Embed Size (px)
Citation preview

Universidade de Lisboa
Faculdade de Medicina
Ana Rita Cascão Rodrigues
Doutoramento em Ciências Biomédicas
Especialidade em Imunologia
2012


Universidade de Lisboa
Faculdade de Medicina
Ana Rita Cascão Rodrigues
Tese orientada por:
Professor Doutor João Eurico Fonseca
Professor Doutor Luís Ferreira Moita
Doutoramento em Ciências Biomédicas
Especialidade em Imunologia
2012




Todas as afirmações efectuadas no
presente documento são da
exclusiva responsabilidade do seu
autor, não cabendo qualquer
responsabilidade à Faculdade de
Medicina da Universidade de
Lisboa pelos conteúdos nele
apresentados.


A impressão desta dissertação
foi aprovada pela Comissão
Coordenadora do Conselho
Científico da Faculdade de
Medicina da Universidade de
Lisboa em reunião de 15 de
Maio de 2012.


Dissertação apresentada à
Faculdade de Medicina da
Universidade de Lisboa, para
obtenção do grau de Doutor em
Ciências Biomédicas.


To Science
“The important thing in science is not so much to obtain new facts
as to discover new ways of thinking about them.”
Sir William Bragg (1862 - 1942)
Nobel Prize in Physics


XIV
Table of Contents
Index of Figures XVIII
Index of Tables XX
Acknowledgments XXII
List of Abbreviations XXIV
Sumário XXVI
Summary XXX
Chapter I Introduction -1-
1. Rheumatoid arthritis - 3 -
1.1 Definition - 3 -
1.2 Pathogenesis - 5 -
1.3 Immune cells - 8 -
1.3.1 Macrophages - 8 -
1.3.2 Neutrophils - 8 -
1.3.3 T cells - 9 -
1.3.4 B cells - 10 -
1.4 Cytokines - 10 -
1.4.1 IL-1β - 10 -
1.4.2 TNF - 12 -

XV
1.4.3 IL-6 - 13 -
1.4.4 IL-17 - 14 -
1.5 Treatment options - 14 -
1.5.1 Glucocorticoids - 15 -
1.5.2 Conventional DMARDs - 15 -
1.5.3 Biologic DMARDs - 15 -
2. IL-1β Immunological and inflammatory functions - 18 -
2.1 IL-1 family - 18 -
2.2 IL-1β pathway - 18 -
2.2.1 Priming - 18 -
2.2.2 Processing - 19 -
2.2.3 Secretion - 21 -
2.2.4 Signaling - 21 -
2.3 IL-1β and autoinflammatory diseases - 22 -
2.4 IL-1β and rheumatoid arthritis - 24 -
2.5 Targeting IL-1β in rheumatoid arthritis: the case of anakinra - 27 -
Chapter II Aims -31-
Chapter III Results -35-
Cytokine pattern in very early rheumatoid arthritis favours B cell activation and survival
- 39 -

XVI
Identification of a cytokine network sustaining neutrophil and Th17 activation in untreated
early rheumatoid arthritis - 41 -
Caspase-1 is active since the early phase of rheumatoid arthritis - 43 -
Effective treatment of rat adjuvant-induced arthritis by celastrol - 45 -
Gambogic acid is a potent anti-inflammatory and anti-proliferative drug in a rat model of
adjuvant-induced arthritis - 47 -
Chapter IV Discussion -49-
Chapter V Conclusions -67-
References -71-

XVII

XVIII
Index of Figures
Figure 1 – Representative scheme illustrating immune cells and cytokine networks in
rheumatoid joints. - 7 -
Figure 2 – An overview on the multiple roles of IL-1β in rheumatoid arthritis physiopathology.
- 12 -
Figure 3 – A simplified scheme showing main pathophysiologic pathways in rheumatoid arthritis
and the effects of some currently available biologic agents. - 17 -
Figure 4 – IL-1β processing pathways. - 19 -
Figure 5 – Proposed model for cytokine networks in the very early phase of rheumatoid arthritis.
- 54 -
Figure 6 – Proposed mechanism for excessive IL-1β production since early rheumatoid arthritis.
- 57 -
Figure 7 – Proposed mechanism for celastrol and gambogic acid anti-inflammatory effects.
- 62 -

XIX

XX
Index of Tables
Table 1 – The 2010 American College of Rheumatology / European League Against
Rheumatism classification criteria for rheumatoid arthritis. - 4 -
Table 2 – The 1987 American College of Rheumatology criteria for the classification of
rheumatoid arthritis. - 5 -

XXI

XXII
Acknowledgments
First and foremost, I would like to acknowledge both my supervisors, Professor
Doutor João Eurico Fonseca and Professor Doutor Luís Ferreira Moita, for having
accepted me as a PhD student, for all the scientific meetings and discussions, for all
the guidance and opportunities. Thank you both for all the support.
Financial support was generously granted by Sociedade Portuguesa de
Reumatologia, Pfizer and Fundação para a Ciência e a Tecnologia through the PhD
fellowship (SFRH/BD/40513/2007).
My thanks also go to my PhD thesis committee, Professor Doutor Luís Graça,
Professor Doutor Carlos São José and Doutora Margarida Carneiro, for all the
meetings, discussions and suggestions that improved the work. But most of all, thank
you for your unconditional support, motivation and trust.
I would like also to thank all the members from Rheumatology Department,
Hospital de Santa Maria, all the medical doctors and nurses, for the effort to collaborate
in the development of my work, especially in sample collection and in the organization
of clinical data. In particular, a thanking note to Enfª. Lurdes Narciso, Doutora Helena
Canhão, Drª. Ana Maria Rodrigues, Dr. Joaquim Polido Pereira, Drª. Elsa Sousa, Drª.
Cristina Ponte, Drª. Raquel Marques, Drª. Ana Filipa Mourão (Rheumatology
Department, Hospital Egas Moniz) and Dr. Márcio Navalho (Radiology Department,
Hospital da Luz) with whom I have worked closer.
I am also very grateful to all the patients and healthy volunteers for their
willingness in providing us with samples making this project possible.
I wish to acknowledge Doctor Vineet Gupta from the Department of Medicine,
University of Miami for giving us the library of drugs used in this work.
Many thanks to all my colleagues from Instituto de Medicina Molecular that have
directly worked with me, especially Rita Moura, Bruno Vidal, Ana Lopes, Helena
Raquel, Ana Neves Costa, Ana Luísa Caetano and Saba Abdulghani. This work greatly
benefited from the close collaboration and invaluable friendship over the years.
Many thanks also to all my friends for everyday support and friendship.
Finally, I would like to deeply thank my parents and Zé for their unconditional love
and guidance in life.
Thank you all for turning this challenging journey into an enjoyable one!
Obrigada!

XXIII

XXIV
List of Abbreviations
ACPA Anti-citrullinated protein antibodies
ACR American College of Rheumatology
AIA Adjuvant-induced arthritis
APRIL A proliferation-inducing ligand
ASC Adaptor molecule apoptosis associated speck-like protein containing a
caspase recruitment domain
BLyS B-lymphocyte stimulator
CAPS Cryopyrin-associated periodic fever syndromes
CARD Caspase recruitment domain
CINCA/NOMID Chronic infantile neurological cutaneous and articular syndrome/neonatal
onset multisystem inflammatory disease
DAMP Danger associated molecular pattern
DMARD Disease-modifying anti-rheumatic drug
EA Early arthritis
EAE Experimental autoimmune encephalomyelitis
ERA Early rheumatoid arthritis
EULAR European League Against Rheumatism
FCAS Familial cold-induced autoinflammatory syndrome
FcγR Fc gamma-receptor
HLA Human leukocyte antigen
IFNγ Interferon gamma
IL Interleukin
IL-1R IL-1 receptor
IL-1Ra IL-1 receptor antagonist
IL-1RAcP IL-1 receptor accessory protein
IL-1RI IL-1 receptor type I
IL-1RII IL-1 receptor type II
LRR Leucine rich repeat
MAP Mitogen activated protein
MCP-1 Monocyte chemotactic protein-1
MHC Major histocompatibility complex

XXV
MMP Metalloproteinase
MTX Methotrexate
MWS Muckel-Wells syndrome
MyD88 Myeloid differentiation primary response gene 88
NACHT Nucleotide-binding domain or NAIP, CIITA, HET-E and TP1
NF-kB Nuclear factor kappa-light-chain-enhancer of activated B cells
NLR NOD-like receptor
NOD Nucleotide-binding oligomerization domain
NSAID Non-steroid anti-inflammatory drug
PAMP Pathogen associated molecular pattern
PRR Pattern recognition receptor
PYD Pyrin domain
RANK Receptor activator of nuclear factor
RANKL RANK ligand
RF Rheumatoid factor
RORγ Retinoic acid receptor-related orphan receptor gamma
ROS Reactive oxygen species
VEA Very early arthritis
VEGF Vascular endothelial growth factor
VERA Very early rheumatoid arthritis
TCR T cell receptor
TfR1 Transferrin receptor 1
Th T helper
TIR Toll-IL-1 receptor domain
TLR Toll-like receptor
TNF Tumor necrosis factor

XXVI
Sumário
A artrite reumatóide é uma doença inflamatória crónica imunomediada, com
envolvimento predominante da membrana sinovial das pequenas articulações e
destruição progressiva de cartilagem e osso, que induz incapacidade funcional e
aumento da morbilidade e mortalidade dos doentes. O diagnóstico precoce da artrite
reumatóide e a administração imediata de uma terapêutica adequada são cruciais para
a prevenção da progressão da doença e da destruição articular, a qual pode ocorrer
numa fase muito inicial. A característica mais debilitante da artrite reumatóide é a
hiperplasia sinovial. Esta inflamação sinovial é mediada pela interação entre diversas
células do sistema imunitário (macrófagos, neutrófilos, células T e B) e complexas
redes de citocinas (particularmente interleucina (IL)-1β, fator de necrose tumoral (TNF)
e IL-17).
A IL-1β é considerada um dos elementos centrais na patologia da artrite
reumatóide, por induzir um aumento na secreção de citocinas e quimiocinas e
produção de metaloproteinases (MMPs) bem como expansão e sobrevivência de
células T, que por sua vez conduzem a um aumento na produção de autoanticorpos
pelas células B, à diferenciação de células T auxiliares (Th)17 e à osteoclastogénese,
com consequente erosão óssea. É importante salientar que a produção de IL-1β é
fortemente regulada quer a nível da transcrição, quer a nível da pós-tradução,
envolvendo o factor nuclear kappa B (NF-kB), o inflamassoma e a caspase-1. É
interessante notar que estudos anteriores documentaram que a desregulação destes
mecanismos resulta numa resposta inflamatória exacerbada e desnecessária,
característica de doenças autoinflamatórias e autoimunes, como é o caso da artrite
reumatóide. Estes mecanismos poderão ser particularmente relevantes na fase muito
inicial da doença. Contudo, a maioria dos estudos relativos à fisiopatologia da artrite
reumatóide têm sido focados na fase estabelecida da doença, sendo o conhecimento
acerca das interações celulares e dos mecanismos regulatórios imunológicos que
participam na fase inicial da artrite reumatóide ainda muito limitado.
O principal objectivo deste trabalho foi o estudo dos elementos iniciadores do
processo inflamatório na artrite reumatóide, incluindo aqueles que estão relacionados
com a atividade do inflamassoma. Assim, a primeira tarefa realizada neste estudo foi a
análise da libertação de citocinas e quimiocinas na fase muito inicial da artrite
reumatóide. Para tal, um grupo de doentes com poliartrite com menos de seis
semanas de evolução, não tratada, foi prospectivamente seguido. Estes doentes foram

