Embed Size (px)
Citation preview

Activins A and B Regulate Fate-Determining GeneExpression in Islet Cell Lines and Islet Cells From MaleMice
Danielle Andrzejewski, Melissa L. Brown, Nathan Ungerleider, Amy Burnside,and Alan L. Schneyer
Departments of Veterinary and Animal Science (D.A., A.B., A.L.S.) and Nutrition (M.L.B.), and Molecularand Cellular Biology Graduate Program (N.U.), University of Massachusetts-Amherst, Amherst,Massachusetts 01003
TGF� superfamily ligands, receptors, and second messengers, including activins A and B, have beenidentified in pancreatic islets and proposed to have important roles regulating development,proliferation, and function. We previously demonstrated that Fstl3 (an antagonist of activin ac-tivity) null mice have larger islets with �-cell hyperplasia and improved glucose tolerance andinsulin sensitivity in the absence of altered �-cell proliferation. This suggested the hypothesis thatincreased activin signaling influences �-cell expansion by destabilizing the �-cell phenotype andpromoting transdifferentiation to �-cells. We tested the first part of this hypothesis by treating �-and �-cell lines and sorted mouse islet cells with activin and related ligands. Treatment of the�TC1-6 � cell line with activins A or B suppressed critical �-cell gene expression, including Arx,glucagon, and MafB while also enhancing �-cell gene expression. In INS-1E �-cells, activin A treat-ment induced a significant increase in Pax4 (a fate determining �-cell gene) and insulin expression.In sorted primary islet cells, �-cell gene expression was again suppressed by activin treatment in�-cells, whereas Pax4 was enhanced in �-cells. Activin treatment in both cell lines and primary cellsresulted in phosphorylated mothers against decapentaplegic-2 phosphorylation. Finally, treat-ment of �TC1-6 cells with activins A or B significantly inhibited proliferation. These results supportthe hypothesis that activin signaling destabilized the �-cell phenotype while promoting a �-cellfate. Moreover, these results support a model in which the �-cell expansion observed in Fstl3 nullmice may be due, at least in part, to enhanced �- to �-cell transdifferentiation. (Endocrinology 156:2440–2450, 2015)
Diabetes, a disease affecting nearly 10% of Americans,results from loss of functional insulin-producing
�-cells, a condition that results in elevated serum glucoseand eventually life-threatening morbidities. Increasedfunctional �-cell mass usually compensates for enhancedinsulin demand, suggesting that local and/or systemic fac-tors can influence �-cell survival, proliferation, neogene-sis, and/or insulin secretion (1). Identification and func-tional characterization of these factors may lead to targetsfor developing novel diabetes therapies.
The list of candidate regulatory factors includes hor-mones such as insulin, IGF-1, glucagon-like peptide 1, and
estrogen as well as nutrients and metabolic factors such asglucose and cAMP (2, 3). In addition, numerous membersof the TGF� superfamily of growth factors have been at-tributed a variety of roles in islet biology including devel-opment of the pancreas and islets (4), regulation of �-cellfunction including insulin production and secretion (5, 6),and modulating �-cell proliferation (7). TGF� superfam-ily ligands typically bind specific type II receptors, whichcomplex with and activate specific type 1 receptors. Ac-tivated type 1 receptors then phosphorylate subsets ofmothers against decapentaplegic (Smad) second messen-
ISSN Print 0013-7227 ISSN Online 1945-7170Printed in USACopyright © 2015 by the Endocrine SocietyReceived February 18, 2015. Accepted May 6, 2015.First Published Online May 11, 2015
Abbreviations: BMP, bone morphogenetic protein; FSTL3, follistatin like-3; MSTN, myo-statin; p, phosphorylated; PBST, PBS with Tween 20; PC, prohormone convertase; PDX-1,pancreatic duodenal homeobox-1; qPCR, quantitative PCR; Smad, phosphorylated moth-ers against decapentaplegic; �TC1-6, �TC1 clone 6 cells.
O R I G I N A L R E S E A R C H
2440 press.endocrine.org/journal/endo Endocrinology, July 2015, 156(7):2440–2450 doi: 10.1210/en.2015-1167
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.

gers, including Smad2 and Smad3 for activins, TGF�,myostatin (MSTN), and growth and differentiation factor11 and Smad1, Smad5, and Smad8/9 for bone morpho-genetic proteins (BMPs) and related ligands (8–10), al-though exceptions to this canonical signaling pathwayhave been described. Inhibition of activin/TGF� signalingusing dominant-negative activin type II receptors resultedin �-cell hypoplasia and hyperglycemia, suggesting thatthis pathway may regulate �-cell proliferation or survival(11). Inhibition of this pathway in adult islets by inducibleoverexpression of the Smad2/3 antagonist Smad7 also re-sulted in �-cell hypoplasia and diabetes, which was reversedby the removal of the Smad7 inducer (12).
On the other hand, it was recently demonstrated thatincreased Smad7 expression reduced Smad2/3 signalingbut enhanced �-cell proliferation (7), suggesting that ac-tivin and TGF� inhibit �-cell proliferation in some cir-cumstances. Activin was also found to regulate �-cell ma-turity through the decreased expression of insulin, MafA,and Glut2 (also called Slc2a2) mRNA (6). In addition,activins A and B were recently suggested to have opposingeffects in insulin secretion by selectively using Smad2 (ac-tivin A) or Smad3 (activin B) second messengers, whereasinactivation of Inhbb (activin B subunit) enhanced insulinrelease (13), although this finding has not been universallyobserved (14). TGF�, acting through Smad3, inhibitedinsulin biosynthesis and glucose-stimulated insulin secre-tion in rat islets, whereas the suppression of Smad3 ex-pression increased �-cell function in mice (15). Loss ofBMP4 signaling suppressed �-cell function in mice,whereas the injection of BMP4 enhanced insulin secretion(16). Taken together, these studies demonstrate importantintraislet communication roles for numerous TGF� su-perfamily ligands that are important for maintaining glu-cose homeostasis in adults.
Activin bioactivity is antagonized by follistatin and fol-listatin like-3 (FSTL3) (17, 18). We previously demon-strated that Fstl3-null mice have 2-fold larger islets with�-cell hyperplasia in the context of improved glucose tol-erance and insulin sensitivity (19). Importantly, �-cell pro-liferation was not different between WT and Fstl3 knock-out littermates (20), suggesting that the additional �-cellsin Fstl3-null mice might be derived from alternative pro-cesses. The transcription factors Arx and Pax4 are re-quired to specify fates for �- and �-cells, respectively (21).Misexpression of Arx in �-cells or Pax4 in �-cells resultedin the transdifferentiation to the opposite cell type (22,23), whereas merely reducing Arx expression in �-cellscaused them to transdifferentiate into �-cells (24), dem-onstrating a previously underappreciated plasticity in isletcell type fate. Taken together with our demonstration thatloss of Fstl3 leads to increased activin action in Fstl3-null
mice (25), these transdifferentiation studies suggest amodel whereby �-cell expansion in Fstl3 null mice resultsfrom increased activin signaling within islets that influ-ences islet cell fate to favor �- to �-cell transdifferentiation.One hypothesis derived from this model is that activintreatment of �-cells will destabilize or suppress the �-cellphenotype and promote a �-cell phenotype.
