Embed Size (px)
Citation preview

Allineamento di sequenze di DNA e proteine: possibilità, limiti ed interpretazione
Database e motori di ricerca consentono il deposito ordinato, la gestione e il recupero delle sequenze di DNA o proteine, mentre l'allineamento è il metodo più efficace per confrontare tali sequenze. Non si deve pensare che si tratti di “lavoro bioinformatico”: il confronto per allineamento può rappresentare la routine per laboratori sperimentali; ad es., una volta clonato un frammento di DNA o identificato un trascritto o un gene, per verificarne il contenuto informativo, si paragona la sua sequenza con quelle presenti nelle banche dati, attraverso programmi di allineamento. Se la sequenza in questione risulta molto simile a quella di un gene o di una proteina di funzioni note, è probabile (ma mai sicuro al 100%) che possa avere la stessa funzione o una funzione correlata. Queste analisi sono comuni alla maggior parte dei laboratori in cui si svolgono analisi molecolari (e quindi sono molto utilizzate anche nelle Biotecnologie), ma nella maggior parte dei casi sono svolte in modo superficiale o improprio, portando a risultati incompleti e poco informativi o, ancor peggio, all'erronea interpretazione degli stessi e quindi a seguire false piste. La marcia in più che può fornire un corso di bioinformatica è la capacità di eseguire correttamente le analisi, eventulamente ampliandole e complementandole, andando oltre la semplice analisi con i preset e la lettura superficiale dei risultati.
ATTENZIONE: per interpretare correttamente i risultati non bisogna mai essere "passivi", ovvero mai accettare i risultati in quanto tali senza un controllo critico. Infatti, i programmi di allineamento si limitano a confrontare le sequenze, ma non sono "responsabili" del deposito delle sequenze nei database. Nei database possono essere trovate molte sequenze con errori, o male assemblate o annotate, dal momento che le informazioni (corrette o errate) associate alle sequenze sono fornite da chi le sottomette ai database (che si tratti di gruppi di ricerca o di algoritmi predittivi o d'annotazione). I programmi di allineamento, basando il confronto solo sulle sequenze, estraggono allo stesso modo tanto quelle le cui schede recano informazioni utili e corrette, quanto sequenze con errori (di sequenziamento o di predizione), oppure sequenze corrette ma associate nella scheda ad informazioni scorrette (ad esempio, nella definizione del tipo di molecola, spesso vengono erroneamente definiti mRNA le sole sequenze codificanti o CDS, che invece non sono mRNA completi in quanto prive delle regioni UTR al 5' e al 3'). Sta dunque alla competenza di chi di volta in volta valuta i risultati, e quindi ad analisi sempre prudentie critiche, utilizzare i risultati validi e significativi evitando di farsi portare fuori pista da informazioni fuorvianti.
L'allineamento tra due biosequenze consente di determinare se tra esse vi è una relazione di similarità. Se la sequenza di un gene clonato è molto simile a quella di un gene noto, è probabile (mai "certo") che esso abbia una funzione identica o simile. L'allineamento di acidi nucleici è effettuato sempre utilizzando sequenze di DNA, poichè gli mRNA sono riportati nelle banche dati come cDNA ed anche rRNA, tRNA, miRNA sono rappresentati dalle corrispondenti sequenze geniche.
Da un punto di vista funzionale, relativo ai prodotti proteici, il confronto tra le sequenze aminoacidiche è più informativo di quello tra le corrispondenti sequenze codificanti o tra trascritti o geni. Infatti, soprattutto negli eucarioti, differenze nelle regioni UTR (mRNA) e nelle sequenze introniche (geni), incluse inserzioni e delezioni, possono pesare notevolmente sul livello di similarità. Tuttavia, molte mutazioni introniche, se non riguardano i consensus di splicing, possono non avere alcun effetto sulla maturazione del trascritto. Inoltre, nelle regioni codificanti, residui identici a livello aminoacidico possono essere codificati da codoni differenti (18 dei 20 aminoacidi sono codificati da 2, 4 o più codoni), cosicchè ciò che è identico a livello proteico (stesso aminoacido) non sempre lo è a livello nucleotidico (codoni differenti per lo stesso aminoacido), ma le sostituzione di singole basi, che portino a mutazione silente (l'aminoacido non cambi) o non silente (l'aminoacido cambi) hanno lo stesso peso nel calcolo della similarità nucleotidica, alterando quindi l'associazione della variabilità della sequenza alla possibile vicinanza o lontananza funzionale.

ATTENZIONE: quanto appena esposto non intende assolutamente creare l'equivoco che le anailsi funzionali riguardino solo il confronto tra sequenze aminoacidiche. Il riferimento è solo relativo ai prodotti proteici. Il conceto di "funzione" tuttavia è più vasto e, per studiare i meccanismi regolativi, è importante invece confrontare i trascritti (ad esempio per studiarne la variabilità per splicing o predirne la stabilità dalla variazione delle regioni UTR, siti di riconoscimento per miRNA ecc.) e le regioni genomiche (per individuare elementi promotoriali, ehancer, consensus di splicing, possibili esoni criptici ecc.).
Ad un essere umano può apparire talora banale definire cosa è simile e cosa no tra due sequenze, ma un allineamento manuale è facile solo quando le sequenze da allineare sono palesemente simili. Quando invece i confini tra regioni conservate e divergenti non sono immediatamente individuabili, poichè ad esempio le due sequenze, pur correlate, mostrano similarità bassa o molto bassa, diventa molto difficile se non impossibile scegliere arbitrariamente tra i possibili allineamenti alternativi. E' necessario quindi stabilire un criterio per individuare l'allineamento "migliore" tra quelli possibili: gli algoritmi di allineamento prevedono che sia individuato il sistema per rendere minimo il numero delle differenze.
Le analisi di allineamento possono rivelare l'omologia tra geni e tra proteine, ma è scorretto parlare di "grado di omologia" o "percentuale di omologia". Infatti l'omologia è un carattere qualitativo ed esprime solo la correlazione evolutiva tra sequenze che derivano da un ancestore comune e si sono differenziate attraverso un processo di speciazione molecolare. In pratica, il concetto non quantitativo di omologia è paragonabile a concetti come sterile, vivo, morto, infinito, non quantificabili per gradi. Un animale o è vivo o è morto, una siringa o è sterile o no, un insieme o è infinito o finito: non può essere "molto infinito", "poco infinito" o "più infinito di"..... parimenti, non si può definire una proteina "più omologa di" e, se ci si vuole esprimere in termini quantitativi, è corretto parlare di percentuali di "identità" e/o di "similarità".
Il DNA è rappresentato da 4 lettere che corrispondono alle basi azotate, il cui ruolo biologico prevalente è la conservazione dell'informazione genetica che poi si trasforma in informazione funzionale attraverso l'espressione: in pratica, la successione delle basi nel DNA consente eventi di codifica e riconoscimento necessari per specificare "prodotti" (in particolare proteine, attraverso il codice genetico, ma anche molecole di rRNA, tRNA, snRNA, miRNA) ed individuare sequenze di riconoscimento per le proteine che interagiscono, a vario titolo, con il DNA stesso (componenti della cromatina, enzimi, fattori trascrizionali....). Nel DNA le sostituzioni non sono equivalenti (le transizioni sono più frequenti delle transversioni), ma si preferisce non distinguere, perchè il rapporto transizioni/trasversioni può variare da caso a caso. Insomma, per il DNA non ha molto senso parlare di residui "simili" ed in genere si preferisce fare riferimento all'identità.
L’alfabeto degli aminoacidi, molto più articolato e simile a un vero alfabeto (ha 20 lettere, contro le 4 del DNA) consente alle proteine di avere sequenze complesse, in grado di mediare un'infinità di meccanismi molecolari grazie alla maggiore eterogeneità delle proprietà chimico-fisiche degli aminoacidi, che possono essere polari o idrofobici, avere catene laterali molto grandi o piccole, cariche, aromatiche... inoltre, la combinazione di tali proprietà li differenzia ulteriormente (idrofobico piccolo, idrofobico grande ecc.). Da un punto di vista funzionale, il confronto tra le sequenze di due proteine è dunque più informativo del confronto tra le rispettive sequenze codificanti e non sbilanciato dalle mutazioni silenti, come spiegato in precedenza. Poichè la memoria delle mutazioni silenti è persa dopo la traduzione del trascritto in proteina, si deve comunque ricordare che non è possibile risalire correttamente dalla sequenza proteica a quella del suo trascritto. Infatti, in una sequenza proteica si può risalire ai codoni solo nel caso dei due aminoacidi metionina e triptofano (ciascuno è codificato da un singolo codone). Per gli altri 18 aminoacidi - codificati da due o più codoni - è spesso

impossibile risalire a quale specifico codone fosse presente nel trascritto. Infatti, anche nel caso si applichino le regole di codon usage dello specifico organismo, si potranno assegnare ulteriori codoni solo nei casi di associazione unvivoca (uso di un solo codone per aminoacido), ma non in quelli in cui il numero di codoni utilizzabili, pur ridotto dal codon usage, resti pari a due o più per aminoacido. Come si vedrà più avanti, questa impossibilità di retro-traduzione ha riflessi importanti sulle applicazioni di BLAST, il più importante ed utilizzato software di allineamento. Se il confronto non avviene tra tracritti maturi ma tra interi geni, il rumore di fondo causato da mutazioni funzionalmente irrilevanti è ancora più alto. Infatti, soprattutto negli eucarioti, un gran numero di mutazioni nei geni, incluse inserzioni e delezioni, riguarda le sequenze introniche, spesso molto più grandi degli esoni. Questa frazione, quasi sempre ampiamente maggioritaria (soprattutto in Homo sapiens e negli eucarioti più complessi), delle mutazioni geniche può pesare notevolmente sul livello di similarità tra due geni, anche quando i corrispondenti trascritti sono invece molto simili. Negli introni, non tutte le mutazioni hanno lo stesso peso: quelle che riguardano i consensus di splicing, ad esempio, possono avere drastici effetti sulla maturazione dei trascritti e quindi sulla sequenza finale dei prodotti proteicim mentre in larghe regioni introniche, le mutazioni non hanno alcun effetto sul trascritto e possono accumularsi, contribuendo a mostrare divergenza tra sequenze pur non avendo alcun effetto sui prodotti finali, talora identici o quasi.
Per le proteine l'indicazione della percentuale di aminoacidi identici è importante ma non esaustiva, poiché è fortemente indicativa anche la percentuale di residui che in qualche modo sono "simili" o "correlati" (positives). I venti aminoacidi che compongono l'alfabeto delle proteine (quelli modificati, come l'idrossiprolina, non sono indicati da simboli specifici nelle sequenze, trattandosi di modificazioni post-traduzionali) sono raggruppabili per condivisione di proprietà chimico-fisiche (idrofobicità, polarità, carica, ingombro sterico....) o perchè i codoni che li specificano sono più o meno "vicini" nel codice genetico (ovvero, condividono più o meno basi).
Quali sono le possibilità offerte dall'allineamento delle sequenze? Quali sono i limiti? Per comprendere bene il problema, è necessario tornare alla complessità delle informazioni disponibili. Il confronto tra le sequenze di DNA o proteine può fornire indicazioni preziose, attraverso l'individuazione di regioni "conservate" o "divergenti", ovvero parti più o meno lunghe della sequenza, in cui le molecole comparate mostrano notevole condivisione o non condivisione dei residui costituenti. Infatti, l'evoluzione dei viventi è mediata dall'evoluzione continua dei loro geni e proteine; la speciazione molecolare è a sua volta mediata prevalentemente da eventi di mutazione, ricombinazione e trasposizione delle sequenze di DNA, i cui prodotti sono successivamente sottoposti a meccanismi di selezione e deriva.
Quando le mutazioni in una regione di DNA sono poco tollerate, in quanto lesive della funzione specificata, la selezione provvede a "conservare" quella regione (ovvero, a limitarne la variabilità), tanto tra le specie quanto tra i membri di una famiglia genica. Quando, invece, le mutazioni sono tollerate in quanto non influiscono pesantemente sulla funzione codificata, la variabilità osservabile dal confronto tra le sequenze è più alta. Infine, talora la variazione della sequenza può riflettere una specializzazione funzionale, ovvero la creazione di nuove funzioni a partire da una molecola progenitrice.
Nota di genomica funzionale: La specializzazione funzionale per divergenza parziale della sequenza è un fenomeno spesso associato agli eventi di duplicazione e amplificazione genica in un organismo (creazione di paraloghi, ovvero geni omologhi derivanti, nello stesso organismo, da un ancestore comune). Infatti, la duplicazione di un gene consente alla sua "copia" un'evoluzione più libera, dal momento che il gene doppio può permettersi di accumulare mutazioni che transientemente portano a una perdita o a un peggioramento di alcuni parametri funzionali, fino al raggiungimento di una funzione nuova, senza ridurre la fitness dell'organismo nelle fasi "sfavorevoli", poichè il gene master

