Embed Size (px)
Citation preview

Article
AID Recognizes Structure
d DNA for Class SwitchRecombinationGraphical Abstract
Highlights
d Structured substrates, such as G4 substrates, are preferred
AID targets in vitro
d A bifurcated substrate-binding surface supports structured-
substrate recognition
d G4 substrates induce AID oligomerization upon binding
d Disrupting structured-substrate recognition or AID
oligomerization compromises CSR
Qiao et al., 2017, Molecular Cell 67, 361–373August 3, 2017 ª 2017 Elsevier Inc.http://dx.doi.org/10.1016/j.molcel.2017.06.034
Authors
Qi Qiao, Li Wang, Fei-Long Meng,
Joyce K. Hwang,
Frederick W. Alt, Hao Wu
In Brief
Qiao et al. demonstrated that structured
substrates, like G4 and branched DNA,
are preferred AID targets in vitro. A
bifurcated substrate-binding surface in
AID structure supports structured-
substrate recognition. G4 substrates
mimicking the Ig S regions also induce
cooperative AID oligomerization.
Disrupting structured-substrate
recognition or AID oligomerization both
compromise CSR.

Molecular Cell
Article
AID Recognizes Structured DNAfor Class Switch RecombinationQi Qiao,1,2,5 Li Wang,1,2,5 Fei-Long Meng,1,2,3,4 Joyce K. Hwang,1,2,3 Frederick W. Alt,1,2,3 and Hao Wu1,2,6,*1Program in Cellular and Molecular Medicine, Boston Children’s Hospital2Department of Biological Chemistry and Molecular Pharmacology3Howard Hughes Medical InstituteHarvard Medical School, Boston, MA 02115, USA4Present address: Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320
Yue-yang Road, Shanghai 200031, China5These authors contributed equally6Lead Contact
*Correspondence: [email protected]
http://dx.doi.org/10.1016/j.molcel.2017.06.034
SUMMARY
Activation-induced cytidine deaminase (AID) initiatesboth class switch recombination (CSR) and somatichypermutation (SHM) in antibody diversification.Mechanisms of AID targeting and catalysis remainelusive despite its critical immunological rolesand off-target effects in tumorigenesis. Here, we pro-duced active human AID and revealed its preferredrecognitionanddeaminationof structuredsubstrates.G-quadruplex (G4)-containing substrates mimickingthe mammalian immunoglobulin switch regions areparticularly good AID substrates in vitro. By solvingcrystal structures of maltose binding protein (MBP)-fused AID alone and in complex with deoxycytidinemonophosphate, we surprisingly identify a bifurcatedsubstrate-binding surface that explains structuredsubstrate recognition by capturing two adjacent sin-gle-stranded overhangs simultaneously. Moreover,G4 substrates induce cooperative AID oligomeriza-tion. Structure-based mutations that disrupt bifur-cated substrate recognition or oligomerization bothcompromise CSR in splenic B cells. Collectively, ourdata implicate intrinsic preference of AID for struc-tured substrates and uncover the importance of G4recognition and oligomerization of AID in CSR.
INTRODUCTION
Antibody diversification is a central process in adaptive immunity
that produces antigen-specific, high-affinity antibodies to com-
bat millions of antigens. In B cells, immunoglobulin (Ig) genes un-
dergo two DNA-alteration events to enhance the specificity and
functionality of antibodies: somatic hypermutation (SHM) and
class switch recombination (CSR). Until now, activation-induced
cytidine deaminase (AID) is the single enzyme that is known to
initiate both SHM and CSR. AID belongs to the APOBEC cytidine
M
deaminase family. It converts deoxycytidine into deoxyuridine
on single-stranded DNA (ssDNA) substrates in vitro and in vivo
but does not show detectable activity on RNA substrates (Bran-
steitter et al., 2003). Although AID exhibits sequence similarity
to APOBEC cytidine deaminases, its critical function in antibody
diversification, especially in CSR, cannot be substituted by
APOBEC proteins.
Across the genome, AID predominantly mutates immunoglob-
ulin genes at the variable (V) and switch (S) regions. In SHM, V re-
gion sequencing data showed that a WRCH (W = A/T, R = A/G,
and H = A/C/T) hotspot motif exhibits higher AID-induced muta-
tion probability than average, by about 2- to 4-fold (Larijani et al.,
2005; Rogozin and Diaz, 2004). In CSR, mammalian S regions lie
between sets of constant region exons and are enriched in the
AGCT sequence, a palindromic example of WRCH. Deamination
at both strands of DNA may contribute to double-strand breaks
(DSBs) by co-opted repair pathways (Han et al., 2011; Yeap
et al., 2015). Joining of AID-initiated DSBs replaces the IgM
heavy chain constant region with that of other isotypes to
achieve class switching (Matthews et al., 2014a). Patients with
AID mutations produce mainly low-affinity IgM antibodies with
impairment in CSR and/or SHM, a syndrome known as hyper-
IgM immunodeficiency (Xu et al., 2012). AID off-target mutations
as well as the subsequent DSBs and chromosomal transloca-
tions often promote tumorigenesis, in particular for many types
of leukemia and lymphoma (Xu et al., 2012).
The mechanism of AID targeting has been a long-standing
mystery. Genome sequencing data showed that AID mutates
Ig regions at a 10�4–10�3 per base, per generation frequency
(McKean et al., 1984; Rajewsky et al., 1987), which is a million-
fold higher than the 10�9 genomic basal mutation frequency.
Numerous models of AID targeting have been proposed, which
both propel the field forward and leave remaining questions un-
answered. Chromatin accessibility and transcription that pro-
vides ssDNA substrates for AID to act is a known prerequisite,
but it does not explain AID targeting specificity to limited
genomic regions. Convergently transcribed regions in certain su-
per-enhancer loci appear to correlate with enhanced AID
off-target mutations (Meng et al., 2014), but it is not clear
whether Ig regions undergo convergent transcription, and not
olecular Cell 67, 361–373, August 3, 2017 ª 2017 Elsevier Inc. 361

all super-enhancer loci with convergent transcription are AID tar-
gets (Qian et al., 2014). The originally proposed hotspot model
(Pham et al., 2003) also does not appear sufficient, as ssDNA
substrates containing hotspot motifs do not show advantage in
recruiting AID in vitro (Larijani et al., 2007), and hotspot motif dis-
tribution in the genome does not correlate with the AID off-tar-
geting spectrum in vivo (Duke et al., 2013). In AID recruiter
models, various proteins, including replication protein A (RPA)
(Chaudhuri et al., 2004), Spt5 (Pavri et al., 2010), and 14-3-3
(Xu et al., 2010), have been proposed in guiding AID to genes.
However, the genome distribution of these recruiters is not
unique to Ig regions. A more recent recruiter model proposed
that transcribed G-repeat rich switch RNAs form G-quadruplex
(G4) structures, bind AID, and guide AID to genes for CSR (Zheng
et al., 2015). However, themechanism of transfer of AID between
G4 RNA and target DNA has not been addressed.
Here, we present a comprehensive biochemical and structural
study on AID, which provides unexpected insights on AID tar-
geting specificity. Through protein engineering, we produced a
fully functional monomeric AID with intact activity in vitro and in
cells. Using various forms of DNA, we found that structured
substrates containing multiple ssDNA overhangs, like G4 and
branched DNA, are preferred by AID in binding and deamination
over linear ssDNA substrates in vitro. This observation may form
the basis for the frequent targeting of AID tomammalian Ig switch
regions containing high density ofG-repeat sequences.Wedeter-
mined the crystal structures of maltose binding protein (MBP)-
fused AID and its complex with cytidine (C), deoxycytidine (dC),
and deoxycytidine monophosphate (dCMP). These structures
not only explain the discrimination between DNA and RNA in
AID catalysis but also reveal a bifurcated substrate-binding sur-
face, which strongly supports that one AID recognizes two adja-
cent ssDNA overhangs from one structured substrate to achieve
high affinity. In addition, we observed that G4 structured sub-
strates induce AID cooperative oligomerization, which may pro-
mote clustered mutations in Ig S regions. Structure-guided muta-
genesis revealed that both the bifurcated substrate binding
surface and the putative oligomerization interface are essential
for CSR, elucidating recognition of structured substrates as an
important AID-targeting mechanism to Ig S regions.
RESULTS
Active AID Monomer from Protein EngineeringAlthough endogenous AID extracted from B cells exhibited near-
monomer molecular mass (Chaudhuri et al., 2003), previously
published data (Larijani et al., 2007) and our results showed
largely heterogeneous and poorly active aggregates of recombi-
nant wild-type (WT) AID (Figures 1A, 1B, S1A, and S1B). To
obtain physiologically relevant recombinant AID for functional
elucidation, we performed rounds of protein engineering. We
found that the His-MBP-fused double-mutant H130A/R131E
produced a low amount of monomeric AID, and further N-
and C-terminal tail (CTT) truncations improved the monomer
yield, resulting in constructs that we named AID.mono+CTT
and AID.mono, respectively (Figures 1A, 1B, S1A, and S1B).
Both monomeric and aggregated AID fractions were purified
and compared in deamination assays, revealing that monomeric
362 Molecular Cell 67, 361–373, August 3, 2017
AID with and without CTT exhibited much higher deamination
activity on ssDNA than aggregated fractions of WT AID,
AID.mono+CTT, and AID.mono (Figure 1C). Compared to pre-
viously reported AID turnover rates (�10 fmol/min/mg; King
et al., 2015), the activities of monomeric AID.mono+CTT and
AID.mono were roughly 1,000 fold higher (�104 fmol/min/mg).
These data suggest that aggregation renders AID largely inac-
tive. Consistent with the lack of sequence conservation of
H130 and R131 among AID species (Figure S1C), the H130A/
R131E mutant behaved as WT in mutation frequency by an
SHM-mimic rifampicin resistance (RifR) assay (Petersen-Mahrt
et al., 2002; Figure 1D) and fully rescuedCSRwhen reconstituted
into AID-deficient ex vivo CSR-activated splenic B cells (Figures
1E and S1F). Notably, all AID constructs tested in RifR and CSR
assays contain only the internal mutations, with no fusion tag,
and no truncations of the nuclear transportation signals at the
N or C terminus. Because AID.mono+CTT and AID.mono both
showed robust deamination activity and the His-MBP tag did
not affect this activity (Figures S1D and S1E), we mainly used
the His-MBP-fused AID.mono with higher yield in subsequent
biochemical characterizations.
