Embed Size (px)
Citation preview

An Efficient Fluctuating Charge Model for Transition MetalComplexes
Peter Comba,* Bodo Martin, and Avik Sanyal
A fluctuating charge model for transition metal complexes,
based on the Hirshfeld partitioning scheme, spectroscopic
energy data from the NIST Atomic Spectroscopy Database and
the electronegativity equalization approach, has been devel-
oped and parameterized for organic ligands and their high-
and low-spin FeII and FeIII, low-spin CoIII and CuII complexes,
using atom types defined in the Momec force field. Based on
large training sets comprising a variety of transition metal
complexes, a general parameter set has been developed and
independently validated which allows the efficient computa-
tion of geometry-dependent charge distributions in the field
of transition metal coordination compounds. VC 2013 Wiley
Periodicals, Inc.
DOI: 10.1002/jcc.23297
Introduction
Although partial atomic charges are not physical observables,
and therefore, are not real, they are an important concept in
chemistry and specifically in molecular mechanics. They are
fundamental for the accurate description of molecules when
electrostatic effects are not implicitly included in other force
field terms. The electrostatic interaction comprises a significant
part of nonbonded interactions between polar species (atoms
or atom groups). Traditionally, molecular mechanics force fields
assign fixed charges to specific sites within a molecule and
allow them to interact via Coulomb interactions.[1–8] These
fixed charges suffer from the disadvantage that they cannot
readjust to changes in the molecular structure or chemical
environment, for example, in geometry optimizations or mo-
lecular dynamics simulations. In some cases, the inclusion of a
dielectric constant as a damping factor in the electrostatic
equation can improve results, but for a generally applicable
force field this must remain a limited approach.
To improve on computed atomic charges, geometry-depend-
ent fluctuating charge schemes were developed based on the
electronegativity equalization principle,[9] which states that
when atoms with an infinite separation come together to form
a molecule, there is rearrangement of electronic charges, and
this continues until the electronegativities of the constituent
atoms become equal. Based on this idea, a charge model was
constructed[10] and two decades later it was the Partial Equaliza-
tion of Orbital Electronegativity (PEOE) method[11] that intro-
duced geometry-dependent charges in molecular mechanics.
PEOE-calculated charges were found to yield good results for
such charge-sensitive properties as core electron binding ener-
gies (ESCA shifts), NMR resonance shifts,[11,12] and gas-phase
proton affinities of amines.[13] Other notable examples of fluctu-
ating charge models are the electronegativity equalization
method (EEM),[14] the charge equilibration method (QEq),[15] the
atom-bond electronegativity equalization method,[16] and
charge-density based equalization schemes.[17,18]
The EE and QEq methods have found wide acceptance in
the area of organic and biomolecular force fields due to the
simplicity of the equations involved and also due to their rig-
orous theoretical basis in conceptual density functional theory
(DFT).[19–21] EEM[14] allows the direct calculation of partial
atomic charges (Qi) and molecular electronegativity veq by
using eq. (1)
veq 5 v�i 12h�i Qi1
Xj>i
Qj
rij(1)
where vi* and gi
* are the “effective” electronegativity and hard-
ness of atom i. The summation in eq. (1) represents the energy
due to interatomic interactions in the molecule. In the litera-
ture, values of vi* and gi
* for several elements can be found,
and these are calibrated against different quantum chemical
charge schemes and levels of theory.[22–25]
In the QEq method,[15] the charge distribution is calculated
by an analogous eq. (2):
veq 5 v0i 1 J0
ii Qi1
Xj>i
QjJij (2)
where vi0 and Jii
0 are the electronegativity and idempotential,
respectively, of atom i and the interatomic interaction term Jij
is calculated as a two-center Coulomb integral. Atomic ioniza-
tion energies and electron affinities (corrected for inclusion of
P. Comba, B. Martin and A. Sanyal
Anorganisch-Chemisches Institut, Universit€at Heidelberg, INF 270, D-69120,
Heidelberg, Germany
Fax: (149) 6226 546617
E-mail: [email protected]
Contract grant sponsor: German Science Foundation (DFG).
Contract grant sponsor: Heidelberg Graduate School of Mathematical and
Computational Methods for the Sciences (HGS MathComp).
VC 2013 Wiley Periodicals, Inc.
Journal of Computational Chemistry 2013, 00, 000–000 1
FULL PAPERWWW.C-CHEM.ORG

an atom in a molecule) provide a measure of vi0 and Jii
0; Jij’s
are calculated from orbital exponents of Slater-type orbitals.
The orbital exponents are taken to be charge independent
except for hydrogen, which necessitates an iterative approach
for the QEq scheme.
In transition metal chemistry, the central metal ion is usually
positively charged which makes it difficult to apply conven-
tional EEM or QEq in the majority of cases. In the QEq scheme,
the correction to ionization energies and electron affinities
due to inclusion of an atom in a molecule requires either
extensive quantum chemical calculation of the electron corre-
lation or a thorough optimization of the model parameters.
Also, the iterative nature of the method makes it computation-
ally demanding. EEM is simpler and noniterative, yet it suffers
from the drawback of using an unscreened Coulomb potential
to model the interatomic interaction term. This gives rise to
incorrect behavior of the total electrostatic energy function at
short distances. Modifications have been proposed to rectify
these deficiencies but the scope of the extensions have been
limited to organic and biomolecules, and some special zeolites
and catalysts.[26,27]
Here, we present a parameterized fluctuating charge model
that is fast, devoid of the problems of EEM, QEq and related
schemes and can be used with comparable accuracy for main
group compounds as well as transition metal complexes. Our
model contains two optimizable parameters per atom type
and is implemented in the framework of the molecular
mechanics program Momec[28–32] developed in our group.
Theory
The NIST Atomic Spectroscopy Database[33] contains data on
spectroscopic energy levels of isolated atoms and ions dis-
played in order of energy relative to the electronic ground
state. A relative energy (Erel) vs. ionization state/charge (Q) plot
can be constructed for elements from the NIST database. The
“zero” on the x-axis corresponds to a neutral atom (Q 5 0),
“11” to a singly ionized species (Q 5 11), and so on. The rela-
tive energy plotted on the y-axis is calculated by taking the
ground state (with a specific term symbol SLJ) of a particular
charged species to be of “zero” energy and using the valence
state ionization energy (VSIE) of that species as the value of
Erel, for example, when Q 5 0, the VSIE of the ground state is
taken as the value of Erel, when Q 5 11, the sum of the first
and second VSIE is taken as Erel, and so on. Using term ener-
gies to arrive at the relative energy values is essential to distin-
guish between the many possible spin states of transition
metal ions. Such fits are constructed from NIST data for the
elements carbon, nitrogen, oxygen, sulfur, chlorine, phospho-
rus, and hydrogen, and for the transition metals iron in both
low- and high-spin states, cobalt and copper (cf. Supporting
Information). It was found that in the case of all the above
elements the relative energy can be approximately expressed
as a quadratic function of the ionization state/charge as in
eq. (3).
Eat Qð Þ5Eat 0ð Þ1Aat Q1Bat Q2 (3)
In the above equation, the superscript “at” denotes isolated
atom properties, and A and B are two energy level dependent
fit parameters.
The quadratic relationship between atomic energy and ioni-
zation state has been suggested before[34] while defining the
electronegativity v of an atomic species with charge Qi as the
slope of its total electronic energy vs. ionization state curve at
Q 5 Qi, as shown in eq. (4).
v Qið Þ5oE
oQ
� �Qi
(4)
It is reasonable to assume that a similar quadratic relation-
ship exists between Erel and Q when we consider atoms in
molecules instead of isolated atoms, as shown in eq. (5).