mantiene comunque la funzione originale. La possibilità quindi di "modulare" la funzione base di un gene e del suo prodotto proteico, riscontrata in natura, è alla base dei progetti di ingegnerizzazione biotecnologica di geni e proteine. Per questo, imparare a individuare con la bioinformatica le regioni di modulazione funzionale, fornisce al biotecnologo la capacità di smart design dell'ingegnerizzazione per mutagenesi mirata.
In tutti i casi, poichè nessuno era presente al momento in cui avvenivano gli eventi di speciazione molecolare, l'allineamento delle sequenze permette di dedurre sia eventi di filogenesi molecolare, atti a ricostruire la storia evolutiva di molecole ed organismi, sia meccanismi alla base delle funzioni e delle loro specializzazioni. Residui o regioni di sequenze sempre identici o altamente conservati in geni o proteine di specie evolutivamente distanti, sono probabilmente cruciali per la (o per una) funzione mediata; i residui o le regioni che variano sono forse meno importanti, oppure hanno consentito di modulare la funzione originale, consentendone una specializzazione.
Oltre a ricostruire, sulla base del grado di similarità, relazioni filogenetiche e funzionali, l'allineamento può servire anche per ricostruire la sequenza di interi cromosomi attraverso l'individuazione di regioni identiche "sovrapposte", condivise dai frammenti sequenziati (sequence assembly). Inoltre, il confronto tra la sequenza di un cDNA e quella del DNA genomico della stessa specie può consentire di ricostruire la struttura della sequenza codificante, individuandone gli esoni e, quindi, anche gli introni e la regione di regolazione trascrizionale al 5' del primo esone. L'interpretazione dei risultati di un'analisi di allineamento ha dei limiti ed è importante conoscerli per evitare deduzioni errate. Ad esempio, solo quando tutte le sequenze da confrontare sono già note (sperimentalmente determinate), non sono in gioco variabili dipendenti dal grado di precisione degli algoritmi di predizione e l'accuratezza del confronto dipende interamente dagli algoritmi di allineamento e dai criteri utilizzati per valutare la similarità.
Molto spesso l'allineamento avviene tra sequenze o frammenti di sequenze utilizzate come "sonde" (query) e sequenze presenti in una o più banche dati. In tal caso, i risultati dell'analisi non dipendono solo dagli algoritmi di allineamento e dai parametri e set di dati utilizzati per valutare il grado di similarità, ma anche dalla precisione con cui sono stati compilati i database e dall'accuratezza delle predizioni relative a (tante) sequenze geniche e proteiche "ipotetiche", depositate nelle banche dati alla stessa stregua di quelle "reali".
Ulteriori fattori limitanti sono la completezza dei dati disponibili e la selezione operata. In altri termini, quando si indica ad un software di allineamento in quali banche dati andare a cercare le sequenze che saranno confrontate con la sonda, ci si deve rendere conto che il programma può trovare (e ci mostrerà nei risultati) sequenze omologhe solo nei database selezionati e che quindi tutte le ulteriori sequenze omologhe, esistenti ma depositate in banche dati non selezionate (dall'operatore, o non preselezionate come preset) non saranno estratte. Errori di valutazione possono essere commessi anche valutando quelli che si ritengono i “risultati” dell’allineamento e che invece, molto spesso, rappresentano solo la sottoselezione dei risultati presentata in output. Ad esempio, se il preset del programma di allineamento è impostato su “mostra i primi 100 risultati” e l'operatore non è consapevole di tale preset, nel caso le sequenze allineate siano 1430, l'operatore osserverà "i risultati" credendo che corrispondano solo a quanto sta visualizzando, ma le sequenze dalla 101 alla 1430 non saranno visibili, sebbene pienamente parte de "i risultati". Ciò può far perdere preziose informazioni.
Esempio: immaginiamo di utilizzare come sonda la sequenza di una proteina conservata in tutti gli organismi, ma non ancora caratterizzata funzionalmente. Per ottenere predizioni funzionali, l'allineamento con proteine omologhe degli altri organismi è in questo caso inutile (perchè sono altrettanto non caratterizzate), mentre sono molto più utili gli allineamenti con proteine meno simili, ma già completamente o parzialmente caratterizzate e che quindi potrebbero suggerire, sulla base della conservazione di una o più regioni con la proteina d'interesse, una o più possibili funzioni.

Supponiamo ora che nel database preselezionato le proteine omologhe di altri organismi siano 134. Poichè gli allineamenti mostrati per primi sono quelli con maggiore similarità, con il preset “mostra i primi 100 risultati” l'operatore inconsapevole visualizzerà solo i primi 100 allineamenti (inutili) con proteine omologhe in altri organismi e dedurrà che l'analisi per allineamento non gli è servita a niente. Erroneamente, perchè sarebbe stato sufficiente modificare il preset in “mostra i primi 250 risultati”, per ottenere, a partire dall'allineamento 135, preziose informazioni in grado di suggerirci gli esperimenti corretti per scoprire e dimostrare la funzione della proteina....
Se si somma l'assenza delle sequenze non ancora determinate e/o depositate agli errori di predizione, che sono numerosi, ci si rende conto che qualsiasi dato deve essere considerato con molta cautela. Resta sempre valida l'asserzione: "assenza di evidenza non è evidenza di assenza": il non trovare sequenze omologhe ad una sonda, infatti, non vuol dire che esse non esistano e può darsi che non siano state ancora sequenziate. Alternativamente, la mancata estrazione potrebbe dipendere da errori di predizione genica e/o dell'annotazione del database, o dalle impostazioni di "stringenza" relative al software di allineamento o di visualizzazione dei risultati. Dunque, l'interpretazione dei risultati non deve essere frettolosa e deve maturare dopo un processo di verifica dei parametri impostati, dei set di dati analizzati, ecc. E' inoltre prudente non fidarsi eccessivamente degli accordi internazionali tra le banche dati e svolgere ricerche ampie. Il prezzo da pagare è l'estrazione di dati ridondanti, ma le analisi effettuate su più banche dati possono individuare, proprio nelle aree di non condivisione delle sequenze depositate, preziose informazioni.
Criteri per la valutazione della similarità
I metodi di allineamento consentono di formulare ipotesi la cui "robustezza" dipende non solo dal set di dati disponibili per il confronto delle sequenze, ma anche dai criteri adottati per valutare la similarità, che fondamentalmente sono i seguenti:
(1) identità / non identità, che attribuisce un valore costante alle coppie di residui identici;
(2) criterio del codice genetico, in cui è attribuito un punteggio che dipende dal numero di sostituzioni nucleotidiche per passare dai codoni che codificano un aminoacido a quelli che codificano l'altro;
(3) criterio (2) soppesato in relazione alle proprietà strutturali;
(4) matrici di punteggi basate sull'interconvertibilità osservata in un dato set di proteine omologhe: matrici PAM e BLOSUM.
Per valutare la similarità tra sequenze di DNA il criterio più appropriato è quello di identità / non identità; il motivo è che l'alfabeto del DNA è troppo semplice (solo 4 lettere) per consentire "sfumature" ovvero stabilire relazioni di compatibilità tra le sue lettere. In pratica, il DNA trasmette l'informazione come codice (sequenza specifica delle lettere) piuttosto che attraverso proprietà chimico-fisiche più o meno simili delle basi.
Per le proteine, invece, è meglio valutare anche il grado di correlazione tra aminoacidi. Infatti, negli output di allineamento tra proteine è solitamente riportata sia l'indicazione delle "identities" (I) che degli aminoacidi compatibili ("positives", P). Possono essere sviluppate numerose scale arbitrarie per soppesare la similarità tra i 20 aminoacidi; ad esempio, si può tentare di valutare insieme sia la somiglianza strutturale che l'interconvertibilità per mutazione (Feng et al., 1985):

La matrice in figura esemplifica - indipendentemente dal criterio con cui sono attribuiti i punteggi - la struttura delle matrici di sostituzione dove sono rappresentati i 20 aminoacidi sia in ascissa che in ordinata (matrici A x A) e quindi le celle di intersezione riportano i punteggi per ciascuna possibile sostituzione di un aminoacido con un altro (ovvero, per tutte le possibili coppie di aminoacidi allineati). Si può notare come le celle della diagonale, che riportano i casi di identità, mostrino sempre lo stesso punteggio, che è anche il massimo, mentre le altre celle, corrispondenti a coppie di residui differenti, mostrino valori più alti o più bassi a seconda della "vicinanza" tra aminoacidi, sulla base del criterio adottato per costruire la tabella. In questa tabella, che tiene conto sia delle proprietà degli aminoacidi che della vicinanza tra codoni codificanti (e quindi maggiore o minore interconvertibilità per mutazione) si può notare come, ad esempio, alla coppia D-E (Asp e Glu) sia assegnato il valore più alto dopo l'identità, essendo i due residui entrambi acidi e codificati da due codoni che differiscono solo per una base. Lo stesso accade per i residui Ile e Leu, mentre per coppie come Cys e Glu il valore è zero, essendo sia le proprietà che i codoni molto differenti. I criteri per valutare correlazioni dal punto di vista prettamente evolutivo o principalmente funzionale non sempre sono coincidenti. Ad esempio, il criterio del codice genetico si presta bene a studi evoluzionistici, mentre per analisi funzionali è fondamentale considerare le proprietà chimico-fisiche degli aminoacidi.
In ogni caso, per valutare correttamente le relazioni tra sequenze è necessario valutare la significatività statistica della similarità rilevata in un allineamento ed evincere il rumore di fondo, prodotto dalla similarità casuale. Il rumore di fondo può essere dedotto dall'analisi comparativa con sequenze casuali aventi la medesima composizione in basi o aminoacidi. Per generare un certo numero di sequenze casuali aventi la medesima composizione è sufficiente adottare un generatore Montecarlo; ne esistono alcuni localizzati su server bioinformatici, ma il generatore di sequenze casuali non è certo un'invenzione bioinformatica. Tuttavia, poiché le sequenze di DNA e proteine non rispettano distribuzioni stocastiche dei residui, è necessario introdurre fattori correttivi. Sono state ottenute varie relazioni empiriche alla base di metodiche per la valutazione della significatività statistica della similarità.
La metodica di Smith e collaboratori si basa sull'osservazione di una serie di allineamenti ed in particolare della correlazione tra i punteggi di similarità massima associati a subsequenze e la loro lunghezza. Ciò ha consentito di ricavare il valore medio di massima similarità e calcolare in modo empirico la significatività di quella osservata. La metodica di Karlin e Altschul prevede che i segmenti allineati siano privi di interruzioni ed è stata inclusa nella formulazione originaria dell'algoritmo di BLAST, tuttavia superata dalla formulazione di metodi che consentono di valutare la significatività anche in presenza di interruzioni (Gapped BLAST). I punteggi relativi alle regioni di massima similarità (MSP) seguono una distribuzione non normale che dipende dalla matrice utilizzata (PAM o, più frequentemente, BLOSUM-62). Dal confronto di due sequenze emergono, oltre alla regione di massima similarità, anche regioni di similarità elevata (HSP); la metodica di Karlin e