Linear and G4 Structured Substrates from Ig S RegionsGenome sequencing has shown that themammalian Ig S regions
contain abundant tandem G repeats interspersed by AGCT hot-
spots and are heavily targeted by AID during CSR (Figure S2A;
Yu et al., 2003). It has been proposed that, during transcription,
G4 structures form on the G-repeat non-template strand and
contribute to R-loop stability (Duquette et al., 2004). Consis-
tently, we found that authentic non-template Sm fragment (64
nt) and a single G-repeat substrate both spontaneously assem-
bled into G4 structures (Figures 2A and 2B). The G4 assembly
was validated by the fluorescence enhancement of a G4-specific
dye and the disruptive effect of LiCl (Figure 2B; Bardin and Leroy,
2008). Gel electrophoresis showed that the 64-nt Sm fragment
formed heterogeneous oligomers, likely representing mixed
inter- and intramolecular G4s (Figure S2B). Differently, the single
G-repeat substrate formed homogeneous intermolecular G4
that could be separated from linear ssDNA by size-exclusion
chromatography and gel electrophoresis (Figures 2C, 2D, and
S2B). Dimethyl sulfate protection footprinting indicated that
all Gs in the GGGGTG motif were involved in the G4 assembly
(Figure S2C; Sun and Hurley, 2010). Circular dichroism (CD)
spectroscopy showed that the G-repeat substrates were pre-
dominantly parallel, instead of anti-parallel, G4 structures (Fig-
ure S2D; Vorlı�ckova et al., 2012). Notably, the purified linear
and G4 fractions of the single G-repeat substrate share the iden-
tical DNA sequence and only differ in structure. The same size-
exclusion purification procedure was used in preparing other
linear and G4 structured substrates containing a single G repeat
for in vitro studies.
AID, but Not APOBECs, Prefers Binding andDeaminating G4 Structured Substrates over LinearSubstratesDespite the identical DNA sequence, the purified linear and G4
substrates exhibited distinct behavior in AID binding and deam-
ination assays. The G4 structured substrates displayed�10-fold
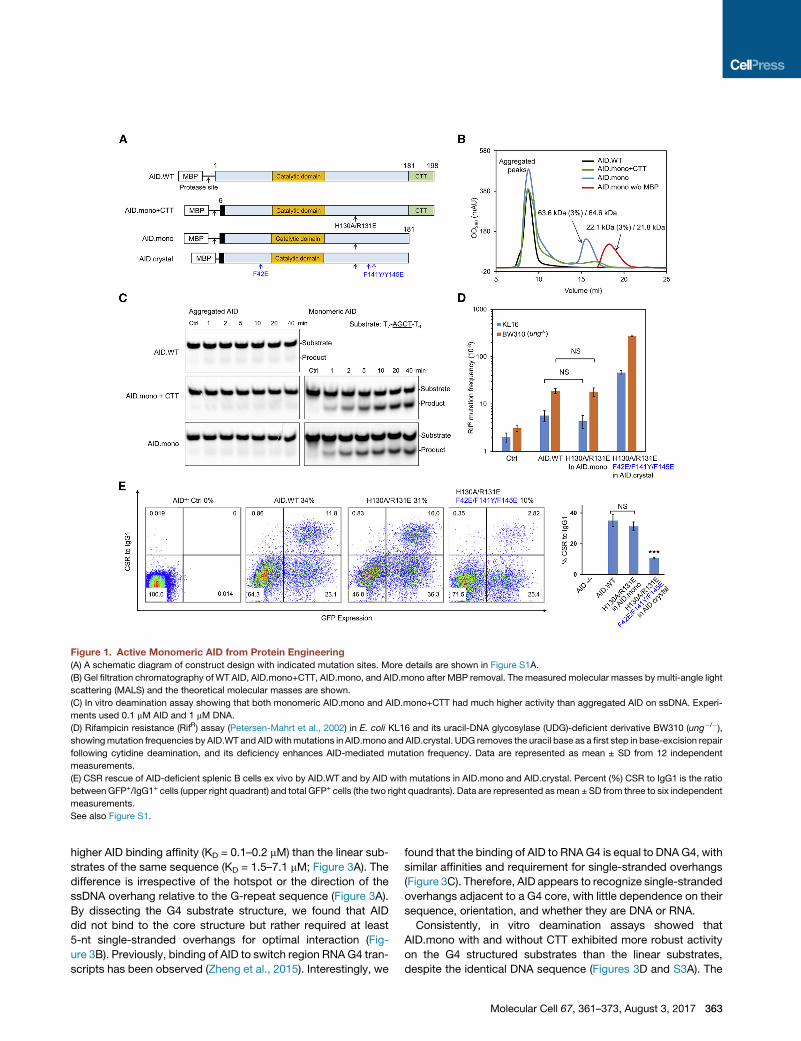
Figure 1. Active Monomeric AID from Protein Engineering
(A) A schematic diagram of construct design with indicated mutation sites. More details are shown in Figure S1A.
(B) Gel filtration chromatography of WT AID, AID.mono+CTT, AID.mono, and AID.mono after MBP removal. The measured molecular masses by multi-angle light
scattering (MALS) and the theoretical molecular masses are shown.
(C) In vitro deamination assay showing that both monomeric AID.mono and AID.mono+CTT had much higher activity than aggregated AID on ssDNA. Experi-
ments used 0.1 mM AID and 1 mM DNA.
(D) Rifampicin resistance (RifR) assay (Petersen-Mahrt et al., 2002) in E. coli KL16 and its uracil-DNA glycosylase (UDG)-deficient derivative BW310 (ung�/�),showingmutation frequencies by AID.WT and AIDwithmutations in AID.mono and AID.crystal. UDG removes the uracil base as a first step in base-excision repair
following cytidine deamination, and its deficiency enhances AID-mediated mutation frequency. Data are represented as mean ± SD from 12 independent
measurements.
(E) CSR rescue of AID-deficient splenic B cells ex vivo by AID.WT and by AID with mutations in AID.mono and AID.crystal. Percent (%) CSR to IgG1 is the ratio
between GFP+/IgG1+ cells (upper right quadrant) and total GFP+ cells (the two right quadrants). Data are represented asmean ± SD from three to six independent
measurements.
See also Figure S1.
higher AID binding affinity (KD = 0.1–0.2 mM) than the linear sub-
strates of the same sequence (KD = 1.5–7.1 mM; Figure 3A). The
difference is irrespective of the hotspot or the direction of the
ssDNA overhang relative to the G-repeat sequence (Figure 3A).
By dissecting the G4 substrate structure, we found that AID
did not bind to the core structure but rather required at least
5-nt single-stranded overhangs for optimal interaction (Fig-
ure 3B). Previously, binding of AID to switch region RNA G4 tran-
scripts has been observed (Zheng et al., 2015). Interestingly, we
found that the binding of AID to RNA G4 is equal to DNA G4, with
similar affinities and requirement for single-stranded overhangs
(Figure 3C). Therefore, AID appears to recognize single-stranded
overhangs adjacent to a G4 core, with little dependence on their
sequence, orientation, and whether they are DNA or RNA.
Consistently, in vitro deamination assays showed that
AID.mono with and without CTT exhibited more robust activity
on the G4 structured substrates than the linear substrates,
despite the identical DNA sequence (Figures 3D and S3A). The
Molecular Cell 67, 361–373, August 3, 2017 363

Figure 2. Linear and G4 Structured Substrates
(A) Two types of AID substrates: one with multiple G-repeats and hotspots as in a mouse Sm fragment and the other with a single G repeat and hotspot. The
potential ability of these substrates to form either intramolecular or intermolecular G4 structure is illustrated.
(B) G4 structure formation confirmed by the G4-specific dye N-methyl mesoporphyrin IX (NMM). LiCl is known to inhibit G4 structures. In total, 10 mM DNA and
20 mMNMMwere used in each experiment. Heat denaturationwas done by 95�C incubation for 10min followed by flash cooling to eliminate DNA structures. Data
are represented as mean ± SD from three independent measurements.
(C) Separation of intermolecular G4 and linear fractions of single G-repeat substrate using a Superdex 75 gel filtration column.
(D) Native gel showing the clear separation of linear and G4 structured substrate from Superdex 75 gel filtration purification in (C).
See also Figure S2.
preference remained irrespective whether the substrates contain
a hotspot (AGCT) or cold spot (TTCT; Figures 3D and S3A).
Remarkably, in a competition assay, linear ssDNAwith or without
hotspots did not affect G4 structured substrate deamination
even at 100-fold molar excess but severely inhibited linear sub-
strate deamination at 10-fold molar excess (Figure 3E). In com-
parison, two AID homologs APOBEC3A and APOBEC3G did
not show a catalytic preference for G4 structured substrate (Fig-
ure 3F), suggesting that the G4 structure preference may be a
unique feature for AID specific functions, like CSR.
Location-Dependent Deamination by AID on G4Structured SubstrateTo probe how AID performs catalysis on G4 structured sub-
strates, we designed a series of G4 substrates with a hotspot
(AACT) or a cold spot (TTCT) located at different positions of
an ssDNA overhang. We found that the peak deamination activ-
ity was achieved when the target deoxycytidine was placed at
the third position 30 to the G4 core (Figures 3G and S3B). AID-
mediated deamination steadily decayed when the deoxycytidine
was moved away from the third position, albeit still significantly
364 Molecular Cell 67, 361–373, August 3, 2017
higher than that for the linear substrate (Figure 3G). Peak activ-
ities at the third position were similar between hotspot- and
cold-spot-containing substrates, suggesting that the G4 struc-
ture might override the hotspot preference in AID targeting. In
positions away from the third position, deamination activity on
hotspot substrates roughly doubled that on cold spot sub-
strates, recapitulating the observed hotspot preference in vitro
and in cells (Larijani et al., 2005; Pham et al., 2003). Notably, in
Ig S regions, a deoxycytidine often exists exactly at the third po-
sition from the G-repeat motif (GGGGTG; Figure S2A), an ideal
site for AID deamination, as suggested by our results.
Because of the significant cooperativity observed in binding
assays using G4 structured substrates (Hill coefficient n R 2;
Figure 3A), we suspected that AID-AID interaction might
occur uponG4 binding. The hypothesis was confirmed by recon-
stituting AID.mono/G4 complex in vitro, which mainly eluted
from the void position of a size-exclusion column with an
apparent measured molecular mass of �1.3 MDa (Figure S3C)
andwas observed as large oligomers under electronmicroscopy
(Figure S3D). To discern the consequence of AID oligomeriza-
tion, we designed a similar series of G4 substrates with the target
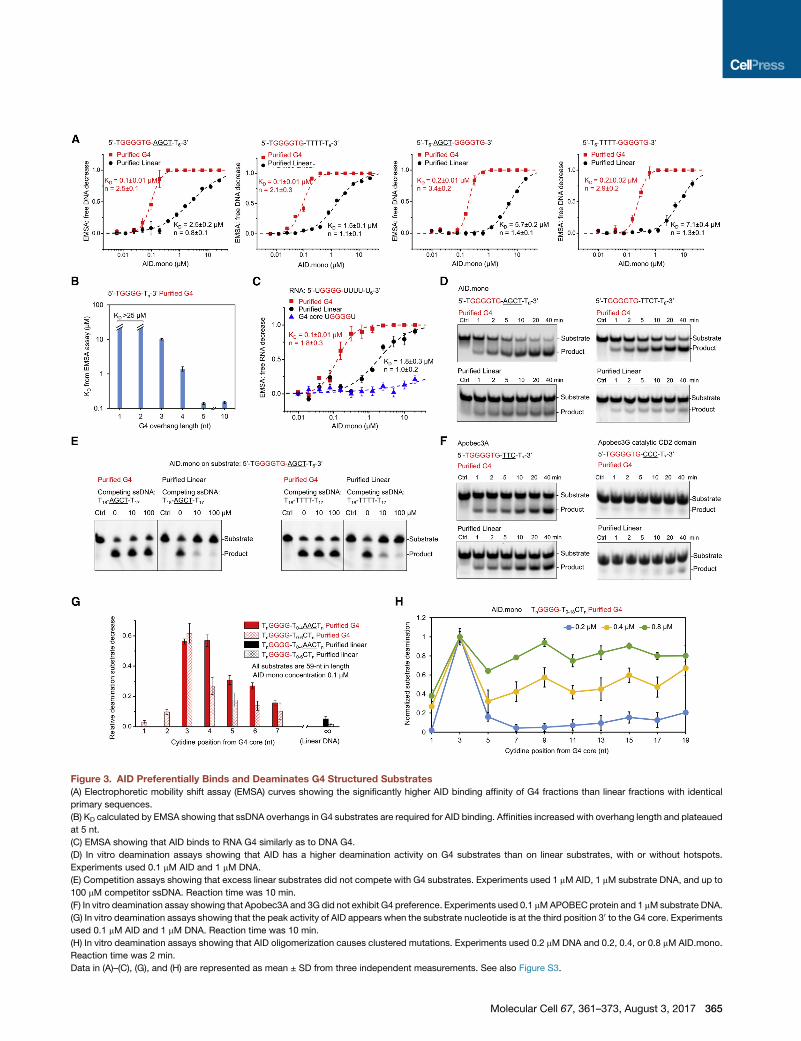
Figure 3. AID Preferentially Binds and Deaminates G4 Structured Substrates
(A) Electrophoretic mobility shift assay (EMSA) curves showing the significantly higher AID binding affinity of G4 fractions than linear fractions with identical
primary sequences.