Emol Qð Þ5Emol 0ð Þ1Amol Q1Bmol Q2 (5)
where the superscript “mol” denotes atom in molecule proper-
ties. When forming a molecule, the shape and size of the
atomic charge cloud is modified and it is expected that a sub-
sequent change in the energy-level parameters A and B
occurs. To account for this modification, it is assumed that a
simple linear relationship exists between the two sets of pa-
rameters [eqs. (6) and (7)].
Amol 5 Aat 1 DA (6)
Bmol 5 Bat 1 DB (7)
where DA and DB are corrections to the respective isolated
atom values due to incorporation in a molecule. Taking first
and second derivatives of eq. (3) with respect to charge leads
to eqs. (8) and (9).
dEmol Qð ÞdQ
5Amol 12Bmol Q (8)
d2Emol Qð ÞdQ2
52Bmol (9)
In a molecule, the interactions between constituent atoms
give rise to an additional electrostatic term in the total energy
expression. Thus, for an N-atomic molecule, the total electro-
static energy can be expressed as a function of partial atomic
charges as in eq. (10).[15,34]
E Q1::QNð Þ5XN
i51
E 0ð Þmoli 1
oEmol
oQ2
� �Qi
Qi11
2
o2Emol
oQ2
� �Qi
Q2i
!
1
XN
i51
Xj>i
QiQjDij (10)
Equation (10) consists of two parts—the first is the elec-
tronic energy of the individual atoms constituting the mole-
cule and the second is the interatomic interaction or external
FULL PAPER WWW.C-CHEM.ORG
2 Journal of Computational Chemistry 2013, 00, 000–000 WWW.CHEMISTRYVIEWS.COM

potential. The interatomic interaction term Dij is defined as the
electrostatic interaction between two unit charges separated
by a distance rij.
Use of eqs. (8) and (9) in eq. (10) yields eq. (11):
E Qi::QNð Þ5XN
i51
E 0ð Þmoli 1 Amol
i Qi13Bmoli Q2
i
� �
11
2
XN
i51
Xj 6¼i
QjDij (11)
Taking the first derivative of eq. (11) with respect to the par-
tial charges Qi gives vi, the Iczkowski–Margrave electronegativ-
ity of atom i in a molecule, as shown in eq. (12).
vi5Amoli 16Bmol
i Qi11
2
Xj 6¼i
QjDij (12)
An equation such as eq. (12) can be set up for each of the
atoms constituting the molecule, thereby yielding N simultane-
ous equations. Sanderson’s electronegativity equalization
principle[9] can now be applied to simulate a flow of charges
from high density to low and subsequent equalization of the
individual atomic electronegativities to a molecular value veq,
as given in eq. (13).
v15v25:::5vN5veq (13)
The conservation of the total charge in the molecule adds
another equation to this set. The (N 1 1) simultaneous equa-
tions can be solved for the (N 1 1) unknowns (N Qi and veq)
by standard linear algebraic methods. In matrix form, this
equation can be expressed as eq. (14).
6Bmol1
1
2D12 :::
1
2D1N 21
1
2D21 6Bmol
2 :::1
2D2N 21
: : ::: : :1
2DN1
1
2DN2 ::: 6Bmol
N 21
1 1 ::: 1 0
0BBBBBBB@
1CCCCCCCA
Q1
Q2
:::QN
veq
0BBBB@
1CCCCA5
2Amol1
2Amol2
:::2Amol
N
Qtot
0BBBB@
1CCCCA (14)
where Qtot is the total charge of the molecule.
The interatomic interaction term Dij deserves some consider-
ation. If atoms are assumed to be point charges, then the
potential due to two interacting atoms can be expressed
according to Coulomb’s inverse square law. This is a reasona-
ble approximation when electrostatic interactions between
nonbonded atoms at medium to large separation are consid-
ered, but when dealing with atoms that are bonded or in each
other’s van der Waals region, the effect due to overlap of dif-
fuse charge clouds (screening) must be taken into account.
There are two more or less obvious constraints that have to
be considered when modeling the interaction between atomic
charge clouds: (1) at large interatomic distances the functional
should behave like Coulomb’s inverse square law and (2) as
the separation approaches zero, Dij should attain a finite value.
A simple screened Coulomb-type equation that satisfies the
above constraints is eq. (15).
Dij51ffiffiffiffiffiffiffiffiffiffiffiffiffi
r2ij 1g2
ij
q (15)
where cij is a measure of the amount of screening for atom
pair ij.
Similar expressions for interatomic interaction in a mole-
cule have been proposed previously.[35,36] These methods
are aimed at rapid calculation of two-center Coulomb inte-
grals in the framework of semiempirical electronic structure
theory.
It is interesting to note that as rij tends to zero, eq. (15)
simplifies to eq. (16).
Dij rij ! 0� �
51
gij
(16)
A distance of zero between atoms i and j corresponds to an
unphysical situation, where atom i is placed on top of atom j.
This is comparable to the interaction of two electrons con-
tained in the same valence orbital of an atom. The repulsive
energy in that case is called the self-repulsion integral in semi-
empirical electronic structure theory. It has been argued that
the second derivative of atomic energy with respect to charge
can be approximately equated to this self-repulsion inte-
gral.[20],[37–39] This is shown in eq. (17).
d2E
dQ2
� �Qi
� Dij rij ! 0� �
51
gi
(17)
Comparing eq. (9) with eq. (17) gives eq. (18).
gi51
2Bmoli
(18)
Equation (15) can now be expressed in the form of eq. (19).
Dij51ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
r2ij 1
12Bij
� �2r (19)
The atom-pair-based parameter Bij can be simplified to an
atom-type-based parameter by assuming a simple geometric
mean combining rule, as shown in eq. (20).
Bij5ffiffiffiffiffiffiffiffiBiBj
p(20)
Equation (19) is used as the expression for Dij in our fluctu-
ating charge model.
FULL PAPERWWW.C-CHEM.ORG
Journal of Computational Chemistry 2013, 00, 000–000 3

Computational Details
We have shown above that one can calculate geometry-de-
pendent partial atomic charges from a knowledge of the mo-
lecular geometry (rij), the total formal charge of the molecule
(Qtot), experimental spectroscopic energies (Aat and Bat), and
the energy correction due to incorporation of an atom in a
molecule (DA and DB). However, DA and DB cannot be
obtained in a straightforward way from experimental or theo-
retical data and hence are used as optimizable parameters in
our model.
Note that in the present implementation in Momec, energy
gradients due to the fluctuating charges are not included in
the structure optimization (energy minimization) procedure as
in the molecular mechanics concept used in Momec, intramo-
lecular electrostatic terms are not considered explic-
itly.[28,30,31,40–43] For intermolecular interactions (solvation,
crystal lattices), electrostatic potentials are of importance, and
one of the next steps clearly will be to include the correspond-
ing energy gradients.
Choice of reference data
Our aim was to develop a fluctuating charge model applicable
to compounds of the main group elements as well as transi-
tion metal complexes. As such, four training sets were devel-
oped, the first containing 28 small organic molecules (set 1),
the second containing 21 six-coordinate complexes of iron in
different oxidation and spin states (set 2), the third containing
28 six-coordinate complexes of low-spin CoIII (set 3), and the
fourth containing 19 complexes of four- and six-coordinate
CuII (set 4) (for a complete list cf. the Supporting Information).
The geometries of all molecules in set 1 were optimized at the
B3LYP/6–31G* level using Gaussian 09.[44] The structures of the
transition metal complexes in sets 2, 3, and 4 were taken from
the Cambridge Crystallographic Structure Database (CCSD).[45]
Force-field atom-type assignments within Momec are also pro-
vided in Supporting Information.
Choice of reference charges from Quantum Mechanical
methods
Atomic charges are not physical observables, and conse-
quently cannot be obtained directly from experiments. How-
ever, charges are accessible from quantum chemical
calculations. Several methods exist, such as the Mulliken Popu-
lation Analysis (MPA),[46] the Natural Population Analysis
(NPA),[47] the “Atoms in a Molecule” scheme (QTAIM),[48] Hirsh-
feld partitioning,[49] or electrostatic potential fitting.[50–53] Each
of these schemes partition the molecular electron density dif-
ferently to arrive at atomic contributions, but there is no
unique method that works equally well for all classes of com-
pounds and for all purposes.
