Embed Size (px)
Citation preview

AMPHIPHILIC FULLERENES FOR
B IOMEDICAL AND OPTOELECTRONICAL
APPLICATIONS
Den Naturwissenschaftlichen Fakultäten
der Friedrich-Alexander-Universität Erlangen-Nürnberg
zur
Erlangung des Doktorgrades
vorgelegt von
Patrick Witte
aus Nürnberg

Als Dissertation genehmigt von den Naturwissenschaftlichen Fakultäten der
Universität Erlangen-Nürnberg
Tag der mündlichen Prüfung: 25.04.2008
Vorsitzender
der Prüfungskommission: Prof. Dr. Eberhard Bänsch
Erstberichterstatter: Prof. Dr. Andreas Hirsch
Zweitberichterstatter: Prof. Dr. Tim Clark

Meinem Doktorvater, Prof. Dr. A. Hirsch, gilt mein besonderer Dank für sein reges
Interesse am Fortgang dieser Arbeit sowie für seine Anregungen und die Diskussionen
mit ihm.
Die vorliegende Arbeit wurde in der Zeit zwischen Dezember 2003 bis Dezember
2007 am Institut für Organische Chemie der Friedrich-Alexander-Universität Erlangen-
Nürnberg durchgeführt.

Dedication
- Science is facts;just as houses are made of stones, so is science made of facts;
but a pile of stones is not a house and a collection of facts is not necessarily science
Henri Poincare (1854 - 1912)
For my Parents and Kati

Index of Abbreviations
tBu . . . . . . . . . . . . . . . . . tert-Butyl
BAM . . . . . . . . . . . . . . . Brewster Angle Microscopy
Boc . . . . . . . . . . . . . . . . tert-Butoxycarbonyl
CV . . . . . . . . . . . . . . . . . Cyclic Voltammetry
DBU . . . . . . . . . . . . . . . 1,8-Diaza-bicyclo[5.4.0]undecen-7-en
DCE . . . . . . . . . . . . . . . 1,2-Dichloroethane
DCU . . . . . . . . . . . . . . . Dicyclohexylurea
DMA . . . . . . . . . . . . . . . 9,10-Dimethylanthracene
DMAP . . . . . . . . . . . . . . 4-Dimethylaminopyridine
DMSO . . . . . . . . . . . . . Dimethyl Sulfoxide
dpf . . . . . . . . . . . . . . . . . Days Post Fertilization
EA . . . . . . . . . . . . . . . . . Elemental Analysis
EDC . . . . . . . . . . . . . . . 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Hydrochloride
eq . . . . . . . . . . . . . . . . . . Equivalent
FAB . . . . . . . . . . . . . . . . Fast Atom Bombardment
FC . . . . . . . . . . . . . . . . . Flash Column Chromatography
HOBt . . . . . . . . . . . . . . . 1-Hydroxybenzotriazole
hpf . . . . . . . . . . . . . . . . . Hours Post Fertilization
HPLC . . . . . . . . . . . . . . High Performance Liquid Chromatography
IPR . . . . . . . . . . . . . . . . Isolated Pentagon Rule
IR . . . . . . . . . . . . . . . . . . Infrared Spectroscopy
LB . . . . . . . . . . . . . . . . . Langmuir-Blodgett
I

MW . . . . . . . . . . . . . . . . Molecular Weight
NBA . . . . . . . . . . . . . . . 3-Nitrobenzylalcohol
NMR . . . . . . . . . . . . . . . Nuclear Magnetic Resonance
PBS . . . . . . . . . . . . . . . . Phosphate Buffered Saline
PCBM . . . . . . . . . . . . . . [6,6]-Phenyl-C61 Butyric Acid Methyl Ester
pf . . . . . . . . . . . . . . . . . . Post Fertilization
ppm . . . . . . . . . . . . . . . . Parts per Million
ROS . . . . . . . . . . . . . . . Reactive Oxygen Species
RT . . . . . . . . . . . . . . . . . Room Temperature
STM . . . . . . . . . . . . . . . Scanning Tunneling Microscopy
SWCNT . . . . . . . . . . . . Single Walled Carbon Nanotube
TFA . . . . . . . . . . . . . . . . Trifluoroacetic Acid
THF . . . . . . . . . . . . . . . . Tetrahydrofuran
TLC . . . . . . . . . . . . . . . . Thin Layer Chromatography
UV/Vis . . . . . . . . . . . . . Ultraviolet-Visible Spectroscopy
XPS . . . . . . . . . . . . . . . . X-ray Photoelectron Spectroscopy
II

Table of Contents
1 Introduction 1
1.1 Nanostructured Materials . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.2 The Discovery of Fullerenes . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.3 The Structure of Fullerenes . . . . . . . . . . . . . . . . . . . . . . . . . 5
1.4 Physical Properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
1.4.1 Thermodynamic and Kinetic Stability of C60 . . . . . . . . . . . . 7
1.4.2 Solubility of C60 . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
1.5 Spectroscopic Properties . . . . . . . . . . . . . . . . . . . . . . . . . . 9
1.5.1 UV/Vis-Spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . 9
1.5.2 Mass Spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . . 10
1.5.3 NMR Spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . . 11
1.5.3.1 3He and 1H Spectroscopy . . . . . . . . . . . . . . . . . 11
1.5.3.2 13C Spectroscopy . . . . . . . . . . . . . . . . . . . . . 13
1.6 Electronic Structure and Reactivity of Fullerenes . . . . . . . . . . . . . 14
1.7 Spherical Aromaticity of C60 . . . . . . . . . . . . . . . . . . . . . . . . . 15
1.8 Chemistry of C60 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2 Proposal 20
3 Results and Discussion 22
3.1 Water-soluble Amphiphilic Fullerene-Monoadducts . . . . . . . . . . . . 22
3.1.1 Synthesis of Anionic Amphiphilic Monoadducts . . . . . . . . . . 24
III

Table of Contents
3.1.2 Synthesis of an Anionic Amphiphilic Monoadduct Carrying an
Unsaturated Fatty Acid . . . . . . . . . . . . . . . . . . . . . . . . 33
3.1.3 Synthesis of a Cationic Amphiphilic Monoadduct . . . . . . . . . 39
3.1.4 Amphiphilic Fullerenes as Potential Drug Candidates . . . . . . . 44
3.1.4.1 Introduction and Background . . . . . . . . . . . . . . . 44
3.1.4.2 Antioxidant Activity . . . . . . . . . . . . . . . . . . . . 47
3.1.4.3 Cytochrome C Binding . . . . . . . . . . . . . . . . . . 52
3.1.4.4 In vivo Studies of the Amphiphilic Fullerenes using Ze-
brafish (Danio Rerio) Embryos as Model System . . . . 57
3.1.5 Mechanistic Aspects of the Reaction of Fullerenes with Superoxide 69
3.1.5.1 Cyclic Voltammetry Measurements of Amphiphilic Mono-
adducts . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
3.1.5.2 Kinetic Measurements of Amphiphilic Monoadducts . . 73
3.1.6 Amphiphilic Fullerenes in Material Science Applications . . . . . 77
3.1.6.1 Formation of LANGMUIR-Films with Amphiphilic Fullerene-
Monoadducts . . . . . . . . . . . . . . . . . . . . . . . . 79
3.1.6.2 Incorporation of the Amphiphilic Fullerene-Monoadducts
in Organic Solar Cell Devices . . . . . . . . . . . . . . . 85
3.2 Triazole Dendrimers Based Fullerenes via "Click Chemistry" . . . . . . . 89
3.2.1 Synthesis of Novel Dendritic Triazol-Fullerenes . . . . . . . . . . 92
3.3 Synthesis of Novel Fullerene-SWCNT Hybrids . . . . . . . . . . . . . . 102
3.3.1 Covalent Sidewall Functionalization of SWCNT’s with a Fullerene-
Monocarboxylic Acid Derivative . . . . . . . . . . . . . . . . . . . 103
3.3.2 Non-Covalent Functionalization of SWCNT’s with a Fullerene-
Pyrene Dyad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 106
3.4 Supramolecular Approach for the Formation of C60-Bisadducts . . . . . 116
3.4.1 Metallomacrocycles as Tethers for Regioselective Cyclopropana-
tion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
3.4.2 Hydrogen-bonded Dimers as Tethers for Regioselective Cyclo-
propanation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
IV

Table of Contents
3.5 Synthesis of Novel Multiple Fullerene Arrays Consisting of Mixed C60-
Hexakisadduct Units . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
3.5.1 Synthesis of Bisfunctionalized Janus-Type Fullerene-Dimers . . 127
3.5.2 Synthesis of a Fullerene-Rich Nanocluster . . . . . . . . . . . . . 137
4 Summary 142
4 Zusammenfassung 146
5 Experimental Part 151
5.1 Chemicals and Instrumentation . . . . . . . . . . . . . . . . . . . . . . . 151
5.2 Synthetic Procedures and Spectroscopic Data . . . . . . . . . . . . . . 154
Appendices 224
A Materials and Methods for the Determination of Biological Activity in vivo 224
B Materials and Methods for the Preparation and Examination of SWCNT-
Fullerene-Hybrid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 229
References 231
V

CHAPTER 1
1 Introduction
1.1 Nanostructured Materials
Although the idea of carrying on manipulations at smaller and smaller scales has been
around for quite some time the birth of nanotechnology, at least on an ideological level,
is usually traced back to a speech by RICHARD FEYNMAN at the December 1959 meet-
ing of the American Physical Society. In his speech, he challenged his fellow scientists
to find ways by which to create manufacturing, storage, and retrieval systems that are
as efficient as DNA and to contain such systems in a submicroscopic, self-contained
unit with the size of a cell. It would be over two decades before the first recognized
paper on molecular nanotechnology was published.[1]
The challenge in nanoscience is to understand how materials behave when sample
sizes are close to atomic dimensions. Figure 1.1 for example shows an overview of
artificial nanostructures, being of the same size as biological entities, which allows
them to interact with biomolecules on the surface of the cell and inside it. When the
characteristic length scale of the structure is in the 1- 100 nm range, it becomes com-
parable with the critical length scales of physical phenomena, resulting in the so-called
"size and shape effects". This leads to unique properties and the opportunity to use
1

Chapter 1 Introduction
Figure 1.1: Artificial (top) and biological (bottom) nanostructures.
such nanostructured materials in novel applications and devices. Phenomena occur-
ring on this length scale are of interest to chemists, physicists, biologists, electrical
and mechanical engineers, and computer scientists, making research in nanotech-
nology a frontier activity in materials science. Nanomaterials, which can be classi-
fied as carbon-based nanomaterials, nanocomposites, biological nanomaterials, nano-
polymers, nano-glasses and nano-ceramics find and promise applications in a wide
range of fields such as medicine (therapeutic agent, sensors, labelling), device tech-
nology (nanophotonics, solar energy conversion, opto-electronics) and chemical syn-
thesis (catalysis). This thesis deals with the design and synthesis of functionalized
carbon-based nanomaterials, to get more insight in structure-function relationships and
2

Chapter 1 Introduction
to provide a predictive mechanism that will allow chemists to efficiently design nano-
materials that perform exactly as desired.
3

Chapter 1 Introduction
1.2 The Discovery of Fullerenes
Figure 1.2: Leonardo da Vinci‘s
"Truncated Icosahedron".[2]
The discovery of C60 has a long and very in-
teresting history.[3] The structure of truncated
icosahedron was already known about more
than 500 years ago. ARCHIMEDES is credited
for discovering the structure and LEONARDO
DA VINCI included it in one of his drawings.
At the end of 1960’s, scientists were increas-
ingly interested in non-planar aromatic struc-
ture, and thereafter the bowl-shaped corannu-
lene was synthesized.[4] In 1970, EIJI OSAWA
realized that a molecule made up of sp2 hy-
bridized carbons could have a spherical struc-
ture. He therefore made the first proposal for
C60.[5] Then, a group of Russian scientists in-
dependently proposed the C60 structure, the
paper published by BOCHVAR and GAL’PERN
in 1973 not only predicted some properties of
C60, but also of C20 (the smallest fullerene) as well.[6] The first spectroscopic evidence
for C60 and other fullerenes was published in 1984 by ROHLFING and coworkers.[7]
Eventually in 1985 ROBERT CURL and RICHARD SMALLEY from Rice University, and
HAROLD KROTO from the University of Sussex discovered the fullerenes while doing
experiments with a laser-vaporization supersonic cluster beam apparatus developed by
SMALLEY. Upon vaporizing graphite from the disk with high-power laser pulses, they
found in their data, to their surprise, an indication of what appeared to be a cluster con-
sisting of 60 carbon atoms. After furiously debating, building models, and consulting
the literature, they theorized that the new 60-carbon structure had the form of a sphere
comprising 20 hexagons and 12 pentagons, known to mathematicians as the before
mentioned truncated icosahedron and familiar to the form of a soccer ball. The struc-
4

Chapter 1 Introduction
ture reminded KROTO of the geodesic dome in Montreal, so the group decided to name
the new molecule buckminsterfullerene after the architect R. BUCKMINSTER FULLER,
who popularized the geodesic dome. Eleven years later, in 1996, they were awarded
for the NOBEL PRIZE in chemistry for their accomplishments. However, the evidence for
the existence of these molecules remained indirect until 1990, when the researchers
KRÄTSCHMER and HUFFMAN at the Max Planck Institute for Nuclear Physics in HEIDEL-
BERG, GERMANY used a carbon-arc plasma to produce the first macroscopic quantities
of them.[8]
Since then fullerenes were extensively investigated and are constantly attracting great
amounts of attention. In 1991, SCIENCE MAGAZINE named C60 "Molecule of the Year",
professing it "the discovery most likely to shape the course of scientific research in the
years ahead".
1.3 The Structure of Fullerenes
Fullerenes are all-carbon molecules which have the form of hollow, closed nets com-
posed of 12 pentagons and n hexagons and the composition C20+2n (Euler‘s Theo-
rem).[9] A second empirical rule that governs fullerene-type structures is the Isolated
Pentagon Rule (IPR). This rule, based on both steric and electronic considerations,
states that two pentagons may never share a common edge. Indeed, among the 1812
distinct fullerene isomers of buckminsterfullerene, only Ih-C60 containing 12 pentagons
isolated by 20 hexagons (soccer ball structure) is formed in accordance with the IPR.
The precise geometric structure of this isomer was determined by X-Ray analysis of
pristine C60 at low temperature [10–12], C60 derivatives [13], C60 solvates [14,15], and solid-
state 13C NMR measurements [16]. Such experimental findings definitively confirmed
the postulated Ih-symmetry with a mean diameter of ∼ 7.1 Å for the sphere. Its VAN DER
WAALS diameter is ∼ 10.4 Å, and the distance across the cavity is ∼ 3.5 Å. As a result
of the presence of both five- and sixmembered rings within the structure of C60, there
are two types of bonds – namely bonds at the junction between two sixmembered rings
([6,6-bonds], mean distance = 1.391 Å), and bonds at the junction between a five- and
a six-membered ring ([6,5-bonds], mean distance = 1.449 Å).[10, 17] The electronic
5

Chapter 1 Introduction
structure [18–20] of the fullerenes is such that bonds at [6,6]-ring junctions have much
double bond character, while bonds at [6,5]-ring junctions are essentially single bonds.
This arrangement results in C60 having a strongly bond-alternated structure which can
be best described as a spherical tessellation of [5]radialene and 1,3,5-cyclohexatriene
subunits (figure 1.3).
(a) (b) (c)
Figure 1.3: Illustration of (a) a [6,6]-bond; (b) a [6,5]-bond; and (c) the [5]radialene and 1,3,5-cyclohexatriene substructures of C60
The chemical behavior of C60 mainly depends on these structural properties:
• The 30 bonds at the junctions of two hexagons ([6,6]-bonds) are shorter than the
60 bonds at the junctions of a hexagon and a pentagon ([5,6]-bonds).
• The highly pyramidalized sp3 C-Atoms in C60 cause a large amount of strain en-
ergy within the molecule. The pyramidalization angle, defined by HADDON and
RAGHAVACHARI, of carbon atom orbitals in C60 structure is 11.6,[21,22] an angle
between sp2 and sp3 hybridization, which are 0 and 19.47 respectively (see fig-
ure 1.4).
6

Chapter 1 Introduction
sp2 sp3
θ σπ = 90° θ σπ = 109.47°
Figure 1.4: Pyramidalization of the carbon atom orbitals in C60. The angle of pyramidalizationχ = θ σπ - 90° is between the angles for sp 2 and sp3 hybridization, which causesthe high strain energy in C60.
1.4 Physical Properties
1.4.1 Thermodynamic and Kinetic Stability of C 60
The heat of formation of pristine C60 have been determined theoretically and experi-
mentally by calorimetry to be 10.16 kcal mol−1 per C-atom (relative to graphite with 0.0
kcal mol−1 per C-atom).[23] Also in the comparison to diamond (0.4 kcal mol−1 per C-
atom), C60 is a energy-rich carbon allotrope. This comparatively high energy content is
originated in the high strain energy of C60 due to pyramidalization of the sp2 orbitals and
accounts for about 80 % of the heat of formation.[24] This makes C60 to one of the most
strained molecule, which is stable under standard conditions. In addition, the fullerene-
cluster gets over the extremely high temperatures within the fullerene production. Un-
der the conditions of ion beam experiments, other stable aromatic molecules like ben-
zene instantly decompose.[25] This extraordinary kinetic stability of C60 can also be
seen in the FAB-mass-spectroscopic characterization of fullerene-derivatives, where
the fragmentation is leading to the dominating molecular peak of unfunctionalized C60.
1.4.2 Solubility of C 60
The solubility of C60 in organic solvents is important to enable purification and chemical
modification. In general, the solubility in the majority of solvents is very low, because
C60 exhibit a high tendency for aggregation. On the other hand the interaction between
7

Chapter 1 Introduction
the solvent molecules and C60 is usually very weak, since the fullerene is a nonpolar
molecule, which is hardly polarizable due to the large HOMO-LUMO gap. In summary,
the solubility of C60 in polar solvents such as methanol and water is nearly zero. This
low solubility can also be seen in the case of alkanes as solvents, whereas the solu-
bility in chloroalkanes is slightly better. The best solubilities are obtained in aromatic
solvents and carbon disulfide, which makes these solvents to the standard solvents
for preparative use. Table 1.1 summarizes the solubilities of C60 in the most common
solvents.[26–29]
Solvents Solubility [mg/mL]
acetonitrile 0.000THF 0.00methanol 3.5 · 10−5
water 1.3 · 10−11
n-pentane 0.005cyclohexane 0.036chloroform 0.16dichloromethane 0.26tetrachloromethane 0.32pyridine 0.89benzene 1.70toluene 2.801,1,2,2-tetrachloroethane 5.30anisole 5.60chinoline 7.20carbon disulphide 7.901,2,4-trichlorobenzene 8.501,2-dichlorobenzene 27.001-chloronaphthalene 51.00
Table 1.1: Solubilities of C60 in the most common solvents.[30]
8

Chapter 1 Introduction
1.5 Spectroscopic Properties
1.5.1 UV/Vis-Spectroscopy
The UV/Vis spectrum of C60 shows intense absorption bands between 190 and 410 nm
(maxima at 326, 253 and 208 nm). These bands are due to symmetry-allowed singlet-
singlet transitions from the HOMO to the LUMO+1 (see figure 1.8). In the visible region,
the spectrum is characterized by a weak broad band between 440 and 620 nm with
two maxima located at 598 and 543 nm [31–33] which correspond to symmetry-forbidden
singlet-singlet transitions from the HOMO to the LUMO and LUMO+1. Chemical func-
tionalization of C60 modifies the electronic structure of the fullerene chromophore. This
is strongly reflected in the UV-Vis spectra of its derivatives. The degree of variation
is dependent on i) the number of addends, ii) the geometric addition pattern in multi-
adducts, and iii) the electronic structure of the functional group.[31,34] The derivatization
of the fullerene core reduces its symmetry, thereby enhancing transition probabilities.
Consequently, C60 derivatives show stronger absorptions in the visible region with re-
spect to pristine C60. The absorptions at 257 and 329 nm are hardly shifted as a
result of the functionalization but less intense, which is consistent with the transition
from a 60- to a 58-π- electron system.[31] Very characteristic for all [6-6]-closed (see
chapter 1.8) monoadducts is the absorption at ∼ 425 nm.[35–37] The influence of subse-
quent functionalization can be nicely followed by the characteristic color of the corre-
sponding derivatives in solution. The introduction of additional groups reduces the π-
system of the C60-core, which changes the color from red (monoadducts) over orange
(pentaadducts) to yellow (hexaadducts). In the case of hexaadducts the remaining π-
system is significantly reduced, whereas the remaining π-electrons are located within
a cubic, cyclophane-like substructure of eight benzenoid rings.[38]
9

Chapter 1 Introduction
200 300 400 500 600 700 800
abso
rbance
wavelenght [nm]
400 500 600 700 800
Figure 1.5: UV/Vis spectrum of C60 in heptane. Inset: Region between 420 - 470 nm.
1.5.2 Mass Spectroscopy
Mass spectrometric characterization of fullerenes has been vital since the first carbon
clusters were produced.[39,40] Mass spectrometry has played a key role in the discov-
ery of fullerenes, and continues to reveal the structures and properties of these unique
molecules.
In mass spectroscopy , C60 and its derivatives can be measured with different ioniza-
tion methods (EI, FAB, MALDI) depending on the nature of the addends. The molecular
peak of C60 can be easily identified at m/z = 720 by the accompanied series of frag-
ment ions (M+-24, M+-48, M+-72, etc.) due to so-called "shrink wrapping", where the
molecule subsequently loses C2 units.[41,42]
10

Chapter 1 Introduction
1.5.3 NMR Spectroscopy
1.5.3.1 3He and 1H Spectroscopy
Already early in the discovery history of the fullerenes, the question about the aromatic-
ity of these new molecules appeared. While in former times aromaticity was defined
by the smell and later by the reactivity of certain molecular structures, today physical
characteristics are consulted for the definition of aromaticity. Next to structural pa-
rameters like delocalized atomic bonds, magnetic characteristics serve to classify a
molecule as aromatic. Cyclic aromatic molecules exhibit a diamagnetic ring current,
which manifests itself in high magnetic susceptibilities and "abnormal" chemical shifts
in the NMR spectrum. The first forecasts of KROTO, SMALLEY, CURL and coworkers
[43] predicted C60 as an aromatic molecule, whose π-electrons would move freely on
the fullerene surface. As a consequence C60 would be highly diamagnetic [44,45] and
would have to exhibit a particularly high magnetic susceptibility, which could not be
confirmed experimentally.[46,47] The determined magnetic susceptibility χ of C60 with χ
= -260 CGS ppm mol−1 (=̂ -4.3 CGS ppm per mol of C) is smaller than those from
diamond with χ = -5.5 CGS ppm per mol of C.
Nevertheless there are ring currents present in C60. The working groups HADDON [48,49]
as well as ZAMESI and BOWLER [50] could show that in the pentagons strong paramag-
netic ring currents are present, while weaker diamagnetic ring currents circulate over
larger areas of the fullerene surface (figure 1.6).
Figure 1.6: Left: Diamagnetic (blue) and paramagnetic (red) ring current in the corannulenesubstructure of C60. Right: Ring current contour map of C60.[50]
The paramagnetic and diamagnetic ring currents neutralize each other almost, which
explains the small macroscopic magnetic susceptibility of C60.[48] For the experimen-
11

Chapter 1 Introduction
tal proof of the ring currents a method had to be found, which can measure the local
magnetic properties. For this purpose NMR spectroscopy is a useful tool. Fullerenes
have internal cavities, large enough to encapsulate atoms [51,52] and so it is possible to
achieve noble-gas doped endohedral compounds. Because 3He has a spin of 1/2 and
is an excellent NMR nucleus, it can be used as a probe for the magnetic shielding envi-
ronment inside the fullerene cavity.[53] It was found, that the 3He nucleus encapsulated
in C60 is shielded by δ = -6.36 ppm, relative to free 3He. This chemical shift is a result
of the ring currents in the π-system and the shielding effects of the sigma framework.
Measurements on helium complexes of fullerene derivatives showed, that the chemi-
cal shift of the helium atom in homofullerenes with [5,6]-opened structure is almost the
same shift as obtained with pure C60. In derivatives with [6,6]-closed structure, the He-
signal is shifted highfield for about 3 ppm, with the exception of methanofullerenes. The
reason for the different relative shifts is to be seen in the respective electronic struc-
tures of the fullerene derivatives. While the electronic structure of a homofullerene
agrees to a large extent with that of unfunctionalized C60, the π-electronic structure
in a [6,6]-closed structure is distinctly disturbed in relation to C60. Methanofullerenes
with [6,6]-closed structure stand structurally between the homofullerenes and the other
[6,6]-closed structures. Through to addition of a cyclopropane ring, whose bondings
possess partial double bond character, the 60 π-electron structure of C60 is disturbed,
but the single bond located between the hexagons still possesses partial double bond
character and locates the methanofullerenes between a 60 π-electron and a 58 π-
electron system.
H OEt
OH
O
EtO
3.32 ppm 6.79 ppm
1 2
Figure 1.7: Chemical Shifts for protons in homofullerenes.[54]
12

Chapter 1 Introduction
Beside helium atoms in the inside of the fullerene cage, also hydrogens attached close
to the fullerene surface can serve as probe for local ring currents (figure 1.7). In the
case of the homofullerenes 1 and 2 the π-electron system is hardly disturbed in relation
to C60, what can be directly seen in the UV/Vis-, NMR-spectra and the electrochem-
istry of the 3He doped complexes. In compound 1 the hydrogen atom is located over
a hexagon and calculations indicate, that, because of the diamagnetic ring currents
in the hexagon, the signal for the proton is shifted high-field, as it is known for the
[5]paracyclophane. On the other hand, paramagnetic ring currents, that are present in
the pentagons, should magnetically shield the hydrogen atom in 2, which was indeed
experimentally proven by different groups.[54–56]
1.5.3.2 13C Spectroscopy
In pure C60 all C-atoms are chemically and magnetically equivalent and therefore only
one resonance signal is to be expected. In deuterated benzene, the resonance signal
appears at 143.2 ppm and is at the low end of the range, where resonances for qua-
ternary C-atoms of unsubstituted polycyclic aromatics are expected.
The qualitative estimation of the chemical shift for 13C-NMR-signals in fullerenes is not
simple, since apart from the local curvature also ring current effects are responsible
for the observed chemical shift.[57,58] As general trend it can be stated however that a
pyramidalization of the unsaturated system leads to a low field shift.[59] This trend is
also recognizable in the case of the aromatics corannulene, fluoranthene and pyracy-
lene, where the C-atom of the curved corannulene shows a remarkable low-field shift,
compared to the planar molecules like fluoranthene and pyracylene. With this strong
dependence of the chemical shifts from local parameters, the 13C-NMR-spectroscopy
is an important method for the characterization and determination of the symmetry of
fullerene derivatives.
13

Chapter 1 Introduction
1.6 Electronic Structure and Reactivity of Fullerenes
C60 exhibits highly interesting electrochemical properties which are related to its elec-
tronic configuration. The before mentioned rehybridization is in part responsible for
the high electron affinity of C60 since it considerably reduces the energy of the lowest
unoccupied molecular orbital (LUMO). A complete picture of the electronic structure of
C60 is obtained by Hückel Molecular Orbital (HMO) theory which predicts an electronic
configuration with a five-fold degenerate HOMO (hu) and the three-fold degenerate
low-lying molecular orbitals LUMO (t1u) and LUMO+1 (t2u) separated by a moderate
energy gap of 0.757 β.[60–62]
0
-1
LUMO
HOMO
HOMO
LUMO
LUMO+1
hu
t1u
e2u
En
erg
y (
)β
(0.38)
(0.14)
(-0.62)
(+1)
(-1)
-0.5
0.5
1
e1u
t2u
0.757 β
2.00 β
Figure 1.8: Hückel Molecular Orbital (HMO) diagram of C60 and benzene.
In accordance with the degeneration of the LUMO level, the redox chemistry demon-
strates the ability of [60]fullerene to accept up to six electrons.[63] The systematic proof
for the triple degeneracy of the LUMO level of C60 came with the detection of fullerene
anions Cn−60 (n = 1 - 6), which can be generated by subsequent reversible one-electron
reductions. The separation between any two successive reductions is relatively con-
stant and amounts to ∼ 450 ± 50 mV.[64] This correlates well with the triple degeneracy
14

Chapter 1 Introduction
of the LUMO level. Considering the HMO diagram (figure 1.8), the oxidation of C60 con-
sists in the removal of an electron from the low-lying HOMO, leading to an important
destabilization of the π-electron system. Correspondingly, the first one-electron oxida-
tion of C60 occurs at a highly positive potential, 1.26 V vs Fc/Fc+ in tetrachloroethane.
The difference in potential between the first oxidation and the first reduction of C60
(E1/ 2ox − E1/ 2
ox = 2.32 V) is a good measure of the HOMO-LUMO gap in solution and
correlates well with the calculated value (1.5 - 2.0 eV).[65]
Since C60 exhibits a highly rigid framework in the ground state as well as in the excited
state, the reorganization energies are very low.[66] The reducibility of fullerenes together
with their small reorganization energy associated with nearly all their reactions, make
them especially interesting building blocks.
1.7 Spherical Aromaticity of C 60
The magnetic properties of fullerenes clearly show, that the delocalized character of
the conjugated π-electrons can cause the establishment of diamagnetic or paramag-
netic ring currents within the loops of the hexagons and pentagons. Neutral C60, for in-
stance, exhibits no pronounced overall aromaticity since it contains diatropic hexagons
and paratropic pentagons and was labeled ambiguously aromatic. BÜHL and HIRSCH
have showed that the treatment of the π-electrons system as a spherical electron gas
allows to determine the nature of three-dimensional aromaticity of fullerenes and other
cage compounds.[61]
The aromaticity of two-dimensional annulenes in singlet ground states follows the Hückel
rule, i. e. annulenes with 4N+2 π-electrons are aromatic, those with 4N π-electrons are
antiaromatic. Due to their closed-shell structures, annulenes with 4N + 2 π-electrons
are not distorted (Dnh-symmetry) and show strong diamagnetic ring currents. The rule
is reversed for triplet open-shell analogues.
The aromaticity of icosahedral fullerenes (C20, C60 and C80) and their cluster distortions
depends on the number of delocalized electrons in the valence shell. The maximum of
spherical aromaticity of a cluster can only be achieved with 2(N + 1)2 electrons filling
the shell completely. The 2(N +1)2 rule represents the spherical analogue of the Hückel
15

Chapter 1 Introduction
rule for planar annulenes.
1.8 Chemistry of C 60
Since the discovery of fullerenes, a broad variety of chemical modifications with C60
were performed. In general these modifications can be classified in five different topics
(figure 1.9):[67–73]
Figure 1.9: Overview of the possible modifications of C60. a) Exohedral functionalization; b)Heterofullerenes; c) Endohedral functionalization; d) Cage-opening modificationsand d) Alkali metal fullerides.
• a) Exohedral addition reactions, including nucleophilic- and radical additions, cy-
cloadditions, hydrogenations, oxygenation and halogenation
• b) Substitution of carbon atoms in the fullerene framework with different atoms,
e.g. boron or nitrogen, leading to heterofullerenes
16

Chapter 1 Introduction
• c) Encapsulation of one or more atoms inside the fullerene cage, yielding endo-
hedral fullerenes
• d) Ring-opening and fragmentation reactions, which could be used for subse-
quent endohedral functionalization
• e) Reduction reactions with electropositive metals, e.g. alkali- and alkaline earth
metals, yielding alkali metal fullerides
Considering the previously mentioned properties of C60, the following general reactivity
patterns can be emerged:
• Due to the low-lying degenerate LUMO level the chemical reactivity of the formal
double bonds is close to that of a strained, electron deficient polyolefin.
• The relief of strain due to a change in hybridization at the reacting carbons on the
spherical surface is the driving force for addition reactions.
• The regiochemistry of all addition reactions is governed by the minimization of
[5,6]-double-bonds within the fullerene cage. The relocation of each double-bond
into the [5,6]-bond costs about 8.5 kcal/mol. Therefore addition reactions nor-
mally take place at [6,6]-bonds, whereby 1,2-additions (preferred addition) intro-
duce eclipsing interactions between the addends. For a combination of sterically
demanding addends, [1,4]- and [1,6]-addition can take place.
RR
RR R
RR
R
[6,5]-open [6,5]-closed [6,6]-open [6,6]-closed
+ 6 kcal/mol + 21 kcal/mol not a minimum energy structure 0 kcal/mol
Figure 1.10: Possible isomeric methanofullerenes and their relative energies from PM3 calcu-lations (methanoannulene-type subunits highlighted in red).[74]
17

Chapter 1 Introduction
The following itemization lists the mainly used reactions for the chemical exohedral
functionalization of C60:
• Cyclopropanation with carbon nucleophiles [75,76]
• DIELS-ALDER-type [4+2]-cycloadditions [77–79]
• Photochemical [2+2]-cycloaddition of α, β-unsaturated ketones [80,81]
• [3+2]-cycloaddition using diazo compounds, azomethine ylides and azides [82–89]
• Nucleophilic addition of Grignard or organolithium compounds
• Complexation of transition metal complexes [90]
• Oxygenation, osmylation, halogenation and hydrogenation
Since the work described in this thesis is mainly based on methanofullerenes, a brief
account of the synthetic approaches toward methanofullerenes is given in the following
section. For more details about the other reactions, as well as the different modification
possibilities in figure 1.9, the reader is referred to the following reviews [67,91–93] and
books.[68,94,95]
O O
O O
Br
NaH or DBUtoluene
O O
O O
Br
COOEt
BrEtOOC
COOEt
COOEt
3
4
5
6
Scheme 1.1: Cyclopropanation of C60 by nucleophilic addition of α-bromomalonate carbanions(BINGEL-reaction).
18

Chapter 1 Introduction
The cyclopropanation of C60 with carbon nucleophiles is one of the most commonly
used reaction in fullerene derivatization due to the high yields and variability. The orig-
inal conditions for the BINGEL reaction involve the treatment of bromomalonates with
NaH in the presence of C60.[75] From a mechanistic point of view a two-step process
is involved at this type of reaction. Deprotonation of the α-bromomalonate 3 affords
the α-bromo carbanion 4 that adds to C60, giving the anionic fullerene intermediate
5. In a second step, the displacement of bromide by an intramolecular nucleophilic
substitution gives the methanofullerene 6 (scheme 1.1). Since the synthesis of more
complex bromomalonate precursors is often rather difficult, HIRSCH modified the re-
action protocol by preparing the α-halomalonate in situ from the corresponding mal-
onate in the presence of CBr4 and a non-nucleophilic base (DBU) (so called BINGEL-
HIRSCH reaction).[96] In some cases the use of iodine as halogenation reagent can be
preferable.[97] These reactions proceed under mild conditions and can be adopted for a
wide range of functional groups. The corresponding methanofullerenes are in general
thermally stable, with a well-defined directionality of the ester groups due to the highly
rigid cyclopropane ring.
19

CHAPTER 2
2 Proposal
The aim of this work is the synthesis and characterization of novel fullerene archi-
tectures, which are accessible through exohedral functionalization of parent C60. The
application of these compounds in material science and biomedicine should be consid-
ered, and the derivatives should be specifically designed to provide properties, which
make the implementation possible.
In the first instance a series of anionic and cationic amphiphilic fullerene derivatives
(amphifullerenes) should be synthesized. To minimize the disruption of the fullerene
π-system with increasing number of addends, these amphiphiles should be C60-mono-
adducts. Therefore the amphiphilic character has to be introduced by the attachment of
an asymmetric malonate, which carries a hydrophilic (dendritic) and a lipophilic (alkyl
chain) part. This concept enables the fine-tuning of the ratio between hydrophilic and
lipophilic part within the molecule, which is of importance for the bioavailability of these
substances. In cooperation with C-SIXTY Inc. and PHYLONIX Pharmaceuticals Inc., the
amphifullerenes should be investigated concerning their antioxidant activity against re-
active oxygen species (ROS) in vitro and their ability to protect cells and tissue from ox-
idative injury and cell death in vivo. Furthermore the amphifullerenes should be utilized
for the construction of nanostructured films by the Langmuir-Blodgett (LB) technique.
20

Chapter 2 Proposal
In this context, the ability to prepare homogeneous layers with control over molecular
organization, film thickness and architecture should be studied.
In the second part of this work a novel concept for the efficient synthesis of neutral
and charged water-soluble fullerene structures should be developed. Particularly the
copper(I)-catalyzed HUISGEN 1,3-dipolar cycloaddition should be examined to its ap-
plicability in fullerene chemistry and the influence of the multiple triazole linkages on
the water-solubility should be studied.
In line of this work novel hybrid nanomaterials should be prepared, consisting of
fullerenes covalently and non-covalently attached to the outside surface of single wall
carbon nanotubes (SWCNTs). This should be accomplished by the introduction of suit-
able functionalities onto the fullerene sphere, which allows to immobilize these deriva-
tives onto the SWCNT surface. The structural investigation of these hybrids should
include microscopic techniques, to determine the degree of functionalization and the
constitution of the supramolecular hybrids.
The regioselective bisfunctionalization of C60 is generally accomplished by the use of
macrocyclic or open-chain malonates as tethers for the subsequent nucleophilic cy-
clopropanation. In a new approach the preorganization of the malonates should be
obtained by complexation with different metall ions and by dimerization via hydrogen-
bonding.
Furthermore a new synthetic strategy should be developed, which enables the syn-
thesis of fullerene-rich macromolecules with different functionalities within the same
molecule. This should lead to aesthetically pleasing architectures, where two ore more
C60 cages are covalently connected by bridging organic addends.
21

CHAPTER 3
3 Results and Discussion
3.1 Water-soluble Amphiphilic Fullerene-Monoadducts
Amphiphile is a term describing a chemical compound possessing both hydrophilic and
hydrophobic properties. Such a compound is called amphiphilic or amphipathic. This
forms the basis for a number of areas of research in chemistry and biochemistry, no-
tably that of lipid polymorphism. As mentioned before, to obtain amphiphilic properties,
the structure has to consist of two different functional groups. The hydrophobic group
is typically a large hydrocarbon moiety, such as long saturated or polyunsaturated alkyl
chains. The hydrophilic group falls into one of the following categories:
• charged groups, such as anionic groups (carboxylates, sulfates, sulfonates, phos-
phates) and cationic groups (ammonium and pyridinium salts)
• polar, uncharged groups, such as polyalcohols or polyethers
• amphoteric groups
In the last few years HIRSCH and coworkers succeeded in synthesizing a broad vari-
ety of amphiphilic [60]fullerene derivatives, which differ in the number and the addition
22

Chapter 3 Results and Discussion
pattern of the attached functional groups. Exemplary representatives of these deriva-
tives are for example [5:1] hexakisadducts with an octahedral addition pattern, using a
Newkome-type amide dendron as a hydrophilic addend and five didodecyl malonates
as lipophilic addends. This globular amphiphile dissolves in water, forming unilamellar
vesicles with diameters typically between 100 and 400 nm, and reveals a very small
critical micelle concentration (cmc).[98,99] Latest results deal with the synthesis of am-
phiphilic [3:3] hexakisadducts, using a trisadduct precursor with an e,e,e-addition pat-
tern as starting material. For the completion of the octahedral addition pattern, DMA
template-mediated cyclopropanation was accomplished with different polar and den-
dritic ionic malonates. These new compounds were examined in detail for their ten-
dency to form micelles and to build stable monolayers.[100]
All these examples use C60 as structure-forming core, systematically functionalized
with different addends. It is well known, that the chemical reactivity and physical prop-
erties change, when the conjugated π-electron chromophore of the fullerene is reduced
as a result of increasing functionalization. It has been shown, that in the series of mono-
through hexakisadducts reductions become increasingly difficult and more irreversible
with increasing number of addends. Hence, with incremental functionalization of the
fullerene, the LUMO of the remaining conjugated framework is raised in energy. [101]
In order to almost retain the unique properties of pristine C60, but to improve the water-
solubility and bioavailability, the first part of this thesis deals with the synthesis of a
series of amphiphilic [60]fullerene monoadducts.
23

Chapter 3 Results and Discussion
3.1.1 Synthesis of Anionic Amphiphilic Monoadducts
In the first instance anionic amphiphiles were synthesized, which carry alkyl chains,
differing in number and length, as apolar addends. NEWKOME-type dendrons of differ-
ent generations provide the polar part and serve for the solubility in aqueous solutions.
The crucial point in the synthesis of amphiphilic monoadducts is the facile generation of
unsymmetrical malonate precursors that serve as addends. Previously, unsymmetrical
malonates have been prepared by the successive esterification of malonic acid with
suitable alcohols (scheme 3.1).[102,103]
HO OH
O O
DCC,DMAP
O O OO O
OO
TFA
O O OHO O
O
HO
O
O O OHO O
O
R1O
O
HO O OO O
O
R1O O OO O
O
DCC,DMAP
R1O O OHO O
O
R1O O OR2O O
OO O OR2
O O
O
R1O
O
DCC,DMAP
2 eq 1 eq
R1OH
TFADCC,DMAPR1OH
DCC,DMAPR2OH
DCC,DMAPR2OH
O
OHO O
OHO
O
77
8
9
10
11
12
13
14
15
Scheme 3.1: Different pathways for the synthesis of unsymmetrical malonates.
24

Chapter 3 Results and Discussion
In principle, the asymmetry can be obtained in different states of the reaction sequence.
One possibility, as described in scheme 3.1 (left side), is the formation of a symmetric
malonate 9, which can be monofunctionalized by a stoichiometrical controlled reaction
with alcohols or amines. The sequence on the right side in scheme 3.1 starts with
the formation of the asymmetric malonic derivative 12, which can be further bisfunc-
tionalized by sequential reaction with different alcohols or amines. However, these ap-
proaches led in a number of cases to separation problems and unsatisfactory yields. In
a new approach, MELDRUM’S acid (2,2-dimethyl-1,3-dioxane-4,6-dione) was allowed to
react with a long chain alcohol to give the monoalkyl malonate (see scheme 3.2). This
reaction can be carried out in large scale with almost quantitative yields and without
extensive purification.
O O
O O+ R1OH
R1O OH
O O
R1 = (CH2)5CH3
R1 = (CH2)17CH3
R1 = CH((CH2)7CH3)2
R1 = (CH2)5CH3 R1 = (CH2)17CH3 R1 = CH((CH2)7CH3)2
130 °C, 3 h
16
171819
Scheme 3.2: New approach for the synthesis of unsymmetrical malonates, using MELDRUM’Sacid.
It is important to notice, that these monoalkyl malonates can only be obtained by the
use of primary and secondary alcohols. Tertiary alcohols do not react with MELDRUM’S
acid, presumably to their decreased nucleophilicity. As the second terminus, an alcohol
is required that contains a protected carboxylic group in its periphery serving as anchor
point for the introduction of the dendritic building blocks. This alcohol also serves as
a spacer, by increasing the distance between the malonate and the dendritic group.
This is an essential requirement to obtain adequate yields in the subsequent BINGEL-
HIRSCH reaction. The synthesis of the spacer was accomplished using a modified
literature method [104] by treating ε-caprolactone with benzyl bromide leading to the
formation of 20 (scheme 3.3).
The protection of the terminal carboxylic groups was carried out by reaction with iso-
butene to give the tert-butyl ester 21. After removal of the benzyl group by catalytic
hydrogenation with Pd/C as catalyst, the deprotected alcohol 22 was obtained in 71
25

Chapter 3 Results and Discussion
O
O
O OR
OKOH, BzBr R = H
R = tBuH2SO4,
Pd/C,H2
HO O
O
DCC,DMAP
O OR
OR1O
OOR1 = (CH2)5CH3
R1 = (CH2)17CH3
R1 = CH((CH2)7CH3)2
R = tBuR = H
TFA
OO
R1O
OOO
OR1O
OO HN
O
O
O
O
O O
EDC, DMAP, 1-HOBt
EDC, DMAP, 1-HOBt
HN
HN
OHN
O
O
O
O
O
O
OONH
OO
OO
O
O
OO
O O O
O
C60, CBr4, DBU C60, CBr4, DBU
OO
R1O
OO HN
OR
O
OR
O
O OR
OO
R1O
OO HN
HN
OHN
O
O
OR
O
OR
O
ORONH
ORO
ORO
RO
O
ROO
RO O OR
O
R = tBuR = H
TFAR = tBuR = H
TFA
2021
22
17, 18, 19
23, 24, 2526, 27, 28
29 30
31, 32, 33 34, 35, 36
37, 38, 39 40, 41, 4243, 44, 45 46, 47, 48
Scheme 3.3: Optimized multistep synthesis of the amphiphilic monoadducts 1st generation43, 44, 45 and 2nd generation 46, 47, 48.
26

Chapter 3 Results and Discussion
% overall yield. The coupling of the spacer unit 22 with the monoalkyl malonates
17, 18, 19 via the STEGLICH coupling procedure [105–107] gave the protected unsym-
metrical malonates 23, 24, 25 in reasonable yields. After deprotection of the tert-butyl
protection group with TFA in chloroform, the corresponding carboxylic acids 26, 27, 28
were coupled with the dendritic building blocks 29 and 30 via the in situ activation with
EDC.[108] The dendrimers are based on the iterative architecture principle of the branch-
ing of branches and represent NEWKOME-type dendrimers, which were synthesized
following literature procedures.[109,110] The use of EDC instead of DCC as activation
reagent for the formation of the first generation dendritic malonates 31, 32, 33 and the
second generation malonates 34, 35, 36 turned out to be more effective. The higher
yields (about 10 % more yield) of the coupling reaction with EDC and the saving of time
during the purification progress justifies the higher price of EDC compared to DCC. The
monoadducts 37, 38, 39 and 40, 41, 42 can be obtained via the BINGEL-HIRSCH re-
action with CBr4 in good yields. However the use of iodine as halogenation reagent
was not appropriate in this case, leading to decreased conversion rates and increased
formation of side products. For the deprotection of the peripheral tert-butyl esters the
monoadducts 37, 38, 39, 40, 41, 42 were dissolved in formic acid and stirred at rt for
48 h. Purification of the crude deprotected monoadducts can be obtained by reprecipi-
tation from THF/diethyl ether in the case of 43, 44, 45 and from methanol/diethyl ether
in the case of 46, 47, 48 (scheme 3.3). All monoadducts were isolated as red brownish
solids. Surprisingly the amphifullerenes 43, 44, 45, where the hydrophilic part is repre-
sented by the 1st generation dendron, consisting of three carboxylic acids, showed no
significant water solubility at pH = 7.2 or higher pH values. In previously synthesized
trisadducts, where carboxylic acids were introduced by a tether controlled synthesis,
three ionic groups were sufficient enough to promote a remarkable water-solubility.[111]
This leads to the conclusion, that not only the number of carboxylic groups is impor-
tant, but also the arrangement over the fullerene core plays an essential role for the
solubility. However the water-solubility can be promoted by the use of DMSO as co-
solvent. For the aqueous solutions the monoadducts were dissolved in a small amount
of DMSO followed by the addition of water (pH = 7.2). Such solutions are remarkable
27

Chapter 3 Results and Discussion
100
80
60
40
20
0
4.6E3
3.7E3
2.7E3
1.8E3
9.1E2
0.0E0
1900 2000 2100 2200 2300 2400 2500 2600 2700 2800
1902 1970
2107
21612214 2273
2387 2483 2548 261226762734 2858
Figure 3.1: FAB mass spectrum of 2nd generation amphiphile 47.
stable and can be stored for several weeks.
The 2nd generation analogs 46, 47, 48 on the other hand showed very good solubility
in water at pH = 7.2. The characterization of the amphifullerenes was carried out by
means of 1H- and 13C-NMR, by mass spectrometry and UV/Vis spectroscopy. Due to
the similar spectroscopic properties of the amphiphiles the following figures show the
characterization of 44, representative for the 1st generation amphiphiles and 47 for the
2nd generation amphiphiles.
Figure 3.1 shows the FAB mass spectrum of 47. The dominating molecular peak at
2107 and the absence of fragments with higher molecular weight clearly indicates the
complete deprotection. The quantitative conversion into the corresponding carboxylic
acids can also be followed in the 1H-NMR spectrum by the disappearance of the char-
acteristic signal for the tert-butyl groups at about 1.41 ppm.
The 13C-NMR spectra in figure 3.2 show the spectroscopic changes within the conver-
sion of the malonate 32, via the monoadduct 38 to the amphifullerene 44. The splitting
of the signals for the carbonyl-C-atoms (3,5) at 164 ppm clarifies the asymmetry of
the molecules. The comparison of the spectra of 38 and 44 show an abundantly clear
low-field shift for the carbon atom 4. The sp2-region of 38 exhibits 21 carbon signals,
whereas 9 signals show double intensity. This is in accordance with the 31 expected
signals for a monoadduct with Cs-symmetry. In the case of the Cs-symmetrical am-
phifullerene 44, the sp2-region contains 28 signals, whereas 3 signals show double
28

Chapter 3 Results and Discussion
0102030405060708090100110120130140150160170180
0102030405060708090100110120130140150160170180
0102030405060708090100110120130140150160170180
Chemical Shift (ppm)
12
8
13
2,69 4
7
1
12
8
13
2,6
9
4 7
10
1
11
sp -2C
sp -3 C
- H -C 2
14
14
10 11
12
8
9
4
71
sp -C2
sp -3 C
- H -C 2
10 11
- H -C 2
1
2
3
4
5
6 7 89
10
11 12
1314
1
2
3
4
5
6 7 89
10
11 12
1314
3 5
3 5
3 5
* *
*
*
167.2 167.0
164.3 164.1 163.9
164.2 164.1 164.0
167.1
1
2
3
4
5
6 7 8910 11
12
32
38
44
Figure 3.2: 13C NMR of malonate 32 (100.5 MHz, RT, CDCl3), protected monoadduct 38(100.5 MHz, RT, CDCl3) and deprotected monoadduct 44 (100.5 MHz, RT, THF-d8).
29

Chapter 3 Results and Discussion
intensity. The disappearance of the signals for the tert-butyl groups at 80.91 ppm and
28.31 ppm in the spectrum of 44 shows the complete cleavage of the ester groups.
Figure 3.3 represents the spectroscopic changes within the conversion of the malonate
35, via the monoadduct 41 to the amphifullerene 47 in the case of the 2nd generation
systems and show almost the same chemical shifts as observed in the 1st genera-
tion systems. The signals for the amide and ester carbon atoms are furthest shifted
to low-field and can be detected according to their number in the molecule in the in-
tensity ratio 1:3:9. In contrast to the spectrum of 44, the spectrum of the deprotected
monoadduct 47 shows a very bad signal to noise ratio, even at very high scan rates
(≈ 20000 scans). Furthermore a broadening of the signals can be detected, indicating
the aggregation tendency in polar solvents. Figure 3.4 shows the UV/Vis-spectra of
the protected monoadduct 38 and deprotected monoadduct 44, exemplary for the 1st
generation amphifullerenes. The UV/Vis spectrum of 38 reveals the unique features
of a monoadduct, notably the small characteristic absorption peak at 425 nm. In the
case of the deprotected monoadduct 44 the UV/Vis spectrum in DMSO indicates the
formation of micellar organization in solution, whereas the slightly shifted absorption
peak at 428 nm is still detectable. In buffered aqueous solution at pH = 7.2 (solubility
was promoted by the addition of a small amount of DMSO), the aggregation behavior is
clearly observable, which results in a characterless slope with a broadened maximum
at 328 nm. As expected, the similar trend can be followed in the UV/Vis spectra of the
2nd generation amphifullerene 47 (figure 3.5). Contrary to the 1st generation analog
even in polar-aprotic solvents the aggregation is highly facilitated, which can be seen
in the almost identical slope of the spectrum in DMSO and buffered H2O, respectively.
An effect of the different alkyl chains within the series of amphifullerenes could not
be explored by UV/Vis spectroscopy, in fact the measured spectra show the identical
characteristics as mentioned above. More sensitive methods, like cryo-transmission
electron microscopy (cryo-TEM) would be useful to study the effect of the alkyl chain
on the size and shape of these micellar structures.
30

Chapter 3 Results and Discussion
0102030405060708090100110120130140150160170180
0102030405060708090100110120130140150160170180
0102030405060708090100110120130140150160170180
173.5 173.0 172.5 167.1 167.0 166.9
173.4 173.0 164.2 163.8
sp -2C
812
163 5
17
2,6
9,13
47
10,11
14,15
- H -C 2
1
18
*
812
163 5
17
2,6
9,13
4 7
- H -C 2
1
18*
sp -2 C
12
3
4
5
6 78 9 10
11 1213 14
15 16
sp -2C
2,6
9,13
4 7
1sp -3 C
16
8,12 3,5
- H -C 2
10,11
14,15
10,11
14*
12
3
4
5
6 78 9 10
11 1213 14
15 16
1718
12
3
4
5
6 78 9 10
11 1213 14
15 16
1718
15
35
41
47
Figure 3.3: 13C NMR of malonate 35 (100.5 MHz, RT, CDCl3), protected monoadduct 41(100.5 MHz, RT, CDCl3) and deprotected monoadduct 47 (100.5 MHz, RT, DMSO-d6).
31

Chapter 3 Results and Discussion
300 350 400 450 500 550 600
ab
sorb
an
ce
wavelength [nm]
44
38 in CH2Cl244 in DMSO44 in H2O at pH = 7.2
Figure 3.4: UV/Vis spectra of protected monoadduct 38 (black) and deprotected monoadduct44 in DMSO (red) and H2O (pH = 7.2) (blue).
300 350 400 450 500 550 600
ab
sorb
an
ce
wavelength [nm]
47
41 in CH2Cl247 in DMSO47 in H2O at pH = 7.2
Figure 3.5: UV/Vis spectra of protected monoadduct 41 (black) and deprotected monoadduct47 in DMSO (red) and H2O (pH = 7.2) (blue).
32

Chapter 3 Results and Discussion
3.1.2 Synthesis of an Anionic Amphiphilic Monoadduct Carry ing
an Unsaturated Fatty Acid
The blood-brain barrier (BBB) is the specialized system of capillary endothelial cells
that protects the brain from harmful substances in the blood stream, while supplying
the brain with the required nutrients for proper function. Unlike peripheral capillaries
that allow relatively free exchange of substance across / between cells, the BBB strictly
limits transport into the brain through both physical (tight junctions) and metabolic (en-
zymes) barriers. Thus the BBB is often the rate-limiting factor in determining perme-
ation of therapeutic drugs into the brain. Overcoming the difficulty of delivering thera-
peutic agents to specific regions of the brain presents a major challenge to treatment
of most brain disorders. Therapeutic molecules and genes that might otherwise be ef-
fective in diagnosis and therapy do not cross the BBB in adequate amounts. Given the
benefits and substantial properties of C60 as therapeutic drugs, we considered that it
may be feasible to increase the efficiency by modifying its access to brain target sites.
As a strategy aimed at testing this hypothesis, we followed previous findings with ester
or amide derivatives of fatty acids important in the composition of brain tissue, includ-
ing some that are effectively transported into the central nervous system (CNS). Such
derivatives can markedly enhance entry of some compounds into the brain, including
for example hydrophilic agents such as GABA and dopamine.[112,113] Accordingly, we
considered preparing derivatives of [60]fullerene and fatty acids that are abundant in
brain tissue but must be supplied exogenously and transported through the blood-brain
barrier. We hypothesized that such compounds might increase and prolong the uptake
of water-soluble fullerenes into the brain. Docosahexaenoic acid (DHA) is a particularly
interesting candidate for attachment to C60 because it is vigorously transported into the
CNS and accounts for a high proportion of cerebral fatty acid content.[114,115] Moreover,
in earlier work, DHA proved to be particularly effective in facilitating entry of hydrophilic
compounds into the brain.[113] DHA is a 22-carbon chain, omega-3 unsaturated fatty
acid, containing six cis-double bonds. It is present in neuronal tissue, nerve termi-
nals and synapses, predominantly within membrane phospholipid constituents phos-
33
Chapter 3 Results and Discussion
O
OH
+ HO OH
O
O OH
HO O
O+
HO OH
OO
O O
OHO
O O
=̂ R1
R1O O
O OOR
O
OO
O
OO HN
HN
OHN
O
O
O
O
O
O
OONH
OO
OO
O
O
OO
O O O
O
C60, CBr4, DBU
OO
O
OO HN
HN
OHN
O
O
OR
O
OR
O
ORONH
ORO
ORO
RO
O
ROO
RO O OR
O
O
O
R1
R = tBuR = H
TFA
R = tBuR = H
TFA
EDC, DMAP, 1-HOBt
EDC, DMAP
EDC, DMAP
EDC, DMAP
49 50
22 51
50
5253
30
54
5556
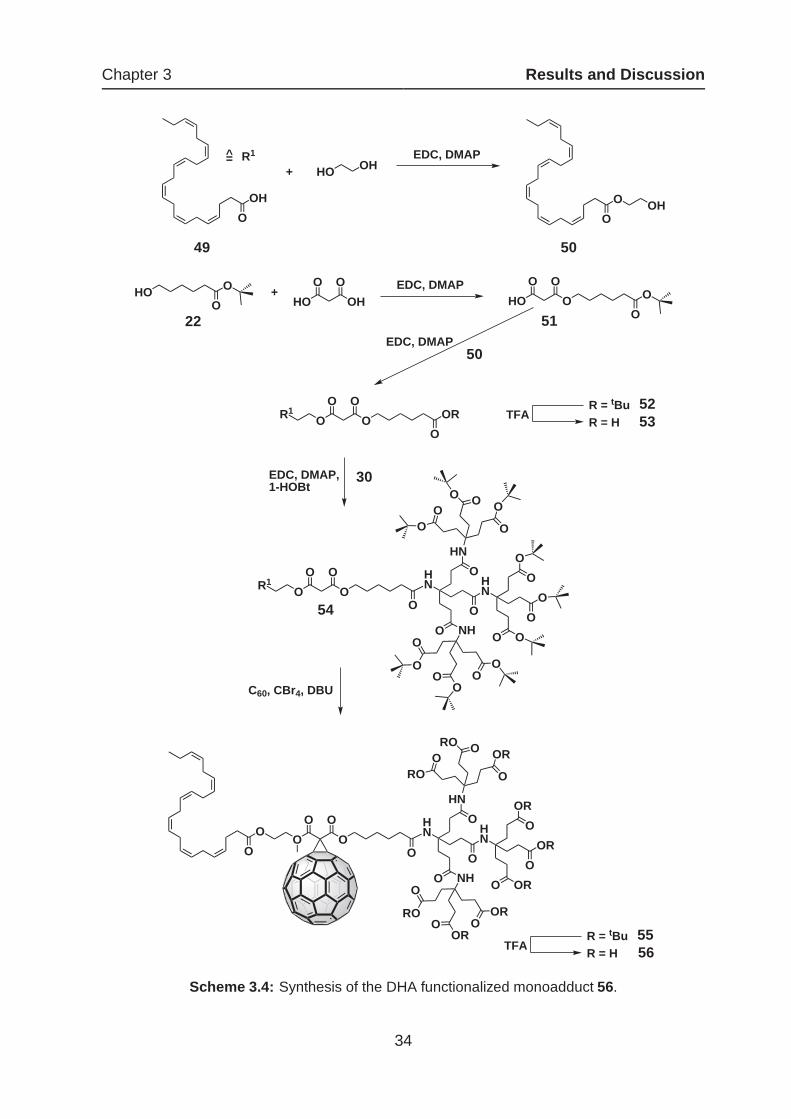
Scheme 3.4: Synthesis of the DHA functionalized monoadduct 56.
34

Chapter 3 Results and Discussion
phatidylethanolamine, phosphatidylserine, and phosphatidylcholine.[115,116] The fatty
acids are transported into the CNS by incompletely defined mechanisms that may in-
clude receptor-mediation.[115,117,118] DHA can be obtained by extraction from naturally
occurring oils, such as marine animal oil and various vegetable oils. The resulting
mixture of pure fatty acids is then subjected to separation by means of urea complex-
ing to remove saturated fatty acids and most mono-unsaturated fatty acids. The urea
is then removed from the filtrate which is subsequently subjected to low temperature
fractional crystallization in the presence of an organic solvent such as acetone to give
substantially pure polyunsaturated fatty acid.[119] Based on the preceding considera-
tions, a water-soluble, DHA-functionalized fullerene monoadduct was synthesized.
Scheme 3.4 shows the multistep synthesis of the DHA functionalized water-soluble
monoadduct. In the first step the all-cis-docosahexaenoic acid was esterificated with
glycol in order to receive the alcohol 50. Construction of the asymmetric malonate
via the MELDRUM’S acid method failed in this case, leading to a decomposition of
the starting material, probably by the thermal instability of the unsaturated fatty ester.
To overcome this problem, the asymmetric malonate unit 51 was synthesized using
the procedure described in scheme 3.1, followed by the coupling with 50 to yield the
malonate 52. After deprotection of the tert-butyl ester with TFA in CH2Cl2, the corre-
sponding carboxylic acid 53 was amidated with the 2nd generation dendron 30 by the
use of EDC. The cyclopropanation of 54 with C60 gave the monoadduct 55 with traces
of a less polar impurity (maybe side reactions with the DHA group). The repeated sep-
aration of these impurities with flash column chromatography failed in this case and
made further purification by preparative HPLC necessary. Deprotection to 56 was ac-
complished in a TFA/CHCl3 mixture and was completed after 48 h in quantitative yield.
The characterization of 55 is given in the following figures. The 1H NMR spectrum of
55 shows the characteristic resonances at 5.29 and 2.75 ppm for the polyunsaturated
acid chain. The methylene groups 4 and 5 are shifted low-field by the fullerene core,
resulting in the superimposed signal at 4.41 ppm for 3 and 5. This can be clearly iden-
tified by comparison of the spectra of the malonate 54 and the adduct 55. The signals
for the dendritic branch and the spacer unit are in the typical range and are comparable
35