(B) KD calculated by EMSA showing that ssDNA overhangs in G4 substrates are required for AID binding. Affinities increased with overhang length and plateaued
at 5 nt.
(C) EMSA showing that AID binds to RNA G4 similarly as to DNA G4.
(D) In vitro deamination assays showing that AID has a higher deamination activity on G4 substrates than on linear substrates, with or without hotspots.
Experiments used 0.1 mM AID and 1 mM DNA.
(E) Competition assays showing that excess linear substrates did not compete with G4 substrates. Experiments used 1 mM AID, 1 mM substrate DNA, and up to
100 mM competitor ssDNA. Reaction time was 10 min.
(F) In vitro deamination assay showing that Apobec3A and 3G did not exhibit G4 preference. Experiments used 0.1 mMAPOBEC protein and 1 mMsubstrate DNA.
(G) In vitro deamination assays showing that the peak activity of AID appears when the substrate nucleotide is at the third position 30 to the G4 core. Experiments
used 0.1 mM AID and 1 mM DNA. Reaction time was 10 min.
(H) In vitro deamination assays showing that AID oligomerization causes clustered mutations. Experiments used 0.2 mM DNA and 0.2, 0.4, or 0.8 mM AID.mono.
Reaction time was 2 min.
Data in (A)–(C), (G), and (H) are represented as mean ± SD from three independent measurements. See also Figure S3.
Molecular Cell 67, 361–373, August 3, 2017 365

C located at up to 18 nt away from the G4 core (Figure S3E),
which showed a similar peak of deamination at the third position
at a low AID concentration (Figure 3H, blue trace). However,
when we increased the AID concentration to induce oligomeriza-
tion, we observed that the cytidine sites away from the G4 core
were more efficiently deaminated (Figure 3H, green and yellow
traces), suggesting that AID oligomerization on G4 may spread
the mutations to more distal sites. The peaks of deamination
are separated by �6 nt (Figure 3H), which is consistent with
the 5-nt minimal ssDNA length for AID binding that we identified
earlier (Figure 3B).
Structures of AID and Its Complex with SubstratesBecause oligomerization of AID.mono upon binding to a G4
substrate resulted in a heterogeneous oligomeric complex,
we screened additional mutations on surface hydrophobic resi-
dues that could disrupt AID oligomerization to facilitate crystalli-
zation. We found that the F42E/F141Y/F145E triple mutation
plus shortening of the linker to the MBP tag rendered AID.mono
entirely monomeric as measured by multi-angle light scattering
(MALS) (Figures 1A and S4A). The new construct that we named
AID.crystal maintained G4 preference in vitro (Figure S4B)
and formed a homogeneous AID2/G4 complex without further
oligomerization, as determined byMALS (Figure 4A). The stoichi-
ometry of the complex suggested that each AID is capable of
binding two ssDNA overhangs in the G4 substrate. Confirma-
tively, a designed branched substrate with only two ssDNA over-
hangs displayed a 1:1 interaction with AID.crystal (Figure 4A).
Interestingly, AID.mono bound and deaminated better on the
two-overhang branched substrate in comparison with a single-
overhang substrate, but the binding did not show cooperativity
(Hill coefficient nz1.0; Figures 4B and S4C) as for G4 substrates
(Figure 3A).
The MBP-fused AID.crystal and its catalytically dead mutant
E58A were crystallized in complex with G4 DNA or branched
DNA but did not crystallize alone. However, despite confirmed
presence of DNA in the crystals (Figure S4D), only fragmented
DNA density was visible, which appeared to mediate crystal
packing. Indeed, AID also co-crystallized with blunt-ended dou-
ble-stranded DNA (dsDNA) that it did not bind in solution (Fig-
ure 4B); the dsDNA stacked in the crystal lattice and likely
neutralized repulsion between highly positively charged AID
with isoelectric point (PI) of �9.0 (Figure S4E). Upon trying
many different substrates (Table S1), we obtained in total
seven structures of AID, alone and in complex with cytidine
(C), 20-deoxycytidine (dC), and 20-deoxycytidine-50-monophos-
phate (dCMP) at a highest resolution of 2.4 A (Figures 4C; Table
S2). Importantly, mutations in AID.crystal at residues F42, F141,
and F145 all localize on the opposite side of the active site
defined by the bound dCMP (Figure 4D).
In contrast to the recently reported crystal structure of an AID-
APOBEC3A hybrid (AIDv) alone (Pham et al., 2016), our AID/
dCMP complex captured using the E58A mutant revealed a
deep substrate channel and the direction of ssDNA binding
(Figure 4E). Because of the replacement of residues 7–36 of
AID with those of APOBEC3A, the AIDv structure exhibits dis-
rupted shape and charge distribution at the substrate channel
(Figures 4F and S4F). The active site is comprised of the catalytic
366 Molecular Cell 67, 361–373, August 3, 2017
proton-donating residue E58 and the Zn2+ ion coordinated by
H56, C87, C90, and usually a fourth ligand, e.g., a water in the
dCMP complex or a cacodylic acid from an Apo-AID crystalliza-
tion condition (Figures 4G and S4G). No significant conforma-
tional changes were observed at the active site between WT
and E58A structures (Figure S4H).
Mechanisms of Cytosine Recognition and DNA/RNADifferentiationThe dCMP-defined substrate channel is mainly formed by the
a1-b1, b2-a2, and the b4-a4 loops (Figure 4E), among which the
b4-a4 loop was previously designated as the recognition loop
important for hotspot specificity (Kohli et al., 2009; Wang et al.,
2010). Although there are limited global conformational differ-
ences between structures of AID alone and its complexes, the
side chain of F115 in the b4-a4 recognition loop is antiparallel to
theconservedY114 inall complexstructures (FigureS4I),whereas
a stacking conformation between F115 and Y114 was also
observed in Apo-AID structures (Figure S4I), suggesting that the
flip of F115 may be induced or stabilized by substrate binding.
The cytosine is cradled by aromatic residues H56, W84, and
Y114 (Figure 4G) and precisely positioned by interactions with
surrounding residues. The atom N4 forms hydrogen bonds to
the Zn2+-coordinating water and the carbonyl oxygen of S85,
whereas the atom O2 hydrogen bonds with the hydroxyl of T27
(Figure 4G). If we superimpose the E58-containing Apo-AID
structure, N4 also interacts with the side chain of E58 (Figure 4G).
In contrast, the product uracil possesses an O4 instead of an N4
and cannot form the stabilizing hydrogen bonds, consistent
with the lack of ligand density in AID co-crystallized with uridine
(Table S1). The core arrangement of the AID active site is akin to
that of human cytidine deaminase (CDA) despite a different
structural fold (Figure S4J), with similar Zn2+ coordination and
location of the catalytic Glu (Figure S4K). This structural observa-
tion suggests that AID uses a similar deamination mechanism, in
which the E58 side chain interacts with N3 of the pyrimidine ring
to facilitate nucleophilic attack at C4 by the Zn2+-activated water
for deamination (Chaudhuri and Alt, 2004).
Previous data showed that AID does not deaminate RNA
substrates (Bransteitter et al., 2003). Particularly, replacing
the target dC with C on an otherwise ssDNA substrate abol-
ished AID-induced deamination (Nabel et al., 2013). Support-
ing this observation, we only captured significant electron
density of dCMP, but not CMP, in the catalytic center (Fig-
ure S4L), in spite of similar binding behaviors of AID for DNA
and RNA in vitro (Figures 3A and 3C). In the AID/dCMP com-
plex, R25 interacts with the 50-phosphate and Y114 interacts
with the O50, whereas N51 hydrogen bonds with the 30-OH
(Figure 4G). Compared to the Apo-AID structure, both R25
and N51 undergo significant side chain adjustments upon sub-
strate binding (Figure 4H). Proximity of the carbonyl oxygen of
R25 to the C20 of the deoxyribose indicates a steric hindrance
if the deoxyribose is replaced by a ribose (Figures 4G and 4H).
Without the 50-phosphate, the AID structure in complex with
C or dC either showed different sugar position or much
weaker density (Figures S4L and S4M), suggesting that the
50-phosphate is essential for fixing dCMP orientation in the
catalytic center for DNA/RNA differentiation. The interactions

Figure 4. Structures of AID and Its Complex with dCMP
(A) Gel filtration chromatography with in-line MALS showing that AID.crystal binds G4 DNA in 2:1 ratio and branched DNA in 1:1 ratio. Measured and calculated
molecular masses are labeled.
(B) EMSA curves showing enhanced AID binding affinity for branched substrate with two overhangs (red), in comparison to that with one overhang (linear
substrate, black) or no overhang (dsDNA, blue). Data are represented as mean ± SD from three independent measurements.
(C) Ribbon diagram of human AID in rainbow color showing the secondary structures and catalytic residues near active site Zn2+. W, water.
(D) Locations of F42, F141, and F145 mutated in AID.crystal on the face of the crystal structure opposite to the active site.
(E) Surface charge distribution of the AID/dCMP structure and pocket prediction revealed a substrate binding channel that passes through the active site
(green mesh).
(F) Comparison between the AID-APOBEC3A hybrid AIDv (PDB: 5JJ4) and AID.crystal showed distinct surface charge distribution at the substrate channel.
(G) Substrate dCMP in AID (E58A) catalytic center showing the interactionswith surrounding residues. The E58 side chain is taken from theWTApo-AID structure.
(H) Alignment between Apo- and dCMP-bound AID structures showing the movement of R25 and N51 upon substrate recognition.