XXVII
avaliados antes do tratamento, após terapêutica de curta duração com dose baixa de
corticosteróides e após metotrexato (MTX). O acompanhamento clínico destes
doentes permitiu a identificação de dois subgrupos: um que evoluiu para artrite
reumatóide e um outro que evoluiu para outras doenças crónicas inflamatórias
articulares ou que incluiu formas autolimitadas de artrite. Adicionalmente, foi analisado
um segundo grupo de doentes com artrite reumatóide na fase estabelecida da doença,
sob terapêutica com MTX. É interessante notar que, na fase muito inicial da artrite
reumatóide, foram observados no soro dos doentes níveis aumentados de citocinas
relacionadas com o recrutamento (IL-6), maturação e sobrevivência das células B (a
proliferation-inducing ligand (APRIL) e B-lymphocyte stimulator (BLyS)), bem como um
perfil de citocinas relacionadas com o recrutamento de neutrófilos (IL-8) e com a
polarização (IL-1β e IL-6) e ativação (IL-17A e IL-22) de células Th17,
comparativamente com doentes que não evoluem para artrite reumatóide e com
doentes com artrite reumatóide estabelecida. Além disso, a análise de amostras de
líquido sinovial de doentes tratados com MTX na fase estabelecida da artrite
reumatóide revelou também um perfil de citocinas semelhante ao da fase muito inicial
desta doença. De salientar que o tratamento com corticosteróides e MTX, embora
clinicamente eficaz na redução das manifestações inflamatórias, não pareceu afetar o
padrão de citocinas em circulação.
A elevada concentração sérica e sinovial de IL-1β observada desde a fase muito
inicial da artrite reumatóide pode ser explicada pelo aumento nos níveis de caspase-1
ativada que foi também observado, quer em doentes com artrite reumatóide com
menos de 1 ano de evolução e não tratados, quer nos doentes com artrite reumatóide
estabelecida, tratados com MTX. O papel central da IL-1β faria supor que esta citocina
seria um alvo ideal na terapêutica da artrite reumatóide. Contudo, os resultados dos
ensaios clínicos em doentes com artrite reumatóide estabelecida com um bloqueador
do recetor da IL-1β (anakinra) e com um anticorpo monoclonal (canakinumab) não
demonstraram o mesmo nível de eficácia dos antagonistas do TNF. Colocámos a
hipótese de que a IL-1β desempenhe um papel complementar ao do TNF e mais
relevante na fase inicial da doença. Por este motivo, a interferência na fase inicial da
artrite com a via reguladora da libertação de IL-1β e de TNF poderia constituir uma
opção terapêutica promissora. Para testar esta hipótese, usou-se uma biblioteca de
drogas na linha celular macrofágica THP1 no sentido de identificar as drogas que
simultaneamente diminuam a produção de ambas as citocinas. As drogas
selecionadas foram posteriormente estudadas in vivo num modelo de rato Wistar de
artrite induzida por adjuvante para analisar as suas propriedades anti-inflamatórias.

XXVIII
Nestas experiências in vivo foi observado que duas das drogas selecionadas, o
celastrol e o ácido gambógico, têm propriedades anti-inflamatórias e anti-proliferativas,
tratando eficazmente os ratos artríticos devido ao seu efeito inibitório sobre a IL-1β e o
TNF, induzido pela diminuição da ativação quer da caspase-1, quer do NF-kB. Por fim,
com o intuito de distinguir entre um papel direto da IL-1β como elemento efetor ou
como iniciador do processo inflamatório, pela indução da diferenciação das células
Th17, foi também testada in vivo uma droga inibitória da atividade transcricional do
retinoic acid receptor-related orphan receptor gamma (RORγ)T (digoxina) no mesmo
modelo animal, permitindo a análise do seu efeito anti-inflamatório. Após o tratamento
com a digoxina foi observada uma melhoria nos sinais inflamatórios dos ratos artríticos
quando a droga foi administrada na fase inicial do desenvolvimento da artrite, tendo-se
verificado um efeito menos rápido e menos eficiente na progressão da doença
comparativamente com as drogas acima mencionadas. Contrariamente à digoxina, o
celastrol e o ácido gambógico são também eficientes quando administrados em fases
tardias do desenvolvimento da artrite, sendo este resultado muito importante no
contexto da prática clínica. Estes resultados sugerem que a inibição da polarização
das células Th17 per se parece ser uma estratégia com uma eficácia limitada, pelo
menos no que diz respeito à fase estabelecida da doença.
No seu conjunto, os resultados da presente tese suportam a hipótese de que a IL-
1β desempenha um papel mais relevante na fase inicial do que na fase tardia da artrite
reumatóide. Estes novos dados têm assim uma implicação clínica importante, ao
sugerirem que a introdução precoce de terapêuticas direcionadas às vias reguladoras
de IL-1β poderão ser particularmente eficazes na indução da remissão na fase inicial
da doença, devendo esta estratégia ser avaliada em doentes com artrite reumatóide.
Palavras-chave: Artrite reumatóide, Inflamassoma, Caspase-1, NF-kB, IL-1β, TNF, IL-
17A, Modelo de rato AIA, Drogas

XXIX

XXX
Summary
Rheumatoid arthritis is a chronic inflammatory and immuno-mediated disease with
a significant involvement of the synovial membrane of multiple small joints and
progressive destruction of cartilage and bone, which leads to functional impairment and
a significant increase of both morbidity and mortality of patients. Early diagnosis and an
accurate prompt therapeutic strategy are crucial to prevent rheumatoid arthritis
progression and joint destruction that can occur immediately after its onset. The most
debilitating characteristic of rheumatoid arthritis is the synovial hyperplasia of joints,
which is mediated by the interplay of several immune cells (macrophages, neutrophils,
T and B cells) and complex cytokine networks (particularly interleukin (IL)-1β, tumor
necrosis factor (TNF) and IL-17).
IL-1β is considered to be a crucial element in rheumatoid arthritis pathology due to
its ability to enhance cytokine and chemokine secretion, metalloproteinases (MMPs)
production, T cell expansion and survival which further increases antibody production
by B cells, T helper (Th)17 cells differentiation, and osteoclastogenesis with
consequent bone loss. Importantly, the production of IL-1β is tightly regulated both at
the transcriptional and post-translational levels, involving nuclear factor kappa-light-
chain-enhancer of activated B cells (NF-kB), inflammasomes and caspase-1.
Interestingly, it has been shown that perturbation of these regulatory mechanisms
results in an exaggerated inflammatory response that underlies autoinflammatory and
autoimmune diseases, such as rheumatoid arthritis. These mechanisms might be
particularly relevant in the early phase of the disease. Nevertheless, the majority of the
studies concerning rheumatoid arthritis physiopathology have focused on the
established phase of this disease, while the knowledge regarding the cellular
interactions and the immunologic regulatory cascades involved in its onset are still
scarce.
The main goal of this work was to study the early triggers of the inflammatory
process in rheumatoid arthritis, including those involved in inflammasome activation.
Accordingly, the first task in this study was the analysis of the early cytokine and
chemokine environment in the early phase of rheumatoid arthritis. To that end, a cohort
of untreated polyarthritis patients with less than six weeks of disease duration was
prospectively followed up. These patients were evaluated at baseline (without
treatment) and after short-term therapy with low dose of corticosteroids and
methotrexate (MTX). The clinical follow up of these patients allowed the identification of
two subgroups of patients: one that evolved into rheumatoid arthritis and another which

XXXI
evolved into other chronic inflammatory joint diseases or had self-limited forms of
arthritis. Additionally, a second cohort of rheumatoid arthritis patients with established
disease and under MTX therapy was also analyzed. Interestingly, we found that in the
very early phase of rheumatoid arthritis there are increased levels of circulating
cytokines related with the recruitment (IL-6), maturation and survival (a proliferation-
inducing ligand (APRIL) and B-lymphocyte stimulator (BLyS)) of B cells, as well as a
cytokine profile that supports neutrophil recruitment (IL-8) and Th17 cells polarization
(IL-1β and IL-6) and activation (IL-17A and IL-22) when compared with other very early
chronic inflammatory joint diseases and with the established phase of the disease.
Furthermore, the analysis of synovial fluid samples from established rheumatoid
arthritis patients treated with MTX also revealed a pattern of cytokines similar to that of
the very early phase of the disease. Of note, treatment with corticosteroids and MTX,
although clinically effective in reducing inflammatory manifestations, did not seem to
affect the cytokine content in circulation.
The increased concentration of IL-1β observed since the early phase of the
disease both in peripheral blood and in the synovial fluid may be explained by the
increased levels of active caspase-1 that were also observed both in early untreated
rheumatoid arthritis patients with less than 1 year of disease evolution and in
established rheumatoid arthritis patients treated with MTX. The crucial role of IL-1β
suggests that this cytokine could be a promising target for rheumatoid arthritis
treatment. However, clinical trials with IL-1 receptor targeting (anakinra) and with IL-1
monoclonal antibody (canakinumab) have not revealed encouraging results in the
established phase of rheumatoid arthritis compared with TNF antagonists. Therefore,
we raised the hypothesis that IL-1β plays a complementary role to that of TNF and
more relevant in the early stage of the disease rather than in the established phase.
Consequently, interference, during the early phase of arthritis, with pathways regulating
both IL-1β and TNF could constitute a promising therapeutic option. To test this
hypothesis, a drug screen was performed in a THP1 macrophage-like cell line in order
to identify those that simultaneously down-regulate the production of both cytokines.
The drugs that were selected were further studied in vivo in a wistar rat model of
adjuvant-induced arthritis to analyze their potential anti-inflammatory properties. The in
vivo experiments revealed that two of the selected drugs, celastrol and gambogic acid,
have anti-inflammatory and anti-proliferative properties and are able to effectively treat
arthritic rats, by an inhibitory effect of IL-1β and TNF, induced by a down-regulation of
caspase-1 and NF-kB activation. Finally, to distinguish between the direct role for IL-1β
as an effector or as an upstream initiator, by driving the differentiation of Th17 cells in