Chapter 3 Results and Discussion
00.511.522.533.544.555.566.577.58
4 3,5
2,91
Chemical Shift (ppm)
*
1
2
3
4 5
6
7
8
9
6,8 7
55
Figure 3.6: 1H NMR of protected monoadduct 55 (300 MHz, RT, CDCl3).
with the amphiphiles described in chapter 3.1.1. In the carbonyl region of the 13C NMR
spectrum of 55 all non-equivalent C-atoms could be resolved as single signals. The
resonances for the amide and ester carbon atoms are the furthest low-field shifted sig-
nals and could be detected according to their quantity in the molecule in the intensity
ratio 1:3:9. The splitting of the carbonyl C-atoms at 163.5 ppm into two signals clarify
the asymmetry in the molecule. The sp2 area of the spectrum exhibits 26 of the 31
signals, as expected for the Cs-symmetry, whereas 5 signals show the double intensity
(figure 3.7 (b)). The 12 C-atoms located at the double bonds of the fatty acid could
be detected in the area from 127 to 132 ppm. The signals for the C-atoms located
between the double bonds overlap to three different signals at about 25.7 ppm. The
OCH2 units from the ethylene glycol and spacer moiety are positioned between the
sp3-signal at 71.5 ppm and the quaternary C-atoms of the dendron at 57.5 ppm. The
signals for the dendritic branch can be taken from figure 3.7 (c).
In analogy to chapter 3.1.1, 56 reveals a good solubility in buffered aqueous solution
at pH = 7.2. The UV/Vis spectra of 56 follows the same trend as previously described.
The amphiphilic character of the molecule leads to a significant line broadening in the
absorption spectrum (see figure 3.8). Compared to the spectrum for 55, the charac-
teristic small absorption peak at 425 nm is not identifiable any more (figure 3.8, inset).
36

Chapter 3 Results and Discussion
0102030405060708090100110120130140150160170180
Chemical Shift (ppm)
16,20,24
8,10
sp -3 C
6711
17,21
9 15123
1
2
34
6
7
8910
11
12
13
14
15
16
5 17 18
19 20
21 22
23 24
2526
25
*
55
(a)
139140141142143144145146
(b)
25262728293031323334
4 18,19
22,23
26
(c)
Figure 3.7: 13C NMR of protected monoadduct 55 (75 MHz, RT, CDCl3) (a) and zoom intospecific regions (b,c).
The investigation of 56 concerning the biological activity (chapter 3.1.4) is currently
under progress. The findings will clarify, if the polyunsaturated fatty acid chain directly
affects the transport properties of 56 to specific tissue regions.
37

Chapter 3 Results and Discussion
300 400 500 600
abso
rptio
n
wavelength [nm]
350 375 400 425 450 475 500
55 in CH2Cl256 in buffered H2O (pH = 7.2)
Figure 3.8: UV/Vis spectra of protected precursor 55 (black) and deprotected water-solubleamphiphile 56 (red).
38

Chapter 3 Results and Discussion
3.1.3 Synthesis of a Cationic Amphiphilic Monoadduct
In the last years the research on anionic fullerene-derivatives proceeded rapidly and
numerous compounds were tested for their efficiency as antioxidants and for their po-
tential to inhibit neurodegenerative diseases. In the field of cationic fullerene-derivatives
amazingly only moderate progress was made. From a synthetic point of view anionic
water-soluble fullerene derivatives can be efficiently prepared by the unleash of car-
boxylic acids from the corresponding tert-butyl esters as described in the previous
sections. As the structural motive, NEWKOME-type dendrons are an appropriate way to
vary the numbers of carboxylic functions by simply adding higher generations. Never-
theless, only a few cationic dendritic systems are literature known so far [120–123], prob-
ably by the missing of a suitable protection group and difficult purification procedures.
In line with this thesis a cationic amphiphilic fullerene derivative should be prepared,
which could be used as reference substances to the known anionic compounds, to
give more detailed insight into the structure-efficiency relationship for the pharmaco-
logical activity. These comparison measurements could help to clarify the question, if
the biological activity of these compounds substantially depends on facts like addition
pattern or the chemical nature of the addends. Another questions concerns, whether
the charges only enable the water-solubility or if specific COULOMB-interactions with
complementary charged proteins or other receptor-type molecules play an important
role here.
To avoid any pH-value problems, we decided to synthesize a permanently charged
cationic derivative. Therefore an appropriate precursor had to be found, which could
be transformed quantitatively into the corresponding charged derivative. This should
be done in the last synthesis step, since charges complicate the purification rigorously.
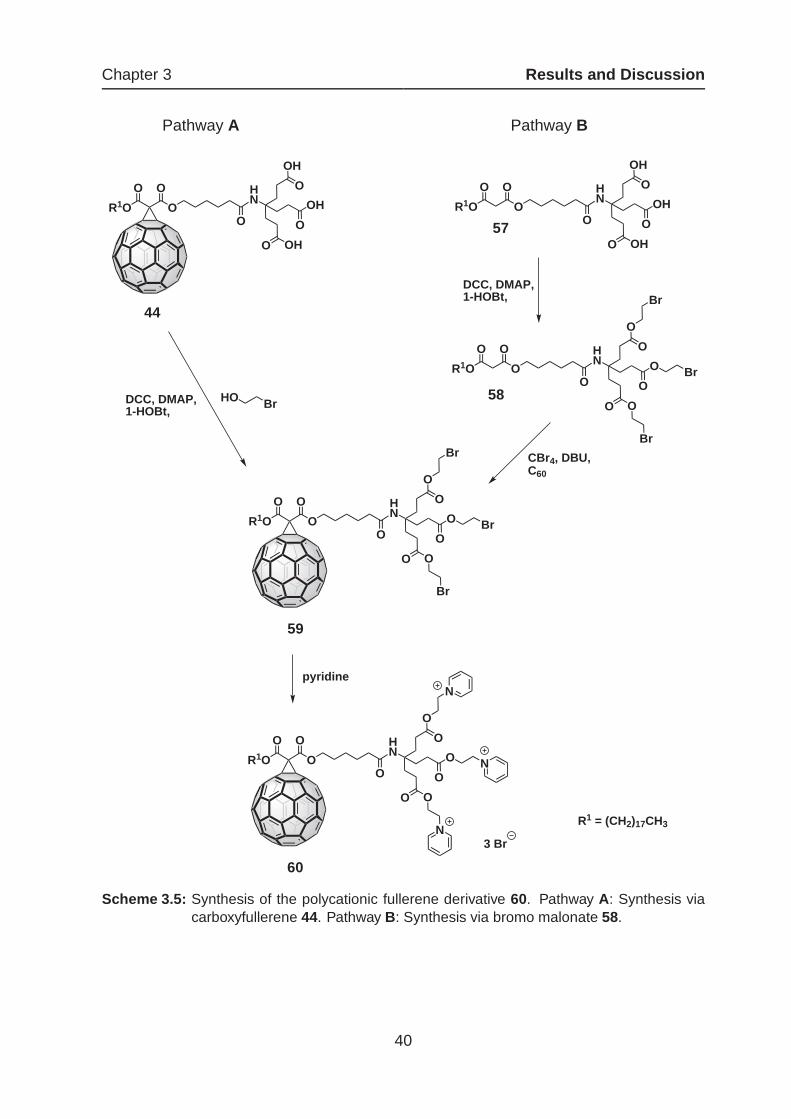
In scheme 3.5 the two possible synthetic pathways for the synthesis of the polycationic
derivative 60 is shown.
Pathway A takes advantage of the already synthesized amphiphilic carboxyfullerene
44 as starting material, which was esterificated with 2-bromoethanol in a threefold
STEGLICH-coupling. The introduction of the bromoethyl group displays a versatile tool
for the initiation of cationic charges, as the halogen atom could be easily displaced by
39
Chapter 3 Results and Discussion
OO
R1O
OO HN
OH
O
OH
O
OHO
OO
R1O
OO HN
O
O
O
OH
OH
OH
OO
R1O
OO HN
O
O
O
O
OO
Br
Br
Br
OO
R1O
OO HN
O
O
O
O
OO
Br
Br
Br
OO
R1O
OO HN
O
O
O
O
OO
N
N
N
3 Br
DCC, DMAP,1-HOBt,
BrHODCC, DMAP,1-HOBt,
CBr4, DBU,C60
pyridine
R1 = (CH2)17CH3
Pathway A Pathway B
44
57
58
59
60
Scheme 3.5: Synthesis of the polycationic fullerene derivative 60. Pathway A: Synthesis viacarboxyfullerene 44. Pathway B: Synthesis via bromo malonate 58.
40

Chapter 3 Results and Discussion
substitution with a variety of different nucleophiles. As already mentioned, the coupling
to the carboxy functionality under STEGLICH-conditions in the presence of the C60-core
is often afflicted with intricateness. Also in this case the reaction did not lead to a sat-
isfying result and even after long reaction times (120 h), remarkable amounts of mono
and bis-esterificated byproducts could be detected. In order to obviate this problem,
the malonate 57 was used as basic module for the synthesis via Pathway B. The depro-
tected malonate 57 could be easily achieved from the precursor 32 by acidic cleavage
of the tert-butyl esters. Reaction with 2-bromoethanol gave the malonate 58, which
exhibits three peripheral bromo groups. In contrast to the reaction described above
the bromo malonate 58 could be obtained in relatively high yields (78 %) and only a
small amount of fragmentary byproducts had to be separated by flash column chro-
matography. Subsequent cyclopropanation afforded the monoadduct 59, which should
be expeditiously further converted, since decomposition could be observed. Quater-
nization of the pyridine nitrogen via nucleophilic substitution reaction was achieved by
stirring 59 in dry pyridine at 60°C and was completed after 48 h in quantit ative yield.
The crude product 60 was purified by repeated precipitation from methanol with diethyl
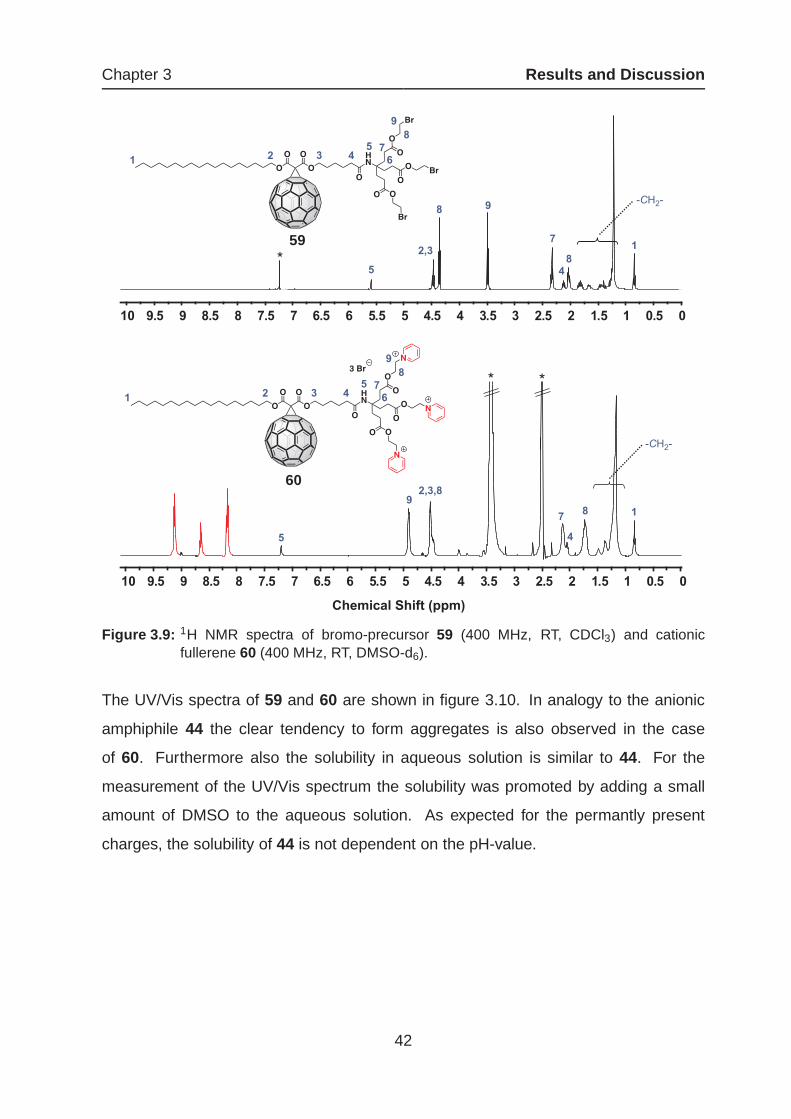
ether. The quantitative quaternization can be nicely followed in the 1H NMR spectrum
(figure 3.9).
The signals between 8.20 and 9.13 ppm correspond to the pyridinium rings and show
the characteristic splitting pattern. The relation of the integral for the pyridinium rings
and for the methyl group 1 with the ratio 5:1 verifies the complete quaternization. Com-
pared to the spectrum of the bromo derivative 59, the introduction of the pyridinium
groups results in obvious shifting of the resonances. Particularly strong is the effect in
the case of the methylene groups 8 and 9, with a low-field shift of about 1 ppm.
41
Chapter 3 Results and Discussion
00.511.522.533.544.555.566.577.588.599.510
00.511.522.533.544.555.566.577.588.599.510
Chemical Shift (ppm)
* *
*
1 2 3 4 67
8
9
5
12,3
8 9
4
7
8
- H -C 2
5
1 2 3 4 67
8
9
5
1
2,3,89
4
78
5
- H -C 2
59
60
Figure 3.9: 1H NMR spectra of bromo-precursor 59 (400 MHz, RT, CDCl3) and cationicfullerene 60 (400 MHz, RT, DMSO-d6).
The UV/Vis spectra of 59 and 60 are shown in figure 3.10. In analogy to the anionic
amphiphile 44 the clear tendency to form aggregates is also observed in the case
of 60. Furthermore also the solubility in aqueous solution is similar to 44. For the
measurement of the UV/Vis spectrum the solubility was promoted by adding a small
amount of DMSO to the aqueous solution. As expected for the permantly present
charges, the solubility of 44 is not dependent on the pH-value.
42

Chapter 3 Results and Discussion
300 400 500 600
abso
rbance
wavelength [nm]
59 in CH2Cl260 in H2O
60
Figure 3.10: UV/Vis spectra of bromo-precursor 59 and cationic fullerene 60.
43

Chapter 3 Results and Discussion
3.1.4 Amphiphilic Fullerenes as Potential Drug Candidates
3.1.4.1 Introduction and Background
Within a few years after the first production of fullerenes in macroscopic quantities,[8]
it was recognized that the extended conjugated π-system of C60 exhibits unusual po-
tency for scavenging radicals including reactive oxygen species (ROS).[68] The major
impediment for the development of fullerene-based biological antioxidants has been
the relative insolubility of C60 in either aqueous or lipid-based solvents. Early attempts
to modify the surface of the fullerene by polyhydroxylation and hexasulfobutylation pro-
duced water-soluble fullerenes with good biological distribution and cell penetration.
[124–131] These early classes of water-soluble fullerenes exhibited surprisingly potent
antioxidant and cytoprotective activities in vivo, including:
• significant reduction of death and permanent tissue loss associated with severe
ischemia/reperfusion injury resulting from complete blockage and subsequent re-
opening of coronary [124] and carotid [125] vasculature
• protection of cultured neurons from glutamate excitotoxicity [126,127] and peroxide-
induced injury to rat hippocampal slices [128]
• protection of small intestine from ischemia/reperfusion injury [129] and in intestinal
grafts after transplantation [130]
• protection of pulmonary tissue from pulmonary hypertension induced by chronic
hypoxia [131] and bronchoconstriction due to exsanguination and acute blood loss
One major drawback in the development of pharmaceutical applications for polyhydrox-
ylated and hexasulfobutyl fullerenes is that they have highly heterogeneous structures
with respect both to number of addends (polyhydroxyfullerenes) as well as the regioi-
somerism of the addends attached to the fullerene core (both classes). From a phar-
maceutical perspective, the most significant advance in the development of fullerene-
based antioxidants took place in the mid-1990s with the synthesis and characterization
of methanofullerenes bearing terminal carboxy groups (carboxyfullerenes), a class of
44

Chapter 3 Results and Discussion
exohedral fullerene adducts that could be synthesized in a highly purified and chem-
ically homogeneous form.[109,132–135] Furthermore, since the carboxyfullerenes can be
generated as single regioisomers, it is possible to design and synthesize a wide variety
of different three-dimensional structures and charge distributions to optimize structure-
function relationships and minimize "off target" binding and toxicity. So far different car-
boxyfullerenes have been synthesized and investigated with respect to their antioxidant
properties: carboxyfullerenes C3-[e,e,e-C63(COOH)6] 61 and D3-[trans3,trans3,trans3-
C63(COOH)6] 62.[134,135]
HO
OH
O
O
OHHO
O O
OH
OHO
O
HO
OH
O
O
OHHO
O O
OH
OHO
O
O
O
O
O
O
HN
HN
OHN
O
O
OH
O
OH
O
OHONH
OHO
OHO
HO
O
HOO
HO O OH
O
O
HN
NH
OHN
O
O
OH
O
HO
O
HO O HN
HOO
HOO
OH
O
OHO
OHOHO
O
61 62
63
Figure 3.11: C3-[e,e,e-C63(COOH)6] 61, D3-[trans3,trans3,trans3-C63(COOH)6] 62 and den-drofullerene 63.
Another prominent carboxyfullerene is the dendrofullerene 63, which is highly soluble in
water and has been investigated with respect to a variety of biomedical properties.[109]
In experiments similar to those described for fullerenols and hexasulfobutylated fullerenes,
carboxyfullerenes 61 and 62 have been shown to exhibit the following therapeutic ben-
efits linked to their antioxidant activities:
• carboxyfullerenes locate preferentially to mitochondria [136] and can reconstitute
45

Chapter 3 Results and Discussion
mitochondrial superoxide dismutase protection from superoxide radicals in SOD2
(superoxide dismutase 2) genetically deficient mice [137]
• carboxyfullerenes are highly potent neuroprotective agents, preventing cell death
across a variety of different neuronal types in disease models as diverse as
Parkinson’s, Alzheimer’s, ALS (amyotrophic lateral sclerosis), excitotoxicity, mac-
ular degeneration and stroke [138–145]
• carboxyfullerenes protect cells from damaging effects of UV and gamma-irradiation
[146]
• carboxyfullerenes protect from morbidity and mortality in the presence of over-
whelming infection with both, gram-positive and gram-negative organisms [147–150]
• carboxyfullerenes protect a variety of different cell types, including leukocytes,
hepatocytes and renal tubular epithelial cells from oxidative injury associated with
chemical and biological agents [151–153]
Although these surface-modified, water-soluble fullerenes have been shown to exhibit
strong antioxidant activity against reactive oxygen species (ROS), the progress in de-
veloping fullerenes as bona fide drug candidates has been hampered by three de-
velopment issues: 1) Lack of methods for scalable synthesis; 2) inability to produce
highly-purified, single-species regioisomers compatible with pharmaceutical applica-
tions; and 3) inadequate understanding of structure-function relationships with respect
to various surface modifications (e.g., anionic versus cationic versus charge-neutral po-
larity, degree of lipophilicity in the molecule). Also the stability of the derivatives is an
important factor. In the case of the carboxyfullerenes 61, 62 for instance, it turned out,
that these compounds are not stable in solution. HPLC analysis of the degradation
products indicates that decarboxylation reactions of the malonyl adducts represents
the major pathway for degradation of 61. Three major decomposition products, namely
the mono-, bis- and tris-decarboxylation products 64, 65 and 66 could be identified
(figure 3.12). The initial breakdown product is mostly 64, but with continued degrada-
tion, significant amounts of 65 and 66 are observed as well. In the presence of DMSO,
complete degradation to 66 appears complete within 1-2 minutes at room temperature.
46

Chapter 3 Results and Discussion
HO
H
O
OHHO
O O
OH
OHO
O
HO
H
O
OHHO
OH
OHO
O
HO
H
O
OHHO
OH
HO
64 65 66
Figure 3.12: Schematical representation of the C3-[e,e,e-C63(COOH)6] 61 decarboxylationproducts 64, 65 and 66. Only one stereoisomer each is represented althoughthey were formed as mixtures of isomers (NMR).
Toxicological investigations disclosed, that these decomposition products exhibit a sig-
nificantly increased toxicity, compared to the carboxyfullerene 61 (see chapter 3.1.4.4).
On the other hand the dendrofullerene 63 showed no instability in solution; aqueous
solutions of 63 were stable at room temperature for several weeks. In spite of the
high stability and the good water-solubility of 63, the highly charged and voluminous
dendritic branch, together with the absence of an additional lipophilic part, could be
problematic concerning the membrane permeability. To combine the benefits of the
dendrofullerene 63 with new structural advantages, concerning the lipophilicity, we de-
cided to synthesize a series of novel amphiphilic fullerenes, which differ in the number
and type of charge as well as in the lipophilicity (see chapter 3.1 and figure 3.13).
The following chapters describe the examination of these compounds, with respect of
their antioxidant activity against superoxide anions and the cytoprotective activity in
zebrafish models.
3.1.4.2 Antioxidant Activity
Reactive oxygen species (ROS) are derived from the metabolism of molecular
oxygen.[154] ROS include superoxide anion radical (O−·2 ), singlet oxygen (1O2), hydro-
gen peroxide (H2O2), and the highly reactive hydroxyl radical (·OH). The deleterious
effects of oxygen results from its metabolic reduction to these highly reactive and toxic
species.[155] In living cells, the major source of endogenous ROS are hydrogen peroxide
and the superoxide anion, which are generated as by-products of cellular metabolism
such as mitochondrial respiration.[156] Alternatively, hydrogen peroxide may be con-
47

Chapter 3 Results and Discussion
OO
R1O
OO HN
OH
O
OH
O
O OH
OO
R1O
OO HN
HN
OHN
O
O
OH
O
OH
O
OHONH
OHO
OHO
HO
O
HOO
HO O OH
O
R1 = (CH2)5CH3
R1 = (CH2)17CH3
R1 = CH((CH2)7CH3)2
R1 = (CH2)5CH3
R1 = (CH2)17CH3
R1 = CH((CH2)7CH3)2
OO
O
OO HN
O
O
O
O
O O
N
N
N
3 Br
43
44
45
46
47
48
60
Figure 3.13: Overview over the amphiphilic monoadducts examined in chapter 3.1.4.
verted into water by the enzymes catalase or glutathione peroxidase. Variability or
inductive changes in the expression of these enzymes can significantly influence cel-
lular redox potential. ROS can cause tissue damage by reacting with lipids in cellu-
lar membranes, nucleotides in DNA,[157] sulfhydryl groups in proteins [158] and cross-
linking/fragmentation of ribonucleoproteins.[159] Oxidative stress is caused by an imbal-
ance between the production of reactive oxygen and a biological system’s ability to
readily detoxify the reactive intermediates or easily repair the resulting damage. In the
recent years it has been shown, that this oxidative stress plays a role in various clinical
conditions such as malignant diseases, diabetes, atherosclerosis, chronic inflamma-
tion, viral infection, and ischemia-reperfusion injury.[160–162] Diseases associated with
48

Chapter 3 Results and Discussion
oxidative stress such as diabetes mellitus and cancer show a pro-oxidative shift in the
redox state and impaired glucose clearance suggesting that muscle mitochondria is
the major site of elevated ROS production. Because of its high metabolic rate and
relatively reduced capacity for cellular regeneration, the brain is believed to be par-
ticularly susceptible to the damaging effects of ROS. In neurodegenerative diseases
like Parkinson’s, Alzheimer’s and amyotrophic lateral sclerosis (ALS), ROS damage
has been reported within the specific brain region that undergo selective neurodegen-
eration. Protein oxidation has been reported in the hippocampus and neocortex of
patients with Alzheimer’s disease, Lewy bodies in Parkinson’s disease and within the
motor neurons in ALS.[163]
An antioxidant is a molecule capable of deactivating these ROS. Well-known antioxi-
dants include a number of enzymes (catalase, superoxide dismutase) and other sub-
stances such as vitamin C, vitamin E and beta-carotene (which is converted to vitamin
A) that are capable of counteracting the damaging effects of oxidation. Fullerene-
derivatives are able to permeate into cells and to scavenge ROS in a highly effective
mechanism. The following properties are generally agreed to make fullerenes uniquely
effective as intracellular antioxidants:
• The energy level of the LUMO fullerene orbital is comparable to the involved
orbitals of superoxide radical anion
• The fullerene antioxidant quenching process appears to be an effective catalytic
mechanism (see chapter 3.1.5)
• Fullerene antioxidants localize within cells to mitochondria and other sites where
excess free radical production occurs
The standard assay to investigate the superoxide quenching ability of an antioxidant is
based on xanthine/xanthine oxidase to generate superoxide causing the reduction of
cytochrome C.[164] This process is followed photometrically by looking at the changes
in intensity of the absorption band of reduced cytochrome C at 550 nm (see scheme
3.6).
49

Chapter 3 Results and Discussion
Xanthine
H2O2+
Uric acid
XOD
2O2
2O2-
Cytochrome C
OD 550nm
Antioxidant
O2 + H2O2
Scheme 3.6: Schematic representation of reactions involved in the xanthine/xanthine oxidaseassay.
Using this protocol we compared the new amphiphilic fullerene structures with trisadduct
61 as standard. The results are summarized in table 3.1 and allow to draw conclusions
concerning a structure-function relationship.
fullerene derivatives IC50 [µM]
1st generation (anionic)43 –44 24.045 26.1
2nd generation (anionic)46 13.347 15.447 14.7
1st generation (cationic) 60 3.1
Trisadduct 61 and decompositionproducts
61 18.564 6265 4866 44
Dendrofullerene 63 11.0
Table 3.1: IC50 values for the superoxide quenching activities of fullerene derivatives in xan-thine/xanthine oxidase assays.
Within the class of polycarboxyfullerenes, the best activities were reached for the den-
drofullerene 63 with an IC50 value of 11.0 µM. Also the trisadduct 61 showed a re-
markable good scavenging activity with an IC50 value of 18.5 µM. The decarboxylation
50

Chapter 3 Results and Discussion
products themselves are less effective. With rising the degree of decarboxylation in
64, 65, 66, the IC50 values decrease from 62 to 44 µM. In the case of the amphiphilic
monoadducts the second generation compounds 46, 47, 48 exhibit significant lower
inhibition concentrations with IC50 between 13.3 and 15.5 µM than the first genera-
tion ones (44, 45), possessing values of 24.0 and 26.1 µM. A possible explanation
for these observations may be the different aggregation states of these molecules in
aqueous solutions due to their intrinsic structures. Whereas the dendrofullerene 63 is
water-soluble in an almost monomeric form the amphiphilic strutures have a strong ten-
dency to form aggregates in water as it is concerned by line broadening in the UV/Vis
and NMR spectra. Building up such clusters decreases the actual concentration of
active fullerene species in solution leading perhaps to lower quenching activities com-
pared with monomers. Because of their considerable lower net charge and smaller
hydrophilic head group together with a comparable lipophilic moiety and the essential
presence of polar surfactants like DMSO the supramolecular superstructures of the
first generation derivatives might be even greater resulting in lower fullerene availabil-
ity comparing to the second generation derivatives. These considerations might be
a possible explanation for the different superoxide scavenging activities. In the direct
comparison the cationic compound 60 showed significant better antioxidant properties.
This may result from the positive charge of the molecule, which is expected to be more
likely to capture negatively charged superoxide anions.[165]
Increasing the number of addends on the fullerene surface, which leads to an increased
rupture of the conjugated π-system, causes a decreasing ability of superoxide quench-
ing. For instance, the trisadduct 61 shows a substantially larger IC50 value. The
decarboxylation products of 61 on their part are more effective than 61 by itself. With
rising degree of the decarboxylation 64, 65, 66, the IC50 values decrease.
These structure-function studies indicate that antioxidant properties of water-soluble
fullerenes depend on a number of factors, not all of which are completely understood
at present. However, within closely related structural families, the similarities of behav-
ior are sufficient enough to adopt the following trends:
• Increasing the number of addends on the fullerene causes a decreasing ability of
51

Chapter 3 Results and Discussion
superoxide quenching.
• The antioxidant properties of water-soluble fullerenes depend on a number of fac-
tors, like the structure, size and shape of the addends. The empirical correlations
of the IC50 values with the structures are not completely understood at present.
• The charge seems to play an important role for the activity.
3.1.4.3 Cytochrome C Binding
The problem of the antioxidant studies described above are various influences through
additional components present in the assay matrix. In this regard it is important to
ask whether the activity of the fullerene adducts against the xanthine/xanthine oxi-
dase/cytochrome C assay is exclusively due to the ability of quenching superoxide or
due to additional effects caused by the interplay of the components present in this ma-
trix. One possible interaction, which may play a crucial role is electrostatic interaction
between the charged fullerene derivatives and the positively charged cytochrome C.
Previous investigations in the HIRSCH group studied the electrostatic binding and pho-
toinduced electron transfer between cytochrome C and 63.[166,167] In this case strong
electrostatic binding with an association constant of Ks = 1.7 x 105 M−1 (pH = 6) was ob-
served. To complement the aforementioned results of the assay the different fullerene
derivatives were subjected to binding studies with cytochrome C in cooperation with the
GULDI group at the University Erlangen-Nürnberg. In the case of intact cytochrome C,
the protein shell that surrounds the iron-(II) / iron-(III)-porphyrin hampers the direct
electronic interactions that are readily observed, between fullerenes and unbound por-
phyrins. To avoid this, the excited interactions with zinc-(II)-porphyrin chromophore
rather than the iron-(II) / iron-(III)-porphyrin were studied. This was deemed neces-
sary, because iron-(II) or iron-(III) centers tend to deactivate excited states efficiently.
In the corresponding experiments, aqueous solutions of Zn-(II)-cytochrome C (i.e., 0.9
- 1.4 x 106 M) buffered at pH 7.2 were titrated with variable concentrations of the dif-
ferent amphiphilic fullerene derivatives (i.e., 0.7 - 22.4 x 106 M). Regardless of the
strength of fullerene / Zn-(II)-cytochrome C interactions using other assays, the absorp-
tion spectra were not sufficiently sensitive to detect these interactions, and are best
52

Chapter 3 Results and Discussion
described as the linear superimposition of the component spectra, namely, fullerene
and Zn-(II)-cytochrome C. Fluorescence-quenching experiments provided a more sen-
sitive and consistent assay for fullerene / Zn-(II)-cytochrome C interactions. For Zn-(II)-
cytochrome C, recording the typical fluorescence pattern of a zinc-(II)-porphyrin with
maxima at 585 and 644 nm and with high quantum yields of ca. 0.04, demonstrates
the ability of this method to measure Zn-(II)-porphyrin photoactivity inside of the protein
matrix. In the presence of the fullerene derivatives, a non-linear quenching of the Zn-
(II)-cytochrome C centered fluorescence is clearly discernable (see figure 3.14). It is
important to note that the fluorescence emission spectrum remains constant through-
out the titrations, although the amplitude varies due to quenching by the fullerenes.
wavelength [nm]
em
issio
n [
a.u
.]
47
Figure 3.14: Room temperature fluorescence spectra of 1.4 x 10−6 M Zn-(II)-Cyt C in aqueoussolutions in the presence of variable concentrations of 47 – excitation wavelength:420 nm.
53

Chapter 3 Results and Discussion
0.0 2.0x10-6
4.0x10-6
6.0x10-6
8.0x10-6
1.0x10-5
1.2x10-5
0.5
0.6
0.7
0.8
0.9
1.0
1.1
I/I 0
Cfullerene[M]
Figure 3.15: A plot of the change in fluorescence intensity as a ratio of I/I0 versus 47 con-centration, the solid line represents the estimated curve-fit obtained by non-linearleast-squares analysis.
0.0 2.0x10-6
4.0x10-6
6.0x10-6
8.0x10-6
0.0
0.2
0.4
0.6
0.8
1.0
1.2
Cfullerene[M]
relativeintensities
offluorescingcomponents
Figure 3.16: A plot of the change in relative fluorescence intensities versus 47 concentra-tion, the solid line represents the estimated curve-fit obtained by non-linear least-squares analysis.
54

Chapter 3 Results and Discussion
This nicely mirrors the trend seen in the absorption spectra, where no notable impact
due to fullerene binding was seen on the absorption spectra. It is important in this
context to consider the mechanism of fluorescence quenching of Zn-(II)-cytochrome
C in the presence of fullerenes: past work has provided thermodynamic, kinetic and
spectroscopic evidence in support of an intra-ensemble charge separation that evolves
between the photoexcited Zn-(II)-cytochrome C and the electron accepting fullerene
63.[166] In the next step, the non-linear quenching dependences were used to deter-
mine the binding constants (i.e., Ka) for forming Zn-(II)-cytochrome C / fullerene donor-
acceptor ensembles held together by electrostatic forces (see figure 3.15). In table
3.2 the binding constants of in each case a representative of the different amphiphilic
fullerenes are summarized. At first glance, the positively charged fullerene derivative
60 gives rise to binding constants below the detection limit of ca. 102 M−1. This is
rationalized on the basis of repulsive interactions between equally charged donor and
acceptor constituents. When comparing 44 with 47 a more than 20-fold enhancement
is seen. This observation is in line with the expectations by the presence of eight
positive charges that are located within the docking station of cytochrome C in gen-
eral. Independent confirmation was gathered using time-resolved fluorescence exper-
iments. The Zn-(II)-cytochrome C fluorescence lifetime was measured in the absence
and presence of the electron accepting fullerene derivatives. When no fullerene is
present the recorded fluorescence time profiles are well fitted by a mono-exponential
decay function. A lifetime of 2.2 ± 0.2 ns was determined under such conditions.
Compound K aI [M−1] Fluorescence
lifetimes [ns]K a
II [M−1]
61 1.5 x 106 0.25 8.6 x 105
44 8.2 x 104 0.7647 1.8 x 106 0.97 1.2 x 106
I Steady-state fluorescence measurements.II Time-resolved fluorescencemeasurements
Table 3.2: Binding constants and fluorescence lifetimes of Zn(II)-cyt C/fullerene complexes withthe amphifullerenes 44, 47 and the carboxyfullerene 61.
55

Chapter 3 Results and Discussion
In contrast, adding any of the studied fullerene derivatives led to fluorescence decays
that were only reasonably fitted - with a χ2-value of close to 1 - by a bi-exponential
function. Here, a long-lived component (i.e., 2.2 ± 0.2 ns) and a short-lived component
were registered. Interestingly, the exact value of the short-lived component was found
to be dependent on the structure of the fullerene derivative (see table 3.2). Through-
out the titration experiments the two lifetimes remained constant. The only variable is
the relative ratio between the long-lived and short-lived components. The long-lived
component decreased continuously, while the short-lived component increased simul-
taneously. Similar to the steady-state experiments, the differing ratios were used to
derive the binding constants (see figure 3.16). Notably, an excellent agreement with
the steady-state analysis is observed for the tested systems.
56

Chapter 3 Results and Discussion
3.1.4.4 In vivo Studies of the Amphiphilic Fullerenes using Zebrafish ( Danio
Rerio) Embryos as Model System
The data from the xanthine/xanthine oxidase assay proved the fundamental potency
of fullerenes to work as highly effective antioxidants. To further address the challenge
of developing fullerene-based drugs, the evaluation of the toxicity and effectivity in the
human organism is an important step. In this context the zebrafish embryo has be-
come an important vertebrate model for the preclinical drug screening. It is well suited
for studies in genetics, embryology, development, and cell biology. Zebrafish embryos
exhibit unique characteristics, including the ability to absorb compounds through the
water (ease of drug administration) and short reproductive cycles. Moreover, since
zebrafish embryos are small (they can be studied qualitatively and quantitatively in a
single well of a 96-well plate), and transparent up to ten days post fertilization, the toxic
and cytoprotective effects of small molecules can be studied in situ during this period.
Finally, since the morphological structure of zebrafish embryos has been mapped to
the single-cell level, the toxic, apoptotic and cytoprotective effects on various organs
can be tracked to approximately the single-cell level in situ.[168–171]
The following "zebrafish-tests" were done in cooperation with PHYLONIX Pharmaceuti-
cals Inc., Massachusetts. For the experimental details see appendix A.
4h 24h 5 days
3 months
Figure 3.17: Different stages of zebrafish development.
57

Chapter 3 Results and Discussion
Toxicity of water-soluble fullerenes in zebrafish embryos
LC50 values
To compare the overall toxicity of the amphiphilic fullerenes in zebrafish embryos, the
derivatives were added to the water in single-wells of a 96-well plate, each containing a
single zebrafish embryo at 24 hpf (hours post fertilisation). The lethality along with any
observed morphologic abnormalities were scored at 120 hpf. In most cases, fullerenes
were tested at varying concentrations up to 500 mM, but in some cases the maximum
concentration tested was 250 mM due to solubility issues at higher concentrations. The
LC50 was calculated as the fullerene concentration at which 50 % lethality is observed
at 120 hpf. Table 3.3 summarizes the results for the amphiphilic fullerenes, with the
trisadduct 61 and the decarboxylation products 64, 65, 66 as reference.
Compound Class LC 50 Zebrafish [µM]
44 amphiphilic fullerene ≫ 250 (0 % lethality at 250 µM)45 amphiphilic fullerene ND46 amphiphilic fullerene ND47 amphiphilic fullerene ND48 amphiphilic fullerene ND61 trisadduct 59664 trisadduct 37365 trisadduct 13466 trisadduct 1063 Dendrofullerene > 500 (20 % lethality at 250 µM)
Table 3.3: LC50 of amphiphilic fullerenes in zebrafish.
For all of the amphiphilic fullerenes the LC50 are considerably high, reaching values up
to 0 % lethality at the highest possible concentrations. The breakdown products of 61
represent an important exception with respect to the general low toxicity for the anionic
fullerenes. With progressive decarboxylation steps, each resulting breakdown product
shows increasing toxicity with respect to the parent trisadduct 61. The LC50 for 61 is
596 µM, followed by continuous increase in lethality to 10 µM for 66. The symmetrical
dendrofullerene 63 is located in the range of 20 % lethality at 250 µM.
58

Chapter 3 Results and Discussion
Body Morphology
In specific cases morphological abnormalities such as shortened body length and ab-
normal body curvature were detected in embryos treated with fullerene concentrations
at or near the LC50. In the case of 61 a shortened body length was observed at 250
µM (figure 3.18, panel B). Decarboxylation product 66, which caused an enhancement
in the mortality of the zebrafish embryos, had no influences on the morphology during
embryonic growth. Treatment of the zebrafish embryos with 500 µM of 63 resulted in
a slight curvature of the body (panel C). In contrast, the amphiphilic fullerenes showed
no observable body length or curvature abnormalities (figure 3.18 panel G,H,J,K,L; for
the fullerene concentrations, see table 3.4). This together with the considerable high
LC50 values elucidates the general low toxicity of the amphiphilic fullerenes.
Figure 3.18: Morphological body-length abnormalities caused by doses of fullerenes (for thecompounds to the different entries see table 3.4. For comparison the correspond-ing free G-2 dendron 30 was investigated (panel I).
59

Chapter 3 Results and Discussion
Panel Compound Concentration [µM]
A control –B 61 250C 63 500D control –E 66 10F control –G 45 250H 44 250I 67J 46 500K 47 500L 48 500
Table 3.4: Fullerene concentrations used for the determination of morphological body-lengthabnormalities in figure 3.18.
Internal Organs
The optical transparency of zebrafish embryos permits the visualization of individual
internal organs, which can be examined with high resolution microscopy. Therefore,
compared with laborious histological examination in mouse, examining drug effects on
internal organs in zebrafish is simple. The organ morphology in the development of
liver, intestine and heart caused by doses of fullerenes is shown in figure 3.19. 250
µM 61-treated zebrafish exhibited unfolded cardiac chambers, underdeveloped liver
and intestine. The 500 µM 63 treated zebrafish exhibited enlarged liver and intestine.
For the amphiphilic fullerenes, 47 is exemplary shown. No obvious defects on liver,
intestine, and heart were observed in this case and also for the other amphiphilic com-
pounds 44, 45, 46, 47, 48. In general, for all examined fullerenes, no organ necrosis
could be observed with any of the treatments.
Cardiotoxity
Cardiotoxicity is a major problem with numerous pharmaceutical agents and indus-
trial chemicals. The use of zebrafish as an animal model for cardiotoxicity testing can
provide more accurate results compared to cell-based assays. Drug effects on heart
60

Chapter 3 Results and Discussion
(a) (b) (c)
(d) (e)
(f) (g)
control
control
control 250 µM 61 500 µM 63
10 µM 66
250 µM 47
Figure 3.19: Morphological organ abnormalities in the development of liver, intestine and heartcaused by doses of carboxyfullerenes 61, 63, 66 and amphifullerene 47. The liver,intestine, heart are outlined by magenta, yellow and red lines respectively for easyvisual assessment.
rates, heart beat rhythm, and contractility can be directly assessed in the transparent
zebrafish under a dissecting microscope, providing information on the pharmacologi-
cal effects of drugs on cardiac function. Figure 3.20 displays the bradycardia values
induced by the water soluble fullerenes. Figure 3.20 indicates that decarboxylation
product 66 induces bradycardia at very low doses of 1 mM or less. In contrast, no signif-
icant bradycardia is observed for dendrofullerene 63 or carboxyfullerene 61 at concen-
trations of 100 mM. The extent of bradycardia and associated cardiac abnormalities is
probably sufficient to account for the increased toxicity of the decarboxylation products
of 61 in the LC50 assay. The observed effects on cardiac conduction could presum-
ably be interpreted by the binding to the zERG potassium channel (zebrafish homolog
61
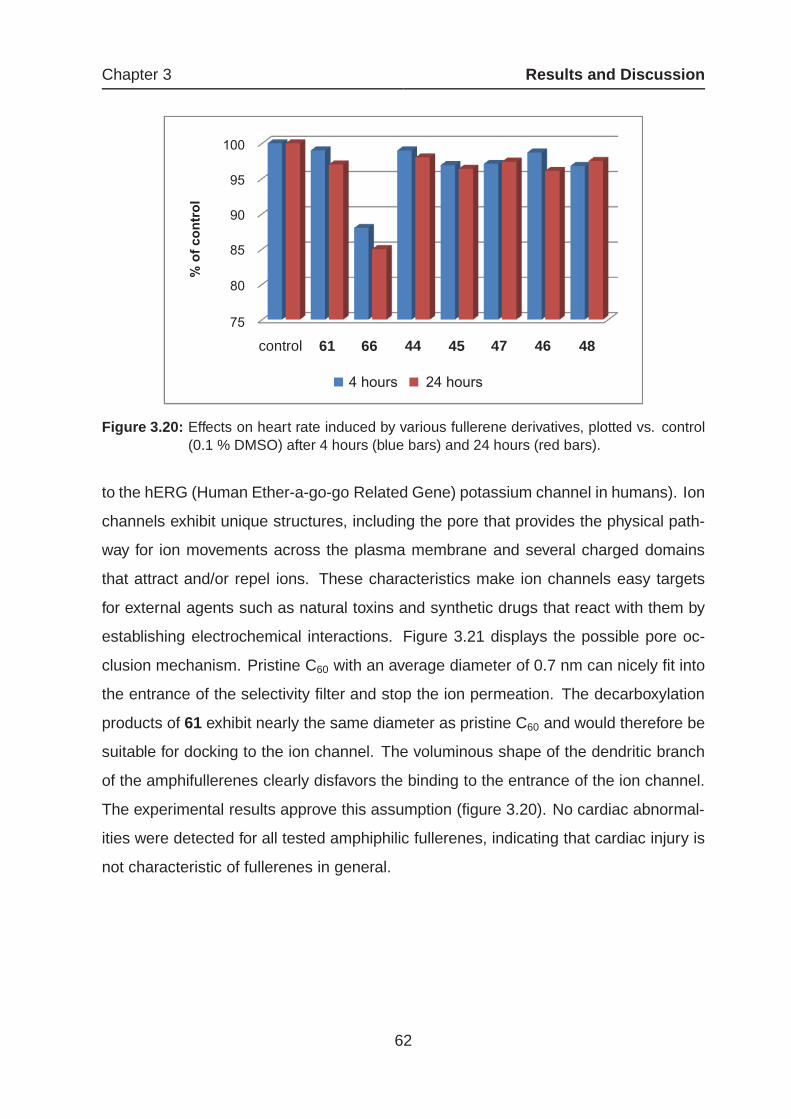
Chapter 3 Results and Discussion
100
95
90
85
80
75
% o
f co
ntr
ol
4 hours 24 hours
control 61 66 44 45 47 46 48
Figure 3.20: Effects on heart rate induced by various fullerene derivatives, plotted vs. control(0.1 % DMSO) after 4 hours (blue bars) and 24 hours (red bars).
to the hERG (Human Ether-a-go-go Related Gene) potassium channel in humans). Ion
channels exhibit unique structures, including the pore that provides the physical path-
way for ion movements across the plasma membrane and several charged domains
that attract and/or repel ions. These characteristics make ion channels easy targets
for external agents such as natural toxins and synthetic drugs that react with them by
establishing electrochemical interactions. Figure 3.21 displays the possible pore oc-
clusion mechanism. Pristine C60 with an average diameter of 0.7 nm can nicely fit into
the entrance of the selectivity filter and stop the ion permeation. The decarboxylation
products of 61 exhibit nearly the same diameter as pristine C60 and would therefore be
suitable for docking to the ion channel. The voluminous shape of the dendritic branch
of the amphifullerenes clearly disfavors the binding to the entrance of the ion channel.
The experimental results approve this assumption (figure 3.20). No cardiac abnormal-
ities were detected for all tested amphiphilic fullerenes, indicating that cardiac injury is
not characteristic of fullerenes in general.
62

Chapter 3 Results and Discussion
Figure 3.21: Left: Crystal structure of the KscA K+ channel. The image was constructed fromthe file 1BL8 from the Protein Data Bank. Right: Docking simulation of a fullerenewith an average diameter of 0.7 nm, which can block the entrance of the selectivityfilter and stop the ion permeation.
Cytoprotective activities
Protection of zebrafish embryos from CNS injury due to 6-hydroxydopamine (6-OHDA)
Programmed cell death is an essential regulator of normal development and homeosta-
sis of the CNS.[172,173] Neuronal apoptosis is thought to occur to eliminate those neu-
ronal precursors that fail to establish appropriate synaptic connections.[174] Inadequate
apoptosis has been found in various human neurodegenerative diseases in which a
massive neuronal loss occurs, such as Alzheimer’s disease or Parkinson’s disease.
CNS dopaminergic neurons are particularly sensitive to the toxic effects of 6-OHDA.
The apoptotic processes in zebrafish and mammals are very similar and therefore the
ability of fullerenes to protect from 6-OHDA-induced apoptosis of CNS dopaminergic
neurons in developing zebrafish was tested. Dopaminergic CNS neurons can be eas-
ily detected in live zebrafish embryos by the presence of tyrosine hydroxylase, which
can be detected by immunohistochemistry. 6-OHDA crosses the blood-brain barrier
and is taken up preferentially by dopaminergic CNS neurons, and to a lesser extent by
non-dopaminergic CNS neurons. Intracellular 6-OHDA induces cellular apoptosis and
death, at least in part through oxidative injury.[175] The amphiphilic fullerene 47 showed
the highest levels of CNS neuroprotection against 6-OHDA injury, and was able to pro-
63

Chapter 3 Results and Discussion
tect 60 % of tyrosine hydroxylase-positive dopaminergic neurons in the diencephalon.
Figure 3.22 shows the results obtained with 47, in protecting CNS dopaminergic neu-
rons against 6-OHDA-induced cell death.
control
6-OHDA
6-OHDA +
250 µM 47
Figure 3.22: Protection of CNS dopaminergic neurons from 6-OHDA-induced apoptosis by 47.
The untreated (top panels), 6-OHDA plus vehicle treatment (middle panels), and 6-
OHDA plus 250 mM 47 (lower panels) zebrafish embryos were stained with anti-tyrosine
hydroxylase antibody. At a time five images of each group were examined and the
number of TH-immunoreactive cells clustered in the diencephalons (between the eyes)
was detected. At untreated animals, 20-40 TH-cells could be observed (blue arrows).
After 6-OHDA treatment, a decrease in the number of TH-immunoreactive cells was
observed (1-17) (red arrows). After 250 mM 47 treatment, three of five 47-treated an-
imals showed a normal number of TH-immunoreactive cells with normal morphology
(black arrows).
Protection of zebrafish embryos from mechanoreceptor hair cell injury and death
induced by cis-platinum and gentamicin
In zebrafish, the dorsal mechanoreceptor hair cells are responsible for sensing posi-
tion, orientation, balance and movement and are biochemically and morphologically
similar to outer hair cells of the inner ear in mammals. These hair cells could be
easily examined by staining with 2-(4-dimethyl-aminostyryl)-N-ethyl pyridinium iodide
(DASEPI), which specifically colorizes the mechanoreceptor cells.
64

Chapter 3 Results and Discussion
control
CDDP
A
B
C
D
Figure 3.23: Protection of hair cells from cis-platinum damage. Hair cells detected by DASEPIstaining (black spots). For the different entries see table 3.5.
Entry Compound EC50 [µM]
A 61 ND (not detected)B 45 285C 47 194D 48 438
Table 3.5: EC50 values of the fullerene derivatives shown in figure 3.23.
65

Chapter 3 Results and Discussion
In this way, the fate of each lateral-line neuromast cell can be tracked in zebrafish em-
bryos after exposure to chemical toxins which induce apoptosis in these cells.[176] Cis-
platinum or cis-diamminedichloroplatinum(II) (CDDP) is a platinum-based chemother-
apy drug, widely used to treat various types of cancers. The drawbacks of cis-platinum
are the extensive side-effects, including damage to renal tubular cells and outer hair
cells of the inner ear, leading to renal failure and loss of hearing acuity or deafness,
respectively. The mechanism of cell damage, induced by CDDP is believed to be pri-
marily direct DNA damage, although perturbation of cellular redox pathways may also
play a role.[177] In zebrafish embryos, cisplatinum rapidly induces apoptotic cell death
in all of the dorsal mechanoreceptor hair cells detectable by DASPEI staining. Figure
3.23 shows the ability to block cis-platinum induced mechanoreceptor cell apoptosis, by
comparison of the amphiphilic fullerenes to carboxyfullerene 61. Fullerene 61, which
was highly effective in the superoxide assay (see table 3.1), was almost completely
unable to block apoptosis induced by cisplatinum, achieving only 5 % protection at
500 µM. In contrast to 61, the amphiphiles 45, 47, 48 show an efficient protection at
quite low concentrations (table 3.5). To further evaluate the otoprotective activities of
fullerenes, the protection of gentamicin treated zebrafish was tested. Gentamicin is an
aminoglycoside antibiotic, and can treat many types of bacterial infections, particularly
Gram-negative infection. All aminoglycosides are toxic to the sensory cells of the ear,
but they vary greatly in their relative effects on hearing versus balance. Gentamicin is
a vestibulotoxin, and can usually lead to permanent loss of equilibrioception, caused
by damage to the vestibular apparatus of the inner ear.[178–181] In zebrafish embryos,
exposure to gentamicin or the related antibiotic neomycin also induces apoptosis and
complete loss of DASPEI-stained dorsal mechanoreceptor hair cells in a pattern indis-
tinguishable for that described for cis-platinum. Surprisingly, the results for 61 contrast
substantially with those observed in the same cells using cis-platinum-induced cell loss
(see figure 3.23). In this case 61 is highly effective with an EC50 values of 34 µM (ta-
ble 3.6). This indicates, that the protection ability of fullerenes from different chemical
toxins could not be generalized and should be tested for each toxin separately.
66

Chapter 3 Results and Discussion
control
gentamicin
GSH
A
B
C
D
E
Figure 3.24: Protection of hair cells from gentamicin damage. Hair cells detected by DASEPIstaining (black spots). For the different entries see table 3.6.
Entry Compound EC 50 [µM]
GSH glutathione 438A 61 34B 47 29C 48 124D 44 35E 45 47
Table 3.6: EC50 values of the fullerene derivatives shown in figure 3.24.
67

Chapter 3 Results and Discussion
The tested amphiphiles 44, 45, 47, 48, which showed to be highly efficient against cis-
platinum induced hair cell loss (table 3.5), represent moderate EC50 values, ranking
between 29 for 47 and 124 µM for 48. In all cases, it should be mentioned that even
the least effective fullerenes in this group are 3-4 fold more effective than glutathione
(EC50 438 µM),[176,182,183] which has been used successfully in animal models to pro-
tect against aminoglycoside induced hearing loss and renal injury.
The results described above, showed that water-soluble fullerenes are able to block
apoptosis of specific cell types induced by chemical toxins and commonly used drugs
in a highly accessible zebrafish embryo model system. It has been also shown, that the
mechanism of protecting cells from apoptosis is not monolithic and depends both upon
the specific mechanism of toxicity, even in a single cell type. The differing effectivities
in the protection from gentamicin and cis-platinum may reflect, that the intracellular lo-
calization of each fullerene, the chemical reactivity and the interaction with biological
molecules, involved in apoptosis regulation, plays an important role. The introduc-
tion of a highly hydrophilic dendritic branch together with a lipophilic side chain, which
should increase the cell tissue accessibility and biodistribution within lipid-rich regions,
turned out to be an efficient tool to fulfill these issues. The overall high cytoprotective
activity against both cis-platinum and gentamicin induced apoptosis may prove these
amphiphiles to be useful as candidates for anti-apoptosis drugs.
68

Chapter 3 Results and Discussion
3.1.5 Mechanistic Aspects of the Reaction of Fullerenes wit h
Superoxide
Chapter 3.1.4 describes the capability of a series of functionalized fullerenes to serve
as highly efficient antioxidants. However the mechanism behind the reaction of C60
with reactive oxygen species is still not definitely clarified. On the one hand it is well
known, that irridation of solubilized [60]fullerene and its derivatives generate super-
oxide (O2·−) in aqueous solutions, under aerobic conditions and in the presence of
reducing agents.[184–187] This, besides the production of singlet oxygen (1O2), hydroxyl
radical (·OH) and hydrogen peroxide (H2O2), may be the primary factor for the photo-
toxicity of fullerenes,[185,188–191] on which their potential application as photosensitizers
in the photodynamic therapy is based. The generation of superoxide is based on the
outer-sphere electron transfer from reduced C60 to O2 (eq. 3.1).
C60·− + O2 −→ C60 + O2
·− (3.1)
On the other hand the antioxidant activity of fullerenes by scavenging superoxide anion
radicals could be clearly demonstrated (chapter 3.1.4.2). From a mechanistic point of
view, DUGAN et al. offered the evidence for a catalytic superoxide dismutation mecha-
nism, instead of direct radical attack on the C60 moiety.[137] She found that trisadduct 61
treatment of SOD2 −/− mice, which lack expression of mitochondrial manganese super-
oxide dismutase (MnSOD), significantly increased their life span, by the replacement of
MnSOD. The catalytic promoted dismutation was proposed to result from complexation
between C60 and O2·−, which electrostatically guide and stabilize the superoxide.
The observed high antioxidant efficiencies also suggests, that the reaction with super-
oxide is not based on stoichiometric scavenging. To further determine the mechanistic
pathway, we studied in cooperation with the IVANOVIC-BURMAZOVIC group the redox
properties of the amphiphilic fullerene derivatives (chapter 3.1.1, 3.1.3 and figure 3.25)
and their reactivity towards superoxide in DMSO and aqueous solutions.
69