To test this hypothesis, we analyzed gene expression in�- and �-cells in response to treatment with activins A orB. We found that activin suppressed expression of impor-tant �-cell genes, including Arx, and promoted the expres-sion of �-cell genes in the �TC1-6 �-cell line and enhancedthe expression of �-cell genes, including Pax4, in theINS-1E �-cell line. Activin similarly suppressed �-cell geneexpression in sorted mouse �-cells. In both cell lines andprimary cells, activins A and B signaled through phos-phorylated (p) Smad2, whereas pSmad3 was undetect-able. These results indicate that activin signaling inducedalterations in gene expression that would favor �- to �-celltransdifferentiation and thus support the hypothesis that�- to �-cell transdifferentiation is involved in the �-cellhyperplasia identified in the Fstl3-null mice.
Materials and Methods
Tissue culture�TC1 clone 6 cells (�TC1-6; CRL-2934), a mouse line se-
lected for lack of insulin production (26), and HEP3B cells (HB-8064), a human liver line, were obtained from American TypeCulture Collection. INS-1E cells (C0018009), a rat pancreatic�-cell line, were obtained from AddexBio. �TC1-6 cells weregrown in low-glucose DMEM containing L-glutamine and 110mg/L sodium pyruvate (Life Technologies) and were supple-mented with 15 mM HEPES, 0.1 nM nonessential amino acids,1.5 g/L sodium bicarbonate, 2 g/L D-glucose, 10% heat inacti-vated fetal bovine serum, and 1% penicillin-streptomycin.HEP3B cells, which were used as controls for Smad activity, weregrown in high-glucose DMEM containing L-glutamine and 110mg/L sodium pyruvate (Life Technologies) and were supple-mented with 10% heat-inactivated fetal bovine serum and 1%penicillin-streptomycin. INS-1E cells were grown in RPMI 1640medium (Sigma) containing L-glutamine and sodium bicarbon-ate supplemented with 10 mM HEPES, 50 �M �-mercaptoetha-nol, 10% heat-inactivated fetal bovine serum, and 1% penicillin-streptomycin. All cell lines were incubated in 5% CO2-95% airat 37°C and passaged at 70% confluence via trypsinization with0.05% trypsin and 0.53 mM EDTA.
Islet isolationAnimals were used in accordance with all regulations and
guidelines, and research protocols were approved by the BaystateMedical Center’s Institutional Animal Care and Use Committee.Islets were isolated from 10- to 12-week-old C57BL6 male miceas previously described (27). After isolation, islets were culturedfor 24 hours in RPMI 1640 (11 mM glucose; Mediatech) sup-
doi: 10.1210/en.2015-1167 press.endocrine.org/journal/endo 2441
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.

plemented with 10% heat inactivated fetal bovine serum and 1%penicillin-streptomycin and then cultured with or without 3 nMactivin A for 60 minutes, after which proteins were extracted asdescribed below for cell lines. Additional islets were used for cellsorting as described below.
Cell sortingEnriched populations of �- and �-cells were obtained using
the method of Kohler et al (28) adapted for mouse islets. Briefly,isolated mouse islets were dispersed into single cells using tryp-sin-EDTA solution (450 mL, 37°C, 7–9 min), washed, and thenplaced in a low-glucose sorting buffer (Hanks’ balanced salt so-lution plus 2.5 mM glucose; 1% BSA; 100 U/mL penicillin G; 100�g/mL streptomycin-sulfate; and 25 mM HEPES, pH 7.4) andfiltered through a 0.2-�m mesh screen. The dispersed cells werethen sorted into primary �-cells and �-cells by fluorescence-ac-tivated cell sorter (BD FACSAria; BD Biosciences) using auto-fluorescence induced by low-glucose solution and gating strat-egies as previously described (28). Cells were collected in �TC1-6culture medium and placed into culture with �-cells cultured in�TC1-6 medium and �-cells cultured in islet medium. Some cellswere fixed after 48 hours for immunocytochemistry, and otherswere cultured in the presence or absence of activin A or B (3 nM)for 24, 48, or 96 hours, after which the RNA was extracted andanalyzed as described for the cell lines.
Gene expression�TC1-6 and/or INS-1E cells were plated on 24-well plates
with 100 000 cells/well and treated 72 hours later with 3 nMactivin A, 3 nM activin B, 3 nM BMP7, 3 nM TGF�1, or 3 nMMSTN. RNA was extracted after 24, 48, and 96 hours (�TC) or24 hours (INS-1E) using the QIAGEN RNEasy microkit andreverse transcribed using SuperScript III reverse transcriptase(Invitrogen). Specific gene expression was determined by SYBRquantitative PCR (qPCR) using the QuantiTect SYBR GreenPCR kit (QIAGEN), except for Pax4, which was assessed usingQuantitect Taqman PCR kit and self-designed probe/primers.All PCRs were performed using an MX3005 qPCR machine(Agilent Technologies), and primers are listed in SupplementalTable 1. A standard was created by reverse transcribing multiplealiquots of RNA pooled from islets and cell lines for mouse (�TCcells and islet cells) or rat (INS-1E) cells and run at three dilutionsin each PCR for each target. The result for each sample wasinterpolated from the standard curve and then normalized to theRpl19 housekeeping gene, also interpolated off the standardcurve. Because all PCR targets are expressed relative to the samestandard, results for each target are comparable for a givenspecies.
ImmunocytochemistrySorted �- and �-cells were cultured for 48 hours, after which
they were fixed in 4% glutaraldehyde, washed in PBS, treatedwith 0.2% Triton X-100 for 20 minutes, washed in PBS con-taining 0.5% donkey serum, blocked in PBS containing 5% don-key serum, incubated with antiinsulin (number A0564; Dako)and antiglucagon (number G2654; Sigma) primary antibodies(both at 1:150) overnight at 4ºC, washed with PBS containing0.5% donkey serum, and incubated with Cy3 donkey antiguineapig (number 706-165-148; Jackson ImmunoResearch) and goatantimouse Alexa Fluor 647 (number A21236; Life Technolo-
gies), both at 1:150, for 1 hour at room temperature. The cellswere then washed in PBS and photographed.
Western blottingCells were plated on six-well plates until 90% confluent and
then treated with 1 nM of activin A, activin B, TGF�1, BMP4, orMSTN for 45–60 minutes (or 24 h activin A treatment for pan-creatic duodenal homeobox-1 (PDX-1) and Aristaless relatedhomeobox [ARX]). Cells were washed with cold PBS on ice andthen lysed with radioimmunoprecipitation assay protein extrac-tion buffer containing 0.1% sodium dodecyl sulfate, 0.5% so-dium deoxycholate, and 1% Triton X-100 in PBS with protease(number 11836170001; Roche), and phosphatase (numberP5726; Sigma) inhibitors.
Collected protein was centrifuged for 15 minutes at 12 000rpm and the supernatant collected. Protein was quantified usingthe BCA protein assay kit (Thermo Scientific). Proteins (17–30�g of cell line and 20–30 �g of mouse islet) were electrophoresedusing 10% miniprotean TGX gels and then transferred to aPVDF membrane using the Mini Trans-Blot Cell box (all SDS-PAGE and transfer materials are from Bio-Rad Laboratories).The blot was blocked in 3% milk (or 3% BSA in the case ofpSmad1/5/9) in PBS with 0.05% Tween 20 (PBST) for 1 hour andthen incubated with the primary antibody (pSmad2 and pSmad3together, pSmad1/5/9, ARX, or PDX-1; see Table 1 for dilutions)in PBST/1% milk (2% BSA for pSmad1/5/9). After 24 hoursrotating at 4º, the blot was put into a 1:5000 donkey antirabbit(number 711-035-152; Jackson ImmunoResearch) or antimouse(number 715-035-150; Jackson ImmunoResearch) antibody in1% milk in PBST for 1 hour at room temperature. Blots werewashed and the image was acquired using a G:BOX system (Syn-geneD). After exposure of primary proteins, blots were strippedusing a pH 2.2 buffer containing 0.2% sodium dodecyl sulfate,1.5% glycine, 1% Tween 20, and 0.05% dithiothreitol in waterreblocked in 3% milk, and put into a total Smad2/3, total Smad1,or �-actin (for ARX and PDX-1). Protein quantification wasdone using ImageJ (National Institutes of Health), and the blotimages were subject to background subtraction prior toquantification.