XXXII
the inflammatory process, a drug which inhibits retinoic acid receptor-related orphan
receptor gamma (RORγ)T transcriptional activity (digoxin) was also tested in vivo in the
same animal model in order to observe its anti-inflammatory effects. We found that
digoxin was able to ameliorate the inflammatory signs of the arthritic rats only if
administered in the early phase of arthritis development, albeit with a slower and less
efficient effect on disease progression compared with the drugs above mentioned. In
contrast to digoxin, celastrol and gambogic acid are also effective if administered in the
later phase of arthritis development, which is crucial for rheumatoid arthritis
management. These results suggest that the isolated inhibition of Th17 cells
polarization might be a strategy with limited efficacy, at least in the established phase
of the disease.
Overall, the results of the present thesis support the hypothesis that IL-1β plays a
more relevant role in the early rather than late phase of rheumatoid arthritis. Thus,
these original results have important clinical implications, by suggesting that an earlier
intervention of therapies targeting IL-1β pathways might be of beneficial clinical use to
induce early remission and this strategy should be evaluated in rheumatoid arthritis
patients.
Key-words: Rheumatoid arthritis, Inflammasome, Caspase-1, NF-kB, IL-1β, TNF, IL-
17A, AIA rat model, Drugs

XXXIII

Chapter I
Introduction

- 2 -

Introduction
- 3 -
1. Rheumatoid arthritis
1.1 Definition
Rheumatoid arthritis is a chronic, systemic, autoimmune inflammatory disease that
mainly affects the synovium of multiple joints, leading to the progressive destruction of
cartilage and bone. This disease is often associated with other co-morbidities, such as
cardiovascular diseases, infections and cancer 1, 2. Rheumatoid arthritis causes
considerable personal and economic costs as it is associated with serious disability,
reduced quality of life, loss of work capacity 3 and diminished life expectancy 4.
Epidemiological studies have shown that rheumatoid arthritis affects about 1% of the
worldwide population and 75% of those afflicted are women 5, suggesting an influence
of hormonal factors in the occurrence of the disease. Moreover, its incidence peaks
among those aged between 30 and 50 years old and because it is a chronic condition,
its prevalence increases with age.
Importantly, the clinical presentation of rheumatoid arthritis can be elusive and
insidious and therefore several months can pass before the clinician makes a definitive
diagnosis. The predominant symptoms are symmetrical pain, stiffness and swelling of
peripheral joints. The clinical course of the disease is also variable, ranging from mild
to severe and highly destructive arthritis. The analysis of the clinical course and of
laboratorial and radiological parameters have defined prognostic factors related to
progressive joint destruction, such as the presence of rheumatoid factor (RF) and anti-
citrullinated protein antibodies (ACPA) as well as the early development of joint
erosions. Such prognostic factors can influence treatment decisions, particularly in the
early phase of the disease. However, they do not drive treatment decisions by
themselves. In fact, tight disease control and treatment driven by a predefined disease
activity target are crucial for an effective long-term management of rheumatoid arthritis.
A prompt diagnosis and an accurate early therapeutic strategy are essential to prevent
rheumatoid arthritis progression and joint erosions, which can occur as early as 4
months after the onset of the disease 6, 7, highlighting the importance of diagnosing
rheumatoid arthritis in the earliest possible phase of its course. To contribute to that
important goal, the American College of Rheumatology and the European League
Against Rheumatism (ACR/EULAR) published the reviewed Rheumatoid Arthritis
Classification Criteria in 2010, which refocuses on the number of involved joints,
serologic abnormalities, elevated acute-phase proteins and symptoms duration (Table
1) 8.

Introduction
- 4 -
Table 1 – The 2010 American College of Rheumatology / European League Against Rheumatism classification criteria for rheumatoid arthritis. * The criteria are aimed at classification of newly
presenting patients; ** Differential diagnoses vary among patients with different presentations. If it is unclear about the relevant differential diagnoses to consider, an expert rheumatologist should be consulted; ‡ Although patients with a score of < 6/10 are not classifiable as having rheumatoid arthritis, their status can be reassessed and the criteria might be fulfilled cumulatively over time; § Joint involvement refers to any swollen or tender joint on examination, which may be confirmed by imaging evidence of synovitis; # “Large joints” refers to shoulders, elbows, hips, knees, and ankles; ## “Small joints” refers to the metacarpophalangeal joints, proximal interphalangeal joints, second through fifth metatarsophalangeal joints, thumb interphalangeal joints and wrists; †† Negative refers to international unit (IU) values that are less than or equal to the upper limit of normal (ULN) for the laboratory and assay; low-positive refers to IU values that are higher than the ULN but ≤ 3 times the ULN for the laboratory and assay; high-positive refers to IU values that are > 3 times the ULN for the laboratory and assay. Where rheumatoid factor (RF) information is only available as positive or negative, a positive result should be scored as low-positive for RF. ACPA – anti-citrullinated protein antibody; ‡‡ Normal/abnormal is determined by local laboratory standards. CRP – C-reactive protein, ESR – erythrocyte sedimentation rate; §§ Duration of symptoms refers to patient self-report of the duration of signs or symptoms of synovitis (e.g. pain, swelling, tenderness) of joints that are clinically involved at the time of assessment, regardless of treatment status.
However, since this new revision occurred after the completion of the experimental
procedures of this thesis, the criteria used for rheumatoid arthritis classification were
the 1987 ACR criteria (Table 2) 9.

Introduction
- 5 -
Table 2 – The 1987 American College of Rheumatology criteria for the classification of rheumatoid arthritis. A patient is said to have rheumatoid arthritis if he or she meets at least four criteria. Patients with
two clinical diagnoses are not excluded. Designation as classic, definite, or probable rheumatoid arthritis is not to be made.
1.2 Pathogenesis
The etiopathogenesis of rheumatoid arthritis is not yet well understood. The
current lack of knowledge is partially due to the fact that rheumatoid arthritis is a
complex multifactorial process, which involves an interplay among genotype,
environmental triggers and chance. Among the genetic factors, the strongest
association with the occurrence of the disease has been established with the major
histocompatibility complex (MHC) class II human leukocyte antigen (HLA)-DRB1*0404
and DRB1*0401 alleles 10. Specifically, in the Portuguese population and in some other
Mediterranean populations, an association with the DRB1*1001 allele was also found
11. HLA-DRB1 alleles are involved in MHC molecule-based antigen presentation and
are responsible for self-peptide selection and T cell repertoire. As T cell specificity is
mediated by an interaction between the T cell receptor (TCR) and the peptide
presented by MHC molecules, it has been suggested that rheumatoid arthritis is
caused by the presentation of an unidentified arthritogenic antigen 12, either exogenous
or endogenous 13. Smoking is the most established environmental risk factor for
rheumatoid arthritis 14. Several studies have shown that smoking is a risk factor for the
presence of autoantibodies, RFs or ACPAs 15, which are predictor factors for a severe
form of rheumatoid arthritis. Interestingly, the observation that autoantibodies may be

Introduction
- 6 -
present for several years before the clinical onset of rheumatoid arthritis suggests that
these risk factors are acting several years before the disease development. Findings
from studies of gene-environmental factors interaction showed that smoking increases
the risk of rheumatoid arthritis in individuals with susceptible HLA-DRB1 alleles 16.
Moreover, smoking and HLA-DRB1 alleles synergistically increase the risk of having
ACPA 17. Therefore, the prevailing view is that gene-environment interactions may lead
to altered post-translational modifications, such as citrullination, which consequently
promote a loss of tolerance to these altered self-proteins. However, there has been no
consistent evidence that a single environmental factor could explain per se the onset of
this disease.
The most debilitating feature of rheumatoid arthritis is joint destruction, which is
derived from an uncontrolled inflammatory process. Inflammation is usually tightly
regulated, involving both the mediators which initiate and stop it. In the case of chronic
inflammation, such as rheumatoid arthritis, there is an unbalance between the two
opposing groups of mediators resulting in the destruction of cartilage and bone.
The articular cartilage is composed of specialized cells, the chondrocytes,
embedded in a structured extracellular matrix of proteoglycans and collagen type II.
The destruction of cartilage is a consequence of two major events: altered synthesis of
matrix components by chondrocytes and increased enzymatic degradation of the
matrix, with both mechanisms being influenced by the ongoing inflammatory process.
In addition, the formation of a highly invasive synovial tissue, the pannus, leads to
further cartilage destruction and bone erosion. The pannus evolves throughout the
course of the disease. Initially, it is characterized by the presence of activated
mononuclear cells and fibroblasts 18 and increased production of metalloproteinases
(MMPs) by synovial-lining cells 19, 20. Subsequently, the cellular pannus can be
replaced by fibrous pannus composed of a residual vascularized layer of pannus cells
and collagen overlying cartilage 21. Synovial fibroblasts, together with macrophages,
also promote bone erosion by the induction of osteoclastogenesis 22. Another
characteristic feature of rheumatoid arthritis is the formation of an inflamed synovium,
which is characterized by cellular hyperplasia, influx of inflammatory leukocytes,
increased angiogenesis and alterations in the expression of adhesion molecules,
proteinases, proteinase inhibitors and cytokines. Furthermore, the characteristics of the
inflamed synovium also vary with disease progression. During the first weeks of the
disease onset, there is a neutrophil influx, tissue oedema and fibrin deposition 23. Next,
the synovial lining becomes hyperplastic as it consists of macrophage-like and
fibroblast-like synoviocytes, and finally in the sublining layer a pronounced infiltration of
Introduction
- 7 -
mononuclear cells occurs, namely macrophages, neutrophils, dendritic cells, T cells, B
cells and plasma cells. Importantly, synovial-vessel endothelial cells are transformed
into high endothelial venules early in the course of the disease, thus promoting the
infiltration of inflammatory leukocytes 24 and facilitating the oxygenation of this hypoxic
environment 25. Interestingly, several innate immune sensing pathways are found in
rheumatoid arthritis joints, namely Toll-like receptors (TLRs), nucleotide-binding
oligomerization domain (NOD)-like receptors (NLRs) and molecular components of the
inflammasome promoting the detection of tissue damage signals 26, 27. In conclusion,
the synovial inflammation characteristic of rheumatoid arthritis is mediated by the
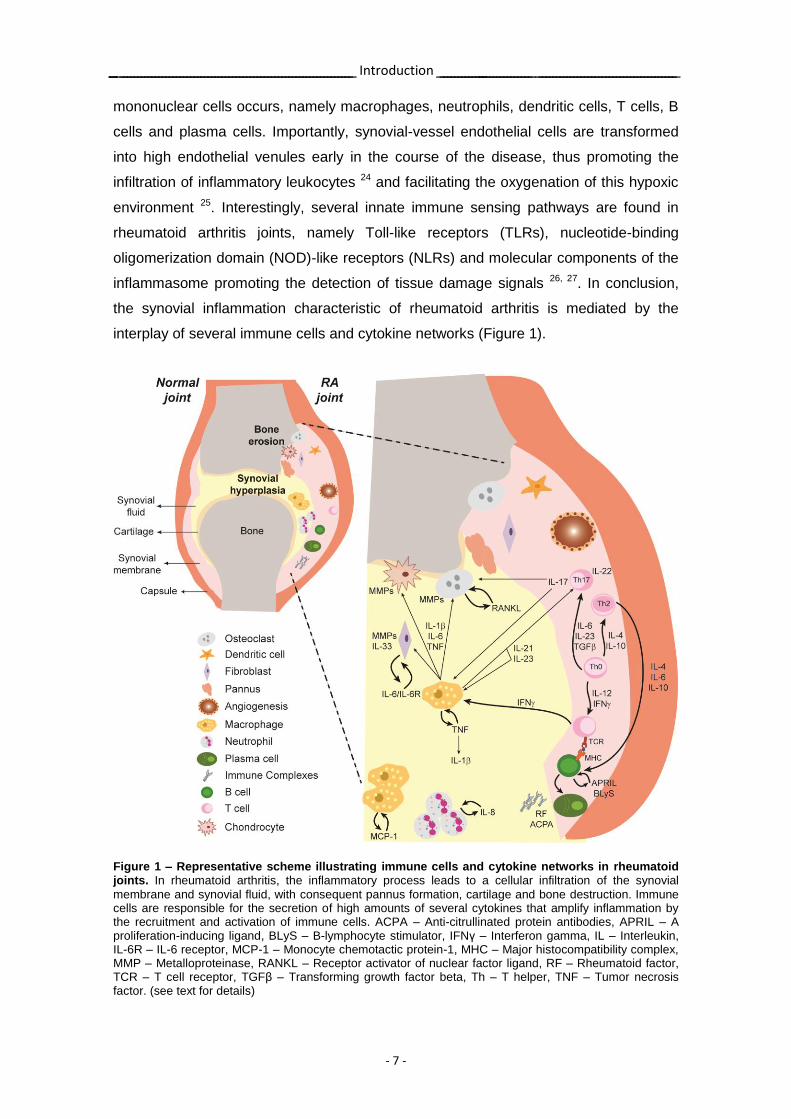
interplay of several immune cells and cytokine networks (Figure 1).
Figure 1 – Representative scheme illustrating immune cells and cytokine networks in rheumatoid joints. In rheumatoid arthritis, the inflammatory process leads to a cellular infiltration of the synovial
membrane and synovial fluid, with consequent pannus formation, cartilage and bone destruction. Immune cells are responsible for the secretion of high amounts of several cytokines that amplify inflammation by the recruitment and activation of immune cells. ACPA – Anti-citrullinated protein antibodies, APRIL – A proliferation-inducing ligand, BLyS – B-lymphocyte stimulator, IFNγ – Interferon gamma, IL – Interleukin, IL-6R – IL-6 receptor, MCP-1 – Monocyte chemotactic protein-1, MHC – Major histocompatibility complex, MMP – Metalloproteinase, RANKL – Receptor activator of nuclear factor ligand, RF – Rheumatoid factor, TCR – T cell receptor, TGFβ – Transforming growth factor beta, Th – T helper, TNF – Tumor necrosis factor. (see text for details)