Mulliken population analysis[46] is conceptually simple and
several fluctuating charge schemes have been calibrated based
on MPA as the method of choice but it suffers from two seri-
ous disadvantages. First, MPA is known to be basis-set de-
pendent which limits its utility, and second, the electron
density is partitioned equally between two neighboring atoms
irrespective of their electronegativity difference which often
leads to charges that do not conform to chemical intuition.
The NPA method[47] is fast, as it requires only matrix diago-
nalization and orthogonalization steps, and accurate. For a
large set of medicinally active compounds containing H, C, N,
O, and F, EEM-derived NPA charges were calculated and found
to be in good agreement with quantum chemical values.[24]
However, from studies on complexes of glyoxal diimine with
21 cations of the first row transition metals, it was concluded
that NPA charges are inconsistent with the electron transfer
scheme obtained from ab initio orbital analyses.[54]
QTAIM[48] is a theoretically robust method but calculation of
atomic charges using this scheme is computationally much
more demanding than the other schemes outlined here, espe-
cially when transition metal complexes are considered.
Electrostatic potential fitted charges, like those obtained by
CHELPG[50] or those used in AMBER,[51] are popular in force
fields as they are designed to reproduce the surface electro-
static potential accurately, but these methods also suffer from
certain inadequacies. The main criticism is that atoms buried
deep inside the molecule contribute less to the surface elec-
trostatic potential and are often assigned ambiguous charges
by the method. This is a serious problem when dealing with
transition metal complexes as a fair amount of the charge on
the complex remains with the embedded central metal ion
and its first coordination sphere. Also, many different sets of
atomic charges may yield the same surface electrostatic poten-
tial and hence, the set of atomic charges obtained by these
methods is often ambiguous and redundant. The RESP
model[52] uses restraints in the potential fitting process to rem-
edy some of these problems and yield better quality atomic
charges. Force fields with RESP charges have been developed
for organic and biological molecules and are known to per-
form well.[53]
Another quantum-chemical method that partitions electron
density into atomic contributions and yields atomic charges is
the Hirshfeld partitioning scheme.[49] The underlying concept
is that the electron density at each point in Cartesian space is
distributed among all constituent atoms. A weighting function
is then defined that is given by eq. (21):
wA rð Þ5 q0A rð Þ
q0mol rð Þ (21)
where q0A(r) is the electron density of the isolated atom A and
q0mol is the “promolecular” density, that is, the density of the
superposition of all isolated atom densities, keeping all atoms
in their original position. The individual electron densities of
the atoms are then computed using eq. (22):
qA rð Þ5 wA rð Þq rð Þ5 q0A rð Þ
q0mol rð Þ qmol rð Þ (22)
where qmol(r) is the original electron density of the molecule.
This method is known to be robust and to produce reactivity
indices that are in agreement with chemical intuition. A major
FULL PAPER WWW.C-CHEM.ORG
4 Journal of Computational Chemistry 2013, 00, 000–000 WWW.CHEMISTRYVIEWS.COM

drawback, however, is that the definition of the promolecule is
arbitrary and, in most cases, derived from the electron density
of isolated atoms.[55,56] This makes the scheme unsuitable for
the application to charged species.
To overcome the problems inherent in the Hirshfeld parti-
tioning scheme, an iterative algorithm has been proposed.[57]
This eliminates the requirement to have a predefined promole-
cule, that is, the method itself determines the promolecule
from the knowledge of the molecular electron density. The
weighting function for the ith iteration is given by eq. (23):
wiA rð Þ5 qi21
A rð Þqi21
mol rð Þ (23)
The aim of this iterative algorithm is to find a converged so-
lution to the electron population. A salient feature of this
approach is that, unlike Hirshfeld’s original scheme, it can be
applied to charged species.
Keeping in mind all the merits and disadvantages associated
with the different quantum chemical charge calculation
schemes, the iterative Hirshfeld method was chosen to gener-
ate reference charges for our training sets. The level of theory
and basis set used for the calibration data sets are set 1:
B3LYP/6–31G*;[58] and sets 2, 3, and 4: B3LYP/TZVP.[59,60] The
HiPart program[61] was used to generate iterative Hirshfeld
charges from electron densities calculated at the above-men-
tioned level of theory by Gaussian 09. For set 1, geometry-
optimized structures at the B3LYP/6–31G* level were used for
the calculation of electron densities and subsequently the iter-
ative Hirshfeld charges. For the other sets, single-point calcula-
tions at the B3LYP/TZVP level were done on crystal structures
taken from the CCSD to obtain the electron densities, and sub-
sequently the iterative Hirshfeld charges.
Erel vs. Q fits from NIST data
VSIE data for the ionization states 0, 11, 12, 13, and 14
were obtained from the NIST Atomic Spectroscopy Data-
base[33] for a continuous quadratic fit of the Erel vs. Q data for
the elements considered here. The quadratic fit was obtained
using the least-squares procedure, as in eq. (3). The case of
hydrogen deserves special mention. The only ionization states
possible for hydrogen are 21, 0, and 11 (Erel for Q 5 21 state
is the electron affinity), and the fit for hydrogen uses only
these three values. The quadratic fit parameters for the ele-
ments considered here are presented in Table 1, and the fits
are given as Supporting Information.
Calibration of parameters
As mentioned earlier, our model contains two optimizable pa-
rameters per atom type, DA and DB. An automatic simplex
optimization algorithm developed in-house (distributed with
Momec) was used to achieve parameter optimizations. The
goal of the procedure was to determine a set of atom-type-
based parameters that when inserted into eq. (14) yields
atomic charges that differ only minimally from the correspond-
ing quantum chemical charges.
Results and Discussion
The process of optimization of parameters proved to be difficult
and laborious for all four reference sets. This may be attributed
to several factors. The presence of many local minima in the
multidimensional parameter space is a major hindrance to effi-
cient convergence. To avoid this, our optimizer carried out 100
random initial Monte Carlo steps to find the best set of starting
values for the parameters. This led to significant improvement
in the optimization process. Another point that deserves men-
tion in this context is the sensitivity of the parameters to the fit-
ness function, a factor that has been reported previously.[24,27]
The presence of many atom types, with some types having
only a limited number of data points, presented another chal-
lenge to the optimization process. Atom types are a fundamen-
tal part of a force field and they are determined on the basis of
element and bonding environment. In the case of transition
metal complexes, a large number of bonding environments are
possible for a particular element and, consequently, a large
number of atom types are generally used. The Momec force
field for transition metal complexes[28] uses 14 atom types each
for carbon and oxygen, 16 for nitrogen, and so on. The large
spread in reference charges for a single element depending on
the chemical environment necessitates the use of a number of
atom types for fluctuating charge calculations. In the course of
our investigations, it was observed that several atom types per
element were necessary for the development of an efficient
scheme (see Supporting Information for the atom types used in
this communication). Although we have adopted the ligand
field molecular mechanics approach in Momec, which allows to
model metal centers of different spin state with a single param-
eter set,[62,63] we have found that to obtain accurate charges,
we need to define various atom types for the four sets of iron
complexes (FeII and FeIII in high- and low-spin electronic
configurations).
Figure 1 shows the reference (iterative Hirshfeld, calculated
at B3LYP/6–31G* level) vs. calculated (by our parameterized
model) partial atomic charges for set 1 with 28 small organic
Table 1. Quadratic fit parameters for Erel vs. Q [see eq. (3)]. Erel is plotted
in eV and Q in atomic units (au).
