Embed Size (px)
Citation preview

American Journal of Medical Genetics 49:344-347 (1994)
Brief Clinical Report
Additional Case of Opsismodysplasia Supporting Autosomal Recessive Inheritance
Frits A. Beemer and Kazimierz S. Kozlowski Clinical Genetics Center Utrecht (F.A.B.), Utrecht, The Netherlands; and Department of Radiology (K.S.K.), The Children’s Hospital, Sydney, Australia
The authors describe clinical and radiological findings in a 2-year-old boy from consan- guineous parents. A diagnosis of opsi(s)mod- ysplasia ( = delayed maturation) had been made (MIM 258480). The purpose of this paper is to draw attention to the striking radiologi- cal manifestations. Consanguinity in the par- ents of our case and occurrence in a brother and sister in a previous report support an au- tosomal recessive transmission. 0 1994 Wiley-Liss, Inc.
KEY WORDS: platyspondyly, retarded bone age, dwarfism
INTRODUCTION The name opsismodysplasia (opsirnos in Greek means
late) was coined by Maroteaux et al. [1982] when they described in 2 patients a condition that was charac- terized by severe delay in skeletal maturation. Subse- quently, Maroteaux et al. [1984] described 3 cases in greater detail and applied the term opsismodysplasia. The first patient, a 21-month-old boy, had been described by Zonana et al. 119771. To date 8 cases have been de- scribed [Zonana et al., 1977; Maroteaux et al., 1982 and 1984; Spranger and Maroteaux, 19911. The diagnosis can be made at birth based on the association of clinical and radiological manifestations. The craniofacial abnor- malities include frontal bossing, short nose, and long philtrum. In addition there is narrow chest and muscu- lar hypotonia, resulting in recurrent respiratory tract infections that often prove to be fatal in the first 3 years of life [Maroteaux, 19901. Rhizomelic micromelia is
Received for publication May 10,1993; revision received August 9, 1993.
Address reprint requests to F’rits A. Beemer, M.D., Clinical Genetics Center Utrecht, P.O. Box 18009, 3501 CA Utrecht, The Netherlands.
0 1994 Wiley-Liss, Inc.
present with short fingers. Radiographically there is a marked delay in ossification, especially noticeable in the carpal bones. The growth cartilage shows a wide hyper- trophic area containing thick connective tissue septa LMaroteaux et al., 19841. Immunohistochemically type I collagen instead of type I1 was detected in this area [Maroteaux, 19901.