(I) Mutations associated with the hyper-IgM syndrome mapped to AID structure.
See also Figure S4 and Tables S1–S3.
Molecular Cell 67, 361–373, August 3, 2017 367
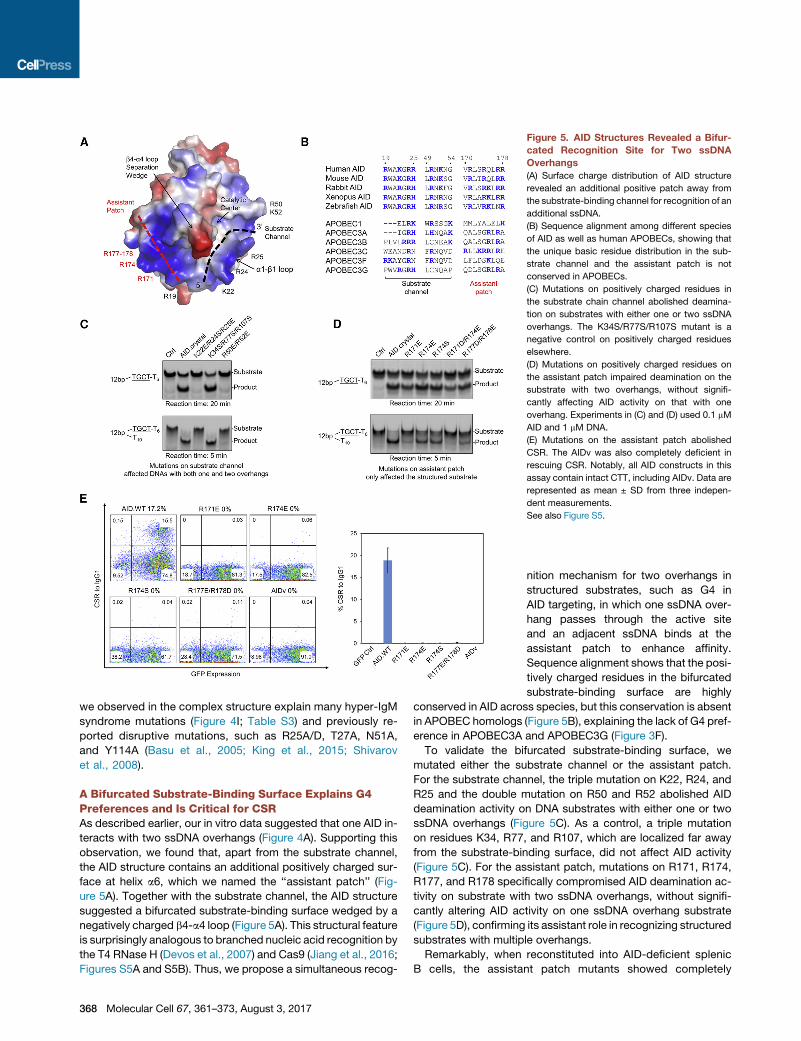
Figure 5. AID Structures Revealed a Bifur-
cated Recognition Site for Two ssDNA
Overhangs
(A) Surface charge distribution of AID structure
revealed an additional positive patch away from
the substrate-binding channel for recognition of an
additional ssDNA.
(B) Sequence alignment among different species
of AID as well as human APOBECs, showing that
the unique basic residue distribution in the sub-
strate channel and the assistant patch is not
conserved in APOBECs.
(C) Mutations on positively charged residues in
the substrate chain channel abolished deamina-
tion on substrates with either one or two ssDNA
overhangs. The K34S/R77S/R107S mutant is a
negative control on positively charged residues
elsewhere.
(D) Mutations on positively charged residues on
the assistant patch impaired deamination on the
substrate with two overhangs, without signifi-
cantly affecting AID activity on that with one
overhang. Experiments in (C) and (D) used 0.1 mM
AID and 1 mM DNA.
(E) Mutations on the assistant patch abolished
CSR. The AIDv was also completely deficient in
rescuing CSR. Notably, all AID constructs in this
assay contain intact CTT, including AIDv. Data are
represented as mean ± SD from three indepen-
dent measurements.
See also Figure S5.
we observed in the complex structure explain many hyper-IgM
syndrome mutations (Figure 4I; Table S3) and previously re-
ported disruptive mutations, such as R25A/D, T27A, N51A,
and Y114A (Basu et al., 2005; King et al., 2015; Shivarov
et al., 2008).
A Bifurcated Substrate-Binding Surface Explains G4Preferences and Is Critical for CSRAs described earlier, our in vitro data suggested that one AID in-
teracts with two ssDNA overhangs (Figure 4A). Supporting this
observation, we found that, apart from the substrate channel,
the AID structure contains an additional positively charged sur-
face at helix a6, which we named the ‘‘assistant patch’’ (Fig-
ure 5A). Together with the substrate channel, the AID structure
suggested a bifurcated substrate-binding surface wedged by a
negatively charged b4-a4 loop (Figure 5A). This structural feature
is surprisingly analogous to branched nucleic acid recognition by
the T4 RNase H (Devos et al., 2007) and Cas9 (Jiang et al., 2016;
Figures S5A and S5B). Thus, we propose a simultaneous recog-
368 Molecular Cell 67, 361–373, August 3, 2017
nition mechanism for two overhangs in
structured substrates, such as G4 in
AID targeting, in which one ssDNA over-
hang passes through the active site
and an adjacent ssDNA binds at the
assistant patch to enhance affinity.
Sequence alignment shows that the posi-
tively charged residues in the bifurcated
substrate-binding surface are highly
conserved in AID across species, but this conservation is absent
in APOBEC homologs (Figure 5B), explaining the lack of G4 pref-
erence in APOBEC3A and APOBEC3G (Figure 3F).
To validate the bifurcated substrate-binding surface, we
mutated either the substrate channel or the assistant patch.
For the substrate channel, the triple mutation on K22, R24, and
R25 and the double mutation on R50 and R52 abolished AID
deamination activity on DNA substrates with either one or two
ssDNA overhangs (Figure 5C). As a control, a triple mutation
on residues K34, R77, and R107, which are localized far away
from the substrate-binding surface, did not affect AID activity
(Figure 5C). For the assistant patch, mutations on R171, R174,
R177, and R178 specifically compromised AID deamination ac-
tivity on substrate with two ssDNA overhangs, without signifi-
cantly altering AID activity on one ssDNA overhang substrate
(Figure 5D), confirming its assistant role in recognizing structured
substrates with multiple overhangs.
Remarkably, when reconstituted into AID-deficient splenic
B cells, the assistant patch mutants showed completely
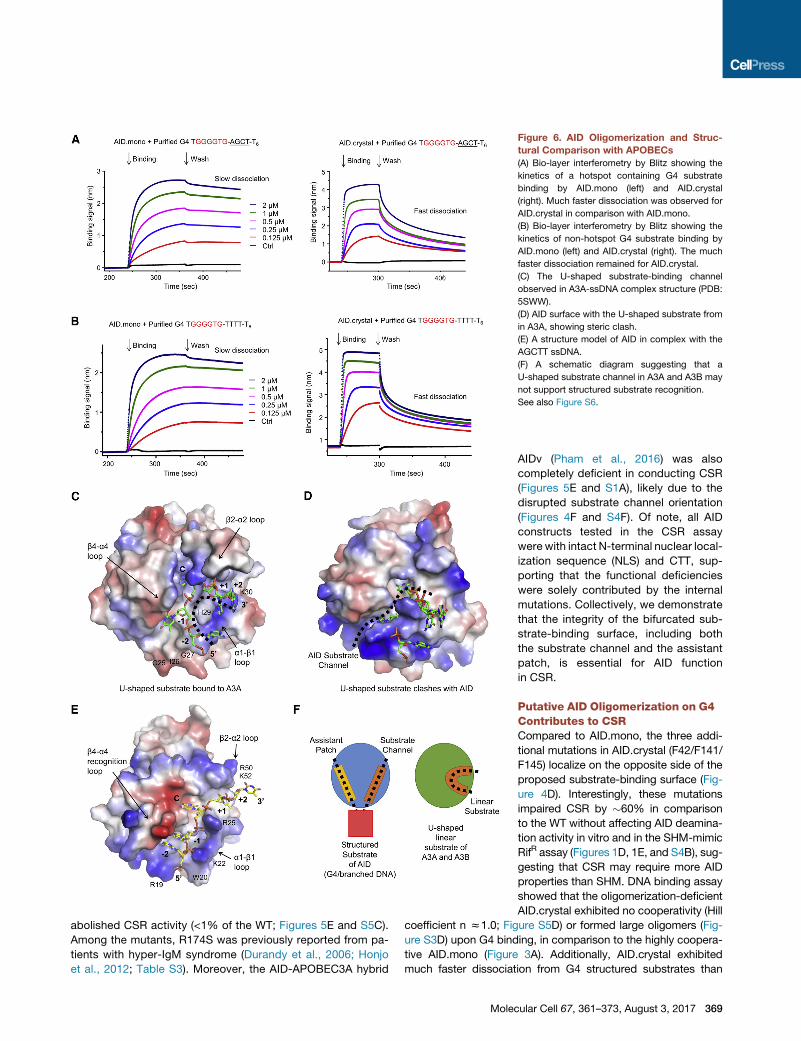
Figure 6. AID Oligomerization and Struc-
tural Comparison with APOBECs
(A) Bio-layer interferometry by Blitz showing the
kinetics of a hotspot containing G4 substrate
binding by AID.mono (left) and AID.crystal
(right). Much faster dissociation was observed for
AID.crystal in comparison with AID.mono.
(B) Bio-layer interferometry by Blitz showing the
kinetics of non-hotspot G4 substrate binding by
AID.mono (left) and AID.crystal (right). The much
faster dissociation remained for AID.crystal.
(C) The U-shaped substrate-binding channel
observed in A3A-ssDNA complex structure (PDB:
5SWW).
(D) AID surface with the U-shaped substrate from
in A3A, showing steric clash.
(E) A structure model of AID in complex with the
AGCTT ssDNA.
(F) A schematic diagram suggesting that a
U-shaped substrate channel in A3A and A3B may
not support structured substrate recognition.
See also Figure S6.
abolished CSR activity (<1% of the WT; Figures 5E and S5C).
Among the mutants, R174S was previously reported from pa-
tients with hyper-IgM syndrome (Durandy et al., 2006; Honjo
et al., 2012; Table S3). Moreover, the AID-APOBEC3A hybrid
Mole
AIDv (Pham et al., 2016) was also
completely deficient in conducting CSR
(Figures 5E and S1A), likely due to the
disrupted substrate channel orientation
(Figures 4F and S4F). Of note, all AID
constructs tested in the CSR assay
were with intact N-terminal nuclear local-
ization sequence (NLS) and CTT, sup-
porting that the functional deficiencies
were solely contributed by the internal
mutations. Collectively, we demonstrate
that the integrity of the bifurcated sub-
strate-binding surface, including both
the substrate channel and the assistant
patch, is essential for AID function
in CSR.