Introduction
- 8 -
1.3 Immune cells
Rheumatoid arthritis pathophysiology is characterized by the presence of several
different interacting immune cells both from the innate immune system, such as
macrophages and neutrophils, as well as from the adaptive immune system, namely T
and B cells.
1.3.1 Macrophages
Macrophages are pivotal cells in the pathophysiology of rheumatoid arthritis and
one of the most abundant cell types in the inflamed synovium. These cells migrate as
monocytes from peripheral blood, infiltrate and accumulate in the synovium.
Macrophages promote the inflammatory process and joint destruction through the
production of several proinflammatory cytokines, such as interleukin (IL)-1β and tumor
necrosis factor (TNF), and through osteoclastogenesis 28. Crucial to the mechanism of
osteoclastogenesis is the differentiation of monocytes into bone-resorbing osteoclasts,
a process mediated by the binding of their receptor activator of nuclear factor (RANK)
with its ligand (RANKL) expressed by osteoblasts and some immune cells.
In the synovium, the activation of macrophages can occur by T and B cells,
through direct cell-cell contact or indirectly by the production of cytokines 28-30.
Moreover, macrophages can also be activated by the immune complexes that deposit
in the joints through the low-affinity IgG receptor Fc gamma receptor (FcγR) IIIa 31.
Activation of macrophages is also driven by TLRs and NLRs that recognize a wide
range of pathogen associated molecular pattern (PAMP) and danger associated
molecular pattern (DAMP) 32.
1.3.2 Neutrophils
Neutrophils are the first to infiltrate the synovium 33. In fact, the synovial tissue of
very early rheumatoid arthritis patients can be heavily infiltrated by neutrophils 23.
Moreover, circulating neutrophils from these patients have decreased levels of
spontaneous apoptosis 34. Synovial neutrophils internalize immune complexes that are
present in the joints and become activated. The activated neutrophils release
damaging proteases and MMPs, in addition to producing high amounts of reactive
oxygen species (ROS), thus contributing to chronic inflammation and host tissue
damage 33. Furthermore, neutrophils secrete high amounts of cytokines and

Introduction
- 9 -
chemokines, such as IL-1β, IL-8, TNF and B-lymphocyte stimulator (BLyS), which are
crucial to amplify inflammation by recruiting and activating more neutrophils and other
immune cells such as macrophages, T and B cells 35-37. The delay observed in the
apoptosis of neutrophils after rheumatoid arthritis onset can be the result of the action
of proinflammatory cytokines, such as TNF, and may create the appropriate conditions
for an inflammatory vicious cycle that might contribute to the self-perpetuation of an
initial acute arthritis episode in these patients 34.
1.3.3 T cells
The participation of T cells in the etiopathogenesis of rheumatoid arthritis has been
supported by several studies 38-43. In fact, the genetic evidence that HLA-DRB1 alleles
contribute to rheumatoid arthritis susceptibility suggests a role for T cells in the
systemic initiation of the inflammatory process. Moreover, T cells can infiltrate the
inflamed synovium and organize themselves into aggregates similar to germinal centre-
like structures 38. However, there is still some controversy regarding their precise role.
Some studies have shown that T cells do not directly cause synovitis in the joint
microenvironment by demonstrating that synovial infiltrating T cells do not show
significant proliferation 44, that the phenotype of these cells suggests a recruitment of
previously stimulated and mature T cells 45, 46, as opposed to an in situ maturation as
previously thought, and also that the synovial cytokine environment has a small
contribution of T cell-derived cytokines 47. Therefore, although T cells probably play a
part in the systemic initiation of rheumatoid arthritis, their direct role in synovitis and
joint destruction is still unclear.
Classically, rheumatoid arthritis has long been classified as a T helper (Th)1 driven
disease 41. However, recent data have pointed out that the Th1 contribution was
relatively small in the global context of rheumatoid arthritis 48. More recently, this Th1
paradigm was redefined by the increasingly substantiated relevance of the IL-17-
secreting Th17 cells subpopulation in the context of this disease. A recent report has
demonstrated that self-reactive T cells stimulate antigen-presenting cells to secrete IL-
6, which in turn is able to induce the differentiation of naïve self-reactive T cells into
Th17 cells 49, via nuclear transcription factor retinoic acid receptor-related orphan
receptor gamma (RORγ)T. Other cytokines also participate in the differentiation of
Th17 cells such as IL-1β and IL-21. In fact, macrophage-derived and dendritic cell-
derived cytokines provide a milieu that supports Th17 differentiation and suppress
regulatory T cells polarization, thus shifting T cell homeostasis towards inflammation.

Introduction
- 10 -
Furthermore, Th17 cells produce IL-17, a cytokine involved in increased production of
other cytokines, cartilage-destructive enzymes and also expression of bone
destruction-related mediators such as RANKL 50, 51.
1.3.4 B cells
Several reports have also highlighted the participation of B cells in the
etiopathogenesis of rheumatoid arthritis 52-57. These cells can contribute to disease
onset and perpetuation through their ability to function as antigen-presenting cells to
secrete cytokines and to produce autoantibodies, such as RF and ACPA, leading to
immune complex formation, which can occur even before the onset of disease
symptoms 52. These phenomena might be explained by a deficient removal of
autoreactive B cells and an abnormal B cell tolerance checkpoint in the bone marrow
that occur in rheumatoid arthritis patients 53. Interestingly, data have shown that
peripheral B cells from rheumatoid arthritis patients have altered expression of key
molecules, such as high co-stimulatory molecule CD86 and low FcγRIIb (inhibitory
receptor for IgG immune complexes required for feedback inhibition), potentially
contributing to tolerance breakdown and development of humoral autoimmunity 58.
1.4 Cytokines
Several cytokines have also been implicated in the pathophysiology of rheumatoid
arthritis. However, it is still difficult to clearly establish their functional hierarchy and
regulatory networks. Specifically, IL-1β, TNF, IL-6 and more recently IL-17 have been
identified as key players in established rheumatoid arthritis.
1.4.1 IL-1β
IL-1β is a pleiotropic proinflammatory cytokine produced mainly by macrophages
that is crucial for the joint pathology and destruction seen in rheumatoid arthritis 51, 59.
Binding of IL-1β to its receptor leads to activation of the mitogen activated protein
(MAP) kinase pathway and the nuclear factor kappa-light-chain-enhancer of activated
B cells (NF-kB) pathway. Both pathways lead to the downstream secretion of several
cytokines, chemokines and MMPs, which are responsible for the inflammatory process
and cartilage destruction 59-61. In addition, IL-1β is able to activate several cell types,