Cell proliferation assayCells were plated on 96-well plates (20 000–40 000 cells/
well) and treated with 4 nM activin A, 4 nM activin B, or 5.7 �Minsulin. The cells were analyzed daily using the CellTiter 96AQueous nonradioactive cell proliferation assay (Promega) ac-cording to the manufacturer’s recommendations.
Statistical analysisExperiments with two groups were compared using t tests.
When more than two treatment groups were analyzed, meanswere analyzed by ANOVA with a Bonferroni post hoc test forsignificance.
Results
Activin regulates gene expression in an �-cell lineWe first used the �TC1-6 cell line that was subcloned
from a mouse glucagonoma tumor cell line [�TC1(26)]
2442 Andrzejewski et al Activin Regulates Islet Cell Gene Expression Endocrinology, July 2015, 156(7):2440–2450
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.
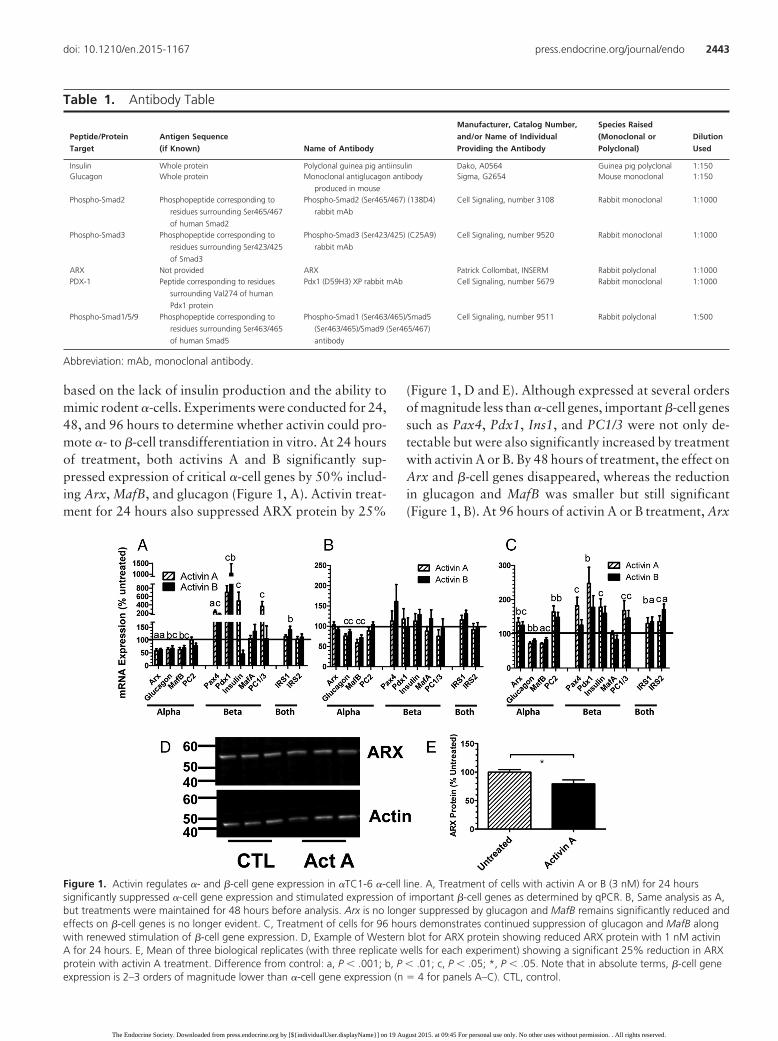
based on the lack of insulin production and the ability tomimic rodent �-cells. Experiments were conducted for 24,48, and 96 hours to determine whether activin could pro-mote �- to �-cell transdifferentiation in vitro. At 24 hoursof treatment, both activins A and B significantly sup-pressed expression of critical �-cell genes by 50% includ-ing Arx, MafB, and glucagon (Figure 1, A). Activin treat-ment for 24 hours also suppressed ARX protein by 25%
(Figure 1, D and E). Although expressed at several ordersof magnitude less than �-cell genes, important �-cell genessuch as Pax4, Pdx1, Ins1, and PC1/3 were not only de-tectable but were also significantly increased by treatmentwith activin A or B. By 48 hours of treatment, the effect onArx and �-cell genes disappeared, whereas the reductionin glucagon and MafB was smaller but still significant(Figure 1, B). At 96 hours of activin A or B treatment, Arx
Table 1. Antibody Table
Peptide/Protein
Target
Antigen Sequence
(if Known) Name of Antibody
Manufacturer, Catalog Number,
and/or Name of Individual
Providing the Antibody
Species Raised
(Monoclonal or
Polyclonal)
Dilution
Used
Insulin Whole protein Polyclonal guinea pig antiinsulin Dako, A0564 Guinea pig polyclonal 1:150Glucagon Whole protein Monoclonal antiglucagon antibody
produced in mouse
Sigma, G2654 Mouse monoclonal 1:150
Phospho-Smad2 Phosphopeptide corresponding to
residues surrounding Ser465/467
of human Smad2
Phospho-Smad2 (Ser465/467) (138D4)
rabbit mAb
Cell Signaling, number 3108 Rabbit monoclonal 1:1000
Phospho-Smad3 Phosphopeptide corresponding to
residues surrounding Ser423/425
of Smad3
Phospho-Smad3 (Ser423/425) (C25A9)
rabbit mAb
Cell Signaling, number 9520 Rabbit monoclonal 1:1000
ARX Not provided ARX Patrick Collombat, INSERM Rabbit polyclonal 1:1000PDX-1 Peptide corresponding to residues
surrounding Val274 of human
Pdx1 protein
Pdx1 (D59H3) XP rabbit mAb Cell Signaling, number 5679 Rabbit monoclonal 1:1000
Phospho-Smad1/5/9 Phosphopeptide corresponding to
residues surrounding Ser463/465
of human Smad5
Phospho-Smad1 (Ser463/465)/Smad5
(Ser463/465)/Smad9 (Ser465/467)
antibody
Cell Signaling, number 9511 Rabbit polyclonal 1:500
Abbreviation: mAb, monoclonal antibody.
Figure 1. Activin regulates �- and �-cell gene expression in �TC1-6 �-cell line. A, Treatment of cells with activin A or B (3 nM) for 24 hourssignificantly suppressed �-cell gene expression and stimulated expression of important �-cell genes as determined by qPCR. B, Same analysis as A,but treatments were maintained for 48 hours before analysis. Arx is no longer suppressed by glucagon and MafB remains significantly reduced andeffects on �-cell genes is no longer evident. C, Treatment of cells for 96 hours demonstrates continued suppression of glucagon and MafB alongwith renewed stimulation of �-cell gene expression. D, Example of Western blot for ARX protein showing reduced ARX protein with 1 nM activinA for 24 hours. E, Mean of three biological replicates (with three replicate wells for each experiment) showing a significant 25% reduction in ARXprotein with activin A treatment. Difference from control: a, P � .001; b, P � .01; c, P � .05; *, P � .05. Note that in absolute terms, �-cell geneexpression is 2–3 orders of magnitude lower than �-cell gene expression (n � 4 for panels A–C). CTL, control.
doi: 10.1210/en.2015-1167 press.endocrine.org/journal/endo 2443
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.