Altschul valuta la significatività statistica tanto in relazione a MSP quanto a HSP. Tuttavia, poichè similarità statisticamente non significative possono essere biologicamente rilevanti, non è corretto scartare ipotesi sulla base dell'assenza di significatività statistica, che perde parte del suo valore analitico, soprattutto in relazione ad analisi di tipo funzionale. Il fatto che similarità statisticamente non significative possono essere biologicamente rilevanti dipende dalla larga diffusione e conservazione evolutiva di regioni proteiche o nucleotidiche altamente degenerate ma funzionalmente conservate. Ne sono un esempio le lunghe regioni ad elica di proteine strutturali, in cui è importante solo che siano mantenute caratteristiche della sequenza compatibili con la struttura ad alfa elica e registri idrofilici o idrofobici atti a coordinare l'assemblaggio tra eliche o il passaggio di ioni. Anche nel DNA sono presenti regioni di ridotta complessità, sebbene il discorso di alta e bassa complessità sia più applicabile alle proteine, per l'intrinseca bassa complessità dell'alfabeto a sole 4 lettere del DNA.
Nelle analisi evoluzionistiche può essere importante ricostruire eventi di filogenesi utilizzando anche molecole che, a causa di mutazioni in determinate regioni della sequenza, abbiano perso l'originale funzione, pur mantenendo una struttura globale quasi inalterata. E' il caso, ad esempio, di alcune proteine che, sebbene fortemente conservate nell'ambito della famiglia delle chinasi, hanno perso l'attività enzimatica a causa di poche o anche di una sola mutazione nel sito catalitico. Per un'analisi di correlazione tra specie può essere irrilevante il fatto che i membri di una "famiglia" proteica abbiano la stessa attività o l'abbiano persa; conta maggiormente il livello globale di divergenza dal comune ancestore. Proteine la cui funzione dipenda da un numero ridotto di residui, ovvero per le quali sia possibile preservare struttura e funzione "conservando" solo gruppi chiave di residui e consentendo una notevole variazione e combinazione degli altri, potranno, al contrario, essere tagliate fuori da analisi di correlazione evolutiva, in quanto mostranti livelli di similarità globale o perfino locale difficilmente distinguibili dal rumore di fondo.
Quando, invece, i metodi di allineamento sono finalizzati ad analisi di tipo funzionale, la valutazione della significatività statistica deve essere integrata (e talora può essere soppiantata) da parametri quali, ad esempio, la conservazione della struttura globale o locale e quella di residui chiave. Può divenire difficile distinguere tra casi di forte divergenza e quelli di convergenza, ovvero tra i casi in cui proteine derivanti (a) da un comune ancestore abbiano conservato solo i residui cruciali per la funzione, divergendo in tutte le altre regioni della sequenza o (b) da differenti ancestori siano giunte a condividere la funzione e, quindi, a mostrare similarità nelle regioni comprendenti i residui chiave. In ogni caso, nella valutazione della similarità ai fini di analisi funzionali, qualsiasi indicazione dalla letteratura scientifica disponibile sul ruolo di uno o più residui deve essere presa in considerazione per "ponderare" localmente la variabilità, ovvero per individuare residui e regioni di sicura importanza per la funzione. Anche le analisi sulla struttura secondaria e quelle di modeling tridimensionale sono fondamentali per integrare la significatività dei dati derivanti da analisi di allineamento.
Allineamento globale e locale
Per tentare di raggiungere il miglior allineamento tra due sequenze si può ricorrere a due strategie:
- allineamento globale (comprende tutti gli elementi delle sequenze allineate)
- allineamento locale (individua le subsequenze con massimo livello di similarità)
L'algoritmo di allineamento globale "classico" proposto inizialmente da Needleman e Wunsch è solitamente basato per gli allineamenti di DNA su matrici di identità / non identità e per quelli di proteine su matrici PAM o BLOSUM (trattate più avanti). L'algoritmo di Smith-Waterman è volto

invece ad individuare regioni di similarità locale, ovvero a determinare il miglior allineamento attraverso subsequenze. Secondo il metodo di Smith-Waterman, la matrice viene inizializzata definendo per ciascuna coppia di residui un punteggio di similarità s. Anche alle inserzioni/delezioni è attribuito un punteggio (gap penalty), determinato dalla somma di una penalità costante (gap open) ed una proporzionale (gap elongation) alla lunghezza della delezione. In questo metodo di allineamento si parte dall'elemento che realizza il massimo punteggio e si procede fino a quando l’allungamento comporta un aumento dello score. Nella comparazione di sequenze di proteine si utilizzano matrici che valutano anche le proprietà strutturali degli aminoacidi.
A parte "come" vengano realizzati gli allineamenti locali o globali, è fondamentale innanzitutto chiarirsi le idee sul perchè/quando utilizzare un approccio o l'altro. Il metodo più utilizzato è l'allineamento locale, in particolare attraverso l'algoritmo BLAST (Basic Local Alignment Search Tool). Il metodo locale permette infatti di evidenziare, alll'interno di due differenti sequenze, le regioni simili, anche nel caso esse rappresentino solo frazioni delle intere sequenze. Ciò è fondamentale, perchè le reali unità funzionali nell'evoluzione non sono solo interi geni e proteine, bensì frammenti rilevanti degli stessi, quali domini proteici capaci di mediare specifiche funzioni all'interno di una proteina e quindi conservati anche in proteine diverse. Tali domini sono spesso codificati, negli eucarioti, da un esone o gruppi di esoni. Grazie al metodo di allineamento locale, se due grandi proteine formate da più domini condividono solo un dominio ed una regione di legame al calcio, BLAST allineerà solo tali regioni e non quelle totalmente differenti rappresentate dai domini non condivisi. Per evitare equivoci, è da chiarire che l'allineamento locale è in grado di individuare frammenti simili, ma non è "obbligato" a limitarsi ai frammenti: quando due proteine (o due geni, poichè il discorso vale anche per le sequenze di DNA) sono simili per l'intera lunghezza, BLAST allineerà per tutta la lunghezza le proteine o i geni. In pratica, BLAST allinea tutto ciò che risulta avere una similarità al di sopra di una certa soglia, quindi, se tutta la sequenza è sopra tale soglia, l'allineamento è completo, se invece è sopra soglia solo un frammento, allinea solo tale frammento.
Ci si potrebbe chiedere: se l'allineamento locale è così efficiente e, nei casi di similarità diffusa su tutta la sequenza, consente anche l'allineamento totale, perchè si utilizzano ancora algoritmi di allineamento globale? La risposta, banalizzando, potrebbe essere: non sempre si cercano e si vogliono valutare solo le similarità. Esistono, infatti, dei casi in cui è importante confrontare anche le parti differenti, al fine di valutarne il peso (appunto, in termini di divergenza): in particolare, in alcune analisi evoluzionistiche, limitarsi alle parti condivise è una forte limitazione, poichè la comparsa (o scomparsa) di nuove regioni in geni/proteine è un marcatore di speciazione molecolare e di separazione evolutiva. Anche per gli allineamenti associati agli studi strutturali si utilizza l'allineamento globale. Infatti, se si confrontano due strutture conservate come fold e divergenti come sequenza, l'allineamento locale rischia di "tagliare" fuori le zone di similarità troppo bassa e ciò è incompatibile con il fatto che nel confronto strutturale, tutte le parti della catena polipeptidica (anche quelle di sequenza divergente) devono essere confrontate. Si può intuire che, nelle analisi biotecnologiche/funzianali è prevalente l'uso degli algoritmi di allineamento locale. Infatti, numerosissimi sono i casi di condivisione di regioni funzionali (siti di legame, esoni, domini o motivi proteici) tra molecole (geni, proteine) che nel resto della sequenza possono divergere fortemente o essere completamente diverse.
Matrici dot-plot (e concetto di finestra)
Le regioni di similarità in un allineamento possono essere visualizzate creando una matrice dot-plot, costituita da celle comprese tra la sequenza orizzontale (inferiore) e quella verticale (a sinistra) e costruita marcando con un punto (dot, in inglese) le celle di intersezione tra residui identici. Le identità casuali producono un rumore di fondo elevato: per ridurlo si può passare dal confronto dei

singoli residui a quello di brevi sequenze (finestre) di più residui. In tal caso, il dot è riportato solo quando in una finestra di w residui, s sono identici. A valori più alti di s corrisponde una stringenza più alta, massima per s = w. Ovviamente, la variazione di w e s ha influenza sul rumore di fondo. I valori di s e w più adatti al confronto di sequenze nucleotidiche e proteiche sono determinati empiricamente. A causa dell'alfabeto di solo 4 lettere, negli allineamenti tra acidi nucleici il rumore di fondo (identità casuale) è elevato:
Per questo motivo è necessario adottare valori più elevati di s: in pratica, è estremamente facile che l’identità tra due nucleotidi (che sono di soli quattro tipi) di due sequenze sia casuale; l’identità casuale tra dinucleotidi (16 combinazioni possibili) è più bassa, ancor più bassa quella tra trinucleotidi ecc. Una matrice dot-plot che consideri solo le identità non fornisce una reale indicazione dei rapporti di similarità per le proteine, ove la “non identità” tra aminoacidi può avere implicazioni biologiche profondamente diverse. Infatti, in alcuni casi la sostituzione di un residuo con un altro non identico, ma con proprietà molto simili (es: leucina ed isoleucina), può essere quasi irrilevante; in altri casi, due residui non identici possono avere proprietà molto diverse.
Matrici PAM (Point Accepted Mutation) e BLOSUM (Block Substitution Matrix)
La matrice è basata su osservazioni "a posteriori", in quanto basata sul concetto di quanto una sostituzione risulti più o meno accettata in natura. Ritornando alla matrice mostrata in precedenza, si può notare che non differisce dalla matrice PAM nella struttura (entrambe le matrici sono A x A), bensì nei criteri di attribuzione del punteggio, assegnato da Feng sulla base di proprietà intrinseche (vicinanza delle proprietà delle catene laterali e delle sequenze dei codoni) e nelle matrici PAM a partire dai dati empirici, ovvero dall'osservazione del reale. La matrice PAM è infatti ottenuta a partire da una collezione di gruppi di proteine con similarità > 85% (per ridurre il peso delle mutazioni occorse più volte sullo stesso sito). Questo gruppo di > 1500 sostituzioni porta ad una matrice in cui, per ciascun aminoacido, è calcolata una "mutabilità relativa" pari al rapporto tra numero di sostituzioni in cui è coinvolto e la sua abbondanza relativa. Sebbene gli aminoacidi siano 20, la loro frequenza non è un costante 5%: per alcuni aminoacidi è 7,5-10% e per altri < 2,5%. La matrice PAM più utilizzata è la PAM-250 (Dayhoff et al., 1978).
Anche le matrici BLOSUM si basano su dati empirici ed esse si rivelano particolarmente efficaci nell'allineamento di proteine evolutivamente distanti. Le matrici BLOSUM sono ottenute a partire da più di 2000 blocchi (non intere proteine, a differenza del dataset di partenza PAM) di allineamento, che consentono di stimare in modo più accurato il grado di similarità. Infatti, allineando proteine intere, si considera la variazione sia nelle parti conservate che in quelle variabili; limitando ai blocchi, la variazione è più significativa nell’ambito della conservazione funzionale (le regioni al di fuori dei blocchi possono mediare funzioni accessorie e talora alternative). Nelle matrici BLOSUM tutte le sequenze con similarità superiore ad una certa soglia sono considerate come una singola sequenza, per ridurre il peso delle coppie di residui che appartengono a proteine stretamente correlate. La matrice più utilizzata (BLOSUM 62, Henikoff and Henikoff, 1992) raggruppa sequenze con similarità > 62%.