Element E(O)at Aat Bat
C 0.5952 20.3026 9.2837
N 1.3615 0.4519 10.2662
O 0.0353 3.1075 10.5289
S 1.3240 0.6113 7.2627
Cl 0.4786 4.3901 7.0115
P 1.7007 20.8285 7.1319
H 0.0 6.4192 7.1792
Fe (low spin) 2.5379 29.5301 8.3271
Fe (high spin) 2.8724 28.2711 8.8076
Co 2.6988 27.4608 8.8364
Cu 1.7732 25.7057 9.0887
Aat and Bat, therefore, have units of eV/au and eV/au2, respectively.
FULL PAPERWWW.C-CHEM.ORG
Journal of Computational Chemistry 2013, 00, 000–000 5
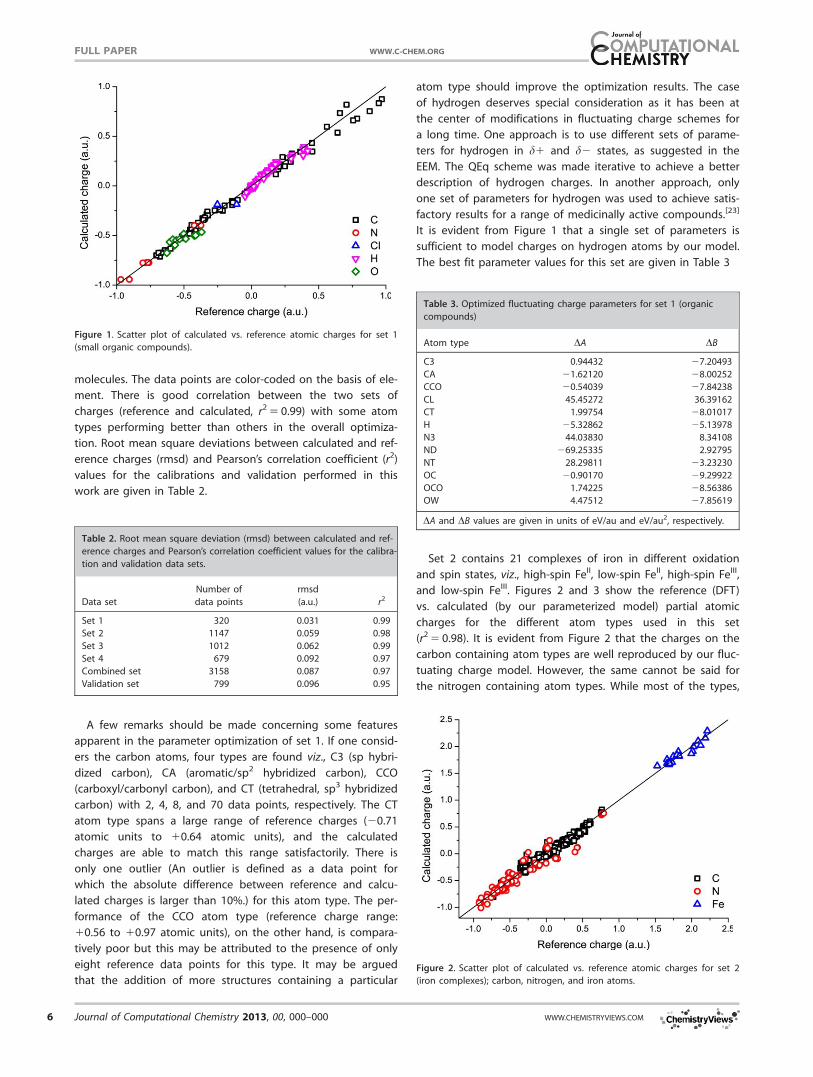
molecules. The data points are color-coded on the basis of ele-
ment. There is good correlation between the two sets of
charges (reference and calculated, r2 5 0.99) with some atom
types performing better than others in the overall optimiza-
tion. Root mean square deviations between calculated and ref-
erence charges (rmsd) and Pearson’s correlation coefficient (r2)
values for the calibrations and validation performed in this
work are given in Table 2.
A few remarks should be made concerning some features
apparent in the parameter optimization of set 1. If one consid-
ers the carbon atoms, four types are found viz., C3 (sp hybri-
dized carbon), CA (aromatic/sp2 hybridized carbon), CCO
(carboxyl/carbonyl carbon), and CT (tetrahedral, sp3 hybridized
carbon) with 2, 4, 8, and 70 data points, respectively. The CT
atom type spans a large range of reference charges (20.71
atomic units to 10.64 atomic units), and the calculated
charges are able to match this range satisfactorily. There is
only one outlier (An outlier is defined as a data point for
which the absolute difference between reference and calcu-
lated charges is larger than 10%.) for this atom type. The per-
formance of the CCO atom type (reference charge range:
10.56 to 10.97 atomic units), on the other hand, is compara-
tively poor but this may be attributed to the presence of only
eight reference data points for this type. It may be argued
that the addition of more structures containing a particular
atom type should improve the optimization results. The case
of hydrogen deserves special consideration as it has been at
the center of modifications in fluctuating charge schemes for
a long time. One approach is to use different sets of parame-
ters for hydrogen in d1 and d2 states, as suggested in the
EEM. The QEq scheme was made iterative to achieve a better
description of hydrogen charges. In another approach, only
one set of parameters for hydrogen was used to achieve satis-
factory results for a range of medicinally active compounds.[23]
It is evident from Figure 1 that a single set of parameters is
sufficient to model charges on hydrogen atoms by our model.
The best fit parameter values for this set are given in Table 3
Set 2 contains 21 complexes of iron in different oxidation
and spin states, viz., high-spin FeII, low-spin FeII, high-spin FeIII,
and low-spin FeIII. Figures 2 and 3 show the reference (DFT)
vs. calculated (by our parameterized model) partial atomic
charges for the different atom types used in this set
(r2 5 0.98). It is evident from Figure 2 that the charges on the
carbon containing atom types are well reproduced by our fluc-
tuating charge model. However, the same cannot be said for
the nitrogen containing atom types. While most of the types,
Figure 1. Scatter plot of calculated vs. reference atomic charges for set 1
(small organic compounds).
Table 2. Root mean square deviation (rmsd) between calculated and ref-
erence charges and Pearson’s correlation coefficient values for the calibra-
tion and validation data sets.
Data set
Number of
data points
rmsd
(a.u.) r2
Set 1 320 0.031 0.99
Set 2 1147 0.059 0.98
Set 3 1012 0.062 0.99
Set 4 679 0.092 0.97
Combined set 3158 0.087 0.97
Validation set 799 0.096 0.95
Table 3. Optimized fluctuating charge parameters for set 1 (organic
compounds)
Atom type DA DB
C3 0.94432 27.20493
CA 21.62120 28.00252
CCO 20.54039 27.84238
CL 45.45272 36.39162
CT 1.99754 28.01017
H 25.32862 25.13978
N3 44.03830 8.34108
ND 269.25335 2.92795
NT 28.29811 23.23230
OC 20.90170 29.29922
OCO 1.74225 28.56386
OW 4.47512 27.85619
DA and DB values are given in units of eV/au and eV/au2, respectively.
Figure 2. Scatter plot of calculated vs. reference atomic charges for set 2
(iron complexes); carbon, nitrogen, and iron atoms.