expression was slightly but significantly enhanced, whereasthat of glucagon and MafB remained significantly sup-pressed (Figure 1, C). Interestingly, the expression of some�-cell genes were again enhanced, whereas others thatwere not altered previously were also enhanced, includingPC2, Irs1, and Irs2.Taken together, activin’s effectson the�TC1-6 �-cell line were consistent with activin enhancinga transition from an �-cell fate to that more resembling a�-cell expression profile.
Regulation of INS-1E gene expression by activin Aand related ligands
We further explored the ability of activin to enhanceexpression of �-cell genes in the INS-1E rat �-cell line andcompared this activity with related TGF� superfamily li-gands that share some of activin’s signaling pathways. Wechose 24 hours because activin up-regulated �-cell geneexpression in this time frame. Consistent with activin’seffects in �TC1-6 cells, activin A induced a 10-fold in-crease in Pax4 expression, a transcription factor that caninduce �- to �-cell transdifferentiation (22) as well as asmaller but significant increase in insulin expression (Fig-ure 2, A). Activin A treatment suppressed MafB, activin Asubunit, Pdx1, and prohormone convertase (PC)-2 ex-pression.MSTNrecapitulatednearlyall of activin’s effectsas expected from its use of the same Smad signaling path-way (Figure 2, A). BMP7 and TGF�1 appeared to have noeffect on expression of analyzed genes with the exceptionthat BMP7 slightly suppressed expression of PC1. Con-sistent with its effects on mRNA, we determined that ac-tivin also suppressed PDX-1 protein in these cells (Figure2, B). These results indicate that the induction of Pax4 andinsulin expression by activin that was observed in the�TC1-6 line is recapitulated by activin and MSTN in the
INS-1E �-cell line, supporting the hypothesis that activinpromotes the adoption of a �-cell identity.
Activin regulated gene expression in sorted mouseislet cells
To determine whether the effects of activin on geneexpression observed in �- and �-cell lines is also valid forprimary islet cells, we adapted the sorting method devel-oped by Kohler et al (28) for human and rat islet cells toprovide enriched populations of mouse �- and �-cells.This method takes advantage of natural fluorescence of�-cells as well as their larger size to separate populationsof �- and �-cells, which can then be recovered for study.Dispersed islet cells from C57BL6 male mice were sortedand gates set as shown in Figure 3, A. For this sort, �-cellscomprised 18.8% of sorted cells, whereas the �-cell frac-tion was 54% (Figure 3, B), indicating that most of the�-cells and a majority of the �-cells were recovered.
To characterize the degree of enrichment for the �- and�-cell populations obtained from cell sorting, we culturedcells for 48 hours to recover from sorting and then ana-lyzed the cells by immunofluorescence. As expected, glu-cagon was primarily observed in the �-cell population,whereas insulin was observed in the �-cell population (Fig-ure 3, C). Contamination with the opposite cell type wasless than 5%, and many of these cells were dual stained forboth insulin and glucagon (Figure 3, C). We further char-acterized these cells by qPCR after a 48-hour recoveryperiod. Expression of �-cell genes in �-cells was increasedover the �-cell pool by more than 20-fold for glucagon,MafB, Arx, and Irx2 (Figure 3, D), whereas the expressionof �-cell genes in �-cells was similarly enhanced comparedwith the �-cell population, including insulin, MafA, Pdx1,Glut2, and PC1 (Figure 3, D). Taken together, these im-
Figure 2. Activin and related TGF� superfamily ligands regulate gene expression in INS-1E �-cells. A, Activin and MSTN significantly stimulatedPax4 and insulin gene expression, whereas BMP7 and TGF�1 had no effect. The same ligands suppressed Pdx1, MafB, Inhba (activin A subunit),and PC2 gene expression, although the MSTN effect on PC2 was not significant. BMP7 suppressed PC1 gene expression (n � 3). B, Western blotshowing reduced PDX1 protein with 1 nM activin treatment (left panel) and composite of three experiments showing significant 55% reduction inPDX1 protein with activin treatment. *, P � .05; **, P � .01; ***, P � .001. CTL, control.
2444 Andrzejewski et al Activin Regulates Islet Cell Gene Expression Endocrinology, July 2015, 156(7):2440–2450
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.
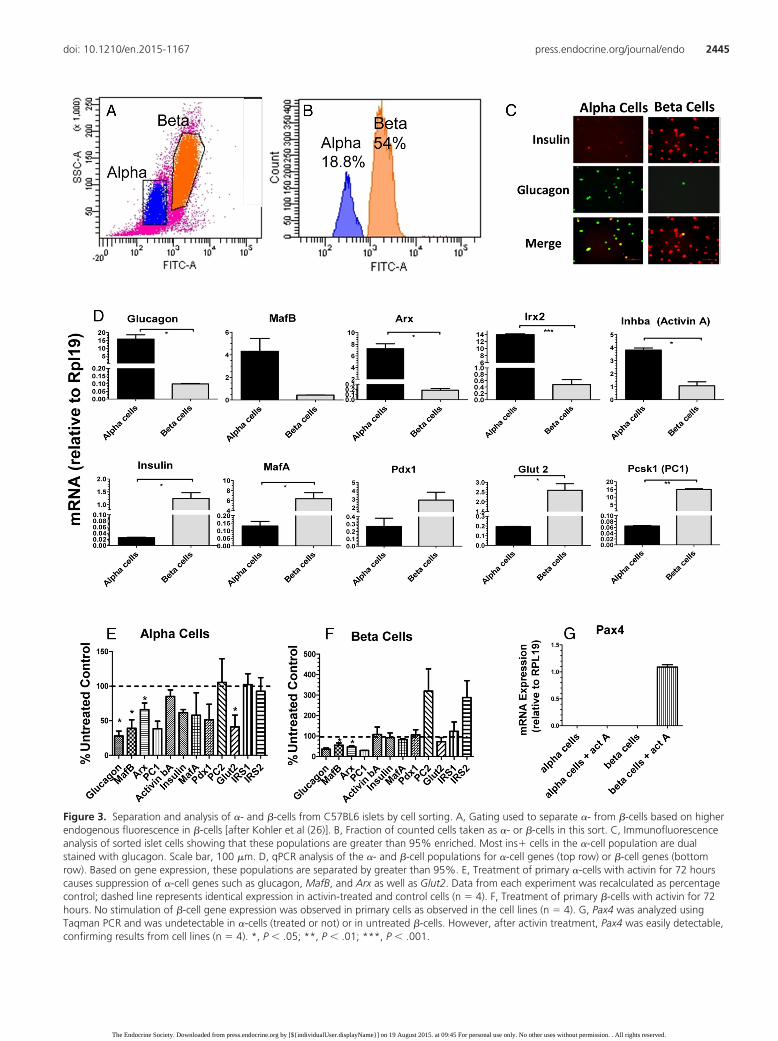
Figure 3. Separation and analysis of �- and �-cells from C57BL6 islets by cell sorting. A, Gating used to separate �- from �-cells based on higherendogenous fluorescence in �-cells [after Kohler et al (26)]. B, Fraction of counted cells taken as �- or �-cells in this sort. C, Immunofluorescenceanalysis of sorted islet cells showing that these populations are greater than 95% enriched. Most ins� cells in the �-cell population are dualstained with glucagon. Scale bar, 100 �m. D, qPCR analysis of the �- and �-cell populations for �-cell genes (top row) or �-cell genes (bottomrow). Based on gene expression, these populations are separated by greater than 95%. E, Treatment of primary �-cells with activin for 72 hourscauses suppression of �-cell genes such as glucagon, MafB, and Arx as well as Glut2. Data from each experiment was recalculated as percentagecontrol; dashed line represents identical expression in activin-treated and control cells (n � 4). F, Treatment of primary �-cells with activin for 72hours. No stimulation of �-cell gene expression was observed in primary cells as observed in the cell lines (n � 4). G, Pax4 was analyzed usingTaqman PCR and was undetectable in �-cells (treated or not) or in untreated �-cells. However, after activin treatment, Pax4 was easily detectable,confirming results from cell lines (n � 4). *, P � .05; **, P � .01; ***, P � .001.
doi: 10.1210/en.2015-1167 press.endocrine.org/journal/endo 2445
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.