Chapter 3 Results and Discussion
OO
R1O
OO HN
OH
O
OH
O
O OH
OO
R1O
OO HN
HN
OHN
O
O
OH
O
OH
O
OHONH
OHO
OHO
HO
O
HOO
HO O OH
O
R1 = (CH2)17CH3
OO
R1O
OO HN
O
O
O
O
O O
N
N
N3 Br
44
60
47
Figure 3.25: Overview over the amphiphilic monoadducts examined in chapter 3.1.5.
3.1.5.1 Cyclic Voltammetry Measurements of Amphiphilic Mo noadducts
A consequence of the unique spherical structure of C60 is the electronic configuration,
which exhibits a five-fold degenerated HOMO (hu) and a triply degenerated LUMO (t1u)
level (figure 1.8). In accord with the degeneration of the lowest unoccupied molecu-
lar orbital, electrochemical investigations, namely cyclic voltammetry studies, demon-
strated unambiguously the ability to reduce C60 up to the corresponding hexaanion, by
reversible one-electron reduction steps. As demonstrated in figure 3.26 (a) the first re-
duction potential is located at -0.98 V vs. Fc/Fc+ with PhMe/MeCN as solvent mixture.
The subsequent reduction steps follow in the typical range of 0.4 and 0.5 V.[63,64]
The cyclic voltammetry measurements [192] of an assortment of amphiphilic mono-
adducts (chapter 3.1.1, 3.1.3) have been carried out in DMSO as solvent, which al-
lows the direct comparison of the data with the results observed for the kinetic studies
(chapter 3.1.5.2). Two reversible electro-reductions in the potential range from 0 to
-1 V (vs. SCE) could be observed in all cases (figure 3.26). Table 3.7 summa-
rizes the measured redox potentials of amphiphile 44, 47 and 60, exposing the much
higher first reduction potentials for 44, 47 and 60, compared to the oxidation poten-
70
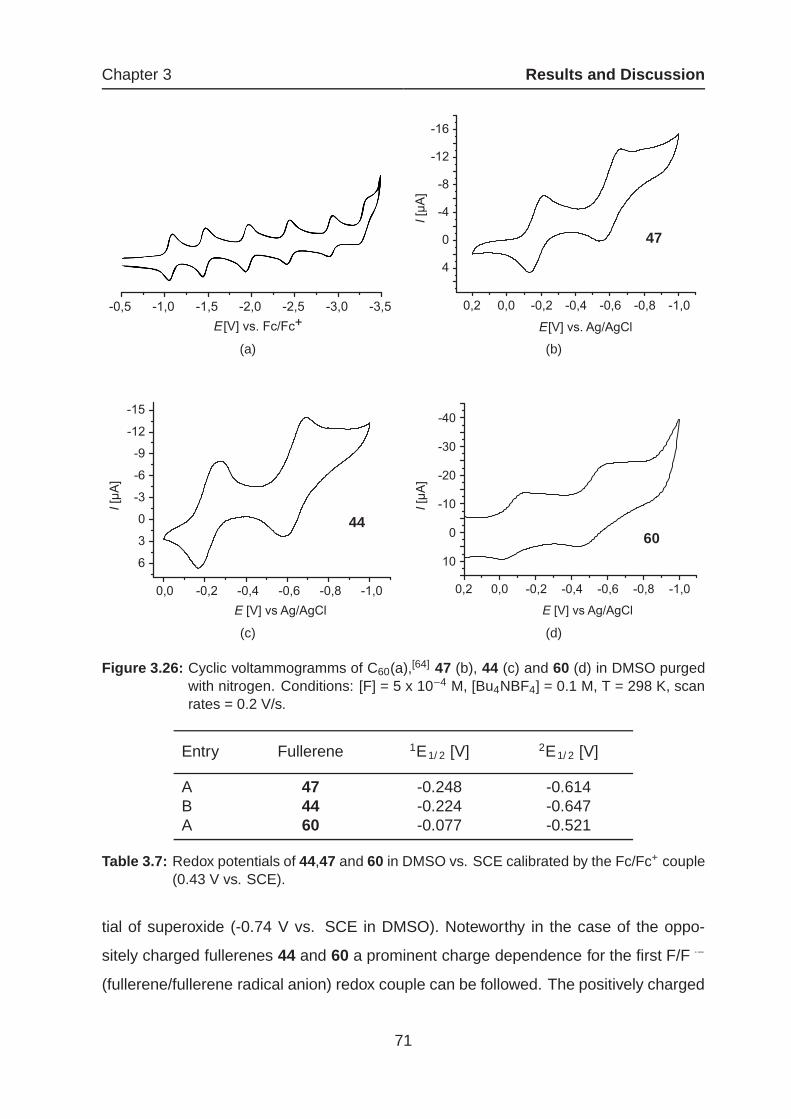
Chapter 3 Results and Discussion
-0,5 -1,0 -1,5 -2,0 -2,5 -3,0 -3,5
E [V] vs. Fc/Fc+
(a)
0,2 0,0 -0,2 -0,4 -0,6 -0,8 -1,0
4
0
-4
-8
-12
-16
E[V] vs. Ag/AgCl
I[µ
A]
47
(b)
0,0 -0,2 -0,4 -0,6 -0,8 -1,0
6
3
0
-3
-6
-9
-12
-15
E [V] vs Ag/AgCl
I[µ
A]
44
(c)
0,2 0,0 -0,2 -0,4 -0,6 -0,8 -1,0
10
0
-10
-20
-30
-40
E [V] vs Ag/AgCl
I[µ
A]
60
(d)
Figure 3.26: Cyclic voltammogramms of C60(a),[64] 47 (b), 44 (c) and 60 (d) in DMSO purgedwith nitrogen. Conditions: [F] = 5 x 10−4 M, [Bu4NBF4] = 0.1 M, T = 298 K, scanrates = 0.2 V/s.
Entry Fullerene 1E1/ 2 [V] 2E1/ 2 [V]
A 47 -0.248 -0.614B 44 -0.224 -0.647A 60 -0.077 -0.521
Table 3.7: Redox potentials of 44,47 and 60 in DMSO vs. SCE calibrated by the Fc/Fc+ couple(0.43 V vs. SCE).
tial of superoxide (-0.74 V vs. SCE in DMSO). Noteworthy in the case of the oppo-
sitely charged fullerenes 44 and 60 a prominent charge dependence for the first F/F ·−
(fullerene/fullerene radical anion) redox couple can be followed. The positively charged
71
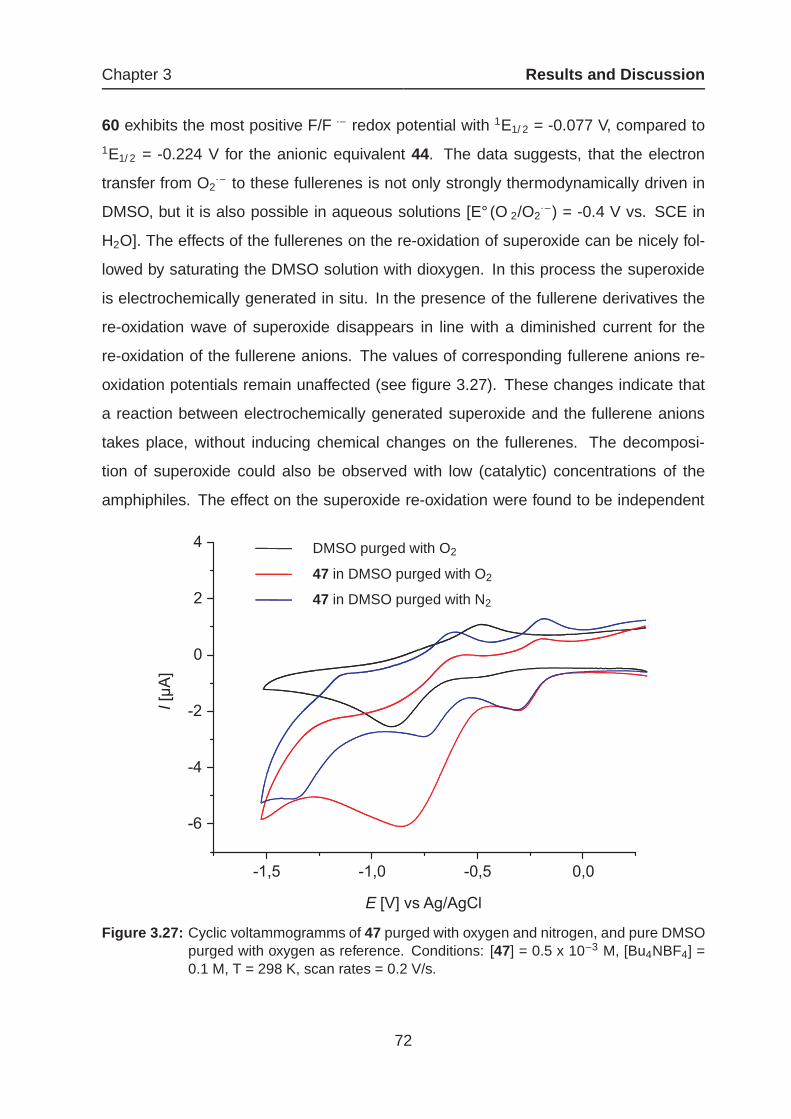
Chapter 3 Results and Discussion
60 exhibits the most positive F/F ·− redox potential with 1E1/ 2 = -0.077 V, compared to
1E1/ 2 = -0.224 V for the anionic equivalent 44. The data suggests, that the electron
transfer from O2·− to these fullerenes is not only strongly thermodynamically driven in
DMSO, but it is also possible in aqueous solutions [E° (O 2/O2·−) = -0.4 V vs. SCE in
H2O]. The effects of the fullerenes on the re-oxidation of superoxide can be nicely fol-
lowed by saturating the DMSO solution with dioxygen. In this process the superoxide
is electrochemically generated in situ. In the presence of the fullerene derivatives the
re-oxidation wave of superoxide disappears in line with a diminished current for the
re-oxidation of the fullerene anions. The values of corresponding fullerene anions re-
oxidation potentials remain unaffected (see figure 3.27). These changes indicate that
a reaction between electrochemically generated superoxide and the fullerene anions
takes place, without inducing chemical changes on the fullerenes. The decomposi-
tion of superoxide could also be observed with low (catalytic) concentrations of the
amphiphiles. The effect on the superoxide re-oxidation were found to be independent
-1,5 -1,0 -0,5 0,0
-6
-4
-2
0
2
4
E [V] vs Ag/AgCl
I[µ
A]
DMSO purged with O2
47 in DMSO purged with O2
47 in DMSO purged with N2
Figure 3.27: Cyclic voltammogramms of 47 purged with oxygen and nitrogen, and pure DMSOpurged with oxygen as reference. Conditions: [47] = 0.5 x 10−3 M, [Bu4NBF4] =0.1 M, T = 298 K, scan rates = 0.2 V/s.
72

Chapter 3 Results and Discussion
from the applied concentrations, which supports the proposed catalytic mechanism.
3.1.5.2 Kinetic Measurements of Amphiphilic Monoadducts
An exclusive method to follow rapid reactions is the stopped-flow spectrophotometry.
In a stopped-flow spectrophotometer, two syringes rapidly inject equal small volumes
of reactant solutions through a mixer and an observation cell, into another syringe.
When this receiving syringe is filled completely, the flow stops and a trigger activates
the computer to acquire data. The photomultiplier output is recorded in digitized form at
an acquisition rate set by the user. Depending on the instrument, mixing is essentially
complete within a matter of a few milliseconds, and reactions that are complete within
a range of milliseconds are amenable to this technique. Compared with the MCCORD-
FRIDOVICH assay, which was used in chapter 3.1.4.2 to determine the antioxidant ef-
fectivities, this method allows the direct measurement of superoxide decomposition
without any possible side effects through matrix components, present in the assays.
Using the stopped-flow method we studied the reactions of 44, 47 and 60 with a large
excess of KO2 in DMSO containing a controlled amount of water (0.06 %), which was
in excess over the superoxide and fullerene concentrations.[192]
300 400 500 600 700 800
0.0
0.5
1.0
1.5
2.0
2.5
Abso
rbance
Wavelength [nm]
A
B
(a)
300 400 500 600 700 800
0.0
0.5
1.0
1.5
2.0
2.5
Ab
sorb
an
ce
Wavelength [nm]
(b)
Figure 3.28: (a) Time resolved UV/vis spectra recorded for the reaction of 47 (5 x 10−5 M) with1 mM KO2 in DMSO at rt. A: spectrum before mixing; B: spectra obtained aftermixing (using a stopped-flow module) with time intervals of 10 s (total observationtime 30 min). (b) Control reaction without addition of 47 followed over 2.5 h.
73

Chapter 3 Results and Discussion
Time-resolved UV/vis spectra using a stopped-flow module (figure 3.28) showed the
rapid decomposition of O2·−, after mixing a superoxide solution with the fullerene so-
lution (decrease of the absorbance in the range of 270 nm). Less prominent spectral
changes at the wavelengths higher than 350 nm, in the case of 44 and 47 were found
to be caused by KOH which is inevitably present in KO2. By using an electrochemically
generated superoxide solution these spectral changes were not observed. The prod-
ucts of superoxide disproportionation, O2 and H2O2, were qualitatively detected in all
four experiments.[192] To verify the effect of the dendritic branches as structural parts
of the amphiphiles, the dendrimere 30 was measured as reference. For 30 itself, no
induced superoxide decomposition could be observed. To access the rate constants
of the rapid reaction, the decrease in the absorbance at 270 nm in the presence of
catalytic concentrations of the fullerenes was followed by stopped-flow measurements
(figure 3.29). Application of a microcuvette accessory (which reduced the dead time of
the instrument down to 0.4 ms) enabled observation of the fast disappearance of the
270 nm absorption, which could be fitted as a first-order process to obtain the char-
acteristic kobs (s−1) value. The plots of kobs vs. the fullerene concentrations showed
the desired linearity, from which the catalytic rate constants k cat can be determined
(see figure 3.30). The summarized data in table 3.8 show a clear dependence of the
fullerenes SOD activity (k cat ) on their redox potentials, namely, the higher the redox
potential (ability to be reduced by O2·−) the higher is the SOD activity. Contrary to
the literature,[193–195] this suggests that the electron transfer from O2·− to the fullerene
indeed is possible and plays an important role for the efficiency in the catalytic dismu-
tation of superoxide.
Entry Fullerene 1E1/ 2 [V] 2E1/ 2 [V] k cat x 106
[M−1s−1]
A 44 -0.224 -0.647 4.29 ± 0.06B 47 -0.248 -0.614 2.64 ± 0.04C 60 -0.077 -0.521 12.02 ± 0.22
Table 3.8: Redox potentials and catalytic rate constants of anionic amphifullerenes 44, 47 andcationic amphifullerene 60 in DMSO obtained by using direct stopped-flow measure-ments (kcat ) in DMSO (0.06 % water).
74

Chapter 3 Results and Discussion
0.00 0.02 0.04 0.06 0.08 0.100.86
0.88
0.90
0.92
0.94
0.96
0.98
1.00A
bso
rba
nce
Time [s]
(a)
0.00 0.02 0.04 0.06 0.08 0.10
1.14
1.16
1.18
1.20
1.22
1.24
1.26
1.28
1.30
Ab
sorb
an
ce
Time [s]
(b)
0.00 0.02 0.040.96
0.98
1.00
1.02
1.04
1.06
1.08
1.10
1.12
1.14
Ab
sorb
an
ce
Time [s]
(c)
Figure 3.29: Stopped-flow kinetics of the reaction of 2nd generation anionic amphifullerene 47(a), 1st generation anionic amphifullerene 44 (b) and cationic amphifullerene 60(c) with superoxide at 270 nm (298 K).
The results in this chapter from electrochemical, spectrophotometrical and submillisec-
ond mixing UV/vis stopped-flow measurements, approved that amphiphilic fullerenes
act as metal free SOD mimetics. The direct measurement using stopped-flow-technique
excluded any possible interferences through additional matrix components and ratified
a catalytic superoxide dismutation mechanism (see figure 3.31). We could also demon-
strate by examining a series of amphiphiles (differing in charge and hydrophilicity) that
the redox and structural properties of the studied fullerenes directly influences their
SOD activity.
75
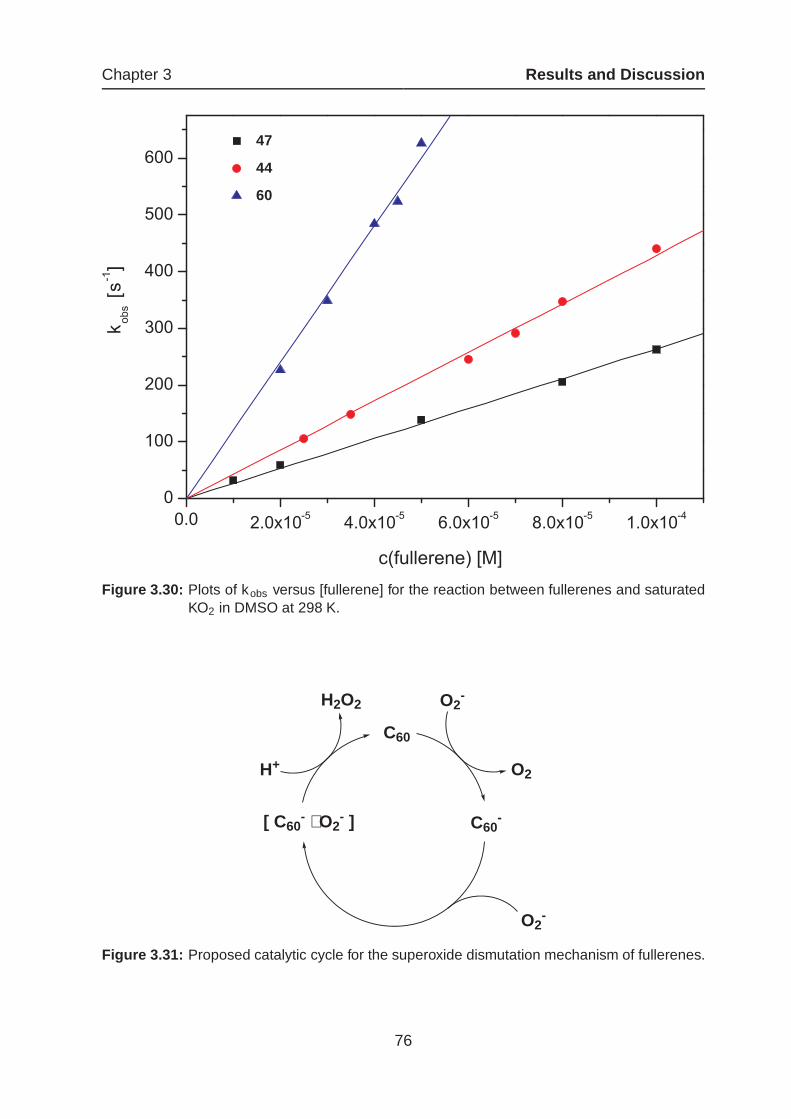
Chapter 3 Results and Discussion
0.0 2.0x10-5
4.0x10-5
6.0x10-5
8.0x10-5
1.0x10-4
0
100
200
300
400
500
600
kobs
[s-1]
c(fullerene) [M]
47
44
60
Figure 3.30: Plots of kobs versus [fullerene] for the reaction between fullerenes and saturatedKO2 in DMSO at 298 K.
C60
C60-
O2-
O2
[ C60- ⋅ O2
- ]
H+
H2O2
O2-
Figure 3.31: Proposed catalytic cycle for the superoxide dismutation mechanism of fullerenes.
76

Chapter 3 Results and Discussion
3.1.6 Amphiphilic Fullerenes in Material Science Applicat ions
With the cost of fossil fuels rising, attention is turning to organic solar cells, which
can produce electricity much more cheaply than conventional silicon based solar cells.
However, organic solar cells have to date been plagued by low efficiency, typically con-
verting the sun’s energy to electricity in the range of 4-5 percent.[196,197] The advantage
of these photovoltaic cells when compared to electro-chemical cells is predominantly
the absence of a liquid electrolyte, which generates problems with sealing against air,
but also the prospect of using flexible substrates.
In recent years, polymer-fullerene composites turned out to be one of the most ef-
fective and therefore extensively studied concepts in device design for organic solar
cells.[198,199] In these composites, the photon absorption takes place in the polymer
and forms bound electron-hole pairs that separate due to the electron-donating nature
of conjugated polymers and the electron-accepting nature of C60. As it is well known,
the precise microstructural features of the films are of crucial importance for the op-
timal device performance, where photogeneration of carriers must be accompanied
by its efficient transport and collection.[200] Although blend structures exhibit the max-
imum interface between donor and acceptor molecules, paths for charge percolation
to the electrodes and ohmic contacts at the film/electrode interfaces become limiting
factors to the device performance. The bilayer structure, on the other hand, has limited
interfacial area, however charge transport through the appropriate phase and charge
collection at the right electrode are intuitively less complicated. Various strategies have
been followed to optimize the blend structures and to control the weak compatibility of
conjugated polymers and C60, including:
• chemical modification of C60
• modification of the conjugated polymer
• addition of plasticizer components
In the typical composite devices, poly(3,4-ethylenedioxythiophene)-poly(styrene-
sulfonate) (PEDOT-PSS) is broadly used to form the ohmic contact with several metals
77

Chapter 3 Results and Discussion
and indium tin oxide (ITO) coated glass substrates. Since it is a water-soluble poly-
thiophene derivative, its polar groups could cause the segregation of the hydrophobic
molecules of fullerene derivatives present in the active bulk layer of the device. An
amphiphilic molecular layer, with the proper electrical and optical properties, seems
adequate to solve this problem.
OO
R1O
OO HN
OH
O
OH
O
O OH
OO
R1O
OO HN
HN
OHN
O
O
OH
O
OH
O
OHONH
OHO
OHO
HO
O
HOO
HO O OH
O
R1 = (CH2)17CH3
OO
R1O
OO HN
O
O
O
O
O O
N
N
N3 Br
O
O OO
O
OO HN
HN
OHN
O
O
OH
O
OH
O
OHONH
OHO
OHO
HO
O
HOO
HO O OH
O
44
6047
56
Figure 3.32: Overview over the amphiphilic monoadducts examined in chapter 3.1.6.
78

Chapter 3 Results and Discussion
3.1.6.1 Formation of L ANGMUIR-Films with Amphiphilic Fullerene-Monoadducts
A possible approach towards structurally ordered fullerene assemblies is the prepa-
ration of LANGMUIR films at the air-water interface and their subsequent transfer onto
solid substrates.[99,201,202] Typically a monomolecular layer is conveniently formed when
a solution of surface-active material (amphiphilic compound), dissolved in a water-
insoluble, volatile organic solvent, is placed drop by drop onto a clean water surface.
The hydrophobic tail of the amphiphiles makes this part of the molecule insoluble in
water which supports the formation of a monolayer, while the hydrophilic head serves
to orient the molecules at the water-air interface. The drops spread and the solvent
evaporates, leaving behind a monomolecular layer of the dissolved material which can
be compressed by moving barriers on the surface. The monolayer surface pressure π
[mNm−1] can be defined by the change in surface tension of the water surface caused
by the addition of the monolayer, where γ0 is the surface tension of the clean water
surface, and γm is the surface tension of the monolayer-covered surface.
π = γ0 − γm (3.2)
It is important to note, that π is a twodimensional pressure, which is generally a function
of many variables such as pH of the water, temperature, type of dissolved compound,
etc. It also varies with the surface concentration of the monolayer forming molecules,
making it possible to plot isotherms of surface pressure versus the average surface
area occupied per molecule in the monolayer. These isotherms describe the different
phase states of the monomolecular films during the compression. In the uncompressed
condition no intermolecular interactions are present, resulting in a gaseous-like state.
Continuous compression of the surface leads to an almost solid-like behaviour arising
from tight molecular packing in the monolayer, by increased intermolecular interactions.
When a monolayer is compressed into this tightly packed, coherent state it is possible,
by passing a carefully prepared solid substrate through it, to transfer the monolayer
intact from the liquid to solid surfaces. Depending on the nature of the solid material,
either the hydrophilic or hydrophobic parts of the amphiphiles adhere on the surface.
79

Chapter 3 Results and Discussion
In cooperation with Dr. GALLANI in Strasbourg, we examined the amphiphilic mono-
adducts (chapter 3.1.1, 3.1.3) for their ability to build LB-films (LANGMUIR-BLODGETT-
films) and the possibility to transfer them onto substrates. The anionic 1st generation
monoadduct 44 forms LANGMUIR films quite easily.
Figure 3.33: BAM-images of LB-films from 1st generation anionic amphifullerene 44 at 200 Å2
(top left), 130 Å2 (top right), 65 Å2 (bottom left) and collapsus (bottom right).
The Brewster-Angle-Microscopy (BAM) images of the film formation (figure 3.33), shows
that the final film at 53 mNm−1 is perfect and solid-like. The examined molecular
area at this pressure is 65 Å2. Continuing compression results in the collapse of the
monomolecular film (long wrinkles), which comes from the evasion of the molecules
into the third dimension.
The cationic 1st generation monoadduct 60 again nicely form films at the air-water in-
terface. The final molecular area is almost pH independent, with 130 Å2 at pH = 7,
110 Å2 at pH = 3.2 and 150 Å2 at pH = 11.7 (see figure 3.34 (a)). The finally formed
film showed a perfect molecular layer under the Brewster-Angle-Microscope (see figure
3.34 (b)).
80

Chapter 3 Results and Discussion
(a) (b)
Figure 3.34: pH dependence of the molecular area of the cationic 1st generation amphi-fullerene 60.
The reversibility of the isotherms on the other hand is much more pH-dependent (see
figure 3.35). Going to higher pH-values significantly reduces the reversibility, which is
nearly lost at pH = 11.7. The reason for this observation is not completely understood
yet. The pH value should not have an influence on the solubility of the permanently
charged 60. Maybe the exchange of the counterions with buffer-ions or the saponifica-
tion of the esters play an important role here. Further experiments will help to clarify
this issue.
For the anionic 2nd generation monoadducts, the amphiphiles 47 and 56 were exam-
ined. In both cases the solubility in CHCl3 was not sufficient enough and therefore
dissolution was improved with the adjunction of a small amount of ethanol. This comes
together with a small error in the measurements of the molecular area, because some
of the compound may dissolve into the water subphase upon spreading. Nevertheless
the obtained values seem to be reasonable in view of the molecular shape. For 47 the
final molecular area at neutral pH is 100 Å2, which would be exactly what is expected if
the compression of the fullerene core is the limiting factor. Since the reversibility is quite
good, the limiting factor cannot be the compression of the fullerene core since C60-C60
interactions would lead to irreversible aggregation (see figure 3.36 (a)). Moreover the
films are stable in the sense that the surface pressure stays constant when the com-
pression is paused. This means that the molecules do not dissolve or spontaneously
form supramolecular architectures. Brewster-Angle-Microscopy (BAM) observations
81

Chapter 3 Results and Discussion
100 150 2000
10
20
30pH=3.2
S
urfa
ce p
ress
ure
[m
N/m
]
Molecular area [Å2]
(a)
100 150 2000
10
20
30pH=6.8
Sur
face
pre
ssur
e [
mN
/m]
Molecular area [Å2]
(b)
100 150 2000
10
20
30pH=11.7
Sur
face
pre
ssur
e [
mN
/m]
Molecular area [Å2]
(c)
Figure 3.35: pH dependence on the reversibility of the isotherms for the cationic amphi-fullerene 60.
show that the molecules experience intermolecular attraction forces (filament-like struc-
tures in the first pictures), but eventually the film is homogeneous and quite nice, with-
out defects. No obvious sign of a collapse, even at the very end of the compression,
could be detected (see figure 3.37). Change of the pH value directly influences the
collapse pressure and the reproducibility. At higher pH values the pressure decreases
significantly to ca. 13 mNm−1, corresponding to the increased solubility by ionization
of the polar head group. At pH = 10.1 the reversibility is obviously lost, meaning that
the water-solubility at this pH value accelerates the dipping into the subphase (figure
3.36 (b,c)). Interestingly this behavior is directly influenced by the nature of the base
(NaOH, KOH) used for changing the pH. Figure 3.36 (d) displays the effects of the base
on the surface pressure. In the case of NaOH as the base, the isotherm is significantly
82

Chapter 3 Results and Discussion
(a)
50 100 150 200 2500
30
60pH=10.5
pH=10.1
pH=9.7other curves : pH 4.4 -> 9.3
Surf
ace
pre
ssure
[m
N/m
]
Molecular area [Å2
]
(b)
100 150 200 2500
10
20
30
40
pH=9.3
pure water
pH=10.1
Surf
ace
pre
ssure
[m
N/m
]
Molecular area [Å2]
(c)
0 100 200 300 4000
10
20
30
40
50
pH=10.7 KOH
pH=10.5 NaOH
Su
rfa
ce p
ress
ure
[m
N/m
]
Molecular area [Å2]
(d)
Figure 3.36: Reversibility of 2nd generation anionic amphifullerene 47 in water – 3 compres-sion expansion cycles with pressure limited to 35 mNm−1 (a); isotherms and re-versibility as a function of the pH in a NaOH subphase (b,c); influence of the ioniccontent of the subphase (d).
flattened resulting in a decreased surface pressure at nearly the same pH. The reason
for this behavior is not clarified yet, but maybe results from a degradation of the com-
pound, which is directly influenced by the nature of the base.
The observed pH dependent tendency can also be seen in the case of the fatty acid
functionalized anionic amphifullerene 56. The final molecular area for 56 ranges be-
tween 87 Å2 at pH = 3.2 to 7 and 140 Å2 at pH = 11.2. The shape of the isotherms
nicely corresponds to the isotherms of 47, although a difference in the final molecu-
lar area can be observed. Possibly the increased rigidity of the unsaturated fatty acid
chain effects a more rod-like structure in the case of 56. In contrast the saturated chain
in 44 is more flexible and therefore tend to increase the molecular area.
83

Chapter 3 Results and Discussion
Figure 3.37: BAM-images of LB-films from anionic 2nd generation amphifullerene 47 at 200Å2 (top left), 150 Å2 (top right), 125 Å2 (bottom left) and 87 Å2 (bottom right).
For compound 47 the ability to transfer LB films onto hydrophilic silicon was tested. It
is possible to transfer at least up to five layers with good quality. The quality of the
LB films can be assessed with grazing incidence X-ray reflectivity. The results are
summarized in Figure 3.38. The left spectra shows the diffracted intensity at different
numbers of monomolecular layers. Although the quality of the films is very good, the
0 2 4 6 8 10 121
100
1E4
1E6
1E8
1E10
1E12
5 layers3 layers1 layer
Diff
ract
ed
inte
nsi
ty [
a.u
.]
2θ [degrees]0 2 4 6 8 10
1
100
1E4
1E6
1E8
Diff
ract
ed
inte
nsi
ty [
a.u
.]
0 5 10 15 20 250
10
20
30
40
50
60
70
80
distance from substrate
ele
ctro
nic
de
nsi
ty
2θ [degrees]
Figure 3.38: X-ray reflectivity of the LB films of 47 at different numbers of layers (left); X-rayscan and "best" fit for 5 layers. Inset is the profile of the electronic density for onelayer.
84

Chapter 3 Results and Discussion
thickness of each sublayer stays not constant, ranging from 30 Å2 for the first layer to
125 Å2 for 5 layers. The right graph in figure 3.38 shows the spectrum of a 5-layer-thick
LB film on silicon. We tried to fit the data with a box model, where each molecular
layer has been divided into three sublayers, corresponding to polar head, C60 and alkyl
chain. Each sublayer could further be ascribed with a specific thickness, electronic
density, and roughness. With this model, the fit of the data for 47 was not acceptable.
If it was assumed, that each molecular layer is not a perfect mono-molecular and has
some "defects", e.g. supramolecular architectures, the fit quality was significantly im-
proved. Nevertheless the data so far can not elucidate if aggregation is the reason
for this behavior, since the quality of the observed LB film is actually immaculate. The
investigation of additional parameters is still under progress and will help to understand
this issue.
3.1.6.2 Incorporation of the Amphiphilic Fullerene-Monoa dducts in Organic
Solar Cell Devices
In the past view years the preparation of photovoltaic devices, made from composite
films of π-conjugated polymers and fullerene derivatives attracted considerable atten-
tion. As a concept a bulk heterojunction is used, which consists of a three dimensional
interpenetrating donor-acceptor network, sandwiched between two electrodes with dif-
ferent work functions to generate an electric field across the organic layer. From photo-
physical studies it has been demonstrated that after absorption of a photon, ultra-fast
electron transfer takes place from the excited state of a conducting polymer to acceptor
molecules such as C60, with a quantum efficiency close to unity.[203,204] Subsequently,
the separated charge carriers are transported via the interpenetrating network to the
electrodes. The photogenerated current is directly governed by the charge carrier mo-
bility, alongside the number of photoexcited charge carriers. For the understanding
of the opto-electronic properties of these photovoltaic devices, knowledge about the
charge transport properties of the individual components is indispensable.
In cooperation with KONARKA Technologies GmbH [205] the amphiphiles 44 and 47
have been incorporated in organic field-effect transistors (OFETs) to derive a value for
85

Chapter 3 Results and Discussion
Drain Source
Gate
Silicon substrate
Insulator
Semiconductor
PCBM
Figure 3.39: Schematic representation of a top contact organic field-effect transistor (left).[6,6]-phenyl C61-butyric acid methyl ester (PCBM) (right).
the mobility of the charge carriers.[206] The field effect transistors were prepared using
highly doped p-Si wafers as the gate electrode. A layer of thermally grown SiO2, with
a thickness of 100-150 nm, served as the gate insulator. After applying the fullerene
layer to the wafer, the source and drain electrodes (LiF/Al) were deposited onto the
semiconducting layer.
The structure of a typical top contact OFET is shown in figure 3.39 (left). The mobilities
can be determined from the transfer characteristics of the OFET (Ids vs. Vgs for a low,
constant Vds) according to the gradual channel approximation, with the equation [207]
∂Ids
∂Vgs= µFE
WCiVds
L(3.3)
where Ids is the current flowing in the channel between the drain and source contacts,
Vgs the gate voltage, µFE the field effect mobility, W the width of the channel, Ci the
capacitance of the insulating layer, Vds the voltage between the drain and source con-
tacts, and L the length of the channel.
In the OFET containing amphiphile 44 the electron mobilities determined according to
eq. 3.3 were found to be in the range of 5 x 10−4 cm2/Vs. The mobility of the OFET with
47 was quite low and could not be determined exactly (experimental resolution limited
to 10−6 cm2/Vs). This large difference in the mobilities maybe results from the larger
hydrophilic head group of amphiphile 47. Presumably the larger dendritic branch di-
rectly influences the morphology of the device films with an increased C60-C60 distance,
which affects the charge transport in the blend. Compared to PCBM ([6,6]-phenyl C61-
86

Chapter 3 Results and Discussion
butyric acid methyl ester) which is widely used as "standard" fullerene derivative in
polymer electronics and photovoltaics, the mobility value for 44 is only about 1 magni-
tude lower (7 x 10−3 cm2/Vs for PCBM). This is one of the highest values observed for
this type of fullerenes, so far.
We also studied the influence of amphiphile 44 on the morphology in blends with
terthiophene polymers.[205] Polyterthiophenes are attractive conducting polymers for
use in photoelectric conversion devices mainly due to their exceptional stability in
air and high charge carrier mobility values.[208] Poly(3,3"-dihexyl-2,2’:5’2"-terthiophene)
(C6-TT) can be synthesized by using Stille copolymerization [209] with an average molec-
ular weight of 42000 g/mol. Pristine C6-TT shows a distinctive shoulder at 600 nm
indicative for the aggregation of the thiophene backbone. This behavior can be used
to determine the morphology properties in corresponding composites via optical ab-
sorption spectroscopy. The polymer was utilized to build up devices, containing 44
as electron-accepting moiety which were examined concerning the blend morphology.
Figure 3.40 (a) shows the UV/vis spectrum of the terthiophene in the presence of 44.
The characteristic signal at 600 nm indicates the packaging of the polyterthiophene
backbone in presence of the fullerene. In contrast blends containing PCBM/C6-TT as
active layer, the aggregation is clearly hindered (figure 3.40 (b)). This suggests, that
PCBM could fill the side-chain spacing of the C6-TT backbone, as suggested in fig-
ure 3.40 (c), which would suppress the cofacial packing of the parallel chains. The
asymmetric monoadduct 44 is too big to fit nicely in this spacing, which leads to ad-
vantageous morphology forming properties. Further investigations are currently being
carried out and will verify the efficiency of this promising class of materials for photo-
voltaic operation.
87

Chapter 3 Results and Discussion
1.0
0.8
0.6
0.4
0.2
0.0
OD
[a
.u.]
400 600 800 1000
Wavelenght [nm]
before treatment1 min 140 °C
4 min 140 °C4 min 165 °C4 min 190 °C
(a)
1.0
0.8
0.6
0.4
0.2
0.0400 500 600 700
OD
[a.u
.]
C6-TTC6-TT ppt
C6-TT/PCBMC6-TT/PCBM ppt
Wavelenght [nm]
(b)
(c)
Figure 3.40: (a) Absorption spectrum of 44/C6-TT blend. (b) Absorption spectrum of aPCBM/C6-TT blend. (c) Comparison of the side-chain distance with the diam-eter of C60. Pristine C60 would fit nicely into this gap (as well as PCBM).[210]
88

Chapter 3 Results and Discussion
3.2 Triazole Dendrimers Based Fullerenes via "Click
Chemistry"
"Click chemistry" is a new approach that takes advantage of chemical building blocks
with built-in high-energy content to drive a spontaneous and irreversible linkage reac-
tion with appropriate complementary sites in other blocks. SHARPLESS and coworkers
defined what makes a click reaction as one that is wide in scope and easy to perform,
uses only readily available reagents, and is insensitive to oxygen and water.[211] In fact,
in several instances water is the ideal reaction solvent, providing the best yields and
highest rates. Reaction work-up and purification uses benign solvents and avoids chro-
matography. Carbon-heteroatom bond forming reactions comprise the most common
examples for this type of reaction, including the following classes of chemical transfor-
mations:
• cycloadditions of unsaturated species, especially 1,3-dipolar cycloaddition reac-
tions, but also the Diels-Alder family of transformations [212]
• nucleophilic substitution chemistry, particularly ring-opening reactions of strained
heterocyclic electrophiles such as epoxides, aziridines, aziridiniumions, and episul-
foniumions [213]
• carbonyl chemistry of the "non-aldol" type, such as formation of ureas, thioureas,
aromatic heterocycles, oxime ethers, hydrazones, and amides
• additions to carbon-carbon multiple bonds, especially oxidative cases such as
epoxidation, dihydroxylation, aziridination, and sulfenyl halide addition, but also
MICHAEL additions of Nu-H reactants [214]
These click reactions achieve their required characteristics by having a high thermody-
namic driving force, usually greater than 20 kcal mol−1.
Of the reactions comprising the click universe, the "perfect" example is the HUISGEN
1,3-dipolar cycloaddition of alkynes to azides, which gives acess to 1,2,3-triazoles as
89

Chapter 3 Results and Discussion
R1 + R2N380 - 120 °C N
N N
R1
R2
N
N NR2
+ R1
1,4-regioisomer 1,5-regioisomer
Scheme 3.7: HUISGEN cyclization of azides and alkynes.
a mixture of 1,4- and 1,5-regioisomers, which can be explained by the similarity in ac-
tivation energies for both processes (see scheme 3.7).[215,216] Such intrinsic features
of the HUISGEN cycloaddition make it unsuitable for being considered as a click re-
action. Nevertheless, the observation that copper(I) salts promote faster (up to 107
times) and regiospecific couplings between terminal alkynes and azides allowed for
the rapid development of this reaction; these results were reported independently by
the groups of SHARPLESS [217] and MELDAL.[218] The mechanistic outline of the Cu[I]-
catalyzed reaction is depicted in scheme 3.8. The stepwise cycloaddition catalyzed
by a monomeric Cu[I] species lowers the activation barrier relative to a uncatalyzed
process by as much as 11 kcal/mol, which explains the incredible rate enhancement
observed under CuI catalysis.[219] The catalytic cycle begins with the formation of a
Cu[I] acetylide species 70 via the π-complex 68, which is proposed based on earlier
results of CuI insertion into terminal alkynes [220,221] and the fact, that internal alkynes
show no activity in this type of reaction.[217,218]. This active species reacts further by
the azide displacement of one ligand, generating a copper acetylide-azide complex,
such as the dicopper species 71. The following metallocycle 72 is generated by the
nucleophilic attack of the acetylide carbon to the azide, which is activated therefore by
copper complexation.[222,223] In this way the bound azide is perfectly situated for the
subsequent ring contraction by a transannular association of the nitrogen lone pair of
electrons with the C-Cu π∗orbital.[219] Protonation of triazole-copper derivative 73 fol-
lowed by dissociation of the product ends the reaction and regenerates the catalyst.
90

Chapter 3 Results and Discussion
R1 H
LnCu [LnCu]2
+
R1 H
CumLn
LnCu2 R2
LnCu2 R2
Cu acetylide
R1N3
R2
CuCuL
L
NN
NR1N
N N CuL
Cu
R1
R2N
NN
R2LnCu2
R2
R1
NN
N
R2LnCu2
R1
B
B-H
NN
N
R2H
R1
B
B-H
Cu catalyst
2
L
68
69
70
7172
73
74
Scheme 3.8: Proposed outline of species involved in the catalyzed cycle of the copper cat-alyzed HUISGEN cyclization.
In supramolecular chemistry dendrimeric materials are an extremely attractive class
of substances. SHARPLESS and coworkers introduced a highly efficient and prepara-
tively simple approach for the generation of diverse dendritic structures of high purity
and in excellent yield.[224] The topological and electronic similarities of 1,2,3-triazoles
R1 NH
O
R2NNN R2R1
mimicked by
R1 to R2 distance:3.9 Å
R1 to R2 distance:5.0 Å
Figure 3.41: Topological and electronic similarities of amides and 1,2,3-triazoles
91

Chapter 3 Results and Discussion
to amides (figure 3.41), together with the fact, that the susceptibility toward hydrolytic
cleavage is considerably decreased, make them promising candidates for biological
applications.[225–227] Anti-HIV activity,[228] selective β3 adrenergic receptor inhibition,[229]
anti-bacterial activity,[230] and potent anti-histamine activity [231,232] are examples re-
ported in the literature so far.
3.2.1 Synthesis of Novel Dendritic Triazol-Fullerenes
To combine the unique properties of 1,2,3-triazoles and [60]fullerene, we decided
to synthesize triazole dendrimers by the copper(I)-catalyzed ligation of azides and
alkynes and attach these moieties to the C60 core. An additional advantage of the
triazole scaffold is a reasonable solubility in alcohols and even aqueous mixtures and
could be therefore a perfect building block for the structural modification of fullerenes,
to produce charged and primarily neutral water-soluble derivatives. For the topic of
fullerenes in drug design (see chapter 3.1), such neutral compounds could exhibit to
all intents and purposes perspicuous advantages, compared to charged systems. Neu-
tral fullerene derivatives should increase the bioavailability and pharmacokinetics, by
a demanding membrane permeability. Preexaminations on polyether and oligohydroxy
functionalized fullerenes showed, that these polar groups were not sufficient to promote
an appropriate water-solubility.[233] Through the introduction of the triazole functionality,
combined with polar groups like polyethers we expected to obtain a significantly higher
water-solubility.
For the synthesis of the dendritic triazoles, the divergent approach was utilized (scheme
3.9).[234,235] The AB2 monomer unit 77 can be easily synthesized by etherification of
methyl 3,5-dihydroxybenzoate 75 with propargyl bromide, yielding the benzoate 76.
Reduction of the methyl ester using LiAlH4 gave the benzyl alcohol 77 in 83 % yield.
For the attachment to the C60-core a convenient functional group has to be introduced.
In order to deploy the advantages of the BINGEL-HIRSCH reaction (high yields, high re-
action rates) the benzyl alcohol 77 was further allowed to react with malonyl dichloride
to give the benzyl malonate 78, which was purified by flash column chromatography.
For the synthesis of dendritic triazol-fullerene derivatives, in principle two different syn-
92

Chapter 3 Results and Discussion
O O
O OO
O
O
O
OHHO
O O
K2CO3,
18-crown-6
BrOO
O O
LiAlH 2OO
OH
malonyl dichloride,pyridine
OO
OO
O
O O
O
NN
NR1
NNN
R1 N NN
R1
N NN
R1
OO
OO
O
O O
O
NN
NR1
NNN
R1 N NN
R1
N NN
R1
OO
OO
O
O O
O
C60, CBr4,DBU
5 mol % CuSO 4,10 mol % sodium ascorbat
C60, CBr4,DBU
5 mol % CuSO 4,10 mol % sodium ascorbat
NH
O
O
N3 O O
R1:
R2: O O
NH
O
ON3
N3 O O
NH
O
ON3
NH
O
OR1:
R2: O O
75 76
77
78
79
81
81
82
82
83
84
85
86
Pathway A Pathway B
Scheme 3.9: Synthesis of the dendritic triazol-fullerenes 85 and 86 via 1,3-dipolar cycloaddi-tion of azides on alkyne-malonates (Pathway A) and alkyne-monoadducts (Path-way B).
93
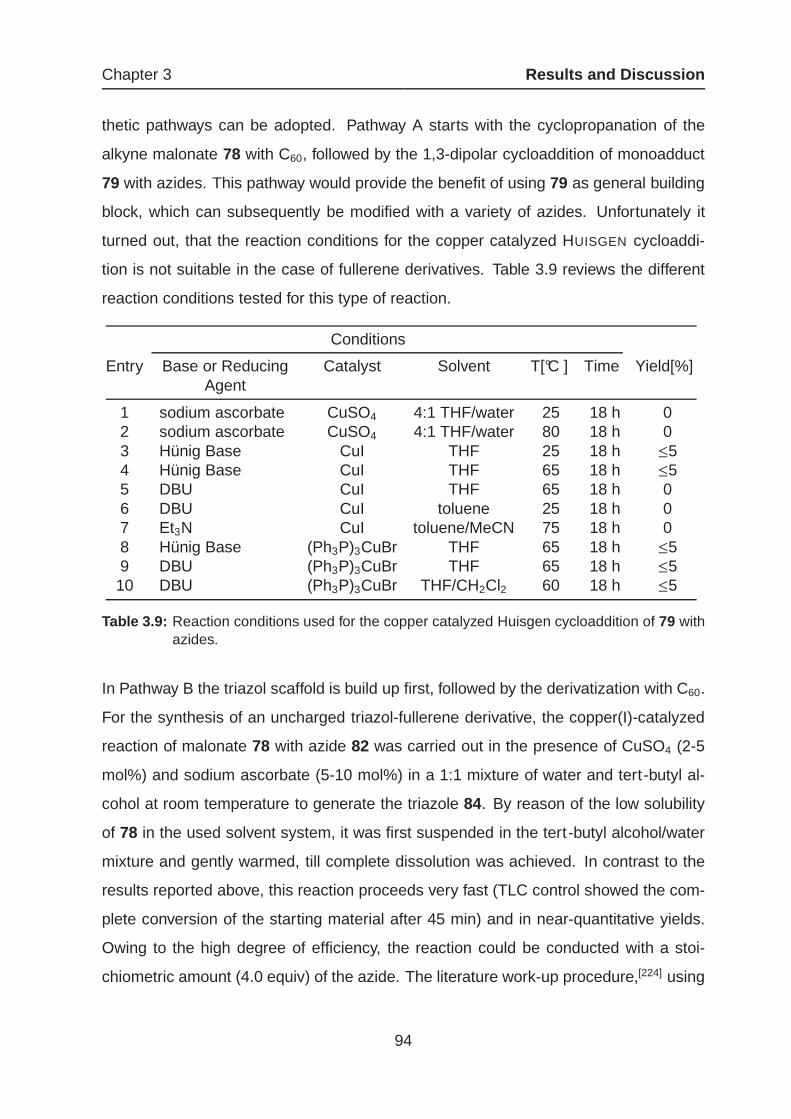
Chapter 3 Results and Discussion
thetic pathways can be adopted. Pathway A starts with the cyclopropanation of the
alkyne malonate 78 with C60, followed by the 1,3-dipolar cycloaddition of monoadduct
79 with azides. This pathway would provide the benefit of using 79 as general building
block, which can subsequently be modified with a variety of azides. Unfortunately it
turned out, that the reaction conditions for the copper catalyzed HUISGEN cycloaddi-
tion is not suitable in the case of fullerene derivatives. Table 3.9 reviews the different
reaction conditions tested for this type of reaction.
Conditions
Entry Base or ReducingAgent
Catalyst Solvent T[°C ] Time Yield[%]
1 sodium ascorbate CuSO4 4:1 THF/water 25 18 h 02 sodium ascorbate CuSO4 4:1 THF/water 80 18 h 03 Hünig Base CuI THF 25 18 h ≤54 Hünig Base CuI THF 65 18 h ≤55 DBU CuI THF 65 18 h 06 DBU CuI toluene 25 18 h 07 Et3N CuI toluene/MeCN 75 18 h 08 Hünig Base (Ph3P)3CuBr THF 65 18 h ≤59 DBU (Ph3P)3CuBr THF 65 18 h ≤510 DBU (Ph3P)3CuBr THF/CH2Cl2 60 18 h ≤5
Table 3.9: Reaction conditions used for the copper catalyzed Huisgen cycloaddition of 79 withazides.
In Pathway B the triazol scaffold is build up first, followed by the derivatization with C60.
For the synthesis of an uncharged triazol-fullerene derivative, the copper(I)-catalyzed
reaction of malonate 78 with azide 82 was carried out in the presence of CuSO4 (2-5
mol%) and sodium ascorbate (5-10 mol%) in a 1:1 mixture of water and tert-butyl al-
cohol at room temperature to generate the triazole 84. By reason of the low solubility
of 78 in the used solvent system, it was first suspended in the tert-butyl alcohol/water
mixture and gently warmed, till complete dissolution was achieved. In contrast to the
results reported above, this reaction proceeds very fast (TLC control showed the com-
plete conversion of the starting material after 45 min) and in near-quantitative yields.
Owing to the high degree of efficiency, the reaction could be conducted with a stoi-
chiometric amount (4.0 equiv) of the azide. The literature work-up procedure,[224] using
94

Chapter 3 Results and Discussion
100
80
60
40
20
0
200 300 400 500 600 700 800 900 1000 1100 1200 1300 1400 1500 1600
1.2E6
9.7E5
7.3E5
4.9E5
2.4E5
0.0E0
201 276621
791
898
1081
391
489
715 121312901384 1539
1143
Figure 3.42: FAB mass spectrum of the crude reaction mixture of 84 after 45 min.
ammonium hydroxide for the removal of trace amounts of copper salts, could not be
adopted in this case, since saponification of the malonic esters occured. Therefore
the crude product was washed several times with a highly diluted ammonium hydrox-
ide/citrate aqueous buffer. Purifications were greatly simplified due to the absence of
side products. In the case of the triazol dendron 84 purification was obtained via flash
column chromatography through a short silica plug. Figure 3.42 shows the FAB-MS of
84 recorded from the crude reaction mixture after 45 min reaction time. The dominat-
ing peak at 1081 belongs to the molecular peak of 84, whereas the missing molecular
peak for the starting material 78 shows the high reaction rates for this conversion.
The 1H NMR spectroscopy is an especially useful tool for illustrating the growth of the
dendrimer generation, as shown from the stacked plots in Figure 3.43. The new res-
onances at 7.7 ppm correspond to the unique triazol protons and are in the typical
range for 1,4-disubstituted 1,2,3-triazoles. Missing of a high-field shifted signal at 7.4
ppm (typical proton signal for 1,5-disubstituted 1,2,3-triazoles), confirms the exclusive
formation of the 1,4-isomer. The splitting of the aromatic protons 3 and 4 with the
integration ratio of 2:1 in the case of the alkyne malonate 78 could not be obtained
in the case of the triazol dendron 84, resulting in a superimposed broad signal. The
signals for the polyether chain are shifted low-field, whereas the signals for the methyl
groups next to the triazol ring show the largest shift. The solubility of dendron 84 was
expectable high in polar solvents, like THF, DMSO and alcoholic solutions as well as
in chlorinated hydrocarbons. Unfortunately the solubility in aromatic organic solvents
95
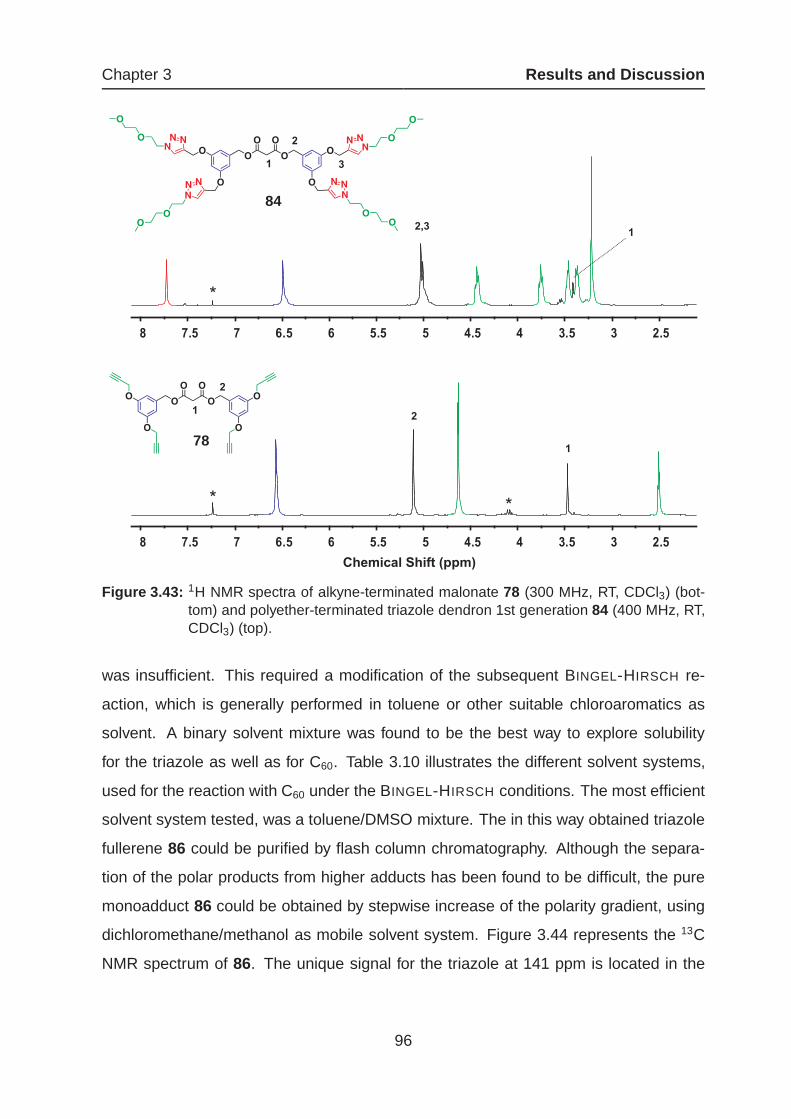
Chapter 3 Results and Discussion
2.533.544.555.566.577.58
2.533.544.555.566.577.58
12,3
1
2
3
1
2
2
1
*
* *
Chemical Shift (ppm)
84
78
Figure 3.43: 1H NMR spectra of alkyne-terminated malonate 78 (300 MHz, RT, CDCl3) (bot-tom) and polyether-terminated triazole dendron 1st generation 84 (400 MHz, RT,CDCl3) (top).
was insufficient. This required a modification of the subsequent BINGEL-HIRSCH re-
action, which is generally performed in toluene or other suitable chloroaromatics as
solvent. A binary solvent mixture was found to be the best way to explore solubility
for the triazole as well as for C60. Table 3.10 illustrates the different solvent systems,
used for the reaction with C60 under the BINGEL-HIRSCH conditions. The most efficient
solvent system tested, was a toluene/DMSO mixture. The in this way obtained triazole
fullerene 86 could be purified by flash column chromatography. Although the separa-
tion of the polar products from higher adducts has been found to be difficult, the pure
monoadduct 86 could be obtained by stepwise increase of the polarity gradient, using
dichloromethane/methanol as mobile solvent system. Figure 3.44 represents the 13C
NMR spectrum of 86. The unique signal for the triazole at 141 ppm is located in the
96

Chapter 3 Results and Discussion
Conditions
Entry Solvent Base Brominating Agent Yield[%]
1 toluene DBU CBr4 ≤52 toluene/CH2Cl2 = 2:1 DBU CBr4 53 toluene/CH2Cl2 = 1:1 DBU CBr4 54 toluene/THF = 3:1 DBU CBr4 85 toluene/DMSO = 3:1 DBU CBr4 32
Table 3.10: Tested binary solvent mixtures for the cyclopropanation of triazole dendron 84 withC60 at rt after 24 h.
range of the fullerene sp2 signals, but could be clearly associated through the C-H cor-
related HETCOR-spectrum. The signal for the methylene group 8 could be detected
at 62.1 ppm and shows the typical high-field shift, caused by the electron-rich triazole
102030405060708090100110120130140150160170 0
Chemical Shift (ppm)
12
3 4 5
6
78
9 1011
1213
14
15
2
6
94
10
5
7
11
15
8
3
14
13
12
sp -3 C
1
sp -2 C
*
86
Figure 3.44: 13C NMR spectrum of polyether-terminated triazole monoadduct 86 (100.5 MHz,RT, CDCl3).
97

Chapter 3 Results and Discussion
OO
OO
O
O O
ONNN
NNN
N NN
N NN
NH
HNO
HNO
NHO
OO
O
O
O
TFA,CH2Cl2
OO
OO
O
O O
ONNN
NNN
N NN
N NN
NH3
NH3
H3N
H3N
4 CF3COO
85
87
Scheme 3.10: Synthesis of charged triazole-fullerene 87 through acidic cleavage of the carba-mate groups in triazole-fullerene 85.
unit. The signals for the peripheral polyether-groups remain nearly unaffected from the
C60-core and show no obvious shifts, compared to the malonate 84. Triazole-fullerene
86 exhibited solubility in alcoholic solutions, THF and chlorinated hydrocarbons. Dis-
solution in water could not be obtained.
Since the solubility of 86 did not fulfill the expectations, we decided to introduce ter-
minal amino groups to elevate the water-solubility. Although there are a variety of dif-
ferent amino-fullerenes known in the literature,[236,237] these compounds combine the
property of only being soluble in acidic aqueous solutions. Therefore the incorpora-
tion of triazole linkages could promote the solubility over an extended pH-range. The
basic chemical 2-aminoethanol was used for the synthesis of the precursor. The nu-
cleophilic amino-functionality of 2-aminoethanol was protected with BOC-anhydride as
carbamate 80,[238] followed by the nucleophilic substitution with sodium azide to ob-
tain the corresponding azide 81.[224] The 1,3-dipolar cycloaddition with the acetylenic
98

Chapter 3 Results and Discussion
malonate 78, as depicted in scheme 3.9 (Pathway B), gave the corresponding triazole
dendron 83 by following the synthetic procedure described for 84. The reaction suc-
ceeded with the same reaction rates and in nearly quantitative yield, as mentioned
before in the case of 84. Triazole dendron 83 could be easily purified by recrystalliza-
tion from methanol. The yield of monoadduct 85 after nucleophilic cyclopropanation
could be again clearly increased by the use of a toluene/DMSO mixture as solvent.
The purification via flash column chromatography emerged to be facile in this case.
Stirring of 85 in a TFA/CH2Cl2-mixture resulted in the complete cleavage of the carba-
mates (scheme 3.10). The adduct 87 could be isolated after reprecipitation in nearly
quantitative yield in the form of the corresponding ammonium salts. The purity of the
precursors and the adducts 85, 87 could be confirmed by the means of FAB-MS, 1H
and 13C NMR-spectroscopy. The observed spectra show the same characteristica as
reported for 86. As expected, the introduction of the triazole groups results in a pH
independent solubility behavior of 87 in aqueous solutions.
250 300 350 400 450 500 550 600
abso
rbance
wavelength [nm]
pH = 4.0
pH = 7.0
pH = 11.0
Figure 3.45: UV/Vis spectra of amino-terminated triazole-fullerene 87 at pH = 4.0 (red), pH =7.0 (grey) and pH = 11.0 (blue).
99

Chapter 3 Results and Discussion
The stacked graphs in figure 3.45 represent the UV/Vis spectra of 87 at different pH
values. In buffered alkaline and neutral solution the typical flattened spectra for ag-
gregated monomers can be observed. Although the slope of the spectrum at pH =
4 does not differ substantially from the spectra at higher pH values, the appearance
of the characteristic monoadduct absorption at about 428 nm indicates the decreased
aggregation tendency in acidic solution. Since all amine groups are completely pro-
tonated at this pH value, this result is consistent with repulsive interactions between
the corresponding ammonium groups, which should clearly disfavor the formation of
supramolecular aggregates.
OO
OH
OO
Cl
OO
Cl
N NN R1
NNNR1
OO
N3
N NN R1
NNNR1
O
OO
O
O
NN N
N NN
O
O
NN
N R1
NN N R1
O
O
NNNR1
N NN
R1
O
O
O
NNN
NNN
O
O
NNNR1
NN
NR1
O
O
NNN
R1
NN N R1
SOCl2
Cu(I), baseR1-N3
NaN3
Cu(I), base
77 88
8990
78
91
Scheme 3.11: Possible synthetic strategy for the preparation of higher generation triazole-dendrons.
100

Chapter 3 Results and Discussion
The near-perfect reliability of the copper(I) catalyzed synthesis of 1,2,3-triazoles from
azides and alkynes should facilitate the synthesis of higher generation triazole den-
drimers. A possible synthetic strategy, as outlined in scheme 3.11, could be the intro-
duction of a chloromethyl group in the AB2 monomer 77, which allows the facile acti-
vation of the focalpoint group during dendrimer construction by the convergent-growth
approach. The attachment of higher generation dendrons to the fullerene core could
enhance the solubility in aqueous solutions and can be considered in future investiga-
tions.
101

Chapter 3 Results and Discussion
3.3 Synthesis of Novel Fullerene-SWCNT Hybrids
Carbon nanotubes (CNTs) are an exciting allotrope of carbon. A single-walled carbon
nanotube is a one-atom thick sheet of graphite (called graphene) rolled up into a seam-
less cylinder with a diameter in the order of a nanometer.
This results in a nanostructure where the length-to-diameter ratio exceeds 10,000.
Such cylindrical carbon molecules have exciting novel physical properties [239–242] that
make them potentially useful in many applications in nanotechnology, electronics, op-
tics and other fields of materials science.[243–249] They exhibit extraordinary strength
and unique electrical properties, and are efficient conductors of heat. Nanotubes are
members of the fullerene structural family, which also includes fullerenes. Whereas
C60 is spherical in shape, a nanotube is a cylindrical and hollow tube shape carbon
material. Their name is derived from their size, since the diameter of a nanotube is
in the order of a few nanometers (approximately 50,000 times smaller than the width
of a human hair), while they can be up to several millimeters in length. There are two
main types of nanotubes: single-walled carbon nanotubes (SWCNTs) and multi-walled
carbon nanotubes (MWCNTs). The nature of the bonding of a nanotube is described
by applied quantum chemistry, specifically orbital hybridization. The chemical bond-
ing of nanotubes is composed entirely of sp2 bonds, similar to those of graphite. This
bonding structure, which is stronger than the sp3 bonds found in diamond, provides
the molecules with their unique strength. Nanotubes naturally align themselves into
"ropes" held together by van der Waals forces. Under high pressure, nanotubes can
merge together, trading some sp2 bonds for sp3 bonds, giving great possibility for pro-
ducing strong, unlimited-length wires through high-pressure nanotube linking.[250]
In the last years the functionalization of carbon nanotubes became a topic of interests
for chemists and material researchers. Due to the insolubility of carbon nanotubes in
both water and organic solvents, the enhancement of the solubility [251] is a major task
for the application in materials and devices. Additionally the chemical functionalization
allows the unique properties of SWCNT´s to be coupled to those of other types of mate-
rials. Figure 3.46 shows the four different approaches for the chemical functionalization
102

Chapter 3 Results and Discussion
SWCNT
A
B
CD
Figure 3.46: Schematic representation of the different possibilities for the functionalization ofcarbon nanotubes. A) endohedral functionalization; B) defect functionalization;C) non covalent functionalization; D) covalent sidewall functionalization.
of carbon nanotubes such as, endohedral functionalization (A), defect functionalization
(B), non covalent functionalization (C) and covalent sidewall functionalization (D). The
following chapters describe the synthesis and characterzation of novel [60]fullerene-
SWCNT hybrids via the covalent and non-covalent pathway.
3.3.1 Covalent Sidewall Functionalization of SWCNT’s with a
Fullerene-Monocarboxylic Acid Derivative
With the chemical sidewall functionalization it is possible to link a broad variety of func-
tional groups onto the sidewall of the nanotube. It is accompanied by a change of
hybridization from sp2 to sp3 and a simultaneous loss of conjugation. Due to the lower
reactivity of CNT’s compared to fullerenes,[252] the functionalization requires very re-
103