FULL PAPER WWW.C-CHEM.ORG
6 Journal of Computational Chemistry 2013, 00, 000–000 WWW.CHEMISTRYVIEWS.COM
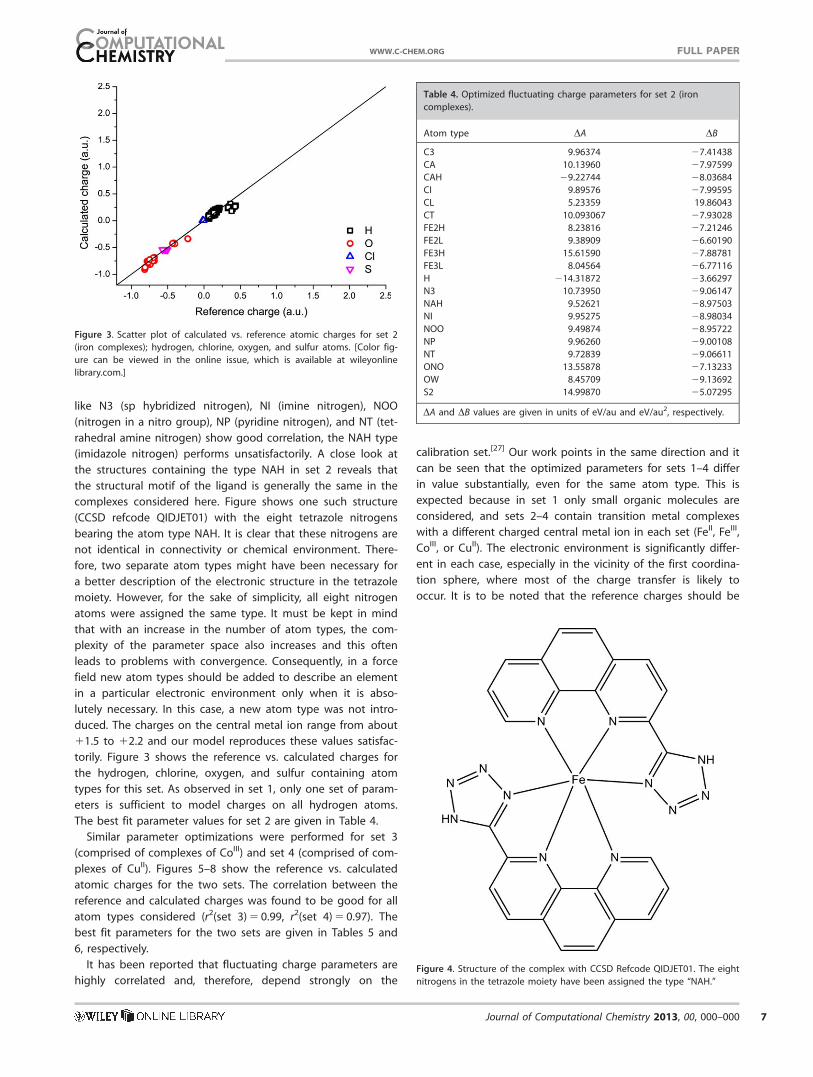
like N3 (sp hybridized nitrogen), NI (imine nitrogen), NOO
(nitrogen in a nitro group), NP (pyridine nitrogen), and NT (tet-
rahedral amine nitrogen) show good correlation, the NAH type
(imidazole nitrogen) performs unsatisfactorily. A close look at
the structures containing the type NAH in set 2 reveals that
the structural motif of the ligand is generally the same in the
complexes considered here. Figure shows one such structure
(CCSD refcode QIDJET01) with the eight tetrazole nitrogens
bearing the atom type NAH. It is clear that these nitrogens are
not identical in connectivity or chemical environment. There-
fore, two separate atom types might have been necessary for
a better description of the electronic structure in the tetrazole
moiety. However, for the sake of simplicity, all eight nitrogen
atoms were assigned the same type. It must be kept in mind
that with an increase in the number of atom types, the com-
plexity of the parameter space also increases and this often
leads to problems with convergence. Consequently, in a force
field new atom types should be added to describe an element
in a particular electronic environment only when it is abso-
lutely necessary. In this case, a new atom type was not intro-
duced. The charges on the central metal ion range from about
11.5 to 12.2 and our model reproduces these values satisfac-
torily. Figure 3 shows the reference vs. calculated charges for
the hydrogen, chlorine, oxygen, and sulfur containing atom
types for this set. As observed in set 1, only one set of param-
eters is sufficient to model charges on all hydrogen atoms.
The best fit parameter values for set 2 are given in Table 4.
Similar parameter optimizations were performed for set 3
(comprised of complexes of CoIII) and set 4 (comprised of com-
plexes of CuII). Figures 5–8 show the reference vs. calculated
atomic charges for the two sets. The correlation between the
reference and calculated charges was found to be good for all
atom types considered (r2(set 3) 5 0.99, r2(set 4) 5 0.97). The
best fit parameters for the two sets are given in Tables 5 and
6, respectively.
It has been reported that fluctuating charge parameters are
highly correlated and, therefore, depend strongly on the
calibration set.[27] Our work points in the same direction and it
can be seen that the optimized parameters for sets 1–4 differ
in value substantially, even for the same atom type. This is
expected because in set 1 only small organic molecules are
considered, and sets 2–4 contain transition metal complexes
with a different charged central metal ion in each set (FeII, FeIII,
CoIII, or CuII). The electronic environment is significantly differ-
ent in each case, especially in the vicinity of the first coordina-
tion sphere, where most of the charge transfer is likely to
occur. It is to be noted that the reference charges should be
Figure 3. Scatter plot of calculated vs. reference atomic charges for set 2
(iron complexes); hydrogen, chlorine, oxygen, and sulfur atoms. [Color fig-
ure can be viewed in the online issue, which is available at wileyonline
library.com.]
Table 4. Optimized fluctuating charge parameters for set 2 (iron
complexes).
Atom type DA DB
C3 9.96374 27.41438
CA 10.13960 27.97599
CAH 29.22744 28.03684
CI 9.89576 27.99595
CL 5.23359 19.86043
CT 10.093067 27.93028
FE2H 8.23816 27.21246
FE2L 9.38909 26.60190
FE3H 15.61590 27.88781
FE3L 8.04564 26.77116
H 214.31872 23.66297
N3 10.73950 29.06147
NAH 9.52621 28.97503
NI 9.95275 28.98034
NOO 9.49874 28.95722
NP 9.96260 29.00108
NT 9.72839 29.06611
ONO 13.55878 27.13233
OW 8.45709 29.13692
S2 14.99870 25.07295
DA and DB values are given in units of eV/au and eV/au2, respectively.
Figure 4. Structure of the complex with CCSD Refcode QIDJET01. The eight
nitrogens in the tetrazole moiety have been assigned the type “NAH.”
FULL PAPERWWW.C-CHEM.ORG
Journal of Computational Chemistry 2013, 00, 000–000 7
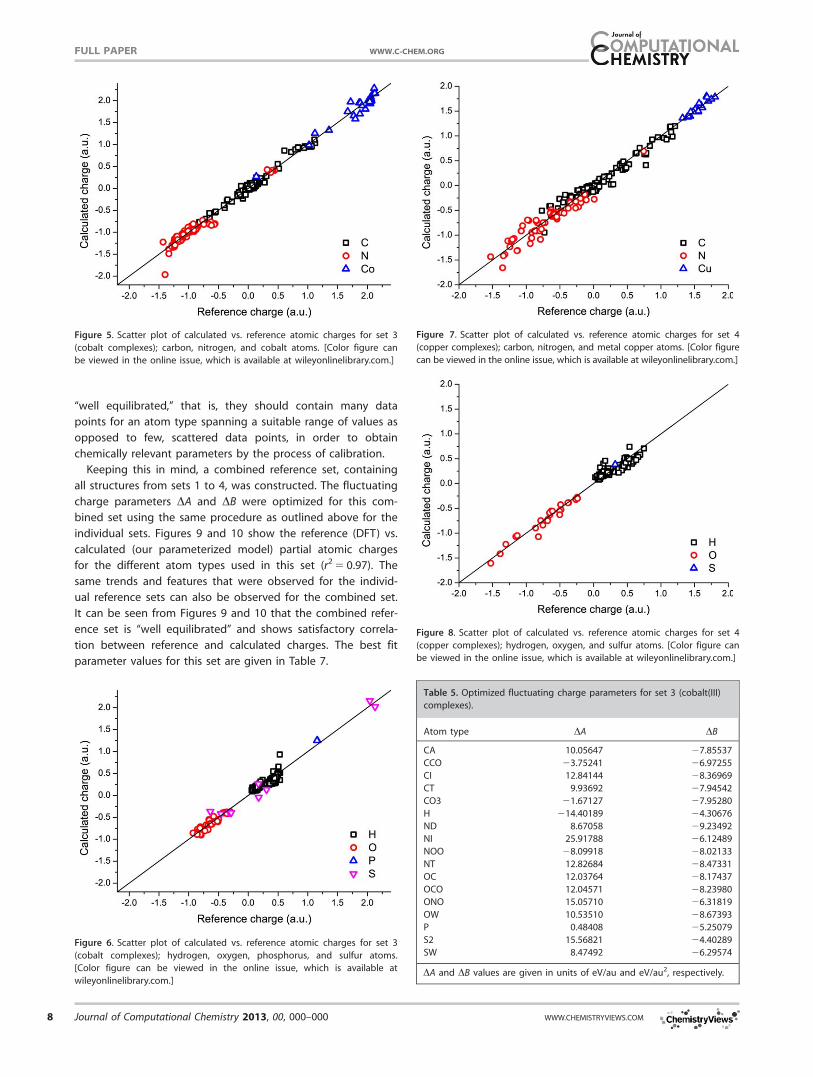
“well equilibrated,” that is, they should contain many data
points for an atom type spanning a suitable range of values as
opposed to few, scattered data points, in order to obtain
chemically relevant parameters by the process of calibration.