Allineamento locale: BLAST
BLAST (Basic Local Alignment Search Tool) è il più diffuso programma di allineamento locale delle sequenze. Per vari anni il metodo FASTA (da non confondere con l’omonimo formato) e BLAST sono stati utilizzati in alternativa o in modo complementare; progressivamente, però, la maggiore velocità, il continuo lavoro di sviluppo e di ottimizzazione hanno reso l’uso di BLAST pressocché universale. FASTA esiste ancora ma, come altri validissimi programmi, è utilizzato in ambito più ristretto. Pertanto la descrizione di FASTA, non più inclusa nel programma d’esame (che in compenso riporta una più completa descrizione di BLAST e consigli per la sua fruibilità), è tuttavia ospitata sul sito del corso tra la documentazione di supporto (sezione “fuori programma”), per gli eventuali interessati (non sarà, comunque, oggetto di domande d’esame o di valutazione).
Coerentemente con quanto già spiegato per l'allineamento locale in genere, le sequenze individuate in ricerche BLAST possono rivelarsi lunghe (esattamente o approssimativamente) quanto l'intera sonda, o corrispondere a regioni più o meno estese della stessa. Infine, le sequenze individuate possono, a loro volta, rappresentare intere proteine o solo frammenti delle stesse (probabilmente, domini o insieme di domini). Infatti, non si deve mai dimenticare che BLAST individua per allineamento regioni di similarità locale e quindi la natura locale dell'allineamento può riguardare sia la sequenza query (sottomessa dall'operatore) che la sequenza subject (trovata nel database). In pratica, tutte le combinazioni possibili possono verificarsi: (a) l'intera sequenza query si allinea con un'intera sequenza subject (stessa lunghezza e similarità sopra soglia su tutte le regioni); (b) la sequenza query si allinea per intero con una regione omologa di una sequenza subject più lunga; (c) un frammento della sequenza query si allinea con un'intera sequenza subject (in questo caso è più lunga la query); (d) un frammento della sequenza query si allinea con un frammento della sequenza subject (evidentemente, una regione conservata presente in sequenze per il resto diverse). Poichè inoltre BLAST allinea tutti i frammenti di query e subject che risultano sopra soglia, può anche accadere che più frammenti di query e subject siano allineati. Ad esempio, se la sequenza di una proteina query è composta da tre regioni corrispondenti ai domini conservati A, B e C e la sequenza subject comprende le regioni A', D e C', BLAST allineerà le regioni omologhe N-terminale A-A' e C-terminale C-C', mentre le regioni centrali non omologhe B e D non risulteranno allineate. Talora BLAST può individuare una o più sequenze identiche alla sonda, malgrado nome e numero d'accesso risultino diversi. Ciò non è sorprendente, poiché non sempre i compilatori dei database si accorgono della ridondanza tra sequenze, ed alcune (o anche molte) sono depositate come nuove, senza verificarne la coincidenza totale con quelle già note. A volte l'identità può riguardare una sequenza codificante predetta a partire da dati genomici e quella reale di un cDNA. In tal caso si ottengono la verifica della predizione e dati sull'espressione (stadio o tessuto-specifica, stimolata da induttori, associata a patologie ecc.).
Basi algoritmiche
BLAST si basa sull'algoritmo sviluppato da Altschul e collaboratori e sull'assunto che in sequenze omologhe - anche notevolmente divergenti - possono essere individuate regioni che mostrano una similarità statisticamente significativa. Nel confronto tra sequenze nucleotidiche è attribuito punteggio positivo alle identità e negativo alle non identità. Invece, per confrontare sequenze proteiche, sono adottate le matrici di punteggi PAM o BLOSUM. In BLAST è definita Maximal Score segment Pair (MSP) la regione di massima similarità, la cui significatività statistica è calcolata secondo il metodo di Karlin ed Altschul. Tutte le altre regioni con punteggio di similarità statisticamemente significativo in quanto superiore ad una soglia S sono definite High Score segment Pair (HSP). BLAST estrae dai database, ove trova le sequenze da confrontare con la sonda, solo regioni il cui punteggio di similarità sia superiore ad una determinata soglia. L'abbassamento della soglia aumenta la sensibilità, ma riduce la velocità. Il valore della soglia è determinato in modo che risulti automaticamente E = 10, ove E (Expected) rappresenta il numero atteso di regioni non correlate condivise tra sonda e sequenze

del database. In considerazione di quanto già illustrato in precedenza sul reale valore della significatività statistica, sono particolarmente apprezzabili le possibilità di modulare i parametri di BLAST (matrici, valori soglia, filtri ecc.) poichè, come al solito, solo la valutazione critica dei risultati ottenuti con ricerche reiterate e modulate può portare a conclusioni attendibili.
Per capire come funziona BLAST si deve partire dalla constatazione che, quando si confrontano sequenze ad esempio di lunghezza diversa e non chiaramente omologhe, tanto l'occhio umano quanto l'algoritmo non saprebbero "da dove partire" per trovare regioni allineabili. Per questo motivo, si può immaginare di "fare a pezzi" la sequenza e - tenendo traccia delle posizioni relative dei frammentini - provare ad allinearne ciascuno con tutti gli altri. A tutti i possibili allineamenti si dovranno attribuire punteggi e ne risulterà una graduatoria in cui verranno identificate regioni MSP e HSP. A partire da tali frammenti si potranno "estendere" gli allineamenti aggiungendo al 5' (o Nter) e al 3' (o Cter) altri frammenti, se quelli contigui nelle sequenze originali risulteranno sopra soglia.
Applicazioni di BLAST
Il metodo BLAST, dall'originaria formulazione ad oggi, ha rappresentato un validissimo strumento d'analisi, che numerosi server bioinformatici consentono di utilizzare on-line per ricerche nelle principali banche dati. Sono state sviluppate numerose applicazioni, basate sul metodo BLAST ma ottimizzate per il tipo di ricerca, sonda e database nei quali si intenda cercare sequenze omologhe. Gli algoritmi di BLAST sono stati progressivamente potenziati, implementando nuove funzioni che consentono, ad esempio, di adottare matrici definite sulla base del set di dati in analisi o di integrare l'analisi di similarità con quella per pattern.
I principali server che ospitano pagine dedicate a BLAST sono quelli dell'NCBI e dell'EBI, ma BLAST è utilizzabile on-line anche collegandosi ad altri server bioinformatici oppure localmente, scaricando via ftp database e programmi nella versione adeguata al sistema operativo utilizzato (Unix, Windows, MacOSX, Linux). Presso il server dell’NCBI è possibile usufruire di applicazioni BLAST per screening on-line di vari database; esse sono divise, rispecchiando la logica del browser Entrez gestito dallo stesso server, in sezioni principali. Nella prima sezione (BLAST Assembled RefSeq Genomes) BLAST è ottimizzato per la ricerca in specifici genomi. Nella seconda sezione (Basic BLAST) sono presenti le applicazioni principali per l’analisi per allineamento delle sequenze proteiche e nucleotidiche. Nella terza sezione (Specialized BLAST) sono presenti applicazioni basate su BLAST ed ottimizzate per usi specifici.
Basic BLAST
La sezione Basic BLAST si presenta con le sottosezioni nucleotide BLAST e protein BLAST che confrontano set di dati omogenei (query sequence e sequenze nel database sono dello stesso tipo).
Le applicazioni nella sezione nucleotide BLAST servono a confrontare una query nucleotidica con le sequenze contenute nei db nucleotidici, utilizzando l’algoritmo più diffuso, blastn, oppure gli algoritmi Megablast e discontiguous Megablast, ottimizzati per l’allineamento di lunghe sequenze di DNA. Megablast è ottimizzato per allineare sequenze quasi uguali, le cui differenze possano derivare da errori di sequenziamento o polimorfismi. Pertanto, Megablast o suoi derivati sono utilizzati nell'assemblaggio di frammenti genomici o nel clustering di trascritti. La differenza fondamentale tra Megablast e blastn è nella scelta della "word size", ovvero la lunghezza minima della stringa di residui contigui considerata ai fini della valutazione di identità, che in Megablast è ottimale con valori maggiori o uguali a 16 (multipli di 4). Ciò rende più selettiva l'accettazione di identità e fino a 10 volte più veloce Megablast che, quindi, si presta meglio all'uso con sequenze molto lunghe. I default settings di Megablast richiedono, attualmente, una word lunga 28. Megablast è utile per l'analisi di sequenze molto lunghe ed estremamente simili; per lo studio di sequenze lunghe e

divergenti è invece da utilizzare "discontiguous Megablast ", in cui il valore di word size torna a livelli più bassi. In particolare, i "gap costs" sono pari a 5 per l'apertura di un gap e pari a 2 per la sua estensione. Si può comprendere come, accettando un numero di sostituzioni ancora basso, ma più alto di quello settato per Megablast, discontiguous Megablast sia utilizzato soprattutto per progetti di genomica comparata, in cui si confrontano sequenze di specie affini, le cui sequenze non divergono quanto quelle di specie evolutivamente lontane, ma più delle sequenze identiche (a parte i polimorfismi) della stessa specie.
Le applicazioni nella sezione protein BLAST servono a confrontare una query proteica con le sequenze contenute nei db di proteine, utilizzando l’algoritmo più diffuso, blastp, oppure gli algoritmi psi-blast e phi-blast, ottimizzati per l’ottenimento di matrici di posizione o per ricerche centrate su un pattern. La ricerca con blastp è in genere più veloce che con blastn, perchè paragona sequenze omogenee e complesse in un insieme ridotto di dati. Ciò dipende dal fatto che le sequenze proteiche contengono un numero minore di residui rispetto alle regioni codificanti (per ciascun aminoacido, rappresentato dal codice ad una lettera, vi sono le tre basi di un codone), ma anche dalla presenza, nei database di acidi nucleici, delle sequenze di introni, UTR, trasposoni, regioni mediamente ed altamente ripetute. Inoltre, nelle banche dati nucleotidiche possono coesistere dati genomici grezzi ed annotati, predizioni geniche e cDNA, EST e STS ecc..... La sigla "psi", che contraddistingue l'algoritmo psi-blast, significa "position-specific iterated", perchè realizza ricerche iterative, in cui le sequenze estratte sono usate per costruire una matrice, che sarà utilizzata nella reiterazione della ricerca e così via. In pratica, la matrice che va a definirsi in PSI-BLAST è del tipo QxA, ove Q è la lunghezza della sequenza sonda ed A quella dell'alfabeto (in termini di simboli dei residui). La matrice costruita in tal modo è definita "Position Specific Score Matrix" o PSSM e può essere salvata come file di testo per l'uso in screening successivi di differenti database. Una serie di applicazioni BLAST, inoltre, implementa algoritmi ed esplora database in modo da individuare regioni di similarità con domini proteici.
Quando invece è necessario confrontare la query di un tipo con database contenenti sequenze di tipo diverso, per consentire il corretto confronto tra i differenti alfabeti di DNA e proteine si ricorre alle applicazioni translated BLAST, che sono blastx, tblastn e tblastx. Quando si dispone di una sonda nucleotidica e si vuole confrontarla con sequenze proteiche (poichè è noto o si ipotizza possa contenere una sequenza codificante), l'applicazione blastx è appropriata, in quanto traduce la sequenza nucleotidica sonda nei sei possibili registri di lettura e confronta le sequenze tradotte con i database di proteine selezionati. Semplicisticamente, è come se si sottoponessero contemporaneamente sei sonde proteiche con blastp. L'applicazione tblastn, invece, confronta una sonda proteica con la traduzione dinamica nei sei registri di lettura dei database nucleotidici selezionati. Ovviamente i database non vengono tradotti ogni volta che si lancia tblastn; essi sono già presenti sul server che fornisce l'applicazione blast e sono periodicamente rigenerati in occasione di ciascun aggiornamento del database nucleotidico. tblastn può servire a risalire dalla sequenza proteica alle sequenze dei trascritti in grado di codificare la proteina; in tal modo si ottengono informazioni sull'espressione. Ottenuto il trascritto codificante, inoltre, si possono ottenere anche le regioni non codificanti 5'-UTR e 3'-UTR ed il trascritto completo, confrontato con le sequenze genomiche, consente di risalire al locus codificante ed alla struttura del gene. Ad esempio, negli eucarioti il confronto tra il trascritto completo e il genoma risulta in una serie di allineamenti di frammenti del trascritto a frammenti di un cromosoma, corrispondenti agli esoni. Le regioni tra esoni sono quindi identificate quali introni e la regione al 5' del primo esone può essere analizzata per cercare regioni promotoriali e siti di legame per fattori di trascrizione. L'applicazione che impiega il tempo maggiore, per ovvie maggiori esigenze di calcolo, è tblastx, che confronta una sequenza nucleotidica, dinamicamente tradotta nei sei registri di lettura, con le sequenze proteiche derivanti dalla traduzione dinamica nei sei registri dei database nucleotidici selezionati. A cosa serve? Si potrebbe pensare, infatti, che tblastx sia un'inutile e macchinoso doppione di blastn. Invece, le deduzioni che si ottengono confrontando ad esempio due genomi attraverso blastn o tblastx sono