Introduction
- 11 -
such as macrophages, T and B cells, fibroblasts, osteoblasts, endothelial and epithelial
cells (Figure 2). IL-1β has been known for a long time to promote T cell responses. For
example, IL-1β derived from antigen-presenting cells acts directly on both naïve and
memory T cells to enhance their expansion and survival 62. Moreover, increased T cell
numbers due to IL-1β also result in an enhanced help for antibody production by B cells
62, thus contributing to rheumatoid arthritis perpetuation. Additionally, recent findings
have also identified a key role for IL-1β in the differentiation of Th17 cells 63, 64, which is
an important T cell subset in rheumatoid arthritis physiopathology. Furthermore, IL-1β
together with TNF have been implicated in bone loss by the induction of RANKL
expression contributing to osteoclast differentiation via RANK/RANKL interaction 65 66.
Therefore, IL-1β can be viewed not only as a regulator of innate immune cells but also
as a major amplifier of adaptive immune cells.
Importantly, IL-1β is present in high levels in the synovial fluid and tissue of
rheumatoid arthritis patients. In fact, some studies suggest that the synovial membrane
mRNA levels of IL-1β, TNF and IL-17 are predictors of damage progression 67. In
addition, animal models have demonstrated that targeting of IL-1β and components of
its receptor is effective in reducing inflammation, particularly in articular cartilage and
bone damage 68, 69. Of note, when transgenic mice expressing human TNF were
crossed with IL-1β deficient mice, inflammation still occurred but bone erosions were
dramatically reduced 70. Additional studies performed in TNF transgenic mice have also
shown that treatment with IL-1 receptor (IL-1R) antagonist fully abolished arthritis even
with confirmed high TNF levels, arguing for a possible dominant role for IL-1β 71, 72.
Introduction
- 12 -
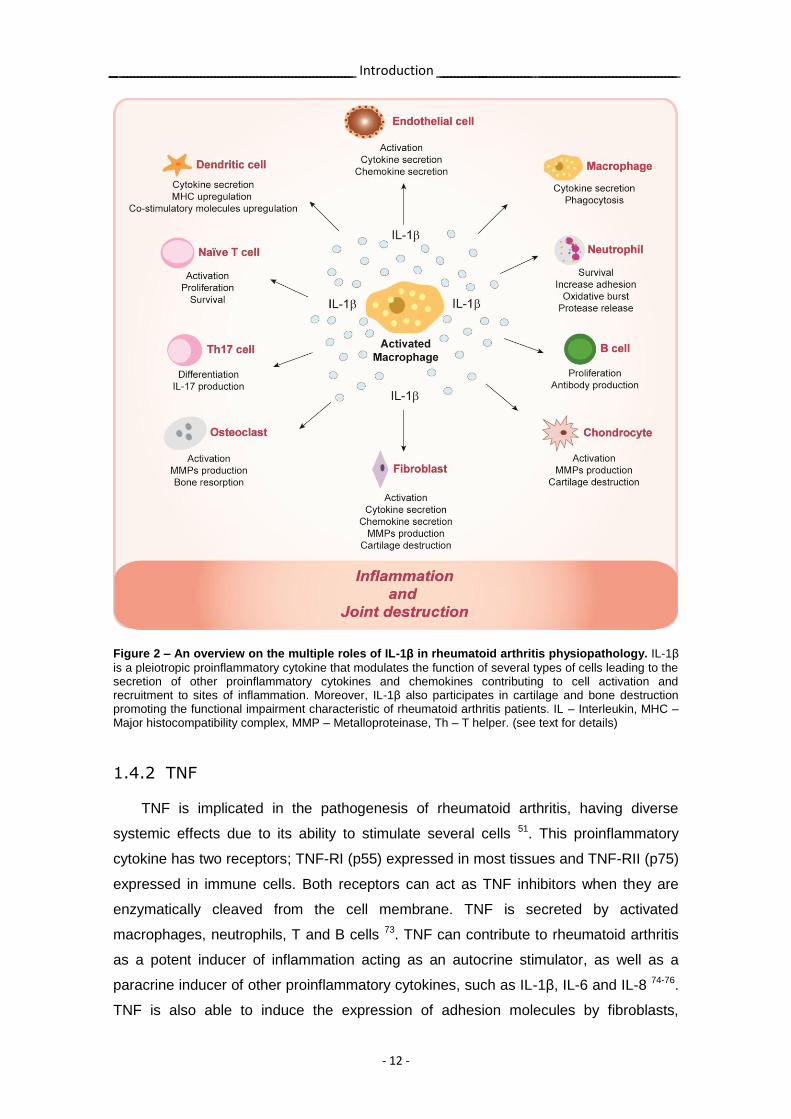
Figure 2 – An overview on the multiple roles of IL-1β in rheumatoid arthritis physiopathology. IL-1β
is a pleiotropic proinflammatory cytokine that modulates the function of several types of cells leading to the secretion of other proinflammatory cytokines and chemokines contributing to cell activation and recruitment to sites of inflammation. Moreover, IL-1β also participates in cartilage and bone destruction promoting the functional impairment characteristic of rheumatoid arthritis patients. IL – Interleukin, MHC – Major histocompatibility complex, MMP – Metalloproteinase, Th – T helper. (see text for details)
1.4.2 TNF
TNF is implicated in the pathogenesis of rheumatoid arthritis, having diverse
systemic effects due to its ability to stimulate several cells 51. This proinflammatory
cytokine has two receptors; TNF-RI (p55) expressed in most tissues and TNF-RII (p75)
expressed in immune cells. Both receptors can act as TNF inhibitors when they are
enzymatically cleaved from the cell membrane. TNF is secreted by activated
macrophages, neutrophils, T and B cells 73. TNF can contribute to rheumatoid arthritis
as a potent inducer of inflammation acting as an autocrine stimulator, as well as a
paracrine inducer of other proinflammatory cytokines, such as IL-1β, IL-6 and IL-8 74-76.
TNF is also able to induce the expression of adhesion molecules by fibroblasts,

Introduction
- 13 -
facilitating the infiltration of leukocytes into the joints of rheumatoid arthritis patients 77.
Furthermore, in vitro studies have already shown that TNF is able to degrade cartilage
78 and bone 79, thus contributing to the debilitating loss of articular joint function.
The serum and synovial fluid concentration of TNF is increased in rheumatoid
arthritis patients with active disease 80. It was demonstrated through in vitro studies that
cultures of rheumatoid arthritis synovial mononuclear cells are able to spontaneously
and chronically produce TNF and other proinflammatory cytokines such as IL-1β, IL-6
and IL-8 81, 82. Transgenic mice expressing a modified human TNFA gene
spontaneously develop peripheral arthritis and show a significant improvement after
administration of antibodies specific for human TNF 71. Of note, animal studies have
also revealed that blocking TNF is more effective in early stages of arthritis, in contrast
to IL-1β, for which the treatment outcome was equally effective in early and established
arthritis 69, 83.
1.4.3 IL-6
IL-6 is a multifactorial cytokine that also plays a relevant role in the pathogenesis
of rheumatoid arthritis. This cytokine is produced by monocytes, macrophages, T cells
and synovial fibroblasts 84. IL-6 possesses diverse biological activities, which include
the regulation of immune responses, inflammation, hematopoiesis, hypothalamus-
pituitary-adrenal gland axis activity, and the increase of the adrenocorticotropic
hormone and cortisol production. The proinflammatory properties of IL-6 comprise the
induction of acute-phase proteins in liver cells 85, the increase of neutrophil blood
counts 86, the participation in Th17 polarization 49 and plasma B cells differentiation 87,
88, the increase in the production of chemokines, adhesion molecules 89, and vascular
endothelial growth factor (VEGF), which is thought to be the most important angiogenic
factor in rheumatoid arthritis 90, as well as the induction of RANKL expression 91 and
the production of MMPs and their tissue inhibitors 92-94. These evidences support the
concept that IL-6 aggravates the local inflammatory reaction by amplifying inflammatory
cell infiltration and by inducing osteoclastogenesis and extracellular matrix turnover.
The serum and synovial fluid concentration of IL-6 is increased in rheumatoid
arthritis patients and correlate to disease activity 95, 96. Synovial fibroblastic cells
produce augmented levels of IL-6 upon stimulation with inflammatory cytokines, such
as IL-1β, TNF and IL-17. Subsequently IL-6 is able by itself to increase the proliferation
of these synovial fibroblastic cells 91, 97. The recently shown effective treatment of
rheumatoid arthritis patients with the humanized anti-IL-6 receptor antibody

Introduction
- 14 -
(tocilizumab) 98, 99, which binds to both the soluble and the membrane-bound IL-6
receptors 100, reinforces the important role of IL-6 in the context of this disease. In fact,
it has already been observed that tocilizumab treatment not only significantly improves
inflammation but also halts radiographic progression 101 in rheumatoid arthritis
patients 102.
1.4.4 IL-17
The IL-17 family is composed of 6 members (IL-17A to F) with distinct biological
roles. IL-17A (IL-17) is however the most studied one and has long been implicated in
several autoimmune diseases. IL-17, produced by Th17 cells, is an important cytokine
in the physiopathology of rheumatoid arthritis, mainly due to its participation in joint
destruction 103, 104. This cytokine promotes the activation of diverse cells, such as
macrophages, synovial fibroblasts, chondrocytes and osteoblasts, and also induces the
secretion of proinflammatory cytokines such as IL-1β, TNF, IL-6 and IL-23, which
amplify positive feedback loops leading to Th17 cells differentiation 105-107. IL-17 also
stimulates the production of chemokines responsible for the recruitment of
macrophages, neutrophils, T and B cells to the arthritic joints 108. Additionally, it has
been shown that IL-17 can synergize with IL-1β and TNF promoting cartilage and bone
damage 109. In fact, this cytokine induces MMPs production by synoviocytes, which are
crucial for cartilage destruction 110. Furthermore, in addition to the stimulation of
osteoclast-mediated bone resorption, it was recently shown that IL-17 is able to inhibit
collagen type I synthesis, further increasing bone destruction 111.
Importantly, IL-17-positive cells were observed in synovial biopsies from
rheumatoid arthritis patients 112 and increased levels of IL-17, which correlated with
disease activity, were also observed in the serum and synovial fluid samples from
these patients 104. Furthermore, in vivo results have also highlighted the relevance of
IL-17 in rheumatoid arthritis by showing that the neutralization of IL-17 in mice
decreased the severity of antigen-induced and collagen-induced arthritis and that IL-
17-deficient mice presented reduced disease severity 113, 114.
1.5 Treatment options
The presentation and the clinical course of rheumatoid arthritis patients as well as
the response to the available treatment options are highly variable. Despite the