Chapter 3 Results and Discussion
active reagents, like elemental fluorine,[253] GRIGNARD-reagents,[254] radicals [255] or
nitrenes.[256] Recently the HIRSCH group succeeded in the synthesis of hydroxy func-
tionalized SWCNT’s [257] through reductive alkylation. For this purpose, the SWCNTs
were first reduced using sodium and naphthalene in absolute THF. The following alky-
lation was accomplished by the use of 2-(3-bromopropoxy)-tetrahydropyran as alkyl
halide. The cleavage of the ether moieties could be obtained with a THF/HClconc. solu-
tion in quantitative yields. In order to functionalize the remaining hydroxy groups with
suitable fullerene-derivatives, we had to apply methods leading to a complete conver-
sion, since the separation of a reaction mixture is impossible because of the presence
of multiple addends on a molecular SWCNT. We therefore decided to synthesize a
sterically unhindered fullerene monoadduct, which holds a carboxylic acid functional-
ity that can easily be transferred to the corresponding acid chloride. In scheme 3.12
the synthetic pathway for the carboxyfullerene 94 is shown. To avoid cross-linking
between the fullerene and the SWCNTs the asymmetric malonate 92 was obtained
by reaction of 22 with methylmalonyl chloride. The following cyclopropanation via the
BINGEL/HIRSCH reaction yielded monoadduct 93 in high yields. Deprotection following
standard procedures with TFA in toluene gave 94 in quantitative yields. By reason of
the impossibility to isolate the corresponding acyl chloride, it was generated in situ,
with the use of SOCl2 in a mixture of TCE and CHCl3 at 60°C followed by the addition
of the hydroxy-SWCNTs [257] in TCE. The reaction mixture was stirred for additional 4
d at rt. After evaporation of the solvents under reduced pressure, the crude product 95
was resuspended in a mixture of 50 ml water and 50 ml ethanol. The suspension was
filtered through a 0.2 µm PTFE membrane filter, washed with ethanol and THF. The re-
sulting black solid was dried in a vacuum oven at 50°C overnig ht. The characterization
of 95 by standard spectroscopic methods fails in this case, therefore X-ray photoelec-
tron spectroscopy (XPS) [258,259] was used to investigate the atomic composition of the
material and to determine the degree of functionalization.
The measured XPS spectra showed a significant increase of the amount of oxygen in
the survey spectrum of 95 compared to the starting material. This increase can be as-
signed to the introduction of the fullerene-malonate moiety by esterification. Recorded
104

Chapter 3 Results and Discussion
HO O
O pyridine
MeO Cl
OO
O O
OO
OO
OO
OOO
OOO
OOOH
O
C60,CBr4, DBU
TFA
OO
O O
O
O
1. SOCl2, TCE/CHCl3, 60 °C2. Hydroxy-SWCNT, rt
22 92
9394
95
Scheme 3.12: Synthetic pathway for the fullerene/SWCNT-hybrid 95.
TEM images of the fullerene-SWCNTs allowed the direct proof of the presence of
fullerene molecules on the sidewalls of the SWCNT’s.[257] Figure 3.47 represents the
comparison of the hydroxy-SWCNTs and the fullerene-SWCNT-hybrids, showing the
unique structure of the new compound 95. Through the combination of the outstanding
properties of fullerenes and SWCNTs, this new compound may show novel electron
transfer properties and would be a interesting candidate for new material applications.
105

Chapter 3 Results and Discussion
(a) (b)
Figure 3.47: TEM images of 3-hydroxypropyl-SWCNTs (a) and fullerene-SWCNT-hybrid 95(b).
3.3.2 Non-Covalent Functionalization of SWCNT’s with a
Fullerene-Pyrene Dyad
The extended π-system of the carbon nanotubes sidewall allows the functionalization
with various groups through π-π-stacking interactions. This has for example a special
interest for the non destructive purification process of carbon nanotubes by increased
solubilization. In this context the pyrene-group showed to be a promising candidate
for the effective assembling to the graphitic sidewall via π-stacking interactions.[260,261]
The pyrene moiety was therefore specifically designed in such a way that a variety of
molecules with intriguing properties could be further coupled via amide linkages.[260]
With this aproach, the immobilization of proteins, DNA, and other smaller biomolecules
on the side-walls of SWNT’s could be achieved. Similarly, pyrene-carrying ammonium
ions were used to help solubilize CNT’s in water. This chapter reports on a new type
of [60]fullerene-hybridized SWCNT material, in which a symmetrically covalently linked
[60]fullerene-pyrene conjugate was immobilized onto the surface of SWCNTs by using
a noncovalent method. The preparation of the conjugate 99 is depicted in scheme 3.13
106

Chapter 3 Results and Discussion
O
OH
O
OOH
O
O OO
OO
O O
O
O OO
OO
O O
EDC, DMAP
HO OH
Cl
O
Cl
O
pyridine
C60,CBr4, DBU
96 97
98
99
Scheme 3.13: Synthesis of the pyrene-fullerene-conjugate 99.
and starts with the reaction of 1-pyrenebutyric acid 96 with 1,2-ethanediol. The incor-
poration of this C2-unit should increase the flexibility of the conjugate and therefore a
higher reciprocal effect with the sidewall of the SWCNT’s is expected. The esterifica-
tion of the alcohol 97 with malonyl dichloride yielded the symmetric malonate 98 in 67
% yield. Coupling of the malonate 98 to C60 was sucessfully obtained by the use CBr4
and DBU. Separation of the monoadduct 99 from higher adducts was accomplished
by flash column chromatography using toluene/ethyl acetate = 10/1 to 8/1 as eluent.
To prevent any impurities (e.g remaining silica) the conjugate 99 was further purified
by HPLC (Nucleosil 5 µm) using CH2Cl2/methanol = 98/2 as eluent. Monoadduct 99
could be characterized by the standard methods. In figure 3.48 the 1H-NMR spectra
is shown. The protons of the C2-spacer unit (marked green) are shifted lowfield, due
to the influence of the fullerene core. The remaining signals could be detected in al-
most unaltered position in the spectrum. The zoom into the pyrene region of 98 and
107

Chapter 3 Results and Discussion
1.522.533.544.555.566.577.588.5
1.522.533.544.555.566.577.588.5
Chemical Shift (ppm)
1
b
aa
b
a b
b
a a
b
a b
1
*
*
*
98
99
(a)
7.657.757.857.958.058.158.25
Chemical Shift (ppm)
7.657.757.857.958.058.158.25
Chemical Shift (ppm)
(b)
Figure 3.48: (a) 1H-NMR spectra of pyrene-malonate 98 (400 MHz, RT, CDCl3) and pyrene-fullerene-conjugate 99 (400 MHz, RT, CDCl3); (b) zoom into the pyrene region of98 (left) and 99 (right).
108

Chapter 3 Results and Discussion
99 (see figure 3.48 (b)) show no significant changes in the observed chemical shifts,
and indicates no significant backbinding of the pyrene-units to the fullerene-core via
π-π-interactions, which can be explained on the basis of the spherical shape of C60.
Prior investigations on the interaction of a water-soluble pyrene derivative [262] with C60
restated this result. For the cylindrical shape of the SWCNTs we expected much higher
interactions. The UV/Vis spectrum of 99 exhibits the characteristic slope of a pyrene
derivative. The characteristic absorptions of the C60 moiety significantly increases the
intensity in the UV region between 260 and 400 nm. The absorption maximum at 425
nm shows the characteristic feature of a C60 monoadduct (figure 3.49).
300 350 400 450 500
0
20
40
60
80
100
120
140
160
180
200
ε[M
-1cm
-1x
10
3]
wavelength [nm]
9899
99
98
Figure 3.49: UV/Vis spectra of bispyrenylmalonate 98 and bispyrenylmonoadduct 99.
For the preparation of the fullerene-SWCNT-hybrid, a solution of 99 in 1,2-dichloro-
ethane (DCE) was added to a suspension of SWCNT’s in DCE. The suspension was
stirred for several minutes, till decolorization of the supernatant occurred (for experi-
mental details see appendix B). The disappearance of the typical reddish color for the
109

Chapter 3 Results and Discussion
Figure 3.50: Images of conjugate 99 in DCE (left), after addition of SWCNT’s in DCE andstirring (right) and after sedimentation of the suspension (middle).
monoadduct 99 gave the first indication of the successful immobilization of conjugate
99 on the SWCNT surface (figure 3.50). To verify the degree of functionalization, we
decided to use the Scanning Tunneling Microscopy (STM) technique.[263,264] The STM
has been shown to be a valuable tool for viewing the surface morphology down to
the atomic scale. Against the expectations, the STM measurements indicate, that the
SWCNTs reveal very low amount of functional moieties. Figure 3.51 left shows an
unfunctionalized SWCNT. This individual SWCNT crosses a monoatomic step of the
substrate surface. As expected for the π-stacking functionalization, the SWCNT side-
wall remained intact, there are no additional defects introduced by the functionaliza-
tion. The herringbone reconstruction appearing on the substrate surface surrounding
this SWCNT indicates that the surface did not suffer from contamination, but remained
mainly clean on the atomic scale. We expect this result as the preparation method is
carried out in ultra high vacuum. Due to their little corrugation, the reconstruction is
not visible in the current color scale. However, no bundles of SWCNTs were observed,
which on the other hand indicates a debundeling effect through the functionalization
process, as well as the preparation method via dry transfer printing which is believed
to peel the SWCNTs out of bundles. The relatively big particle reflects the content of
residual catalyst material from the synthesis process.
Figure 3.51 right shows an STM image of typically protruding functional molecules on
110

Chapter 3 Results and Discussion
5nm
5nm
2.0
1.5
1.0
0.5
0.0
Heig
ht [n
m]
35302520151050
Position [nm]
Figure 3.51: Left: STM topograph of an individual unfunctionalized SWCNT. The surroundingAu surface is not further contaminated, the herringbone reconstruction conserved.Right top: STM image of a functionalized SWCNT. The fullerene derivative 99 isplaced on top of the SWCNT. Right bottom: Line profile over the SWCNT and themoiety.
the SWCNTs. The height of this protrusion with respect to the SWCNT reaches up
to 9 Å. The diameter of the moiety appears 5 nm which seems to be surprisingly big.
But as the STM topography resembles the convolution of sample and tip geometry, the
lateral size thus is expected to be overestimated. An additional enlarging effect is that
the functional molecule could rotate around the SWCNT during sampling. This might
be induced by the attractive VAN DER WAALS interaction with the STM tip. As conju-
gate 99 is bound to the SWCNT via the relatively weak π-stacking interaction, this force
could be sufficient to make the molecule hopping to neighboring SWCNT lattice sites.
Therefore, the lateral dimensions of the moiety clearly are difficult for quantitative iden-
tification purposes. We can only conclude rotational symmetry reliably. Fortunately, the
height information does not suffer from the mentioned effects and a height of 9 Å nicely
fits the assumption of a π-stacking bound 99 molecule on top of the SWCNT (figure
3.52).
But although the geometry fits nicely, of course these protrusions have not necessarily
to be fullerene-derivatives. Catalyst particles could fit the size as well and are present
in every SWCNT material. For further identifaction, it is compulsory to have a look on
the molecular substructure of the moieties. Figure 3.53 shows the atomically resolved
111

Chapter 3 Results and Discussion
Figure 3.52: Representation of the possible π complex between 99 and the SWCNT.
3nm
(a)
3nm
(b)
Figure 3.53: STM topography (a) and current (b) image of 99 functionalized SWCNT’s. TheSWCNT configuration is irregular, there are various noisy regions indicating mo-bile particles or insufficiently stabilized SWCNT’s. Marked is a 99 molecule whichis suspected to functionalize the SWCNT.
STM topography (a) and current image (b) of functionalized SWCNT’s. Here several
SWCNTs are alinged near each other. Only one relatively dense layer of SWCNT’s
appears, it is no bundle as it might appear on the first look. The marked monoadduct
99 has flipped beside so that it is not only bound to the corresponding SWCNT but also
is mechanically supported by the neighboring SWCNT as well as by the substrate sur-
112

Chapter 3 Results and Discussion
3
2
1
0
dI/
dV
[a
rb.
un
its]
10-1
Bias Voltage [V]
SWCNT PTFE @ SWCNT
Figure 3.54: STS measurement on 99 functionalized SWCNT.
face. This allows to resolve it atomically. The revealed rich substructure seems to be
nearly spherical and resembles that of fullerenes. Although 99 is a fullerene derivate,
it has not necessarily to reveal fullerene substructure. One clearly can see something
which can be associated with the additional pyrene sidegroup pointing to the SWCNT.
To verify that a chemical interaction is formed and the 99 molecules are not only lo-
cated occasionally nearby the SWCNTs, scanning tunneling spectroscopy (STS) [265]
measurements on the functional molecules of which figure 3.54 shows an example,
were carried out. Three different measuring points were performed at one time. The
surface state of Au(111) has been measured to line up the measuring setup, mainly the
tip configuration concerning adsorbates and particles. Figure 3.54 illustrates the mea-
surements of the DOS (Electronic Density of States) of the SWCNT a few nm beside
the moiety (solid line) and on the functional molecule itself (dotted line). The SWCNT
DOS reflects a metallic SWCNT and even reveals the pseudo gap arising from the
bending of the graphene states. The VAN HOOVE singularities however appear more
steplike. In the case of the functional molecule, a variety of additional states arise and
also the underlying SWCNT DOS is modified.
After having proven that indeed functionalization of the SWCNT’s occured, it had to
be tested, whether functional molecules can be removed from the surface. Interest-
ingly, the substrate surface nearby the SWCNT’s not always but usually reveals the
coverage with removed functional molecules of which one is marked with an arrow in
113

Chapter 3 Results and Discussion
5nm
(a) (b)
Figure 3.55: STM topography (a) and current (b) image of connected SWCNT fragments and99 molecules on the neighboring substrate surface.
10nm
(a)
3nm
(b)
Figure 3.56: (a): C60 nanowire at a relatively large scan size. The unaffected monoatomicgold step edges indicate clearly, that the nanowires assembly is not substratemediated. (b): close-up of a C60 nanowire. Little interruptions makes evident thatthere is no nanotube underneath the fullerene chain.
figure 3.55. Especially in the current image, there are several fullerene-like spheres
which seem to stand on two sail-like aligned and flat protrusions pointing towards the
gold surface. The SWCNT itself seem to consist of several fragments. Surprisingly,
the removed 99 molecules seemed to be in a kind preorientated, using the SWCNT as
a template. Figure 3.56 shows this result. On the left, a large section of a nanowire
114

Chapter 3 Results and Discussion
is shown. The length of those wires reach up to 300 nm or even more (larger scans
were not performed). As the surface area provides a variety of different step edges
and different orientations of them, it can be excluded that the wire simply follows one
of the step edges. This can also be seen from the fact that the wire is not exactly
straight but somehow bent. The typical height of those wires is in the range of 4 Å,
so we can exclude the presence of a SWCNT under the fullerenes to possibly cause
the wirelike structure. Figure 3.56 right shows a close up of a nanowire. Clearly the
wire consists of aligned spherical particles, the functional fullerenes. The height of the
wire measures 0.4 nm. This can be attributed to the lower transmission coefficient of
electron tunneling through the wire. Nearby the wire, there are contaminations of the
gold surface visible, e.g. residual solvents or mechanically crushed 99 molecules could
be possible.
To handle, operate and manipulate nanoscopic devices, selforganization of molecular
modules on solid surfaces and interfaces, is one of the main challenges in nanotech-
nology and nanoscience. The results demonstrate the ability to fine-tune interfacial
and intermolecular interactions, which permits the building of well-defined, ordered
structures over large areas, which could be visualized and characterized by scanning
tunneling spectroscopy. By using SWCNTs as templates, it is possible to transfer preor-
ganized fullerene structures to suitable substrates in well-defined strands of molecules
by stamping-like printing onto a surface. These one-dimensional nanostructures of or-
ganic semiconductors can be attractive candidates as active components in electronic
nanodevices.
115

Chapter 3 Results and Discussion
3.4 Supramolecular Approach for the Formation of
C60-Bisadducts
Chemical functionalization of fullerenes is the first step to investigate their intriguing
properties and applications. With higher adducts of C60 it is possible to generate
unique architectures, in which the fullerene core can be modified with different func-
tional groups. By controlling the degree of addition and the addition patterns, it is
possible to adjust the physical and chemical properties directly to possible applications
in nanomaterials. In contrast to the mono-functionalization of C60, which is straight-
forward, the multiple functionalization is afflicted with many problems. The stepwise
multiple additions to C60 yield a mixture of geometric isomers with low yield and low
selectivity for respective isomers, and tedious chromatographic separations are re-
quired. A general solution to overcome these problems is to find a generic linker which
can direct the following additions to expected positions on the C60 surface. DIEDERICH
and coworkers reported the first important approach in this content, by the tether di-
rected functionalization of C60 with open-chain bismalonates. With the subsequent
cyclopropanation of these sterically rigid and predefined malonates with C60, a broad
variety of different bisadducts with defined addition pattern have been synthesized so
far.[97,266–268] (For a detailed description of the possible regioisomeric bisadducts see
[68,269]). Although this method represents a particularly suitable method for the regio-
selective cyclopropanation, it takes disadvantage from poor yields, extensive synthetic
procedures and the lack of possible further functionalization. To circumvent this diffi-
culties, HIRSCH and coworkers introduced a modified concept. Thereby macrocyclic
cyclo-[n]-malonates are used, to build the suitable tether. Variation of the length of the
flexible alkylchain between the malonate groups allows to tune the regioselectivity of
the tether. In contrast to the method described above, the regioselectivity is not based
on steric preorganization, but on the avoidance of unequal strain in the alkyl chains.
Within this approach it was possible to synthesize a diversity of highly regioselective
bis-, tris- and tetrakisadducts of C60.[132,269–271] Although the selectivity and yields of
the cyclopropanation with this cyclic tethers is more than satisfying, the corresponding
116

Chapter 3 Results and Discussion
OOO O
OO O O
Tether
OO
O O
O
O
O
O
Tether
O
O
O
O
(CH2)n
(CH2)n O
O
O
O
O
O
O
O
O
O
O
O
(CH2)n
(CH2)n
C60, DBU, CBr 4
toluene
C60, DBU, CBr 4
toluene
Scheme 3.14: Regioselective bisfunctionalization of C60 with open-chain malonate tethers(top) and macrocyclic malonate tethers (bottom).
malonates are often hardly accessible.
The standard method for their preparation is the reaction of malonyl dichloride with
alkanediols. This reaction can only be accomplished by the dropwise addition of mal-
onyl dichloride to a highly diluted solution of the diol.[132] Nevertheless side products
like higher macrocycles and polymeric material require an extensive chromatographic
purification process, which is aggravated by the similar polarities of product and side
products. The following chapter describes a new approach for multiple C60 adducts,
where the malonates are linked via supramolecular interactions.
3.4.1 Metallomacrocycles as Tethers for Regioselective
Cyclopropanation
The strategy to ligate metal ions as building blocks for the construction of cyclic mal-
onate structures is depicted in scheme 3.15. The symmetric malonate 100 can be
easily obtained by reaction of malonyl dichloride with an 2-fold excess of the alco-
hol 22. Deprotection of the tert-butyl esters with TFA in chloroform yielded the dicar-
boxylic malonate 101 in quantitative yield. For the preparation of the bridged dinuclear
117

Chapter 3 Results and Discussion
Cl Cl
O O
O O
O OOR
O
RO
O
R = tBuR = H
TFA
pyridine HO O
O
M: metalL: co-ligand
O O
O OO
OO
O O
O OO
O
O
O
M L
O
ML
22
100101
Scheme 3.15: Concept for the metal ion mediated cyclization of dicarboxylic malonates.
metal complexes, the dicarboxylate ligand 101 was generally treated with an equimolar
amount of the metal salt in the presence of a suitable co-ligand. The following table
3.11 summarizes the different reagents and reaction conditions used for this type of
dimerization.
Entry Metal salt Co-ligand Reaction conditions
A Zn(OH)2 none hexane, reflux, 3 hB MnCl2 · 4 H2O none H2O, pH = 5.5, reflux, 8 hC CuCl2 · 2 H2O 1,10-phenanthroline
monohydrateMeOH/H2O = 1/1, 65°C , 3days
Table 3.11: Reagents and reaction conditions for the metallomacrocyclization of dicarboxylicmalonate 101.
118

Chapter 3 Results and Discussion
Complexation with Zn [272]
For the complexation with Zn, Zn(OH)2 was freshly precipitated from a ZnSO4 solution
with NaOH, extensively washed (to avoid any NaOH residues, which could lead to the
decomposition of the malonesters) and dried. Zn(OH)2 was further refluxed with an
equimolar amount of 101 in hexane. After 3 h a transparent solution was obtained.
The reaction mixture was allowed slowly to cool to room temperature, whereas the
precipitation of a white solid could be observed. The solid material was filtered off,
washed with hexane and dried in vacuum. Although the dissolution of the malonate
indicates the neutralization reaction between Zn(OH)2 and 101, IR and NMR studies
showed no evidence for the formation of the bis(carboxylato)zinc complex. However,
most of the precipitate was identified as unconverted starting material 101.
Complexation with Mn [273]
For the complexation with Mn, MnCl2 · 4 H2O was refluxed with an equimolar amount
of 101 in destilled water at pH = 5.5 for 8 h. The reaction mixture was allowed to cool to
room temperature, whereas no precipitation of solid material could be observed. Evap-
oration of the solvent and subsequent IR and NMR studies again showed no evidence
for the formation of the bis(carboxylato)manganese complex.
Complexation with Cu [274]
For the complexation with Cu, CuCl2 · 2 H2O was added to an equimolar amount of
101 in a mixture of MeOH/H2O = 1/1. Treatment with an equimolar amount of 1,10-
phenanthroline monohydrate (C12H8N2 · H2O) as co-ligand resulted in a blue solution,
which was filtered. Na2CO3 (1 M) was dropwise added, leading to a fine, pale-blue
precipitate which was separated. The filtrate (pH = 5.5) was kept at 65°C for 3 days
and after cooling to room temperature, blue crystals could be obtained. X-ray analysis
of the crystals showed not the formation of the azelaato-bridged dinuclear copper(II)
complex. Instead of, the aqua-(malonato-O,O’)-(1,10-phenanthroline-N,N’)-copper(II)-
complex 102 was identified, indicating the metal induced hydrolysis of the malonesters
(figure 3.57). The X-ray sample contained two different crystal species, which ex-
119

Chapter 3 Results and Discussion
Figure 3.57: Crystal structure of (malonato-O,O’)-(1,10-phenanthroline-N,N’)-copper(II) 102(thermal ellipsoids at 50 % probability level).
hibited the complex 102 in the two different packing motives 2[Cu(H2O)(mal)(phen)]
[Cu(mal)(phen)] x 11 H2O and [Cu(H2O)(mal)(phen)] x 1.5 H2O.[275]
3.4.2 Hydrogen-bonded Dimers as Tethers for Regioselectiv e
Cyclopropanation
Hydrogen bonding displays a very attractive interaction for the formation and organiza-
tion of supramolecular architectures. One of the most astonishing example is offered by
nature, where multiple hydrogen bonds and hydrophobic interactions form the excep-
tionally stable double-stranded DNA. Such biological examples have provided a power-
ful incentive for researchers to explore molecular association more broadly, and a vari-
ety of hydrogen bonding motifs were discovered by different groups so far. In figure 3.58
the most relevant hydrogen-bonding arrays are summarized. In the case of the simplest
donor-acceptor (DA) arrays, association constants (K a) of around 10 M−1 could be ob-
served in CHCl3. Subsequent increase of the number of donor-acceptor units to triple
hydrogen bonding and quadruple hydrogen bonding (figure 3.58 b) and c)), significantly
increases the K a values to 103 M−1 and 105 M−1 (CHCl3), respectively.[276–278] If hydro-
gen bonds are combined with other noncovalent interactions, such as complementary
amidinium-carboxylate bridges (figure 3.58 a)), association constants greater than 106
120

Chapter 3 Results and Discussion
R2N N
R2R1
O O
R3
H HN
N
OBu
O
H NN
N
N
Et
NH
H
NH
H
N
NNR1
O
N O
NR2H
HH
N
N N R1
O
NO
NR2 H
H H NO
NO
N
O
H
H
R2
ON
H
NN
H
O
ON
H
NN
H
O
R1
a)b)
c)
d)
Figure 3.58: Relevant hydrogen-bonding motifs with high association constants.
M−1 can be obtained.[279,280] One of the most stable arrays discovered so far, is the
HAMILTON motif (figure 3.58 d)), where a receptor, which contains 2,6-diaminopyridine
units could efficiently bind targets like barbiturates and their derivatives through six
hydrogen bonds.[281] This motif was assimilated by HIRSCH and coworkers for the self-
assembly of extremely stable supramolecular structures.[282–284]
By taking advantage of the unique features of hydrogen-bonding we decided to uti-
lize them for the building of novel tether systems for multiple functionalization of C60.
Thereby the tether is primarily formed by dimerization through multiple hydrogen-
bonding, followed by the subsequent reaction with C60 (see scheme 3.16). The de-
pendence of the association constant upon the solvent, allows directly to influence the
hydrogen bond equilibria between the monomeric and dimeric constitution. As bond-
ing motif we decided to use a system, similar to the array described in figure 3.58
c), composed from self-complementary 2-ureido-4-pyrimidinones units which have a
121

Chapter 3 Results and Discussion
R1O
O
OO
R1O
O
OO
(CH2)n
(CH2)nR1
O
O O
O (CH2)n
O
O
O
O
O
O
O
O
(CH2)n
(CH2)n
R1
R1
C60,CBr4, DBU
Scheme 3.16: Schematic representation for the building of hydrogen-bonded tethers.
donor donor acceptor acceptor (DDAA) hydrogen bonding motif. This motif gives rise
to a very high dimerization constant (K d ≥ 6 · 10 M7), as a result of attractive secondary
interactions.[285,286]
To overcome solubility problems of the sparely soluble ureidopyrimidines, the asym-
metric carboxy malonate 27 was used. The lipophilic alkyl chain in 27 should provide
sufficient solubility in most organic solvents. The carboxylic acid 27 was converted into
the corresponding acid chloride 103, after which reaction with sodium azide yielded the
acyl azide 104. Heating 104 in the presence of 6-methylisocytosine at 85°C afforded
106 in good yield (76 %, four steps, starting from 27 via isocyanate 105, scheme 3.17).
122

Chapter 3 Results and Discussion
O O
O OR2
R1
R1 = COOH
R1 = COCl
R1 = CON3
100 °C,pyridine
N
NH2NHO
O O
O OR2
NH
O
NH
N
HN
O
O O
O OR2
N
O
N N
HN
O
OO
OOR2N
O
NN
NH
O
H HH H
O
O
O
O
O
O
O
O
NO
N N
HN
O
NO
NN
NH
O
H HH H
R2
R2O
O
O
O
O
O
O
O
R2
R2
N O
N
N
NH
O
H
H
N
O NNHN
O
HH
C60,I2, DBU
R1 = NCO
R2 = (CH2)17CH3
NaN3
C2Cl2O2
∆
27
103
104
105
106
106
107107
Scheme 3.17: Reaction sequence for bisadduct 107 via dimeric hydrogen-bonded malonate106.
The 1H NMR shows the definite evidence for the dimeric structure of 106. In CDCl3
large lowfield shifts were found for the protons of the hydrogen-bonding motif. The
signals of the urea NH protons are observed at δ = 11.79 and 10.31 ppm and the
intramolecularly chelated pyrimidinone NH at δ = 13.23 ppm. This observation is
123
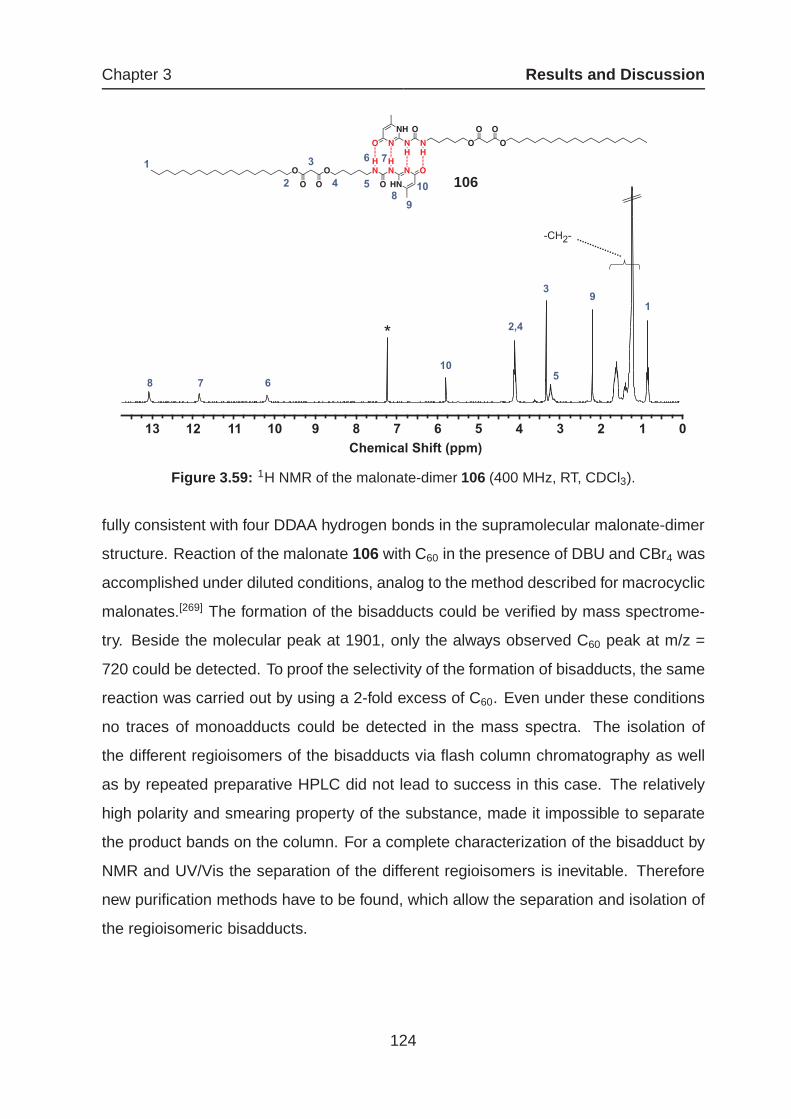
Chapter 3 Results and Discussion
Chemical Shift (ppm)
1
2
3
4 5
6 7
89
10
19
5
3
2,4
10
8 7 6
-CH -2
*
012345678910111213
106
Figure 3.59: 1H NMR of the malonate-dimer 106 (400 MHz, RT, CDCl3).
fully consistent with four DDAA hydrogen bonds in the supramolecular malonate-dimer
structure. Reaction of the malonate 106 with C60 in the presence of DBU and CBr4 was
accomplished under diluted conditions, analog to the method described for macrocyclic
malonates.[269] The formation of the bisadducts could be verified by mass spectrome-
try. Beside the molecular peak at 1901, only the always observed C60 peak at m/z =
720 could be detected. To proof the selectivity of the formation of bisadducts, the same
reaction was carried out by using a 2-fold excess of C60. Even under these conditions
no traces of monoadducts could be detected in the mass spectra. The isolation of
the different regioisomers of the bisadducts via flash column chromatography as well
as by repeated preparative HPLC did not lead to success in this case. The relatively
high polarity and smearing property of the substance, made it impossible to separate
the product bands on the column. For a complete characterization of the bisadduct by
NMR and UV/Vis the separation of the different regioisomers is inevitable. Therefore
new purification methods have to be found, which allow the separation and isolation of
the regioisomeric bisadducts.
124

Chapter 3 Results and Discussion
3.5 Synthesis of Novel Multiple Fullerene Arrays
Consisting of Mixed C 60-Hexakisadduct Units
Multiple additions to the C60 core enable the facile access to highly symmetrical, poly-
functional macromolecules. By controlling the addition pattern, these three-dimensional
nanostructures with defined symmetry and shape could target interesting topics in
supramolecular chemistry and advanced materials. One of the most important re-
actions in this context is the template mediated sixth-fold cyclopropanation using 9,10-
dimethylanthracene (DMA).[287,288] The thermal reversibility of the Diels-Alder reaction
between C60 and 9,10-dimethylanthracene creates an equilibrium mixture containing
e,e-C60(DMA)2 and e,e,e-C60(DMA)3 templates that direct subsequent cycloadditions
to the remaining equatorial sites.[289] The method allows the synthesis of a number of
BINGEL-type Th symmetrical hexakisadducts, C66(COOR)12, in yields up to 50 %. This
mediation strategy was also adopted for the synthesis of mixed hexakisadducts. These
structures contain two different types of addends in octahedral positions, which allows
the synthesis of new bifunctional macromolecules, with unique properties.[290]
In principle two different synthetic strategies can be used for the synthesis of [5,1]-
hexakisadducts. The best accessible one starts with the synthesis of the monoadduct
108, followed by the formation of the hexakisadduct 109 via the DMA templated re-
action sequence (Pathway A, sheme 3.18).[98,291,292] The alternative route starts with
the synthesis of the triazoline monoadduct 111, followed by the template-mediated cy-
clopropanation to the mixed triazoline hexakisadduct 112. The hexakisadduct could
further be converted into the pentakisadduct 113, by thermally induced [3+2] cyclore-
version of the labile triazoline group. The following cyclopropanation to the mixed
hexakisadducts proceeds with quantitative regioselectivity and high yields. The ma-
jor drawback of this strategy is the thermal decomposition of 112, which also leads to
the formation of the open [5,6]- and closed [6,6]-azahomofullerene as side products
by expulsion of a nitrogen atom. The overall yield for 113 is comparatively low and
purification process of the involved intermediates is laborious.[293]
Substitution of the malonate H2(COOR1)2 with a bis(functional) addend, makes it pos-
125

Chapter 3 Results and Discussion
R1O O
O OR1
N NN MEM
OO
O O
R2R2
R2
R2R2
R2
O
O O
O
O
O
O
O
O
OO
R2
R2
O O
O
R1 R1
O
OO
O
R2
R2
O
O
OO
O O
R2R2
R2
R2R2
R2
O
O O
O
O
O
O
O
O
OO
R2
R2O
OO
O
R2
R2O
N NN
MEM
OO
O O
R2R2
R2
R2R2
R2
O
O O
O
O
O
O
O
O
OO
R2
R2O
OO
O
R2
R2O
O O N3
DMA,H2C(COOR2)2
CBr4, DBU
∆
H2C(COOR1)2,CBr4, DBU
DMA,H2C(COOR2)2,CBr4, DBU
H2C(COOR1)2,CBr4, DBU
Pathway A Pathway B
110
108 111
112
113109
Scheme 3.18: Different strategies for the synthesis of [5,1]-hexakisadducts, using templatemediation with DMA.
sible to achieve fullerene-rich molecular architectures, by the direct coupling of two C60
units. With this method HIRSCH and coworkers recently synthesized a water soluble
bis(fullerene) icosacation, where the fullerenes are connected via a chiral C4-spacer
unit.[294] Since the synthesis of this dumbbell shaped molecule starts with the gener-
ation of a bis(fullerene), followed by the completion of the octahedral addition pattern
126

Chapter 3 Results and Discussion
O
OO
OO
O
O
O
R1
R1
O
OOOR1
R1
O
OOOR1
R1
O
O
OO
R1R1
O
O
OO
R1
R1
O
OO
O O
O
O
O
R1
R1
O
O OO R1
R1
O
O OO R1
R1
O
O
OO
R1R1
O
O
OO
R1
R1
Spacer
(a)
O
OO
OO
O
O
O
R1
R1
O
OOOR1
R1
O
OOOR1
R1
O
O
OO
R1R1
O
O
OO
R1
R1
O
OO
O O
O
O
O
R2
R2
O
O OO R2
R2
O
O OO R2
R2
O
O
OO
R2R2
O
O
OO
R2
R2
Spacer
(b)
Figure 3.60: Illustration of a symmetrical (a) and asymmetrical (b) functionalized fullerenedimer.
(scheme 3.18, pathway A), only symmetrical functionalized fullerene dimers are acces-
sible (see figure 3.60 (a)). For the synthesis of asymmetric fullerene dimers (figure 3.60
(b)) a new concept had to be found, which will be described in the following chapter.
3.5.1 Synthesis of Bisfunctionalized Janus-Type Fulleren e-Dimers
In principle both synthetic strategies for mixed [5,1]hexakisadducts can be adopted
for the synthesis of Janus-type (Janus: Roman good of gates and doors, represented
with a double-faced head) fullerene-dimers. The preparation of two different pentak-
isadducts should enable the combination of two totally different functionalities into one
dimer molecule, by direct bridging of the two fullerene moieties with a bis(functional)
addend. Since the preparation of pentakisadducts via the triazoline route, introduced
in scheme 3.18, is afflicted with laborious synthetic procedures and low yields, we
decided to utilize the retro-cycloaddition reaction of pyrrolidinofullerenes, recently dis-
covered by MARTIN et al., for the synthesis of fullerene pentakisadducts.[295] Pyrrolidi-
nofullerenes are easily accessible by the 1,3-dipolar cycloaddition reaction of in situ
127

Chapter 3 Results and Discussion
generated azomethine ylides to C60. The pyrrolidine ring can be efficiently removed
by a thermal reaction in the presence of an excess of a highly efficient dipolarophile.
In some cases the addition of a metal lewis acid catalyzed this reaction, presumably
by coordination to the nitrogen atom. The pyrrolidinofullerene 114 was synthesized
following literature procedure [296] by the reaction of C60 with benzaldehyde and N-
methylglycine in boiling toluene. The condensation product of the aromatic aldehyde
and N-methylglycine successively undergoes dehydration and decarboxylation to give
the corresponding ylide. Nucleophilic attack by the latter on C60 followed by intramolec-
ular ring closure leads to the fused pyrrolidinofullerene system. The template-mediated
DMA strategy was further used to obtain the mixed hexakisadduct 116 by the nucle-
ophilic cyclopropanation with diethylmalonate 115. The yield in this reaction step was
relatively low, supposably through the different regioselectivity of the pyrrolidine ring in
directing the malonates for the subsequent additions. Compared to methanofullerenes,
where the second attack of a nucleophile is specified at the preferred e and trans-
3 positions,[297] the bisaddition on pyrrolidinofullerenes generally forms the regioiso-
mers with no significant favoritism.[298] Despite the DMA mediation, which should ac-
tivate the equatorial positions, the reaction yield could not be improved. The retro-
cycloaddition reaction was carried out using standard conditions.[295] Hexakisadduct
116 was refluxed in o-DCB with the presence of an excess of maleic anhydride, in
order to trap the corresponding ylide that results from the thermal retro-cycloaddition
reaction. After 12 h, reaction control via TLC and mass spectrometry showed no con-
version of the starting material. Also longer reaction periods gave no evidence for the
formation of the corresponding pentakisadduct. The addition of 1 eq. CuTf2 (cop-
per(II)trifluoromethylsulfonate) as a metal lewis acid, did not improve the efficiency of
the removal of the pyrrolidine ring. In fact, the hydrolysis of the malonesters could be
observed, resulting in a decomposition product, which could not be purified and char-
acterized further more.
Since it has been shown, that the retro-cycloaddition reaction of pyrrolidinofullerenes
suffers from its incompatibility with various functional groups, we decided to use al-
ternatively a synthetic procedure similar to pathway A described in scheme 3.18. In
128

Chapter 3 Results and Discussion
OO
O O
R1R1
R1
R1R1
R1
O
O O
O
O
O
O
O
O
OO
R1
R1O
OO
O
R1
R1O
N PhN Ph
OO
O O
R1R1
R1
R1R1
R1
O
O O
O
O
O
O
O
O
OO
R1
R1O
OO
O
R1
R1O R1 = CH2CH3
benzaldehyde,N-methylglycine
∆
DMA,CBr4, DBU
O O
O O
114 115
116
117
Figure 3.61: Synthesis of the [5,0]pentakisadduct 117 via the retro-cycloaddition reaction ofpyrrolidinofullerenes.
order to have the possibility to introduce different functionalities by this method, a new
hexakisadduct precursor had to be found, which allows the attachment of an additional
C60 molecule that can be itself further functionalized. Therefore the bifunctional macro-
cycle 118 [132] was used for the synthesis of the monofunctionalized fullerene 119,
which still exhibits a free malonic group for further functionalization (scheme 3.19).
cyclo-[2]-Octylmalonate 118 is a suitable building block for this reaction, because the
specific length of the spacer units is not appropriate to the formation of a strain-free
bisadduct, which is normally the competitive reaction pathway. The nucleophilic cy-
clopropanation of 118 was performed with an equimolar amount of C60, in the pres-
ence of CBr4 and DBU. Considering the possible side reactions of the bifunctional
malonate, including the formation of DBU-salts and polymerization, the reaction pro-
ceeded with reasonable yield (28 %). For the synthesis of the corresponding [5,1]hex-
akisadduct, different reaction conditions and malonates were applied and tested for
their efficiency in this type of reaction. All reactions were carried out under DMA-
mediation conditions and the results are summarized in table 3.12. However the use
129
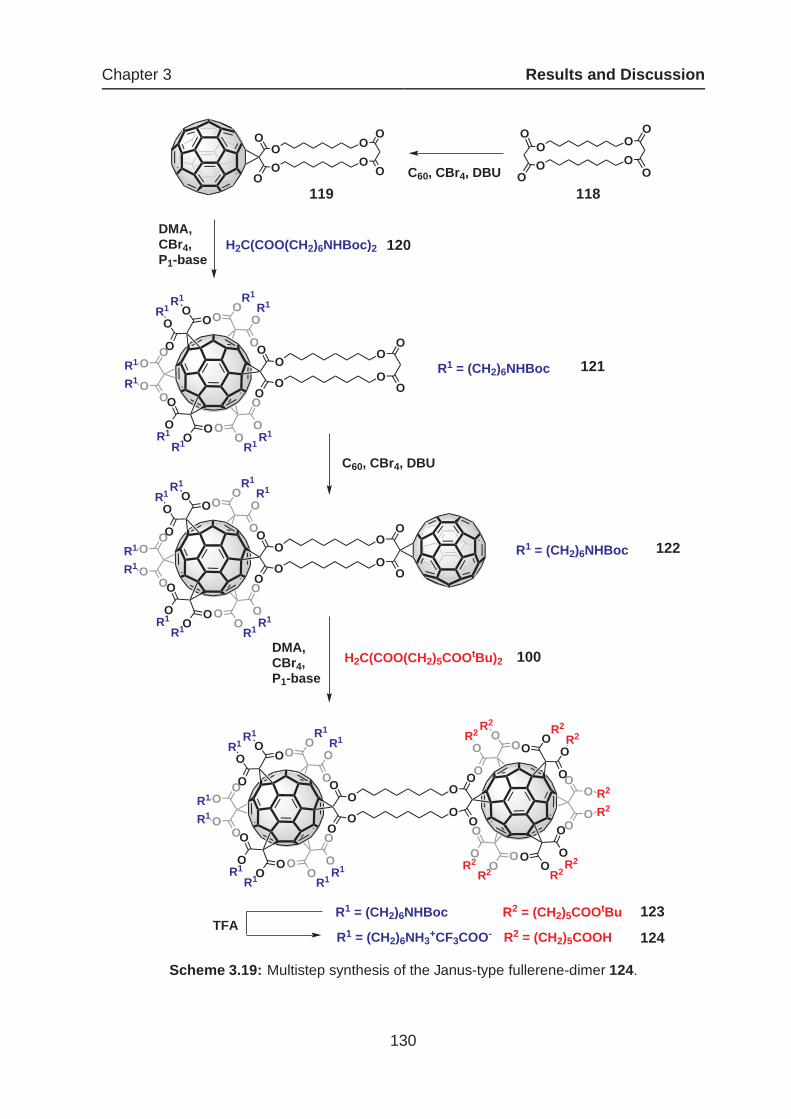
Chapter 3 Results and Discussion
O
O
O
OO
O
O
O
R1 = (CH2)6NHBoc
C60, CBr4, DBU
DMA,CBr4, P1-base
C60, CBr4, DBU
DMA,CBr4, P1-base
H2C(COO(CH2)5COOtBu)2
H2C(COO(CH2)6NHBoc) 2
TFAR1 = (CH2)6NHBoc R2 = (CH2)5COOtBu
R1 = (CH2)6NH3+CF3COO- R2 = (CH2)5COOH
O
OO
OO
O
O
O
O
O
O
O
R1
R1
O
OOOR1
R1
O
OOOR1
R1
O
O
OO
R1R1
O
O
OO
R1
R1
O
OO
OO
O
O
O
O
O
O
O
R1
R1
O
OOOR1
R1
O
OOOR1
R1
O
O
OO
R1R1
O
O
OO
R1
R1
R1 = (CH2)6NHBoc
O
OO
O O
O
O
O
O
O
R1
R1
O
OOOR1
R1
O
OOOR1
R1
O
O
OO
R1R1
O
O
OO
R1
R1
O
O O
O
O
O
R2
R2
O
O OO R2
R2
O
O OO R2
R2
O
O
OO
R2R2
O
O
OO
R2
R2
O
O
O
O O
O
O
O
118119
120
121
122
100
123
124
Scheme 3.19: Multistep synthesis of the Janus-type fullerene-dimer 124.
130

Chapter 3 Results and Discussion
Entry Malonate Base Halogenatingreagent
Yield
A 10 eq. 120 10 eq. DBU CBr4 ≤ 5 %B 10 eq. 120 10 eq. P1-tBu CBr4 11 %C 10 eq. diethyl
α-bromomalonate125
10 eq. DBU — —
D 10 eq. diethylα-bromomalonate125
10 eq. P1-tBu — ≤ 5 %
E 20 eq. 120 10 eq. DBU CBr4 14 %F 20 eq. 120 10 eq. P1-tBu CBr4 25 %
Table 3.12: Reaction conditions for the synthesis of mixed [5,1]hexakisadduct precursor 121.
of α-bromomalonic esters, which should suspend possible side reactions on the free
malonic group through the absence of a halogenating reagent, were not sufficient in
this case. The best result could be obtained by the use of a 20-fold excess of malonate
120 in the presence of the phosphazene base P1-tBu. Non converted malonate 120
could be recovered by preliminary flash column chromatography. Final purification of
the [5,1]hexakisadduct 121 was achieved by preparative HPLC. The attachment of an
additional C60 molecule to the free malonic group in 121 was accomplished by the cy-
clopropanation with a 1.5-fold excess of C60 in the presence of CBr4 and P1-tBu-base.
The isolation of the [5,1]hexakis-monoadduct 122 via flash column chromatography
was straight forward and no additional purification via preparative HPLC had to be
applied. The UV/Vis spectrum of 122 can be interpreted as a superposition of the indi-
vidual spectra of its components. Figure 3.62 shows the spectra of monoadduct 119,
[5,1]hexakisadduct 121, the parent [5,1]hexakis-monoadduct 122 and the calculated
product spectrum, which was obtained by addition of the monoadduct spectrum and
the [5,1]hexakisadduct spectrum. The UV/Vis spectra of 119 and 121 itself show the
typical characteristic slope of a C60-monoadduct and a C60-hexakisadduct respectively.
The measured spectrum of [5,1]hexakis-monoadduct 122 fits the calculated prediction
quite well, whereas the typical monoadduct maximum at 425 nm is clearly recogniz-
able (figure 3.62, inset). The presence of a less functionalized C60 hemisphere in
the [5,1]hexakis-monoadduct 122 opens the possibility to introduce further functional
131

Chapter 3 Results and Discussion
250 300 350 400 450 500 550 600
0
20
40
60
80
100
120
140
160
180
200ε
= M
-1cm
-1x
10
3
wavelength [nm]
400 425 450
119121122calculated (119 + 121)
Figure 3.62: UV/Vis spectra of monoadduct 119 (black), [5,1]hexakisadduct 121 (red),[5,1]hexakis-monoadduct 122 (blue) and the calculated spectrum for 122 (green).
groups into the molecule. Similar to the reaction described for the synthesis of 121, the
completion of the octahedral addition pattern with malonate 100 results in the dimeric
structure 123, whereas two different functionalized [5,1]hexakisadducts are bridged via
a macrocyclic addend. Compared to the above mentioned reaction, the synthesis of
123 only requires a 10-fold excess of malonate 100 in the presence of P1-tBu. Also
in this case the separation of 123 from lower adducts via preparative HPLC was in-
evitable.
Figure 3.63 shows the comparison of the 13C NMR spectra of [5,1]hexakisadduct 121,
[5,1]hexakis-monoadduct 122 and [5,1]hexakis-dimer 123. The resonances of the C60
sp2-carbons in the case of 121 can be clearly assigned to a pseudo Th symmetrical hex-
akisadduct. The resonance at 42.42 ppm and the signals for the malonester carbons
at 166.63 ppm, indicates the existence of the free malonic group, which was further
manifested by the C-H correlated HETCOR-spectrum. The conversion of 121 into the
132

Chapter 3 Results and Discussion
2030405060708090100110120130140150160170180
2030405060708090100110120130140150160170180
2030405060708090100110120130140150160170180
1
23
4
5
6
7
8
9
10
11
1213
14
15
16
17
18
19
2021
22
23
21
22 10,15
12,13 1
20
19,18,17,16
4,5,6,7,8,9
3
23
2
11,14sp -2
C
sp -3C
*
1
23
4
5
6
7
8
9
10
11
1213
14
15
16
17
18
19
2021
22
23
14,11,2
21
sp -2C (hexa)
sp -2C (mono)
22
*
sp -3C (mono)
sp -3C (hexa)
3,10,15
112,13
20
23
4,5,6,7,8,9
19,18,17,16
1
23
4
5
6
7
8
9
10
11
1213
14
15
16
17
18
19
2021
22
23
24
25
26
27
28
29
30
3132
33
311,11,
2514,21 sp -2
C 32
22
sp -3C
3,10,2615,
1,12,2413,
20
30
23
33
4,5,6,7,8,9
19,18,17,16
27,28,29
Chemical Shift (ppm)
*
121
122
123
Figure 3.63: Comparison of the 13C NMR spectra of [5,1]hexakisadduct 121 (100.5 MHz, RT,CDCl3), [5,1]hexakis-monoadduct 122 (75 MHz, RT, CDCl3) and [5,1]hexakis-dimer 123 (75 MHz, RT, CDCl3).
133

Chapter 3 Results and Discussion
corresponding [5,1]hexakis-monoadduct 122 results in a spectrum, which in principle
shows the characteristics of a hexakisadduct as well as a monoadduct. This can be
clearly seen in the sp2-carbon region of 122. Beside the dominating signals at 145.92
and 141.32 ppm for the hexa-substituted molecule part, the additional resonances
between 145.45 and 139.32 ppm can be clearly allocated to a mono-substituted C60
molecule. Also the sp3-carbon signal at 71.79 ppm exhibits the typical low-field shift
of a C60-monoadduct, compared to the signal at 69.28 ppm for the hexakisadduct sp3-
carbons. This trend can be further followed for the malonate bridge carbon atoms,
whereas the chemical shift amounts to almost 7 ppm. The 13C NMR spectrum of pro-
tected [5,1]hexakis-dimer 123 is similar to the spectrum observed for 121. Due to the
large distance of the functional groups in the malonate addends 100 and 120 to the
C60 cores, no influence on the chemical shift can be detected. On account of this, the
high local symmetry of both fullerene moieties results in only two signals for all 96 sp2
carbon atoms. Furthermore, all 24 sp3-carbons can be detected as one signal at 69.28
ppm. The additional signals compared to the spectrum of 122 can be clearly assigned
to the malonate addend 100.
In order to achieve water solubility, the deprotection was carried out by treatment of 123
with pure TFA (scheme 3.19). Next to the quantitative conversion of the Boc-groups
into the corresponding ammonium salts, the simultaneous cleavage of the acid labile
tert-butyl esters took place as demonstrated by the absence of any signals for both
functional groups in the 1H- and 13C NMR spectra of 123. Beside pure water, the am-
monium salt of the asymmetric dumbbell shaped bis(fullerene) 123 is very soluble in
methanol. Due to the presence of ten amino groups and ten carboxylic acid groups
within the structure of 124, it can be best described as amphoteric oligoelectrolyte.
Contrary to the natural occurring polyampholytes (proteins), the potential charge carri-
ers in macromolecule 124 are allocated and separated over the two C60hemispheres in
a well-defined way, comparable to a Janus medal. The pH dependent solubility behav-
ior of 124 in aqueous solutions is mainly influenced by this amphoteric character. At
a lower pH value (pH = 3), both the carboxylates and amine functions are protonated,
resulting in a positive net charge and a good water-solubility. At higher pH value (pH
134

Chapter 3 Results and Discussion
= 10), the amines exist as neutral bases and the carboxylic acids as the conjugate
bases, giving the molecule a negative net charge and a good water-solubility. At both
mentioned pH values, a clear yellow aqueous solution was obtained (figure 3.65, left
and right). The UV/Vis spectra of dimer 124 in acidic and basic aqueous solution are
almost similar (figure 3.64). However, compared to the spectrum of the corresponding
ammonium salt in methanol, the distinct shoulders at about 317 and 335 nm flatten
out. Supposably the bolaamphiphilic character of dimer 124 causes the agglomeration
of the hydrophobic bridging moieties, which could facilitate the formation of oligomers
with a characterless slope of the spectrum in this region.
250 300 350 400 450 500 550 600
abso
rbance
wavelength [nm]
124 in methanol
124 in water (pH = 3)
124 in water (pH = 10)
Figure 3.64: UV/Vis spectra of 124 as ammonium salt in methanol (black),
At intermediate pH the zwitterion concentration increases, and at a characteristic pH,
called the isoelectric point (IEP), the negatively and positively charged molecular species
are present in equal concentration. In the case of 124 the IEP ranges from approxi-
mately 6 - 6.5. In the space of this pH-region, dimer 124 is not soluble in water any
135

Chapter 3 Results and Discussion
Figure 3.65: Aqueous solutions of deprotected asymmetric [5,1,1,5]-dimer 124 at pH = 3 (left),pH = 6.5 (middle) and pH = 10 (right).
more, which causes the precipitation as pale orange solid, which is completely insolu-
ble in methanol (figure 3.65, middle).
These observations indicate the formation of highly stable supramolecular aggregates
through attractive electrostatic interactions between the charged terminal groups near
the IEP.
136

Chapter 3 Results and Discussion
3.5.2 Synthesis of a Fullerene-Rich Nanocluster
In the last years only a few fullerene-rich nanostructures have been reported so far,
which were prepared as dendritic structures with fullerene units at their surface or with
C60 spheres in the dendritic branches.[299–301] This is mainly associated with the diffi-
culties related to the synthesis of such fullerene-rich macromolecules.[302] The major
problems for the preparation are the low solubility of C60 and its chemical reactivity lim-
iting the range of reactions that can be used for the synthesis of branched structures
bearing multiple C60 units.
In a new approach we decided to synthesize a monodisperse fullerene-rich macro-
molecule with a C60 core as well as peripheral C60 subunits. The [5,1]hexakisadduct
121 could be therefore used as precursor, which provides high solubility in most organic
solvents. Treatment of 121 with pure C60 in the presence of DMA, CBr4 and phosp-
hazene base in toluene gave the highly substituted macromolecular structure 126. In
this star-type architecture the octahedral substituted C60-core ([6,0]hexakisadduct) is
surrounded by six [5,1]hexakisadducts. The major difficulty in the purification of 126 is
the similar polarity of adducts with lower addition degree and the overall high polarity
of 126. Therefore it was necessary to pre-clean compound 126 twice via flash column
chromatography, followed by purification via preparative HPLC. The high molecular
weight of 126 (22622.26 g/mol) made it impossible to characterize the macromolecule
by FAB mass spectrometry. Additionally, attempts for MALDI-TOF mass spectrome-
try with 126 gave no evidence for the molecule ion peak. The laser bombardment in
MALDI-MS caused the fragmentation of 126, with detectable ions in the region of 2840-
4330 mass/charge, which could not be clearly allocated to specific molecule fragments.
Despite of the missing mass spectrum, the NMR spectroscopy shows the evidence for
the formation of 126. The 13C NMR spectrum is greatly simplified through the high
symmetry (pseudo-Th) of 126. As would be expected, two resonances are observed
for the carbonyl C atoms (164.06 and 156.25 ppm), two for the tert-butyl units (79.13
and 28.68 ppm), one for the OCH2 units (67.11 ppm) and five for the CH2 groups of the
Nboc-malonates (40.71, 30.18, 28.57, 26.61 and 25.80 ppm). The signals of the cy-
clo-[2]-octylmalonates show less intensities, whereas two of the three expected signals
137

Chapter 3 Results and Discussion
+
DMA,CBr4, P1-base
O
OO
OO
O
O
O
O
O
O
O
R
R
O
O OO R
R
O
O OO R
R
O
O
OO
RR
O
O
OO
RR
O
OO
OO
O
O
O
O
O
O
O
R
R
O
OOOR
R
O
OOOR
R
O
O
OO
RR
O
O
OO
RR
OO
O
O
O
OOO
O
OOOR
R
OO
O O
R R
O
O
O
O
R
R
O
O
O
O
R
R
OOO
OR R
OO
O
O
O
O OO
O
O OO R
R
OO
OO
RR
O
O
O
O
R
R
O
O
O
O
R
R
OO O
O RR
OO
O
O
O
O
OO
O
O
OO
RR
O
O
O
O
R
R
O
O
OO
RR
O
OOOR
R
O
O
O
O
R
R
OO
O
O
O
O
OO
O
O
OO
RR
O
O
O
O
R
R
O
O
OO
RR
O
O OO R
R
O
O
O
O
R
R
O
O
HNR =
TFANH3
+CF3COO-R =
O
OO
O O
O
O
O
O
O
O
O
R
R
O
OOOR
R
O
OOOR
R
O
O
OO
RR
O
O
OO
RR
O
O
HNR =
121
126
127
Scheme 3.20: Synthesis of the fullerene-rich nanocluster 127 by sixfold addition of the[5,1]hexakisadduct precursor 121 to parent C60.
138

Chapter 3 Results and Discussion
Chemical Shift (ppm)
020406080100120140160180
1
2
3
4
5
6
7
8
9
1011
1213
14
15
16
17
18
19
202122
23
2,11,1421
sp -2 C
*
*
22
sp -3 C
3,10,15
1,12,13
20
16
138140142144146148150 24.525.526.527.528.529.530.531.5
sp -2 C
23,5,8
191718
4,9 6,7
**
126
Figure 3.66: 13C NMR spectrum of [5,1]hexakisfullerenyl-[6,0]hexakisadduct 126 (100.5 MHz,RT, CDCl3) (* = solvent + toluene impurities).
could be clearly identified (29.79 and 26.28 ppm). The missing signal is located under
the tert-butyl resonance. The C60-sp2 region of the spectrum simply consists of two
signals (145.93 and 141.34 ppm), which is characteristic for a pseudo-Th-symmetrical
addition pattern (broadening of these typical signals would indicate the presence of
adducts with lower addition degree). Furthermore all C60-sp3 carbon atoms could be
detected as one signal at 69.29 ppm. Beside the 13C NMR spectrum, also the UV/Vis
spectrum exhibits the characteristic slope of a hexakisadduct. The distinct shoulders at
316 and 335 nm do not flatten out, which would indicate the presence of pentakisadduct
impurities. The cleavage of the carbamates in 126 could be obtained in pure TFA. The
139

Chapter 3 Results and Discussion
250 300 350 400 450 500 550 600
0
100
200
300
400
500
600
700
800
900ε
= M
-1cm
-1x
10
3
wavelength [nm]
Figure 3.67: UV/Vis spectrum of [5,1]hexakisfullerenyl-[6,0]hexakisadduct 127 in H2O (pH =4).
NMR spectra of 127 after reprecipitation from MeOH/Et2O exhibited no carbamate res-
onances any more. The water-solubility of 127 strongly depends on the pH value of
the solution. In neutral and slightly acidic solution, the solubility of 127 is comparatively
low. If the solutions are further acidified, a significant increase of the solubility can be
observed, resulting in a bright yellow solution. Comparable to Nboc-protected struc-
ture 126, the UV/Vis spectrum of 127 shows the typical distinct double-bands at 271
nm, 280 nm and 316 nm, 335 nm. The sum of seven hexasubstituted C60-cores within
the structure of 127 accounts for a remarkable extinction coefficient of approximately
550000 M−1cm−1 at 245 nm (figure 3.67). The geometry optimized (MMFF) gas phase
structure shown in figure 3.68 (left) illustrates the highly symmetric star-shaped struc-
ture of 127 in the neutral constitution with peripheral amino groups. The side chains
of the malonate units are thereby parallel aligned to each other with a distance of the
amino group to the opposite one of approximately 68 Å. The protonation of the amino
140

Chapter 3 Results and Discussion
Figure 3.68: MMFF [303] geometry optimized gas phase structure of [5,1]hexakisfullerenyl-[6,0]hexakisadduct 126 in the neutral (left) and charged (right) constitution.
groups disrupt the compact alignment and the repulsion of the corresponding charged
ammonium end groups results in a more spread out structure (figure 3.68 (right)).
Beside the aesthetically pleasing architecture of 127, the presence of sixty amino
groups in a well-defined, monodisperse macromolecule could make it a promising vec-
tor for nonviral gene delivery. The spherical shape of 127 would be able to mimic
the globular shape of the natural DNA-histone complex, and fits the size-range of the
natural protein complex (around 8 nm) quite well.[304]
141