diverse. Il confronto basato su blastn pesa allo stesso modo le mutazioni che non hanno alcun effetto sulle sequenze codificanti (mutazioni silenti, che trasformano il codone per un aminoacido in un altro codone che codifica lo stesso aminoacido) e le mutazioni che invece determinano sostituzioni aminoacidiche. In pratica, viene pesata correttamente la divergenza evolutiva, ma non la reale divergenza funzionale. Quest'ultima è meglio pesata da tblastx, poichè il passaggio di traduzione elimina le differenze silenti e sono pesate quindi solo le sostituzioni che alterano i prodotti finali, i quali in ultima analisi mediano le funzioni codificate.
Per questi motivi, blastn richiede tempi di esecuzione maggiori e può essere utile limitare le ricerche ad un subset delle sequenze (ad es., solo quelle di un organismo o di un raggruppamento tassonomico, oppure solo EST o DNA genomico, o infine limitazioni combinate).
Specialized BLAST
Negli ultimi anni sono state implementate numerose versioni "speciali" di BLAST che, in pratica, differiscono dalle principali per l'ottimizzazione di algoritmi di ricerca, matrici, funzioni, filtri e database utilizzati, pur restando sostanzialmente basate sull'architettura fondamentale di BLAST.
Una delle applicazioni specializzate più utili per complementare il lavoro molecolare è "Primer-BLAST", che consente di inserire la sequenza del templato, dei primer forward e reverse per la PCR, nonchè parametri che riguardano i primer. Aprendo la sezione "Advanced parameters"si può notare come in Primer-BLAST siano elevati i valori di "Expect" e bassi quelli di "Word Size". Esistono molti programmi per la progettazione dei primer che, più che validi per valutare la compatibilità della melting temperature, i rischi di appaiamento tra primer o di formazione di forcine, tuttavia non valutano la specificità di appaiamento. Il priming aspecifico infatti è un fattore che spesso crea problemi in PCR. Ad esempio, se progettiamo una coppia di primer sulla base della sola sequenza del gene del quale vogliamo amplificare, ad esempio, una regione, può darsi che il risultato dell’amplificazione fornisca più bande e non solo quella attesa. Ciò facilmente può accadere se non abbiamo valutato la specificità della coppia di primer. Infatti, se ad esempio il gene di nostro interesse ha una serie di paraloghi (geni omologhi nello stesso organismo) ed i primer sono progettati in parti della sequenza conservate anche negli altri geni, essi si appaieranno in più punti nel genoma e produrranno più amplificati. Primer-BLAST, quindi, è da utilizzare per predire e quindi evitare amplificazioni aspecifiche, che possono sottrarre tempo alle analisi sperimentali o perfino portare fuori strada.
Un ulteriore tool bioinformatico sviluppato per le analisi molecolari ed in particolare per il clonaggio è VecScreen, utile per "decontaminare" le sequenze genomiche o di cDNA da quelle di vettori. Se, infatti, per sequenziare dei frammenti di DNA clonati si utilizzano un vettore ricombinante come stampo e dei primer per PCR che riconoscono regioni del vettore a monte ed a valle dell'inserto, la sequenza riassemblata conterrà l'inserto, ma anche le parti del vettore comprese nell'amplificato. Depositando sequenze di questo tipo in banca dati si produce, a causa della ripetizione delle sequenze spurie del vettore, un elevato rumore di fondo, che disturba le ricerche e confonde i risultati. Ormai gli esperti dei server bioinformatici che immettono nelle banche dati le nuove sequenze ricevute provvedono, sistematicamente, a verificarne eventuali contaminazioni da vettore. Tuttavia, qualche sequenza di vettore può sfuggire ed, in ogni caso, è opportuno (e corretto) che, prima di sottomettere una sequenza, si provveda ad analizzarla (ad esempio, con VecScreen, o mediante confronto contro database di vettori disponibili presso altri server).
Poichè sempre più rilevante per la farmacogenomica la conoscenza dei polimorfismi genici (Single Nucleotide Polymorphism), nella sezione "Specialized BLAST" è disponibile SNP-BLAST, che consente di utilizzare geni o cDNA come sonde per la ricerca dei relativi polimorfismi non solo in Homo sapiens, ma anche in altri organismi.

Chi analizza l'espressione genica può verificare la disponibilità per una sequenza nucleotidica di profili di espressione genica Gene Expression Omnibus (GEO) e dati su tessuto e stadio specificità; per chi lavora nell'immunologia molecolare è disponibile Ig-BLAST, limitato a human, mouse, rat e rabbit.
BLAST e database
I database associabili a ricerche con applicazioni NCBI-BLAST sono numerosi. In particolare, quelli per la sezione Nucleotide BLAST consentono ricerche in un ambito ampio, selezionando l'insieme delle sequenze non ridondanti o "nr" (che comprende le tre principali banche dati di DNA mondiali) o set più ristretti, quali ad esempio i database di sole EST (con sottosezioni est_human, est_mouse ed est_others) o di sequenze brevettate (Patent). Alcuni database sono riservati alle sequenze di uno specifico organismo, primo tra tutti l'uomo, ma anche lievito (database yeast, figura in alto) o altre specie modello. Attualmente presso l'EBI è implementato WU-Blast2, una versione di BLAST ottimizzata dal pool bioinformatico della Washington University e dall'NCBI. Selezionando la finestra dei database in cui http://www.ebi.ac.uk/blast2/index.htmlWU-Blast2 può effettuare ricerche, si può avere un'idea del gran numero di database proteici e nucleotidici nei quali possono effettuare ricerche le applicazioni BLAST, nonchè del tipo di applicazioni utilizzabili per database.
Scelta delle applicazioni in funzione delle ricerche.
Per ottenere risultati significativi in una ricerca per allineamento è necessario selezionare opportunamente applicazioni e database; per trarne informazioni rilevanti è inoltre fondamentale sapere interpretare correttamente i risultati e rimodulare le richieste a seconda delle necessità. Nel caso non si abbiano le idee chiare o non si rammenti più quanto appreso, si potrà ricorrere agli schemi di selezione presenti sulle pagine guida (sezione Help) di NCBI-BLAST. Guide simili sono presenti anche presso altri server che forniscono servizi BLAST.
In genere, la scelta di sito, applicazioni e database, nonchè dei parametri di ricerca ed output, non richiede studi approfonditi e, nella maggior parte delle ricerche di routine, i valori preimpostati possono essere lasciati inalterati, se è corretta la combinazione tra applicazione da utilizzare e database in cui effettuare la ricerca. Inoltre, in molti casi ormai, modifiche nella selezione dell'applicazione comportano il passaggio automatico al set di database compatibili, evitando ad utenti distratti di inoltrare richieste errate. E' esperienza comune che si ottengano risultati differenti effettuando analisi su server differenti; questo dipende sia dal differente tipo di database interrogabili, le cui sequenze non sono condivise al 100%, sia dal fatto che i parametri preimpostati di query ed output possono essere diversi. E' pertanto sempre opportuno, prima di passare ad una fase di analisi dei risultati e di elaborazione delle ipotesi, procedere in una serie di richieste su più database, reiterate con o senza filtri e con parametri più o meno stringenti. In genere è conveniente effettuare una medesima ricerca con BLAST con e senza filtro per le regioni a bassa complessità, in modo da non perdere informazioni che potrebbero essere mascherate dal rumore di fondo provocato da tali regioni, ma anche da rilevarne la presenza, talora biologicamente significativa. Ad esempio, alcune regioni ricche in prolina contraddistinguono particolari famiglie di proteine, regioni ricche in aminoacidi acidi spesso sono presenti in domini leganti cationi e regioni ricche in aminoacidi basici possono essere presenti in proteine nucleari. Tutto ciò può essere inutile, tuttavia, quando i risultati sono estremamente ed univocamente indicativi, come nel caso di identificazione di sequenze identiche al 100% o comunque altamente similari.
BLAST ed i preset
In molti laboratori BLAST è utilizzato ampiamente, poiché apparentemente l’interfaccia è semplice: si inserisce la sequenza nella finestra di sottomissione, si schiaccia il pulsante si submission e si