Introduction
- 15 -
existence of several treatment options for the management of rheumatoid arthritis, this
is still an incurable disease with enormous personal and economic costs. The strategy
of disease treatment relies on an early diagnosis and a prompt adequate treatment with
the goal of inducing remission in order to preserve joint function and maintain quality of
life 115-117. Drug-free remission is at this moment impossible for most patients.
Rheumatoid arthritis treatments can be divided into the following categories:
glucocorticoids, conventional disease-modifying anti-rheumatic drugs (DMARDs) and
biologic DMARDs.
1.5.1 Glucocorticoids
Glucocorticoids (namely prednisone) act rapidly to suppress inflammation and,
consequently, pain and swelling 118. Glucocorticoids are useful to control symptoms in
the first weeks of disease diagnosis, until slower-action DMARD can start to have an
effect. Glucocorticoids can have an effect in limiting joint damage, but long term
adverse effects, particularly at higher doses, limits their usefulness 119.
1.5.2 Conventional DMARDs
Conventional DMARDs, which include for instance methotrexate (MTX),
hydroxychloroquine, sulfasalazine and leflunomide, have long been used as the main
therapeutic option for newly diagnosed rheumatoid arthritis 118, 120. Among the different
DMARDs, MTX is the most frequently used and the most likely to induce a long-term
response 121-123. DMARDs have a relatively slow onset of action (1-6 months) but they
are effective in the long term and are considered to have an acceptable safety 118, 124
either as monotherapy or in combination therapy. However, around 30% of patients are
either non-responsive to DMARDs or lose their response secondarily or even develop
adverse effects that are incompatible with their use 125, 126 127. Therefore, the recent
development of biologic DMARDs is a relevant therapeutic alternative for these
DMARD refractory patients.
1.5.3 Biologic DMARDs
Nine different biologic DMARDs are currently approved by the European
Medicines Agency and many others are being tested in clinical trials for the treatment
of rheumatoid arthritis patients with inadequate response to one or more non-biologic

Introduction
- 16 -
DMARDs (Figure 3). In rheumatoid arthritis, biologic DMARDs have shown efficacy in
improving clinical, functional and radiographic outcomes 117, 128, especially if
administrated early in the disease course 116.
The first line of biologic DMARDs is the TNF antagonist class. TNF antagonists
approved for the treatment of rheumatoid arthritis include monoclonal antibodies
against TNF (infliximab, adalimumab, certolizumab pegol and golimumab) and a
soluble TNF receptor (etanercept). All TNF antagonists have similar efficacy and the
combination therapy with a TNF antagonist plus MTX has been proven to be superior
to the TNF antagonist alone 116, 129, 130. When patients fail to respond to a TNF
antagonist they have approximately a 50% chance of responding to a second one, but
the probability of efficacy of a third switch is very low 119. There are also approved
biologic DMARDs that target specific immune cells such as abatacept (T-cell co-
stimulation blocker) and rituximab (anti-CD20 monoclonal antibody for B-cell depletion).
Both these drugs, given in combination with MTX, may be useful alternatives for
patients who are non-responders to conventional DMARDs (rituximab is not formally
approved in that indication) or TNF antagonists 131, 132 and have already shown the
ability to inhibit progression of structural damage 133-135. Treatment of rheumatoid
arthritis patients with tocilizumab (humanized anti-IL-6 receptor monoclonal antibody) is
highly effective 98, 99, with reduced systemic inflammation, synovitis and radiographic
progression. This biologic DMARD can be used in the setting of DMARD or TNF
antagonist failure 101. There are also biologic agents targeting the IL-1β receptor
(anakinra, an IL-1β receptor antagonist), blocking the circulating IL-1β (rilonacept, a
fusion protein composed of the IL-1β receptor and IgG) and binding to IL-1β directly
(canakinumab, a monoclonal antibody against IL-1β) 136. Specifically, anakinra is
approved for rheumatoid arthritis treatment and it was shown to be effective albeit at a
lower efficacy than a TNF antagonist 137, 138. Rilonacept and canakinumab are still
undergoing clinical trials but they seem to have also lower efficacy as compared to TNF
antagonists 139. Interestingly, denosumab (antibody against RANKL) was also recently
shown to significantly reduce structural damage, but had no effect on the clinical signs
and symptoms of rheumatoid arthritis 140.
Switching among biologic DMARDs is often considered in patients with inadequate
response to initial treatment and the selection of the second biologic DMARD depends
in most cases on the reason of the first failure (e.g. a second TNF antagonist is less
effective when the first TNF antagonist failed due to lack of efficacy instead of adverse
events), but this rationale is somehow empirical and needs further studies 141, 142.
Overall, biologic DMARDs have been well tolerated by patients, despite the occurrence
Introduction
- 17 -
of infections and injection-site reactions, which are among the most common adverse
effects. Curiously, these types of problems occur with a similar frequency among the
different biologics 143. Moreover, the formation of antibodies to biologic agents is a
great concern since it can decrease the efficacy and increase the adverse effects of the
treatment. Of note, biologic therapies do not achieve optimal response in all patients
and even those who initially respond to treatment may experience diminished efficacy
with time. In addition, remission is only possible in 30% of the patients. Therefore,
searching for new, more effective and safer treatments for rheumatoid arthritis is still a
current medical priority.
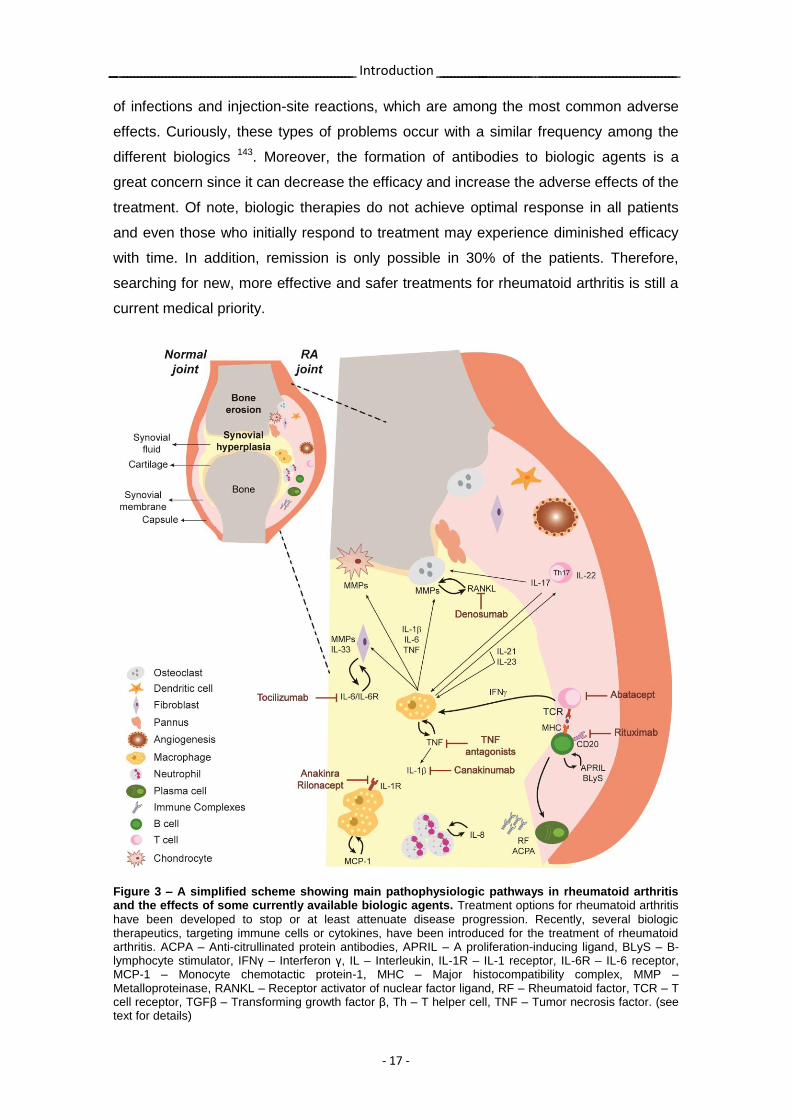
Figure 3 – A simplified scheme showing main pathophysiologic pathways in rheumatoid arthritis and the effects of some currently available biologic agents. Treatment options for rheumatoid arthritis
have been developed to stop or at least attenuate disease progression. Recently, several biologic therapeutics, targeting immune cells or cytokines, have been introduced for the treatment of rheumatoid arthritis. ACPA – Anti-citrullinated protein antibodies, APRIL – A proliferation-inducing ligand, BLyS – B-lymphocyte stimulator, IFNγ – Interferon γ, IL – Interleukin, IL-1R – IL-1 receptor, IL-6R – IL-6 receptor, MCP-1 – Monocyte chemotactic protein-1, MHC – Major histocompatibility complex, MMP – Metalloproteinase, RANKL – Receptor activator of nuclear factor ligand, RF – Rheumatoid factor, TCR – T cell receptor, TGFβ – Transforming growth factor β, Th – T helper cell, TNF – Tumor necrosis factor. (see text for details)