Keeping this in mind, a combined reference set, containing
all structures from sets 1 to 4, was constructed. The fluctuating
charge parameters DA and DB were optimized for this com-
bined set using the same procedure as outlined above for the
individual sets. Figures 9 and 10 show the reference (DFT) vs.
calculated (our parameterized model) partial atomic charges
for the different atom types used in this set (r2 5 0.97). The
same trends and features that were observed for the individ-
ual reference sets can also be observed for the combined set.
It can be seen from Figures 9 and 10 that the combined refer-
ence set is “well equilibrated” and shows satisfactory correla-
tion between reference and calculated charges. The best fit
parameter values for this set are given in Table 7.
Figure 7. Scatter plot of calculated vs. reference atomic charges for set 4
(copper complexes); carbon, nitrogen, and metal copper atoms. [Color figure
can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 5. Scatter plot of calculated vs. reference atomic charges for set 3
(cobalt complexes); carbon, nitrogen, and cobalt atoms. [Color figure can
be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 6. Scatter plot of calculated vs. reference atomic charges for set 3
(cobalt complexes); hydrogen, oxygen, phosphorus, and sulfur atoms.
[Color figure can be viewed in the online issue, which is available at
wileyonlinelibrary.com.]
Table 5. Optimized fluctuating charge parameters for set 3 (cobalt(III)
complexes).
Atom type DA DB
CA 10.05647 27.85537
CCO 23.75241 26.97255
CI 12.84144 28.36969
CT 9.93692 27.94542
CO3 21.67127 27.95280
H 214.40189 24.30676
ND 8.67058 29.23492
NI 25.91788 26.12489
NOO 28.09918 28.02133
NT 12.82684 28.47331
OC 12.03764 28.17437
OCO 12.04571 28.23980
ONO 15.05710 26.31819
OW 10.53510 28.67393
P 0.48408 25.25079
S2 15.56821 24.40289
SW 8.47492 26.29574
DA and DB values are given in units of eV/au and eV/au2, respectively.
Figure 8. Scatter plot of calculated vs. reference atomic charges for set 4
(copper complexes); hydrogen, oxygen, and sulfur atoms. [Color figure can
be viewed in the online issue, which is available at wileyonlinelibrary.com.]
FULL PAPER WWW.C-CHEM.ORG
8 Journal of Computational Chemistry 2013, 00, 000–000 WWW.CHEMISTRYVIEWS.COM
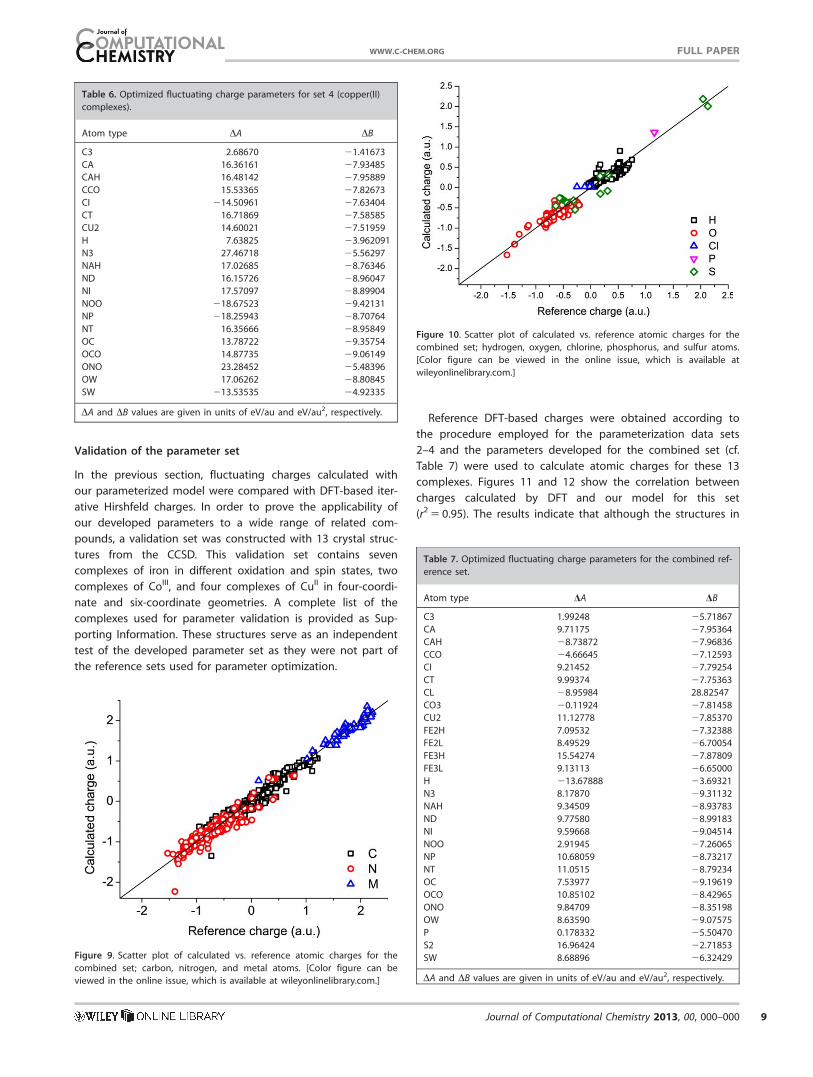
Validation of the parameter set
In the previous section, fluctuating charges calculated with
our parameterized model were compared with DFT-based iter-
ative Hirshfeld charges. In order to prove the applicability of
our developed parameters to a wide range of related com-
pounds, a validation set was constructed with 13 crystal struc-
tures from the CCSD. This validation set contains seven
complexes of iron in different oxidation and spin states, two
complexes of CoIII, and four complexes of CuII in four-coordi-
nate and six-coordinate geometries. A complete list of the
complexes used for parameter validation is provided as Sup-
porting Information. These structures serve as an independent
test of the developed parameter set as they were not part of
the reference sets used for parameter optimization.
Reference DFT-based charges were obtained according to
the procedure employed for the parameterization data sets
2–4 and the parameters developed for the combined set (cf.
Table 7) were used to calculate atomic charges for these 13
complexes. Figures 11 and 12 show the correlation between
charges calculated by DFT and our model for this set
(r2 5 0.95). The results indicate that although the structures in
Table 6. Optimized fluctuating charge parameters for set 4 (copper(II)
complexes).