CHAPTER 4
4 Summary
Fullerenes have an enormous potential in applications to physics and biology. Specif-
ically [60]fullerene with its unique electronic, optical and structural properties has at-
tracted considerable attention for its application in biomedical materials and optoelec-
tronic devices. In this context the selective functionalization of C60, which allows to
combine the parent properties with new attributes like water-solubility or amphiphilicity
is still a challenging topic for the synthetic chemist.
In the first part of the present work a series of water-soluble anionic and cationic
amphiphilic fullerene monoadducts 43, 44, 45, 46, 47, 48, 56, 60 were synthesized.
The amphifullerenes are basically composed of a hydrophilic dendritic branch and a
lipophilic moiety, e.g. alkyl chains or unsaturated fatty acids. By the fine tuning of the
lipophilic side chain and the size of the dendritic part, a better cell tissue accessibility
and biodistribution within lipid rich regions or easier crossing of the blood-brain barrier
is expected. The synthesis of these amphifullerenes starts with the reaction of primary
and secondary alcohols with MELDRUM’S acid to the corresponding mono carboxylic
malonates, which are the precursor compounds for the asymmetric malonates. Unlike
previous reports on the synthesis of asymmetric malonates, this reaction proceeds in
almost quantitative yield and can be easily scaled up. By using these building-blocks,
142

Chapter 4 Summary
a straightforward synthesis procedure could be evaluated, which allows the synthesis
of the anionic amphifullerenes 43, 44, 45, 46, 47, 48 and the cationic amphifullerene
60 in gram scale. The availability of larger amounts enabled detailed examinations
of these amphiphilic fullerene structures as bioactive materials. In cooperation with
C-SIXTY Inc. and PHYLONIX Pharmaceuticals Inc. the antioxidative activity could be
determined applying the xanthine/xanthine oxidase/cytochrome c assay. The overall
low IC50 values for all tested amphifullerenes showed the pronounced ability of these
structures to efficiently deactivate reactive oxygen species. In line with this cooperation
zebrafish embryos as a model system were applied to investigate the toxicity and the
cytoprotective activities of the new compounds. Unlike previous results on specific car-
boxyfullerenes, it could be demonstrated, that none of the amphifullerenes exhibited
significant toxicity even at very high concentrations. By the comparison of the cytopro-
tective activities in the series of amphifullerenes, it could be shown, that the anionic
amphiphilic derivative 47 exhibit cytoprotective activity against both cisplatinum and
gentamicin- induced apoptosis and represents a promising platform for the develop-
ment of new potent anti-apoptosis drugs.
In cooperation with the IVANOVIC-BURMAZOVIC group the ability of the amphifullerenes
to act as metal free SOD mimetics was investigated by electrochemical, spectrophoto-
metrical and submillisecond mixing UV/Vis stopped-flow measurements. For the first
time it could be demonstrated that there is a direct correlation between SOD activity
of the water soluble fullerenes and molecular properties such as i) reduction potential,
ii) charge and iii) molecular structure. The positively charged monoadduct 60 could
be identified as very active new lead structure, which approaches the performance of
natural Mn- and Fe-SODs.
Furthermore the capability of amphifullerenes 44, 47, 56, 60 as materials for the forma-
tion of monomolecular films was examined. The LANGMUIR films were characterized by
film balance experiments and BAM microscopy. The 1st generation amphiphilic mono-
adducts 44, 60 as well as the 2nd generation amphiphilic monoadducts 47, 56 were
able to form stable monolayers. In all cases a dependence of the reversibility for the
film formation on the pH value could be detected. In addition, up to five layers made
143

Chapter 4 Summary
from amphifullerene 47 could be successfully transferred onto hydrophilic silicon.
In the second part of this work the copper(I)-catalyzed HUISGEN 1,3-dipolar cycload-
dition was adopted for the synthesis of novel neutral and charged fullerene structures
containing multiple triazol moieties. It could be demonstrated that this reaction is very
efficient for the synthesis of triazol malonates, which can be easily modified with differ-
ent azides. By modification of the solvent system for the nucleophilic cyclopropanation
reaction, the novel dendritic triazol-fullerenes 86 and 87 have been prepared. The
amine terminated triazol-fullerene 87 exhibits a pronounced solubility in water. Con-
trary to other literature known amine functionalized fullerenes the solubility of 87 is not
limited to acidic aqueous solutions and can be observed over the complete range from
pH = 4 to 11.
The third part describes the covalent and non-covalent sidewall functionalization of
SWCNTs with different fullerene derivatives. For the covalent functionalization the
monocarboxylic fullerene derivative 94 was synthesized. Monoadduct 94 was further
esterificated with functional groups on the SWCNT surface, which were introduced via
reductive alkylation. This transformation resulted in carbon nanotubes decorated with
fullerene molecules on the surface and makes it possible to combine the properties of
both carbon allotrops in new functional materials. Among the available non-covalent
functionalization approaches, the π − π stacking of aromatic compounds on the nano-
tube surface is found to be a viable approach. Since the spherical shape of C60 is not
suitable for the formation of strong π−π interactions, the pyrene-fullerene conjugate 99
was synthesized. STM investigations could show that the π − π interactions between
the SWCNT and the pyrene moiety govern the association of 99 with the sidewalls
of the SWCNT. Furthermore it is possible to remove the pyrene-fullerenes from the
SWCNT surface, which results in structures of aligned functional fullerenes. Thereby
the SWCNTs act as templates, which preorientate the fullerene molecules and make
it possible to transfer these supramolecular structures to substrates in well-defined
strands of aligned molecules.
Chapter 3.4 was mainly focused on the synthesis of new tether-based malonate sys-
tems for the regioselective bisfunctionalization of C60. Therefor it was tried to coordi-
144

Chapter 4 Summary
nate the dicarboxylic malonate 101 to different metal ions, which however did not lead
to the desired metallomacrocycles. In a new approach the ureido-pyrimidinone deriva-
tive 106 was synthesized from the monocarboxylic malonate 27. Through the self-
complementary quadruple hydrogen bonding between the 2-ureido-4(1H)-pyrimidinone
moieties dimeric structures are formed, which were utilized as tethers for the bisfunc-
tionalization of C60. Although it could be demonstrated that the reaction of 106 with C60
almost exclusively led to the formation of C60-bisadducts, the separation of the different
regioisomers could not be achieved.
In the last part of this work a new concept for the synthesis of fullerene-rich nanostruc-
tures is described. For this purpose the cyclo-[2]-octylmalonate monoadduct 119 was
synthesized and successfully transferred into the corresponding [5,1]hexakisadduct
121, applying the DMA mediation method. Due to the presence of a free malonic group
within the hexakisadduct 121, this structure serves as perfect building block for the syn-
thesis of multiple C60-arrays. By means of this precursor compound it was possible to
accomplish the synthesis of the dimeric fullerene structure 124, where two different
functional groups are allocated and separated over two C60-spheres in a well-defined
way. Beside the unique properties of this amphoteric oligoelectrolyte, this structure is
to the best of our knowledge the only example for an asymmetric fullerene dimer so far.
Furthermore precursor 121 was used for the synthesis of a star-shaped fullerene-rich
nanocluster. The linkage of six [5,1]hexaksiaddducts to one C60-core gave the nan-
ocluster 126, which combines a globular structure with an exceptionally high charge
density.
145

CHAPTER 4
4 Zusammenfassung
Fullerene, die neben Graphit und Diamant die dritte Verbindungsgruppe sind, zu der
sich reiner Kohlenstoff zusammenschließen kann, besitzen enormes Potential für An-
wendungen im Bereich der Physik und Biologie. Speziell Buckminster-Fulleren C60 mit
seinen einzigartigen elektronischen, optischen und strukturellen Eigenschaften sorgt
für hohe Aufmerksamkeit in biomedizinischen Materialien und optoelektronischen Ap-
plikationen. In diesem Zusammenhang spielt die selektive Funktionalisierung von C60
immer noch eine große Rolle, die es erlaubt, neue Attribute wie Wasserlöslichkeit oder
Amphiphilie mit den ursprünglichen Eigenschaften von C60 zu verbinden.
Im ersten Teil dieser Arbeit wurde eine Serie wasserlöslicher, anionischer und kation-
ischer amphiphiler Fulleren Monoaddukte 43, 44, 45, 46, 47, 48, 56, 60 synthetisiert.
Diese Amphifullerene bestehen generell aus einem hydrophilen dendritischen Teil und
einer lipophilen Einheit, wie z.B. Alkylketten oder ungesättigte Fettsäuren. Durch die
stufenweise Modulierung der lipophilen Gruppe, sollte es möglich sein die Biokom-
patibilät dieser Verbindungen zu steigern, um auch schwer passierbare physiologis-
che Barrieren wie die Blut-Hirn-Schranke zu durchdringen. Die Synthese der Am-
phifullerene startet mit der Umsetzung von primären und sekundären Alkoholen mit
MELDRUM’S Säure zu den entsprechenden Mono-Carbonsäure Malonaten, die als
146

Chapter 4 Zusammenfassung
Vorstufe für die unsymmetrischen Malonate dienen. Im Gegensatz zu früheren Meth-
oden für die Synthese von unsymmetrischen Malonaten, erfolgt diese Reaktion mit
nahezu quantitativer Ausbeute und kann leicht in großem Ansatz durchgeführt wer-
den. Mit Hilfe dieser Bausteine konnte nun ein effizientes Synthesekonzept entwick-
elt werden, was es erlaubte die anionischen Amphifullerene 43, 44, 45, 46, 47, 48
und das kationische Amphifulleren 60 im Gramm-Maßstab herzustellen. Durch die
Verfügbarkeit von größeren Mengen, konnte somit die Untersuchung der Verbindun-
gen auf deren biomedizinische Eigenschaften bewerkstelligt werden. Die Evaluierung
der antioxidativen und neuroprotektiven Eigenschaften und die Toxizitätsuntersuchun-
gen dieser Substanzen wurden dabei in Kooperation mit C-SIXTY Inc. und PHY-
LONIX Pharmaceuticals Inc. durchgeführt. Durch die Bestimmung der IC50 Werte
mittels Xanthin/Xanthinoxidase/Cytochrom C - Assay, konnte gezeigt werden, dass
alle Amphifullerene ausgeprägte antioxidative Eigenschaften besitzen. Weiterhin kon-
nten Tierversuche an Zebrafischen zeigen, dass im Gegensatz zu früher untersuchten
Verbindungen, keinerlei toxische oder organtoxische Nebenwirkungen zu beobachten
sind. Im Hinblick auf die neuroprotektiven Eigenschaften stellte sich heraus, dass
das anionische Amphifulleren 47 äußerst wirkungsvoll sowohl Cisplatin als auch Gen-
tamizin induzierte Apoptose verhindern kann.
Um zu prüfen, ob die Amphifullerene das katalytische Potenzial von SOD-Mimetika
besitzen, wurde in Kooperation mit dem Arbeitskreis IVANOVIC-BURMAZOVIC elektro-
chemische, spektrophotometrische und Stopped-Flow Untersuchungen durchgeführt.
Zum ersten Mal konnte dadurch gezeigt werden, dass ein direkter Zusammenhang
zwischen der SOD Aktivität der wasserlöslichen Amphifullerene und den Moleküleigen-
schaften, wie i) Reduktionspotenzial, ii) Ladung und iii) Molekülstruktur besteht. Das
positiv geladene Monoaddukt 60 stellte sich in diesem Zusammenhang als äußerst
wirkungsvolle Verbindung heraus, die nahezu die katalytischen Umsätze von natür-
lichen Mn- und Fe-SOD-Enzymen erreicht.
Im Folgenden wurde die Eignung der Amphifullerene 44, 47, 56, 60 als Materalien für
die Bildung von monomolekularen Filmen untersucht. Die LANGMUIR-Filme wurden
durch Filmwaagenexperimente und Brewster-Angle-Microscopy näher charakterisiert.
147

Chapter 4 Zusammenfassung
Sowohl die amphiphilen Monoaddukte erster Generation 44, 128 als auch die am-
phiphilen Monoaddukte zweiter Generation 47, 56 bilden dabei stabile Monomoleku-
lare Filme aus. In allen Fällen wurde dabei eine pH Abhängigkeit der Reversibilität
der Filmbildung detektiert. Zudem lassen sich im Fall des Amphifullerens 47 durch die
LANGMUIR-BLODGETT-Methode diese Filme wiederholt auf hydrophiles Silizium über-
tragen, so dass dieses mit bis zu fünf Schichten überzogen werden konnte.
Im zweiten Kapitel wurde mittels der Kupfer(I) katalysierten HUISGEN 1,3-dipolaren
Cycloaddition neuartige neutrale und geladene Fulleren Strukturen synthetisiert, die
mehrere Triazol Einheiten als Strukturmerkmal aufweisen. Es konnte dabei gezeigt
werden, dass mit dieser Reaktion äußerst effizient Triazol-Malonate dargestellt wer-
den können und diese durch Variation der verwendeten Azide leicht modifiziert werden
können. Durch eine Änderung des Lösungsmittelsystems bei der nukleophilen cyclo-
propanierung mit C60, konnten somit die Triazol-Fullerene 86 und 87 hergestellt wer-
den. Durch die terminalen Aminogruppen im Triazol-Fulleren 87 konnte zudem eine
Löslichkeit in Wasser erreicht werden, die im Gegensatz zu bisher bekannten Amino-
fullerenen nahezu pH unabhängig ist.
Der dritte Teil dieser Arbeit fokussiert auf die kovalente und nicht-kovalente Funk-
tionalisierung von SWCNTs mit verschiedenen Fulleren Derivaten. Um eine kova-
lente Verknüpfung mit den SWCNTs zu erreichen, wurde dafür das Monocarbonsäure-
Fulleren 94 synthetisiert, welches erfolgreich mit funktionellen Gruppen auf der SWCNT
Oberfläche umgesetzt werden konnte, die zuvor über reduktive Alkylierung eingeführt
wurden. Auf diese Weise wurden SWCNT-Derivate dargestellt, an deren Seitenwände
Fullerenmoleküle kovalent gebunden sind, was es ermöglicht, die Eigenschaften bei-
der Kohlenstoff Allotrope in neuen funktionalen Materialien zu vereinigen. Unter den
veschiedenen Möglichkeiten der nicht-kovalenten Funktionalisierung, stellen vor allem
π−π-Wechselwirkungen zwischen aromatischen Verbindungen und der SWCNT Ober-
fläche einen brauchbaren Ansatz dar. Da die spherische, dreidimensionale Struktur
von C60 nicht für die Ausbildung starker π − π-Wechselwirkungen geeignet ist, wurde
als Modifikation das Pyren-Fulleren Konjugat 99 dargestellt. STM Untersuchungen
zeigten dabei, dass aufgrund der aromatischen Wechselwirkungen zwischen Pyren
148

Chapter 4 Zusammenfassung
Einheit und SWCNT, die Immobilisierung des Fullerens 99 auf der SWCNT Oberfläche
erfolgte. Weiterhin konnte gezeigt werden, dass ein Entfernen der Fulleren Moleküle
von der Oberfläche möglich war und sich dadurch perfekt angeordnete Fulleren- Struk-
turen realisieren lassen. Dabei agieren die SWCNTs als Strukturtemplate, die eine
Vororientierung der Fulleren Moleküle ermöglichen. Die entstehenden supramoleku-
laren Überstrukturen können anschließend auf Substrate übertragen werden, was zur
Bildung von hochgeordneten "Fulleren-Drähten" führt.
Das vierte Kapitel der vorliegenden Arbeit beschäftigt sich mit der Synthese von neuar-
tigen Malonat-Systemen, die als Template für die regioselektive Bisfunktionalisierung
von C60 dienen. Dabei wurde zunächst untersucht, ob durch die Koordination des
Dicarbonsäure Malonats 101 an verschiedene Metallionen, die Bildung von Metall
Makrozyklen zu realisieren ist, was nicht zu dem erwünschten Ergebnis führte. Des-
halb wurde in einem neuen Ansatz, ausgehend von dem unsymmetrischen Malonat
27 das Ureido-Pyrimidon Derivat 106 dargestellt. Durch die vier Wasserstoffbrücken-
bindungen vermittelte Selbstkomplementarität der Ureido-Pyrimidon Einheiten, bilden
sich dabei äußerst stabile dimere Strukturen aus, die als Template für die Bisfunktion-
alisierung von C60 eingesetzt wurden. Obwohl bewiesen werden konnte, dass sich bei
der Umsetzung von 106 mit C60 fast ausschließlich C60-Bisaddukte bilden, konnte eine
Trennung der verschiedenen Regioisomere nicht erreicht werden.
Im letzten Teil dieser Arbeit wurde ein neues Konzept für die Synthese von Nanostruk-
turen mit hohem C60-Gehalt entwickelt. Zu diesem Zweck wurde ausgehend von Cy-
clo-[2]-octylmalonat, das entsprechende Monoaddukt 119 synthetisiert, welches erfol-
greich mittels DMA zu dem entsprechenden [5,1]Hexakisaddukt 121 umgesetzt werden
konnte. Durch die Anwesenheit einer freien Malonat Gruppe in Verbindung 121 eröffnet
dieser Baustein die Möglichkeit, Strukturen mit mehreren C60-Kernen herzustellen.
Somit war es möglich, die dimere Fulleren Struktur 124 darzustellen, in der zwei un-
terschiedlich funktionalisierte C60 Sphären miteinander verbrückt sind. Diese Struktur,
die die Eigenschaften eines amphoteren Oligoelektrolyten besitzt, ist nach unserem
Wissen bisher die einzige hochsubstituierte, unsymmetrische, dimere Fulleren Struk-
tur. Desweiteren wurde ausgehend von Verbindung 121 der "sternförmige" Fulleren-
149

Chapter 4 Zusammenfassung
Nanocluster 126 synthetisiert. In dieser Struktur sind sechs [5,1]Hexakisaddukte mit
einem C60 Kern verbunden, was zu einer äußerst sphärischen Struktur mit extrem ho-
her Ladungsdichte führt.
150

CHAPTER 5
5 Experimental Part
5.1 Chemicals and Instrumentation
Chemicals:
Most reagents were purchased from Aldrich, Fluka and Acros Organics and, if not oth-
erwise noted, used as purchased. C60 was obtained by the Hoechst AG/AVENTIS and
separated from higher fullerenes by a plug filtration.[54,305] All analytical-reagent grade
solvents were purified by distillation.
Thin Layer Chromatography (TLC):
Riedel-de-Haën silica gel F254 and Merck silica gel 60 F254, 0.2 mm on aluminum
foil. Detection: UV lamp (254 or 366 nm), H3[P(Mo3O10)4]/Ce(SO4)2/H2SO4/H2O bath,
KMnO4/H2O bath or iodine chamber.
Flash Chromatography (FC):
MERCK silica gel 60 (230 - 400 mesh, 0.04 - 0.063 nm). ICN silica gel 32 - 63, 60 Å.
151

Chapter 5 Experimental Part
Analytical High Performance Liquid Chromatography (HPLC) :
Shimadzu Class-LC10: Liquid Chromatograph LC-10AT, Communication Bus Module
CBM-10A, Diode Array Detector SPD-M10A, Auto Injector SIL-10A, Refractive Index
Detector RID-10A and Selection Valve FCV-10AL. Columns: Nucleosil 200 x 4 mm, 5
µm, Nucleosil 200 x 4 mm, 3 µm, Macherey-Nagel; Solvents were purchased in HPLC
grade from Acros Organics.
Preparative High Performance Liquid Chromatography (HPLC ):
Shimadzu Class-LC10: System Controller SCL-10AVP, Preparative Liquid Chromato-
graph LC-8A, Communication Bus Module CBM-10A, UV/Vis Detector SPD-10A, Auto
Injector SIL-10A and Fraction Collector FRC-10A. Columns: Nucleosil 250 x 21 mm, 5
µm, Macherey-Nagel; Solvents were purified by distillation.
UV/Vis Spectroscopy:
Shimadzu UV-3102 PC, UV-VIS-NIR Scanning Spectrophotometer. The absorption
maxima λmax are given in [nm], the extinction coefficients ǫ in [M−1cm−1], shoulders are
indicated as sh.
IR Spectroscopy:
FT-IR Vector 22 from Bruker Analytische Messtechnik GmbH. The spectra were mea-
sured as KBr pellets. React-IRTM from ASI Applied Systems with ATR DiComp-Detector.
The spectra were measured as powders or liquid films. Absorptions are given in
wavenumbers ν̃ [cm−1].
Mass Spectrometry:
Micromass Zabspec FAB+ mode (3-Nitrobenzylalcohol as matrix).
NMR Spectroscopy:
Jeol JNM EX 400 and Jeol JNM GX 400 (1H: 400MHz, 13C: 100,5 MHz), Bruker Avance
300 (1H: 300 MHz, 13C: 75,4 MHz), Bruker Avance 400 (1H: 400 MHz, 13C: 100,5 MHz).
152

Chapter 5 Experimental Part
The chemical shifts are given in [ppm]. The resonance multiplicities are indicated as s
(singlet), d (doublet), t (triplet), q (quartet) and m (multiplet), broad resonances as br.
The raw data were processed using the program MestReNova 5.1.0 from Mestrelab
Research S.L.
Elementary Analysis:
EA 1110 CHNS from CE Instruments. The results are given as percentage.
153

Chapter 5 Experimental Part
5.2 Synthetic Procedures and Spectroscopic Data
Remarks
The compound names are generated with the algorithm used in the program Chem-
DrawUltra 8.0 from CambridgeSoft.
The following compounds have been synthesized according to the literature. The syn-
thetic procedures and the characterization of these compounds are not described in
detail in this section.
• 9-Heptadecanol 16 [306]
• Newkome-type dendrimers [G-1] 29 and [G-2] 30 [109,110]
• (all cis-4,7,10,13,16,19) Docosahexaenoic acid 49 [307]
• Methyl 3,5-bis(propargyloxy)benzoate 76 [224]
• 3,5-Bis(propargyloxy)benzyl alcohol 77 [224]
• 1-Azido-2-(2-methoxyethoxy)ethane 82 [30]
• N-(tert-Butoxycarbonyl)-2-hydroxyethylamine 80 [308]
• N-(2-Azidoethyl)-N-(tert-butoxycarbonyl)amine 81 [309]
• cyclo-[2]-Octylmalonate 118 [132]
• bis(6-(tert-butoxycarbonylamino)hexyl) malonate 129 [310]
General Procedures for Ester- and Amide-formation
a) Formation of esters and amides by reaction of the corresponding acyl chlorides
(GP 1):
The reaction is carried out under inert gas conditions and with dried glassware.
A solution of the alcohol/amine (1 eq.) in an adequate solvent (normally dry CH2Cl2)
and dry pyridine (1 eq.) were cooled in an ice bath. The acyl chloride (1 eq.) was
154

Chapter 5 Experimental Part
diluted in dry CH2Cl2 and added dropwise over a period of 1h via a dropping funnel. [In
very small-scale reactions the diluted acyl chloride (less than 1 mL) was added slowly
via a syringe over a period of 15 minutes.] The reaction mixture was stirred for 20 h at
room temperature and then washed with saturated NH4Cl-solution and H2O. After dry-
ing over MgSO4 and rotary evaporation of the solvent, FC on silica yielded the desired
product.
b) Formation of ester and amide bonds via activation of the carboxyl group with DCC
(STEGLICH -reaction) (GP 2):
The reaction is carried out under inert gas conditions and with dried glassware.
A solution of the carboxylic acid (1 eq.) and the alcohol/amine (1 eq.) in an ade-
quate dry solvent were cooled in an ice bath. DMAP (20 mol%, 0.2 eq.) and DCC
(1.1 eq.) were added subsequently. After stirring the solution for 2 h at 0°C and 20 h
at room temperature, the resulting dicyclohexylurea as side product precipitated dur-
ing the reaction and was filtered off. Traces of the urea which remained in solution,
were removed by repeated precipitation from ethyl acetate. Purification by FC on silica
yielded the desired product.
The rearrangement to the unreactive N-acylurea as an undesirable side-reaction (no-
tably when DMF is used as solvent) can be prevented by cooling and by the addition
of DMAP and 1-HOBt, which accelerate the respective alcoholysis or aminolysis of the
intermediate O-acylisourea formed by DCC and the carboxyl group, by forming stabile
active esters. 1-HOBt also prevents racemization in peptide synthesis.
c) Formation of ester and amide bonds via activation of the carboxyl group with EDC
(STEGLICH -reaction) (GP 3):
A solution of the carboxylic acid (1 eq.) and the alcohol/amine (1 eq.) in an adequate
solvent were cooled in an ice bath. DMAP (20 mol%, 0.2 eq.), 1-HOBt (1 eq.) and
EDC (1.2 eq.) were added subsequently. After stirring the solution for 2h at 0 °C and
20 h at room temperature, the reaction mixture was washed with H2O and saturated
NaCl-solution. After drying over MgSO4 and rotary evaporation of the solvent, FC on
155

Chapter 5 Experimental Part
silica yielded the desired product. The advantage of this method is the facile separation
of the urea side product, which can be easily extracted with water.
General Procedure for the Formation of C60 Monoadducts (GP 4)
The synthesis of monoadducts of C60 was performed according to the modified BIN-
GEL reaction.[96] However, to obtain higher yields of the desired monoadduct and less
multiple adducts an excess of C60 is applied. The unreacted C60 can be recovered
easily by FC on silica with toluene. C60 (1.5 eq. up to 2 eq.) was dissolved in dry,
degassed toluene (ca. 0.5 mL toluene per mg C60) resulting in a dark purple solution.
For reasons of solubility of the malonate or the desired monoadduct other solvents like
DMSO/toluene mixtures were also used. The solution was stirred until complete disso-
lution was obtained. Afterwards, CBr4 (1.1 eq.) and the malonate (1 eq.) were added.
DBU (1.1 eq.) was diluted in toluene and added dropwise over a period of 1 h to the
stirred solution at room temperature. After the solution was stirred for 2 h TLC control
showed the remaining C60, the monoadduct and traces of bis- and trisadducts. When
unreacted malonate was detected, additional CBr4 and diluted DBU were added. If
the retention factor of the product in pure toluene was small, the reaction mixture was
transferred directly to the FC column. It has been shown, that preliminary rotary evapo-
ration of the solvent induced a decrease of the yield. The yield of the pure monoadduct
was typically 35-50 % depending on the attached malonate. The monoadducts were
typically obtained as black solids or black, highly viscous oils.
General Procedure for the Deprotection of tert-Butylesters (GP 5)
The tert-butyl protected carboxylic acid (1 eq.) was dissolved in CH2Cl2 and treated
with TFA (10-20 eq.) under vigorous stirring at room temperature, till TLC control indi-
cated the complete deprotection of the starting material. Excess of TFA was removed
in vacuo, followed by repeated coevaporation with appropriate solvents like CHCl3 or
toluene.
When the starting material contains substantial amounts of tert-butylesters, pure formic
acid should be preferred, to avoid precipitation of the partially deprotected compound.
156

Chapter 5 Experimental Part
General Procedure for the Formation of Mono-Carboxylic Malonates (GP 6)
A mixture of the alcohol (1 eq.) and MELDRUM’s acid (1 eq.) was heated at 115°C
under vigorous stirring for 3 h. After cooling to room temperature, the product was
washed three times with pentane and dried in vacuo to afford the desired product.
General Procedure for the Formation of [5,1] and [6,0]-Hexakisadducts of C60
(GP 7)
The starting material (C60 or monoadducts of C60) determines the addition pattern ([6,0]
or [5,1]) of the obtained hexakisadducts. C60 (1 eq.) was dissolved completely in thor-
oughly degassed, dry toluene under nitrogen (in the case of monoadducts dry CH2Cl2
was used as well). High dilutions should be avoided. An excess of DMA (10 -12 eq.)
was added to the solution and stirred for 2 h at ambient temperature. Subsequently
the malonate (10 - 20 eq.) and CBr4 (10 - 12 eq.) was added and stirred to allow com-
plete dissolution. The base (10 - 12 eq.) (in this work the P1-tBu-base turned out to
be more efficiently than DBU) was diluted in the used dry solvent and added dropwise
over a period of 1 h. The solution was stirred for 2 - 6 days at room temperature un-
der N2, till TLC control remains unchanged (to exclude light, the flasks were wrapped
with aluminum foil). The unpolar DMA and highly polar by-products were separated
by preliminary flash column chromatography, followed by purification via preparative
HPLC. The essential characteristics for C60 hexakisadducts can be found in the UV/Vis
spectrum and 13C NMR spectrum. The UV/Vis spectra exhibit typical absorptions at
approximately 245, 271, 281, 316 and 335 nm and are unaffected by the addition pat-
tern. The 13C NMR spectra exhibit typical resonances between 146 and 141 ppm (all
hexakisadducts in this work exhibit two characteristic signals for a pseudo Th symmet-
rical hexakisadduct).
157

Chapter 5 Experimental Part
Malonic acid monohexylester (17)
O OH
OO
17
Compound 17 was synthesized according to general proce-
dure GP 6 from 1-hexanol (1.42 g, 13.9 mmol) and MEL-
DRUM’s acid (2 g, 13.9 mmol) and purified by elutriation with
pentane.
Yield: 2.54 g, 13.5 mmol, 97 %, light yellow oil.
1H-NMR (300 MHz, RT, CDCl3): δ = 11.30 (s, br, 1H, COOH), 4.08 (t, 3J = 6.8 Hz, 2H,
OCH2), 3.36 (s, 2H, OCCH2CO), 1.57 (m, 2H, CH2), 1.22 (m, 6H, CH2), 0.80 (t, 3J =
6.9 Hz, 3H, CH3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 171.53 (1C, CO), 166.57 (1C, CO), 65.72 (1C,
OCH2), 40.78 (1C, OCCH2CO), 31.06, 28.06, 25.12, 22.21 (4C, CH2), 13.63 (1C, CH3)
ppm.
MS (FAB, NBA): m/z = 189 [M+H]+; MWcalc.: 188.22 g/mol.
IR (ATR): ν̃ = 3146, 2901, 2733, 1723, 1693, 1468, 1431, 1384, 1234, 1187, 1058,
936, 693 cm−1.
EA: C9H16O4: calcd. C 57.43, H 8.57, O 34.00; found C 57.09, H 8.63.
Malonic acid monooctadecylester (18)
O
O
OH
O
18
Compound 18 was synthesized accord-
ing to general procedure GP 6 from 1-
Octadecanol (4.69 g, 17.3 mmol) and
MELDRUM’s acid (2.5 g, 17.3 mmol) and
purified by elutriation with pentane.
Yield: 6.11 g, 17.1 mmol, 99 %, white solid.
1H-NMR (300 MHz, RT, CDCl3): δ = 10.08 (s, br, 1H, COOH), 4.18 (t, 3J = 6.8 Hz, 2H,
OCH2), 3.44 (s, 2H, OCCH2CO), 1.66 (m, 2H, CH2), 1.26 (m, 30H, CH2), 0.88 (t, 3J =
6.8 Hz, 3H, CH3) ppm.
158

Chapter 5 Experimental Part
13C-NMR (75 MHz, RT, CDCl3): δ = 170.65 (1C, CO), 167.34 (1C, CO), 66.22 (1C,
OCH2), 40.47 (1C, OCCH2CO), 31.91 (1C, CH2), 29.69 (5C, CH2), 29.65 (2C, CH2),
29.63, 29.55, 29.48, 29.35, 29.16, 28.36, 25.73, 22.68, (8C, CH2), 14.11 (1C, CH3)
ppm.
MS (FAB, NBA): m/z = 357 [M+H]+; MWcalc.: 356.54 g/mol.
IR (ATR): ν̃ = 3126, 2920, 2850, 2742, 1746, 1692, 1468, 1429, 1344, 1282, 1244,
1166, 1058, 927, 726, 680 cm−1.
EA: C21H40O4: calcd. C 70.74, H 11.31, O 17.95; found C 70.55, H 11.59.
Malonic acid mono(1-octylnonyl)ester (19)
O
O
OH
O
19
Compound 19 was synthesized according to general pro-
cedure GP 6 from heptadecan-9-ol (3 g, 11.7 mmol) and
MELDRUM’s acid (1.69 g, 11.7 mmol) and purified by elu-
triation with pentane.
Yield: 3.85 g, 11.2 mmol, 96 %, white solid.
1H-NMR (300 MHz, RT, CDCl3): δ = 9.01 (s, br, 1H, COOH), 4.96 (quin, 3J = 6.7 Hz,
1H, OCH), 3.42 (s, 2H, OCCH2CO), 1.54 (m, 4H, CHCH2), 1.25 (m, 24H, CH2), 0.88
(t, 3J = 6.7 Hz, 6H, CH3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 171.07 (1C, CO), 167.08 (1C, CO), 76.78 (1C,
CH), 40.70 (1C, OCCH2CO), 33.85 (2C, CH2), 31.82 (2C, CH2), 29.42 (4C, CH2),
29.20 (2C, CH2), 25.15 (2C, CH2), 22.63 (2C, CH2), 14.09 (2C, CH3) ppm.
MS (FAB, NBA): m/z = 343 [M+H]+; MWcalc.: 342.51 g/mol.
IR (ATR): ν̃ = 3113, 2958, 2919, 2850, 1730, 1684, 1468, 1413, 1321, 1274, 1182,
1128, 1081, 1004, 958, 888, 757 cm−1.
EA: C20H38O4: calcd. C 70.13, H 11.18, O 18.68; found C 69.88, H 11.21.
159

Chapter 5 Experimental Part
6-(Benzyloxy)hexanoic acid (20)
O OH
O
20
Freshly crushed 85 % KOH (49.2 g, 0.745 mol) was added
to a solution of benzyl bromide (120 g, 0.7 mol) and ǫ-
caprolactone (20.06 g, 0.176 mol) in 300 mL toluene. The
mixture was heated at reflux for 48 h using a Dean-Stark
apparatus, then diluted with 200 mL of Et2O and washed with 300 mL of H2O. The
aqueous layer was extracted twice with 200 mL Et2O. The combined organic layers
were concentrated in vacuo to 150 mL. The aqueous layer was cooled in an ice bath
and 2M H2SO4 (180 mL) was added. The turbid aqueous solution was extracted with
CH2Cl2 (3 x 200 mL), dried over anhydrous MgSO4 and concentrated in vacuo to afford
20 as a pale yellow oil. To the residue of the toluene/Et2O layers NaOH (16 g, 0.4 mol)
and H2O (80 mL) was added and the resulting mixture was heated at reflux for 24 h.
The aqueous layer was separated, diluted to 250 mL with H2O and washed with Et2O
(3 x 100 mL). The aqueous layer was acidified with a slurry of 25 mL conc. H2SO4 in
100 mL of ice and extracted with CH2Cl2 (3 x 100 mL). The organic layers were dried
over anhydrous MgSO4 and the solvent was removed in vacuo to afford 20 as a pale
yellow oil.
Yield: 36.3 g, 0.16 mmol, 92.7 %, pale yellow oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 10.41 (s, br, 1H, COOH), 7.34 (m, 5H, arom.H),
4.51 (s, 2H, ArCH2O), 3.48 (t, 3J = 6.5 Hz, 2H, OCH2), 2.37 (t, 3J = 7.6 Hz, 2H,
CH2COOH), 1.66 (m, 4H, CH2), 1.45 (m, 2H, CH2) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 179.02 (1C, COOH), 138.47 (1C, ArC), 128.31
(2C, ArC), 127.59 (2C, ArC), 127.47 (1C, ArC), 72.91 (1C, ArCH2O), 70.06 (1C, OCH2),
33.89 (1C, CH2COOH), 29.42 (1C, CH2CH2COOH), 25.75 (1C, CH2), 24.54 (1C, CH2).
MS (FAB, NBA): m/z = 223 [M+H]+; MWcalc.: 222.28 g/mol.
IR (ATR): ν̃ = 3065, 2933, 2876, 1717, 1614, 1598, 1467, 1443, 1365, 1244, 1201,
1145, 1098, 987, 902, 865, 748, 718 cm−1.
EA: C13H18O3: calcd. C 70.24, H 8.16, O 21.59; found: C 70.09, H 8.18.
160

Chapter 5 Experimental Part
tert-Butyl 6-(benzyloxy)hexanoate (21)
OO
O
21
A solution of 20 (30 g, 0.135 mol) in dry CH2Cl2 (30 mL)
was treated with condensed isobutene (100 mL at -60°C )
and conc. H2SO4 (2 mL). The resulting solution was
stirred for three days at room temperature, then neutral-
ized with a solution of K2CO3 (25 g) in H2O (300 mL). The organic layer was washed
with K2CO3 (saturated solution), citric acid (10 wt % in H2O), and H2O. After the mix-
ture was dried over anhydrous MgSO4, filtered, and concentrated, a pale yellow oil was
obtained.
Yield: 33.6 g, 0.12 mmol, 89.3 %, pale yellow oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 7.34 (m, 5H, arom.H), 4.51 (s, 2H, ArCH2O), 3.48
(t, 3J = 6.6 Hz, 2H, OCH2), 2.22 (t, 3J = 7.5 Hz, 2H, CH2COOtBu), 1.62 (m, 4H, CH2),
1.45 (s, 9H, C(CH3)3), 1.42 (m, 2H, CH2) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.07 (1C, COOtBu), 138.54 (1C, ArC),
128.28 (2C, ArC), 127.54 (2C, ArC), 127.43 (1C, ArC), 79.97 (1C, C(CH3)3), 72.86 (1C,
ArCH2O), 70.19 (1C, OCH2), 35.55 (1C, CH2COOtBu), 29.48 (1C, CH2CH2COOtBu),
28.15 (3C, C(CH3)3), 25.75 (1C, CH2), 24.97 (1C, CH2).
MS (FAB, NBA): m/z = 278 [M]+; MWcalc.: 278.39 g/mol.
IR (ATR): ν̃ = 3033, 2986, 2901, 2865, 1745, 1463, 1364, 1254, 1165, 1121, 863, 768,
689 cm−1.
EA: C17H26O3: calcd. C 73.34, H 9.41, O 17.24; found: C 73.14, H 9.45.
tert-Butyl 6-hydroxyhexanoate (22)
HOO
O
22
A solution of 21 (26 g, 93 mmol) in dry methanol (300 mL) was
treated with 10% palladium on carbon (3 g) and H2 at room
temperature. After 48 h, the suspension was filtered through
celite and concentrated under reduced pressure to give 22 as
an oily product in quantitative yield.
161

Chapter 5 Experimental Part
Yield: 17.6 g, 93 mmol, 100 %, pale yellow oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 3.63 (t, 3J = 6.5 Hz, 2H, OCH2), 2.22 (t, 3J = 7.4
Hz, 2H, CH2COOtBu), 1.81 (s, br, 1H, OH), 1.58 (m, 4H, CH2), 1.43 (s, 9H, C(CH3)3),
1.40 (m, 2H, CH2) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.19 (1C, COOtBu), 80.06 (1C, C(CH3)3),
62.54 (1C, HOCH2), 35.41 (1C, CH2COOtBu), 32.29 (1C, CH2CH2OH), 28.05 (3C,
C(CH3)3), 25.15 (1C, CH2), 24.68 (1C, CH2).
MS (FAB, NBA): m/z = 188 [M]+; MWcalc.: 188.26 g/mol.
IR (ATR): ν̃ = 2998, 2941, 2876, 1793, 1743, 1465, 1432, 1387, 1264, 1161, 1065,
1028, 989, 963, 901, 874, 799, 735 cm−1.
EA: C10H20O3: calcd. 63.80, H 10.71, O 25.50; found: C 63.53, H 10.59.
5-(tert-Butoxycarbonyl)pentyl hexyl malonate (23)
OO
O
OOO
23
Compound 23 was synthesized according to
general procedure GP 2 from 17 (1 g, 5.31
mmol), 22 (1 g, 5.31 mmol), DMAP (64.9 mg,
0.53 mmol) and DCC (1.1 g, 5.31 mmol). The
crude product was purified by column chromatography (SiO2; hexane/ethyl acetate,
100:30) to give 23 as a colorless oil.
Yield: 1.43 g, 3.98 mmol, 75 %, colorless oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 3.89 (m, 4H, OCH2), 3.12 (s, 2H, OCCH2CO),
1.97 (t, 3J = 7.5 Hz, 2H, CH2CO), 1.41 (m, 6H, CH2), 1.20 (s, 9H, CH3), 1.08 (m, 8H,
CH2), 0.66 (t, 3J = 6.4 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 172.08 (1C, CO), 165.99 (2C, CO), 79.18 (1C,
C(CH3)3), 64.81 (1C, OCH2), 64.52 (1C, OCH2), 40.89 (1C, OCCH2CO), 34.59 (1C,
CH2CO), 30.78, 27.84, 27.60 (3C, CH2), 27.38 (3C, C(CH3)3), 24.84, 24.70, 24.00,
21.88 (4C, CH2), 13.29 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 358 [M]+; MWcalc.: 358.47 g/mol.
162

Chapter 5 Experimental Part
IR (ATR): ν̃ = 2935, 2866, 1730, 1460, 1367, 1329, 1259, 1151, 1043, 950, 896, 850,
757, 687 cm−1.
EA: C19H34O6: calcd. C 63.66, H 9.56, O 26.78; found: C 63.91, H 9.78.
5-(tert-Butoxycarbonyl)pentyl octadecyl malonate (24)
OO
O
OOO
24
Compound 24 was synthe-
sized according to general
procedure GP 2 from 18
(3.79 g, 10.64 mmol), 22
(2.00 g, 10.64 mmol), DMAP (130 mg, 1.06 mmol) and DCC (2.20 g, 10.64 mmol).
The crude product was purified by column chromatography (SiO2; hexane/ethyl ac-
etate, 60:15) to give 24 as a colorless oil.
Yield: 4.54 g, 8.62 mmol, 81 %, colorless oil.
1H-NMR (300 MHz, RT, CDCl3): δ = 4.13 (t, 3J = 6.7 Hz, 2H, OCH2), 4.12 (t, 3J = 6.8
Hz, 2H, OCH2), 3.35 (s, 2H, OCCH2CO), 2.20 (t, 3J = 7.4 Hz, 2H, CH2CO), 1.63 (m,
6H, CH2), 1.43 (s, 9H, CH3), 1.25 (m, 32H, CH2), 0.87 (t, 3J = 6.4 Hz, 3H, CH3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 172.77 (1C, CO), 166.57 (2C, CO), 80.00 (1C,
C(CH3)3), 65.62 (1C, OCH2), 65.27 (1C, OCH2), 41.58 (1C, OCCH2CO), 35.29 (1C,
CH2CO), 31.88, 29.65, 29.61, 29.53, 29.47, 29.31, 29.16, 28.42, 28.15 (15C, CH2),
28.05 (3C, C(CH3)3), 25.74, 25.27, 24.60, 22.64 (4C, CH2), 14.06 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 526 [M]+; MWcalc.: 526.79 g/mol.
IR (ATR): ν̃ = 2927, 2858, 1738, 1460, 1367, 1329, 1259, 1151, 1043, 896, 850, 726,
687 cm−1.
EA: C31H58O6: C 70.68, H 11.10, O 18.22. found: C 70.32, H 11.31.
163

Chapter 5 Experimental Part
5-(tert-Butoxycarbonyl)pentyl 1-octylnonyl malonate (25)
O
O
O
O
O
O
25
Compound 25 was synthesized according to
general procedure GP 2 from 19 (2.25 g,
6.57 mmol), 22 (1.24 g, 6.57 mmol), DMAP
(81 mg, 0.66 mmol) and DCC (1.36 g, 6.57
mmol). The crude product was purified by
column chromatography (SiO2; hexane/ethyl acetate, 90:12) to give 25 as a colorless
oil.
Yield: 2.84 g, 5.39 mmol, 82 %, colorless oil.
1H-NMR (300 MHz, RT, CDCl3): δ = 4.91 (quin, 3J = 6.3 Hz, 1H, OCH), 4.14 (t, 3J =
6.7 Hz, 2H, OCH2), 3.35 (s, 2H, OCCH2CO), 2.22 (t, 3J = 7.4 Hz, 2H, CH2CO), 1.64
(m, 6H, CH2), 1.55 (m, 2H, CH2), 1.45 (s, 9H, CH3), 1.26 (m, 26H, CH2), 0.81 (t, 3J =
6.6 Hz, 6H, CH3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 172.86 (1C, CO), 166.74 (1C, CO), 166.35 (1C,
CO), 80.08 (1C, C(CH3)3), 75.97 (1C, CH), 65.28 (1C, OCH2), 41.94 (1C, OCCH2CO),
35.33 (1C, CH2CO), 33.93 (2C, CH(CH2)2), 31.84, 29.48, 29.47, 29.23, 28.20 (9C,
CH2), 28.09 (3C, C(CH3)3), 25.30, 25.18 (3C, CH2), 24.65 (1C, CH2CH2CO), 22.65
(2C, CH2CH3), 14.10 (2C, CH3) ppm.
MS (FAB, NBA): m/z = 512 [M]+; MWcalc.: 512.76 g/mol.
IR (ATR): ν̃ = 2927, 2858, 1738, 1460, 1367, 1321, 1259, 1151, 1043, 989, 896, 850,
803, 726, 680 cm−1.
EA: C30H56O6: calcd. C 70.27, H 11.01, O 18.72; found: C 69.89, H 11.10.
164

Chapter 5 Experimental Part
Hexanoic acid hexyl malonate (26)
OO
O
OOOH
26
Compound 26 was synthesized according to gen-
eral procedure GP 5 from 23 (1.1 g, 3.07 mmol)
and trifluoroacetic acid (2.4 mL, 31.0 mmol) in 50
mL CH2Cl2.
Yield: 919 mg, 3.04 mmol, 99 %, white solid.
1H-NMR (300 MHz, RT, CDCl3): δ = 7.91 (s, br, 1H, COOH), 4.15 (t, 3J = 6.7 Hz, 2H,
OCH2), 4.14 (t, 3J = 6.8 Hz, 2H, OCH2), 3.38 (s, 2H, OCCH2CO), 2.37 (t, 3J = 7.4 Hz,
2H, CH2CO), 1.65 (m, 6H, CH2), 1.43 (m, 2H, CH2), 1.30 (m, 6H, CH2), 0.89 (t, 3J =
6.9 Hz, 3H, CH3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 179.42 (1C, COOH), 166.75 (1C, CO), 166.72
(1C, CO), 65.74 (1C, OCH2), 65.23 (1C, OCH2), 41.58 (1C, OCCH2CO), 33.76 (1C,
CH2CO), 31.32, 28.36, 28.07, 25.40, 25.24, 24.15, 22.48 (7C, CH2), 13.94 (1C, CH3)
ppm.
MS (FAB, NBA): m/z = 303 [M+H]+; MWcalc.: 302.36 g/mol.
IR (ATR): ν̃ = 2935, 2866, 1730, 1460, 1413, 1320, 1274, 1151, 1043, 1004, 950, 904,
796, 734 cm−1.
EA: C15H26O6: calcd. C 59.58, H 8.67, O 31.75. found: C 59.17, H 8.96.
Hexanoic acid octadecyl malonate (27)
OO
O
OOOH
27
Compound 27 was synthe-
sized according to general
procedure GP 5 from 24 (2.00
g, 3.80 mmol) and trifluo-
roacetic acid (2.9 mL, 38.0 mmol) in 100 mL CH2Cl2.
Yield: 1.77 g, 3.76 mmol, 99 %, white solid.
1H-NMR (300 MHz, RT, CDCl3): δ = 10.10 (s, br, 1H, COOH), 4.15 (t, 3J = 6.6 Hz, 2H,
OCH2), 4.13 (t, 3J = 6.7 Hz, 2H, OCH2), 3.37 (s, 2H, OCCH2CO), 2.37 (t, 3J = 7.4 Hz,
165

Chapter 5 Experimental Part
2H, CH2CO), 1.66 (m, 6H, CH2), 1.42 (m, 2H, CH2), 1.26 (m, 30H, CH2), 0.87 (t, 3J =
6.7 Hz, 3H, CH3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 179.49 (1C, COOH), 166.66 (1C, CO), 166.65
(1C, CO), 65.71 (1C, OCH2), 65.19 (1C, OCH2), 41.58 (1C, OCCH2CO), 33.77 (1C,
CH2CO), 31.90, 29.67, 29.65, 29.64, 29.62, 29.56, 29.49, 29.34, 29.19, 28.43, 28.10,
25.76, 25.26 (17C, CH2), 24.17 (1C, CH2CH2CO), 22.67 (1C, CH2), 14.09 (1C, CH3)
ppm.
MS (FAB, NBA): m/z = 471 [M+H]+; MWcalc.: 470.68 g/mol.
IR (ATR): ν̃ = 2920, 2850, 1746, 1707, 1468, 1406, 1352, 1313, 1259, 1182, 1050,
1004, 942, 726, 680 cm−1.
EA: C27H50O6: calcd. C 68.90, H 10.71, O 20.40; found: C 69.07, H 10.77.
Hexanoic acid 1-octylnonyl malonate (28)
O
O
O
O
O
OH
28
Compound 28 was synthesized according to
general procedure GP 5 from 25 (2.00 g, 3.80
mmol) and trifluoroacetic acid (2.9 mL, 38.0
mmol) in 100 mL of CH2Cl2.
Yield: 1.70 g, 3.72 mmol, 98 %, white solid.
1H-NMR (300 MHz, RT, CDCl3): δ = 10.82 (s, br, 1H, COOH), 4.91 (quin, 3J = 6.3 Hz,
1H, OCH), 4.15 (t, 3J = 6.7 Hz, 2H, OCH2), 3.36 (s, 2H, OCCH2CO), 2.37 (t, 3J = 7.4
Hz, 2H, CH2CO), 1.68 (m, 4H, CH(CH2)2), 1.53 (m, 4H, CH2), 1.43 (m, 2H, CH2), 1.26
(m, 24H, CH2), 0.88 (t, 3J = 6.8 Hz, 6H, CH3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 178.90 (1C, COOH), 166.75 (1C, CO), 166.38
(1C, CO), 76.03 (1C, CH), 65.14 (1C, OCH2), 41.93 (1C, OCCH2CO), 33.93 (2C,
CH(CH2)2), 33.68 (1C, CH2CO), 31.84, 29.49, 29.48, 29.23, 28.14, 25.27, 25.19 (12C,
CH2), 24.20 (1C, CH2CH2CO), 22.65 (2C, CH2CH3), 14.09 (2C, CH3) ppm.
MS (FAB, NBA): m/z = 457 [M+H]+; MWcalc.: 456.66 g/mol.
166

Chapter 5 Experimental Part
IR (ATR): ν̃ = 2927, 2858, 1730, 1460, 1413, 1321, 1274, 1159, 1043, 981, 896, 842,
796, 726 cm−1.
EA: C26H48O6: calcd. C 68.38, H 10.59, O 21.02; found: C 68.36, H 10.80.
Asymmetric dendritic malonate [C 6-G1] (31)
OO
O
OO HN
O
O
O
O
O O31
Compound 31 was synthesized accord-
ing to general procedure GP 3 from 26
(500 mg, 1.65 mmol), 29 (688 mg, 1.65
mmol), DMAP (41 mg, 0.33 mmol), 1-
HOBt (223 mg, 1.65 mmol) and EDC
(317 mg, 1.65 mmol) in dry CH2Cl2/THF
= 1/1 (100 mL). The crude product was purified by column chromatography (SiO2; hex-
ane/ethyl acetate, 20:15) to give 31 as a colorless oil.
Yield: 716 mg, 1.02 mmol, 62 %, colorless oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 6.01 (s, br, 1H, CONH), 4.16 (t, 3J = 6.8 Hz, 2H,
OCH2), 4.14 (t, 3J = 6.9 Hz, 2H, OCH2), 3.32 (s, 2H, OCCH2CO), 2.34 (t, 3J = 7.5
Hz, 6H, CH2COOtBu), 2.18 (t, 3J = 7.2 Hz, 2H, CH2CO), 1.99 (t, 3J = 7.6 Hz, 6H,
NHC(CH2)3), 1.62 (m, 6H, CH2), 1.45 (m, 2H, CH2), 1.41 (s, 27H, C(CH3)3), 1.28 (m,
6H, CH2), 0.89 (t, 3J = 6.5 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 172.98 (3C, COOtBu), 172.14 (1C, CONH),
166.75 (1C, CO), 166.69 (1C, CO), 80.44 (3C, C(CH3)3), 65.77 (1C, OCH2), 65.25
(1C, OCH2), 57.34 (1C, NHC(CH2)3), 41.58 (1C, OCCH2CO), 36.99 (1C, CH2CO),
31.33 (1C, CH2), 29.87 (3C, NHC(CH2)3), 29.63 (3C, CH2COOtBu), 28.36, 28.07 (2C,
CH2), 28.01 (9C, C(CH3)3), 25.98 (1C, CH2CH2CO), 25.40, 24.19, 22.55 (3C, CH2),
13.95 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 722 [M+Na]+, 699 [M]+, 532 [M+H-3C4H8]+; MWcalc.: 699.91
g/mol.
IR (ATR): ν̃ = 3349, 3001, 2936, 2886, 1716, 1654, 1563, 1501, 1476, 1432, 1401,
1366, 1318, 1253, 1114, 987, 865, 807, 759, 723, 695 cm−1.
167

Chapter 5 Experimental Part
EA: C37H65NO11: calcd. C 63.49, H 9.36, N 2.00, O 25.15; found: C 63.12, H 9.44, N
2.03.
Asymmetric dendritic malonate [C 18-G1] (32)
OO
O
OO HN
O
O
O
O
O O32
Compound 32 was
synthesized accord-
ing to general pro-
cedure GP 3 from
27 (1.5 g, 3.19
mmol), 29 (1.32 g,
3.19 mmol), DMAP (78 mg, 0.64 mmol), 1-HOBt (431 mg, 3.19 mmol) and EDC (673
mg, 3.51 mmol) in dry CH2Cl2/THF = 1/1 (150 mL). The crude product was purified by
column chromatography (SiO2; hexane/ethyl acetate, 15:10) to give 32 as a colorless
oil.
Yield: 1.88 g, 2.17 mmol, 68 %, colorless oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 5.90 (s, br, 1H, CONH), 4.13 (t, 3J = 6.6 Hz, 2H,
OCH2), 4.12 (t, 3J = 6.8 Hz, 2H, OCH2), 3.36 (s, 2H, OCCH2CO), 2.22 (t, 3J = 7.7
Hz, 6H, CH2COOtBu), 2.11 (t, 3J = 7.4 Hz, 2H, CH2CO), 1.95 (t, 3J = 7.8 Hz, 6H,
NHC(CH2)3), 1.63 (m, 6H, CH2), 1.43 (s, 27H, C(CH3)3), 1.25 (m, 32H, CH2), 0.87 (t,
3J = 6.7 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 172.87 (3C, COOtBu), 172.07 (1C, CONH),
166.66 (1C, CO), 166.61 (1C, CO), 80.63 (3C, C(CH3)3), 65.67 (1C, OCH2), 65.28 (1C,
OCH2), 57.28 (1C, NHC(CH2)3), 41.56 (1C, OCCH2CO), 37.22 (1C, CH2CO), 31.89
(1C, CH2), 29.94 (3C, NHC(CH2)3), 29.79 (3C, CH2COOtBu), 29.66, 29.64, 29.62,
29.55, 29.48, 29.33, 29.18, 28.43, 28.18 (14C, CH2), 28.03 (9C, C(CH3)3), 25.75 (1C,
CH2), 25.48 (1C, CH2CH2CO), 25.23, 22.65 (2C, CH2), 14.09 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 891 [M+Na]+, 868 [M]+, 700 [M+H-3C4H8]+; MWcalc.: 868.23
g/mol.
IR (ATR): ν̃ = 3321, 2920, 2858, 1730, 1645, 1545, 1460, 1421, 1367, 1321, 1251,
168

Chapter 5 Experimental Part
1151, 1020, 950, 850, 803, 757, 718, 680 cm−1.
EA: C49H89NO11: calcd. C 67.78, H 10.33, N 1.61, O 20.27; found: C 67.93, H 10.58,
N 1.64.
Asymmetric dendritic malonate [(C 8)2-G1] (33)
O
O
O
O HN
O
O
O
O
O O
O33
Compound 33 was synthesized ac-
cording to general procedure GP
3 from 28 (1 g, 2.19 mmol), 29
(0.91 g, 2.19 mmol), DMAP (54
mg, 0.44 mmol), 1-HOBt (296 mg,
2.19 mmol) and EDC (462 mg, 2.41
mmol) in dry CH2Cl2/THF = 1/1 (100 mL). The crude product was purified by column
chromatography (SiO2; hexane/ethyl acetate, 20:10 to 15/10) to give 33 as a colorless
oil.
Yield: 1.20 g, 1.40 mmol, 64 %, colorless oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 5.91 (s, br, 1H, CONH), 4.91 (quin, 3J = 6.2
Hz,1H, OCH), 4.13 (t, 3J = 6.7 Hz, 2H, OCH2), 3.36 (s, 2H, OCCH2CO), 2.22 (t, 3J =
7.8 Hz, 6H, CH2COOtBu), 2.11 (t, 3J = 7.6 Hz, 2H, CH2CO), 1.97 (t, 3J = 7.9 Hz, 6H,
NHC(CH2)3), 1.64 (m, 4H, CHCH2), 1.52 (m, 2H, CH2), 1.43 (s, 27H, C(CH3)3), 1.41
(m, 2H, CH2), 1.26 (m, 26H, CH2), 0.88 (t, 3J = 6.8 Hz, 6H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.40 (3C, COOtBu), 172.59 (1C, CONH),
167.20 (1C, CO), 166.85 (1C, CO), 80.69 (3C, C(CH3)3), 75.96 (1C, CH), 65.19 (1C,
OCH2), 57.20 (1C, NHC(CH2)3), 41.72 (1C, OCCH2CO), 37.04 (1C, CH2CO), 33.70
(2C, CH(CH2)2), 31.61, 30.04 (3C, CH2), 29.71 (3C, NHC(CH2)3), 29.55 (3C,
CH2COOtBu), 29.43, 29.24, 29.22, 28.98, 27.98 (6C, CH2), 27.80 (9C, C(CH3)3),
25.26 (1C, CH2CH2CO), 25.00, 24.91, 22.37 (5C, CH2), 13.78 (2C, CH3) ppm.
MS (FAB, NBA): m/z = 877 [M+Na]+, 854 [M]+, 686 [M+H-3C4H8]+; MWcalc.: 854.20
g/mol.
IR (ATR): ν̃ = 3383, 3329, 2935, 2858, 1730, 1661, 1537, 1460, 1367, 1321, 1259,
169