munofluorescent and qPCR results indicate that �- and�-cells were enriched to greater than 95% using thismethod and that these cells can be recovered for furtheranalysis.
We then analyzed the effects of activin treatment onsorted islet cells with results expressed as fold differencefrom untreated to combine results from multiple experi-ments. Immediately after sorting and at 24 hours, wefound that gene expression was significantly suppressedfor all genes including the Rpl19 housekeeping gene (datanot shown). By 48 hours mRNA expression levels re-turned and appeared to be leveling off, so we gave them 1more day of exposure to ensure we could see differenceswith activin treatment. Activin treatment of �-cells sup-pressed glucagon mRNA expression by 66% as well asMafB and Arx by 30%, all similar to changes observed inthe �TC1-6 cell line (Figure 3, E). Activin had no effect onthe expression of Irs1 and Irs2 or PC2. Although the ex-pression of �-cell genes was detectable in the �-cell pop-ulation (Figure 3, F), it was vastly reduced compared withthe expression in the �-cell population (see Figure 3, D).Nevertheless, MafB and Arx were significantly suppressedby activin treatment, suggesting that this expression mightbe from residual �-cells in this population. Activin had nosignificant effect on the expression of most �-cell genes,although PC2 and Irs2 tended to be elevated (Figure 3, F).
However, treatment of �-cells with activin greatly en-hanced Pax 4 expression from levels that were undetect-able in untreated �-cells as well as treated or untreated�-cells (Figure 3, G). Therefore, suppression of �-cellgenes such as Arx and enhancement of Pax4 expressionsupport the hypothesis that activin destabilizes the �-cellphenotype and promotes a �-cell fate.
Activin signals through Smad2 in cell lines andislet cells
Activin typically signals through the second messengersSmad2 and Smad3 that are phosphorylated by activin re-ceptors after ligand binding (10). We therefore examinedwhether activin signaled through this canonical pathwayin islet cells and cell lines. Both activins A and B inducedSmad2 phosphorylation in �TC1-6 cells, althoughpSmad3 was undetectable (Figure 4, A) using an antibodythat has been used successfully by others (29). Similarly,both activins induced phosphorylation of Smad2 inINS-1E cells but again, pSmad3 was undetectable (Figure4, C). Hep3B human hepatoma cells have previously beenreported to contain both Smad2 and Smad3, so we ana-lyzed Hep3B extracts as a control for both �TC1-6 cellsand mouse islet extracts (Figure 4, E). Although activininduced the phosphorylation of Smad2 in all three cell
Figure 4. Activin and related ligands signal through Smad2 but not Smad1/5/8. A, Western blot analysis of �TC1-6 �-cells after treatment with 1nM activin A or B showing a representative blot and graph of multiple blot analyses. For both ligands, pSmad2 is significantly increased, butneither pSmad3 nor total Smad3 was detectable. B, Western blot analysis of �TC1-6 cells treated with activin A or B, TGF�1, BMP-4, or MSTN.Only BMP-4 stimulated pSmad1 significantly. C, Western blot analysis of activin A or B treatment of INS-1E �-cells showing significantly increasedpSmad2 by both ligands. Again, no pSmad3 or total Smad3 was detected. D, Western blot analysis of INS-1E cells treated with activin A or B,TGF�1, BMP-4, or MSTN. Only BMP-4 increased pSmad1 significantly, although MSTN treatment inhibited pSmad1 accumulation, suggestingsome cross talk between the MSTN and BMP pathways. E, Western blot analysis of Hep3B cells, �TC1-6 cells, and mouse islets treated with activinA showing that, like the cell lines, islets respond to activin with significantly increased pSmad2, whereas pSmad3 was undetectable. Total Smad3was detected in Hep3B and maybe �TC1-6 cells, but the ratio of smad2 to smad3 was markedly reduced in islet cells. CTL, control.
2446 Andrzejewski et al Activin Regulates Islet Cell Gene Expression Endocrinology, July 2015, 156(7):2440–2450
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.
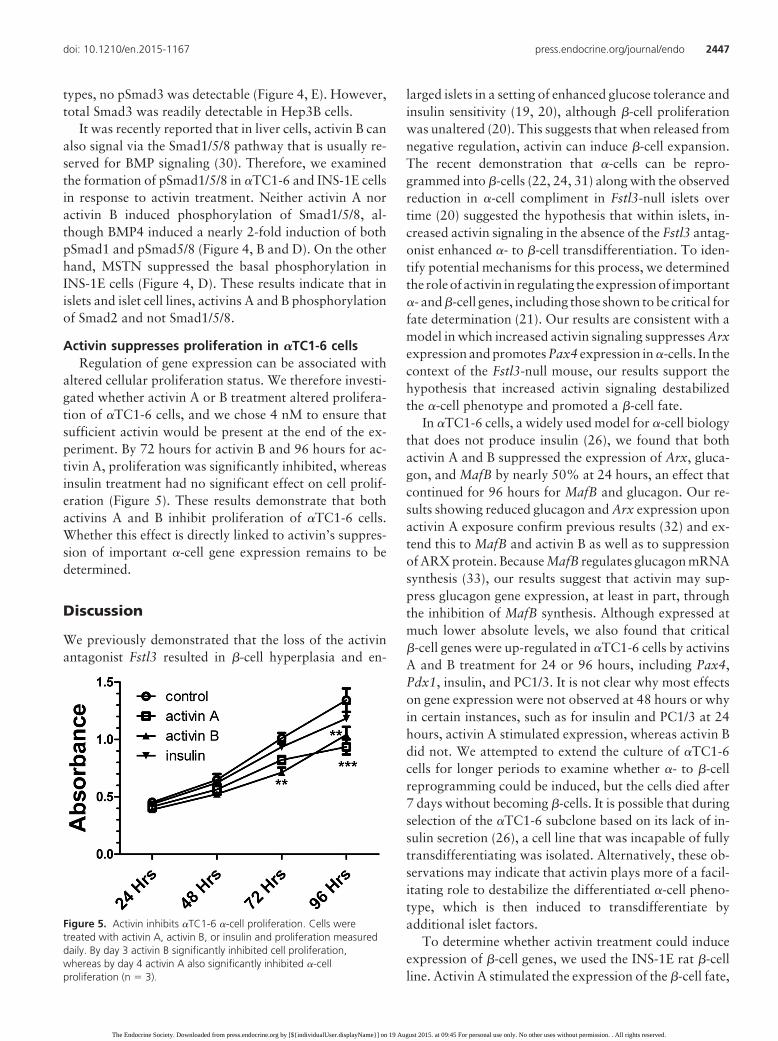
types, no pSmad3 was detectable (Figure 4, E). However,total Smad3 was readily detectable in Hep3B cells.