Introduction
- 18 -
2. IL-1β Immunological and inflammatory functions
2.1 IL-1 family
IL-1β is the most well characterized cytokine among the 11 members of the IL-1
family. The IL-1 family also comprises IL-1α, IL-1 receptor antagonist (IL-1Ra), IL-18,
IL-33 and IL-1F5 to IL-1F10 144. IL-1β, as discussed above, is a pleiotropic
proinflammatory cytokine with immuno- and inflammatory-modulating activities that are
crucial for host responses to infection and also to injuries and immunological
challenges 145. Several cell types produce this cytokine, although its main source are
cells from the innate immune system, such as monocytes and macrophages 145, 146.
Under normal conditions IL-1β is expressed at low levels, however, in the presence of
an inflammatory stimulus, its secretion increases. The production of IL-1β requires
induction in both transcriptional and post-translational steps 145, 146. This dual
mechanism responsible for the production of IL-1β is tightly regulated and ensures that
its secretion only occurs when it is required, thus preventing an unnecessary
exacerbated inflammatory response. The loss of these regulatory steps results in fever,
rash and arthritis, characteristic of autoinflammatory syndromes, such as
cryopyrinopathies 148. Gout is also associated with altered IL-1β regulation, induced by
the stimulus of uric acid crystals 147. Some autoimmune diseases are also likely to be at
least partially dependent on IL-1β 149.
2.2 IL-1β pathway
2.2.1 Priming
The production of IL-1β requires two signals, one for IL-1β gene expression and
the second for the completion of protein synthesis 150 (Figure 4). IL-1β is expressed as
an inactive 31 kDa precursor, designated as pro-IL-1β, in response to PAMPs or
cytokines, such as TNF and IL-1β itself, which signal through pattern recognition
receptors (PRRs) present in macrophages 151. These signaling pathways lead to the
activation of NF-kB, which induces the transcription of IL-1β promoter 152. This
induction of pro-IL-1β gene expression is commonly referred to as the priming step.
However, it does not act as a secretion stimulus. Instead, the primed cell must
encounter a new PAMP or DAMP to induce the processing and secretion of the active
IL-1β.
Introduction
- 19 -
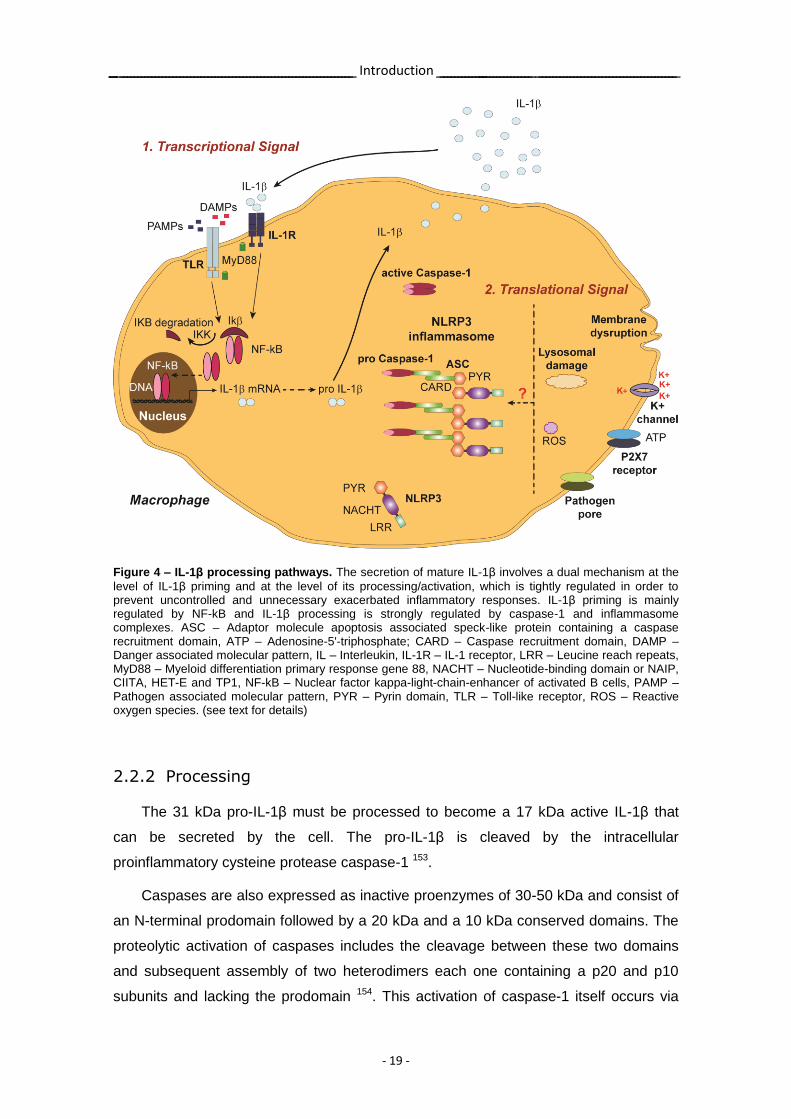
Figure 4 – IL-1β processing pathways. The secretion of mature IL-1β involves a dual mechanism at the
level of IL-1β priming and at the level of its processing/activation, which is tightly regulated in order to prevent uncontrolled and unnecessary exacerbated inflammatory responses. IL-1β priming is mainly regulated by NF-kB and IL-1β processing is strongly regulated by caspase-1 and inflammasome complexes. ASC – Adaptor molecule apoptosis associated speck-like protein containing a caspase recruitment domain, ATP – Adenosine-5'-triphosphate; CARD – Caspase recruitment domain, DAMP – Danger associated molecular pattern, IL – Interleukin, IL-1R – IL-1 receptor, LRR – Leucine reach repeats, MyD88 – Myeloid differentiation primary response gene 88, NACHT – Nucleotide-binding domain or NAIP, CIITA, HET-E and TP1, NF-kB – Nuclear factor kappa-light-chain-enhancer of activated B cells, PAMP – Pathogen associated molecular pattern, PYR – Pyrin domain, TLR – Toll-like receptor, ROS – Reactive oxygen species. (see text for details)
2.2.2 Processing
The 31 kDa pro-IL-1β must be processed to become a 17 kDa active IL-1β that
can be secreted by the cell. The pro-IL-1β is cleaved by the intracellular
proinflammatory cysteine protease caspase-1 153.
Caspases are also expressed as inactive proenzymes of 30-50 kDa and consist of
an N-terminal prodomain followed by a 20 kDa and a 10 kDa conserved domains. The
proteolytic activation of caspases includes the cleavage between these two domains
and subsequent assembly of two heterodimers each one containing a p20 and p10
subunits and lacking the prodomain 154. This activation of caspase-1 itself occurs via

Introduction
- 20 -
recruitment to a large multiprotein complex, termed inflammasome, which is present in
the cytosol 155.
The inflammasome is a molecular scaffold composed of a cytosolic NLR, adaptor
molecules and pro-caspase-1 156. The most well characterized inflammasome is the
NLRP3 (also known as cryopyrin or NALP3) inflammasome. The NLRP3 receptor is
comprised of a C-terminal PAMP/DAMP sensing leucine rich repeat (LRR), a central
nucleotide binding NACHT domain and an N-terminal pyrin domain (PYD) 156. The
NACHT (nucleotide-binding domain or NAIP, CIITA, HET-E and TP1) domain is
responsible for the oligomerization and, consequently, activation of the inflammasome.
The PYD domain of the NLRP3 is responsible for the recruitment of the adaptor
molecule, apoptosis associated speck-like protein containing a caspase recruitment
domain (ASC) or CARDINAL, by homotypic interactions between their PYD domains.
The NLRP3 inflammasome cannot recruit pro-caspase-1 without the adaptor molecule
ASC, because it lacks a caspase recruitment domain (CARD). Therefore, pro-caspase-
1 is recruited to ASC, by a homotypic interaction of CARD domains, facilitating
caspase-1 autocatalytic activation. The activation of the inflammasome itself requires
two signals. The first signal is the priming with TLR and NLR ligands or
proinflammatory cytokines that enhance NF-kB-driven transcription of inflammasome
components 157-159. The NLRP3 is expressed in myeloid cells and is upregulated in
response to a diverse panel of agonists. These agonists include exogenous / foreign
stimulus, such as asbestos, silica, alum, pathogens and pore-forming toxins, and also
endogenous / self-stimulus, such as monosodium urate crystals and calcium
pyrophosphate crystals 147, 160-162. The second signal is the activation and assembling of
the inflammasome, where ROS 163, 164, K+ efflux 165, pore formation at the cellular
membrane and lysosomal disruption 166, 167 seem to play a role. Despite the current
limited insight into the precise mechanism of NLRP3 inflammasome activation, it is
becoming increasingly evident that one signal alone is insufficient to induce its
activation. Also crucial for the regulation of inflammasome activation is its auto-
inhibition, which limits the occurrence of an aberrant inflammatory response. To date,
some mechanisms have been pointed out as possible pathways to inhibit
inflammasome activation, such as NLRs alternative splicing, ASC isoforms 168 and
pyrin 169.
Interestingly, there is also evidence that IL-1β activation can be mediated by
pathways independent of NLRP3 and caspase-1. For example, it was already
demonstrated that neutrophil proteases, such as proteinase 3, are able to cleave pro-
IL-1β, in the absence of NLRP3 and caspase-1 activation 170. Data from animal models

Introduction
- 21 -
have shown that when monosodium urate crystals were injected intraperitoneally in
caspase-1 deficient mice, there was a moderate reduction of cell influx in the peritoneal
cavity. This observation indicates that other mechanisms besides caspase-1 can also
participate in IL-1β processing and mediate the inflammatory response 147, 171. Such
distinct enzymes able to cleave pro-IL-1β may have different kinetics and sites of
cleavage, resulting in the formation of various peptides with variable biological
activities, and with an in vivo role that is yet poorly understood.
Thus, the primary role of caspase-1 and NLRP3 inflammasome is the tight
regulation of IL-1β activation.
2.2.3 Secretion
After caspase-1 processing of pro-IL-1β, mature IL-1β is rapidly secreted from the
cell 172 by a mechanism still incompletely known. There are already some evidences
that the secretion of this cytokine does not follow the conventional route of endoplasmic
reticulum and Golgi apparatus 173. Some authors believe instead that IL-1β can be
exported out of the cell via distinct non-conventional routes, such as lysosomes,
exososomes, microvesicles and membrane pores, depending on the type and duration
of the inflammatory stimulus and the levels of this cytokine required extracellularly to
mount an effective inflammatory response 174.
2.2.4 Signaling
IL-1β signals by binding first to the ligand-binding chain of the IL-1R type I (IL-1RI).
Subsequently, the co-receptor chain termed IL-1R accessory protein (IL-1RAcP) is
recruited, forming a trimeric functional high affinity complex of IL-1β, IL-1RI and IL-
1RAcP. This binding signal leads to the recruitment of the adaptor myeloid
differentiation primary response gene 88 (MyD88) protein to the Toll-IL-1 receptor (TIR)
cytoplasmic domain. Activation of the MAP kinase and of the NF-kB pathways occurs
next, leading to the NF-kB activation and translocation to the nucleus where it initiates
the expression of many inflammatory genes, such as IL-1 gene itself 175. NF-kB is
inactive in the majority of cells, being located in the cytoplasm in complex with
inhibitory Ikβ proteins. When signaling pathways are activated, the Ikβ protein is
phosphorylated by IKK kinases and degraded and, consequently, NF-kB can enter the
nucleus to modulate target gene expression 176.

Introduction
- 22 -
Surprisingly, a crosstalk mechanism between TLRs and IL-1β/IL-1RI seems to
exist, as during this signalling transduction the TIR domain of the TLRs can also recruit
MyD88 and synergistically contribute to the activation of NF-kB.
There is also a tight regulation at the level of IL-1β signalling. IL-1Ra plays a role in
this regulation since it binds to IL-1RI preventing its activation by IL-1β. The expression
of IL-1Ra is promoted by IL-1β-induced NF-kB signaling, thus providing a negative
feedback loop to control inflammation. Interestingly, mice deficient in IL-1Ra
spontaneously develop diseases such as highly destructive forms of arthritis 177. IL-1R
type II (IL-1RII) also regulates IL-1β signalling since it acts as a decoy receptor, being
unable to signal due to the lack of the cytoplasmic TIR domain. IL-1RII has a greater
affinity than IL-1RI in sequestering IL-1β 178 and is expressed mainly in macrophages
and B cells, being present both in cellular membranes and in a soluble form, acting as
an effective anti-inflammatory agent 179. The accessory protein IL-1RAcP is also
sequestered by IL-1RII, since it is recruited by the IL-1β/IL-1RII complex, thus
preventing its participation in IL-1RI signaling. Of note, both the expression of IL-1RII at
the cellular surface and circulating soluble form appears to affect the degree of
inflammation in several diseases. Importantly, the administration of glucocorticoids
induces the expression IL-1RII at the cell surface and this ability might contribute to
their anti-inflammatory effects 180. Thus, both the balance of IL-1β/IL-1Ra and IL-
1RII/IL-1RI may affect disease severity.
2.3 IL-1β and autoinflammatory diseases
An increased production of IL-1β is necessary for the development of several
autoinflammatory diseases. Autoinflammatory disorders include several heterogeneous
pathologies mainly characterized by fever and inflammation, with local as well as
systemic manifestations, that are often periodic, rather than progressive, and occur in
the absence of infections or autoimmune causes 181.
Some classical autoinflammatory disorders are caused by genetic alterations in
inflammatory pathways and, consequently, are usually manifested early in life. One
classical example of inherited autoinflammatory diseases is cryopyrin-associated
periodic fever syndromes (CAPS). Patients with CAPS often display a broad spectrum
of disease severity. The mildest form is familial cold-induced autoinflammatory
syndrome (FCAS) 182, a more severe form is Muckel-Wells syndrome (MWS) 183 and