attendono ed esaminano i risultati. In realtà, la gran maggioranza degli utilizzatori non è consapevole dei preset impostati e del fatto che quelli che sono considerati “i risultati” in realtà variano al variare dei settings. E’ inoltre importante saper leggere i risultati stessi e reiterare le richieste per ottenere più informazioni. Questa sezione è quindi rivolta a fornire indicazioni per un uso “evoluto” di BLAST.
Esaminiamo l’interfaccia di BLAST disponibile presso il sito dell’NCBI. Nel caso si voglia utilizzare una query sequence proteica per cercare sequenze omologhe, si utilizzerà blastp. Nella sezione Basic BLAST si seleziona protein blast e la pagina che si apre è già impostata su blastp. La pagina consente la sottomissione della sequenza, che può essere sottomessa in formato FASTA oppure come semplice sequenza. In quest’ultimo caso, tuttavia, converrà indicare di cosa si tratta nella finestra Job Title per tenere traccia della query utilizzata. Per una singola ricerca ciò non è molto importante, ma quando si usano più sequenze query, spesso si memorizzano i file con i risultati per poi analizzarli successivamente; in tal caso è opportuno che in ciascun file siano evidenti nome ed identificativo della sequenza query, per evitare confusione.
Chi lancia BLAST subito dopo aver inserito la sequenza opera comunque “scelte inconsapevoli”: BLAST è solo un metodo che per funzionare si basa su molteplici settings variabili; inoltre anche i dataset di confronto sono variabili. Utilizzare i preset di base non significa quindi non avere operato alcuna scelta, quanto piuttosto avere accettato i preset. Può darsi che ciò vada bene, ma è opportuno sapere cosa si sta facendo, perché i risultati e la loro visualizzazione, spesso parziale, dipendono dai settings. Una prima cosa di cui spessso un utente non evoluto di BLAST non è consapevole è la scelta del database. Non ha senso dire “questo è il risultato di BLAST” in senso assoluto, poiché BLAST opera un confronto ed il risultato pertanto dipende tanto dalla query sequence quanto dai database che contengono le subject sequences con cui la query è allineata. Se, ad esempio, utilizziamo due volte la stessa query, mantenendo tutti i parametri dell’algoritmo invariati, ma cambiamo database, i risultati saranno diversi. Per questo motivo la ripetizione delle ricerche con BLAST presso più server (ad es. NCBI ed EBI) consente di ottenere informazioni più complete. A volte la completezza di un dataset può essere un problema, più che una risorsa. In blastp il preset per il database è non-redudant protein sequences (nr). Questo database in pratica mira a fornire un set non ridondante di sequenze da più database. In teoria, è la scelta ottimale ed infatti è il preset. Tuttavia c’è comunque ridondanza nei database e confrontare la query con un gran numero di sequenze può portare alla visualizzazione di risultati non sempre indicativi. Supponiamo, ad esempio, che si cerchino indicazioni sulla struttura di domini di una proteina non ancora caratterizzata. In tal caso desideriamo individuare - grazie alla capacità di allineamento locale di blast - “blocchi” di allineamento tra frammenti della nostra query ed altre proteine (intere o frammenti). Tuttavia, se vi sono in banca dati molte sequenze omologhe alla query, poiché il numero di sequenze mosrate in output è limitato da uno dei preset, c’è il rischio che siano visualizzati solo allineamenti completi, nei quali cioè l’intera query è allineata alle sequenze subject. Selezionare uno specifico database è quindi un’operazione di modifica dei preset che molto spesso ha senso e può velocizzare la ricerca di informazioni. Ad esempio, ridurre la ricerca al solo database SwissProt, che contiene meno sequenze, ma ben annotate, porta a trovare “nei risultati”, ovvero tra le sequenze omologhe estratte, proteine per le quali sono spesso disponibili molte informazioni. E’ più importante infatti esaminare poche sequenze “di qualità” che trovare estratte molte sequenze delle quali alcune ridondanti, altre false predizioni, altre infine prive di annotazione d’interesse.... Tuttavia la modifica dei preset è un’operazione che va fatta con intelligenza, utilizzata per estendere la ricerca.
Meglio reiterare 3-4 volte una ricerca con BLAST ed ottenere informazioni utili per indirizzare il lavoro sperimentale, che “risparmiare” mezz’ora di tempo e perdere informazioni che magari possono far risparmiare settimane o mesi di lavoro al banco..... quindi modificare i preset non significa ad es. cercare solo in SwissProt, quanto piuttosto, dopo aver svolto la ricerca con i preset, modificare il database e reiterare con selezione SwissProt. Non si deve pensare, infatti, che selezionando un solo database o l’insieme di tutti che lo contiene sia inutile: le sequenze di SwissProt omologhe alla sonda
potrebbero essere estratte egualmente con la ricerca impostata su nr, ma non comparire in output per la limitazione nel numero di sequenze mostrate. Ad esempio, supponiamo che una certa sequenza “XYZ” presente in SwissProt ed omologa alla query possa fornirci informazioni utilissime. Utilizzando: database = SwissProt, se la sequenza XYZ è la settantaduesima in elenco, essa è visibile poiché è mantenuto il preset: sequenze in output = prime 100. Se tuttavia manteniamo anche il preset: database = non redudant (nr), la sequenza XYZ, a causa dell’estrazione di molte altre sequenze, potrebbe scivolare oltre il centesimo posto in elenco e non essere più visualizzata. Si potrebbe dedurre che sia opportuno modificare il preset di visualizzazione, portandolo ad es. a mille, ma limitarsi a questo per non cambiare database non è una buona strategia. Infatti, quando la sequenza di interesse è un frammento piccolo (score basso), il rischio che sia non visualizzato resta alto. D’altronde, esaminare centinaia di alllineamenti prende molto tempo. Meglio dunque seguire la strategia di più ricerche, ciascuna in differenti database.
Valutazione preliminare di output grafico ed elenco sequenze
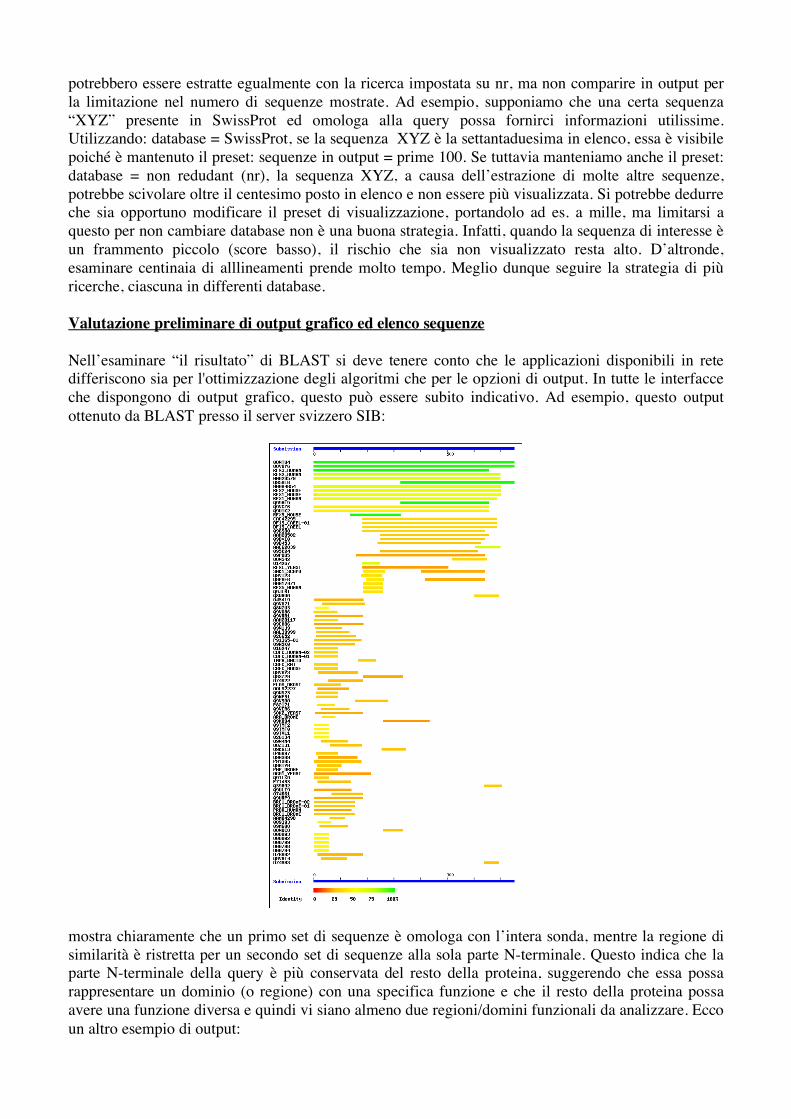
Nell’esaminare “il risultato” di BLAST si deve tenere conto che le applicazioni disponibili in rete differiscono sia per l'ottimizzazione degli algoritmi che per le opzioni di output. In tutte le interfacce che dispongono di output grafico, questo può essere subito indicativo. Ad esempio, questo output ottenuto da BLAST presso il server svizzero SIB:
mostra chiaramente che un primo set di sequenze è omologa con l’intera sonda, mentre la regione di similarità è ristretta per un secondo set di sequenze alla sola parte N-terminale. Questo indica che la parte N-terminale della query è più conservata del resto della proteina, suggerendo che essa possa rappresentare un dominio (o regione) con una specifica funzione e che il resto della proteina possa avere una funzione diversa e quindi vi siano almeno due regioni/domini funzionali da analizzare. Ecco un altro esempio di output:

che mostra l’esistenza di un blocco centrale più conservato delle regioni N- e C-terminale. Quindi, almeno 3 regioni funzionali, dove funzione non vuol dire necessariamente “attività” biochimica, intesa in senso enzimatico, quanto semplicemente regione proposta a mediare un meccanismo (legame di ioni, DNA, altre proteine, attività enzimatica o regolativa ecc.). Poiché la similarità non è alta (lo si deduce dal codice a colori delle barre), le sequenze potrebbero condividere domini specializzatisi per divergenza o essere infine caratterizzate da regioni a bassa complessità, formate per reiterazione e degenerazione di motivi semplici di sequenza, quali, ad esempio, le regioni coiled coil e quelle presenti nelle proteine ricche in un particolare aminoacido:
in cui lo score dipende prevalentemente dalle identities, ovvero dall'allineamento tra gli aminoacidi del tipo iper-rappresentato in entrambe le sequenze. Dopo aver valutato l'output grafico (non sempre disponibile!) si passa all'analisi dell'elenco delle proteine estratte. L'elenco mostrato di seguito è limitato alle prime sequenze per ragioni di spazio:

Le prime tre mostrano i più alti valori di Score e sono rappresentate in verde nell'output grafico. La prima di esse, con numero di accesso Q8WTU4, mostra 100% di identità con la sequenza sonda:
Sebbene (per motivi di spazio) si sia mostrato l'allineamento dei soli primi 60 residui, gli elementi informativi essenziali sono rilevabili: oltre alla descrizione della sequenza estratta "Hypothetical...." sono indicati l'organismo cui appartiene e, subito dopo, la lunghezza. A seguire, i valori di Score (che in questo caso è il più alto e varia da allineamento ad allineamento) e di Expect (pari a zero in questo caso) precedono l'indicazione del numero di residui identici (identities) e dell'insieme "residui identici + compatibili", la cui somma (positives,) che in questo caso coincide con identities perchè le sequenze sono uguali. Quando invece le sequenze sono diverse, il numero di residui "compatibili" (secondo il sistema di valutazione implementato) è pari alla differenza tra positives ed identities.
Infine, è da ricordare che l'allineamento tra le due sequenze viene diviso per motivi di visualizzazione in blocchi di lunghezza prestabilita (in questo caso, 60 residui), in cui la sequenza marcata "Query" è la sonda e quella marcata "Sbjct" è la sequenza estratta. Tra i residui delle due sequenze, la riga intermedia riporta tutti i residui identici e, in caso di residui compatibili, il simbolo "+". Quando i residui allineati non sono compatibili, è lasciato uno spazio vuoto.
Sebbene la prima sequenza Sbjct sia identica alla Query, essa non fornisce indicazioni utili sulla possibile funzione della proteina sonda, in quanto è una "Hypothetical protein" di funzione ignota. Più informativa è la seconda proteina, per la quale è indicata una funzione ("regulatory factor"):
Tuttavia bisogna saper leggere: la Sbjct Q8VBY6 non è il regulatory factor, ma solo una proteina simile ("Similar to") a tale fattore regolativo...... Nell'allineamento dei primi 60 residui mostrati non si rilevano differenze, ma ciò non deve ingannare, poiché è chiaramente indicato che, dei 749 residui allineati, molti ma non tutti (741) sono identici e 4 (ovvero, 745-741) sono "compatibili". La terza proteina estratta, sebbene non omologa nell'estrema regione C-terminale, offre ulteriori informazioni:

Infatti, sebbene abbia un valore di Score più basso della proteina di topo, essa è perfettamente identica alla sonda per i primi 656 residui. Dalla lunghezza indicata (707 AA) si deduce la presenza di una "coda" C-terminale divergente di circa 50 residui. Dalla parte iniziale dell'allineamento tra sonda e sequenza Q9W523 di Drosophila si possono rilevare alcune cose interessanti:
Infatti, a parte il valore di Score più basso (e quello di Expect più alto), si comprende la natura locale dell'allineamento (non per nulla BLAST vuol dire: Basic Local Alignment Search Tool....) tra la regione N-terminale della proteina sonda (ciò è asserito dai numeri "10" e "91" che indicano il primo ed ultimo residuo mostrati della sequenza "Query") ed una regione interna (residui 405-485 di Sbjct) della grande (1624 AA) proteina di Drosophila. Si può inoltre rilevare che la maggior parte degli aminoacidi conservati sono residui di glutamina (simbolo "Q").
Estrazione di informazioni mediante link
L'elenco delle sequenze estratte con BLAST solitamente contiene link (in blu e sottolineati) attraverso i quali si possono ottenere ulteriori informazioni. Tornando alle sequenze estratte nella ricerca "A", un clic sul link rappresentato dal numero di accesso della terza sequenza:
richiama la scheda della proteina stessa (della quale si riportano solo alcune parti):
In ciascuna scheda sono indicate, se disponibili, una o più referenze bibliografiche:

in cui il link finale, rappresentato dalla sigla della rivista e dagli estremi del numero pubblicato, consente di accedere alla pagine web dell'articolo:
in cui sono riportati, ancora una volta, nome della rivista, titolo ed autori e compare un abstract (sommario informativo) dell'articolo. Nella scheda della sequenza estratta e richiamata mediante il link al numero d'accesso sono contenute, sempre o quasi, "Cross-references", ovvero link ad altre schede inerenti, quali ad es. sequenza genica codificante, organismo ed eventuali database relativi a tratti ereditari, malattie genetiche, famiglie di domini ecc.:
Molto spesso, come nella normale ed a volte perfino ludica "navigazione in internet", il passaggio da un link ad un altro, soprattutto se si è abili nel non andare "fuori strada", può in tempi rapidi fornire moltissime informazioni utili e portarci a conoscenza di fenomeni biologici sconosciuti o quasi e possibilmente illuminanti per la nostra ricerca.
Tuning: modifica dei parametri e reiterazione dell'analisi
Nell'uso di BLAST in molti casi ci si limita ad una ricerca effettuata lasciando inalterati i set preimpostati: questo tipo di approccio in alcuni casi è sufficiente a rivelare informazioni abbastanza o completamente indicative; molte volte, tuttavia, fermarsi ai primi risultati ottenuti può essere un errore. Se, ad esempio, l'output di BLAST mostra che la sequenza utilizzata come query si allinea con elevati indici di similarità ad una o più sequenze dei database in cui si è svolta la ricerca, il confronto delle sequenze e l'analisi delle sequenze omologhe può essere sufficientemente informativo. Tuttavia, molto spesso l'output di BLAST mostra la presenza di una o più sequenze omologhe per le quali gli indici di similarità sono più bassi o addirittura molto bassi e l'allineamento è talora limitato a corti frammenti della sequenza query. In questi casi è ragionevole reiterare le analisi modificando i parametri di ricerca. Si definisce gergalmente "fine tuning" uno o più cicli di reiterazione con modifica dei parametri, alla ricerca di risultati non emersi o poco chiari nelle analisi preliminari. Per comprendere come e quando si ricorra al tuning nelle analisi è necessario anche avere un'idea delle molteplici finalità di una ricerca BLAST. Tra i possibili approcci che si avvalgono di BLAST, in