Chapter 5 Experimental Part
1159, 1043, 966, 850, 803, 734 cm−1.
EA: C48H87NO11: calcd. C 67.49, H 10.27, N 1.64, O 20.60; found: C 67.20, H 10.17,
N 1.64.
Asymmetric dendritic malonate [C 6-G2] (34)
OO
O
OO HN
HN
OHN
O
O
O
O
O
O
OONH
OO
OO
O
O
OO
O O O
O
34
Compound 34 was synthe-
sized according to general
procedure GP 3 from 26 (330
mg, 1.09 mmol), 30 (1.57 g,
1.09 mmol), DMAP (27 mg,
0.22 mmol), 1-HOBt (147 mg,
1.09 mmol) and EDC (230 mg,
1.20 mmol) in dry CH2Cl2/THF
= 1/1 (100 mL). The crude
product was purified by col-
umn chromatography (SiO2; hexane/ethyl acetate, 10:10 to 10:15) to give 34 as a
colorless oil.
Yield: 1.38 g, 0.80 mmol, 73 %, colorless oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 7.50 (s, br, 1H, CONH), 6.15 (s, br, 3H, CONH),
3.98 (t, 3J = 6.5 Hz, 2H, OCH2), 3.96 (t, 3J = 6.7 Hz, 2H, OCH2), 3.24 (s, 2H,
OCCH2CO), 2.06 (m, 26H, CH2COOtBu, OCCH2), 1.81 (m, 24H, NHC(CH2)3), 1.50
(m, 6H, CH2), 1.38 (m, 2H, CH2), 1.30 (m, 83H, C(CH3)3, CH2), 1.17 (m, 4H, CH2),
0.71 (t, 3J = 6.8 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 172.87 (1C, CONH), 172.75 (3C, CONH),
172.32 (9C, COOtBu), 166.38 (1C, CO), 166.35 (1C, CO), 80.09 (9C, C(CH3)3), 65.27
(1C, OCH2), 65.04 (1C, OCH2), 57.24 (1C, NHC(CH2)3), 57.07 (3C, NHC(CH2)3),
41.23 (1C, OCCH2CO), 36.76 (1C, CH2CO), 31.53 (3C, NHC(CH2)3), 31.34 (3C,
CH2CON), 31.06 (1C, CH2), 29.49 (9C, NHC(CH2)3), 29.37 (9C, CH2COOtBu), 28.73,
28.54 (2C, CH2), 27.72 (27C, C(CH3)3), 25.27, 25.15 (2C, CH2), 24.97 (1C,
170
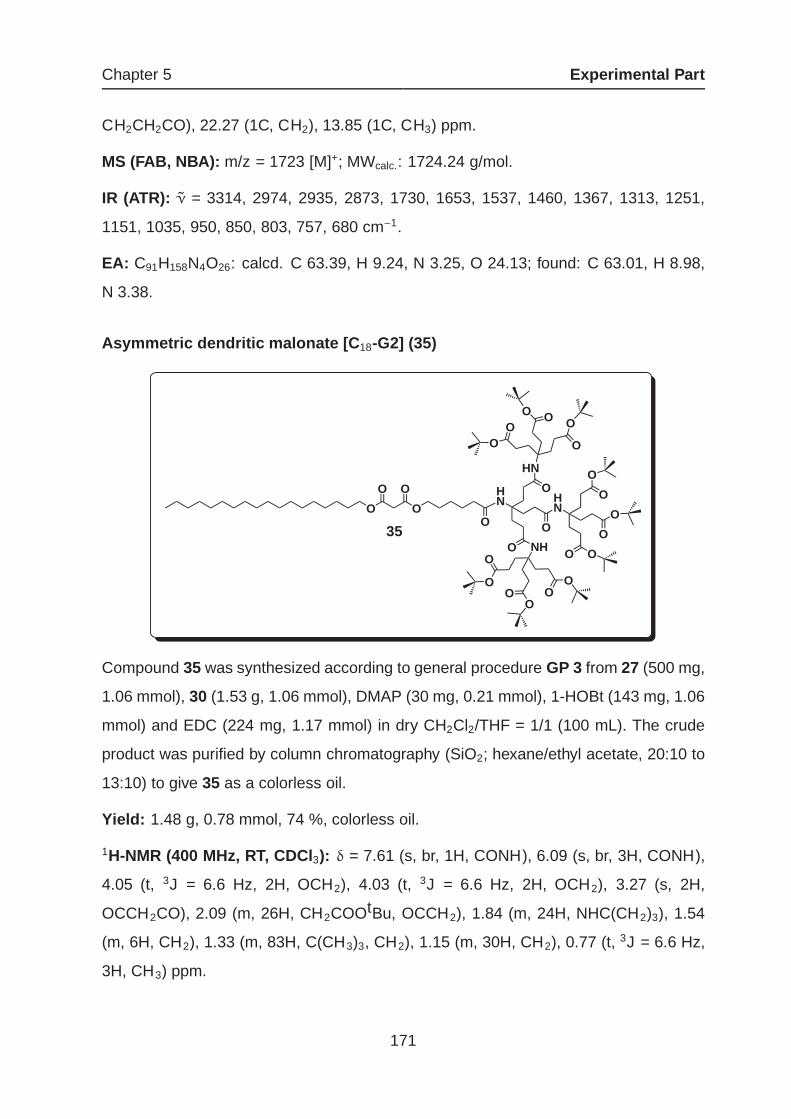
Chapter 5 Experimental Part
CH2CH2CO), 22.27 (1C, CH2), 13.85 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 1723 [M]+; MWcalc.: 1724.24 g/mol.
IR (ATR): ν̃ = 3314, 2974, 2935, 2873, 1730, 1653, 1537, 1460, 1367, 1313, 1251,
1151, 1035, 950, 850, 803, 757, 680 cm−1.
EA: C91H158N4O26: calcd. C 63.39, H 9.24, N 3.25, O 24.13; found: C 63.01, H 8.98,
N 3.38.
Asymmetric dendritic malonate [C 18-G2] (35)
OO
O
OO HN
HN
OHN
O
O
O
O
O
O
OONH
OO
OO
O
O
OO
O O O
O
35
Compound 35 was synthesized according to general procedure GP 3 from 27 (500 mg,
1.06 mmol), 30 (1.53 g, 1.06 mmol), DMAP (30 mg, 0.21 mmol), 1-HOBt (143 mg, 1.06
mmol) and EDC (224 mg, 1.17 mmol) in dry CH2Cl2/THF = 1/1 (100 mL). The crude
product was purified by column chromatography (SiO2; hexane/ethyl acetate, 20:10 to
13:10) to give 35 as a colorless oil.
Yield: 1.48 g, 0.78 mmol, 74 %, colorless oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 7.61 (s, br, 1H, CONH), 6.09 (s, br, 3H, CONH),
4.05 (t, 3J = 6.6 Hz, 2H, OCH2), 4.03 (t, 3J = 6.6 Hz, 2H, OCH2), 3.27 (s, 2H,
OCCH2CO), 2.09 (m, 26H, CH2COOtBu, OCCH2), 1.84 (m, 24H, NHC(CH2)3), 1.54
(m, 6H, CH2), 1.33 (m, 83H, C(CH3)3, CH2), 1.15 (m, 30H, CH2), 0.77 (t, 3J = 6.6 Hz,
3H, CH3) ppm.
171

Chapter 5 Experimental Part
13C-NMR (100.5 MHz, RT, CDCl3): δ = 172.88 (1C, CONH), 172.66 (3C, CONH),
172.38 (9C, COOtBu), 166.46 (1C, CO), 166.43 (1C, CO), 80.25 (9C, C(CH3)3), 65.39
(1C, OCH2), 65.13 (1C, OCH2), 57.26 (1C, NHC(CH2)3), 57.16 (3C, NHC(CH2)3),
41.30 (1C, OCCH2CO), 36.85 (1C, CH2CO), 31.65 (4C, NHC(CH2)3, CH2), 31.47 (3C,
CH2CON), 29.54 (1C, CH2), 29.52 (9C, NHC(CH2)3), 29.42 (9C, CH2COOtBu), 29.41,
29.38, 29.32, 29.25, 29.09, 28.96, 28.21, 27.99 (13C, CH2), 27.83 (27C, C(CH3)3),
25.53 (1C, CH2), 25.32 (1C, CH2CH2CO), 25.02, 22.42 (2C, CH2), 13.88 (1C, CH3)
ppm.
MS (FAB, NBA): m/z = 1891 [M]+; MWcalc.: 1892.56 g/mol.
IR (ATR): ν̃ = 3321, 2981, 2927, 2857, 1730, 1653, 1545, 1460, 1367, 1313, 1251,
1213, 1151, 1035, 958, 850, 757, 680 cm−1.
EA: C103H182N4O26: calcd. C 65.37, H 9.69, N 2.96, O 21.98; found: C 65.47, H 9.79,
N 3.04.
172

Chapter 5 Experimental Part
Asymmetric dendritic malonate [(C 8)2-G2] (36)
O
O
O
O HN
HN
OHN
O
O
O
O
O
O
OONH
OO
OO
O
O
OO
O O O
O
O36
Compound 36 was synthe-
sized according to general
procedure GP 3 from 28
(350 mg, 0.77 mmol), 30
(1.10 g, 0.77 mmol), DMAP
(19 mg, 0.15 mmol), 1-
HOBt (104 mg, 0.77 mmol)
and EDC (162 mg, 0.85
mmol) in dry CH2Cl2/THF
= 1/1 (100 mL). The crude
product was purified by column chromatography (SiO2; hexane/ethyl acetate, 13:10 to
10:10) to give 36 as a colorless oil.
Yield: 998 mg, 0.53 mmol, 69 %, colorless oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 7.57 (s, br, 1H, CONH), 6.09 (s, br, 3H, CONH),
4.84 (quin, 3J = 6.3 Hz, 1H, CH), 4.08 (t, 3J = 6.7 Hz, 2H, OCH2), 3.29 (s, 2H,
OCCH2CO), 2.13 (m, 26H, CH2COOtBu, OCCH2), 1.88 (m, 24H, NHC(CH2)3), 1.60
(m, 4H, CH(CH2)2), 1.46 (m, 4H, CH2), 1.37 (m, 83H, C(CH3)3, CH2), 1.19 (m, 24H,
CH2), 0.81 (t, 3J = 6.8 Hz, 6H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.11 (1C, CONH), 172.90 (3C, CONH),
172.64 (9C, COOtBu), 166.75 (1C, CO), 166.36 (1C, CO), 80.40 (9C, C(CH3)3), 75.72
(1C, CH), 65.18 (1C, OCH2), 57.28 (1C, NHC(CH2)3), 57.23 (3C, NHC(CH2)3), 41.64
(1C, OCCH2CO), 36.88 (1C, CH2CO), 33.70 (2C, CH2), 31.66 (3C, NHC(CH2)3), 31.62
(2C, CH2), 31.53 (3C, CH2CON), 29.60 (9C, NHC(CH2)3), 29.56 (9C, CH2COOtBu),
29.25, 29.23, 29.00, 28.06 (7C, CH2), 27.85 (27C, C(CH3)3), 25.36 (1C, CH2CH2CO),
25.06, 24.94, 22.41 (5C, CH2), 13.87 (2C, CH3) ppm.
MS (FAB, NBA): m/z = 1877 [M]+; MWcalc.: 1878.53 g/mol.
IR (ATR): ν̃ = 3306, 2981, 2935, 2866, 1730, 1661, 1537, 1460, 1367, 1313, 1259,
1151, 1105, 1035, 958, 850, 803, 734 cm−1.
173

Chapter 5 Experimental Part
EA: C102H180N4O26: calcd. C 65.22, H 9.66, N 2.98, O 22.14; found: C 64.83, H 9.77,
N 3.10.
Asymmetric dendritic monoadduct [C 6-G1] (37)
OO
O
OO HN
O
O
O
O
O O
37
Compound 37 was synthesized accord-
ing to general procedure GP 4 from
malonate 31 (400 mg, 0.57 mmol), C60
(494 mg, 0.69 mmol), CBr4 (209 mg,
0.63 mmol) and DBU (94 µL, 0.63
mmol) in 250 mL toluene. The crude
product was purified by column chro-
matography (SiO2; toluene/ethyl ac-
etate, 80:15 to 80:30) to give 37 as a red brownish solid.
Yield: 396 mg, 0.28 mmol, 49 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 5.99 (s, br, 3H, CONH), 4.54 (t, 3J = 6.7 Hz, 2H,
OCH2), 4.52 (t, 3J = 6.8 Hz, 2H, OCH2), 2.36 (t, 3J = 7.5 Hz, 6H, CH2COOtBu), 2.22
(t, 3J = 7.1 Hz, 2H, CH2), 2.00 (t, 3J = 7.5 Hz, 6H, NHC(CH2)3), 1.86 (m, 2H, CH2),
1.67 (m, 4H, CH2), 1.49 (m, 2H, CH2), 1.41 (s, 27H, C(CH3)3), 1.28 (m, 6H, CH2), 0.91
(t, 3J = 6.5 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 172.93 (3C, COOtBu), 172.03 (1C, CONH),
163.66 (1C, CO), 163.57 (1C, CO), 145.15, 145.14, 145.12, 145.07, 145.05, 145.03,
145.01, 144.76, 144.57, 144.53, 144.52, 144.49, 144.48, 143.75, 142.93, 142.92,
142.88, 142.85, 142.09, 142.05, 141.79, 141.71, 140.86, 140.75, 139.05, 138.89
(58C, C60-sp2), 80.49 (3C, C(CH3)3), 71.78 (2C, C60-sp3), 67.66 (1C, OCH2), 67.28
(1C, OCH2), 57.30 (1C, NHC(CH2)3), 51.38 (1C, OCCCO), 37.09 (1C, CH2CO), 31.29
(1C, CH2), 29.85 (3C, NHC(CH2)3), 29.65 (3C, CH2COOtBu), 28.36, 28.12 (2C, CH2),
28.02 (9C, C(CH3)3), 25.96 (1C, CH2CH2CO), 25.41, 24.20, 22.43 (3C, CH2), 13.95
(1C, CH3) ppm.
MS (FAB, NBA): m/z = 1440 [M+Na]+, 1417 [M]+, 720 [C60]+; MWcalc.: 1418.54 g/mol.
174

Chapter 5 Experimental Part
IR (ATR): ν̃ = 2989, 2945, 1801, 1732, 1664, 1535, 1456, 1418, 1396, 1356, 1254,
1201, 1165, 1101, 1065, 989, 963, 841, 787, 723, 701, 687, 645 cm−1.
UV/Vis (CH 2Cl2): λmax = 254, 323, 425, 475 nm.
Asymmetric dendritic monoadduct [C 18-G1] (38)
OO
O
OO HN
O
O
O
O
O O
38
Compound 38 was
synthesized accord-
ing to general pro-
cedure GP 4 from
malonate 32 (500
mg, 0.58 mmol),
C60 (498 mg, 0.69
mmol), CBr4 (212
mg, 0.64 mmol) and DBU (96 µL, 0.64 mmol) in 250 mL toluene. The crude product
was purified by column chromatography (SiO2; toluene/ethyl acetate, 80:10 to 80:15)
to give 38 as a red brownish solid.
Yield: 488 mg, 0.31 mmol, 53 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 5.90 (s, br, 1H, CONH), 4.50 (t, 3J = 6.6 Hz,
2H, OCH2), 4.49 (t, 3J = 6.7 Hz, 2H, OCH2), 2.22 (t, 3J = 7.8 Hz, 6H, CH2COOtBu),
2.13 (t, 3J = 7.6 Hz, 2H, CH2CO), 1.98 (t, 3J = 7.8 Hz, 6H, NHC(CH2)3), 1.85 (m, 4H,
OCH2CH2), 1.70 (m, 2H, CH2), 1.44 (s, 27H, C(CH3)3), 1.25 (m, 32H, CH2), 0.88 (t, 3J
= 6.8 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 172.88 (3C, COOtBu), 171.96 (1C, CONH),
163.67 (1C, CO), 163.59 (1C, CO), 145.37, 145.33, 145.25, 145.16, 145.14, 144.67,
144.63, 144.59, 143.86, 143.09, 143.06, 143.07, 143.00, 142.97, 142.19, 141.88,
140.93, 138.98, 138.93 (58C, C60-sp2), 80.69 (3C, C(CH3)3), 71.65 (2C, C60-sp3), 67.50
(1C, OCH2), 67.13 (1C, OCH2), 57.34 (1C, NHC(CH2)3), 49.87 (1C, OCCCO), 37.22
(1C, CH2CO), 31.92 (1C, CH2), 30.00 (3C, NHC(CH2)3), 29.83 (3C, CH2COOtBu),
29.72, 29.70, 29.67, 29.62, 29.37, 29.22, 28.60, 28.35 (14C, CH2), 28.06 (9C, C(CH3)3),
175

Chapter 5 Experimental Part
26.00 (1C, CH2), 25.64 (1C, CH2CH2CO), 25.24, 22.69 (2C, CH2), 14.14 (1C, CH3)
ppm.
MS (FAB, NBA): m/z = 1609 [M+Na]+, 1586 [M]+, 720 [C60]+; MWcalc.: 1586.86 g/mol.
IR (ATR): ν̃ = 3001, 2953, 1823, 1801, 1743, 1674, 1553, 1467, 1421, 1388, 1327,
1299, 1207, 1163, 1089, 1045, 993, 942, 868, 734, 712, 679, 663 cm−1.
UV/Vis (CH 2Cl2): λmax = 254, 323, 425, 475 nm.
Asymmetric dendritic monoadduct [(C 8)2-G1] (39)
O
O
O
O HN
O
O
O
O
O O
O
39
Compound 39 was synthesized ac-
cording to general procedure GP 4
from malonate 33 (300 mg, 0.35
mmol), C60 (304 mg, 0.42 mmol),
CBr4 (130 mg, 0.39 mmol) and
DBU (59 µL, 0.39 mmol) in 175 mL
toluene. The crude product was
purified by column chromatography
(SiO2; toluene/ethyl acetate, 90:10 to 70:10) to give 39 as a red brownish solid.
Yield: 310 mg, 0.20 mmol, 56 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 5.89 (s, br, 1H, CONH), 5.25 (m,1H, OCH), 4.48
(t, 3J = 6.8 Hz, 2H, OCH2), 2.22 (t, 3J = 7.8 Hz, 6H, CH2COOtBu), 2.13 (t, 3J = 7.7 Hz,
2H, CH2CO), 1.97 (t, 3J = 7.8 Hz, 6H, NHC(CH2)3), 1.87 (m, 2H, CH2), 1.78 (m, 2H,
CH2), 1.68 (m, 6H, CHCH2, CH2), 1.44 (m, 29H, C(CH3)3, CH2), 1.27 (m, 22H, CH2),
0.88 (t, 3J = 6.8 Hz, 6H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.01 (3C, COOtBu), 172.08 (1C, CONH),
163.70 (1C, CO), 163.29 (1C, CO), 145.61, 145.46, 145.33, 145.25, 144.93, 144.77,
144.75, 144.65, 143.95, 143.94, 143.18, 143.09, 143.05, 142.28, 141.98, 141.00,
139.11, 138.89 (58C, C60-sp2), 80.69 (3C, C(CH3)3), 78.56 (1C, CH), 71.79 (2C, C60-
sp3), 67.12 (1C, OCH2), 57.31 (1C, NHC(CH2)3), 52.74 (1C, OCCCO), 37.16 (1C,
CH2CO), 33.87 (2C, CH(CH2)2), 31.83, 30.11 (3C, CH2), 29.92 (3C, NHC(CH2)3),
176

Chapter 5 Experimental Part
29.76 (3C, CH2COOtBu), 29.63, 29.55, 29.48, 29.21, 28.35 (6C, CH2), 28.01 (9C,
C(CH3)3), 25.59 (1C, CH2CH2CO), 25.32, 25.18, 22.60 (5C, CH2), 14.08 (2C, CH3)
ppm.
MS (FAB, NBA): m/z = 1595 [M+Na]+, 1572 [M]+, 720 [C60]+; MWcalc.: 1572.83 g/mol.
IR (ATR): ν̃ = 2996, 2932, 1866, 1813, 1796, 1732, 1671, 1543, 1446, 1432, 1388,
1346, 1263, 1197, 1134, 1073, 1036, 988, 916, 763, 703, 683, 655 cm−1.
UV/Vis (CH 2Cl2): λmax = 254, 324, 425, 475 nm.
Asymmetric dendritic monoadduct [C 6-G2] (40)
OO
O
OO HN
HN
OHN
O
O
O
O
O
O
OONH
OO
OO
O
O
OO
O O O
O
40
Compound 40 was synthe-
sized according to general
procedure GP 4 from mal-
onate 34 (260 mg, 0.15 mmol),
C60 (144 mg, 0.2 mmol),
CBr4 (56 mg, 0.17 mmol) and
DBU (25 µL, 0.17 mmol) in
100 mL toluene. The crude
product was purified by col-
umn chromatography (SiO2;
toluene/ethyl acetate, 30:10 to 15:10) to give 40 as a red brownish solid.
Yield: 171 mg, 0.07 mmol, 47 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 7.57 (s, br, 1H, CONH), 6.14 (s, br, 3H, CONH),
4.50 (t, 3J = 6.5 Hz, 2H, OCH2), 4.49 (t, 3J = 6.4 Hz, 2H, OCH2), 2.19 (m, 26H,
CH2COOtBu, OCCH2), 1.95 (m, 24H, NHC(CH2)3), 1.84 (m, 4H, CH2), 1.69 (m, 2H,
CH2), 1.47 (m, 2H, CH2), 1.44 (m, 83H, C(CH3)3, CH2), 1.26 (m, 4H, CH2), 0.85 (t, 3J
= 6.7 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.29 (1C, CONH), 173.15 (3C, CONH),
172.80 (9C, COOtBu), 163.81 (1C, CO), 163.74 (1C, CO), 145.67, 145.53, 145.43,
145.32, 145.26, 145.12, 144.95, 144.83, 144.76, 144.68, 144.65, 143.95, 143.91,
177

Chapter 5 Experimental Part
143.53, 143.13, 143.05, 142.26, 141.98, 141.00, 139.12, 139.03, 139.00 (58C, C60-
sp2), 80.59 (9C, C(CH3)3), 71.65 (2C, C60-sp3), 67.47 (1C, OCH2), 67.33 (1C, OCH2),
57.58 (1C, NHC(CH2)3), 57.42 (3C, NHC(CH2)3), 52.36 (1C, OCCCO), 37.01 (1C,
CH2CO), 31.68 (3C, NHC(CH2)3), 31.30 (3C, CH2CON), 30.75 (1C, CH2), 29.72 (9C,
NHC(CH2)3), 29.61 (9C, CH2COOtBu), 28.47, 28.32 (2C, CH2), 28.01 (27C, C(CH3)3),
25.55, 25.23 (2C, CH2), 24.38 (1C, CH2CH2CO), 22.53 (1C, CH2), 14.00 (1C, CH3)
ppm.
MS (FAB, NBA): m/z = 2466 [M+Na]+, 2443 [M]+, 720 [C60]+; MWcalc.: 2442.87 g/mol.
IR (ATR): ν̃ = 3278, 3265, 3002, 2945, 2937, 1801, 1768, 1586, 1467, 1413, 1395,
1346, 1274, 1201, 1166, 1114, 1065, 1004, 901, 857, 765, 694 cm−1.
UV/Vis (CH 2Cl2): λmax = 254.5, 324, 424.5, 476 nm.
Asymmetric dendritic monoadduct [C 18-G2] (41)
OO
O
OO HN
HN
OHN
O
O
O
O
O
O
OONH
OO
OO
O
O
OO
O O O
O
41
Compound 41 was synthesized according to general procedure GP 4 from malonate
35 (600 mg, 0.32 mmol), C60 (302 mg, 0.42 mmol), CBr4 (117 mg, 0.35 mmol) and
DBU (52 µL, 0.35 mmol) in 150 mL toluene. The crude product was purified by col-
umn chromatography (SiO2; toluene/ethyl acetate, 70:20 to 70:50) to give 41 as a red
brownish solid.
Yield: 366 mg, 0.14 mmol, 44 %, red brownish solid.
178

Chapter 5 Experimental Part
1H-NMR (400 MHz, RT, CDCl3): δ = 7.63 (s, br, 1H, CONH), 6.04 (s, br, 3H, CONH),
4.51 (t, 3J = 6.4 Hz, 2H, OCH2), 4.50 (t, 3J = 6.8 Hz, 2H, OCH2), 2.20 (m, 26H,
CH2COOtBu, OCCH2), 1.96 (m, 24H, NHC(CH2)3), 1.83 (m, 4H, CH2), 1.71 (m, 2H,
CH2), 1.44 (m, 83H, C(CH3)3, CH2), 1.25 (m, 30H, CH2), 0.88 (t, 3J = 6.8 Hz, 3H, CH3)
ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.07 (1C, CONH), 172.99 (3C, CONH),
172.77 (9C, COOtBu), 163.75 (1C, CO), 163.68 (1C, CO), 145.48, 145.26, 145.19,
145.16, 145.11, 145.09, 144.80, 144.63, 144.62, 144.59, 144.55, 144.53, 143.83,
143.81, 143.02, 142.95, 142.93, 142.90, 142.15, 142.13, 141.85, 140.88, 139.16,
139.10, 138.75 (58C, C60-sp2), 80.43 (9C, C(CH3)3), 71.66 (2C, C60-sp3), 67.44 (1C,
OCH2), 67.32 (1C, OCH2), 57.57 (1C, NHC(CH2)3), 57.39 (3C, NHC(CH2)3), 52.42
(1C, OCCCO), 37.02 (1C, CH2CO), 31.84 (3C, NHC(CH2)3), 31.73 (1C, CH2), 31.63
(1C, CH2), 31.62 (3C, CH2CON), 29.74 (3C, CH2), 29.71 (9C, NHC(CH2)3), 29.64
(9C, CH2COOtBu), 29.61, 29.59, 29.56, 29.55, 29.29, 29.27, 29.14, 28.53, 28.28 (9C,
CH2), 28.02 (27C, C(CH3)3), 27.97, 25.51 (2C, CH2), 25.23 (1C, CH2CH2CO), 24.84,
22.61 (2C, CH2), 14.04 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 2634 [M+Na]+, 2611 [M]+, 720 [C60]+; MWcalc.: 2611.18 g/mol.
IR (ATR): ν̃ = 3337, 3267, 2974, 2927, 2858, 1730, 1661, 1537, 1460, 1423, 1367,
1305, 1244, 1151, 1105, 950, 850, 757, 711, 684 cm−1.
UV/Vis (CH 2Cl2): λmax = 254, 324, 424, 476 nm.
179

Chapter 5 Experimental Part
Asymmetric dendritic monoadduct [(C 8)2-G2] (42)
O
O
O
O HN
HN
OHN
O
O
O
O
O
O
OONH
OO
OO
O
O
OO
O O O
O
O
42
Compound 42 was synthe-
sized according to general
procedure GP 4 from mal-
onate 36 (250 mg, 0.13
mmol), C60 (122 mg, 0.17
mmol), CBr4 (48 mg, 0.14
mmol) and DBU (22 µL,
0.14 mmol) in 100 mL
toluene. The crude prod-
uct was purified by column
chromatography (SiO2; toluene/ethyl acetate, 70:20 to 70:50) to give 42 as a red brown-
ish solid.
Yield: 159 mg, 0.06 mmol, 47 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 7.59 (s, br, 1H, CONH), 6.04 (s, br, 3H, CONH),
5.24 (quin, 3J = 6.4 Hz, 1H, CH), 4.48 (t, 3J = 7.1 Hz, 2H, OCH2), 2.19 (m, 26H,
CH2COOtBu, OCCH2), 1.95 (m, 24H, NHC(CH2)3), 1.69 (m, 4H, CH(CH2)2), 1.59 (m,
4H, CH2), 1.43 (m, 83H, C(CH3)3, CH2), 1.25 (m, 24H, CH2), 0.87 (t, 3J = 6.9 Hz, 6H,
CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 172.82 (3C, CONH), 172.74 (1C, CONH),
172.62 (9C, COOtBu), 163.54 (1C, CO), 163.12 (1C, CO), 145.62, 145.36, 145.24,
145.20, 145.15, 145.13, 145.10, 144.80, 144.68, 144.64, 144.56, 144.53, 143.85,
143.82, 143.02, 142.98, 142.96, 142.94, 142.91, 142.17, 141.86, 140.89, 139.10,
138.72 (58C, C60-sp2), 80.55 (9C, C(CH3)3), 77.20 (1C, CH), 67.32 (1C, OCH2), 57.42
(3C, NHC(CH2)3), 57.38 (1C, NHC(CH2)3), 49.08 (1C, OCCCO), 37.02 (1C, CH2CO),
33.91 (2C, CH2), 31.88 (2C, CH2), 31.85 (3C, NHC(CH2)3), 31.70 (3C, CH2CON),
29.79 (9C, NHC(CH2)3), 29.76 (9C, CH2COOtBu), 29.65, 29.57, 29.48, 29.23, 28.44
(8C, CH2), 28.07 (27C, C(CH3)3), 25.58 (1C, CH2CH2CO), 25.34, 24.91, 22.64 (6C,
CH2), 14.13 (2C, CH3) ppm.
180

Chapter 5 Experimental Part
MS (FAB, NBA): m/z = 2620 [M+Na]+, 2597 [M]+, 720 [C60]+; MWcalc.: 2597.16 g/mol.
IR (ATR): ν̃ = 3301, 3293, 2974, 2864, 2803, 1763, 1702, 1678, 1565, 1487, 1443,
1353, 1312, 1266, 1207, 1168, 1121, 963, 882, 757, 703, 693 cm−1.
UV/Vis (CH 2Cl2): λmax = 254.5, 324, 424.5, 476 nm.
Deprotected asymmetric dendritic monoadduct [C 6-G1] (43)
OO
O
OO HN
OH
O
OH
O
O OH
43
Compound 43 was synthesized accord-
ing to general procedure GP 5 from
monoadduct 37 (200 mg, 0.14 mmol) in
formic acid (20 mL). The crude prod-
uct was purified by reprecipitation from
THF/Et2O to give 43 as a red brownish
solid.
Yield: 170 mg, 0.14 mmol, 97 %, red brownish solid.
1H-NMR (400 MHz, RT, THF-d8): δ = 11.09 (s, br, 3H, COOH), 7.01 (s, br, 1H,
CONH), 4.49 (m, br, 4H, OCH2), 2.05 (m, br, 6H, CH2COOH, CH2), 1.79 (m, br, 8H,
NHC(CH2)3, CH2), 1.51 (m, br, 2H, CH2), 1.39 (m, br, 8H, CH2), 1.19 (m, br, 4H, CH2),
0.88 (m, br, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, THF-d8): δ = 174.97 (3C, COOH), 171.95 (1C, CONH),
162.87 (1C, CO), 162.79 (1C, CO), 145.48, 145.01, 144.79, 144.26, 143.56, 143.01,
142.59, 141.71, 141.43, 140.69, 140.42, 139.12, 137.91 (58C, C60-sp2), 71.59 (2C,
C60-sp3), 67.53 (2C, OCH2), 56.99 (1C, NHC(CH2)3), 52.03 (1C, OCCCO), 37.43 (1C,
CH2CO), 31.01 (1C, CH2), 30.01 (3C, NHC(CH2)3), 29.78 (3C, CH2COOH), 28.44,
28.22 (2C, CH2), 26.01 (1C, CH2CH2CO), 25.44, 24.26, 22.39 (3C, CH2), 13.98 (1C,
CH3) ppm.
MS (FAB, NBA): m/z = 1250 [M]+, 720 [C60]+; MWcalc.: 1250.22 g/mol.
IR (ATR): ν̃ = 3231, 2989, 2673, 1756, 1674, 1635, 1555, 1432, 1401, 1265, 1144,
1087, 1001, 836, 796, 712, 674 cm−1.
181

Chapter 5 Experimental Part
UV/Vis (DMSO): λmax = 252, 322, 424 nm.
Deprotected asymmetric dendritic monoadduct [C 18-G1] (44)
OO
O
OO HN
OH
O
OH
O
O OH
44
Compound 44 was
synthesized accord-
ing to general pro-
cedure GP 5 from
monoadduct 38 (300
mg, 0.19 mmol) in
formic acid (25 mL).
The crude product was purified by reprecipitation from THF/Et2O to give 44 as a red
brownish solid.
Yield: 259 mg, 0.18 mmol, 96 %, red brownish solid.
1H-NMR (400 MHz, RT, THF-d8): δ = 12.05 (s, br, 3H, COOH), 7.14 (s, br, 1H,
CONH), 4.39 (m, br, 4H, OCH2), 2.09 (m, br, 8H, CH2COOH, CH2CO), 1.82 (m, br,
8H, NHC(CH2)3, CH2), 1.51 (m, br, 4H, OCH2CH2), 1.36 (m, br, 32H, CH2), 0.85 (m,
br, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, THF-d8): δ = 174.42 (3C, COOH), 171.81 (1C, CONH),
163.63 (1C, CO), 163.52 (1C, CO), 144.71, 144.13, 143.40, 142.53, 141.69, 141.30,
140.61, 140.35, 139.10, 138.85 (58C, C60-sp2), 71.42 (2C, C60-sp3), 67.12 (1C, OCH2),
67.02 (1C, OCH2), 56.34 (1C, NHC(CH2)3), 52.32 (1C, OCCCO), 38.13 (1C, CH2CO),
31.73 (1C, CH2), 29.97 (3C, NHC(CH2)3), 29.88 (3C, CH2COOH), 29.69, 29.70, 29.60,
29.62, 29.33, 29.28, 28.54, 28.37 (14C, CH2), 25.99 (1C, CH2), 25.42 (1C, CH2CH2CO),
25.06, 22.57 (2C, CH2), 14.23 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 1419 [M]+, 720 [C60]+; MWcalc.: 1418.54 g/mol.
IR (ATR): ν̃ = 3221, 2920, 2850, 2647, 1692, 1537, 1452, 1228, 1182, 1105, 1058,
881, 788, 703, 664 cm−1.
UV/Vis (DMSO): λmax = 254, 323.5, 424.5 nm.
182

Chapter 5 Experimental Part
Deprotected asymmetric dendritic monoadduct [(C 8)2-G1] (45)
O
O
O
O HN
OH
O
OH
O
O OH
O
45
Compound 45 was synthesized ac-
cording to general procedure GP 5
from monoadduct 39 (250 mg, 0.16
mmol) in formic acid (25 mL). The
crude product was purified by repre-
cipitation from THF/Et2O to give 45 as
a red brownish solid.
Yield: 218 mg, 0.16 mmol, 97 %, red brownish solid.
1H-NMR (400 MHz, RT, THF-d8): δ = 11.89 (s, br, 3H, COOH), 7.04 (s, br, 1H, CONH),
5.18 (m, br, 1H, OCH), 4.41 (m, br, 2H, OCH2), 2.18 (m, br, 8H, CH2COOH, CH2CO),
1.91 (m, br, 8H, NHC(CH2)3, CH2), 1.65 (m, br, 8H, CHCH2, CH2), 1.41 (m, br, 2H,
CH2), 1.21 (m, br, 22H, CH2), 0.85 (m, br, 6H, CH3) ppm.
13C-NMR (100.5 MHz, RT, THF-d8): δ = 175.02 (3C, COOH), 172.00 (1C, CONH),
163.59 (1C, CO), 163.51 (1C, CO), 144.88, 144.76, 144.36, 144.01, 143.76, 143.35,
143.08, 142.28, 141.88, 141.00, 139.11, 138.79 (58C, C60-sp2), 78.01 (1C, CH), 71.59
(2C, C60-sp3), 67.54 (1C, OCH2), 56.69 (1C, NHC(CH2)3), 52.56 (1C, OCCCO), 37.99
(1C, CH2CO), 33.98 (2C, CH(CH2)2), 31.89, 30.13 (3C, CH2), 29.89 (3C, NHC(CH2)3),
29.71 (3C, CH2COOH), 29.63, 29.48, 29.01, 28.37 (6C, CH2), 25.61 (1C, CH2CH2CO),
25.37, 22.60 (5C, CH2), 14.11 (2C, CH3) ppm.
MS (FAB, NBA): m/z = 1405 [M]+, 720 [C60]+; MWcalc.: 1404.51 g/mol.
IR (ATR): ν̃ = 3199, 2974, 2603, 1815, 1767, 1656, 1599, 1531, 1433, 1403, 1301,
1265, 1141, 1078, 967, 844, 797, 703, 688 cm−1.
UV/Vis (DMSO): λmax = 254, 323.5, 424.5 nm.
183

Chapter 5 Experimental Part
Deprotected asymmetric dendritic monoadduct [C 6-G2] (46)
OO
O
OO HN
HN
OHN
O
O
OH
O
OH
O
OHONH
OHO
OHO
HO
O
HOO
HO O OH
O
46
Compound 46 was synthesized
according to general procedure
GP 5 from monoadduct 40 (150
mg, 0.06 mmol) in formic acid
(35 mL). The crude product was
purified by reprecipitation from
MeOH/Et2O to give 46 as a red
brownish solid.
Yield: 112 mg, 0.06 mmol, 97 %, red brownish solid.
1H-NMR (400 MHz, RT, DMSO-d6): δ = 12.01 (s, br, 9H, COOH), 8.05 (s, br, 1H,
CONH), 7.16 (s, br, 3H, CONH), 4.51 (m, br, 4H, OCH2), 2.11 (m, br, 26H, CH2COOH,
CH2CO), 1.91 (m, br, 28H, NHC(CH2)3, CH2), 1.51 (m, br, 6H, CH2), 1.26 (m, br, 4H,
CH2), 0.84 (m, br, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, DMSO-d6): δ = 174.97 (9C, COOH), 172.39 (4C, CONH),
163.31, 163.28 (2C, CO), 145.55, 145.46, 145.26, 145.09, 144.78, 144.72, 144.65,
144.62, 144.31, 144.21, 143.96, 143.78, 143.39, 143.163, 142.50, 142.47, 141.99,
141.77, 141.05, 139.78, 139.41, 139.19, 138.58 (58C, C60-sp2), 71.34 (2C, C60-sp3),
67.43, 67.39 (2C, OCH2), 56.67 (4C, NHC(CH2)3), 52.91 (1C, OCCCO), 36.84 (1C,
CH2CO), 30.51 (6C, NHC(CH2)3, CH2CON), 30.68, (1C, CH2), 29.33 ((9C, NHC(CH2)3),
28.65, 28.19 (2C, CH2), 28.24 (9C, CH2COOH), 25.98, 25.55, 24.50, 22.19 (4C, CH2),
14.11 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 1938 [M+H]+, 720 [C60]+; MWcalc.: 1937.91 g/mol.
UV/Vis (DMSO): λmax = 264, 319 nm.
184

Chapter 5 Experimental Part
Deprotected asymmetric dendritic monoadduct [C 18-G2] (47)
OO
O
OO HN
HN
OHN
O
O
OH
O
OH
O
OHONH
OHO
OHO
HO
O
HOO
HO O OH
O
47
Compound 47 was synthesized according to general procedure GP 5 from monoadduct
41 (300 mg, 0.11 mmol) in formic acid (50 mL). The crude product was purified by
reprecipitation from MeOH/Et2O to give 47 as a red brownish solid.
Yield: 237 mg, 0.11 mmol, 98 %, red brownish solid.
1H-NMR (400 MHz, RT, DMSO-d6): δ = 12.03 (s, br, 9H, COOH), 8.12 (s, br, 1H,
CONH), 7.20 (s, br, 3H, CONH), 4.46 (m, br, 4H, OCH2), 2.09 (m, br, 26H, CH2COOH,
CH2CO), 1.81 (m, br, 30H, NHC(CH2)3, CH2), 1.21 (m, br, 32H, CH2), 0.82 (m, br, 3H,
CH3) ppm.
13C-NMR (100.5 MHz, RT, DMSO-d6): δ = 174.66 (9C, COOH), 172.49 (4C, CONH),
163.28 (2C, CO), 145.67, 145.61, 145.14, 144.94, 144.85, 144.77, 144.46, 144.38,
144.25, 143.56, 143.48, 142.74, 142.67, 141.89, 141.81, 141.52, 140.66, 140.60,
139.20, 138.14 (58C, C60-sp2), 71.56 (2C, C60-sp3), 67.42 (2C, OCH2), 56.34 (4C,
NHC(CH2)3), 52.86 (1C, OCCCO), 36.05 (1C, CH2CO), 31.45, 30.76, 30.26 (5C, CH2),
29.22 (6C, NHC(CH2)3, CH2CON), 29.07 ((9C, NHC(CH2)3), 28.89, 28.66 (5C, CH2),
28.09 (9C, CH2COOH), 25.70, 25.34, 25.17, 24.50, 22.25 (9C, CH2), 14.06 (1C, CH3)
ppm.
MS (FAB, NBA): m/z = 2106 [M+H]+, 720 [C60]+; MWcalc.: 2106.23 g/mol.
IR (ATR): ν̃ = 3356, 3101, 2968, 2865, 2664, 1714, 1654, 1552, 1464, 1424, 1278,
1231, 1201, 1104, 912, 878, 801, 769, 703, 685 cm−1.
185

Chapter 5 Experimental Part
UV/Vis (DMSO): λmax = 263, 320 nm.
Deprotected asymmetric dendritic monoadduct [(C 8)2-G2] (48)
O
O
O
O HN
HN
OHN
O
O
OH
O
OH
O
OHONH
OHO
OHO
HO
O
HOO
HO O OH
O
O
48
Compound 48 was synthe-
sized according to general
procedure GP 5 from mono-
adduct 42 (150 mg, 0.06
mmol) in formic acid (35 mL).
The crude product was pu-
rified by reprecipitation from
MeOH/Et2O to give 48 as a
red brownish solid.
Yield: 117 mg, 0.06 mmol, 97 %, red brownish solid.
1H-NMR (400 MHz, RT, DMSO-d6): δ = 12.11 (s, br, 9H, COOH), 8.07 (s, br, 1H,
CONH), 7.01 (s, br, 3H, CONH), 5.20 (m, br, 1H, CH), 4.41 (m, br, 2H, OCH2), 2.13
(m, br, 26H, CH2COOH, CH2CO), 1.89 (m, 28H, NHC(CH2)3, CH(CH2)2), 1.51 (m, br,
6H, CH2), 1.259 (m, br, 24H, CH2), 0.87 (m, br, 6H, CH3) ppm.
13C-NMR (100.5 MHz, RT, DMSO-d6): δ = 174.71 (9C, COOH), 172.44 (4C, CONH),
163.31, 163.01 (2C, CO), 145.63, 145.59, 145.24, 145.19, 145.01, 144.97, 144.80,
144.69, 144.64, 144.41, 144.22, 143.87, 143.82, 143.01, 142.77, 142.69, 141.86,
141.44, 140.89, 139.11, 138.73 (58C, C60-sp2), 77.55 (1C, CH), 71.66 (2C, C60-sp3),
67.39 (1C, OCH2), 56.46 (4C, NHC(CH2)3), 52.01 (1C, OCCCO), 36.44 (1C, CH2CO),
33.87, 32.01 (4C, CH2), 29.63 (6C, NHC(CH2)3, CH2CON), 29.24 ((9C, NHC(CH2)3),
29.78, 29.14, 29.06, 29.01, 28.94 (6C, CH2), 28.22 (9C, CH2COOH), 25.11 (1C,
CH2CH2CO), 24.99, 24.89, 22.66 (6C, CH2), 14.21 (2C, CH3) ppm.
MS (FAB, NBA): m/z = 2092 [M+H]+, 720 [C60]+; MWcalc.: 2092.20 g/mol.
IR (ATR): ν̃ = 3343, 3153, 2987, 2878, 2666, 1801, 1732, 1701, 1634, 1549, 1501,
1498, 1433, 1263, 1203, 1144, 1103, 987, 953, 871, 771, 734, 691 cm−1.
UV/Vis (DMSO): λmax = 264, 318 nm.
186

Chapter 5 Experimental Part
2-Hydroxyethyl all-cis-docosa-4,7,10,13,16,19-hexaenoate (50)
O
O OH
50
Compound 50 was synthesized
according to general procedure
GP 3 from all-cis-docosa-
4,7,10,13,16,19-hexaenoic acid 49
(2.5 g, 7.61 mmol), ethane-1,2-diol (710 mg, 11.42 mmol), EDC (1.90 g, 9.89 mmol)
and DMAP (140 mg, 1.14 mmol) in dry CH2Cl2. The crude product was purified by
flash column chromatography (SiO2; hexane/ethyl acetate, 50:10 to 50:30) to give 50
as yellow oil.
Yield: 2.51 g, 6.74 mmol, 89 %, yellow oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 5.33 (m, 12H, CH), 4.16 (t, 3J = 4.8 Hz, 2H,
OCH2), 3.75 (t, 3J = 4.8 Hz, 2H, CH2OH), 2.80 (m, 10H, CH2), 2.41 (s, br, 1H, OH),
2.36 (m, 4H, CH2)), 2.03 (m, CH2CH3), 0.92 (t, 3J = 7.5 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.50 (1C, CO), 132.07, 129.50, 128.62,
128.37, 128.33, 128.31, 128.06, 127.94, 127.86, 127.09 (12C, CH), 66.07 (1C, OCH2),
61.12 (1C, CH2OH), 34.14 (1C, CH2CO), 25.72, 25.67, 25.62 (5C, CH2), 22.82
(CH2CH2CO), 20.64 (1C, CH2CH3), 14.35 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 373 [M+H]+; MWcalc.: 372,54 g/mol.
IR (ATR): ν̃ = 3360, 2981, 2935, 1722, 1676, 1537, 1452, 1367, 1321, 1251, 1151,
1043, 958, 850, 757 cm−1.
EA: C24H36O3: calcd. C 77.38, H 9.74, O 12.88; found: C 76.58, H 9.53.
187

Chapter 5 Experimental Part
3-(6-tert-Butoxy-6-oxohexyloxy)-3-oxopropanoic acid (51)
HO O
O OO
O51
Malonic acid (1.11 g, 10.62 mmol) and 22 (2.0 g, 10.62
mmol) were dissolved in dry CH2Cl2 and cooled to 0°C.
DCC (2.19 g, 10.62 mmol) was dissolved in dry CH2Cl2
and added dropwise over a period of 2 h via a dropping
funnel. After stirring the solution for additional 1 h at 0°C and 8 h at rt, TLC control
showed the complete conversion of the starting material. The resulting dicyclohexy-
lurea was filtered off and traces which remained in solution were removed by repeated
precipitation from ethyl acetate. The filtrate was extracted with saturated NaHCO3-
solution (3 x 200 mL), followed by acidification with 5 N HCl. The aqueous solution was
extracted with Et2O (3 x 200 mL) and dried over anhydrous MgSO4. Removal of the
solvent under reduced pressure gave 51 as pale yellow oil.
Yield: 1.01 g, 3.68 mmol, 35 %, pale yellow oil.
1H-NMR (300 MHz, RT, CDCl3): δ = 11.19 (s, br, 1H, COOH), 4.13 (t, 3J = 6.4 Hz,2H,
OCH2), 3.38 (s, 2H, OCCH2CO), 2.19 (t, 3J = 7.1 Hz, 2H, CH2CO), 1.59 (m, 4H, CH2),
1.40 (s, 9H, C(CH3)3), 1.34 (m, 2H, CH2) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 173.53 (1C, CO), 170.89 (1C, CO), 167.09 (1C,
CO), 80.59 (1C, C(CH3)3), 65.90 (1C, OCH2), 41.05 (1C, OCCH2CO), 35.53 (1C,
CH2CO), 28.25 (3C, C(CH3)3), 25.47, 24.77 (3C, CH2) ppm.
MS (FAB, NBA): m/z = 274 [M]+, 217 [M-tBu]; MWcalc.: 274.31 g/mol.
EA: C13H22O6: calcd. C 56.92, H 8.08, O 35.00; found: C 56.56, H 7.98.
188

Chapter 5 Experimental Part
6-tert-Butoxy-6-oxohexyl(2- all-cis-docosa-4,7,10,13,16,19-hexaenoyloxy)ethyl
malonate (52)
O
O O O
O OO
O52
Compound 52 was synthesized according to general procedure GP 3 from 50 (2.26 g,
6.07 mmol), 51 (1.51 g, 5.52 mmol), EDC (1.38 g, 7.17 mmol) and DMAP (101 mg, 0.83
mmol) in dry CH2Cl2. The crude product was purified by flash column chromatography
(SiO2; hexane/ethyl acetate, 65:10) to give 52 as yellow oil.
Yield: 2.52 g, 4.01 mmol, 66 %, yellow oil.
1H-NMR (300 MHz, RT, CDCl3): δ = 5.34 (m, 12H, CH), 4.28 (m, 4H, OCH2, CH2O),
4.11 (t, 3J = 6.7 Hz, 2H, OCH2), 3.36 (s, 2H, OCCH2CO), 2.81 (m, 10H, CH2), 2.36 (m,
4H, CH2)), 2.18 (t, 3J = 7.4 Hz, 2H, CH2CO), 2.04 (m, CH2CH3), 1.60 (m, 4H, CH2),
1.40 (s, 9H, C(CH3)3), 1.34 (m, 2H, CH2), 0.94 (t, 3J = 7.5 Hz, 3H, CH3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 172.99 (1C, CO), 172.91 (1C, CO), 166.50 (1C,
CO), 166.43 (1C, CO), 132.18, 129.57, 128.73, 128.46, 128.44, 128.41, 128.26, 128.24,
128.19, 128.03, 127.91, 127.18 (12C, CH), 80.25 (1C, C(CH3)3), 65.62 (1C, OCH2),
63.29 (1C, OCH2), 61.94 (1C, CH2O), 41.48 (1C, OCCH2CO), 35.49 (1C, CH2CO),
34.10 (1C, CH2CO), 28.35 (1C, CH2), 28.28 (3C, C(CH3)3), 25.80, 25.76, 25.71 (5C,
CH2), 24.80, 22.81 (2C, CH2), 20.72 (1C, CH2CH3), 14.43 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 629 [M]+; MWcalc.: 628,84 g/mol.
IR (ATR): ν̃ = 3165, 2976, 2932, 2868, 1731, 1654, 1458, 1404, 1399, 1367, 1301,
1256, 1203, 1152, 1101, 1055, 989, 956, 887, 848, 763, 701 cm−1.
EA: C37H56O8: calcd. C 70.67, H 8.98, O 20.35; found: C 70.17, H 8.57.
189

Chapter 5 Experimental Part
6-(3-(2-(all-cis-Docosa-4,7,10,13,16,19-hexaenoyloxy)ethoxy)-3- oxop ropanoyl-
oxy)hexanoic acid (53)
O
O O O
O OOH
O53
Compound 53 was synthesized according to general procedure GP 5 from 53 (1.5 g,
2.39 mmol) and trifluoroacetic acid (1.8 mL, 24 mmol) in CHCl3.
Yield: 1.36 g, 2.37 mmol, 99 %, yellow oil.
1H-NMR (300 MHz, RT, CDCl3): δ = 9.76 (s, br, 1H, COOH), 5.34 (m, 12H, CH), 4.28
(m, 4H, OCH2, CH2O), 4.11 (t, 3J = 6.9 Hz, 2H, OCH2), 3.37 (s, 2H, OCCH2CO), 2.81
(m, 10H, CH2), 2.34 (m, 4H, CH2), 2.01 (m, CH2CH3), 1.63 (m, 4H, CH2), 1.37 (m, 2H,
CH2), 0.93 (t, 3J = 7.5 Hz, 3H, CH3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 179.65 (1C, CO), 173.32 (1C, CO), 166.84 (1C,
CO), 166.78 (1C, CO), 132.41, 129.81, 128.94, 128.68, 128.65, 128.63, 128.47,
128.45, 128.40, 128.25, 128.09, 127.39 (12C, CH), 65.78 (1C, OCH2), 63.55 (1C,
OCH2), 62.21 (1C, CH2O), 41.69 (1C, OCCH2CO), 34.32 (1C, CH2CO), 34.16 (1C,
CH2CO), 28.49 (1C, CH2), 26.02, 25.97, 25.93 (5C, CH2), 24.58, 23.01 (2C, CH2),
20.95 (1C, CH2CH3), 14.69 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 573 [M+H]+; MWcalc.: 572,73 g/mol.
190

Chapter 5 Experimental Part
Asymmetric dendritic malonate [DHA-G2] (54)
O
O OO
O
OO HN
HN
OHN
O
O
O
O
O
O
OONH
OO
OO
O
O
OO
O O O
O
54
Compound 54 was
synthesized according
to general procedure
GP 3 from 53 (1.31
g, 2.29 mmol), 30
(3.95 g, 2.74 mmol),
EDC (526 mg, 2.74
mmol), DMAP (140
mg, 1.14 mmol) and
1-HOBt (371 mg, 2.74
mmol) in dry CH2Cl2. The crude product was purified by flash column chromatography
(SiO2; hexane/ethyl acetate, 50:40) to give 54 as yellow oil.
Yield: 2.42 g, 1.21 mmol, 53 %, yellow oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 7.53 (s, br, 1H, CONH), 6.00 (s, br, 3H, CONH),
5.25 (m, 12H, CH), 4.21 (m, 4H, OCH2, CH2O), 4.03 (t, 3J = 6.7 Hz, 2H, OCH2),
3.29 (s, 2H, OCCH2CO), 2.72 (m, 10H, CH2), 2.28 (m, 4H, CH2), 2.07 (m, 24H,
CH2COOtBu), 1.84 (m, 24H, NHC(CH2)3), 1.54 (m, 4H, CH2), 1.31 (m, 83H, C(CH3)3,
CH2), 0.85 (t, 3J = 7.5 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.09 (1C, CONH), 172.90 (3C, CONH),
172.63 (9C, COOtBu), 166.41 (1C, CO), 166.32 (1C, CO), 131.96, 129.35, 128.52,
128.24, 128.23, 128.20, 128.07, 128.05, 128.01, 127.84, 127.75, 127.00 (12C, CH),
80.51 (9C, C(CH3)3), 65.50 (1C, OCH2), 63.10 (1C, OCH2), 61.82 (1C, CH2O), 57.49
(1C, NHC(CH2)3), 57.42 (3C, NHC(CH2)3), 41.27 (1C, OCCH2CO), 37.10 (1C, CH2CO),
33.91 (1C, CH2CO), 31.88 (3C, NHC(CH2)3), 31.73 (3C, CH2CON), 29.82 (9C,
NHC(CH2)3), 29.78 (9C, CH2COOtBu), 28.23 (1C, CH2), 28.08 (27C, C(CH3)3), 25.62,
25.57, 25.52 (5C, CH2), 25.26, 22.63 (2C, CH2), 20.54 (1C, CH2CH3), 14.27 (1C, CH3)
ppm.
MS (FAB, NBA): m/z = 1993 [M]+, 2016[M+Na]+; MWcalc.: 1994.61 g/mol.
191

Chapter 5 Experimental Part
IR (ATR): ν̃ = 3244, 3165, 2976, 1766, 1701, 1653, 1488, 1399, 1363, 1356, 1301,
1278, 1213, 1188, 1094, 1046, 976, 914, 855, 8418, 743, 684 cm−1.
EA: C109H180N4O28 · 1/3 C4H8O2: calcd. C 65.47, H 9.10, N 2.77, O 22.66; found: C
65.09, H 8.99, N 2.54.
Asymmetric dendritic monoadduct [DHA-G2] (55)
O
O OO
O
OO HN
HN
OHN
O
O
O
O
O
O
OONH
OO
OO
O
O
OO
O O O
O
55
Compound 55 was syn-
thesized according to
general procedure GP
4 from malonate 54
(866 mg, 0.43 mmol),
C60 (376 mg, 0.52
mmol), CBr4 (173 mg,
0.52 mmol) and DBU
(78 µL, 0.52 mmol) in
175 mL toluene. Pre-
cleaning by flash column chromatography (SiO2; toluene/ethyl acetate, 70:40 to 60:40)
and subsequent purification by preparative HPLC (Nucleosil 5 µm; toluene/ethyl ac-
etate 68:32) gave 55 as a red brownish solid.
Yield: 443 mg, 0.16 mmol, 38 %, yellow oil.
1H-NMR (300 MHz, RT, CDCl3): δ = 7.61 (s, br, 1H, CONH), 6.02 (s, br, 3H, CONH),
5.29 (m, 12H, CH), 4.65 (m, 2H, CH2O), 4.41 (m, 4H, OCH2), 2.75 (m, 10H, CH2),
2.30 (m, 4H, CH2), 2.12 (m, 24H, CH2COOtBu), 1.90 (m, 24H, NHC(CH2)3), 1.63 (m,
4H, CH2), 1.36 (m, 83H, C(CH3)3, CH2), 0.89 (t, 3J = 7.4 Hz, 3H, CH3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 173.03 (1C, CONH), 172.98 (3C, CONH), 172.71
(9C, COOtBu), 163.52 (1C, CO), 163.38 (1C, CO), 145.44, 145.31, 145.24, 145.21,
145.16, 145.08, 144.94, 144.74, 144.72, 144.69, 144.65, 144.61, 143.95, 143.90,
143.13, 143.08, 143.06, 142.97, 142.25, 142.22, 141.90, 141.02, 140.96, 139.39,
138.75 (58C, C60-sp2), 132.06, 129.50, 128.60, 128.35, 128.31, 128.28, 128.16, 128.13,
192

Chapter 5 Experimental Part
128.09, 127.92, 127.77, 127.07 (12C, CH), 80.46 (9C, C(CH3)3), 71.49 (2C, C60-sp3),
67.50 (1C, OCH2), 64.77 (1C, CH2O), 61.88 (1C, OCH2), 57.50 (4C, NHC(CH2)3),
52.08 (1C, OCCCO), 37.14 (1C, CH2CO), 33.97 (1C, CH2CO), 31.80 (6C, NHC(CH2)3,
CH2CON), 29.82 (18C, NHC(CH2)3, CH2COOtBu), 28.40 (1C, CH2), 28.16 (27C,
C(CH3)3), 25.71, 25.61, 25.38 (5C, CH2), 22.69 (1C, CH2), 20.64 (1C, CH2CH3), 14.41
(1C, CH3) ppm.
MS (FAB, NBA): m/z = 2712 [M]+, 720 [C60]+; MWcalc.: 2713.23 g/mol.
IR (ATR): ν̃ = 3320, 2981, 2943, 1722, 1668, 1529, 1460, 1421, 1367, 1313, 1267,
1151, 1105, 958, 896, 850, 734 cm−1.
UV/Vis (CH 2Cl2): λmax = 258, 325, 425, 495 nm.
Deprotected asymmetric dendritic monoadduct [DHA-G2] (56 )
O
O OO
O
OO HN
HN
OHN
O
O
OH
O
OH
O
OHONH
OHO
OHO
HO
O
HOO
HO O OH
O
56
Compound 56 was syn-
thesized according to
general procedure GP
5 from monoadduct 55
(250 mg, 0.09 mmol) in
formic acid (15 mL). The
crude product was pu-
rified by reprecipitation
from MeOH/Et2O to give
56 as a red brownish
solid.
Yield: 185 mg, 0.08 mmol, 91 %, red brownish solid.
1H-NMR (400 MHz, RT, DMSO-d6): δ = 11.99 (s, br, COOH), 7.97 (s, br, 3H, CONH),
6.84 (s, br, 1H, CONH), 5.44 (m, br, 12H, CH), 4.43 (m, br, 2H, CH2O), 4.33 (m, br, 4H,
OCH2), 2.81 (m, br, 10H, CH2), 2.22 (m, br, 4H, CH2), 2.09 (m, br, 24H, CH2COOH),
1.86 (m, br, 24H, NHC(CH2)3), 1.55 (m, br, 4H, CH2), 1.29 (m, br, 2H,CH2), 0.83 (m,
br, 3H, CH3) ppm.
193

Chapter 5 Experimental Part
13C-NMR: the spectrum was of low quality, due to the broadening of the signals through
aggregation. Nevertheless no signals for the tert-butyl groups were observed.
MS (FAB, NBA): m/z = 2208 [M+H]+, 720 [C60]+; MWcalc.: 2208.28 g/mol.
UV/Vis (H 2O pH = 7.2): λmax = 257, 325 nm.
Deprotected asymmetric dendritic malonate [C 18-G1] (57)
OO
O
OO HN
OH
O
OH
O
O OH57
Compound 57 was
synthesized accord-
ing to general pro-
cedure GP 5 from
malonate 32 (1.37 g,
1.58 mmol) in formic acid (25 mL). The product was repeated evaporated with CHCl3
as entraining solvent under reduced pressure to give 57 as white solid.
Yield: 1.09 g, 1.58 mmol, 99 %, white solid.
1H-NMR (300 MHz, RT, THF-d8): δ = 10.33 (s, br, 3H, COOH), 6.21 (s, br, 1H,
CONH), 4.17 (t, 3J = 6.6 Hz, 2H, OCH2), 4.14 (t, 3J = 6.8 Hz, 2H, OCH2), 3.32 (s, 2H,
OCCH2CO), 2.22 (t, 3J = 7.7 Hz, 6H, CH2COOH), 2.11 (t, 3J = 7.4 Hz, 2H, OCCH2),
1.98 (t, 3J = 7.8 Hz, 6H, NHC(CH2)3), 1.67 (m, 6H, CH2), 1.25 (m, 32H, CH2), 0.91 (t,
3J = 6.7 Hz, 3H, CH3) ppm.
13C-NMR (75 MHz, RT, THF-d8): δ = 174.88 (3C, COOH), 172.12 (1C, CONH), 166.87
(1C, CO), 166.79 (1C, CO), 65.66 (1C, OCH2), 65.31 (1C, OCH2), 57.19 (1C,
NHC(CH2)3), 41.44 (1C, OCCH2CO), 37.19 (1C, CH2CO), 31.86 (1C, CH2), 29.89 (3C,
NHC(CH2)3), 29.68, 29.64, 29.61, 29.49, 29.48, 29.37, 29.19 (10C, CH2), 28.71 (3C,
CH2COOH), 28.39, 28.18, 25.71 (5C, CH2), 25.48 (1C, CH2CH2CO), 25.18, 22.66 (2C,
CH2), 14.12 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 699 [M]+; MWcalc.: 699.91 g/mol.
IR (ATR): ν̃ = 3402, 2963, 2745, 2612, 2545, 1871, 1648, 1507, 1482, 1467, 1457,
1432, 1324, 1266, 1117, 950, 791, 686, 624 cm−1.
194

Chapter 5 Experimental Part
EA: C37H65NO11 · C2HF3O2: calcd. C 57.55, H 8.17, F 7.00, N 1.72, O 25.55; found: C
57.99, H 8.52, N 2.01.
Asymmetric dendritic malonate [C 18-G1-(Br)3] (58)
OO
O
OO HN
O
O
O
O
O O
Br
Br
Br
58
Compound 58 was synthesized according to general procedure GP 2 from malonate 57
(1.09 g, 1.56 mmol), 2-bromoethanol (0.79 g, 6.32 mmol), DMAP (193 mg, 1.58 mmol),
1-HOBt (747 mg, 5.53 mmol) and DCC (1.14 g, 5.53 mmol) in dry THF (150 mL). The
crude product was purified by column chromatography (SiO2; dichloromethane/ethyl
acetate, 15:1 to 5:1) to give 58 as pale yellow oil.
Yield: 1.22 g, 1.20 mmol, 76 %, pale yellow oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 5.68 (s, br, 1H, CONH), 4.39 (t, 3J = 6.2 Hz, 6H,
CH2CH2Br), 4.15 (t, 3J = 6.7 Hz, 2H, OCH2), 4.13 (t, 3J = 6.7 Hz, 2H, OCH2), 3.52 (t,
3J = 6.1 Hz, 6H, CH2Br), 3.37 (s, 2H, OCCH2CO), 2.36 (t, 3J = 7.8 Hz, 6H, CH2COO),
2.14 (t, 3J = 7.6 Hz, 2H, OCCH2), 2.06 (t, 3J = 7.2 Hz, 6H, NHC(CH2)3), 1.64 (m, 6H,
CH2), 1.25 (m, 32H, CH2), 0.88 (t, 3J = 6.9 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 172.73 (3C, COO), 172.35 (1C, CONH),
166.69 (1C, CO), 166.62 (1C, CO), 65.69 (1C, OCH2), 65.21 (1C, OCH2), 64.01 (3C,
CH2CH2Br), 57.24 (1C, NHC(CH2)3), 41.54 (1C, OCCH2CO), 37.11 (1C, CH2CO),
31.86 (1C, CH2), 29.81 (7C, NHC(CH2)3, CH2), 29.64, 29.47, 29.34 (6C, CH2), 28.77
(3C, CH2COO), 28.58 (3C, CH2Br), 28.53, 28.29 (4C, CH2), 25.91 (1C, CH2CH2CO),
25.63, 25.29, 22.80 (3C, CH2), 14.22 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 1020 [M]+; MWcalc.: 1020.76 g/mol.
195