Putative AID Oligomerization on G4Contributes to CSRCompared to AID.mono, the three addi-
tional mutations in AID.crystal (F42/F141/
F145) localize on the opposite side of the
proposed substrate-binding surface (Fig-
ure 4D). Interestingly, these mutations
impaired CSR by �60% in comparison
to the WT without affecting AID deamina-
tion activity in vitro and in the SHM-mimic
RifR assay (Figures 1D, 1E, and S4B), sug-
gesting that CSR may require more AID
properties than SHM. DNA binding assay
showed that the oligomerization-deficient
AID.crystal exhibited no cooperativity (Hill
coefficient n z1.0; Figure S5D) or formed large oligomers (Fig-
ure S3D) upon G4 binding, in comparison to the highly coopera-
tive AID.mono (Figure 3A). Additionally, AID.crystal exhibited
much faster dissociation from G4 structured substrates than
cular Cell 67, 361–373, August 3, 2017 369

Figure 7. A Model of G4-Structure-Medi-
ated AID Recruitment and Oligomerization
that Create Mutation Clusters and DSB
WT-like AID.mono as measured by bio-layer interferometry, irre-
spective of the DNA sequence (Figures 6A and 6B). Based on
these data, we hypothesize that AID oligomerization upon G4
structure binding contributes to CSR by enhancing AID recruit-
ment and accumulation to Ig S regions. It should be noted that
AID.mono only cooperatively oligomerizes on G4, but not on a
simpler structured substrate, such as branched DNA, that may
be formed by local secondary structures (Figure 4B). The AID
oligomerization may be further facilitated by the CTT (Mondal
et al., 2016) and result in previously observed processivity
(Pham et al., 2003).
Comparison of Substrate Recognition by AID andAPOBECsTwo recent papers reported crystal structures of APOBECs
in complex with ssDNAs, including those of the non-catalytic
APOBEC3G N-domain (A3G-N) (Xiao et al., 2016), APOBEC3A
(A3A), and APOBEC3B containing a replaced a1-b1 loop from
A3A (A3B-A chimera; Shi et al., 2017). Consistent with its lack
370 Molecular Cell 67, 361–373, August 3, 2017
of an active site, A3G-N interacts with
ssDNA at a surface shifted from the anal-
ogous AID active site (Figure S6A). The
A3A and A3B-A chimera structures
displayed the same binding mode to
U-shaped ssDNA (Figure 6C), among
which the cognate C at the center binds
similarly as dCMP in AID (Figure 6D).
However, the remaining ssDNA clashes
with the AID surface and does not follow
the observed substrate channel of AID
(Figures 6D and S6B).
To further validate the observed sub-
strate channel in AID, we modeled the
binding of an AGCTT hotspot substrate
using the bound dCMP position as an an-
chor (Figure 6E). We found that the b4-a4
recognition loop stayed in the conforma-
tion observed in the dCMPcomplex struc-
ture (Figure S4I) and that conformation re-
arrangement is only required at the a1-b1
loop to accommodate the substrate at
the �1 and �2 positions (Figures 6E and
S6C). No gross conformational changes
are needed for the superficial binding
at +1 and +2 positions (Figure 6E). The
modeling exercise therefore supports the
observed substrate channel. We suspect
that, if a U-shaped substrate channel ex-
ists in AID as in A3A and A3B, it will likely
not be able to support simultaneous bind-
ing of another ssDNA at the assistant
patch,whichmaybe the reason for the dif-
ference between substrate recognition by AID and APOBECs
(Figure 6F) and for the CSR deficiency of AIDv (Figure 5E).
DISCUSSION
In this study, we combined biochemical, biophysical, and
structural approaches to elucidate AID targeting mechanisms,
especially in CSR. By using the fully functional AID.mono, we
clearly demonstrated that G4 substrates mimicking the Ig S
regions are preferred AID targets in vitro. Different from the pre-
viously proposed hotspot hypothesis, our data indicate that
the AID preference for G4 substrates is predominantly due to
their bundled ssDNA overhangs structure rather than any pri-
mary sequence motif. By solving structures of the MBP-fused
AID.crystal alone and in complex with dCMP, we observed a
bifurcated substrate-binding surface, which strongly supports
a model of one AID recognizing two adjacent ssDNA overhangs
in a structured substrate, such as G4, to achieve high affinity
(Figure 7). Although AID can recruit either DNA or RNA in a

sequence-independent manner, the positioning of dCMP in the
active site suggests that only deoxycytidine, but not cytidine,
can flip into the catalytic center to be deaminated.
Other than Ig S regions, it has been shown that G4 structures
maybeprevalent in other regions ofmammalian genomes (Cham-
bers et al., 2015; Maizels and Gray, 2013). Especially, G-rich re-
gions in AID off-target genes, like c-MYC and BCL6, have been
proposed to be targeted byAID (Duquette et al., 2005, 2007). After
inspecting many identified AID off-target genes, we found that
these genes often contain G-repeat (GGG) enriched regions,
particularly in their non-template strands, which may lead to G4
assembly during transcription and direct AID recruitment (Fig-
ure S6D; Table S4). However, more experimental data and
genome sequence analysis may be required to establish the cor-
relation between G-repeat motif distribution and AID targeting.
Additionally, our data suggest that G4 DNA-binding-induced
cooperative AID oligomerization may contribute to its accumula-
tion in Ig S regions, promoting high-density mutations, DSBs, and
their joining during CSR (Figure 7). In comparison, SHM can be
induced by CSR-deficient AID variants in cells (Hwang et al.,
2015;Phamet al., 2016) andbyAPOBEC3homologsduring retro-
viral infection (Halemano et al., 2014), suggesting that mutations
in Ig V regions may not require all the AID features employed in
CSR. Because chromatin immunoprecipitation sequencing data
showed that Ig V regions may not bind AID as stably as S regions
(Matthews et al., 2014b), and the assistant patch mutant R174S
also causes SHM deficiency (Durandy et al., 2006; Honjo et al.,
2012), we speculate that Ig V regions may only transiently recruit
AID using local secondary structures like branched DNA, which
do not induce AID oligomerization and stable association.
Although our biochemical data and the bipartite DNA binding
surface in AID structure strongly suggest a model of how AID rec-
ognizes structured substrates (Figure 6F), a definitive complex
structure that contains fully characterized substrate conformation
is still lacking, despite extensive efforts using both crystallography
and electron microscopy (EM). Co-crystallization of purified AID/
G4, AID/branched DNA, and AID/linear DNA complexes did not
yield visible density for the substrates, likely due to crystal packing
effects. Alternatively, we tried co-crystallization using 1- to 5-nt
ssDNA fragments together with a 12-bp dsDNA (Table S1), but
any ssDNA fragment longer than 1 nt did not reveal any density
in the active site. Neither did soaking of various substrates into
pre-formed AID crystals (Table S1). We further used EM on the
AID/G4 complex, but the flexibility of the complex caused issues
in the reconstruction (Figure S6E). Thus, the exactmolecular basis
for ourmodel remains tobedeterminedby futurestructural studies
of AID/DNAcomplexes. Due to the close correlation of AID activity
with B cell lymphoma and other types of cancers, the AID struc-
tureswill alsoprovide templates for potential therapeutic interven-
tion against this important cytidine deaminase.
STAR+METHODS
Detailed methods are provided in the online version of this paper
and include the following:
d KEY RESOURCES TABLE
d CONTACT FOR REAGENT AND RESOURCE SHARING
d METHOD DETAILS
B Protein Engineering and Purification
B Ex Vivo CSR Assay
B Rifampicin Resistance (RifR) Assay
B In Vitro Deamination Assay
B In Vitro EMSA
B G4 and Branched DNA Purification and Character-
ization
B AID/DNA Complex Crystallization and Structure Deter-
mination
B Molecular Mass Measurement by Multi-Angle Light
Scattering (MALS)
B Electron Microscopy
d QUANTIFICATION AND STATISTICAL ANALYSES
B In Vitro EMSA and Deamination Assay Quantification
B Data Analysis in Flow Cytometry
d DATA AND SOFTWARE AVAILABILITY
B Accession Numbers
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and four tables can be found
with this article online at http://dx.doi.org/10.1016/j.molcel.2017.06.034.
AUTHOR CONTRIBUTIONS
H.W. supervised the project. Q.Q. and L.W. designed the experiments and
performed protein engineering, in vitro assays, structure determination, and
analysis. F.-L.M. performed ex vivo CSR assay, and F.W.A. supervised the
effort. H.W. and Q.Q. wrote most of the manuscript with help from J.K.H.
and F.W.A. for the introduction. All authors contributed to data analysis and
critical interpretation of results and approved the manuscript.
ACKNOWLEDGMENTS
We thank Ermelinda Damko and Devendra Srivastava for their earlier work on
this project; Ming Tian, Zhou Du, Leng Siew Yeap, Jiazhi Hu, and Junchao
Dong for discussions; Yang Li for EM data analysis; Xia Xie for assistances
in CSR assay; Rida Mourtada in Dr. Loren D. Walensky’s lab and Kelly Arnett
in Harvard Medical School Center for Macromolecular Interactions for their
assistance with CD spectroscopy; Sukumar Narayanasami and Surajit Bane-
rjee of NE-CAT at the Advance Photon Source for their assistance on data
collection; andMaria Ericsson and Louise Trakimas of HarvardMedical School
EM facility for assistance on EM imaging. This work was supported by a Can-
cer Research Institute Irvington Postdoctoral Fellowship (to Q.Q.), a Lym-
phoma Research Foundation Fellowship (to F.-L.M.), NIH R01 AI077595 (to
F.W.A.), and NIH F30 AI114179 (to J.K.H.). F.W.A. is an Investigator of the Ho-
ward Hughes Medical Institute.
Received: March 1, 2017
Revised: May 3, 2017
Accepted: June 27, 2017
Published: July 27, 2017
REFERENCES
Adams, P.D., Afonine, P.V., Bunkoczi, G., Chen, V.B., Davis, I.W., Echols, N.,
Headd, J.J., Hung, L.W., Kapral, G.J., Grosse-Kunstleve, R.W., et al. (2010).
PHENIX: a comprehensive Python-based system for macromolecular struc-
ture solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221.
Bardin, C., and Leroy, J.L. (2008). The formation pathway of tetramolecular
G-quadruplexes. Nucleic Acids Res. 36, 477–488.
Molecular Cell 67, 361–373, August 3, 2017 371

Basu, U., Chaudhuri, J., Alpert, C., Dutt, S., Ranganath, S., Li, G., Schrum, J.P.,
Manis, J.P., and Alt, F.W. (2005). The AID antibody diversification enzyme is
regulated by protein kinase A phosphorylation. Nature 438, 508–511.
Bransteitter, R., Pham, P., Scharff, M.D., and Goodman, M.F. (2003).
Activation-induced cytidine deaminase deaminates deoxycytidine on single-
stranded DNA but requires the action of RNase. Proc. Natl. Acad. Sci. USA
100, 4102–4107.
Chambers, V.S., Marsico, G., Boutell, J.M., Di Antonio, M., Smith, G.P., and
Balasubramanian, S. (2015). High-throughput sequencing of DNA G-quadru-
plex structures in the human genome. Nat. Biotechnol. 33, 877–881.