alcune analisi evoluzionistiche si usa come sonda "query" un gene o una proteina di un certo organismo per cercarne nei database ortologhi e paraloghi e delinearne la diffusione ed evoluzione negli organismi ed i membri della sua famiglia genica/proteica. E' evidente che, in tal caso, ricerche limitate ai parametri preimpostati, soprattutto per quanto riguarda il tipo di applicazione BLAST (ovvero, blastn, blastp, blastx ecc.) ed il set di database in cui la ricerca è effettuata, forniranno solo una parte, più o meno limitata, dei risultati.
BLAST può essere anche utilizzato per comprendere a quale gene corrisponda un cDNA di un eucariote ottenuto, ad esempio, per RT-PCR. Per ottenere tale informazione potrebbe essere sufficiente, in teoria, utilizzare il cDNA come sonda in un'analisi blastn, deducendo per allineamento tra regioni della query e regioni genomiche esoni ed introni. La realtà, tuttavia, è più complessa: se ci si limita al set di database preimpostato per blastn, non è garantito che sia quello giusto. Infatti, nella maggior parte dei casi, analisi BLAST realizzate presso il server dell'NCBI rimanderanno al set di database NCBI (ad es. Genbank), mentre analisi BLAST presso il server dell'EBI saranno preimpostate in associazione ai database europei (ad es. EMBL). Ciò può far perdere informazioni poiché, malgrado gli accordi per lo scambio reciproco delle sequenze depositate, i database posseggono sequenze "uniche", assenti in altri database. Inoltre, il set di sequenze preimpostato per blastn può contenere, allo stesso tempo, tanto dati genomici "grezzi" (quali le sequenze di "contig" provenienti dai progetti di sequenziamento) quanto geni noti o "predetti", ovvero sequenze di geni "virtualmente esistenti", ovvero sequenze dedotte dai dati grezzi mediante algoritmi di predizione genica.
Nel set di database preimpostati per blastn possono essere incluse anche sequenze di cDNA di banche di EST. E' possibile, in conclusione, che l'output di blastn mostri l'allineamento del cDNA query con numerose sequenze nucleotidiche, tra le quali si dovrà distinguere tra sequenze "reali" e "virtuali" e tra "genomiche" ed "espresse". Anche tra queste ultime, infine, possono essere presenti dei "falsi", dal momento che, sempre più spesso, sono riportate nelle banche dati, con il nome "mRNA", sequenze che non corrispondono a mRNA dimostrati sperimentalmente (ovvero, purificati, retrotrascritti in cDNA e sequenziati) bensì a mRNA "putativi", corrispondenti a sequenze probabilmente codificanti ed immesse automaticamente in banca dati da algoritmi di predizione genica. La reiterazione dell'analisi non sempre è necessaria, ma quasi sempre è utile.
Tornando all'esempio del cDNA, il confronto con i dati genomici puri (ovvero, la sequenza del cromosoma) è quella che consente di individuare locazione cromosomica e struttura di esoni ed introni. Possiamo tuttavia dire di aver stabilito "la" struttura del gene? Quasi certamente no, soprattutto se il cDNA proviene da un eucariote superiore, in cui il fenomeno dello splicing differenziale è estremamente diffuso. Più corretto, quindi, considerare di aver svelato "una" struttura del gene, non escludendo che ne possano esistere altre. Per questo motivo, il confronto del cDNA con altre sequenze espresse presenti n banche dati di EST potrebbe consentire di individuare varianti di splicing dello stesso gene e di associarle ai differenti organi o tessuti.
Anche il confronto con sequenze predette può essere utile, nel suggerire altre "possibili" combinazioni di esoni ed introni. In alcuni casi, anzichè cambiare il database nelle reiterazioni, può essere utile cambiare altri parametri. Uno delle più banali modifiche è la necessità di reiterare un'analisi realizzata con un "filtro", che è preimpostato in quasi tutte le interfacce ai servizi BLAST, per mascherare regioni a bassa complessità. Il filtro, velocizzando la ricerca, altera tuttavia i reali valori di similarità riportati nell'output. Infatti, se nella query sequence sono presenti regioni a basssa complessità ed esse sono mascherate dal filtro, i valori di similarità riportati nell'output sono decurtati dell'apporto dell'eventuale identità o positività dei residui "mascherati" e risultano, quindi, inferiori rispetto ai valori reali. Altri parametri che è opportuno modificare nell'eventuale reiterazione dell'analisi sono le matrici ed il valore di E (Expect), per variare la valutazione dei pesi e la stringenza.

Parametri modificabili, filtri ed opzioni di output
Nel precedente blocco di lezioni si è trattata l'importanza delle matrici per i metodi di allineamento. Solitamente BLAST si avvale della matrice BLOSUM62, ma la selezione Matrix preimpostata può essere modificata, ovviamente in funzione delle necessità di analisi. Tuttavia, lo sviluppo dell'algoritmo di MEGABLAST e di altre applicazioni "ottimizzate" anche attraverso la scelta di matrici differenti, rende meno fondamentale una conoscenza approfondita delle matrici (almeno nel caso della maggior parte delle analisi di routine). Anche il parametro Gap Costs è importante, in quanto influenza la possibilità di ottimizzare (e presentare in output) gli allineamenti mediante l'introduzione di interruzioni (gaps). In particolare, il "peso" che si attribuisce all'inserimento di gaps o al loro allungamento può penalizzare o favorire l'estrazione di hits con gaps; ad esempio, se è noto che le sequenze omologhe rappresentano proteine o domini con regioni divergenti di lunghezza variabile ed è importante estrarne il massimo numero possibile, è opportuno un gap cost basso, mentre se si vuole individuare un blocco con pochi (o nessun) gap, è opportuno che il gap cost sia alto:
Poichè le informazioni sulle matrici possono essere richiamate in linea mediante appositi link a pagine esplicative, può rappresentare un notevole vantaggio impostare correttamente i parametri di una ricerca con BLAST. Infatti, tanto le caratteristiche della sequenza sonda, quanto quelle dei database in cui si ricercano sequenze similari, possono variare notevolmente (lunghezza, numero, ripetizione, conservazione, presenza o meno di regioni a bassa complessità ecc.).
Un altro parametro molto importante (Expect) esprime la soglia di significatività statistica valutata secondo il modello stocastico di Karlin ed Altschul. In pratica, variazioni del valore di Expect modulano la stringenza della ricerca con BLAST: più è basso, minore è il numero delle sequenze estratte. Una soglia di significatività statistica (Inclusion Threshold) può essere introdotta anche nel modello utilizzato da PSI-BLAST per creare per iterazione la matrice PSSM.
Alcune opzioni riguardano applicazioni specifiche di blast; ad esempio, è possibile scegliere il codice genetico da utilizzare (Query Genetic Code) per la traduzione della sonda da parte di blastx. BLAST consente di limitare le ricerche ad un subset di sequenze, utilizzando l'opzione "Limits by Entrez Query", per la quale possono essere utilizzati gli stessi termini ed operatori booleani, nonchè le medesime restrizioni per campo, che accetta il sistema di ricerca Entrez.
L'implementazione di filtri fornisce a BLAST la possibilità di velocizzare o rendere più selettive le ricerche, anche se un uso errato dei filtri può portare, in taluni casi, alla perdita di informazioni importanti. Uno dei filtri più utilizzato è quello per le regioni a bassa complessità (Low-complexity), quali ad esempio regioni ricche in un particolare aminoacido (prolina, lisina...). Il filtro Mask for lookup table only riguarda il passaggio di estensione, che avviene nella fase

computazionale che porta all'estrazione degli allineamenti ritenuti significativi (hits). Questo filtro, correlato al precedente, impedisce che siano considerate le regioni a bassa complessità (LCR) nella fase di costruzione della tabella iniziale (contenenti le regioni "seme" dell'allineamento), ma le LCR non sono escluse dalla fase di estensione. In tal modo il filtro evita che l'estrazione di LCR oscuri quella delle regioni ad alta complessitàk pur non comportando perdita di informazione (LCR associate a regioni ad alta complessità contribuiscono ad allungare l'allineamento).
Il filtro Human repeats è stato sviluppato contro regioni ripetute, quali ad esempio le sequenze LINE e SINE del genoma umano. Utilizzando questo filtro, soprattutto se la sonda è particolarmente lunga o si ricerca in un database con molte sequenze ripetute (htgs), la ricerca procede a velocità maggiore o talora evita di andare in "timeout" (ovvero, fuori tempo massimo per ottenere i risultati in linea). L'opzione Mask Lower Case consente di personalizzare il filtro, inserendo nella finestra di query la sequenza della sonda con caratteri maiuscoli (Upper Case), fatta eccezione per regioni che si vuole siano filtrate, presentate con lettere minuscole (Lower Case). Tale opzione serve a ridurre o eliminare il peso di regioni variabili all'interno di un blocco conservato, o al contrario non pesare una regione conservata per meglio estrarre sulla base della conservazione delle altre regioni. E' molto utile, nelle ricerche con BLAST, che siano modificabili anche le opzioni di output, ovvero di presentazione dei risultati.
Ricerca ampliata o ristretta a specifici set di sequenze
In alcuni casi ampliare o restringere il set di dati iniziali può essere cruciale per ottenere risultati utili. Nella ricerca di ortologhi, realizzare sessioni BLAST presso un server modulando i set di database può non essere sufficiente. In alcuni casi, infatti, le sequenze dei geni o delle proteine di un dato organismo sono presenti solo o in larga parte su database "dedicati" e largamente assenti nei database maggiori, quali quelli di NCBI ed EBI. E' utile, quindi, cercare con pazienza - attraverso motori di ricerca e la "navigazione" via link sui server bioinformatici - anche database minori, nel caso si voglia esser sicuri di reperire il massimo numero possibile di sequenze associate ai vari organismi di un gruppo tassonomico più o meno esteso. Altrettanto logico, nel caso si stia svolgendo tale ricerca per studiare ortologhi e paraloghi in un gruppo tassonomico, è limitare lo screening delle banche dati maggiori al gruppo tassonomico stesso. Ad esempio, se si sta studiando la conservazione e variazione dei membri di una famiglia di geni e/o proteine in piante, è inutile lasciare che la ricerca si svolga anche su sequenze umane, di animali o di lieviti e batteri: ciò rallenta l'esecuzione e comporta anche l'inutile estrazione di sequenze non interessanti ai fini della ricerca impostata. In alcuni casi, inoltre, ortologhi e paraloghi di proteine possono essere "falsi" o "nascosti". Utilizzando la sequenza di una proteina come sonda in una ricerca blastp contro database proteici, possono essere estratte sequenze di proteine "ipotetiche", che "potrebbero" esistere, ma non necessariamente esistono e possono quindi rappresentare dei "falsi", in quanto la sequenza primaria delle stesse è la traduzione di una sequenza codificante dedotta da una predizione genica. La comparazione della stessa sequenza proteica con la traduzione di EST può eventualmente confermare la sequenza predetta o fornire sequenze alternative. Le sequenze proteiche possono anche essere nascoste, nel caso, ad esempio, per un certo organismo gli algoritmi di predizione genica non abbiano individuato la corrispondente sequenza codificante. Infatti gli algoritmi di predizione non producono solo sequenze valide o parzialmente errate; talora non riescono ad individuare, nel DNA, alcune regioni codificanti. Tuttavia, utilizzando tblastn, l'applicazione BLAST che confronta una sonda proteica con i database di acidi nucleici tradotti nei sei registri di lettura, se nel genoma sequenziato dell'organismo o del gruppo di organismi in studio esistono regioni codificanti non individuate, le loro traduzioni in sequenze proteiche saranno estratte da BLAST, che nell'output mantiene l'informazione primaria (ovvero, la sequenza di DNA dalla quale derivano). In tal modo si potrà tentare di ricostruire la sequenza codificante, da confermare, ovviamente, per via sperimentale. Nella caratterizzazione di un gene può essere interessante (o addirittura necessario) studiarne la tessuto-specificità di espressione. In tal caso, l'approccio sperimentale può essere guidato ed affiancato dalla ricerca di cDNA nelle librerie di EST, che