The clinical and radiological findings in one further patient are presented. He is now age 3 years old. Clinical findings are compared between this case and those from the literature. Based on previous observations and on consanguinity of the parents of our patient, autosomal recessive inheritance is most likely.
CLINICAL REPORT This boy was the first and only child of healthy, first-
cousin parents of Moroccan descent. The father was 23 and mother 20 years old. Paternal height was 1.78 m and maternal height 1.60 m. Family history was unremark- able and gestation uneventful. Delivery was sponta- neous at term. There was neither perinatal asphyxia nor early feeding difficulties. Birth weight was unre- ported, the length was 46 cm (-2.5 SD), OFC 34.5 cm (50th centile). Noted at birth were a depressed nasal bridge, small anteverted nose with relatively broad nares, long philtrum, large anterior fontanelle, arched eyebrows, and short hands and feet. Rhizomelic micro- melia was apparent. The thorax was bell shaped. Gen- eral muscle tone was low.
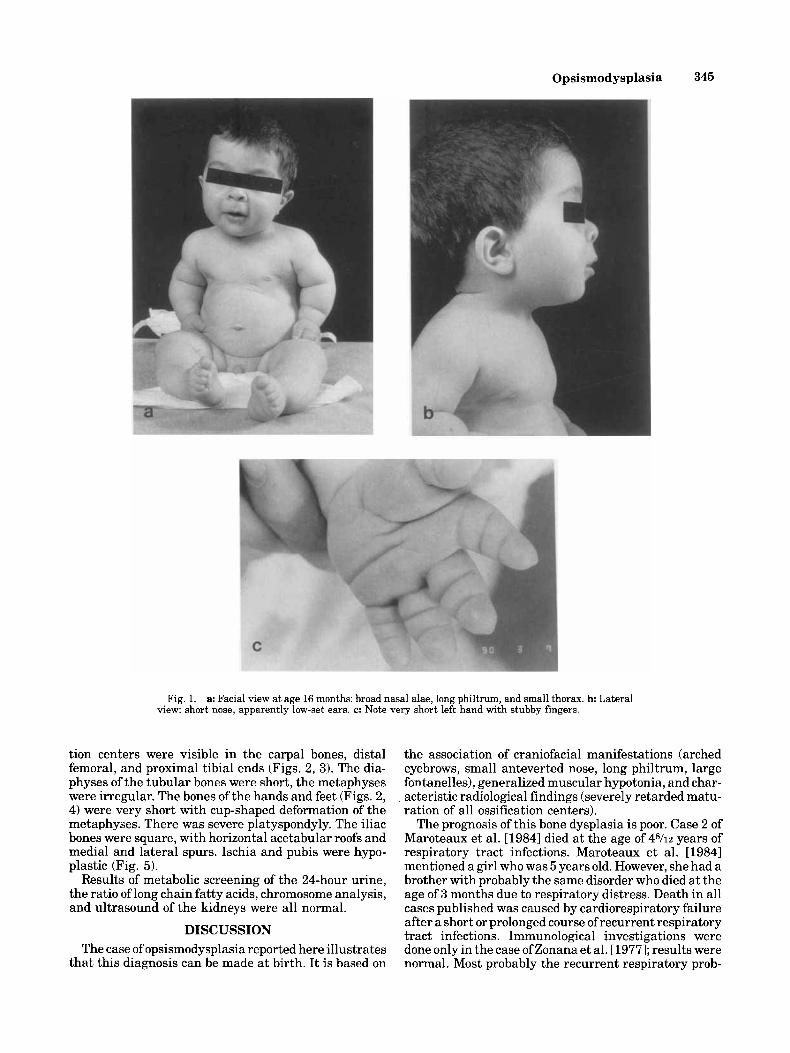
On referral at the age of 16 months the clinical course had been marked by recurrent respiratory tract infections. Motor development was slow and mental development appeared normal. The face was slightly asymmetric and similar to what had been described in the neonatal pe- riod (Fig. la,b). The ears were low set. The thorax was short and bell shaped with a prominent abdomen (Fig. la) . The muscle tone was still low. The limbs were short with deep, almost circumferential skin creases, espe- cially in the upper arms, hands, and feet (Fig. la,c).
Radiologically the skull was normal, with large fon- tanelles. The most striking finding was the severe retar- dation of ossification. At the age of 16 months no ossifica-
Opsismodysplasia 345
Fig. 1. a: Facial view at age 16 months: broad nasal alae, long philtrum, and small thorax. b: Lateral view: short nose, apparently low-set ears. c: Note very short left hand with stubby fingers.
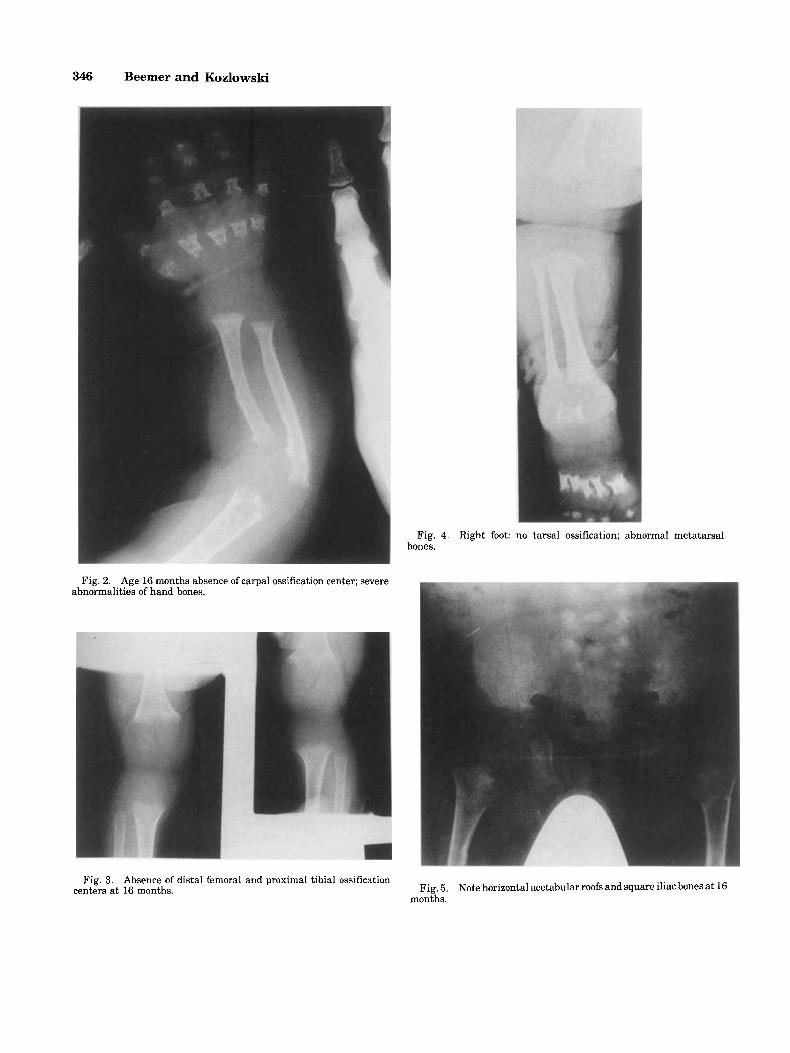
tion centers were visible in the carpal bones, distal femoral, and proximal tibia1 ends (Figs. 2, 3). The dia- physes of the tubular bones were short, the metaphyses were irregular. The bones of the hands and feet (Figs. 2, 4) were very short with cup-shaped deformation of the metaphyses. There was severe platyspondyly. The iliac bones were square, with horizontal acetabular roofs and medial and lateral spurs. Ischia and pubis were hypo- plastic (Fig. 5).