It was recently reported that in liver cells, activin B canalso signal via the Smad1/5/8 pathway that is usually re-served for BMP signaling (30). Therefore, we examinedthe formation of pSmad1/5/8 in �TC1-6 and INS-1E cellsin response to activin treatment. Neither activin A noractivin B induced phosphorylation of Smad1/5/8, al-though BMP4 induced a nearly 2-fold induction of bothpSmad1 and pSmad5/8 (Figure 4, B and D). On the otherhand, MSTN suppressed the basal phosphorylation inINS-1E cells (Figure 4, D). These results indicate that inislets and islet cell lines, activins A and B phosphorylationof Smad2 and not Smad1/5/8.
Activin suppresses proliferation in �TC1-6 cellsRegulation of gene expression can be associated with
altered cellular proliferation status. We therefore investi-gated whether activin A or B treatment altered prolifera-tion of �TC1-6 cells, and we chose 4 nM to ensure thatsufficient activin would be present at the end of the ex-periment. By 72 hours for activin B and 96 hours for ac-tivin A, proliferation was significantly inhibited, whereasinsulin treatment had no significant effect on cell prolif-eration (Figure 5). These results demonstrate that bothactivins A and B inhibit proliferation of �TC1-6 cells.Whether this effect is directly linked to activin’s suppres-sion of important �-cell gene expression remains to bedetermined.
Discussion
We previously demonstrated that the loss of the activinantagonist Fstl3 resulted in �-cell hyperplasia and en-
larged islets in a setting of enhanced glucose tolerance andinsulin sensitivity (19, 20), although �-cell proliferationwas unaltered (20). This suggests that when released fromnegative regulation, activin can induce �-cell expansion.The recent demonstration that �-cells can be repro-grammed into �-cells (22, 24, 31) along with the observedreduction in �-cell compliment in Fstl3-null islets overtime (20) suggested the hypothesis that within islets, in-creased activin signaling in the absence of the Fstl3 antag-onist enhanced �- to �-cell transdifferentiation. To iden-tify potential mechanisms for this process, we determinedthe roleof activin in regulating the expressionof important�- and �-cell genes, including those shown to be critical forfate determination (21). Our results are consistent with amodel in which increased activin signaling suppresses Arxexpression and promotes Pax4 expression in �-cells. In thecontext of the Fstl3-null mouse, our results support thehypothesis that increased activin signaling destabilizedthe �-cell phenotype and promoted a �-cell fate.
In �TC1-6 cells, a widely used model for �-cell biologythat does not produce insulin (26), we found that bothactivin A and B suppressed the expression of Arx, gluca-gon, and MafB by nearly 50% at 24 hours, an effect thatcontinued for 96 hours for MafB and glucagon. Our re-sults showing reduced glucagon and Arx expression uponactivin A exposure confirm previous results (32) and ex-tend this to MafB and activin B as well as to suppressionof ARX protein. Because MafB regulates glucagon mRNAsynthesis (33), our results suggest that activin may sup-press glucagon gene expression, at least in part, throughthe inhibition of MafB synthesis. Although expressed atmuch lower absolute levels, we also found that critical�-cell genes were up-regulated in �TC1-6 cells by activinsA and B treatment for 24 or 96 hours, including Pax4,Pdx1, insulin, and PC1/3. It is not clear why most effectson gene expression were not observed at 48 hours or whyin certain instances, such as for insulin and PC1/3 at 24hours, activin A stimulated expression, whereas activin Bdid not. We attempted to extend the culture of �TC1-6cells for longer periods to examine whether �- to �-cellreprogramming could be induced, but the cells died after7 days without becoming �-cells. It is possible that duringselection of the �TC1-6 subclone based on its lack of in-sulin secretion (26), a cell line that was incapable of fullytransdifferentiating was isolated. Alternatively, these ob-servations may indicate that activin plays more of a facil-itating role to destabilize the differentiated �-cell pheno-type, which is then induced to transdifferentiate byadditional islet factors.
To determine whether activin treatment could induceexpression of �-cell genes, we used the INS-1E rat �-cellline. Activin A stimulated the expression of the �-cell fate,
Figure 5. Activin inhibits �TC1-6 �-cell proliferation. Cells weretreated with activin A, activin B, or insulin and proliferation measureddaily. By day 3 activin B significantly inhibited cell proliferation,whereas by day 4 activin A also significantly inhibited �-cellproliferation (n � 3).
doi: 10.1210/en.2015-1167 press.endocrine.org/journal/endo 2447
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.

determining gene Pax4 more than 10-fold, as did MSTN,which is expressed in mouse islets (27) and uses the samesecond messengers as activin (10). These results confirmand extend observations reported for rat islets (34). Inaddition, activin and MSTN slightly but significantly en-hanced insulin expression and suppressed Pdx1 expres-sion in these cells. Interestingly, we found that INS-1E cellsalso synthesize MafB mRNA, a gene usually expressed in�-cells and is critical for �-cell function, and furthermore,activin suppressed MafB levels as observed in �TC1-6cells. In fact, this suppression of MafB by activin was alsoobserved in primary �-cells, suggesting that MafB can beproduced in both �- and �-cells and is similarly regulated,at least in part, by activin.
To expand these observations to primary islet cells, weadapted a cell-sorting protocol that relied on natural flu-orescence of �-cells and produced enriched �- and �-cellpools to greater than 95% based on gene expression andimmunofluorescence staining of glucagon and insulin. Weconfirmed that activin treatment significantly suppressedexpression of glucagon, MafB, and Arx in sorted �-cells.However, activin treatment did not induce �-cell gene ex-pression as observed in �TC1-6 cells. In sorted �-cells,activin again suppressed �-cell gene expression but hadlittle detectable effect on �-cell gene expression, except forPax4, which was undetectable in �-cells or untreated�-cells but was highly expressed after activin treatment in�-cells. Enhanced Pax4 expression by activin agrees withour observation in INS-1E cells as well as previous obser-vations (34). The effects on �-cell gene expression in thesorted �-cell pool likely reflects residual �-cells in this pop-ulation because the absolute expression levels were muchlower than in the �-cell population, although we cannotexclude reduced �-cell gene expression in �-cells. Never-theless, the suppression of Arx, MafB, and glucagon inprimary �-cells and enhanced Pax4 expression in �-cells isconsistent with our observations in cell lines. Because Arxis required to specify �-cell formation in the pancreas (21)and suppression of Arx expression in �-cells is sufficient toinduce reprogramming of these cells to �-cells (24), ourresults suggest that within adult mouse islet cells, activinmay suppress or destabilize the �-cell phenotype and pro-mote transition to a �-cell phenotype through the sup-pression of Arx biosynthesis.
Although we observed only a 25% decrease in ARXprotein with 24 hours of activin treatment that may beinsufficient to induce transdifferentiation, it is possiblethat the half-life of ARX is longer than its mRNA so thatlonger treatment times simulating the in vivo environmentmight achieve greater ARX reduction. Nevertheless, ourresults are consistent with our model that activin-induced�- to �-cell transdifferentiation is enhanced in Fstl3 null
mice, leading to the observed �-cell hyperplasia and re-duced �-cell compliment (19, 20). Moreover, the decreasein Pdx1 and Glut2 expression in response to activin isinteresting in that it was previously reported that �-cellswithin islets exist in several developmental states and thatreduced Pdx1 and Glut2 were characteristic of the moreimmature �-cell population (6, 35, 36). This might be ex-pected for newly transdifferentiated �-cells and wouldthus be consistent with the proposed model that activinpromotes �- to �-cell transdifferentiation in vivo.