Chaudhuri, J., and Alt, F.W. (2004). Class-switch recombination: interplay of
transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 4,
541–552.
Chaudhuri, J., Tian, M., Khuong, C., Chua, K., Pinaud, E., and Alt, F.W. (2003).
Transcription-targeted DNA deamination by the AID antibody diversification
enzyme. Nature 422, 726–730.
Chaudhuri, J., Khuong, C., and Alt, F.W. (2004). Replication protein A interacts
with AID to promote deamination of somatic hypermutation targets. Nature
430, 992–998.
Cheng, H.L., Vuong, B.Q., Basu, U., Franklin, A., Schwer, B., Astarita, J., Phan,
R.T., Datta, A., Manis, J., Alt, F.W., and Chaudhuri, J. (2009). Integrity of the
AID serine-38 phosphorylation site is critical for class switch recombination
and somatic hypermutation in mice. Proc. Natl. Acad. Sci. USA 106,
2717–2722.
Devos, J.M., Tomanicek, S.J., Jones, C.E., Nossal, N.G., and Mueser, T.C.
(2007). Crystal structure of bacteriophage T4 50 nuclease in complex with a
branched DNA reveals how flap endonuclease-1 family nucleases bind their
substrates. J. Biol. Chem. 282, 31713–31724.
Duke, J.L., Liu, M., Yaari, G., Khalil, A.M., Tomayko, M.M., Shlomchik, M.J.,
Schatz, D.G., and Kleinstein, S.H. (2013). Multiple transcription factor binding
sites predict AID targeting in non-Ig genes. J. Immunol. 190, 3878–3888.
Duquette, M.L., Handa, P., Vincent, J.A., Taylor, A.F., and Maizels, N. (2004).
Intracellular transcription of G-rich DNAs induces formation of G-loops, novel
structures containing G4 DNA. Genes Dev. 18, 1618–1629.
Duquette, M.L., Pham, P., Goodman, M.F., and Maizels, N. (2005). AID binds
to transcription-induced structures in c-MYC that map to regions associated
with translocation and hypermutation. Oncogene 24, 5791–5798.
Duquette, M.L., Huber, M.D., and Maizels, N. (2007). G-rich proto-oncogenes
are targeted for genomic instability in B-cell lymphomas. Cancer Res. 67,
2586–2594.
Durandy, A., Peron, S., Taubenheim, N., and Fischer, A. (2006). Activation-
induced cytidine deaminase: structure-function relationship as based on the
study of mutants. Hum. Mutat. 27, 1185–1191.
Emsley, P., Lohkamp, B., Scott, W.G., and Cowtan, K. (2010). Features and
development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501.
Halemano, K., Guo, K., Heilman, K.J., Barrett, B.S., Smith, D.S., Hasenkrug,
K.J., and Santiago, M.L. (2014). Immunoglobulin somatic hypermutation by
APOBEC3/Rfv3 during retroviral infection. Proc. Natl. Acad. Sci. USA 111,
7759–7764.
Han, L., Masani, S., and Yu, K. (2011). Overlapping activation-induced cytidine
deaminase hotspot motifs in Ig class-switch recombination. Proc. Natl. Acad.
Sci. USA 108, 11584–11589.
Honjo, T., Kobayashi, M., Begum, N., Kotani, A., Sabouri, S., and Nagaoka, H.
(2012). The AID dilemma: infection, or cancer? Adv. Cancer Res. 113, 1–44.
Hwang, J.K., Alt, F.W., and Yeap, L.S. (2015). Relatedmechanisms of antibody
somatic hypermutation and class switch recombination. Microbiol. Spectr. 3,
MDNA3-0037-2014.
Jiang, F., Taylor, D.W., Chen, J.S., Kornfeld, J.E., Zhou, K., Thompson, A.J.,
Nogales, E., and Doudna, J.A. (2016). Structures of a CRISPR-Cas9 R-loop
complex primed for DNA cleavage. Science 351, 867–871.
King, J.J., Manuel, C.A., Barrett, C.V., Raber, S., Lucas, H., Sutter, P., and
Larijani, M. (2015). Catalytic pocket inaccessibility of activation-induced
372 Molecular Cell 67, 361–373, August 3, 2017
cytidine deaminase is a safeguard against excessive mutagenic activity.
Structure 23, 615–627.
Kitamura, S., Ode, H., Nakashima,M., Imahashi, M., Naganawa, Y., Kurosawa,
T., Yokomaku, Y., Yamane, T., Watanabe, N., Suzuki, A., et al. (2012). The
APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat.
Struct. Mol. Biol. 19, 1005–1010.
Kohli, R.M., Abrams, S.R., Gajula, K.S., Maul, R.W., Gearhart, P.J., and
Stivers, J.T. (2009). A portable hot spot recognition loop transfers sequence
preferences from APOBEC family members to activation-induced cytidine
deaminase. J. Biol. Chem. 284, 22898–22904.
Larijani, M., Frieder, D., Basit, W., and Martin, A. (2005). The mutation spec-
trum of purified AID is similar to the mutability index in Ramos cells and in
ung(-/-)msh2(-/-) mice. Immunogenetics 56, 840–845.
Larijani, M., Petrov, A.P., Kolenchenko, O., Berru, M., Krylov, S.N., and Martin,
A. (2007). AID associates with single-strandedDNAwith high affinity and a long
complex half-life in a sequence-independent manner. Mol. Cell. Biol.
27, 20–30.
Maizels, N., and Gray, L.T. (2013). The G4 genome. PLoS Genet. 9, e1003468.
Matthews, A.J., Zheng, S., DiMenna, L.J., and Chaudhuri, J. (2014a).
Regulation of immunoglobulin class-switch recombination: choreography of
noncoding transcription, targeted DNA deamination, and long-range DNA
repair. Adv. Immunol. 122, 1–57.
Matthews, A.J., Husain, S., and Chaudhuri, J. (2014b). Binding of AID to DNA
does not correlate with mutator activity. J. Immunol. 193, 252–257.
McKean, D., Huppi, K., Bell, M., Staudt, L., Gerhard, W., and Weigert, M.
(1984). Generation of antibody diversity in the immune response of BALB/c
mice to influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA 81,
3180–3184.
Meng, F.L., Du, Z., Federation, A., Hu, J., Wang, Q., Kieffer-Kwon, K.R.,
Meyers, R.M., Amor, C., Wasserman, C.R., Neuberg, D., et al. (2014).
Convergent transcription at intragenic super-enhancers targets AID-initiated
genomic instability. Cell 159, 1538–1548.
Mondal, S., Begum, N.A., Hu, W., and Honjo, T. (2016). Functional require-
ments of AID’s higher order structures and their interaction with RNA-binding
proteins. Proc. Natl. Acad. Sci. USA 113, E1545–E1554.
Nabel, C.S., Lee, J.W., Wang, L.C., and Kohli, R.M. (2013). Nucleic acid deter-
minants for selective deamination of DNA over RNA by activation-induced
deaminase. Proc. Natl. Acad. Sci. USA 110, 14225–14230.
Pavri, R., Gazumyan, A., Jankovic, M., Di Virgilio, M., Klein, I., Ansarah-
Sobrinho, C., Resch, W., Yamane, A., Reina San-Martin, B., Barreto, V.,
et al. (2010). Activation-induced cytidine deaminase targets DNA at sites of
RNA polymerase II stalling by interaction with Spt5. Cell 143, 122–133.
Petersen-Mahrt, S.K., Harris, R.S., and Neuberger, M.S. (2002). AIDmutates E.
coli suggesting a DNA deamination mechanism for antibody diversification.
Nature 418, 99–103.
Pham, P., Bransteitter, R., Petruska, J., and Goodman, M.F. (2003).
Processive AID-catalysed cytosine deamination on single-stranded DNA sim-
ulates somatic hypermutation. Nature 424, 103–107.
Pham, P., Afif, S.A., Shimoda, M., Maeda, K., Sakaguchi, N., Pedersen, L.C.,
andGoodman, M.F. (2016). Structural analysis of the activation-induced deox-
ycytidine deaminase required in immunoglobulin diversification. DNA Repair
(Amst.) 43, 48–56.
Qian, J., Wang, Q., Dose, M., Pruett, N., Kieffer-Kwon, K.R., Resch, W., Liang,
G., Tang, Z., Mathe, E., Benner, C., et al. (2014). B cell super-enhancers and
regulatory clusters recruit AID tumorigenic activity. Cell 159, 1524–1537.
Rajewsky, K., Forster, I., and Cumano, A. (1987). Evolutionary and somatic se-
lection of the antibody repertoire in the mouse. Science 238, 1088–1094.
Rogozin, I.B., and Diaz, M. (2004). Cutting edge: DGYW/WRCH is a better pre-
dictor of mutability at G:C bases in Ig hypermutation than the widely accepted
RGYW/WRCY motif and probably reflects a two-step activation-induced cyti-
dine deaminase-triggered process. J. Immunol. 172, 3382–3384.
Shi, K., Carpenter, M.A., Banerjee, S., Shaban, N.M., Kurahashi, K.,
Salamango, D.J., McCann, J.L., Starrett, G.J., Duffy, J.V., Demir, O., et al.

(2017). Structural basis for targeted DNA cytosine deamination and muta-
genesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 24, 131–139.
Shivarov, V., Shinkura, R., and Honjo, T. (2008). Dissociation of in vitro DNA
deamination activity and physiological functions of AID mutants. Proc. Natl.
Acad. Sci. USA 105, 15866–15871.
Sun, D., and Hurley, L.H. (2010). Biochemical techniques for the characteriza-
tion of G-quadruplex structures: EMSA, DMS footprinting, and DNA poly-
merase stop assay. Methods Mol. Biol. 608, 65–79.
Vorlı�ckova, M., Kejnovska, I., Sagi, J., Ren�ciuk, D., Bedna�rova, K., Motlova, J.,
and Kypr, J. (2012). Circular dichroism and guanine quadruplexes. Methods
57, 64–75.
Wang, M., Yang, Z., Rada, C., and Neuberger, M.S. (2009). AID upmutants iso-
lated using a high-throughput screen highlight the immunity/cancer balance
limiting DNA deaminase activity. Nat. Struct. Mol. Biol. 16, 769–776.
Wang, M., Rada, C., and Neuberger, M.S. (2010). Altering the spectrum of
immunoglobulin V gene somatic hypermutation by modifying the active site
of AID. J. Exp. Med. 207, 141–153.
Winn, M.D., Ballard, C.C., Cowtan, K.D., Dodson, E.J., Emsley, P., Evans,
P.R., Keegan, R.M., Krissinel, E.B., Leslie, A.G., McCoy, A., et al. (2011).
Overview of the CCP4 suite and current developments. Acta Crystallogr. D
Biol. Crystallogr. 67, 235–242.
Xiao, X., Li, S.X., Yang, H., and Chen, X.S. (2016). Crystal structures of
APOBEC3G N-domain alone and its complex with DNA. Nat. Commun.
7, 12193.
Xu, Z., Fulop, Z., Wu, G., Pone, E.J., Zhang, J., Mai, T., Thomas, L.M.,
Al-Qahtani, A., White, C.A., Park, S.R., et al. (2010). 14-3-3 adaptor proteins
recruit AID to 50-AGCT-30-rich switch regions for class switch recombination.