Chapter 5 Experimental Part
IR (ATR): ν̃ = 3379, 2972, 1832, 1501, 1455, 1432, 1281, 1268, 1102, 950, 799, 791,
697, 686, 624 cm−1.
EA: C43H74Br3NO11: calcd. C 50.60, H 7.31, Br 23.48, N 1.37, O 17.24; found: C
59.87, H 7.48, N 1.52.
Asymmetric dendritic monoadduct [C 18-G1-(Br)3] (59)
OO
O
OO HN
O
O
O
O
O O
Br
Br
Br
59
Compound 59 was synthesized according to general procedure GP 4 from malonate 58
(750 mg, 0.73 mmol), C60 (741 mg, 1.03 mmol), CBr4 (342 mg, 1.03 mmol) and DBU
(153 µL, 1.03 mmol) in 300 mL toluene. The crude product was purified by column
chromatography (SiO2; toluene/ethyl acetate, 80:5 to 80:25) to give 59 as red brownish
solid.
Yield: 406 mg, 0.28 mmol, 32 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 5.59 (s, br, 1H, CONH), 4.47 (t, 3J = 6.6 Hz, 4H,
OCH2), 4.36 (t, 3J = 6.1 Hz, 6H, CH2CH2Br), 3.49 (t, 3J = 6.1 Hz, 6H, CH2Br), 2.33 (t,
3J = 7.6 Hz, 6H, CH2COO), 2.12 (t, 3J = 7.5 Hz, 2H, OCCH2), 2.03 (t, 3J = 7.3 Hz, 6H,
NHC(CH2)3), 1.83 (m, 4H, CH2), 1.67 (m, 2H, CH2), 1.25 (m, 30H, CH2), 0.85 (t, 3J =
7.0 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.08 (3C, COO), 172.48 (1C, CONH), 164.00
(1C, CO), 163.92 (1C, CO), 145.65, 145.64, 145.55, 145.48, 145.47, 145.42, 145.17,
144.99, 144.96, 144.93, 144.89, 144.16, 143.40, 143.38, 143.31, 143.28, 143.26,
142.49, 142.15, 141.25, 141.22, 139.24, 138.14 (58C, C60-sp2), 71.84 (2C, C60-sp3),
196

Chapter 5 Experimental Part
67.74 (1C, OCH2), 67.28 (1C, OCH2), 64.25 (3C, CH2CH2Br), 57.46 (1C, NHC(CH2)3),
52.61 (1C, OCCCO), 37.32 (1C, CH2CO), 32.08 (1C, CH2), 29.87 (5C, NHC(CH2)3,
CH2), 29.82, 29.78, 29.77, 29.52, 29.38 (7C, CH2), 28.85 (3C, CH2Br), 28.75 (2C,
CH2), 28.56 (3C, CH2COO), 28.47 (2C, CH2), 26.15 (1C, CH2CH2CO), 25.81, 25.27,
22.84, 21.59 (4C, CH2), 14.28 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 1739 [M]+, 720 [C60]+; MWcalc.: 1739.39 g/mol.
IR (ATR): ν̃ = 3332, 3062, 2992, 1832, 1766, 1703, 1654, 1603, 1533, 1478, 1403,
1281, 1235, 1107, 1033, 799, 787, 653 cm−1.
UV/Vis (CH 2Cl2): λmax = 325.5, 425.5, 492 nm.
Cationic asymmetric dendritic monoadduct [C 18-G1-(C5H5N+Br−)3] (60)
OO
O
OO HN
O
O
O
O
O O
N
N
N
3 Br
60
A solution of 38 (150 mg, 0.086 mmol) in 10 mL of dry pyridine was stirred for two days
at 60 °C. After the addition of 10 mL of toluene, the reaction m ixture was filtrated and
the residue was suspended in toluene and distilled under vacuum for several times to
remove traces of pyridine. Reprecipitation from methanol/diethyl ether gave 60 as red
brownish solid.
Yield: 156 mg, 0.079 mmol, 92 %, red brownish solid.
1H-NMR (400 MHz, RT, DMSO-d6): δ = 9.13 (d, 3J = 5.6 Hz, 6H, o-PyrH), 8.65 (t, 3J =
7.8 Hz, 3H, p-PyrH), 8.20 (dd, 3J = 6.0, 7.7 Hz, 6H, m-PyrH), 7.20 (s, br, 1H, CONH),
4.91 (m, 6H, CH2-Pyr), 4.51 (m, 10H, CH2CH2-Pyr, OCH2), 2.13 (m, 6H, CH2COO),
197

Chapter 5 Experimental Part
2.05 (m, 2H, OCCH2), 1.73 (m, 6H, NHC(CH2)3), 1.48 (m, 2H, CH2), 1.35 (m, 4H,
CH2), 1.17 (m, 32H, CH2), 0.84 (t, 3J = 6.2 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, DMSO-d6): δ = 172.59 (3C, COO), 172.56 (1C, CONH),
162.92 (1C, CO), 162.90 (1C, CO), 146.33 (3C, p-PyrC), 145.56 (6C, o-PyrC), 145.51,
145.16, 144.95, 144.94, 144.89, 144.86, 144.80, 144.73, 144.52, 144.36, 144.34,
144.30, 144.25, 143.54, 143.51, 142.81, 142.74, 142.73, 141.85, 141.84, 141.53,
141.46, 140.67, 138.74, 138.22 (58C, C60-sp2), 128.21 (6C, m-PyrC), 71.55 (2C, C60-
sp3), 67.37 (2C, OCH2), 62.56 (3C, CH2CH2-Pyr), 59.76 (3C, CH2-Pyr), 56.29 (1C,
NHC(CH2)3), 52.70 (1C, OCCCO), 35.84 (1C, CH2CO), 31.42 (6C, NHC(CH2)3, CH2),
29.18, 29.11, 29.01 (4C, CH2), 28.84 (3C, CH2COO), 28.65, 28.57, 28.13, 27.80, 27.67
(7C, CH2), 25.69 (1C, CH2CH2CO), 25.08, 24.97, 22.22 (4C, CH2), 14.06 (1C, CH3)
ppm.
MS (FAB, NBA): m/z = 1897 [M]+, 908 [M]2+; MWcalc.: 1976.69 g/mol.
IR (ATR): ν̃ = 3368, 3020, 3001, 2651, 1799, 1765, 1654, 1613, 1546, 1434, 1406,
1256, 1249, 1237, 1111, 1023, 807, 753, 653, 603 cm−1.
UV/Vis (DMSO/H 2O): λmax = 259, 325.5 nm.
3,5-Bis(propargyloxy)benzyl malonate (78)
O O
O OO
O
O
O78
Compound 78 was synthesized according to gen-
eral procedure GP 1 from 3,5-bis(propargyloxy)-
benzyl alcohol 130, 131 (3.5 g, 16.19 mmol),
malonyl dichloride (787 µL, 8.09 mmol) and dry
pyridine (1.31 mL, 16.19 mmol) in 100 mL dry
CH2Cl2. The crude product was purified by re-
crystallization from dichloromethane/hexane to give 78 as white solid.
Yield: 2.79 g, 5.58 mmol, 69 %, white solid.
1H-NMR (300 MHz, RT, CDCl3): δ = 6.57 (m, 6H, ArH), 5.11 (4H, Ar-CH2), 4.63 (d, 4J
= 2.4 Hz, 8H, OCH2), 3.48 (s, 2H, OCCH2CO), 2.51 (t, 4J = 2.5 Hz, 4H, CH) ppm.
198

Chapter 5 Experimental Part
13C-NMR (75 MHz, RT, CDCl3): δ = 166.27 (2C, CO), 159.01 (4C, ArC-O), 137.85 (2C,
ArC-CH2), 107.71 (4C, ArC), 102.37 (2C, ArC), 78.45 (4C, CCH), 76.00 (4C, CCH),
67.06 (2C, Ar-CH2), 56.16 (4C, OCH2), 41.66 (1C, OCCH2CO) ppm.
MS (FAB, NBA): m/z = 501 [M]+; MWcalc.: 500.50 g/mol.
IR (ATR): ν̃ = 2978, 1768, 1720, 1656, 1624, 1576, 1544, 1448, 1368, 1241, 1192,
1160, 1016, 888, 839, 776, 744, 681 cm−1.
EA: C29H24O8: calcd. C 69.59, H 4.83, O 25.57; found C 69.12, H 4.78.
1,2-((3,5-Bis(propargyloxy)benzyloxycarbonyl)-metha no)-1,2-dihydro-
[60]fullerene (79)
OO
OO
O
O O
O
79
Compound 79 was synthesized according to gen-
eral procedure GP 4 from malonate 78 (500 mg,
1.00 mmol), C60 (864 mg, 1.20 mmol), CBr4 (398
mg, 1.20 mmol) and DBU (179 µL, 1.20 mmol) in
400 mL toluene. The crude product was purified by
flash column chromatography (SiO2; toluene/ethyl
acetate, 100:3) to give 79 as red brownish solid.
Yield: 475 mg, 0.39 mmol, 39 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 6.67 (d, 4J = 2.3 Hz, 4H, ArH), 6.58 (t, 4J = 2.3
Hz, 2H, ArH), 5.42 (4H, Ar-CH2), 4.63 (d, 4J = 2.4 Hz, 8H, OCH2), 2.52 (t, (t, 4J = 2.4
Hz, 4H, CH) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 163.58 (2C, CO), 159.17 (4C, ArC-O), 145.54,
145.46, 145.36, 145.20, 145.15, 144.97, 144.88, 144.87, 144.16, 143.34, 143.30,
143.27, 142.48, 142.13, 141.22, 139.33 (58C, C60-sp2), 137.21 (2C, ArC-CH2), 108.37
(4C, ArC), 102.71 (2C, ArC), 78.41 (4C, CCH), 76.19 (4C, CCH), 71.53 (2C, C60-sp3),
68.73 (2C, Ar-CH2), 56.17 (4C, OCH2), 51.82 (1C, OCCCO) ppm.
MS (FAB, NBA): m/z = 1219 [M]+, 720 [C60]+; MWcalc.: 1219.12 g/mol.
IR (ATR): ν̃ = 3290, 1738, 1599, 1452, 1367, 1267, 1228, 1151, 1058, 996, 942, 826,
199

Chapter 5 Experimental Part
742, 672 cm−1.
UV/Vis (CH 2Cl2): λmax = 233, 259, 326, 425, 495 nm.
Bis(3,5-bis((1-(2-( tert-butoxycarbonylamino)ethyl)-1 H-1,2,3-triazol-4-yl)methoxy)
benzyl) malonate (83)
OO
OO
O
O O
O
NN
N
NNN
N NN
N NN
R = NHboc
R
R R
R
83
Malonate 78 (326 mg, 0.72
mmol) and azide 81 (536 mg,
2.88 mmol) were dissolved in 2
mL tert-butanol/H2O =1/1 under
careful heat up. Subsequently,
sodium ascorbate (24 mg, 0.12
mmol) and CuSO4 (18 mg, 0.07 mmol) were added and the reaction mixture was stirred
at rt. After 45 min, TLC control showed almost complete conversion of the starting
material. The light yellow mixture was stirred for additional 2 h, diluted with 2 mL am-
monium hydroxide/citrate buffer (15 %) and stirred for 10 minutes. The aqueous phase
was extracted with ethyl acetate (3 x 25 mL). The organic phase was washed with sat-
urated NaCl (2 x 25 mL), dried over MgSO4 and evaporated under reduced pressure.
The crude product was purified by recrystallization from dichloromethane/pentane to
give 83 as white solid.
Yield: 825 mg, 0.66 mmol, 92 %, white solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 7.64 (s, 4H, triazoleCH), 6.51 (m, 6H, ArH), 5.34
(s, br, 4H, NH), 5.07 (s, 4H, OCH2), 5.04 (s, 8H, OCH2), 4.45 (t, 3J = 5.6 Hz, 8H,
NCH2), 3.60 (m, 8H, HNCH2), 3.49 (s, 2H, OCCH2CO), 1.38 (s, 36H, C(CH3)3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 166.46 (2C, CO), 159.74 (4C, (4C, ArC-O)),
156.25 (4C, COOtBu), 143.86 (2C, ArC-CH2), 137.95 (4C, triazoleC), 124.08 (4C, tri-
azoleC), 107.26 (4C, ArC), 102.03 (2C, ArC), 80.07 (4C, C(CH3)3), 67.02 (2C, OCH2),
62.01 (4C, OCH2), 50.22 (4C, NCH2), 41.83 (1C, OCCH2CO), 40.64 (4C, HNCH2),
28.47 (12C, C(CH3)3) ppm.
MS (FAB, NBA): m/z = 1245 [M+H]+; MWcalc.: 1244.59 g/mol.
200

Chapter 5 Experimental Part
IR (ATR): ν̃ = 3301, 3140, 2978, 2936, 1688, 1640, 1576, 1528, 1448, 1384, 1288,
1240, 1144, 1048, 984, 840, 761, 710, 664 cm−1.
UV/Vis (CH 2Cl2): λmax = 239, 280 nm.
EA: C57H80N16O16 · 3 CH2Cl2: calcd. C 48.04, H 5.78, N 14.94, O 17.06 ; found C
47.65, H 6.01, N 17.17.
Bis(3,5-bis((1-((2-methoxyethoxy)methyl)-1 H-1,2,3-triazol-4-yl)methoxy)benzyl)
malonate (84)
OO
OO
O
O O
O
NN
N
NNN
N NN
N NN
R = O(CH2)2OCH3
R
R R
R
84
Compound 84 was synthesized
following the procedure described
for 83 from malonate 78 (400
mg, 0.80 mmol), azide 82 (464
mg, 3.20 mmol), sodium ascor-
bate (28 mg, 0.14 mmol) and
CuSO4 (20 mg, 0.09 mmol) in 2.5 mL tert-butanol/H2O =1/1. The crude product was
purified by column chromatography (SiO2; dichloromethane/methanol, 100:8) to give
84 as colorless oil.
Yield: 730 mg, 0.71 mmol, 89 %, colorless oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 7.73 (s, 4H, triazoleCH), 6.49 (m, 6H, ArH), 5.03
(s, 8H, OCH2), 5.01 (s, 4H, OCH2), 4.44 (t, 3J = 5.0 Hz, 8H, NCH2), 3.76 (m, 8H,
NCH2CH2), 3.47 (m, 8H, OCH2), 3.42 (s, 2H, OCCH2CO), 3.38 (m, 8H, OCH2), 3.22
(s, 12H, OCH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 166.10 (2C, CO), 159.52 (4C, ArC-O), 143.40
(2C, ArC-CH2), 137.63 (4C, triazoleC), 124.08 (4C, triazoleC), 106.97 (4C, ArC), 101.74
(2C, ArC), 71.62 (4C, OCH2), 70.38 (4C, OCH2), 69.29 (4C, OCH2), 66.77 (2C, OCH2),
61.93 (4C, OCH2), 58.86 (4C, OCH3), 50.17 (4C, NCH2), 41.35 (1C, OCCH2CO) ppm.
MS (FAB, NBA): m/z = 1081 [M+H]+; MWcalc.: 1081.14 g/mol.
IR (ATR): ν̃ = 2996, 2901, 2861, 1592, 1496, 1448, 1352, 1272, 1192, 1144, 1112,
201

Chapter 5 Experimental Part
1064, 984, 904, 856, 808, 776, 711, 668 cm−1.
UV/Vis (CH 2Cl2): λmax = 239.5, 281 nm.
EA: C49H68N12O16 · 1.5 CH3OH: calcd. C 53.71, H 6.61, N 14.88, O 24.80 ; found C
53.24, H 6.63, N 15.07.
1,2-(Bis(3,5-bis((1-(2-( tert-butoxycarbonylamino)ethyl)-1 H-1,2,3-triazol-4-yl)
methoxy)benzyloxycarbonyl)-methano)-1,2-dihydro[60] fullerene (85)
OO
OO
O
O O
O
NN
N
NNN
N NN
N NN
R
R R
R
R = NHboc
85
Compound 85 was synthesized
according to general procedure
GP 4 from malonate 83 (200 mg,
0.16 mmol), C60 (1500 mg, 0.21
mmol), CBr4 (59 mg, 0.18 mmol)
and DBU (27 µL, 0.18 mmol)
in 100 mL toluene/DMSO = 3:1.
The crude product was purified
by flash column chromatography (SiO2; toluene, ethyl acetate/ethanol, 80:15) to give
85 as red brownish solid.
Yield: 98 mg, 0.05 mmol, 29 %, red brownish solid.
1H-NMR (300 MHz, RT, CDCl3): δ = 7.70 (s, 4H, triazoleCH), 6.57 (m, 6H, ArH), 5.55
(s, br, 4H, NH), 5.28 (s, 4H, OCH2), 5.04 (s, 8H, OCH2), 4.45 (m, 8H, NCH2), 3.60 (m,
8H, HNCH2), 1.39 (s, 36H, C(CH3)3) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 163.45 (2C, CO), 159.71 (4C, ArC-O), 156.21 (4C,
COOtBu), 145.47, 145.39, 145.28, 145.10, 145.06, 144.88, 144.82, 144.76,
144.05, 143.57, 143.27, 143.24, 143.18, 142.38, 142.05, 141.17, 139.28 (60C, C60-
sp2, ArC-CH2), 137.05 (4C, triazoleC), 124.17 (4C, triazoleC), 107.69 (4C, ArC), 102.23
(2C, ArC), 80.02 (4C, C(CH3)3), 71.52 (2C, OCH2), 68.72 (2C, C60-sp2), 62.02 (4C,
OCH2), 53.66 (1C, OCCCO), 50.33 (4C, NCH2), 40.74 (4C, HNCH2), 28.57 (12C,
C(CH3)3) ppm.
MS (FAB, NBA): m/z = 1964 [M+H]+, 720 [C60]+; MWcalc.: 1963.97 g/mol.
202

Chapter 5 Experimental Part
IR (ATR): ν̃ = 3268, 3103, 2964, 2902, 1708, 1638, 1586, 1503, 1438, 1401, 1298,
1243, 1163, 1054, 991, 882, 763, 681 cm−1.
UV/Vis (CH 2Cl2): λmax = 232, 257, 324.5, 425.5, 483 nm.
1,2-(Bis(3,5-bis((1-((2-methoxyethoxy)methyl)-1 H-1,2,3-triazol-4-yl)methoxy)
benzyloxycarbonyl)-methano)-1,2-dihydro[60]fulleren e (86)
OO
OO
O
O O
O
NN
N
NNN
N NN
N NN
R
R R
R
R = O(CH2)2OCH3
86
Compound 86 was synthesized
according to general procedure
GP 4 from malonate 84 (200 mg,
0.19 mmol), C60 (200 mg, 0.28
mmol), CBr4 (74 mg, 0.22 mmol)
and DBU (33 µL, 0.22 mmol)
in 100 mL toluene/DMSO = 3:1.
The crude product was purified
by flash column chromatography (SiO2; toluene, dichloromethane/methanol, 100:6) to
give 86 as red brownish solid.
Yield: 109 mg, 0.06 mmol, 32 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 7.77 (s, 4H, triazoleCH), 6.60 (m, 6H, ArH), 5.11
(s, 8H, OCH2), 5.08 (s, 4H, OCH2), 4.50 (t, 3J = 5.0 Hz, 8H, NCH2), 3.82 (t, 3J = 4.6
Hz, 8H, NCH2CH2), 3.53 (m, 8H, OCH2), 3.45 (m, 8H, OCH2), 3.29 (s, 12H, OCH3)
ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 163.36 (2C, CO), 159.81, 159.69 (4C, ArC-
O), 145.27, 145.21, 145.01, 144.76, 144.64, 144.12, 143.89, 143.56, 143.08, 142.28,
141.94, 141.00, 139.14 (60C, C60-sp2, ArC-CH2), 137.10 (4C, triazoleC), 124.21 (4C,
triazoleC), 107.88 (4C, ArC), 102.27 (2C, ArC), 71.84 (4C, OCH2), 70.62 (4C, OCH2),
69.53 (4C, OCH2), 68.77 (2C, C60-sp2), 65.00 (2C, OCH2), 62.15 (4C, OCH2), 59.13
(4C, OCH3), 52.23 (1C, OCCCO), 50.40 (4C, NCH2) ppm.
MS (FAB, NBA): m/z = 1081 [M+H]+; MWcalc.: 1081.14 g/mol.
IR (ATR): ν̃ = 3001, 2913, 2901, 2855, 1601, 1503, 1441, 1415, 1353, 1199, 1151,
203

Chapter 5 Experimental Part
1107, 1071, 1001, 9834, 904, 858, 773, 709, 671 cm−1.
UV/Vis (CH 2Cl2): λmax = 232.5, 258, 325, 425, 486 nm.
Tetraammoniumtrifluoroacetate triazolfullerene (87)
OO
OO
O
O O
O
NN
N
NNN
N NN
N NN
R
R R
R
R = NH3 CF3COO
87
Compound 87 was synthesized
according to general procedure
GP 5 from momoadduct 85 (60
mg, 0.03 mmol) in formic acid
(15 mL). The crude product was
purified by reprecipitation from
MeOH/Et2O to give 87 as a red
brownish solid.
Yield: 36 mg, 0.03 mmol, 91 %, red brownish solid.
1H-NMR (300 MHz, RT, MeOH-d4): δ = 8.10 (s, 4H, triazoleCH), 6.67 (m, 6H, ArH),
5.04 (m, 12H, OCH2), 4.71 (m, 8H, NCH2), 3.49 (m, 8H, NH+3NCH2) ppm.
13C-NMR (75 MHz, RT, MeOH-d4): δ = 161.20 (2C, CO), 158.69 (4C, (4C, ArC-O)),
146.55, 146.53, 146.49, 146.36, 145.80, 145.40, 145.26, 145.14, 144.31, 143.51,
143.17, 142.22, 142.18, 141.47, 141.44, 140.33, 139.17, 139.09, 138.19 (60C, C60-
sp2, ArC-CH2), 138.02 (4C, triazoleC), 126.33 (4C, triazoleC), 106.36 (4C, ArC), 101.89
(2C, ArC), 71.41 (2C, OCH2), 69.97 (2C, C60-sp3), 62.31 (4C, OCH2), 53.04 (1C,
OCCCO), 49.15 (4C, NCH2), 40.39 (4C, HNCH2) ppm.
MS (FAB, NBA): m/z = 1564 [M]+, 720 [C60]+.
IR (ATR): ν̃ = 3314, 3098, 3091, 2971, 2914, 1715, 1651, 1642, 1579, 1511, 1439,
1402, 1301, 1263, 1157, 1101, 993, 969, 878, 755, 689 cm−1.
UV/Vis (DMSO): λmax = 263.5, 325.5, 428 nm.
204

Chapter 5 Experimental Part
6-tert-Butoxy-6-oxohexyl methyl malonate (92)
O O
O OO
O92
Compound 92 was synthesized according to general
procedure GP 1 from 22 (2 g, 10.62 mmol), methyl mal-
onyl chloride (1.14 mL, 10.62 mmol) and dry pyridine
(857 µL, 10.62 mmol) in 150 mL dry CH2Cl2. The crude
product was purified by column chromatography (SiO2; hexane/ethyl acetate, 100:30)
to give 92 as colorless oil.
Yield: 2.35 g, 8.67 mmol, 82 %, colorless oil
1H-NMR (300 MHz, RT, CDCl3): δ = 4.12 (t, 3J = 6.6 Hz, 2H, OCH2), 3.72 (s, 3H,
OCH3), 3.35 (s, 2H, OCCH2CO), 2.19 (t, 3J = 7.4 Hz, 2H, CH2CO), 1.61 (m, 4H, CH2),
1.41 (s, 9H, C(CH3)3), 1.35 (m, 2H, CH2) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 173.09 (1C, COOtBu), 167.21 (1C, CO), 166.73
(1C, CO), 80.31 (1C, C(CH3)3), 65.63 (1C, OCH2), 52.70 (1C, OCH3), 41.57 (1C,
OCCH2CO), 35.55 (1C, CH2CO), 28.37 (1C, CH2), 28.31 (3C, C(CH3)3), 25.51, 24.84
(2C, CH2) ppm.
MS (FAB, NBA): m/z = 288 [M]+, 231 [M-tBu]+; MWcalc.: 288.34 g/mol.
IR (ATR): ν̃ = 3001, 2984, 1736, 1641, 1448, 1368, 1256, 1144, 1016, 936, 856, 776,
728, 696 cm−1.
EA: C14H24O6: calcd. C 58.32, H 8.39, O 33.29; found: C 57.66, H 8.31.
205

Chapter 5 Experimental Part
1,2-(Bis(6- tert-butoxy-6-oxohexyl-methyloxy)-methano)-1,2-dihydro
[60]fullerene (93)
O O
O OO
O
93
Compound 93 was synthesized according to general
procedure GP 4 from malonate 92 (395 mg, 1.37
mmol), C60 (1.48 g, 2.06 mmol), CBr4 (500 mg, 1.51
mmol) and DBU (225 µL, 1.51 mmol) in 750 mL toluene.
The crude product was purified by column chromatog-
raphy (SiO2; toluene/ethyl acetate, 100:5) to give 93 as
a red brownish solid.
Yield: 736 mg, 0.73 mmol, 43 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 4.48 (t, 3J = 6.5 Hz, 2H, OCH2), 4.07 (s, 3H,
OCH3), 2.22 (t, 3J = 7.4 Hz, 2H, CH2CO), 1.85 (m, 2H, CH2), 1.66 (m, 2H, CH2), 1.48
(m, 2H, CH2), 1.43 (s, 9H, C(CH3)3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.14 (1C, COOtBu), 163.44 (1C, CO), 163.91
(1C, CO), 145.57, 145.49, 145.47, 145.41, 145.20, 145.00, 144.94, 144.19, 143.38,
143.32, 143.28, 142.50, 142.23, 142.19, 141.27, 141.26, 139.41, 139.22 (58C, C60-
sp2), 80.43 (1C, C(CH3)3), 71.72 (2C, C60-sp3), 67.41 (1C, OCH2), 54.21 (1C, OCH3),
52.26 (1C, OCCCO), 35.54 (1C, CH2CO), 28.46 (1C, CH2), 28.30 (3C, C(CH3)3),
25.61, 24.75 (2C, CH2) ppm.
MS (FAB, NBA): m/z = 1006 [M]+, 951 [M-tBu]+, 720 [C60]+; MWcalc.: 1000.96 g/mol.
IR (ATR): ν̃ = 2943, 2866, 2016, 1977, 1730, 1537, 1429, 1367, 1236, 1151, 1097,
1058, 1004, 942, 896, 842, 734 cm−1.
UV/Vis (CH 2Cl2): λmax = 259, 326, 426, 493 nm.
206

Chapter 5 Experimental Part
1,2-(Bis(6-(3-methoxy-3-oxopropanoyloxy)hexanoic aci d)-methano)-1,2-dihydro
[60]fullerene (94)
O O
O OOH
O
94
Compound 94 was synthesized according to general pro-
cedure GP 5 from 93 (285 mg, 0.28 mmol) and trifluo-
roacetic acid (2.4 mL, 31.0 mmol) in 50 mL CH2Cl2.
Yield: 257 mg, 0.27 mmol, 96 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 4.49 (t, 3J = 6.4 Hz, 2H, OCH2), 4.07 (s, 3H,
OCH3), 2.38 (t, 3J = 7.3 Hz, 2H, CH2CO), 1.86 (m, 2H, CH2), 1.71 (m, 2H, CH2), 1.52
(m, 2H, CH2) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 179.02 (1C, COOH), 164.44 (1C, CO), 163.92
(1C, CO), 145.58, 145.50, 145.49, 145.46, 145.41, 145.39, 145.21, 145.00, 144.95,
144.94, 144.92, 144.19, 143.39, 143.33, 143.31, 143.28, 142.50, 142.22, 142.17,
141.28, 141.26, 139.44, 139.17 (58C, C60-sp2), 71.69 (2C, C60-sp3), 67.30 (1C, OCH2),
54.23 (1C, OCH3), 52.24 (1C, OCCCO), 33.85 (1C, CH2CO), 28.42 (1C, CH2), 25.64,
24.35 (2C, CH2) ppm.
MS (FAB, NBA): m/z = 951 [M+H]+, 720 [C60]+; MWcalc.: 950.86 g/mol.
IR (ATR): ν̃ = 2991, 2850, 2802, 2645, 2364, 1746, 1699, 1429, 1236, 1182, 1097,
1058, 996, 935, 796, 742, 703 cm−1.
UV/Vis (CH 2Cl2): λmax = 259, 325.5, 425.5, 489 nm.
207

Chapter 5 Experimental Part
2-Hydroxyethyl 4-(pyren-1-yl)butanoate (97)
O
O OH
97
Compound 97 was synthesized according to general pro-
cedure GP 3 from ethane-1,2-diol (1.72 g, 27.75 mmol), 1-
pyrenebutyric acid (2g, 6.94 mmol), EDC (2 g, 10.43 mmol),
DMAP (847 mg, 6.94 mmol) and 1-HOBt (1.41 g, 10.40
mmol) in dry THF/ CH2Cl2 = 2/ 1. The crude product was
purified by column chromatography (SiO2; dichloromethane/acetone, 10:1) to give 97
as yellow oil.
Yield: 1.81 g, 5.45 mmol, 78 %, yellow oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 8.08 (m, 9H, ArH), 4.23 (t, 3J = 4.7 Hz, 2H,
OCOCH2), 3.82 (t, 3J = 4.7 Hz, 2H, CH2OH), 3.40 (t, 3J = 7.7 Hz, 2H, ArCH2), 2.50 (t,
3J = 7.4 Hz, 2H, CH2CO), 2.22 (m, 2H, CH2), 1.26 (s, br, 1H, OH) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.78 (1C, CO), 135.50, 131.36, 130.82,
129.94, 128.70, 127.385, 126.70, 125.82, 125.04, 124.91, 124.75, 123.20 (16C, ArC),
65.98 (1C, CH2OH), 61.18 (1C, OCOCH2), 33.63 (1C, ArCH2), 32.62 (1C, CH2CO),
26.65 (1C, CH2) ppm.
MS (FAB, NBA): m/z = 332 [M]+; MWcalc.: 332.39 g/mol.
IR (ATR): ν̃ = 3461, 2984, 2901, 1736, 1654, 1448, 1368, 1272, 1224, 1160, 1064,
1016, 936, 872, 712, 648 cm−1.
UV/Vis (CH 2Cl2): λmax = 235.5, 244.5, 266, 278, 300 (sh), 314, 328, 344 nm.
208

Chapter 5 Experimental Part
Bis(2-(4-(pyren-1-yl)butanoyloxy)ethyl)malonate (98)
O
OOO
OO
O
O
98
Compound 98 was synthesized ac-
cording to general procedure GP 1
from 97 (750 mg, 2.26 mmol), mal-
onyl dichloride (110 µL, 1.13 mmol)
and dry pyridine (182 µL, 2.26 mmol)
in dry CH2Cl2. The crude product was purified by column chromatography (SiO2;
dichloromethane/acetone, 96:4) to give 98 as yellow oil.
Yield: 553 mg, 0.75 mmol, 67 %, yellow oil.
1H-NMR (400 MHz, RT, CDCl3): δ = 8.06 (m, 18H, ArH), 4.32 (m, 4H, OCOCH2), 4.27
(m, 4H, CH2OCO), 3.44 (s, 2H, OCCH2CO), 3.35 (t, 3J = 7.7 Hz, 4H, ArCH2), 2.46 (t,
3J = 7.3 Hz, 4H, CH2CO), 2.20 (m, 4H, CH2) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.06 (2C, CO), 166.01 (2C, CO), 135.45,
131.27, 130.75, 129.85, 128.59, 127.35, 127.28, 127.18, 126.62, 125.74, 124.93,
124.83, 124.68, 123.12 (32C, ArC), 63.02 (2C, OCOCH2), 61.53 (2C, CH2OCO), 40.86
(1C, OCCH2CO), 33.32 (2C, ArCH2), 32.38 (2C, CH2CO), 26.35 (2C, CH2) ppm.
MS (FAB, NBA): m/z = 733 [M]+; MWcalc.: 732.82 g/mol.
IR (ATR): ν̃ = 2927, 2858, 2116, 1746, 1684, 1452, 1359, 1298, 1244, 1190, 1151,
1043, 950, 888, 842, 788, 718 cm−1.
UV/Vis (CH 2Cl2): λmax = 234, 244, 266, 279, 301 (sh), 314, 327, 344.5 nm.
EA: C47H40O8: calcd. C 77.03, H 5.50, O 17.47; found C 76.77, H 5.63.
209

Chapter 5 Experimental Part
1,2-(Bis(2-(4-(pyren-1-yl)butanoyloxy)ethyloxy-carb onyl)-methano)-1,2-dihydro
[60]fullerene (99)
O
OOO
OO
O
O
99
Compound 99 was synthesized ac-
cording to general procedure GP 4
from malonate 98 (475 mg, 0.65
mmol), C60 (700 mg, 0.97 mmol),
CBr4 (258 mg, 0.78 mmol) and DBU
(117 µL, 0.78 mmol) in 350 mL
toluene. The crude product was pu-
rified by flash column chromatography (SiO2; toluene/ethyl acetate, 10:1 to 8:1) to give
99 as red brownish solid. To prevent any impurities that can distort the STM mea-
surements (e.g. remaining silica in the probe), the product was additionally purified by
preparative HPLC (Nucleosil, 5µm, dichloromethane/methanol = 98/2).
Yield: 452 mg, 0.31 mmol, 48 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 8.02 (m, 18H, ArH), 4.64 (m, 4H, OCOCH2), 4.40
(m, 4H, CH2OCO), 3.35 (t, 3J = 7.7 Hz, 4H, ArCH2), 2.43 (t, 3J = 7.5 Hz, 4H, CH2CO),
2.16 (m, 4H, CH2) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.15 (2C, CO), 163.43 (2C, CO), 145.01,
144.78, 144.66, 144.50, 144.36, 144.21, 143.59, 142.81, 142.68, 142.54, 141.89,
141.52, 140.60, 138.76 (58C, C60-sp2), 135.41, 131.49, 130.95, 130.09, 128.80, 127.56,
127.39, 126.86, 125.95, 125.11, 125.05, 124.90, 124.88, 123.25 (32C, ArC), 70.95
(2C, C60-sp3), 64.78 (2C, OCOCH2), 61.68 (2C, CH2OCO), 51.57 (1C, OCCCO), 33.65
(2C, ArCH2), 32.63 (2C, CH2CO), 26.48 (2C, CH2) ppm.
MS (FAB, NBA): m/z = 1451 [M]+, 720 [C60]+; MWcalc.: 1451.44 g/mol.
IR (ATR): ν̃ = 2934, 2901, 2861, 2109, 1746, 1708, 1693, 1453, 1362, 1297, 1244,
1203, 1188, 1103, 1044, 948, 843, 777, 716, 681 cm−1.
UV/Vis (CH 2Cl2): λmax = 236, 245, 257.5, 266, 278, 315 (sh), 328.5, 345, 425, 495 nm.
The purity (99 %) was determined by HPLC (on Nucleosil MN, 5 µm,
210

Chapter 5 Experimental Part
dichloromethane/methanol = 99/1).
Bis-(5-( tert-butoxycarbonyl)pentyl) malonate (100)
O O
O OO
O
O
O100
Compound 100 was synthesized accord-
ing to general procedure GP 1 from 22 (7
g, 37 mmol), malonyl dichloride (1.81 mL,
18.62 mmol) and dry pyridine (2.99 mL, 37
mmol) in 250 mL dry CH2Cl2. The crude product was purified by column chromatogra-
phy (SiO2; hexane/ethyl acetate, 100:20) to give 100 as white solid.
Yield: 5.5 g, 12.48 mmol, 67 %, white solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 4.11 (t, 3J = 6.7 Hz, 4H, OCH2), 3.33 (s, 2H,
OCCH2CO), 2.19 (t, 3J = 7.5 Hz, 4H, CH2CO), 1.62 (m, 8H, CH2), 1.41 (s, 18H,
C(CH3)3), 1.34 (m, 4H, CH2) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.23 (2C, COOtBu), 166.96 (2C, CO), 80.33
(1C, C(CH3)3), 65.57 (2C, OCH2), 41.73 (1C, OCCH2CO), 35.50 (2C, CH2CO), 28.33
(2C, CH2), 28.24 (6C, C(CH3)3), 25.45, 24.78 (4C, CH2) ppm.
MS (FAB, NBA): m/z = 445 [M]+, 389 [M-tBu]+, 333 [M-2tBu]+; MWcalc.: 444.56 g/mol.
IR (ATR): ν̃ = 2943, 2873, 1730, 1460, 1421, 1367, 1329, 1251, 1151, 1043, 950, 850,
734, 687 cm−1.
EA: C23H40O8 · 1/2 C4H8O2: calcd. C 61.45, H 9.08, O 29.47; found C 60.99, H 9.03.
Bis-(5-(pentylcarbonyl)) malonate (101)
O O
O OOH
O
HO
O101
Compound 101 was synthesized according to
general procedure GP 5 from 100 (2 g, 4.50
mmol) in formic acid (20 mL).
Yield: 1.45 g, 4.36 mmol, 97 %, white solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 10.11 (s, br, 2H, COOH), 4.11 (t, 3J = 6.6 Hz, 4H,
OCH2), 3.34 (s, 2H, OCCH2CO), 2.33 (t, 3J = 7.4 Hz, 4H, CH2CO), 1.63 (m, 8H, CH2),
211

Chapter 5 Experimental Part
1.39 (m, 4H, CH2) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 179.87 (2C, COOH), 166.85 (2C, CO), 65.45
(2C, OCH2), 41.73 (1C, OCCH2CO), 33.93 (2C, CH2CO), 28.26 (2C, CH2), 25.43,
24.34 (4C, CH2) ppm.
MS (FAB, NBA): m/z = 333 [M+H]+; MWcalc.: 332.15 g/mol.
IR (ATR): ν̃ = 3347, 3065, 2963, 1912, 1766, 1761, 1686, 1578, 1563, 1415, 1263,
1193, 1146, 933, 867, 713 cm−1.
5-(3-(6-Methyl-4-oxo-1,4-dihydropyrimidin-2-yl)urei do)pentyl octadecyl malonate
(106)
O O
O O
NH
O
NH
N
HN
O
106
Compound 27 (1.00 g,
2.13 mmol) was dis-
solved in 100 mL dry
CH2Cl2, followed by
the addition of oxalyl chloride (365 µL, 4.25 mmol). The resulting mixture was heated
under reflux for 2 h. After cooling to rt the volatiles were removed in vacuo. The re-
maining acid chloride 103 (1.03 g, 2.11 mmol) was redissolved in 15 mL dry DMF
and sodium azide (275 mg, 4.23 mmol) was added. The resulting mixture was stirred
at rt until TLC control showed complete conversion of the starting material. Subse-
quently, 2-amino-4-hydroxy-6-methylpyrimidine (625 mg, 4.99 mmol) and dry pyridine
(15 mL, 0.19 mol) was added and the reaction mixture was heated to 70 - 80°C, until
TLC control showed complete conversion of the starting material. After cooling to rt
the mixture was partitioned between 250 mL CH2Cl2 and 250 mL H2O. The organic
phase was washed with aqueous HCl (10 %, 2 x 200 mL) and H2O (2 x 200 mL), dried
over anhydrous MgSO4 and concentrated in vacuo. The crude product was purified by
recrystallization from methanol to give 106 as white solid.
Yield: 960 mg, 1.62 mmol, 76 %, white solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 13.08 (1H, NH), 11.84 (1H, NH), 10.18 (1H, NH),
5.79 (1H, CH), 4.11 (t, 3J = 6.5 Hz, 2H, OCH2), 4.09 (t, 3J = 6.5 Hz, 2H, OCH2), 3.33
212

Chapter 5 Experimental Part
(s, 2H, OCCH2CO), 3.22 (m, 2H, CH2NH), 2.21 (s, 3H, CH3), 1.64 (m, 8H, CH2), 1.39
(m, 2H, CH2), 1.22 (m, 28H, CH2), 0.85 (t, 3J = 6.8 Hz, 3H, CH3) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 173.39 (1C, C-Pyrimidine), 167.03 (1C, CO),
167.02 (1C, CO), 156.89 (1C, CO-Pyrimidine), 154.99 (1C, HNCONH), 148.57 (1C,
C-Pyrimidine), 106.96 (1C, CH-Pyrimidine), 65.90 (1C, OCH2), 65.68 (1C, OCH2),
41.78 (1C, OCCH2CO), 39.89 (1C, CH2NH), 32.08, 29.85, 29.83, 29.81, 29.74, 29.67,
29.51, 29.37, 29.24, 28.61, 28.26, 25.94, 23.35, 22.83 (19C, CH2), 19.08 (1C, CH3-
Pyrimidine), 14.26 (1C, CH3) ppm.
MS (FAB, NBA): m/z = 594 [M+H]+, 1188 [2M+2H]+; MWcalc.: 592.81 g/mol.
IR (ATR): ν̃ = 2920, 2850, 1730, 1668, 1583, 1529, 1468, 1406, 1344, 1259, 1182,
1020, 935, 881, 796, 742, 687 cm−1.
EA: C32H56N4O6: calcd. C 64.83, H 9.52, N 9.45, O 16.19; found C 65.40, H 10.01, N
9.58.
cyclo-[2]-Octylmalonyl)-1,2-dihydro[60]fullerene (119)
O
O
O
OO
O
O
O
119
Compound 119 was synthesized according to
general procedure GP 4 from malonate 118 (1 g,
2.33 mmol), C60 (1.34 g, 1.87 mmol), CBr4 (773
mg, 2.33 mmol) and DBU (348 µL, 2.33 mmol) in
750 mL toluene. The crude product was purified
by flash column chromatography (SiO2; toluene;
dichloromethane) to give 119 as red brownish solid.
Yield: 615 mg, 0.54 mmol, 23 %, red brownish solid.
1H-NMR (400 MHz, RT, CDCl3): δ = 4.45 (t, 3J = 6.7 Hz, 4H, OCH2), 4.15 (t, 3J = 6.6
Hz, 4H, OCH2), 3.36 (s, 2H, OCCH2CO), 1.84 (m, 4H, CH2), 1.66 (m, 4H, CH2), 1.49
(m, 4H, CH2), 1.38 (m, 12H, CH2) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 166.82 (2C, CO), 163.95 (2C, CO), 145.55,
145.50, 145.47, 145.18, 144.98, 144.95, 144.90, 144.18, 143.37, 143.31, 143.28,
213

Chapter 5 Experimental Part
142.50, 142.21, 141.23, 139.40 (58C, C60-sp2), 71.78 (2C, C60-sp3), 67.63 (2C, OCH2),
65.74 (2C, OCH2), 52.25 (1C, OCCCO), 42.42 (1C, OCCH2CO), 29.58 , 28.77, 28.73,
26.18, 26.09 (12C, CH2) ppm.
MS (FAB, NBA): m/z = 1147 [M+H]+, 720 [C60]+; MWcalc.: 1147.14 g/mol.
IR (ATR): ν̃ = 2930, 2856, 1741, 1463, 1428, 1390, 1305, 1287, 1235, 1208, 1185,
1154, 1101, 1075, 1019, 946, 884, 737, 706 cm−1.
UV/Vis (CH 2Cl2): λmax = 258.5 (123200), 326 (38500), 425 (2700), 491 (1800) nm.
214

Chapter 5 Experimental Part
(Bis-(6-N- tert-butoxycarbonylamino-1-hexanol)-malonate- cyclo-[2]-octyl
malonate)-[5,1]-hexakisadduct (121)
O
OO
OO
O
O
O
O
O
O
O
NH
O
OOO
NH
O
OOO
NH
O
O
OO
O
O
OO
HNO
O
O O
HN
O
O
OO
NH
OO
HN
OO
O
O
O O
HN
OO
NH
O
OHN
121
Compound 121 was synthesized according to general procedure GP 7 from mono-
adduct 119 (346 mg, 0.30 mmol), malonate 120 (3.04 g, 6.04 mmol), DMA (623
mg, 3.02 mmol), CBr4 (1.00 g, 3.02 mmol) and P1-base (768 µL, 3.02 mmol) in dry
CH2Cl2. Pre-cleaning by flash column chromatography (SiO2; dichloromethane/ethyl
acetate 65:35) and subsequent purification by preparative HPLC (Nucleosil 5 µm;
dichloromethane/ethyl acetate 76:24) gave 121 as yellow solid.
Yield: 256 mg, 0.07 mmol, 23 %, yellow solid.
1H-NMR (300 MHz, RT, CDCl3): δ = 4.75 (s, br, 10H, NH), 4.21 (m, 24H, OCH2), 4.09
(m, 4H, OCH2), 3.32 (s, 2H, OCCH2CO), 3.04 (m, 20H, CH2NH), 1.65 (m, 28H, CH2),
1.39 (s, 90 H, C(CH3)3), 1.31 (m, 76H, CH2) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 166.63 (2C, CO), 164.01 (12C, CO), 156.20
215

Chapter 5 Experimental Part
(10C, COOtBu), 145.88, 141.29 (48C, C60-sp2), 79.07 (10C, C(CH3)3), 69.26 (12C,
C60-sp3), 67.25 (2C, OCH2), 67.03 (10C, OCH2), 65.71 (2C, OCH2), 45.63 (6C,
OCCCO), 42.42 (1C, OCCH2CO), 40.66 (10C, CH2NH), 30.13. 29.57 (16C, CH2),
28.61 (30C, C(CH3)3), 28.52, 26.55, 26.12, 25.74 (36C, CH2) ppm.
MS (FAB, NBA): m/z = 3649 [M]+, 720 [C60]+; MWcalc.: 3650.28 g/mol.
IR (ATR): ν̃ = 3412, 3354, 2964, 2934, 2860, 1741, 1698, 1513, 1455, 1412, 1393,
1366, 1247, 1239, 1170, 1081, 1042, 1019, 865, 799, 733, 664 cm−1.
UV/Vis (CH 2Cl2): λmax = 245.5 (86900), 271 (64200), 281 (66500), 315 (40400), 335
(30800) nm.
Boc-protected [5,1]-hexakisadduct-monoadduct (122)
O
OO
OO
O
O
O
O
O
O
O
NH
O
OOO
NH
O
OOO
NH
O
O
OO
O
O
OO
HNO
O
O O
HN
O
O
OO
NH
OO
HN
OO
O
O
O O
HN
OO
NH
O
OHN
122
Compound 122 was synthesized according to general procedure GP 4 from [5,1]-
hexakisadduct 121 (205 mg, 0.056 mmol), C60 (61 mg, 0.084 mmol), CBr4 (24 mg,
0.073 mmol) and P1-base (16 µL, 0.073 mmol) in 75 mL dry toluene. The crude prod-
uct was purified by flash column chromatography (SiO2; toluene/ethyl acetate 75:35)
216

Chapter 5 Experimental Part
to give 122 as yellow brownish solid.
Yield: 100 mg, 0.023 mmol, 41 %, yellow brownish solid.
1H-NMR (300 MHz, RT, CDCl3): δ = 4.76 (s, br, 10H, NH), 4.46 (t, 3J = 6.4 Hz, 4H,
OCH2-mono), 4.23 (t, 3J = 6.5 Hz, 24H, OCH2-hexakis), 3.06 (m, 20H, CH2NH), 1.86
(m, 4H, CH2), 1.66 (m, 28H, CH2), 1.41, (m, 114H, C(CH3)3, CH2), 1.33 (m, 48H, CH2)
ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 164.05 (12C, CO-hexakis), 163.83 (2C, CO-
mono), 156.23 (10C, COOtBu), 145.92, 141.32 (48C, [5,1]hexakis-C60-sp2), 146.01,
145.97, 145.89, 145.45, 145.41, 145.37, 145.08, 144.87, 144.85, 144.80, 144.08,
143.27, 143.20, 143.18, 142.40, 142.11, 141.25, 141.13, 139.32 (58C, mono-C60-sp2),
79.12 (10C, C(CH3)3), 71.79 (2C, mono-C60-sp3), 69.28 (12C, [5,1]hexakis-C60-sp3),
67.68 (2C, OCH2-mono), 67.09 (12C, OCH2-hexakis), 52.27 (1C, OCCCO-mono),
45.63 (6C, OCCCO-hexakis), 40.69 (10C, CH2NH), 30.17 (10C, CH2), 29.75 (4C,
CH2), 28.66 (34C, C(CH3)3, CH2), 28.55 (10C, CH2), 26.61 (10C, CH2), 26.32, 26.27
(4C, CH2), 25.79 (10C, CH2) ppm.
MS (FAB, NBA): m/z = 4367 [M+H]+, 720 [C60]+; MWcalc.: 4368.90 g/mol.
IR (ATR): ν̃ = 3412, 3350, 2943, 2926, 2856, 1741, 1695, 1517, 1459, 1393, 1366,
1243, 1216, 1170, 1081, 1042, 992, 892, 865, 780, 760, 737, 672 cm−1.
UV/Vis (CH 2Cl2): λmax = 246 (185500), 257 (sh, 175200), 321 (74500), 426 (7500),
488 (5700).
217

Chapter 5 Experimental Part
Asymmetric (Boc-protected) ( tert-butyl-protected) [5,1,1,5]-dimer (123)
O
OO
OO
O
O
O
O
O
O
O
NH
O
OR1OR1O
O
OR1OR1O
O
O
OR1OR1
O
O
OR1
OR1
OO
HN
OO
O
O
O
OO
O OR2
OR2
O
O OR2OR2
O
O
R2OR2O
O
O
R2OR2O
OO
OO
R1 R2123
Compound 123 was synthesized according to general procedure GP 7 from [5,1]-
hexakisadduct-monoadduct 122 (80 mg, 0.018 mmol), malonate 100 (98 mg, 0.22
mmol), DMA (45 mg, 0.22 mmol), CBr4 (73 mg, 0.22 mmol) and P1-base (47 µL, 0.22
mmol) in dry CH2Cl2. Pre-cleaning by flash column chromatography (SiO2; dichloro-
methane/ methanol 100:5) and subsequent purification by preparative HPLC (Nucleosil
5 µm; dichloromethane/methanol 100:4) gave 123 as orange solid.
Yield: 36 mg, 0.0055 mmol, 31 %, orange solid.
1H-NMR (300 MHz, RT, CDCl3): δ = 4.76 (s, br, 10H, NH), 4.22 (t, 3J = 5.9 Hz, 48H,
OCH2), 3.06 (m, 20H, CH2NH), 2.19 (t, 3J = 7.3 Hz, 20H, CH2CO), 1.66 (m, 68H,
CH2), 1.41 (m, 204H, C(CH3)3, CH2), 1.33 (m, 68H, CH2) ppm.
13C-NMR (75 MHz, RT, CDCl3): δ = 172.98 (10C, COOtBu), 164.05 (24C, CO), 156.23
(10C, COOtBu), 145.91, 141.26 (96C, C60-sp2), 80.25 (10C, CH2COOC(CH3)3), 79.11
(10C, HNCOOC(CH3)3), 69.28 (24C, C60-sp3), 67.08, 66.92 (24C, OCH2), 45.64 (12C,
OCCCO), 40.69 (10C, CH2NH), 35.53 (10C, CH2CO), 30.18 (10C, CH2), 29.78 (4C,
CH2), 28.67 (34C, HNCOOC(CH3)3, CH2), 28.56 (10C, CH2), 28.33 (40C,
CH2COOC(CH3)3, CH2), 26.61 (10C, CH2), 26.30 (4C, CH2), 25.80 (10C, CH2), 25.53
(10C, CH2), 24.85 (10C, CH2) ppm.
IR (ATR): ν̃ = 3416, 3370, 3057, 2976, 2937, 2864, 1714, 1513, 1459, 1393, 1366,
1239, 1216, 1154, 1081, 1042, 988, 957, 896, 849, 780, 733, 703 cm−1.
UV/Vis (CH 2Cl2): λmax = 246 (169300), 271 (125300), 281.5 (126600), 316 (78900),
218

Chapter 5 Experimental Part
336 (60100).
Deprotected [5,1,1,5]-dimer (124)
O
OO
OO
O
O
O
O
O
O
O
H3N
O
OR1OR1O
O
OR1OR1O
O
O
OR1OR1
O
O
OR1
OR1
H3N
O
O
O
OO
O OR2
OR2
O
O OR2OR2
O
O
R2OR2O
O
O
R2OR2O
OHO
OHO
R1 R2
CF3COO
CF3COO
124
(N-Boc-protected) (tert-butyl-protected) [5,1,1,5]-dimer 123 (20 mg, 0.003 mmol) was
dissolved in 3 mL pure TFA and stirred at room temperature for 36 h. Excess TFA was
removed in vacuo and coevaporated with methanol for several times to give 124 as
pale orange solid.
Yield: 17 mg, 0.0055 mmol, 92 %, pale orange solid.
1H-NMR (300 MHz, RT, MeOH-d4): δ = 4.30 (m, 48H, OCH2), 2.89 (m, 20H, CH2NH),
2.25 (t, 3J = 7.1 Hz, 20H, CH2CO), 1.69 (m, 48H, CH2), 1.61 (m, 40H, CH2), 1.38 (m,
76H, CH2) ppm.
13C-NMR (75 MHz, RT, MeOH-d4): δ = 177.36 (10C, COOH), 164.77 (24C, CO),
146.77, 142.62 (96C, C60-sp2), 70.56 (24C, C60-sp3), 68.59, 68.23 (24C, OCH2), 47.59
(12C, OCCCO), 40.63 (10C, CH2NH), 34.80 (10C, CH2CO), 30.43 (4C, CH2), 29.67
(4C, CH2), 29.39 (20C, CH2), 28.46 (10C, CH2), 27.00 (14C, CH2), 26.73 (10C, CH2),
26.53 (10C, CH2), 25.67 (10C, CH2) ppm.
IR (ATR): ν̃ = 3308, 2945, 2833, 1745, 1683, 1451, 1420, 1254, 1204, 1139, 1023,
838, 799, 718, 686 cm−1.
UV/Vis (MeOH): λmax = 215.5, 243.5 (sh), 270, 280 (sh), 317.5, 335.5 nm.
219

Chapter 5 Experimental Part
O
OO
OO
O
O
O
O
O
O
O
R
R
O
O OO R
R
O
O OO R
R
O
O
OO
RR
O
O
OO
RR
O
OO
OO
O
O
O
O
O
O
O
R
R
O
OOOR
R
O
OOOR
R
O
O
OO
RR
O
O
OO
RR
OO
O
O
O
OOO
O
OOOR
R
OO
O O
R R
O
O
O
O
R
R
O
O
O
O
R
R
OOO
OR R
OO
O
O
O
O OO
O
O OO R
R
OO
OO
RR
O
O
O
O
R
R
O
O
O
O
R
R
OO O
O RR
OO
O
O
O
O
OO
O
O
OO
RR
O
O
O
O
R
R
O
O
OO
RR
O
OOOR
R
O
O
O
O
R
R
OO
O
O
O
O
OO
O
O
OO
RR
O
O
O
O
R
R
O
O
OO
RR
O
O OO R
R
O
O
O
O
R
R
O
O
HNR = 126
Boc-protected [5,1]-hexakisfullerenyl-[6,0]-hexakisa dduct (126)
Compound 126 was synthesized according to general procedure GP 7 from [5,1]-
hexakisadduct 121 (101 mg, 0.028 mmol), C60 (2 mg, 0.0028 mmol), DMA (6 mg,
0.028 mmol), CBr4 (10 mg, 0.028 mmol) and P1-base (11 µL, 0.028 mmol) in 10 mL
dry toluene. Pre-cleaning by flash column chromatography (SiO2; toluene/ethyl acetate
150:60 to 100:250) and subsequent purification by preparative HPLC (Nucleosil 5 µm;
toluene/ethyl acetate 60:40) gave 126 as yellow solid.
Yield: 18 mg, 0.0008 mmol, 28 %, yellow solid.
220

Chapter 5 Experimental Part
1H-NMR (400 MHz, RT, CDCl3): δ = 4.78 (s, br, 60H, NH), 4.23 (m, 168H, OCH2),
3.06 (m, 120H, CH2NH), 1.67 (m, 168H, CH2), 1.44 (m, 168H, CH2), 1.41 (s, 540H,
C(CH3)3), 1.32 (m, 288H, CH2) ppm.
13C-NMR (100.5 MHz, RT, CDCl3): δ = 164.06 (84C, CO), 156.25 (60C, COOtBu),
145.93, 141.34 (336C, C60-sp2), 79.13 (60C, C(CH3)3), 69.29 (84C, C60-sp3), 67.11
(84C, OCH2), 45.67 (42C, OCCCO), 40.71 (60C, CH2NH), 30.18 (60C, CH2), 29.79
(24C, CH2), 28.68 (180C, C(CH3)3), 28.57 (84C, CH2), 26.61 (60C, CH2), 26.28 (24C,
CH2), 25.80 (60C, CH2) ppm.
IR (ATR): ν̃ = 3412, 3350, 2934, 2860, 1745, 1695, 1521, 1459, 1393, 1366, 1251,
1220, 1170, 1081, 1034, 922, 865, 826, 784, 760, 714, 697 cm−1.
UV/Vis (CH 2Cl2): λmax = 246 (575600), 270 (425600), 280.5 (436600), 317 (282500),
335.5 (231200) nm.
221

Chapter 5 Experimental Part
Deprotected [5,1]-hexakisfullerenyl-[6,0]-hexakisadd uct (127)
O
OO
OO
O
O
O
O
O
O
O
R
R
O
O OO R
R
O
O OO R
R
O
O
OO
RR
O
O
OO
RR
O
OO
OO
O
O
O
O
O
O
O
R
R
O
OOOR
R
O
OOOR
R
O
O
OO
RR
O
O
OO
RR
OO
O
O
O
OOO
O
OOOR
R
OO
O O
R R
O
O
O
O
R
R
O
O
O
O
R
R
OOO
OR R
OO
O
O
O
O OO
O
O OO R
R
OO
OO
RR
O
O
O
O
R
R
O
O
O
O
R
R
OO O
O RR
OO
O
O
O
O
OO
O
O
OO
RR
O
O
O
O
R
R
O
O
OO
RR
O
OOOR
R
O
O
O
O
R
R
OO
O
O
O
O
OO
O
O
OO
RR
O
O
O
O
R
R
O
O
OO
RR
O
O OO R
R
O
O
O
O
R
R
NH3+ CF3COO-
R = 127
Boc-protected [5,1]-hexakisfullerenyl-[6,0]-hexakisadduct 126 (18 mg, 0.0008 mmol)
was dissolved in 2 mL pure TFA and stirred at room temperature for 48 h. The crude
product was purified by reprecipitation from MeOH/Et2O to give 127 as a yellow solid.
Yield: 13 mg, 0.0008 mmol, 98 %, yellow solid.
1H-NMR (400 MHz, RT, MeOH-d4): δ = 4.31 (m, 168H, OCH2), 2.90 (m, 120H, CH2NH),
1.71 (m, 168H, CH2), 1.63 (m, 168H, CH2), 1.39 (m, 288H, CH2) ppm.
13C-NMR (100.5 MHz, RT, MeOH-d4): δ = 164.93 (84C, CO), 146.93, 142.81 (336C,
222

Chapter 5 Experimental Part
C60-sp2), 68.40 (84C, C60-sp3), 67.06 (84C, OCH2), 45.19 (42C, OCCCO), 40.78 (60C,
CH2NH), 29.54 (84C, CH2), 28.62 (84C, CH2), 27.39 (24C, CH2), 27.16 (60C, CH2),
26.69 (60C, CH2) ppm.
IR (ATR): ν̃ = 3416, 3266, 3065, 2941, 2864, 2687, 1745, 1675, 1536, 1467, 1432,
1397, 1355, 1254, 1204, 1135, 1085, 1046, 996, 957, 888, 838, 799, 760, 722, 672
cm−1.
UV/Vis (H 2O/pH 4): λmax = 215 (772100), 242.5 (551900), 271 (422900), 280 (416500),
316 (284700), 334 (228300) nm.
223