Results of metabolic screening of the 24-hour urine, the ratio of long chain fatty acids, chromosome analysis, and ultrasound of the kidneys were all normal.
DISCUSSION The case of opsismodysplasia reported here illustrates
that this diagnosis can be made at birth. It is based on
the association of craniofacial manifestations (arched eyebrows, small anteverted nose, long philtrum, large fontanelles), generalized muscular hypotonia, and char- acteristic radiological findings (severely retarded matu- ration of all ossification centers).
The prognosis of this bone dysplasia is poor. Case 2 of Maroteaux et al. [19841 died at the age of 4%~ years of respiratory tract infections. Maroteaux et al. [ 19841 mentioned a girl who was 5 years old. However, she had a brother with probably the same disorder who died at the age of 3 months due to respiratory distress. Death in all cases published was caused by cardiorespiratory failure after a short or prolonged course of recurrent respiratory tract infections. Immunological investigations were done only in the case of Zonana et al. [ 19771; results were normal. Most probably the recurrent respiratory prob-
346 Beemer and Kozlowski
Fig. 4. Right foot: no tarsal ossification; abnormal metatarsal bones.
Fig. 2. Age 16 months absence of carpal ossification center; severe abnormalities of hand bones.
Fig. 3. Absence of distal femoral and proximal tibia1 ossification centers at 16 months. Fig. 5.
months. Note horizontal acetabular roofs and square iliac bones a t 16

Opsismodysplasia 347
Moreover, this paper also states explicitly that the par- ents were not consanguineous. In our case the parents were first cousins. We think that this finding adds to the observation by Maroteaux et al. [1984] of 2 sibs of differ- ent sex and indicates that an autosomal recessive inher- itance pattern is very likely.
ACKNOWLEDGMENTS The authors are indebted to Mrs. E. van der Ban,
medical student, and to Prof. Dr. D. Sillence for review- ing the data.
lems are caused by the association of thoracic cage de- formities and severe hypotonia.
Chondro-osseous morphology was investigated exten- sively by Maroteaux et al. [1984]. They found a wide and irregular hypertrophic zone in growth cartilage. There were thick connective tissue septa, irregular provisional calcification, and vascular invasion. Type I collagen was detected in the hypertrophic area by immunohistochem- ical and microchemical tests.
Opsismodysplasia has to be differentiated from tho- racic-pelvic-phalangeal dysplasia (asphyxiating thoracic dysplasia; Jeune syndrome), the group of spondylo-epi- metaphyseal dysplasias (including spondylo-epiphyseal dysplasia congenita and spondylo-epi-metaphyseal dys- plasia congenita), thanatophoric dysplasia, and homo- zygous achondroplasia. There is a group of spondylo-epi- metaphyseal dysplasias seen in newborn infants who die early and in whom the disorder cannot be further characterized unless there is distinctive chondro-os- seous histopathology.
Inheritance is most likely autosomal recessive. Maro- teaux et al. [19841 mention 2 sibs of different sex. In the 10th edition of McKusick‘s catalogue [1992] it is stated that “consanguinity in other cases suggested autosomal recessive inheritance” (MIM 258480). However, the in- heritance pattern in these “other cases” could not be verified. Possibly this statement refers to Maroteaux et al. [1982] who mentioned “consanguinity in other cases” and referred to Zonana et al. [1977]. However, this latter paper only described a single case, a 21-month-old boy.
REFERENCES Maroteaux P (1990): Opsismodysplasia. In Buyse ML (ed): “Birth De-
fects Encyclopedia,” Cambridge: Blackwell Scientific Publications, pp 129551296,
Maroteaux P, Stanescu V, Stanescu R (1982): Four recently described osteochondrodysplasias; opsismodysplasia. In Papadatos CJ, Bart- socas C (eds): “Skeletal Dysplasias.” New York: Alan R. Liss, pp 347-348.
Maroteaux P, Stanescu V, Stanescu R, Le Marec €3, Moraine C, Lejar- raga C (1984): Opsismodysplasia: A new type of chondrodysplasia with predominant involvement of the bones of the hand and the vertebrae. Am J Mcd Genet 19:171-182.
McKusick VA (1992): “Mendelian Inheritance in Man: Catalogs of autosomal dominant, autosomal recessive, and X-linked phe- notypes.” 10th ed. Baltimore: The Johns Hopkins University Press, p 1393.
Spranger J , Maroteaux P (1991): Opsismodysplasia. Adv Hum Genet 19:36-37.
Zonana J, Rimoin DL, Lachman RS, Cohen AH (1977): A unique chon- drodysplasia secondary to a defect in chondroosseous transforma- tion. Birth Defects XIII(3D):155-163.





