Atom type DA DB
C3 2.68670 21.41673
CA 16.36161 27.93485
CAH 16.48142 27.95889
CCO 15.53365 27.82673
CI 214.50961 27.63404
CT 16.71869 27.58585
CU2 14.60021 27.51959
H 7.63825 23.962091
N3 27.46718 25.56297
NAH 17.02685 28.76346
ND 16.15726 28.96047
NI 17.57097 28.89904
NOO 218.67523 29.42131
NP 218.25943 28.70764
NT 16.35666 28.95849
OC 13.78722 29.35754
OCO 14.87735 29.06149
ONO 23.28452 25.48396
OW 17.06262 28.80845
SW 213.53535 24.92335
DA and DB values are given in units of eV/au and eV/au2, respectively.
Figure 9. Scatter plot of calculated vs. reference atomic charges for the
combined set; carbon, nitrogen, and metal atoms. [Color figure can be
viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 10. Scatter plot of calculated vs. reference atomic charges for the
combined set; hydrogen, oxygen, chlorine, phosphorus, and sulfur atoms.
[Color figure can be viewed in the online issue, which is available at
wileyonlinelibrary.com.]
Table 7. Optimized fluctuating charge parameters for the combined ref-
erence set.
Atom type DA DB
C3 1.99248 25.71867
CA 9.71175 27.95364
CAH 28.73872 27.96836
CCO 24.66645 27.12593
CI 9.21452 27.79254
CT 9.99374 27.75363
CL 28.95984 28.82547
CO3 20.11924 27.81458
CU2 11.12778 27.85370
FE2H 7.09532 27.32388
FE2L 8.49529 26.70054
FE3H 15.54274 27.87809
FE3L 9.13113 26.65000
H 213.67888 23.69321
N3 8.17870 29.31132
NAH 9.34509 28.93783
ND 9.77580 28.99183
NI 9.59668 29.04514
NOO 2.91945 27.26065
NP 10.68059 28.73217
NT 11.0515 28.79234
OC 7.53977 29.19619
OCO 10.85102 28.42965
ONO 9.84709 28.35198
OW 8.63590 29.07575
P 0.178332 25.50470
S2 16.96424 22.71853
SW 8.68896 26.32429
DA and DB values are given in units of eV/au and eV/au2, respectively.
FULL PAPERWWW.C-CHEM.ORG
Journal of Computational Chemistry 2013, 00, 000–000 9
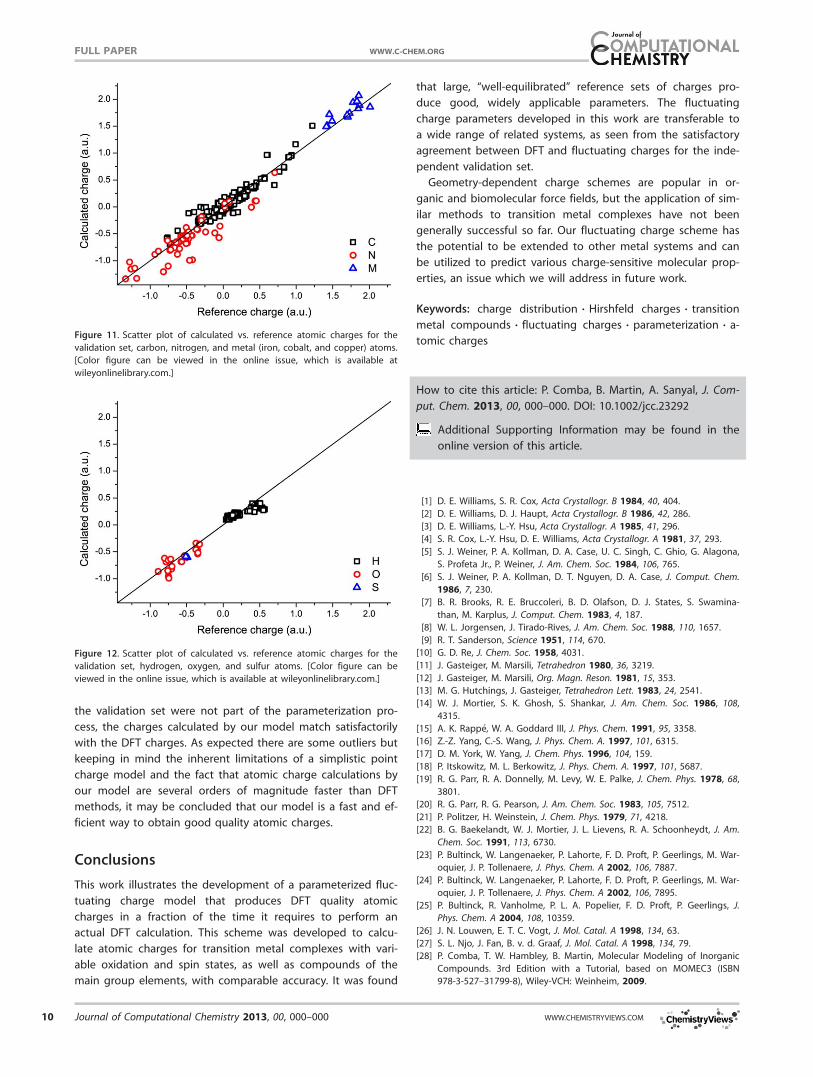
the validation set were not part of the parameterization pro-
cess, the charges calculated by our model match satisfactorily
with the DFT charges. As expected there are some outliers but
keeping in mind the inherent limitations of a simplistic point
charge model and the fact that atomic charge calculations by
our model are several orders of magnitude faster than DFT
methods, it may be concluded that our model is a fast and ef-
ficient way to obtain good quality atomic charges.
Conclusions
This work illustrates the development of a parameterized fluc-
tuating charge model that produces DFT quality atomic
charges in a fraction of the time it requires to perform an
actual DFT calculation. This scheme was developed to calcu-
late atomic charges for transition metal complexes with vari-
able oxidation and spin states, as well as compounds of the
main group elements, with comparable accuracy. It was found
that large, “well-equilibrated” reference sets of charges pro-
duce good, widely applicable parameters. The fluctuating
charge parameters developed in this work are transferable to
a wide range of related systems, as seen from the satisfactory
agreement between DFT and fluctuating charges for the inde-
pendent validation set.
Geometry-dependent charge schemes are popular in or-
ganic and biomolecular force fields, but the application of sim-
ilar methods to transition metal complexes have not been
generally successful so far. Our fluctuating charge scheme has
the potential to be extended to other metal systems and can
be utilized to predict various charge-sensitive molecular prop-
erties, an issue which we will address in future work.
Keywords: charge distribution � Hirshfeld charges � transition
metal compounds � fluctuating charges � parameterization � a-
tomic charges
How to cite this article: P. Comba, B. Martin, A. Sanyal, J. Com-
put. Chem. 2013, 00, 000–000. DOI: 10.1002/jcc.23292
Additional Supporting Information may be found in the
online version of this article.
[1] D. E. Williams, S. R. Cox, Acta Crystallogr. B 1984, 40, 404.
[2] D. E. Williams, D. J. Haupt, Acta Crystallogr. B 1986, 42, 286.
[3] D. E. Williams, L.-Y. Hsu, Acta Crystallogr. A 1985, 41, 296.
[4] S. R. Cox, L.-Y. Hsu, D. E. Williams, Acta Crystallogr. A 1981, 37, 293.
[5] S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona,
S. Profeta Jr., P. Weiner, J. Am. Chem. Soc. 1984, 106, 765.
[6] S. J. Weiner, P. A. Kollman, D. T. Nguyen, D. A. Case, J. Comput. Chem.
1986, 7, 230.
[7] B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swamina-
than, M. Karplus, J. Comput. Chem. 1983, 4, 187.
[8] W. L. Jorgensen, J. Tirado-Rives, J. Am. Chem. Soc. 1988, 110, 1657.