possono fornire indicazioni complementari rispetto a quelle ottenute direttamente in laboratorio. Inoltre, il confronto in silico dei profili di espressioni di due geni consente di stabilire se, ad esempio, essi sono coespressi in un certo organo o tessuto e quindi di predire eventuali, possibili interazioni tra i prodotti proteici dei geni stessi.
Introduzione all'allineamento multiplo
Programmi come BLAST e FASTA sono ampiamente utilizzati per cercare nei database sequenze simili ad una sequenza sonda, ma spesso è necessario disporre di metodi per allineare un insieme di sequenze già disponibili, ad esempio per ricostruire le sequenze dei cromosomi nei progetti di sequenziamento genomico, oppure per evidenziare le parti di sequenza che variano molto o poco tra geni/proteine ortologhi/e. I programmi di multiallineamento sono insostituibili o quasi quando è necessario ottenere informazioni di tipo evoluzionistico (alberi filogenetici) e/o funzionale (identificazione di domini, pattern e profili). Ad esempio, se attraverso una ricerca con BLAST abbiamo individuato numerose sequenze di proteine e vogliano stabilirne livelli di correlazione (ad esempio, appartenenza alla stessa o a differenti sottofamiglie proteiche) o individuarne le caratteristiche condivise e peculiari, sarà necessario allineare le sequenze (o loro frammenti) individuate con BLAST in modo che, per ciascuna posizione dell'allineamento, possano essere valutate conservazione e variazione dei residui (o la loro assenza, pesata grazie all'introduzione di interruzioni o gaps). Dopo una fase iniziale, affidata agli algoritmi del programma utilizzato, sarà necessario ottimizzare il multiallineamento in funzione del tipo di analisi verso cui ci si sta indirizzando. Infatti, se ad es. si sta tentando di stabilire il grado di "parentela" tra le sequenze (ovvero, il livello di correlazione evolutiva), devono essere considerati fattori quali la rappresentazione di un numero adeguato e "pesato" di sequenze appartenenti a specie distanti, nonchè l'eventuale presenza di domini e subdomini differenti, che potrebbero aver seguito, nella loro storia evolutiva, percorsi di mobilità e specializzazione differenti, ecc. Dunque, è fondamentale partire da un set iniziale bilanciato. Per individuare un nuovo dominio/subdominio e definirne le caratteristiche, può essere utile "accorciare" o"allungare" il multiallineamento preliminare, o semplificarlo per renderne più immediata l'interpretazione, ad esempio sostituendo regioni variabili (in composizione e numero dei residui) con simboli (spesso, parentesi con numeri) che mantengono l'informazione sulla variabilità ma limitano la rappresentazione alle regioni di sequenza conservate. Non esistono regole rigide per rappresentare un multiallineamento; i programmi più diffusi, in genere, sono compatibili con numerosi formati di input ed output. Nella letteratura scientifica, a seconda delle esigenze (e della disponibilità di spazio fornita dalle riviste....), la rappresentazione grafica dei multiallineamenti può avvalersi di numerosi formati, variabili soprattutto in relazione alla colorazione dei residui o all'adozione di alfabeti semplificati (ovvero, nei quali residui che condividono una o più proprietà sono rappresentati con un unico simbolo). Un allineamento multiplo può essere realizzato perfino manualmente, purchè si usi un font non proporzionale (ad es. Courier), in modo che ciascun carattere occupi la stessa casella di spazio, ed evitando i font proporzionali (che rappresentano la gran maggioranza di quelli disponibili, ad es. Times), per i quali caratteri più "stretti" come la "i" occupano meno spazio di quelli "larghi" come m, w ecc.
Ecco lo stesso allineamento manuale rappresentato in Times (disallinea) e in Courier (resta corretto):
Ovviamente, i software di multiallineamento non hanno questo problema e ciò potrebbe indurci a pensare che siano da evitare allineamenti "manuali", realizzati operando con un word processor. In realtà, le esigenze di ottimizzazione e rappresentazione grafica spesso portano ad una fase di rielaborazione manuale: basterà non dimenticarsi del problema del font! Non esiste una "regola" per l'adozione di un particolare metodo nella realizzazione di un multiallineamento o per l'impostazione

dei suoi parametri, che sono da scegliere in funzione del tipo di analisi, ovvero quasi sempre sulla base di reiterazioni del multiallineamento e quindi su base empirica. Tuttavia, così come BLAST si è imposto come programma più utilizzato per le ricerche di sequenze similari ad una sonda, attualmente il programma che è divenuto uno"standard" per i multiallineamenti è CLUSTALW (per gli interessati c'è la scheda di approfondimento). In genere, il numero di sequenze da confrontare è abbastanza alto da rendere difficile un multiallineamento manuale, cosicchè inizialmente i multiallineamenti sono ottenuti attraverso programmi come CLUSTALW e successivamente sono "ottimizzati" manualmente e "decorati" con un word processor o appositi programmi di editing.
Individuazione e definizione di domini proteici con PSI-BLAST
PSI-BLAST consente di ampliare il dataset per definire un nuovo motivo/dominio, costruendo per reiterazione una Position Specific Score Matrix (PSSM) "ottimizzata" per la putativa famiglia di proteine di cui si sta definendo un nuovo profilo e che può essere salvata per usi successivi. L'oculata selezione del set di sequenze per le successive iterazioni e la variazione della soglia di inclusione consentono di individuare più sequenze che condividono il motivo o dominio, anche in caso di livelli bassi di similarità. L'output presenta un elenco di sequenze con E-value superiore alla soglia, ma anche un numero di sequenze con E-value inferiore, tra le quali possono essere selezionate sequenze che si vogliono includere per la seconda iterazione e lanciare, quindi, l'iterazione, che estrarrà più sequenze del set di interesse grazie alla matrice ad hoc. Riselezionando tra le sequenze con E-value inferiore alla soglia quelle di interesse e lanciando una terza iterazione, e così via, si può ampliare il set di sequenze fino al punto di convergenza, ovvero il momento in cui non è estratta più alcuna nuova sequenza, essendo state già estratte tutte quelle che la matrice specifica è in grado di estrarre. A tal punto si possono apportare degli aggiustamenti, quali quello di "centrare" correttamente la regione da utilizzare come sonda, in modo da eliminare il peso di regioni estranee al motivo o dominio. Ciò può essere fatto "accorciando" nella parte N-terminale e/o C-terminale la sequenza, o "mascherando" manualmente regioni interne non conservate. Infine, le sequenze raccolte possono essere allineate manuamente o con programmi come CLUSTALW e ciò consente di ottenere un "consensus" da utilizzare, come pattern o profilo, per scanning di database al fine di verificarne la specificità. Nel caso risulti capace di estrarre specificamente le sequenze appartenenti ad una cerca classe proteica, potrà essere sottoposto ad una banca dati funzionale, divenendone un nuovo marcatore.
Analisi funzionali: regioni conservate e specializzate.
Fasi di allineamento con BLAST e, successsivamente, di multiallineamento con CLUSTALW, consentono di ottenere indicazioni di tipo evoluzionistico ma anche di individuare regioni conservate o specializzate e domini putativi, ottenendo indicazioni di tipo funzionale. La ricerca di "tag" (marcatori) funzionali (pattern & profile discovery) è alla base della genomica funzionale; in tale ambito, i multiallineamenti di sequenza permettono di individuare blocchi conservati ed altri variabili, per definire gli insiemi di partenza su cui lavorare per definire nuovi pattern, profili ed individuare domini proteici. A titolo di esempio si presentano le fasi intermedie di un lavoro di identificazione di un dominio proteico. Lo studio è iniziato quando si è trovato che il prodotto del gene umano SYBL1, definito TI-VAMP o VAMP-7, pur essendo correlato alla famiglia delle sinaptobrevine/VAMP, possedeva un'ulteriore estensione N-terminale di circa 110 aminoacidi. Utilizzando come sonda la sequenza di VAMP-7 (220 residui), l'output di BLAST mostrava molte sequenze allineate in corrispondenza della regione C-terminale (ovvero, sinaptobrevine, lunghe 90-110 residui) e poche sequenze in corrispondenza dei 110-120 residui N-terminali. Per scoprire la funzione di questa

regione extra si è riutilizzata come sonda la sola regione N-terminale di VAMP-7 in ricerche reiterate con PSI-BLAST, che produce una matrice "personalizzata" (PSSM). Rendendo meno stringente la ricerca e utilizzando PSI-BLAST è stato dimostrato che tale regione N-terminale è conservata negli eucarioti e mostra un livello variabile di conservazione. Ovviamente, sono state condotte numerose ricerche, usando come sonde le sequenze estratte di volta in volta e confrontando i risultati. Dopo aver identificato varie regioni N-terminali omologhe, un gruppo di sequenze tra quelle identificate, bilanciato per rappresentazione tra specie, è stato sottomesso in formato FASTA al programma CLUSTALW (per ragioni di spazio, si mostrano nella submission solo le prime tre sequenze):
Il programma ha fornito l'allineamento, con gaps, diviso per la rappresentazione in due parti:
In realtà, il lavoro è più complesso e, se si fosse utilizzato solo CLUSTALW o ci si fosse limitati a ricerche BLAST con preset, forse non si sarebbe scoperto alcunchè. In alcune fasi è stato determinante riallineare manualmente le sequenze, allungare o accorciare le sonde, provare l'effetto della modifica di vari parametri, confrontare i dati in letteratura e quelli associati alle schede di ciascuna entry identificata. L'uso di BLAST e PSI-BLAST per la ricerca presso numerosi server ed in diverse banche dati e la verifica di multiallineamento con CLUSTALW hanno consentito di definire il set finale di dati su cui costruire l'ipotesi, pubblicata e poi confermata sperimentalmente, dell'esistenza di un dominio conservato negli eucarioti. Questo è solo un esempio; sono centinaia i domini e motivi proteici scoperti con multiallineamenti, e molti articoli con dati di multiallineamento hanno portato ad

ipotesi funzionali ed a successive verifiche sperimentali "mirate". Molto spesso, in un dominio alcuni residui sono cruciali per struttura e funzione, altri appartengono a regioni "spaziatrici" e possono variare in numero e composizione, o sono caratterizzanti in quanto variano in specifici subdomini, contribuendo a modulare struttura e funzione. La rappresentazione fornita da CLUSTALW non è immediatamente informativa. Pertanto è utile ottimizzare l'output grafico con semplificazioni e colori. Ecco lo stesso allineamento in quella che era la fase quasi finale, pre-pubblicazione:
Si può notare che, per ciascuna sequenza, sono riportati i numeri del primo ed ultimo residuo rappresentato, nonchè il numero di accesso. Inoltre, anzichè presentare tutti i residui, sono stati omessi quelli variabili nell'estremità N-terminale, inutili ai fini dell'illustrazione del blocco conservato, come pure alcuni residui interni, corrispondenti a sottoregioni con numero variabile di residui, che nel multiallineamento originale presentavano gaps. Tali sottoregioni variabili sono state rappresentate da parentesi, al cui interno è stato riportato solo il numero di residui omessi, variabile da zero (nessuna omissione, per sequenze senza "inserzioni") ad un numero n. Ciò ha consentito di focalizzare l'attenzione sulle parti comuni a tutte le sequenze e la colorazione dei residui sulla base di un codice di colore ed ombreggiatura correlato alle proprietà funzionali ha permesso, attraverso un'operazione di semplificazione alfabetica, di giungere all'identificazione di caratteristiche conservate e speciali per definire il blocco di multiallineamento come putativo dominio, senza perdere le informazioni di partenza (la variazione delle sequenze primarie).
© Francesco Filippini, 2009-2019





