Nat. Struct. Mol. Biol. 17, 1124–1135.
Xu, Z., Zan, H., Pone, E.J., Mai, T., and Casali, P. (2012). Immunoglobulin
class-switch DNA recombination: induction, targeting and beyond. Nat. Rev.
Immunol. 12, 517–531.
Yeap, L.S., Hwang, J.K., Du, Z., Meyers, R.M., Meng, F.L., Jakubauskait _e, A.,
Liu, M., Mani, V., Neuberg, D., Kepler, T.B., et al. (2015). Sequence-intrinsic
mechanisms that target AID mutational outcomes on antibody genes. Cell
163, 1124–1137.
Yu, K., Chedin, F., Hsieh, C.L.,Wilson, T.E., and Lieber, M.R. (2003). R-loops at
immunoglobulin class switch regions in the chromosomes of stimulated B
cells. Nat. Immunol. 4, 442–451.
Yu, J., Zhou, Y., Tanaka, I., and Yao, M. (2010). Roll: a new algorithm for the
detection of protein pockets and cavities with a rolling probe sphere.
Bioinformatics 26, 46–52.
Zheng, S., Vuong, B.Q., Vaidyanathan, B., Lin, J.Y., Huang, F.T., and
Chaudhuri, J. (2015). Non-coding RNA generated following lariat debranching
mediates targeting of AID to DNA. Cell 161, 762–773.
Molecular Cell 67, 361–373, August 3, 2017 373
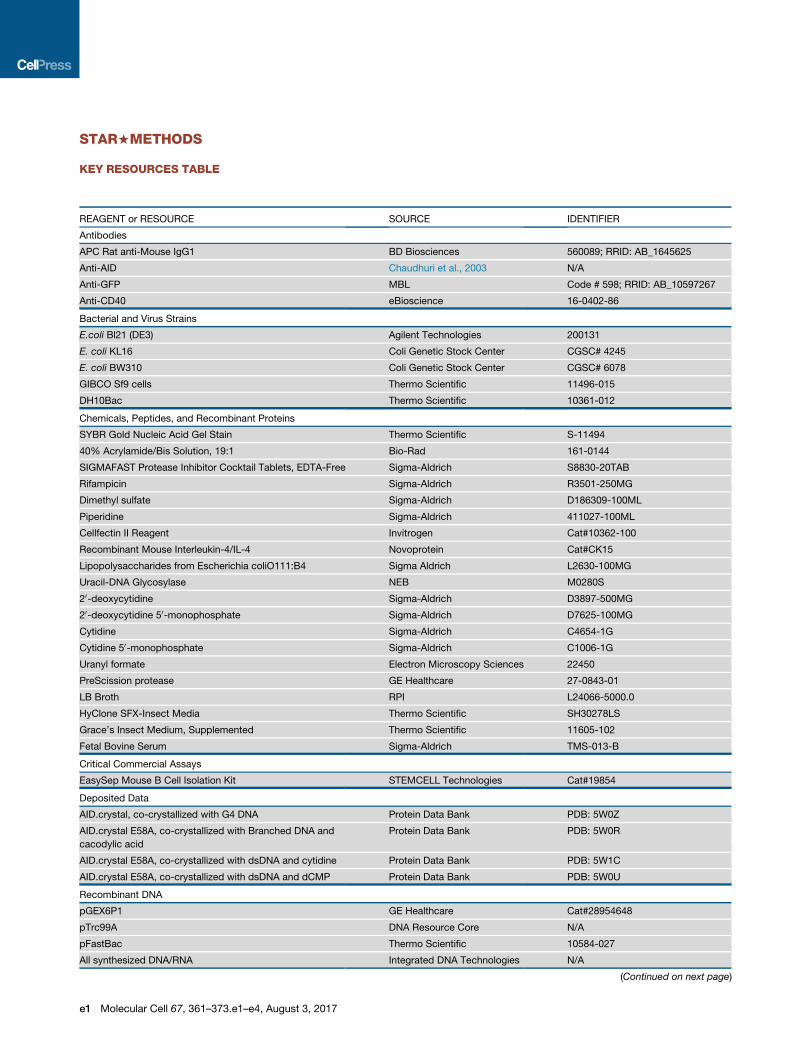
STAR+METHODS
KEY RESOURCES TABLE
REAGENT or RESOURCE SOURCE IDENTIFIER
Antibodies
APC Rat anti-Mouse IgG1 BD Biosciences 560089; RRID: AB_1645625
Anti-AID Chaudhuri et al., 2003 N/A
Anti-GFP MBL Code # 598; RRID: AB_10597267
Anti-CD40 eBioscience 16-0402-86
Bacterial and Virus Strains
E.coli Bl21 (DE3) Agilent Technologies 200131
E. coli KL16 Coli Genetic Stock Center CGSC# 4245
E. coli BW310 Coli Genetic Stock Center CGSC# 6078
GIBCO Sf9 cells Thermo Scientific 11496-015
DH10Bac Thermo Scientific 10361-012
Chemicals, Peptides, and Recombinant Proteins
SYBR Gold Nucleic Acid Gel Stain Thermo Scientific S-11494
40% Acrylamide/Bis Solution, 19:1 Bio-Rad 161-0144
SIGMAFAST Protease Inhibitor Cocktail Tablets, EDTA-Free Sigma-Aldrich S8830-20TAB
Rifampicin Sigma-Aldrich R3501-250MG
Dimethyl sulfate Sigma-Aldrich D186309-100ML
Piperidine Sigma-Aldrich 411027-100ML
Cellfectin II Reagent Invitrogen Cat#10362-100
Recombinant Mouse Interleukin-4/IL-4 Novoprotein Cat#CK15
Lipopolysaccharides from Escherichia coliO111:B4 Sigma Aldrich L2630-100MG
Uracil-DNA Glycosylase NEB M0280S
20-deoxycytidine Sigma-Aldrich D3897-500MG
20-deoxycytidine 50-monophosphate Sigma-Aldrich D7625-100MG
Cytidine Sigma-Aldrich C4654-1G
Cytidine 50-monophosphate Sigma-Aldrich C1006-1G
Uranyl formate Electron Microscopy Sciences 22450
PreScission protease GE Healthcare 27-0843-01
LB Broth RPI L24066-5000.0
HyClone SFX-Insect Media Thermo Scientific SH30278LS
Grace’s Insect Medium, Supplemented Thermo Scientific 11605-102
Fetal Bovine Serum Sigma-Aldrich TMS-013-B
Critical Commercial Assays
EasySep Mouse B Cell Isolation Kit STEMCELL Technologies Cat#19854
Deposited Data
AID.crystal, co-crystallized with G4 DNA Protein Data Bank PDB: 5W0Z
AID.crystal E58A, co-crystallized with Branched DNA and
cacodylic acid
Protein Data Bank PDB: 5W0R
AID.crystal E58A, co-crystallized with dsDNA and cytidine Protein Data Bank PDB: 5W1C
AID.crystal E58A, co-crystallized with dsDNA and dCMP Protein Data Bank PDB: 5W0U
Recombinant DNA
pGEX6P1 GE Healthcare Cat#28954648
pTrc99A DNA Resource Core N/A
pFastBac Thermo Scientific 10584-027
All synthesized DNA/RNA Integrated DNA Technologies N/A
(Continued on next page)
e1 Molecular Cell 67, 361–373.e1–e4, August 3, 2017
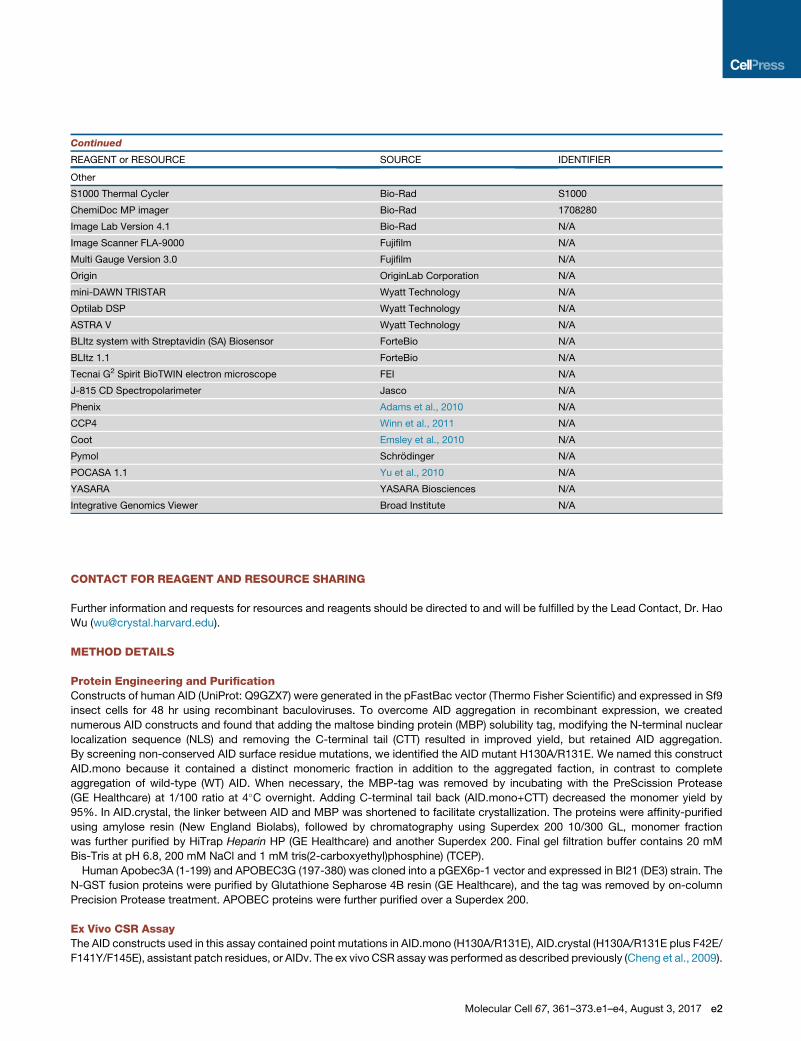
Continued
REAGENT or RESOURCE SOURCE IDENTIFIER
Other
S1000 Thermal Cycler Bio-Rad S1000
ChemiDoc MP imager Bio-Rad 1708280
Image Lab Version 4.1 Bio-Rad N/A
Image Scanner FLA-9000 Fujifilm N/A
Multi Gauge Version 3.0 Fujifilm N/A
Origin OriginLab Corporation N/A
mini-DAWN TRISTAR Wyatt Technology N/A
Optilab DSP Wyatt Technology N/A
ASTRA V Wyatt Technology N/A
BLItz system with Streptavidin (SA) Biosensor ForteBio N/A
BLItz 1.1 ForteBio N/A
Tecnai G2 Spirit BioTWIN electron microscope FEI N/A
J-815 CD Spectropolarimeter Jasco N/A
Phenix Adams et al., 2010 N/A
CCP4 Winn et al., 2011 N/A
Coot Emsley et al., 2010 N/A
Pymol Schrodinger N/A
POCASA 1.1 Yu et al., 2010 N/A
YASARA YASARA Biosciences N/A
Integrative Genomics Viewer Broad Institute N/A
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Hao
Wu ([email protected]).