[9] R. T. Sanderson, Science 1951, 114, 670.
[10] G. D. Re, J. Chem. Soc. 1958, 4031.
[11] J. Gasteiger, M. Marsili, Tetrahedron 1980, 36, 3219.
[12] J. Gasteiger, M. Marsili, Org. Magn. Reson. 1981, 15, 353.
[13] M. G. Hutchings, J. Gasteiger, Tetrahedron Lett. 1983, 24, 2541.
[14] W. J. Mortier, S. K. Ghosh, S. Shankar, J. Am. Chem. Soc. 1986, 108,
4315.
[15] A. K. Rapp�e, W. A. Goddard III, J. Phys. Chem. 1991, 95, 3358.
[16] Z.-Z. Yang, C.-S. Wang, J. Phys. Chem. A. 1997, 101, 6315.
[17] D. M. York, W. Yang, J. Chem. Phys. 1996, 104, 159.
[18] P. Itskowitz, M. L. Berkowitz, J. Phys. Chem. A. 1997, 101, 5687.
[19] R. G. Parr, R. A. Donnelly, M. Levy, W. E. Palke, J. Chem. Phys. 1978, 68,
3801.
[20] R. G. Parr, R. G. Pearson, J. Am. Chem. Soc. 1983, 105, 7512.
[21] P. Politzer, H. Weinstein, J. Chem. Phys. 1979, 71, 4218.
[22] B. G. Baekelandt, W. J. Mortier, J. L. Lievens, R. A. Schoonheydt, J. Am.
Chem. Soc. 1991, 113, 6730.
[23] P. Bultinck, W. Langenaeker, P. Lahorte, F. D. Proft, P. Geerlings, M. War-
oquier, J. P. Tollenaere, J. Phys. Chem. A 2002, 106, 7887.
[24] P. Bultinck, W. Langenaeker, P. Lahorte, F. D. Proft, P. Geerlings, M. War-
oquier, J. P. Tollenaere, J. Phys. Chem. A 2002, 106, 7895.
[25] P. Bultinck, R. Vanholme, P. L. A. Popelier, F. D. Proft, P. Geerlings, J.
Phys. Chem. A 2004, 108, 10359.
[26] J. N. Louwen, E. T. C. Vogt, J. Mol. Catal. A 1998, 134, 63.
[27] S. L. Njo, J. Fan, B. v. d. Graaf, J. Mol. Catal. A 1998, 134, 79.
[28] P. Comba, T. W. Hambley, B. Martin, Molecular Modeling of Inorganic
Compounds. 3rd Edition with a Tutorial, based on MOMEC3 (ISBN
978-3-527–31799-8), Wiley-VCH: Weinheim, 2009.
Figure 12. Scatter plot of calculated vs. reference atomic charges for the
validation set, hydrogen, oxygen, and sulfur atoms. [Color figure can be
viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 11. Scatter plot of calculated vs. reference atomic charges for the
validation set, carbon, nitrogen, and metal (iron, cobalt, and copper) atoms.
[Color figure can be viewed in the online issue, which is available at
wileyonlinelibrary.com.]
FULL PAPER WWW.C-CHEM.ORG
10 Journal of Computational Chemistry 2013, 00, 000–000 WWW.CHEMISTRYVIEWS.COM

[29] Momec is freely available, Available at: http://www.momec.aci.uni-hei-
delberg.de. (14-01-2013)
[30] P. Comba, T. W. Hambley, M. Str€ohle, Helv. Chim. Acta. 1995, 78, 2042.
[31] J. E. Bol, C. Buning, P. Comba, J. Reedijk, M. Str€ohle, J. Comput. Chem.
1998, 19, 512.
[32] P. Comba, N. Okon, R. Remenyi, J. Comput. Chem. 1999, 20, 781.
[33] NIST Atomic Spectroscopy Database, Available at: http://www.physics.-
nist.gov/PhysRefData/ASD. (10-01-2013)
[34] R. P. Iczkowski, J. L. Margrave, J. Am. Chem. Soc. 1961, 83, 3547.
[35] K. Ohno, Theor. Chim. Acta 1964, 2, 219.
[36] G. Klopman, J. Am. Chem. Soc. 1965, 87, 3300.
[37] R. Pariser, J. Chem. Phys. 1953, 21, 568.
[38] R. Pariser, R. G. Parr, J. Chem. Phys. 1953, 21, 466.
[39] R. Pariser, R. G. Parr, J. Chem. Phys. 1953, 21, 767.
[40] P. V. Bernhardt, P. Comba, Inorg. Chem. 1992, 31, 2638.
[41] P. Comba, Coord. Chem. Rev. 1993, 123, 1.
[42] P. Comba, R. Remenyi, J. Comput. Chem. 2002, 23, 697.
[43] P. Comba, R. Remenyi, Coord. Chem. Rev. 2003, 238–239, 9.
[44] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J.
R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H.
Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G.
Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J.
Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T.
Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J.
Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Nor-
mand, K. Raghavachari, A. Rendell, J. C. Burant, S. Iyengar, J. Tomasi,
M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V.
Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O.
Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin,
K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannen-
berg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J.
Cioslowski, D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc.: Wall-
ingford CT, 2009.
[45] F. H. Allen, Acta Cryst. 2002, B58, 380.
[46] R. S. Mulliken, J. Chem. Phys. 1955, 23, 1833.
[47] A. E. Reed, L. A. Curtiss, F. Weinhold, Chem. Rev. 1988, 88, 899.
[48] R. F. W. Bader, Atoms in Molecules—A Quantum Theory, Clarendon
Press, Oxford, 1990.
[49] F. L. Hirshfeld, Theoret. Chim. Acta 1977, 44, 129.
[50] C. M. Breneman, K. B. Wiberg, J. Comput. Chem. 1990, 11, 36.
[51] U. C. Singh, P. A. Kollman, J. Comput. Chem. 1984, 5, 129.
[52] C. I. Bayly, P. Cieplak, W. D. Cornell, P. A. Kollman, J. Phys. Chem. 1993,
97, 10269.
[53] J. Wang, P. Cieplak, P. A. Kollman, J. Comput. Chem. 2000, 21, 1049.
[54] Z. Xiong, Y. Liu, H. Sun, J. Phys. Chem. A. 2008, 112, 2469.
[55] C. F. Guerra, J.-W. Handgraaf, E. J. Baerends, F. M. Bickelhaupt, J. Com-
put. Chem. 2004, 25, 189.
[56] B. Courcot, A. J. Bridgeman, Int. J. Quantum. Chem. 2010, 110, 2155.
[57] P. Bultinck, C. V. Alsenoy, P. W. Ayers, R. Carbo-Dorca, J. Chem. Phys.
2007, 126, 144111.
[58] V. A. Rossolov, M. A. Ratner, J. A. Pople, P. C. Redfern, L. A. Curtiss, J.
Comput. Chem. 2001, 22, 976.
[59] A. Sch€afer, H. Horn, R. Ahlrichs, J. Chem. Phys. 1992, 97, 2571.
[60] A. Sch€afer, C. Huber, R. Ahlrichs, J. Chem. Phys. 1994, 100, 5829.
[61] T. Verstraelen, HiPart program, Available at: http://molmod.unigent.be/
software.
[62] C. M. Handley, R. J. Deeth, J. Chem. Theory Comput. 2012, 8, 194.
[63] A. Bentz, P. Comba, R. J. Deeth, M. Kerscher, H. Pritzkow, B. Seibold, H.
Wadepohl, Inorg. Chem. 2008, 47, 9518.
Received: 22 January 2013Revised: 18 March 2013Accepted: 22 March 2013Published online on
FULL PAPERWWW.C-CHEM.ORG
Journal of Computational Chemistry 2013, 00, 000–000 11





