Although the canonical signaling pathway for activin,TGF�, growth differentiation factor 11, and MSTN usesboth Smad2 and Smad3 second messengers (10), it is notclear that this is always the case or that the two Smads areequally used by all ligands. For example, TGF� was pre-viously reported to induce Smad3 phosphorylation in ratislet cells and the TGF� effects could be inhibited by sup-pressing Smad3 expression (15), although Smad2 was notinvestigated. On the other hand, mouse islets were hyper-trophic and less functional in Smad-2 null heterozygotes(37) and Smad2 disruption in mouse �-cells resulted inimpaired insulin secretion but islet hyperplasia probablydue to compensation for hyperglycemia (38). Wu et al (13)recently reported that activin A more strongly phosphor-ylated pSmad2 relative to Smad3, whereas activin B pre-ferred pSmad3 in INS-1E cells and mouse islets, with over-expression of Smad3 inhibiting insulin release, althoughwhether these differences are functional in vivo remainscontroversial (14). Although we probed blots for bothpSmad2 and pSmad3, we detected only pSmad2 in�TC1-6 cells, INS-1E cells, and mouse islets. Total Smad3was also undetectable in �TC1-6 cells, INS-1E cells, andmouse islets, although we could easily detect total Smad3in control Hep3B hepatoma cells. Both Smad2 and Smad3mRNA were detectable and nearly equal by qPCR in isletcells and cell lines (reference 27 and data not shown). It istherefore unclear why pSmad3 was undetectable underconditions in which pSmad2 was clearly observable, evenin the control Hep3B cell line.
The pSmad3 antibody and blotting conditions we usedhad been previously used for immunoblotting pSmad3 inINS-1E cells in response to Nodal activation, althoughpSmad2 was not investigated in that study (29). We testedsimultaneous or sequential application of pSmad2 andpSmad3 antibodies but never detected a pSmad3 band inany cell line or primary cell. It remains possible thatpSmad3 is present but below the detection threshold forthis antibody at the protein load used, although large dif-ferences in pSmad2 and pSmad3 have not been previouslynoted. Nevertheless, our results demonstrate that both ac-tivins A and B induce their effects in �TC1-6 cells, INS1cells, and mouse islets, at least via pSmad2, although we
2448 Andrzejewski et al Activin Regulates Islet Cell Gene Expression Endocrinology, July 2015, 156(7):2440–2450
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.

cannot exclude signaling through pSmad3 or noncanoni-cal pathways such as the p38 MAPK pathway (39) or theERK1/ERK2 MAPK pathway (40).
Unlike activin and its related TGF� superfamily li-gands, BMPs and their related ligands use one or more ofpSmad1, pSmad5, or pSmad8/9 as second messengers.However, activin B was recently shown to signal viapSmad1/5/8 in human HepG2 liver cells (30), so we ex-tended our investigation to determine whether activin Balso uses Smad1/5/8 in islet cells. Neither activin A noractivin B induced the phosphorylation of these Smads in�TC1-6 or INS1 cells, although phosphorylation wasclearly induced by BMP4. Therefore, it appears that thenoncanonical pSmad1/5/8 pathway is not used by activinin islet cell lines. Interestingly, MSTN significantly inhib-ited basal pSmad1/5/8 phosphorylation, suggesting that inaddition to the use of pSmad2/3 for signaling, MSTN si-multaneously inhibits BMP-mediated activity. TGF� hasbeen shown to inhibit BMP signaling in several bone celllines via at least two different mechanisms including directinteraction of Smad3 and Smad1/5 and specifically notinvolving Smad2 (41, 42). This suggests that studies ofSmad3 involving its knockdown or deletion (13, 15) mayinvolve, at least in part, up-regulation of BMP signaling,which has positive effects on �-cell function (16). Never-theless, our results support Smad2/3 as the primary sig-naling pathway for activins A and B. Additional studies inwhich Smad2 expression is eliminated will be required todetermine whether activin’s effects on gene expression aremediated exclusively via this pathway or include pSmad3.
If activin signaling does in fact induce �- to �-cell trans-differentiation in Fstl3-null mice, then it might be expectedthat activin would also increase �-cell proliferation to re-place �-cells lost via transdifferentiation into �-cells. Incontrast, we found that activin A and B both suppressedproliferation in �TC1-6 cells, although insulin had no ef-fect. This confirms a previous study showing activin Ainhibition of �-cell line proliferation (32) and extends theobservation to activin B as well. However, insulin waspreviously reported to induce proliferation in �TC1 cellsin a dose-dependent manner (43), but the maximal dose inthat study had no effect in �TC1-6 cells in our experi-ments. It is possible that the �TC1-6 subclone, althoughselected based on lack of insulin expression (26), is alsodefective in insulin response because we were also unableto detect pAkt in these cells after 10 minutes of insulintreatment (data not shown). Taken together, these resultssuggest that in islets, activin and insulin may counterregu-late �-cell proliferation. In addition, the reduced �-cellproliferation in response to activin would be consistentwith the decreased �-cell content in Fstl3-null islets (20).
Our results demonstrate that activin treatment sup-pressed the expression of Arx, glucagon, and MafB in�-cells, all of which are required to maintain the �-cellphenotype and function. Because the reduction of Arxalone was sufficient to induce �- to �-cell transdifferen-tiation (24), our results are consistent with the hypothesisthat activin destabilizes the �-cell phenotype, which maythen facilitate, directly or indirectly, induction of trans-differentiation into �-cells. Such a process could explain�-cell expansion observed in Fstl3-null mice (19).
Acknowledgments
Address all correspondence and requests for reprints to: AlanSchneyer, PhD, Department of Veterinary and Animal Science,University of Massachusetts-Amherst, 3601 Main Street, Secondfloor, Springfield MA 01199. E-mail: [email protected].
This work was supported by Grant 17-2012-414 from theJuvenile Diabetes Research Foundation (to A.L.S.).
Disclosure Summary: A.L.S. formed a company, FairbanksPharmaceuticals, to pursue therapeutic aspects of his research.This company provided no financial input to this research, nordoes the company research overlap with that reported here. Theother authors have nothing to disclose.
References
1. Sachdeva MM, Stoffers DA. Minireview: meeting the demand forinsulin: molecular mechanisms of adaptive postnatal �-cell massexpansion. Mol Endocrinol. 2009;23:747–758.
2. Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stew-art AF, Garcia-Ocana A. Human �-cell proliferation and intracel-lular signaling part 2: still driving in the dark without a road map.Diabetes. 2014;63:819–831.
3. Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF. Human �-cellproliferation and intracellular signaling: driving in the dark withouta road map. Diabetes. 2012;61:2205–2213.
4. Oliver-Krasinski JM, Stoffers DA. On the origin of the � cell. GenesDev. 2008;22:1998–2021.
5. Brown ML, Schneyer AL. Emerging roles for the TGF� family inpancreatic �-cell homeostasis. Trends Endocrinol Metab. 2010;21:441–448.
6. Szabat M, Johnson JD, Piret JM. Reciprocal modulation of adult �
cell maturity by activin A and follistatin. Diabetologia. 2010;53:1680–1689.
7. El-Gohary Y, Tulachan S, Wiersch J, et al. A smad signaling networkregulates islet cell proliferation. Diabetes. 2014;63:224–236.
8. Shi Y, Massague J. Mechanisms of TGF-� signaling from cell mem-brane to the nucleus. Cell. 2003;113:685–700.
9. Derynck R, Zhang YE. Smad-dependent and Smad-independentpathways in TGF-� family signalling. Nature. 2003;425:577–584.
10. Massague J, Seoane J, Wotton D. Smad transcription factors. GenesDev. 2005;19:2783–2810.
11. Yamaoka T, Idehara C, Yano M, et al. Hypoplasia of pancreaticislets in transgenic mice expressing activin receptor mutants. J ClinInvest. 1998;102:294–301.