METHOD DETAILS
Protein Engineering and PurificationConstructs of human AID (UniProt: Q9GZX7) were generated in the pFastBac vector (Thermo Fisher Scientific) and expressed in Sf9
insect cells for 48 hr using recombinant baculoviruses. To overcome AID aggregation in recombinant expression, we created
numerous AID constructs and found that adding the maltose binding protein (MBP) solubility tag, modifying the N-terminal nuclear
localization sequence (NLS) and removing the C-terminal tail (CTT) resulted in improved yield, but retained AID aggregation.
By screening non-conserved AID surface residue mutations, we identified the AID mutant H130A/R131E. We named this construct
AID.mono because it contained a distinct monomeric fraction in addition to the aggregated faction, in contrast to complete
aggregation of wild-type (WT) AID. When necessary, the MBP-tag was removed by incubating with the PreScission Protease
(GE Healthcare) at 1/100 ratio at 4�C overnight. Adding C-terminal tail back (AID.mono+CTT) decreased the monomer yield by
95%. In AID.crystal, the linker between AID and MBP was shortened to facilitate crystallization. The proteins were affinity-purified
using amylose resin (New England Biolabs), followed by chromatography using Superdex 200 10/300 GL, monomer fraction
was further purified by HiTrap Heparin HP (GE Healthcare) and another Superdex 200. Final gel filtration buffer contains 20 mM
Bis-Tris at pH 6.8, 200 mM NaCl and 1 mM tris(2-carboxyethyl)phosphine) (TCEP).
Human Apobec3A (1-199) and APOBEC3G (197-380) was cloned into a pGEX6p-1 vector and expressed in Bl21 (DE3) strain. The
N-GST fusion proteins were purified by Glutathione Sepharose 4B resin (GE Healthcare), and the tag was removed by on-column
Precision Protease treatment. APOBEC proteins were further purified over a Superdex 200.
Ex Vivo CSR AssayThe AID constructs used in this assay contained point mutations in AID.mono (H130A/R131E), AID.crystal (H130A/R131E plus F42E/
F141Y/F145E), assistant patch residues, or AIDv. The ex vivo CSR assaywas performed as described previously (Cheng et al., 2009).
Molecular Cell 67, 361–373.e1–e4, August 3, 2017 e2

Briefly, AID-deficient mouse splenic B cells were stimulated with IL-4 (Novoprotein) and anti-CD40 (eBioscience) to induce CSR to
IgG1. One day after stimulation, WT or mutant AID together with GFP via an internal ribosome entry site (IRES) was retrovirally deliv-
ered into the cells. The percentage of GFP-positive cells that underwent switching to IgG1 was taken as the level of CSR rescue. The
data for each mutant were either from 3 or 6 mice.
Rifampicin Resistance (RifR) AssayWT and mutant AID were cloned into the pTrc99A vector (pTrc99A-AID) and the rifampicin resistance (RifR) assay was performed as
previously described (Wang et al., 2009). Briefly, E. coli strain KL16 (Hfr [PO-45] relA1 spoT1 thi-1) and its UDG-deficient derivative
BW310 bacteria transformedwith pTrc99A-AID plasmidswere grown overnight to saturation in LBmedium supplementedwith ampi-
cillin (100 mg/ml) and isopropyl b-D-1-thiogalactopyranoside (IPTG, 1 mM), and plated on LB low-salt agar containing ampicillin
(100 mg/ml) and rifampicin (50 mg/ml). Mutation frequency was measured by determining the median number of colony-forming cells
that survived selection per 107 viable cells plated from 12 independent cultures. The identity of mutations was determined by
sequencing the relevant section of rpoB (typically from 25 to 200 individual colonies) after PCR amplification using oligonucleotides
50-TTGGCGAAATGGCGGAAAACC-30 and 50-CACCGACGGATACCACCTGCTG-30) (synthesized by Integrated DNA Technologies).
In Vitro Deamination AssayEach 10ml reaction contained 0.1 or 1 mMAID, and 1 mMsubstrateDNA labeled by 6-carboxyfluorescein (FAM) at 50 end (synthesized by
Integrated DNA Technologies) in 20 mMHEPES at pH 7.5, 100 mM KCl and 1 mMDTT. Following incubation at 37�C for the indicated
length of time, each reaction was raised to 95�C for 10 min and flash cooled to inactivate AID and resolve DNA structures. Sufficient
amount of UDG (NewEngland Biolabs, 5 unit with each unit catalyzing 60 pmol/min at 37�C) was then added and themixture was incu-
bated at 37�C for 1h. Lastly, NaOH was added to a final concentration of 0.15 M, and treated at 95�C for 15 min to break abasic sites.
Urea gelwasused to separate the product from the substrate. Experiments in Figures 5Gand 5HusedSYBRGold staining to save cost.
Images were taken on Image Scanner FLA-9000 (Fujifilm) using excitation wavelength of 495 nm and emission wavelength of 519 nm.
Quantification was performed using the Image Lab (Bio-Rad) and Multi Gauge (Fujifilm) software.
In Vitro EMSAUp to 25 mMAIDwas titrated into 10 nM FAM labeled DNA followed by 10%–12%acrylamide:bisacrylamide (19:1) native gel to sepa-
rate free DNA and the AID/DNA complex. Quantified free DNA amounts at different AID concentrations were used to calculate the
dissociation constant (KD) and the Hill coefficient (n) of the AID/DNA interaction. 100 nM unlabeled TGGGGT1-5 and TGGGGT10were used in experiments of Figure 3B to eliminate any effect from the fluorophore and its linker to DNA. SYBR Gold was used
for staining. Images were taken on Image Scanner FLA-9000 (Fujifilm). TheMulti Gauge (Fujifilm) software was used for quantification
and the Origin software (OriginLab Corporation) was used in curve fitting.
G4 and Branched DNA Purification and CharacterizationTo generate mostly G4 structures, Sm fragment and G-repeat DNAs were dissolved in 20 mM HEPES at pH 7.5, 100 mM KCl and
1 mM DTT, incubated at 95�C for 5 min and slowly cooled down to room temperature. In contrast, to generate mostly linear DNA,
heat denaturation at 95�C for 10 min followed by flash cooling on ice was used to eliminate DNA structures. A Jasco J-815 Circular
dichroism (CD) spectropolarimeter was used in determining the parallel and anti-parallel conformation of the G4 assembly (Vor-
lı�ckova et al., 2012). When preparing large amounts of G4 structures, up to 1M KCl was used. The size exclusion column Superdex
75 or 200 (GE Healthcare) was used to separate G4 and linear DNA fractions. Notably, the FAM fluorescence intensity was quenched
significantly upon single G-repeat G4 assembly. DMS protection assay was performed as described (Sun and Hurley, 2010).
Branched DNAs were generated by the similar annealing procedure without the further purification steps.
AID/DNA Complex Crystallization and Structure DeterminationGel filtration purified AID.crystal/G4 complex containing G4 DNA with the sequence of TGGGGTTTTTTT was crystallized using
0.26 M NaCl, 0.1 M MES at pH 6.0 and 12% PEG3350 at 25�C. Other AID complexes with G4 substrates including TGGGGTTTTT,
TGGGGTTTTTT, TGGGGTTCTTTT, TGGGGTTCTTT, TGGGGAACTTT, TGGGGAAUTTT and TTTTTTGGGGGTTCTTT were also
crystallized, which showed similar AID structures and no ordered DNA.
Gel filtration purified AID.crystal (WT and E58A)/branched complexes containing DNA with the sequences of GTTCAAGGCCA
GAGCTTT and TTTTTCCTGGCCTTGAAC were crystallized using 0.05 M sodium cacodylate at pH 5.5, 20 mM MgCl2, 10 mM
CaCl2, 10 mM spermidine and 5% PEG3350 at 16�C.When using dsDNA as a crystallization chaperone, AID.crystal (E58A) was mixed with dsDNA with the sequences of GTTCAAGG
CCAG and CTGGCCTTGAAC at 1:1 molar ratio, and crystallized either alone or with various substrate ligands such as 50-dCMP, de-
oxycytidine, and cytidine (Sigma) at 20mM concentration, under 0.1MMES at pH 6.2, 3%PEG3350 and 10mMCaCl2 at 16�C.Many
other short oligos were also tried in co-crystallization, but did not show ordered nucleotide density in the structures (Table S1).
The AID/G4 structure was solved by molecular replacement calculations in Phaser using the crystal structure of MBP (PDB: 3VD8)
and APOBEC3C as the model (PDB: 3VOW) (Kitamura et al., 2012). Subsequent structures were determined by molecular
replacement using the AID structure as a model. Phenix, CCP4 and Coot were used in model building and refinement (Adams
e3 Molecular Cell 67, 361–373.e1–e4, August 3, 2017

et al., 2010; Emsley et al., 2010; Winn et al., 2011). The POCASA 1.1 server was used for pocket prediction (Yu et al., 2010). The
YASARA server was used for energy minimization of the substrate manually docked to AID (YASARA Biosciences). Pymol was uti-
lized for molecular visualization and structure display (Schrodinger).
Molecular Mass Measurement by Multi-Angle Light Scattering (MALS)AID alone and AID-DNA complexes were reconstituted and purified as described above. The complex peak fractions con-
taining �0.2 mg protein were loaded onto a Superdex 200 gel filtration column coupled to a three-angle light scattering detector
(mini-DAWN TRISTAR) and a refractive index detector (Optilab DSP) (Wyatt Technology). Data analysis was carried out using
ASTRA V.
Electron MicroscopyAID and AID/DNA complex were applied to carbon-coated grids and negatively stained with 1%uranyl formate (ElectronMicroscopy
Sciences). Samples were examined in a Tecnai G2 Spirit BioTWIN electron microscope (FEI) at an accelerating voltage of 80 keV and
a nominal magnification of 3 49,000.
QUANTIFICATION AND STATISTICAL ANALYSES
In Vitro EMSA and Deamination Assay QuantificationImages were obtained by instruments described in Method Details. The band areas were boxed and backgrounds were subtracted.
The band quantification results were averaged from three repeats and the error bars were shown as standard deviations.
Data Analysis in Flow CytometryCell populationwas identified in FlowJo. Unpaired t test was used and two-tailed P valuewas calculated usingGraphPad Prism6. The
class switch recombination (CSR) results showed average from three biological replicates, with bars indicated SD.
DATA AND SOFTWARE AVAILABILITY
Accession NumbersThe accession numbers for the data reported in this paper are PDB: 5W0Z (AID.crystal, co-crystallized with G4 DNA), 5W0R
(AID.crystal E58A, co-crystallized with Branched DNA and cacodylic acid), 5W1C (AID.crystal E58A, co-crystallized with dsDNA
and cytidine), and 5W0U (AID.crystal E58A, co-crystallized with dsDNA and dCMP).
Molecular Cell 67, 361–373.e1–e4, August 3, 2017 e4





























