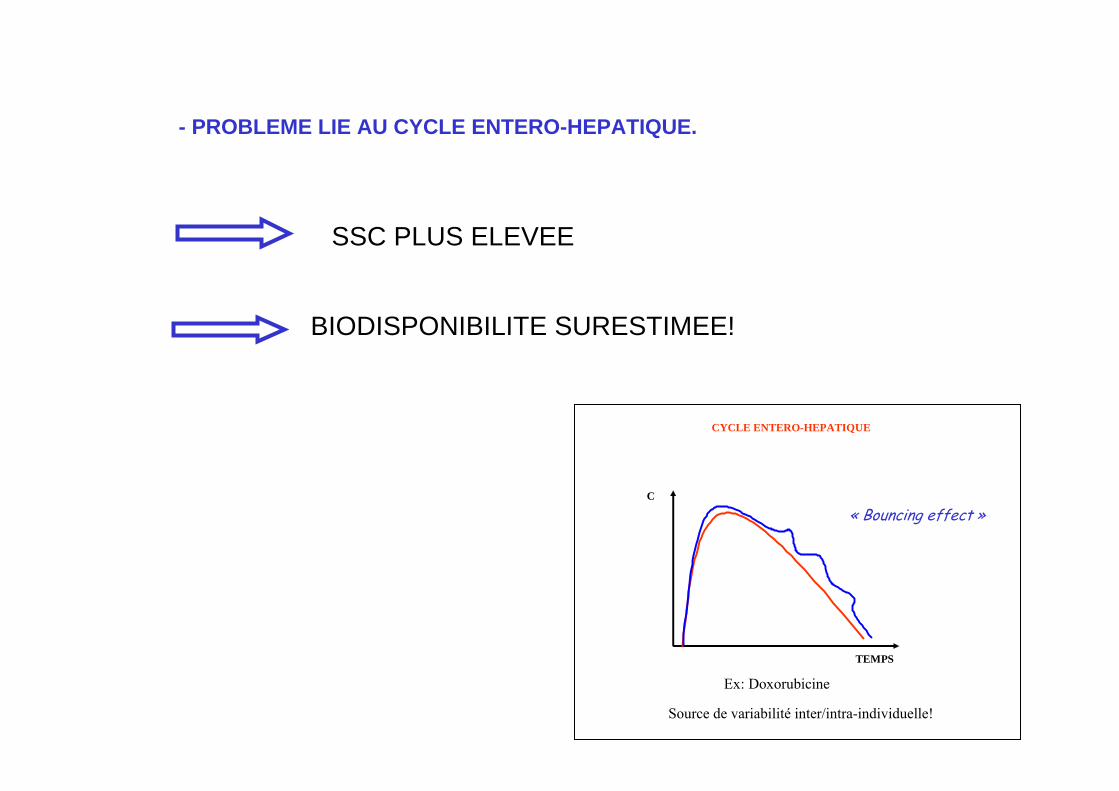
- PROBLEME LIE AU CYCLE ENTERO-HEPATIQUE.
SSC PLUS ELEVEE
BIODISPONIBILITE SURESTIMEE!
C
TEMPS
« Bouncing effect »
CYCLE ENTERO-HEPATIQUE
Ex: Doxorubicine
Source de variabilité inter/intra-individuelle!

2- BIODISPONIBILITE RELATIVE.
A- DEFINITION
« = % de médicament qui, après administration, atteint la circulation générale lors d’une étude comparative entre une nouvelle forme galénique et une forme de référence »

B- NOTION DE BIOEQUIVALENCE
FORMES BIOEQUIVALENTES :
⌦si avec la même posologie chez le même individuon obtient des effets thérapeutiques comparables avec :
- des SSC identiques - mêmes Cmax
- mêmes Tmax
C m a xC m a x
T m a xT m a x
A U CA U C t1 /2t 1 /2
princeps
C m a xC m a x
T m a xT m a x
A U CA U C t1 /2t 1 /2
générique
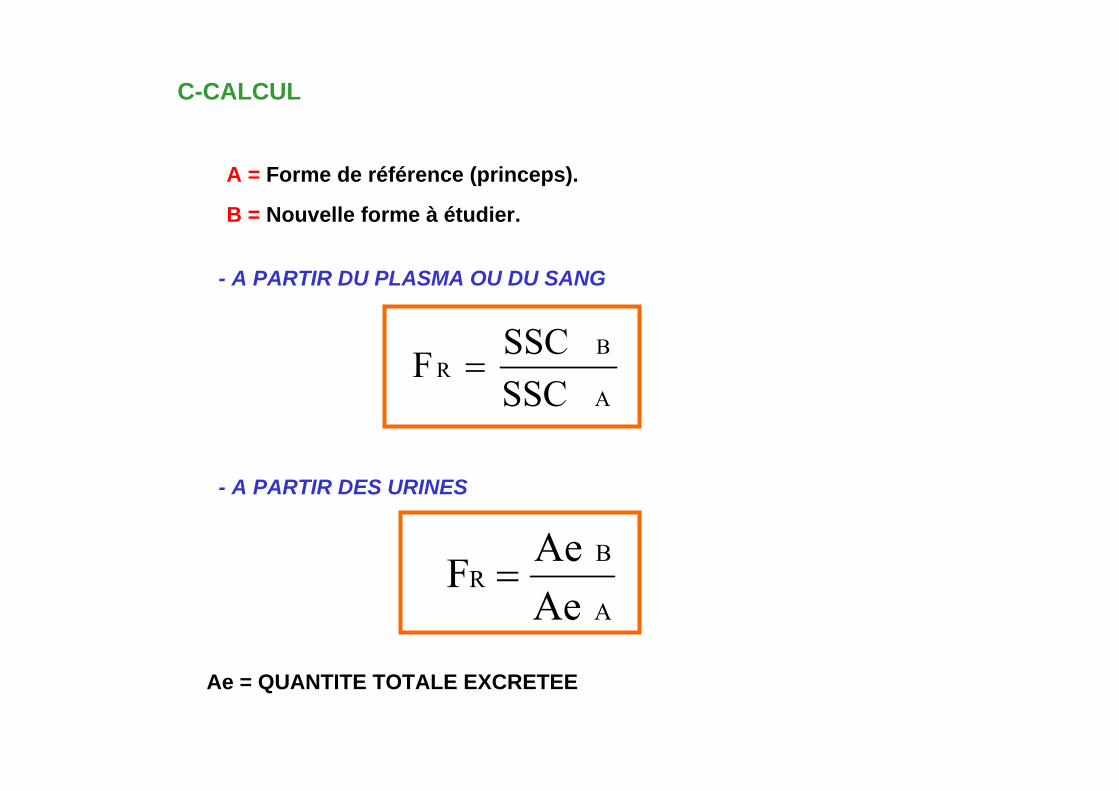
C-CALCUL
A = Forme de référence (princeps).
- A PARTIR DU PLASMA OU DU SANG
Ae = QUANTITE TOTALE EXCRETEE
B = Nouvelle forme à étudier.
A
BR
SSCSSCF =
- A PARTIR DES URINES
A
BR
AeAeF =
ABSORPTION
CONCLUSION
ABSORPTION
CONCLUSION
⌦ Résorption + premiers passages.
⌦ Influencée par de très nombreux facteurs.
⌦ Source de variabilité inter et intra-individuelle!





