Appendices
A Materials and Methods for the Determination of
Biological Activity in vivo
Standard procedures for embryo collection
Embryos were generated by natural pair-wise mating, as described in the Zebrafish
Handbook.[311] Four to five pairs of adult zebrafish were set up for each mating, and,
on average, 100 - 150 embryos per pair were generated. Embryos were maintained
at 28°C in fish water (200 mg Instant Ocean Salt per liter of dei onized water; pH 6.6
- 7.0 maintained with 2.5 mg/liter Jungle pH Stabilizer (Jungle Laboratories Corpora-
tion, Cibolo, TX); conductivity 670 - 760 µS). Embryos were cleaned (dead embryos
removed) and sorted by developmental stage [312] at 6 and 24 hours post-fertilization
(hpf). Because the embryo receives nourishment from an attached yolk sac, no feeding
was required for seven days post fertilization (dpf).
A.1 Determination of LC50 and Organ Toxicity
Treatment of embryos
Zebrafish embryos at 24 hpf were distributed into 96-well cell culture plates, one em-
bryo per well in 100 ml fish water containing the compound. Embryos were exposed
from day 1 to day 5 post-fertilization (pf). Typically, the fish water contains PBS (phos-
phate buffered saline) at a final concentration of 10 %. Treated embryos were com-
pared with untreated and 0.1 % DMSO-treated controls.
224

Appendices
Measurement of LC50 and lethality curves
Mortality was recorded every 24 hrs. At 120 hpf, total mortality was used to gener-
ate the concentration-response curves. The data were averaged from multiple exper-
iments. Best-fit concentration-response curves were generated using KaleidaGraph
software (Synergy Software, Reading, PA, USA) using the equation:
Y = M1 +M2 −M1
1 + ( XM3
)M4(5.1)
where M1 = maximum Y value (100 % in this case), M2 = minimum Y value (0 % in
this case), M3 the concentration corresponding to the value midway between M1 and
M2 (LC50), M4 slope of the curve at M3 (best fit, generating R2 closest to 1), X =
concentration of compound, Y = percent lethality.
Visual toxicity assessment of developing embryos
At 5 dpf, organs in 5 randomly selected embryos were inspected by light microscopy.
Body morphology, liver, intestine, and heart were assessed. Since drug treatment was
initiated at 24 hpf, which is after circulation and heart beat are present, but before the
liver and intestine are developed, observations are for effects on developing organs.
Body morphology
Defects in the development of midline structures, including notochord and floor plate,
often result in abnormal body shape in zebrafish. To assess the potential drug effects
on midline development, abnormalities in body shape, including small body size, bent
or missing tail were examined.
A.2 Determination of Otoprotective Activity
Hair cell assessment
To damage inner hair cells, 5-day zebrafish were treated with gentamicin (2.5 µg/ml)
or cisplatinum (10 µM) for 24 hours to induce apoptosis in inner hair cells. To test drug
effects on protection of hair cell damage, test compounds were administered at 0, 0.1,
1, 10, 100 and 250 µM concentrations with either the gentamicin or cisplatinum treat-
225

Appendices
ment. The inner hair cells in the lateral neuromasts were examined by 2,4-dimethyl-
aminostyryl-N-ethyl pyridinium iodide (DASEPI or 2,4 Di-Asp) staining.
DASPEI staining
Zebrafish were incubated with DASPEI solution (1mM) for 2 hours, then rinsed thor-
oughly in fish water. Zebrafish were anaesthetized with MESAB (0.5 mM 3-aminobenzoic
acid ethyl ester, 2 mM Na2HPO4), and mounted in methylcellulose in depression slide
for observation.
Morphometric analysis
Fluorescent signals (SS) were quantified as: SS = staining area x staining intensity
of neuromasts by particle analysis (Scion Image, Scion Corporation, Frederick, Mary-
land). Images of lateral sides in each animal were obtained by the same exposure time
and fluorescent gain (anterior on the left; posterior on the right; dorsal on the top). The
size of neuromasts were defined and specified for particle analysis. Five animals for
each treatment were quantified and intensity of fluorescent signals was averaged.
Statistics
All data were presented as mean ± standard deviation (SD). Student’s t-Test was used
to compare vehicle-treated and drug treated zebrafish. Significance was defined as P
< 0.05, n = 5.
A.3 Determination of General CNS Neuroprotective Activity
Against 6-Hydroxdopamine (6-OHDA) Induced Neuronal
Apoptosis
Microinjection of 6-OHDA
6-OHDA is extremely unstable in solution, it was prepared fresh for each injection. 3-
day zebrafish were anesthetized in MESAB (0.5 mM 3-aminobenzoic acid ethyl ester,
2 mM Na2HPO4) and 500 mM 6-OHDA was microinjected into the midbrain region.
The estimated amount of injection was about 1-2 pmole per compound. Zebrafish
were rapidly transferred to fresh fish water after injection, allowed to recover for 30
226

Appendices
minutes, and incubated with neuroprotective antioxidants. Water alone was injected
into zebrafish brain region as a vehicle control.
Treatment of embryos
Embryos were exposed to water-soluble fullerenes at 1, 10, 100 and 250 µM for 24
hours, then microinjected with 6-OHDA. Twenty embryos were treated with each con-
centration of each compound.
Morphometric analysis
Fluorescent signals (SS) were quantified as: SS = staining area x staining intensity
of neuromasts by particle analysis (Scion Image, Scion Corporation, Frederick, Mary-
land). Images of dorsal sides in each animal were obtained by the same exposure
time and fluorescent gain. The positive staining was defined by size and fluorescent
intensity evaluation, and specified for particle analysis. Five animals for each treatment
were quantified and intensity of fluorescent signals was averaged.
Fluorescence microscopy and image analyses
All fluorescence microscopy studies were performed using a Zeiss M2Bio fluorescence
microscope (Carl Zeiss Microimaging Inc., Thornwood, NY) equipped with a rhodamine
cube and a green FITC filter (excitation: 488 nm, emission: 515 nm), and a chilled CCD
camera (Axiocam MRM, Carl Zeiss Microimaging Inc., Thornwood, NY). Screens were
routinely done using a 1.6X, 10X and 20X objective archromats and 10X eye pieces.
The system was also equipped with a z-motorized stage, deconvolution software and
4-D reconstruction software (Axiovision, Carl Zeiss Microimaging Inc., Thornwood, NY)
which permits reconstruction of 3-D objects and analyzes Z-stacks. Images were ana-
lyzed with Axiovision software Rel 4.0 (Carl Zeiss Microimaging Inc., Thornwood, NY),
the Adobe Photoshop 6.0 computer program (Adobe, San Jose, CA) and NIH image
software (Bethesda, MD). The patterns and the intensity of staining were recorded and
quantitated.
227

Appendices
Acridine orange staining
At 48, 72 and 120 hpf, five embryos were immersed in 0.5 µg/ml acridine orange
(acridinium chloride hemi-[zinc chloride]) in PBS for 60 minutes and rinsed thoroughly
twice in 10 ml of fresh fish water. Stained embryos are anesthetized with MESAB (0.5
mM 3-aminobenzoic acid ethyl ester, 2 mM Na2HPO4), and mounted in methycellulose
in a depression slide for observation using fluorescent microscopy. Effects of water-
soluble fullerenes apoptosis in the hatching glands, retina, lateral neuromasts, and
olfactory pits were examined.
A.4 Determination of Dopaminergic CNS Neuroprotective Act ivity
Against 6-Hydroxydopamine Induced Apoptosis
Treatment of embryos
2-day zebrafish were treated with 1 % DMSO for vehicle control and treated with 1 %
DMSO plus 250 µM 6-OHDA for DA-loss control. For compound testing, 2-day embryos
were exposed to a mixture of 250 µM 6-OHDA and fullerenes for 72 hours.
Antibody staining
Embryos were fixed in 4 % paraformaldehyde overnight at 4°C. F ixed embryos were
permeabilized with cold acetone at -20°C for 20 minutes and r ehydrated in step-wise
descending ethanol/PBS solutions (95, 79, 50, 25, 0 % ethanol/PBS mix, ten min-
utes for each step). Embryos were then stained with anti-tyrosine hydroxylase an-
tibody (mouse anti-human, Sigma, St Louis, MO) at 4°C overni ght. The next day,
samples were washed with PBS-T (0.1 % Tween 20), incubated with secondary an-
tibody (goat anti-mouse) and color was developed using ABC reagent (Vector Labs,
Burlingame, CA) according to manufacturer’s instructions. Stained embryos were fur-
ther flat-mounted on a glass slide and five randomly selected embryos were examined
for DA neuron loss.
Light microscopy and image analyses
All microscopy studies were performed using a Zeiss light microscope (Carl Zeiss Mi-
croimaging Inc., Thornwood, NY) equipped with a Spot camera (Diagnostic instru-
228

Appendices
ments, Sterling, MI). The patterns and the intensity of staining were recorded and
quantified.
Fluorescence microscopy
All fluorescence microscopy studies were performed using a Nikon Eclipse E600 fluo-
rescence microscope (Nikon Inc., Melville, NY) equipped with a 200 watt mercury/xenon
lamp and a green FITC filter (excitation: 488 nm, emission: 515 nm), and a C5985
chilled CCD camera (Hamamatsu Photonics, Hamamatsu City, Japan). Images were
analyzed with the Adobe Photoshop 6.0 computer program (Adobe, San Jose, CA).
B Materials and Methods for the Preparation and
Examination of SWCNT-Fullerene-Hybrid
The single-walled carbon nanotubes (laser-ablation, University of Karlsruhe) were puri-
fied using an oxidative protocol described elsewhere.[313] The purified nanotubes were
then suspended in 1,2-dichloroethane (DCE) by pulsed high-power ultrasound (Dr.
Hielscher, UP400S, 400W, 24 kHz) to yield a black colored suspension. Bundles of
agglomerated nanotubes were subsequently removed in a centrifugation step for 5
minutes at 5000 rpm. The supernatant was given to a solution of 99 in DCE and stirred
thoroughly for 5 minutes. The fullerene-nanotube hybrid was subsequently removed
from the suspension by centrifugation for 30 minutes at 10000 rpm. The supernatant
was discarded and the precipitate was re-suspended in deionized water. The resulting
suspension was filtered using a microporous membrane (cellulose nitrate, pore-size
0.2 µm) to yield a black-colored solid film (buckypaper).
For the preparation of the fullerene nanowires, the buckypaper consistent of fullerene-
nanotube hybrid is mounted onto a teflon stamp and fixed with a double-sided UHV
compliant scotch tape. Then the configuration is brought into the load lock of the STM
and pumped until the pressure falls below 10−7 mbar. The bucky paper is brought into
contact with the gold substrate in the preparation chamber (10−9 mbar). Therefore sim-
ply the manipulator is lowered via micrometer screws until the bucky paper touches the
surface. This in situ preparation procedure ensures that the surface does not suffer
229

Appendices
from ambient contamination, e.g. hydrocarbons. Prior to STM investigation, the sur-
face is gently annealed to allow possible adsorbates out of the bucky paper to desorb.
We used temperatures of 75◦C to 150◦C applied for several hours. It turned out that
the annealing does not improve the sample quality significantly.
230

References
[1] K. E. Drexler, Proc. Nat. Acad. Sci. U.S.A. 1981, 78, 5275–5278.
[2] L. Reti, The Unknown Leonardo. McGraw-Hill Book Company, Toronto 1974.
[3] M. S. Dresselhaus, G. Dresselhaus, P. C. Eklund, Science of Fullerene and Car-bon Nanotubes. Academic Press 1996.
[4] W. E. Barth, R. G. Lawton, J. Am. Chem. Soc. 1966, 88, 380–381.
[5] E. Osawa, Kagaku 1970, 25, 654–659.
[6] D. A. Bochvar, E. Gal‘pern, Proc. Acad. Sci. 1973, 209, 239–245.
[7] E. A. Rohlfing, D. M. Cox, A. Kaldor, J. Chem. Phys. 1984, 81, 3322–3330.
[8] W. Krätschmer, L. D. Lamb, K. Fostiropoulos, D. R. Huffman, Nature 1990, 347,354–358.
[9] H. Kroto, A. Allaf, S. Balm, Chem. Rev. 1991, 91, 1213–1235.
[10] H.-B. Bürgi, E. Blanc, D. Schwarzenbach, S. Liu, Y. Lu, M. M. Kappes, J. A. Ibers,Angew. Chem. 1992, 104, 667–669; Angew. Chem. Int. Ed. 1992, 31, 640–643.
[11] S. Liu, Y. Lu, M. M. Kappes, J. A. Ibers, Science 1991, 254, 408–410.
[12] W. I. F. David, R. M. Ibberson, J. C. Matthewman, K. Prassides, T. J. S. Dennis,J. P. Hare, H. W. Kroto, R. Taylor, D. R. M. Walton, Nature 1991, 353, 147–149.
[13] J. M. Hawkins, A. Meyer, T. A. Lewis, S. Loren, F. J. Hollander, Science 1991,252, 312–313.
[14] A. L. Balch, J. W. Lee, B. C. Noll, M. M. Olmstead, J. Chem. Soc., Chem. Com-mun. 1993, 56–58.
[15] J. D. Crane, P. B. Hitchcock, H. W. Kroto, R. Taylor, D. R. M. Walton, J. Chem.Soc.,Chem. Commun. 1992, 1764–1765.
[16] C. S. Yannoni, P. P. Bernier, D. S. Bethune, G. Meijer, J. R. Salem, J. Am. Chem.
231

References
Soc. 1991, 113, 3190–3192.
[17] K. Hedberg, L. Hedberg, D. S. Bethune, C. A. Brown, H. C. Dorn, R. D. Johnson,M. D. Vries, Science 1991, 254, 410–412.
[18] H. W. Kroto, Nature 1987, 329, 529–531.
[19] R. Taylor, Tetrahedron Lett. 1991, 32, 3731–3733.
[20] R. Taylor, J. Chem. Soc., Perkin Trans. 2 1992, 4, 453–455.
[21] R. C. Haddon, K. Raghavachari, Buckminsterfullerenes. VCH Publishers Inc.New York 1993, 185–196.
[22] M. Sola, J. Mestres, M. Duran, J. Phys. Chem. 1995, 99, 10758–10762.
[23] H.-D. Beckhaus, C. Rüchardt, M. Kao, F. Diederich, C. S. Foote, Angew. Chem.1992, 104, 69–70; Angew. Chem. Int. Ed. 1992, 31, 63–64.
[24] R. C. Haddon, Science 1993, 261, 1545–1550.
[25] R. D. Beck, P. S. John, M. M. Alvarez, F. Diederich, R. L. Whetten, J. Phys. Chem.1991, 95, 8402–8409.
[26] N. Sivaraman, R. Dhamodaran, I. Kaliappan, T. G. Srinivasan, P. R. V. Rao, C. K.Mathews, J. Org. Chem. 1992, 57, 6077–6079.
[27] R. S. Ruoff, D. S. Tse, R. Malhotra, D. C. Lorents, J. Phys. Chem. 1993, 97,3379–3383.
[28] W. A. Scrivens, J. M. Tour, J. Chem. Soc., Chem. Commun. 1993, 1207–1209.
[29] M. T. Beck, G. Mandi, Fullerene Sci. Technol. 1997, 5, 291–310.
[30] F. Hauke, Chemische Funktionalisierung des Heterofulleren-Dimers (C59N)2,Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2003.
[31] Y.-P. Sun, G. E. Lawson, J. E. Riggs, B. Ma, N. X. Wang, D. K. Moton, J. Phys.Chem. A 1998, 101, 5626–5632.
[32] B. Ma, C. E. Bunker, R. Guduru, X. F. Zhang, Y.-P. Sun, J. Phys. Chem. A 1997,101, 5626–5632.
[33] B. Ma, Y.-P. Sun, J. Chem. Soc., Perkin Trans. 2 1996, 2157–2162.
[34] F. Cardullo, P. Seiler, L. Isaacs, J. F. Nierengarten, R. F. Haldimann, F. Diederich,T. M. Denti, W. Thiel, C. Boudon, J. P. Gisselbrecht, M. Gross, Helv. Chim. Acta1997, 80, 343–371.
[35] A. Curioni, P. Giannozzi, J. Hutter, W. Andreoni, J. Phys. Chem. 1995, 99, 4008–4014.
[36] N. Armaroli, F. Diederich, C. O. D. Buchecker, L. Falmigni, G. Marconi, J.-F.
232

References
Nierengarten, J.-P. Sauvage, Chem. Eur. J. 1998, 4, 406–416.
[37] L. Isaacs, F. Diederich, Helv. Chim. Acta 1993, 76, 2454–2464.
[38] M. Braun, Functionalized Amphiphilic Fullerene Derivatives, Dissertation,Friedrich-Alexander-Universität Erlangen-Nürnberg, 2003.
[39] Y. Wu, J. Wang, H. Fu, Y. Li, D. Yang, Microchem. J. 1993, 48, 29–32.
[40] R. F. Curl, Buckminsterfullerenes. VCH Publishers Inc. New York 1993.
[41] S. C. O’Brien, J. R. Heath, R. F. Curl, R. E. Smalley, J. chem. Phys. 1988, 88,220–230.
[42] R. E. Smalley, ACS Symp. Ser. 1992, 481, 141–159.
[43] H. W. Kroto, J. R. Heath, S. C. O´Brien, R. F. Curl, R. E. Smalley, Nature 1985,318, 162–163.
[44] V. Elser, R. C. Haddon, Nature 1987, 325, 792–794.
[45] R. C. Haddon, Nature 1995, 378, 249–255.
[46] R. C. Haddon, L. F. Schneemeyer, J. V. Waszczak, S. H. Glarum, R. Tycko,G. Dabbagh, A. R. Kortan, A. J. Muller, A. M. Mujsce, M. J. Rosseinsky, S. M.Zahurak, A. V. Makhija, F. A. Thiel, K. Raghavachari, E. Cockayne, V. Elser,Nature 1991, 350, 46–47.
[47] R. S. Ruoff, D. Beach, J. Cuomo, T. McGuire, R. L. Whetten, F. Diederich, J.Phys. Chem. 1991, 95, 3457–3459.
[48] A. Pasquarello, M. Schlüter, R. C. Haddon, Science 1992, 257, 1660–1661.
[49] A. Pasquarello, M. Schlüter, R. C. Haddon, Phys. Rev. A: At. Mol. Opt. Phys.1993, 47, 1783–1789.
[50] R. Zanasi, P. W. Fowler, Chem. Phys. Lett. 1995, 238, 270–280.
[51] J. R. Heath, S. C. O’Brien, Q. Zhang, Y. Liu, R. F. Curl, F. K. Tittel, R. E. Smalley,J. Am. Chem. Soc. 1985, 107, 7779–7780.
[52] D. S. Bethune, R. D. Johnson, J. R. Salem, M. S. de Vries, C. S. Yannoni, NatureNovember (1993), 366, 123–128.
[53] M. Saunders, R. J. Cross, H. A. Jimenez-Vazquez, R. Shimshi, A. Khong, Sci-ence 1996, 271, 1693–1697.
[54] L. Isaacs, A. Wehrsig, F. Diederich, Helv. Chim. Acta 1993, 76, 1231–1250.
[55] T. Suzuki, Q. C. Li, K. C. Khemani, F. Wudl, J. Am. Chem. Soc. 1992, 114, 7301–7302.
[56] M. Prato, T. Suzuki, F. Wudl, V. Lucchini, M. Maggini, J. Am. Chem. Soc. 1993,
233

References
115, 7876–7877.
[57] B. M. Trost, W. B. Herdle, J. Am. Chem. Soc. 1976, 98, 4080–4086.
[58] P. E. Hansen, Org. magn. Res. 1979, 12, 109–142.
[59] J. M. Smith, D. A. Hrovat, W. T. Borden, M. Allan, K. R. Asmis, C. Bulliard,E. Haselbach, U. C. Meier, J. Am. Chem. Soc. 1993, 115, 3816–3817.
[60] R. C. Haddon, L. E. Brus, K. Raghavachari, Chem. Phys. Lett. 1986, 125, 459–464.
[61] M. Bühl, A. Hirsch, Chem. Rev. 2001, 101, 1153–1183.
[62] P. D. Hale, J. Am. Chem. Soc. 1986, 108, 6087–6088.
[63] L. Echegoyen, L. E. Echegoyen, Acc. Chem. Res. 1998, 31, 593–601.
[64] Q. Xie, E. Perez-Cordero, L. Echegoyen, J. Am. Chem. Soc. 1992, 114, 3978–3980.
[65] S. H. Yang, C. L. Pettiette, J. Conceicao, O. Cheshnovsky, R. E. Smalley, Chem.Phys. Lett. 1987, 139, 233–238.
[66] H. Imahori, K. Hagiwara, T. Akiyama, M. Aoki, S. Taniguchi, T. Okada, M. Shi-rakawa, Y. Sakata, Chem. Phys. Lett. 1996, 263, 545–550.
[67] A. Hirsch, Chem. Unserer Zeit 1994, 28, 79–87.
[68] A. Hirsch, M. Brettreich, Fullerenes, Chemistry and Reactions. Wiley-VCH, Weil-heim 2005.
[69] A. Hirsch, Top. Curr. Chem. 1999, 199, 1–65.
[70] A. Hirsch, Synthesis 1995, 8, 895–913.
[71] F. Diederich, R. Kessinger, Acc. Chem. Res. 1999, 32, 537–545.
[72] R. Taylor, D. R. Walton, Nature 1993, 363, 685–693.
[73] F. Diederich, C. Thilgen, Science 1996, 271, 317–323.
[74] F. Diederich, L. Isaacs, D. Philp, Chem. Soc. Rev. 1994, 23, 243–255.
[75] C. Bingel, Chem. Ber. 1993, 126, 1957–1959.
[76] C. Bingel, H. Schiffer, Lieb. Ann. Chem. 1995, 1551–1553.
[77] W. Sliwa, Fullerene Sci. Technol. 1997, 5, 1133–1175.
[78] M. W. J. Beulen, L. Echegoyen, Chem. Commun. 2000, 1065–1066.
[79] S. Eguchi, Fullerene Sci. Technol. 1997, 5, 977–987.
234

References
[80] S. R. Wilson, N. Kaprinidis, Y. Wu, D. I. Schuster, J. Am. Chem. Soc. 1993, 115,8495–8496.
[81] S. R. Wilson, Y. Wu, N. Kaprinidis, D. I. Schuster, J. Org. Chem. 1993, 58, 6548–6549.
[82] T. Suzuki, L. Qi, K. C. Kheimani, F. Wudl, J. Am. Chem. Soc. 1992, 114, 7300–7301.
[83] M. Maggini, G. Scorrano, M. Prato, J. Am. Chem. Soc. 1993, 115, 9789–9799.
[84] M. Prato, M. Maggini, Acc. Chem. Res. 1998, 31, 519–526.
[85] T. D. Ros, M. Prato, V. Lucchini, J. Org. Chem. 2000, 65, 4289–4297.
[86] J. C. Hummelen, B. Knight, J. Pavlovich, R. Gonzalez, F. Wudl, Science 1995,269, 1554–1556.
[87] I. Lamparth, B. Nuber, G. Schick, A. Skiebe, T. Groesser, A. Hirsch, Angew.Chem. 1995, 107, 2473–2476; Angew. Chem. Int. Ed. 1995, 34, 2257–2259.
[88] B. Nuber, A. Hirsch, Chem. Commun. 1996, 1421–1422.
[89] B. Nuber, A. Hirsch, Fullerene Sci. Technol. 1996, 4, 715–728.
[90] A. L. Balch, V. J. Catalano, J. W. Lee, M. M. Olmstead, S. R. Parkin, J. Am.Chem. Soc. 1991, 113, 8953–8955.
[91] A. Hirsch, Angew. Chem. 1993, 105, 1189–1192; Angew. Chem. Int. Ed. 1993,32, 1138–1141.
[92] R. Taylor, D. R. M. Walton, Nature 1993, 363, 685–693.
[93] S. Samal, S. K. Sahoo, Bull. Mater. Sci. 1997, 20, 141–230.
[94] A. Hirsch, The Chemistry of the Fullerenes. Georg Thieme Verlag, Stuttgart, NewYork 1994.
[95] K. M. Kadish, R. S. Ruoff, Fullerenes: Chemistry, Physics and Technology.Wiley-Interscience 2000.
[96] X. Camps, A. Hirsch, J. Chem. Soc., Perkin Trans. 1 1997, 1595–1596.
[97] J.-F. Nierengarten, V. Gramlich, F. Cardullo, F. Diederich, Angew. Chem. 1996,108, 2242–2244; Angew. Chem. Int. Ed. 1996, 35, 2101–2103.
[98] M. Brettreich, S. Burghardt, C. Bottcher, T. Bayerl, S. Bayerl, A. Hirsch, Angew.Chem. 2000, 112, 1915–1918; Angew. Chem. Int. Ed. 2000, 39, 1845–1848.
[99] S. Burghardt, A. Hirsch, B. Schade, K. Ludwig, C. Boettcher, Angew. Chem.2005, 117, 3036–3039; Angew. Chem. Int. Ed. 2005, 44, 2976–2979.
[100] B. Schade, K. Ludwig, C. Boettcher, U. Hartnagel, A. Hirsch, Angew. Chem.
235

References
2007, 119, 4472–4475; Angew. Chem. Int. Ed. 2007, 46, 4393–4396.
[101] C. Boudon, J.-P. Gisselbrecht, M. Gross, L. Isaacs, H. L. Anderson, R. Faust,F. Diederich, Helv. Chim. Acta 1995, 78, 1334–1344.
[102] C. Kovacs, A. Hirsch, Eur. J. Org. Chem. 2006, 15, 3348–3357.
[103] U. Hartnagel, Elektrostatische Hybridisierung von kationischen Metalloporphyri-nen und anionischen Fulleren-Dendrimeren, Diplomarbeit, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2003.
[104] E. Ravanelli, Synthesis and Characterization of Stereochemically de-fined Fullero-Peptides Hybrids, Dissertation, Friedrich-Alexander-UniversitätErlangen-Nürnberg, 2004.
[105] B. Neises, W. Steglich, Angew. Chem. 1978, 90, 556–557; Angew. Chem. Int.Ed. 1978, 17, 522–524.
[106] G. Höfle, W. Steglich, H. Vorbrüggen, Angew. Chem. 1978, 90, 602–615; Angew.Chem. Int. Ed. 1978, 17, 569–583.
[107] J. Otera, Esterification. Wiley-VCH, Weinheim 2003.
[108] J. C. Sheehan, S. L. Ledis, J. Am. Chem. Soc. 1973, 95, 875–879.
[109] M. Brettreich, A. Hirsch, Tetrahedron Lett. 1998, 39, 2731–2734.
[110] M. Brettreich, A. Hirsch, Synlett 1999, 1396–1398.
[111] F. Beuerle. personal communication.
[112] J. N. Jacob, V. E. Shashoua, A. Campbell, R. J. Baldessarini, J. Med. Chem.1985, 28, 106–110.
[113] V. E. Shashoua, G. W. Hesse, Life Sci. 1996, 58, 1347–1357.
[114] M. Söderberg, C. Edlund, K. Kristensson, G. Dallner, Lipids 1991, 26, 421–425.
[115] R. E. Martin, E. B. R. de Turco, N. G. Bazan, J. Nutr. Biochem. 1994, 5, 151–160.
[116] R. E. Martin, N. G. Bazan, J. Neurochem. 1992, 59, 318–325.
[117] B. L. Scott, N. G. Bazan, Proc. Nat. Acad. Sci. U.S.A. 1989, 86, 2903–2907.
[118] N. G. Bazan, Nutrition and the Brain, volume 8. Raven Press, New York 1990,1–24.
[119] D. Rubin, E. J. Rubin. Method of extraction and purification of polyunsaturatedfatty acids from natural sources. Patent Pending, December (1988). 4792418.
[120] R. Engel, K. Rengan, C. S. Chan, Heteroat. Chem. 1993, 4, 181–184.
[121] K. Rengan, R. Engel, J. Chem. Soc., Chem. Commun. 1992, 757–758.
236

References
[122] K. Rengan, R. Engel, J. Chem. Soc., Perkin Trans. 1 1991, 987–990.
[123] P. R. Ashton, K. Shibata, A. N. Shipway, J. F. Stoddart, Angew. Chem. 1997, 109,2902–2905; Angew. Chem. Int. Ed. 1997, 36, 2781–2783.
[124] S. Huang, S. Tsai, L. Chiang, L. Chih, M. Tsai, Drug Dev. Res. 2001, 53, 244–253.
[125] S. S. Huang, S. K. Tsai, C. L. Chih, L. Y. Chiang, H. M. Hsieh, C. M. Teng, M. C.Tsai, Free Radical Biol. Med. 2001, 30, 643–649.
[126] L. L. Dugan, J. K. Gabrielsen, S. P. Yu, T. S. Lin, D. W. Choi, Neurobiol. Dis.1996, 3, 129–135.
[127] H. Jin, W. Q. Chen, X. W. Tang, L. Y. Chiang, C. Y. Yang, J. V. Schloss, J. Y. Wu,J. Neurosci. Res. 2000, 62, 600–607.
[128] M. C. Tsai, Y. H. Chen, L. Y. Chiang, J Pharm Pharmacol 1997, 49, 438–445.
[129] H. S. Lai, W. J. Chen, L. Y. Chiang, World J. Surg. 2000, 24, 450–454.
[130] H. S. Lai, Y. Chen, W. J. Chen, J. Chang, L. Y. Chiang, Transplant. Proc. 2000,32, 1272–1274.
[131] Y. L. Lai, H. D. Wu, C. F. Chen, J. Cardiovasc. Pharmacol. 1998, 32, 714–720.
[132] U. Reuther, T. Brandmuller, W. Donaubauer, F. Hampel, A. Hirsch, Chem. Eur. J.2002, 8, 2261–2273.
[133] S. H. Friedman, P. S. Ganapathi, Y. Rubin, G. L. Kenyon, J. Med. Chem. 1998,41, 2424–2429.
[134] F. Djojo, A. Hirsch, Chem. Eur. J. 1998, 4, 344–356.
[135] I. Lamparth, A. Hirsch, J. Chem. Soc. Chem. Commun. 1994, 1727–1728.
[136] S. Foley, C. Crowley, M. Smaihi, C. Bonfils, B. F. Erlanger, P. Seta, C. Larroque,Biochem. Biophys. Res. Commun. 2002, 294, 116–119.
[137] S. S. Ali, J. I. Hardt, K. L. Quick, J. S. Kim-Han, B. F. Erlanger, T. T. Huang, C. J.Epstein, L. L. Dugan, Free Radical Biol. Med. 2004, 37, 1191–1202.
[138] S. F. Tzeng, J. L. Lee, J. S. Kuo, C. S. Yang, P. Murugan, L. Ai Tai, K. Chu Hwang,Brain Res. 2002, 940, 61–68.
[139] J. M. Rieger, A. R. Shah, J. M. Gidday, Exp. Eye Res. 2002, 74, 493–501.
[140] A. M. Lin, S. F. Fang, S. Z. Lin, C. K. Chou, T. Y. Luh, L. T. Ho, Neurosci. Res.2002, 43, 317–321.
[141] L. L. Dugan, E. G. Lovett, K. L. Quick, J. Lotharius, T. T. Lin, K. L. O’Malley,Parkinsonism Relat. Disord. 2001, 7, 243–246.
237

References
[142] M. Bisaglia, B. Natalini, R. Pellicciari, E. Straface, W. Malorni, D. Monti,C. Franceschi, G. Schettini, J. Neurochem. 2000, 74, 1197–1204.
[143] J. Lotharius, L. L. Dugan, K. L. O’Malley, J. Neurosci. 1999, 19, 1284–1293.
[144] A. M. Lin, B. Y. Chyi, S. D. Wang, H. H. Yu, P. P. Kanakamma, T. Y. Luh, C. K.Chou, L. T. Ho, J. Neurochem. 1999, 72, 1634–1640.
[145] L. L. Dugan, D. M. Turetsky, C. Du, D. Lobner, M. Wheeler, C. R. Almli, C. K.Shen, T. Y. Luh, D. W. Choi, T. S. Lin, Proc. Nat. Acad. Sci. U.S.A. 1997, 94,9434–9439.
[146] H. S. Lin, T. S. Lin, R. S. Lai, T. D’Rosario, T. Y. Luh, Int. J. Radiat Biol. 2001, 77,235–239.
[147] N. Tsao, T. Y. Luh, C. K. Chou, J. J. Wu, Y. S. Lin, H. Y. Lei, Antimicrob. AgentsChemother. 2001, 45, 1788–1793.
[148] N. Tsao, T. Y. Luh, C. K. Chou, T. Y. Chang, J. J. Wu, C. C. Liu, H. Y. Lei, J.Antimicrob. Chemother. 2002, 49, 641–649.
[149] S. Bosi, T. Da Ros, S. Castellano, E. Banfi, M. Prato, Bioorg. Med. Chem. Lett.2000, 10, 1043–1045.
[150] T. Mashino, K. Okuda, T. Hirota, M. Hirobe, T. Nagano, M. Mochizuki, Bioorg.Med. Chem. Lett. 1999, 9, 2959–2962.
[151] S. Kiritoshi, T. Nishikawa, K. Sonoda, D. Kukidome, T. Senokuchi, T. Matsuo,T. Matsumura, H. Tokunaga, M. Brownlee, E. Araki, Diabetes 2003, 52, 2570–2577.
[152] D. Monti, L. Moretti, S. Salvioli, E. Straface, W. Malorni, R. Pellicciari, G. Schet-tini, M. Bisaglia, C. Pincelli, C. Fumelli, M. Bonafe, C. Franceschi, Biochem.Biophys. Res. Commun. 2000, 277, 711–717.
[153] Y. L. Huang, C. K. Shen, T. Y. Luh, H. C. Yang, K. C. Hwang, C. K. Chou, Eur. J.Biochem. 1998, 254, 38–43.
[154] B. Halliwell, Mutat. Res. 1999, 443, 37–52.
[155] D. D. Buechter, Pharm. Res. 1988, 5, 253–260.
[156] H. Nohl, A. V. Kozlov, L. G. K., Staniek, Biochem. Soc. Trans. 2003, 31, 1308–1311.
[157] H. Ahsan, A. Ali, R. Ali, Clin. Exp. Immunol. 2003, 31, 398–404.
[158] J. A. Knight, Ann. Clin. Lab. Sci. 1995, 25, 111–121.
[159] G. Waris, K. Alam, Biochem. Mol. Biol. Int. 1998, 45, 33–45.
[160] K. Apel, H. Hirt, Annu. Rev. Plant Biol. 2004, 55, 373–399.
238

References
[161] C. M. Bergamini, S. Gambetti, A. Dondi, C. Cervellati, Curr. Pharm. Des. 2004,10, 1611–1626.
[162] M. B. Reddy, L. Clark, Nutr. Rev. 2004, 62, 120–124.
[163] J. K. Andersen, Nat. Rev. Neurosci. 2004, 10, 18–25.
[164] J. M. McCord, I. Fridovich, J. Biol. Chem. 1969, 244, 6049–6055.
[165] D. M. Guldi, K. D. Asmus, Radiat. Phys. Chem. 1999, 56, 449–456.
[166] M. Braun, S. Atalick, M. Guldi Dirk, H. Lanig, M. Brettreich, S. Burghardt, M. Hatz-imarinaki, E. Ravanelli, M. Prato, R. Van Eldik, A. Hirsch, Chem. Eur. J. 2003, 9,3867–3875.
[167] U. Hartnagel, D. Balbinot, N. Jux, A. Hirsch, Org. Biomol. Chem. 2006, 4, 1785–1795.
[168] C. Parng, W. L. Seng, C. Semino, P. McGrath, Assay Drug Dev. Technol. 2002,1, 41–48.
[169] L. K. Cole, L. S. Ross, Dev. Biol. 2001, 240, 123–142.
[170] M. Rodriguez, W. Driever, Biochem. Cell Biol. 1997, 75, 579–600.
[171] U. Langheinrich, E. Hennen, G. S. G. Vacun, Current Biology 2002, 12, 2023–2028.
[172] H. Steller, Science 1995, 267, 1445–1449.
[173] M. D. Jacobson, M. Weil, M. C. Raff, Cell 1997, 88, 347–354.
[174] W. R. Oppenheim, Annu. Rev. Neurosci. 1991, 14, 453–501.
[175] R. M. Silva, V. Ries, T. F. Oo, O. Yarygina, V. Jackson-Lewis, E. J. Ryu, P. D. Lu,S. J. Marciniak, D. Ron, S. Przedborski, N. Kholodilov, L. A. Greene, R. E. Burke,J. Neurochem. 2002, 95, 974–977.
[176] C. Ton, C. Parng, Hear. Res. 2005, 208, 79–88.
[177] V. G. Schweitzer, Laryngoscope 1993, 103, 1–52.
[178] G. J. Matz, Otolaryngol. Clin. North Am. 1993, 26, 705–712.
[179] P. Stavroulaki, N. Apostolopoulos, D. Dinopoulou, I. Vossinakis, M. Tsakanikos,D. Douniadakis, Int. J. Pediatr. Otorhinolaryngol. 1999, 50, 177–181.
[180] O. F. Unal, S. M. Ghoreishi, A. Atas, N. Akyurek, G. Akyol, B. Gursel, Int. J.Pediatr. Otorhinolaryngol. 2005, 69, 193–195.
[181] T. T. Yoshikawa, Am. Fam. Physician 1980, 21, 125–129.
[182] J. Lautermann, J. McLaren, J. Schacht, Hear. Res. 1995, 86, 15–24.
239

References
[183] S. L. Garetz, R. A. Altschuler, J. Schacht, Hear. Res. 1994, 77, 81.
[184] A. R. Badireddy, E. M. Hotze, S. Chellam, P. Alvarez, M. R. Wiesner, Environ.Sci. Technol. 2007, 41, 6627–6632.
[185] C. M. Sayes, J. D. Fortner, W. Guo, D. Lyon, A. M. Boyd, K. D. Ausman, Y. J. Tao,B. Sitharaman, L. J. Wilson, J. B. Hughes, J. L. West, V. L. Colvin, Nano Lett.2004, 4, 1881–1887.
[186] Y. Yamakoshi, S. Sueyoshi, K. Fukuhara, N. Miyata, J. Am. Chem. Soc. 1998,120, 12363–12364.
[187] I. Nakanishi, S. Fukuzumi, T. Konishi, K. Ohkubo, M. Fujitsuka, O. Ito, N. Miyata,J. Phys. Chem. B 2002, 106, 2372–2380.
[188] I. Nakanishi, K. Ohkubo, S. Fujita, S. Fukuzumi, T. Konishi, M. Fujitsuka, O. Ito,N. Miyata, J. Chem. Soc., Perkin Trans. 2002, 2, 1829–1833.
[189] Y. Yamakoshi, N. Umezawa, A. Ryu, K. Arakane, N. Miyata, Y. Goda, T. Masum-izu, T. Nagano, J. Am. Chem. Soc. 2003, 125, 12803–12809.
[190] Y. Iwamoto, Y. Yamakoshi, Chem. Commun. 2006, 4805–4807.
[191] P. Mroz, A. Pawlak, M. Satti, H. Lee, T. Wharton, H. Gali, T. Sarna, M. R. Hamblin,Free Radical Biol. Med. 2007, 43, 711–719.
[192] G.-F. Liu, M. Filipovic, F. W. Heinemann, I. Ivanovic-Burmazovic, Inorg. Chem.2007, 46, 8825–8835.
[193] K. K. Jain, Drug Discovery Today 2005, 10, 1435–1442.
[194] R. A. Freitas, J. Comput. Theor. Nanosci. 2005, 2, 1–25.
[195] J. F. Kukowska-Latallo, K. A. Candido, Z. Cao, S. S. Nigavekar, I. J. Majoros,T. P. Thomas, L. P. Balogh, M. K. Khan, J. R. Baker, Cancer Res. 2005, 65,5317–5324.
[196] J. Xue, S. Uchida, B. P. Rand, S. R. Forrest, Appl. Phys. Lett. 2004, 84, 3013–3015.
[197] M. Reyes-Reyes, K. Kim, D. L. Carroll, Appl. Phys. Lett. 2005, 87, 083506/1–083506/3.
[198] G. Yu, J. Gao, J. C. Hummelen, F. Wudl, A. J. Heeger, Science 1995, 270, 1789–1791.
[199] P. Peumans, A. Yakimov, S. R. Forrest, J. Appl. Phys. 2003, 93, 3693–3723.
[200] C. J. Brabec, N. S. Sariciftci, J. C. Hummelen, Adv. Funct. Mater. 2001, 11, 15–26.
[201] C. A. Mirkin, W. B. Caldwell, Tetrahedron 1996, 52, 5113–5130.
240

References
[202] L. Valli, D. M. Guldi, Developments in Fullerene Science, volume 4. SpringerNetherlands 2002, 327–385.
[203] N. S. Sariciftci, L. Smilowitz, A. J. Heeger, F. Wudl, Science 1992, 258, 1474–1476.
[204] C. J. Brabec, G. Zerza, G. Cerullo, S. D. Silvestri, S. Luzzati, J. C. Hummelen,S. Sariciftci, Chem. Phys. Lett. 2001, 340, 232–234.
[205] BMBF Project Ekos. 03N2023E.
[206] W. Geens, D. Tsamouras, J. Poortsman, G. Hadziioannou, Synth. Met. 2001,122, 191–195.
[207] G. Horowitz, R. Hajlaoui, D. Fichou, A. E. Kassmi, J. Appl. Phys. 1999, 85, 3202–3206.
[208] I. McCulloch, C. Bailey, M. Giles, M. Heeney, I. Love, M. Shkunov, D. Sparrowe,S. Tierney, Chem. Mater. 2005, 17, 1381–1385.
[209] S. Tierney, M. Heeney, I. McCulloch, Synth. Met. 2005, 148, 195–198.
[210] M. Koppe, M. Scharber, C. Brabec, W. Duffy, M. Heeney, I. McCulloch, Adv.Funct. Mater. 2007, 17, 1371–1376.
[211] H. C. Kolb, M. G. Finn, K. B. Sharpless, Angew. Chem. 2001, 113, 2056–2075;Angew. Chem. Int. Ed. 2001, 40, 2004–2021.
[212] B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci, A. L. Demirel,Macromolecules 2006, 39, 5330–5336.
[213] I. M. Pastor, M. Yus, Curr. Org. Chem. 2005, 9, 1–29.
[214] T. Siu, A. K. Yudin, J. Am. Chem. Soc. 2002, 124, 530–531.
[215] R. Huisgen, 1,3-Dipolar cycloaddition - introduction, survey, mechanism. Wiley-Interscience, New York 1984.
[216] Q. Wang, S. Chittaboina, H. N. Barnhill, Lett. Org. Chem. 2005, 2, 293–301.
[217] V. V. Rostovtsev, L. G. Green, V. V. Fokin, K. B. Sharpless, Angew. Chem. 2002,114, 2708–2711; Angew. Chem. Int. Ed. 2002, 41, 2596–2599.
[218] C. W. Tornoe, C. Christensen, M. Meldal, J. Org. Chem. 2002, 67, 3057–3064.
[219] F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless,V. V. Fokin, J. Am. Chem. Soc. 2005, 127, 210–216.
[220] K. Sonogashira, Y. Tohda, N. Hagihara, Tetrahedron Lett. 1975, 16, 4467–4470.
[221] P. Siemsen, R. C. Livingston, F. Diederich, Angew. Chem. 2000, 112, 2740–2767; Angew. Chem. Int. Ed. 2000, 39, 2632–2657.
241

References
[222] D. J. Darensbourg, W.-Z. Lee, M. J. Adams, J. C. Yarbrough, Eur. J. Inorg. Chem.2001, 2811–2822.
[223] A. E. Jukes, Adv. Organomet. Chem. 1974, 12, 228–229.
[224] P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun,J. M. J. Frechet, K. B. Sharpless, V. V. Fokin, Angew. Chem. 2004, 116, 4018–4022; Angew. Chem. Int. Ed. 2004, 43, 3928–3932.
[225] H. C. Kolb, K. B. Sharpless, Drug Discovery Today 2003, 8, 1128–1137.
[226] W. S. Horne, C. D. Stout, M. R. Ghadiri, J. Am. Chem. Soc. 2003, 125, 9372–9376.
[227] W. S. Horne, M. K. Yadav, C. D. Stout, M. R. Ghadiri, J. Am. Chem. Soc. 2004,126, 15366–15367.
[228] R. Alvarez, S. Velazquez, A. San-Felix, S. Aquaro, E. De Clercq, C.-F. Perno,A. Karlsson, J. Balzarini, M. J. Camarasa, J. Med. Chem. 1994, 37, 4185–4194.
[229] L. L. Brockunier, E. R. Parmee, H. O. Ok, M. R. Candelore, M. A. Cascieri,L. F. Colwell, L. Deng, W. P. Feeney, M. J. Forrest, G. J. Hom, D. E. MacIntyre,L. Tota, M. J. Wyvratt, M. H. Fisher, A. E. Weber, Bioorg. Med. Chem. Lett. 2000,10, 2111–2114.
[230] M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A.Garmon, D. R. Graber, K. C. Grega, J. B. Hester, D. K. Hutchinson, J. Morris,R. J. Reischer, C. W. Ford, G. E. Zurenko, J. C. Hamel, R. D. Schaadt, D. Stapert,B. H. Yagi, J. Med. Chem. 2000, 43, 953–970.
[231] D. R. Buckle, C. J. M. Rockell, H. Smith, B. A. Spicer, J. Med. Chem. 1986, 29,2262–2267.
[232] D. R. Buckle, D. J. Outred, C. J. M. Rockell, H. Smith, B. A. Spicer, J. Med. Chem.1983, 26, 251–254.
[233] U. Hartnagel, Synthese von molekularen und supramolekularen Fullerenar-chitekturen: Optoelektronische und biomedizinische Eigenschaften, Disserta-tion, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2006.
[234] E. Buhleier, W. Wehner, F. Vögtle, Synthesis 1978, 155–158.
[235] G. R. Newkome, Z. Q. Yao, G. R. Baker, V. K. Gupta, J. Org. Chem. 1985, 50,2003–2004.
[236] O. A. Troshina, P. A. Troshin, A. S. Peregudov, V. I. Kozlovski, R. N. Lyubovskaya,Chem. Eur. J. 2006, 12, 5569–5577.
[237] H. Isobe, W. Nakanishi, N. Tomita, S. Jinno, H. Okayama, E. Nakamura, Chem.Asian. J. 2006, 2, 167–175.
242

References
[238] J.-M. Becht, O. Meyer, G. Helmchen, Synthesis 2003, 2805–2810.
[239] P. M. Ajayan, Chem. Rev. 1999, 99, 1787–1799.
[240] S. S. Xie, B. H. Chang, W. Z. Li, Z. W. Pan, L. F. Sun, J. M. Mao, X. H. Chen,L. X. Qian, W. Y. Zhou, Adv. Mater. 1999, 11, 1135–1138.
[241] L. Dai, A. W. H. Mau, Adv. Mater. 2001, 13, 899–913.
[242] C. N. R. Rao, B. C. Satishkumar, A. Govindaraj, M. Nath, ChemPhysChem 2001,2, 78–105.
[243] R. Martel, T. Schmidt, H. R. Shea, T. Hertel, P. Avouris, Appl. Phys. Lett. 1998,73, 2447–2449.
[244] P. M. Ajayan, O. Z. Zhou, Top. Appl. Phys. 2001, 80, 391–425.
[245] A. Bachtold, P. Hadley, T. Nakanishi, C. Dekker, Science 2001, 294, 1317–1320.
[246] R. Rosen, W. Simendinger, C. Debbault, H. Shimoda, L. Fleming, B. Stoner,O. Zhou, Appl. Phys. Lett. 2000, 76, 1668–1670.
[247] A. A. Mamedov, N. A. Kotov, M. Prato, D. M. Guldi, J. P. Wicksted, A. Hirsch, Nat.Mater. 2002, 1, 190–194.
[248] J.-M. Bonard, T. Stockli, F. Maier, W. A. de Heer, A. Chatelain, J.-P. Salvetat,L. Forro, Phys. Rev. Lett. 1998, 81, 1441–1444.
[249] H. Sugie, M. Tanemura, V. Filip, K. Iwata, K. Takahashi, F. Okuyama, Appl. Phys.Lett. 2001, 78, 2578–2580.
[250] T. Yildirim, O. Gulseren, C. Kilic, S. Ciraci, Phys. Rev. B: Condens. Matter 2000,62, 12648–12651.
[251] J. L. Bahr, E. T. Mickelson, M. J. Bronikowski, R. E. Smalley, J. M. Tour, Chem.Commun. 2001, 193–194.
[252] Y. Chen, R. C. Haddon, S. Fang, A. M. Rao, P. C. Eklund, W. H. Lee, E. C. Dickey,E. C. Grulke, J. C. Pendergrass, A. Chavan, B. E. Haley, R. E. Smalley, J. Mater.Res. 1998, 13, 2423–2427.
[253] E. T. Mickelson, C. B. Huffman, A. G. Rinzler, R. E. Smalley, R. H. Hauge, J. L.Margrave, Chem. Phys. Lett. 1998, 296, 188–191.
[254] J. L. Bahr, J. Yang, D. V. Kosynkin, M. J. Bronikowski, R. E. Smalley, J. M. Tour,J. Am. Chem. Soc. 2001, 123, 6536–6542.
[255] M. Holzinger, O. Vostrowsky, A. Hirsch, F. Hennrich, M. Kappes, R. Weiss,F. Jellen, Angew. Chem. 2001, 113, 4132–4136; Angew. Chem. Int. Ed. 2001,40, 4002–4005.
[256] M. Holzinger, J. Abraham, P. Whelan, R. Graupner, L. Ley, F. Hennrich,
243

References
M. Kappes, A. Hirsch, J. Am. Chem. Soc. 2003, 125, 8566–8580.
[257] J. Abraham, Functionalization of Carbon Nanotubes, Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2005.
[258] J. T. Grant, D. Briggs, Surface Analysis by Auger and X-ray Photoelectron Spec-troscopy. IM Publications, Chichester UK 2003.
[259] J. F. Moulder, W. F. Stickle, P. E. Sobol, K. D. Bomben, Handbook of X-ray Pho-toelectron Spectroscopy. Perkin-Elmer Corp., Eden Prairie, MN, USA 1992.
[260] R. J. Chen, Y. Zhang, D. Wang, H. Dai, J. Am. Chem. Soc. 2001, 123, 3838–3839.
[261] N. Nakashima, Y. Tomonari, H. Murakami, Chem. Lett. 2002, 638–639.
[262] A. Ebel. personal communication.
[263] G. Binnig, H. Rohrer, C. Gerber, E. Weibel, Phys. Rev. Lett. 1982, 49, 57–61.
[264] C. Bai, Scanning Tunneling Microscopy and its Application. Springer Verlag,Stuttgart 1992.
[265] D. A. Bonnell, Scanning Tunneling Microscopy and Spectroscopy: Theory, Tech-niques, and Applications. John Wiley & Sons Inc., New York 1993.
[266] J.-P. Bourgeois, L. Echegoyen, M. Fibbioli, E. Pretsch, F. Diederich, Angew.Chem. 1998, 110, 2203–2207; Angew. Chem. Int. Ed. 1998, 37, 2118–2121.
[267] F. Cardullo, L. Isaacs, F. Diederich, J.-P. Gisselbrecht, C. Boudon, M. Gross,Chem. Commun. 1996, 797–799.
[268] J.-F. Nierengarten, T. Habicher, R. Kessinger, F. Cardullo, F. Diederich, V. Gram-lich, J.-P. Gisselbrecht, C. Boudon, M. Gross, Helv. Chim. Acta 1997, 80, 2238–2276.
[269] T. Brandmüller, Makrocyclische Malonatsysteme und deren Reaktion mit[60]Fulleren, Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg,2005.
[270] A. Hirsch, Chem. Rec. 2005, 5, 196–208.
[271] N. Chronakis, A. Hirsch, C. R. Chimie 2006, 9, 862–867.
[272] O. Berkesi, I. Dreveni, J. A. Andor, Inorg. Chim. Acta 1992, 195, 169–173.
[273] Y. J. Kim, E. W. Lee, D.-Y. Jung, Chem. Mater. 2001, 13, 2684–2690.
[274] Y.-Q. Zheng, J. Sun, J.-L. Lin, Z. Anorg. Allg. Chem. 2000, 626, 1271–1273.
[275] L. Gasque, R. Moreno-Esparza, E. Mollins, J. L. Brianso-Penalva, L. Ruiz-Ramirez, G. Medina-Dickinson, Acta Cryst. 1999, C55, 158–160.
244

References
[276] S. C. Zimmermann, P. S. Corbin, Struct. Bonding (Berlin) 2000, 63–94.
[277] F. H. Beijer, R. P. Sijbesma, J. A. J. M. Vekemans, E. Meijer, H. Kooijman, A. L.Spek, J. Org. Chem. 1996, 61, 6371–6380.
[278] F. H. Beijer, H. Kooijman, A. L. Spek, R. P. Sijbesma, E. W. Meijer, Angew. Chem.1998, 110, 79–82; Angew. Chem. Int. Ed. 1998, 37, 75–78.
[279] C. Schmuck, W. Wienand, Angew. Chem. 2001, 113, 4493–4499; Angew. Chem.Int. Ed. 2001, 40, 4363–4369.
[280] B. P. Orner, X. Salvatella, J. Sanchez-Quesada, J. de Mendoza, E. Giralt, A. D.Hamilton, Angew. Chem. 2002, 114, 125–127; Angew. Chem. Int. Ed. 2002, 41,117–119.
[281] S. K. Chang, A. D. Hamilton, J. Am. Chem. Soc. 1988, 110, 1318–1319.
[282] K. Hager, A. Franz, A. Hirsch, Chem. Eur. J. 2006, 12, 2663–2679.
[283] K. Hager, U. Hartnagel, A. Hirsch, Eur. J. Org. Chem. 2007, 1942–1956.
[284] K. Maurer, K. Hager, A. Hirsch, Eur. J. Org. Chem. 2006, 3338–3347.
[285] A. El-ghayoury, E. Peeters, A. P. H. J. Schenning, E. W. Meijer, Chem. Commun.2000, 1969–1970.
[286] A. P. H. J. Schenning, P. Jonkheijm, E. Peeters, E. W. Meijer, J. Am. Chem. Soc.2001, 123, 409–416.
[287] I. Lamparth, C. Maichle-Mössmer, A. Hirsch, Angew. Chem. 1995, 107, 1755–1757; Angew. Chem. Int. Ed. 1995, 34, 1607–1609.
[288] I. Lamparth, A. Herzog, A. Hirsch, Tetrahedron 1996, 52, 5065–5075.
[289] I. Lamparth, Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg,1996.
[290] A. Hirsch, O. Vostrowsky, Eur. J. Org. Chem. 2001, 829–848.
[291] M. Braun, X. Camps, O. Vostrowsky, A. Hirsch, E. Endreß, T. M. Bayerl, O. Birk-ert, G. Gauglitz, Eur. J. Org. Chem. 2000, 1173–1181.
[292] X. Camps, E. Dietel, A. Hirsch, S. Pyo, L. Echegoyen, S. Hackbarth, B. Roeder,Chem. Eur. J. 1999, 5, 2362–2373.
[293] A. Herzog, Synthese und Charakterisierung von isomerenreinen Mehrfachad-dukten von C60 mit definierten Kombinationen von zwei verschiedenen Adden-den, Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg, 1998.
[294] N. Chronakis, U. Hartnagel, M. Braun, A. Hirsch, Chem. Commun. 2007, 607–609.
245

[295] N. Martin, M. Altable, S. Filippone, A. Martin-Domenech, L. Echegoyen, C. M.Cardona, Angew. Chem. 2006, 118, 116–120; Angew. Chem. Int. Ed. 2006, 45,110–114.
[296] E. N. Durantini, Synth. Commun. 2006, 36, 2135–2144.
[297] A. Hirsch, I. Lamparth, H. R. Karfunkel, Angew. Chem. 1994, 106, 453–455;Angew. Chem. Int. Ed. 1994, 33, 437–438.
[298] Q. Lu, D. I. Schuster, S. R. Wilson, J. Org. Chem. 1996, 61, 4764–4768.
[299] J.-F. Nierengarten, J.-F. Eckert, Y. Rio, M. P. Carreon, J.-L. Gallani, D. Guillon, J.Am. Chem. Soc. 2001, 123, 9743–9748.
[300] M. Gutierrez-Nava, G. Accorsi, P. Masson, N. Armaroli, J.-F. Nierengarten,Chem. Eur. J. 2004, 10, 5076–5086.
[301] U. Hahn, K. Hosomizu, H. Imahori, J.-F. Nierengarten, Eur. J. Org. Chem. 2006,85–91.
[302] J.-F. Nierengarten, Top. Curr. Chem. 2003, 228, 87.
[303] Spartan’04 Wavefunction, Inc. Irvine, CA.
[304] K. Luger, A. W. Mader, R. K. Richmond, D. F. Sargent, T. J. Richmond, Nature1997, 389, 251–260.
[305] W. A. Scrivens, P. V. Bedworth, J. M. Tour, J. Am. Chem. Soc. 1992, 114, 7917–7919.
[306] H. G. O. Becker, W. Berger, G. Domschke, Organikum. Wiley-VCH Verlag GmbHWeinheim 2001.
[307] The compound was provided by C-Sixty Inc.
[308] D. E. Bergbreiter, P. L. Osburn, C. Li, Org. Lett. 2002, 4, 737–740.
[309] P. G. Mattingly, Synthesis 1990, 366–368.
[310] M. Braun, U. Hartnagel, E. Ravanelli, B. Schade, C. Böttcher, O. Vostrowsky,A. Hirsch, Eur. J. Org. Chem. 2004, 9, 1983–2001.
[311] M. Westerfield, The Zebrafish Book: a Guide for The Laboratory use of Zebrafish.The University of Oregon Press 1993.
[312] C. B. Kimmel, W. W. Ballard, S. R. Kimmel, B. Ullmann, T. F. Schilling, Develop-mental Dynamics 1995, 203, 253.
[313] A. Jung, Functional Materials based on Carbon Nanotubes, Dissertation,Friedrich-Alexander-Universität Erlangen-Nürnberg, 2007.

Index of Publications
High catalytic activity of dendritic C 60 monoadducts in metal-free superoxide dis-
mutation.
Liu, Gao-Feng; Filipovic, Milos; Ivanovic-Burmazovic, Ivana; Beuerle, Florian; Witte,
Patrick; Hirsch, Andreas. Angew. Chem. Int. Ed., 2008, 47(21), 3991–3994.
Water solubility, antioxidant activity and cytochrome C bi nding of four families
of exohedral adducts of C 60 and C70.
Witte, Patrick; Beuerle, Florian; Hartnagel, Uwe; Lebovitz, Russell; Savouchkina, Anas-
tasia; Sali, Sevda; Guldi, Dirk; Chronakis, Nikos; Hirsch, Andreas. Org. Biomol.
Chem., 2007, 5(22), 3599–3613.
Cytoprotective activities of water-soluble fullerenes in zebrafish models.
Beuerle, Florian; Witte, Patrick; Hartnagel, Uwe; Lebovitz, Russell; Parng, Chuenlei;
Hirsch, Andreas. J. Exp. Nanosci., 2007, 2, 147–170.
The transition from oxygen chemisorption to oxidation of ul tra-thin Ni layers on
Cu(111).
Domnick, Ralph; Held, Georg; Witte, Patrick; Steinrück, Hans-Peter. J. Chem. Phys.,
2001, 115, 1902–1908.
247

Curriculum Vitae
Personal Data
Name Patrick Sven Witte
Date of birth 09.09.1977
Place of birth Nürnberg
Nationality German
Education and Military Service
09/1983 – 07/1987 Elementary school in Chamerau
09/1987 – 06/1996 Grammar school in Cham
06/1996 School leaving examination (Abitur)
09/1996 – 09/1997 3./ Panzergrenadierbataillon 112, Regen
Higher Education
10/1997 – 09/1999 Basic studies: Chemistry (Diplom), University Regensburg
10/1999 – 02/2003 Main studies: Chemistry (Diplom), Friedrich-Alexander-
University Erlangen-Nürnberg
04/2003 – 10/2003 Diploma thesis at the Institute of Inorganic Chemistry,
Friedrich-Alexander-University Erlangen-Nürnberg
Supervisor: Prof. Dr. R. van Eldik
Title: “Synthese und Reaktivität modifizierter Pt(II) Komplexe”
(Synthesis and Reactivity of Modified Pt(II)-Complexes)
248

12/2004 – 11/2007 PhD studies at the Institute of Organic Chemistry,
Friedrich-Alexander-University Erlangen-Nürnberg
Supervisor: Prof. Dr. A. Hirsch
Title: “Amphiphilic Fullerenes for Biomedical and
Optoelectronical Applications”











![Fullerene Features Great and Small Jack E. Graver · 2013-08-11 · 2 Representing Fullerenes In An Atlas of Fullerenes[10], Fowler and Manolopouls include drawings of all fullerenes](https://img.pdfslide.net/doc/110x75/5f3a80c8fa11f52219316dc9/fullerene-features-great-and-small-jack-e-graver-2013-08-11-2-representing-fullerenes.jpg)

