12. Smart NG, Apelqvist AA, Gu X, et al. Conditional expression of
doi: 10.1210/en.2015-1167 press.endocrine.org/journal/endo 2449
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.

Smad7 in pancreatic � cells disrupts TGF-� signaling and inducesreversible diabetes mellitus. PLoS Biol. 2006;4:e39.
13. Wu H, Mezghenna K, Marmol P, et al. Differential regulation ofmouse pancreatic islet insulin secretion and Smad proteins by activinligands. Diabetologia. 2014;57:148–156.
14. Bonomi L, Brown M, Ungerleider N, Muse M, Matzuk MM, Sch-neyer A. Activin B regulates islet composition and islet mass but notwhole body glucose homeostasis or insulin sensitivity. Am J PhysiolEndocrinol Metab. 2012;303:E587–E596.
15. Lin HM, Lee JH, Yadav H, et al. Transforming growth factor-�/Smad3 signaling regulates insulin gene transcription and pancreaticislet �-cell function. J Biol Chem. 2009;284:12246–12257.
16. Goulley J, Dahl U, Baeza N, Mishina Y, Edlund H. BMP4-BMPR1Asignaling in � cells is required for and augments glucose-stimulatedinsulin secretion. Cell Metab. 2007;5:207–219.
17. Sidis Y, Mukherjee A, Keutmann H, Delbaere A, Sadatsuki M, Sch-neyer A. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificityfor activin, myostatin, and bone morphogenetic proteins. Endocri-nology. 2006;147:3586–3597.
18. Schneyer A, Sidis Y, Xia Y, et al. Differential actions of follistatinand follistatin-like 3. Mol Cell Endocrinol. 2004;225:25–28.
19. Mukherjee A, Sidis Y, Mahan A, et al. FSTL3 deletion reveals rolesfor TGF-� family ligands in glucose and fat homeostasis in adults.Proc Natl Acad Sci USA. 2007;104:1348–1353.
20. Brown ML, Bonomi L, Ungerleider N, et al. Follistatin and follista-tin like-3 differentially regulate adiposity and glucose homeostasis.Obesity (Silver Spring). 2011;19:1940–1949.
21. Collombat P, Hecksher-Sorensen J, Serup P, Mansouri A. Specifyingpancreatic endocrine cell fates. Mech Dev. 2006;123:501–512.
22. Collombat P, Xu X, Ravassard P, et al. The ectopic expression ofPax4 in the mouse pancreas converts progenitor cells into alpha andsubsequently � cells. Cell. 2009;138:449–462.
23. Collombat P, Hecksher-Sorensen J, Krull J, et al. Embryonic endo-crine pancreas and mature � cells acquire � and PP cell phenotypesupon Arx misexpression. J Clin Invest. 2007;117:961–970.
24. Courtney M, Gjernes E, Druelle N, et al. The inactivation of Arx inpancreatic �-cells triggers their neogenesis and conversion into func-tional �-like cells. PLoS Genet. 2013;9:e1003934.
25. Ungerleider NA, Bonomi LM, Brown ML, Schneyer AL. Increasedactivin bioavailability enhances hepatic insulin sensitivity while in-ducing hepatic steatosis in male mice. Endocrinology. 2013;154:2025–2033.
26. Hamaguchi K, Leiter EH. Comparison of cytokine effects on mousepancreatic �-cell and �-cell lines. Viability, secretory function, andMHC antigen expression. Diabetes. 1990;39:415–425.
27. Brown ML, Kimura F, Bonomi LM, Ungerleider NA, Schneyer AL.Differential synthesis and action of TGFss superfamily ligands inmouse and rat islets. Islets. 2011;3:367–375.
28. Kohler M, Dare E, Ali MY, et al. One-step purification of functional
human and rat pancreatic � cells. Integr Biol (Camb). 2012;4:209–219.
29. Zhao F, Huang F, Tang M, et al. Nodal induces apoptosis throughactivation of the ALK7 signaling pathway in pancreatic INS-1�-cells. Am J Physiol Endocrinol Metab. 2012;303:E132–E143.
30. Besson-Fournier C, Latour C, Kautz L, et al. Induction of activin Bby inflammatory stimuli up-regulates expression of the iron-regu-latory peptide hepcidin through Smad1/5/8 signaling. Blood. 2012;120:431–439.
31. Thorel F, Nepote V, Avril I, et al. Conversion of adult pancreatic�-cells to �-cells after extreme �-cell loss. Nature. 2010;464:1149–1154.
32. Mamin A, Philippe J. Activin A decreases glucagon and arx geneexpression in �-cell lines. Mol Endocrinol. 2007;21:259–273.
33. Artner I, Hang Y, Elghazi L, et al. MafB: an activator of the glucagongene expressed in developing islet �- and �-cells. Diabetes. 2006;55:297–304.
34. Brun T, Franklin I, St-Onge L, et al. The diabetes-linked transcrip-tion factor PAX4 promotes �-cell proliferation and survival in ratand human islets. J Cell Biol. 2004;167:1123–1135.
35. Szabat M, Pourghaderi P, Soukhatcheva G, et al. Kinetics andgenomic profiling of adult human and mouse �-cell maturation.Islets. 2011;3:175–187.
36. Katsuta H, Aguayo-Mazzucato C, Katsuta R, et al. Subpopulationsof GFP-marked mouse pancreatic �-cells differ in size, granularity,and insulin secretion. Endocrinology. 2012;153:5180–5187.
37. Goto Y, Nomura M, Tanaka K, et al. Genetic interactions betweenactivin type IIB receptor and Smad2 genes in asymmetrical pattern-ing of the thoracic organs and the development of pancreas islets.Dev Dyn. 2007;236:2865–2874.
38. Nomura M, Zhu HL, Wang L, Morinaga H, Takayanagi R, Tera-moto N. SMAD2 disruption in mouse pancreatic � cells leads to islethyperplasia and impaired insulin secretion due to the attenuation ofATP-sensitive K� channel activity. Diabetologia. 2014;57:157–166.
39. de Guise C, Lacerte A, Rafiei S, et al. Activin inhibits the human Pit-1gene promoter through the p38 kinase pathway in a Smad-indepen-dent manner. Endocrinology. 2006;147:4351–4362.
40. Bao YL, Tsuchida K, Liu B, Kurisaki A, Matsuzaki T, Sugino H.Synergistic activity of activin A and basic fibroblast growth factor ontyrosine hydroxylase expression through Smad3 and ERK1/ERK2MAPK signaling pathways. J Endocrinol. 2005;184:493–504.
41. Gronroos E, Kingston IJ, Ramachandran A, Randall RA, Vizan P,Hill CS. Transforming growth factor beta inhibits bone morphoge-netic protein-induced transcription through novel phosphorylatedSmad1/5-Smad3 complexes. Mol Cell Biol. 2012;32:2904–2916.
42. Ehnert S, Zhao J, Pscherer S, et al. Transforming growth factor �1inhibits bone morphogenic protein (BMP)-2 and BMP-7 signalingvia upregulation of Ski-related novel protein N (SnoN): possiblemechanism for the failure of BMP therapy? BMC Med. 2012;10:101.
43. Liu Z, Kim W, Chen Z, et al. Insulin and glucagon regulate pancre-atic �-cell proliferation. PLoS One. 2011;6:e16096.
2450 Andrzejewski et al Activin Regulates Islet Cell Gene Expression Endocrinology, July 2015, 156(7):2440–2450
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 19 August 2015. at 09:45 For personal use only. No other uses without permission. . All rights reserved.





