Introduction
- 23 -
finally, the most severe form is chronic infantile neurological cutaneous and articular
syndrome/neonatal onset multisystem inflammatory disease (CINCA/NOMID) 184.
CAPS syndromes, despite being phenotypically heterogeneous, are all characterized
by periodic fevers and rashes, arthralgia, conjunctivitis, bone and joint symptoms and
elevated inflammatory markers. Interestingly, blood monocytes from patients with
CAPS secrete more IL-1β spontaneously or in response to low concentration of
inflammatory stimuli than cells from healthy controls 184-187. IL-1β itself, as mentioned
above, can increase the synthesis of NLRP3 components and caspase-1 as well as its
own IL-1β precursor, creating a positive feedback loop which amplifies the
inflammatory process underlying these diseases 188. A significant breakthrough in
understanding the pathogenesis of CAPS was the observation that these patients
possess a gain-of-function mutation in the NLRP3 gene, which promotes
inflammasome assembly and, consequently, leads to the continuous increase of
caspase-1 activation and IL-1β production 189. Currently, more than 70 inherited and de
novo disease associated-mutations have been identified, most of them occurring in the
NACHT domain of the NLRP3 inflammasome 190. Intriguingly, about 50% of the
patients with classic symptoms and biochemical markers of CAPS and other
autoinflammatory diseases do not have these mutations and, thus, the mechanism for
increased production of IL-1β is still not completely understood. Interestingly, some
mice models with knock-in mutations associated with FCAS and MWS exhibit severe
phenotypes 191, 192 and have increased concentration of IL-1β, as well as TNF and IL-17
at sites of inflammation. Myeloid cells derived from these mice do not spontaneously
secrete IL-1β but instead have a lower threshold in response to PAMPs, arguing that
the gain-of-function mutations associated with CAPS affect the myeloid-monocytic
lineage by lowering the threshold of IL-1β production. Moreover, mice deficient in
NLRP3, ASC and caspase-1 are often resistant to IL-1β mediated inflammation. In
general, there is a rapid and sustained resolution of disease severity and reduction of
biochemical and haematological disease markers in these autoinflammatory diseases
after treatment with IL-1β blockers 184, 193, 194.
Interestingly, in acute or chronic gout and pseudogout, the IL-1β pathway also
seems to play a relevant role. In gout, the deposition of monosodium urate crystals
within the joints of patients induces a strong inflammatory response with increased
levels of IL-1β, TNF and IL-8 195 and recurrent attacks of acute painful arthritis 196.
Importantly, some reports have already demonstrated that such monosodium urate
crystals together with free fatty acids are able to activate the NLRP3 inflammasome
and, consequently, account for the increased production of IL-1β 197, 198. Monosodium

Introduction
- 24 -
urate crystals can be phagocytosed by resident macrophages and their internalization
activates the NLRP3 inflammasome leading to neutrophil-dependent inflammation. In
fact, animal studies have shown that injection of crystals in the peritoneum of mice
induces a strong inflammatory response with an increased neutrophil influx, which
depends on both NLRP3 and IL-1R 147. There are other studies, however, which have
revealed that NLRP3 deficient mice exhibited a phenotype similar to the wild-type as
opposed to ASC and caspase-1 deficient mice which exhibited significantly reduced
synovial inflammation in response to monosodium urate crystal-free fatty acid
combination 197. This remaining synovial inflammation observed in caspase-1 deficient
mice might be explained by the high infiltration of neutrophils in gouty joints, which
could contribute to IL-1β precursor processing due to extracellular neutrophilic
enzymes, such as proteinase 3 170. Notably, mice deficient in IL-1β were fully protected
against joint inflammation after intraarticular administration of monosodium urate
crystals 197. This observation is in agreement with recent developments in the clinical
practice, in which some patients with recurrent attacks of gout, which are resistant to
colchicine and other steroid therapies, showed rapid and sustained reduction in pain
and signs of inflammation after IL-1 blockade therapy 199-202. Interestingly, it has been
suggested that the reason for the effectiveness of colchicine is possibly related with its
ability to inhibit microtubules and thus prevent the crystals from entering the cell or
interacting with NLRP3 by a yet unidentified mechanism 203, 204. Likewise, in
pseudogout, which is a similar crystalline-joint disease derived from the deposition of
calcium pyrophosphate crystals, there is a triggering of the NLRP3 inflammasome 147.
Several publications have also reported significant improvement of symptoms of
pseudogout patients with anakinra treatment 205, 206.
2.4 IL-1β and rheumatoid arthritis
Almost all autoimmune diseases, such as rheumatoid arthritis, have an
inflammatory component. In contrast to autoinflammatory disorders, autoimmune
diseases are primarily caused by deregulation of the immune system, especially of the
adaptive immune system, which mistakenly recognizes the body’s own molecular
constituents as foreign antigens. Moreover, these diseases are associated with high-
titer of autoantibodies or antigen-specific T cells.
In addition to the proinflammatory role of NLRP3 inflammasome in innate immune
responses, recent studies strongly suggest that NLRP3 inflammasome-mediated

Introduction
- 25 -
cytokines, such as IL-1β, play an essential role in modulating adaptive immune
responses. As mentioned previously, several studies have indicated that T cell
differentiation into IL-17-producing cells plays a major role in some autoimmune
diseases and that IL-1β appears to be crucial for the polarization of T cells towards
Th17 cells subset 63, 207-209. The role of IL-1β in the generation of Th17 cells is also in
agreement with the inflammatory process developed in mice deficient in IL-1Ra. In this
mouse model the activity of IL-1β is left unopposed and is thus able to drive Th17
differentiation, which results in spontaneous rheumatoid arthritis-like disease
development. In contrast, mice, which are also deficient in IL-17 show no signs of
disease 177, 210, 211. IL-1β also seems to be relevant for B cell activation. In rheumatoid
arthritis patients the depletion of CD20-bearing B cells results in decreased IL-17
production, which may be due to a reduction in IL-1β production by B cells 212. IL-1β/IL-
1R signaling may, therefore, affect the adaptive immune responses and promote
autoreactivity through several pathways: via the production of cytokines by dendritic
cells and consequent T cell priming 149, via induction of T and B cell interaction 213 and
by directly affecting autoreactive effector T cells 214, contributing to rheumatoid arthritis
physiopathology.
Another interesting point is that immunocomplexes can stimulate RF producing B
cells by concomitant B cell receptor and TLR9 stimulation 215. It has also been
demonstrated that innate immune responses mediated by TLRs may regulate
inflammation in rheumatoid synovial tissue, as supported by the fact that TLR2
deficient mice have reduced disease severity 216 and that TLRs are abundantly
expressed in the synovial tissue from rheumatoid arthritis patients 217. These data
reinforce the possible role of inflammasome/caspase-1/IL-1β pathway in rheumatoid
arthritis since it is known that there is a significant crosstalk between TLRs and NLRs.
Importantly, some recent studies have highlighted the importance of
inflammasome pathways as prominent participants in rheumatoid arthritis
physiopathology. For instance, Kastbom et al have shown that genetic variants of
components of NLRP3 inflammasome are associated with a higher susceptibility,
disease severity and a higher requirement for anti-TNF therapies 218, 219. Additionally,
Fontalba et al also revealed that polymorphisms in components of the NLRP3
inflammasome, namely in CARD8, influence NF-kB transcriptional activity and are
associated with rheumatoid arthritis severity 220. Interestingly, Rosengren et al have
demonstrated that the levels of NLRP3 mRNA are increased in rheumatoid arthritis
synovium, specifically in synovial macrophages, which further increase the levels of
NLRP3 mRNA after TNF stimulation. These authors concluded that there are increased

Introduction
- 26 -
levels of NLRP3 in rheumatoid synovium in later phases of the disease due to the
higher number of infiltrated macrophages, which are the main source of IL-1β 221.
However, another study from Kolly et al raised some controversy by showing that,
despite the increase in NLRP3 mRNA levels in rheumatoid arthritis synovium
comparing with osteoarthritis, the levels of NLRP3 protein were similar between the two
diseases 222. Additionally, the authors also determined the levels of caspase-1 and
found that they were increased in rheumatoid arthritis 222. Nonetheless, these authors
have not studied the levels of activated caspase-1, which is physiologically more
relevant than the total amount of the protein. Kolly et al also showed that mice deficient
in IL-1β presented reduced severity of antigen-induced arthritis, while NLRP3 deficient
animals had a severe phenotype similar to wild-type mice 222.
As described earlier, the potent proinflammatory effects of IL-1β in rheumatoid
arthritis physiopathology are mediated not only by the modulation of the adaptive
immune cells but also by activation of several types of cells accompanied by the
production of several inflammatory mediators, such as IL-1β itself, TNF, IL-6, IL-8,
cyclooxygenase 2 and prostaglandin E2, which account for the pain, swelling and
tenderness of joints. In addition, IL-1β contributes to the formation of pannus, leading
to cartilage and bone destruction which directly contribute to patients’ disability.
Interestingly, the levels of IL-1β are increased in rheumatoid arthritis synovial fluid and
correlate with disease severity 223, 224. Moreover, in rheumatoid arthritis patients, IL-1β
is overexpressed in inflamed synovial tissue, in particular in the lining and sublining
cells 225. Furthermore, IL-1β is localized in the synovial pannus, in close proximity to
bone and cartilage 226, and this cartilage exhibits up-regulation of IL-1β mRNA when
compared with normal tissue 227.
Therefore, although the regulatory cascades that mediate the initiation and
escalation of rheumatoid arthritis into a self-sustained immune response are not yet
completely understood, recent studies described herein have outlined the role of
inflammasome pathways as prominent participants in joint inflammation. However, their
exact role in rheumatoid arthritis, particularly in the initial phase of the disease, remains
largely unknown.

Introduction
- 27 -
2.5 Targeting IL-1β in rheumatoid arthritis:
the case of anakinra
Anakinra was approved in 2001 for the treatment of rheumatoid arthritis 228. As
mentioned before, anakinra is an injectable recombinant humanized form of the
endogenous IL-1Ra. It acts by blocking IL-1β/IL-1R binding and, thus, inhibits the
proinflammatory effects of this signal transduction 229-232.
Several trials have shown the benefit of anakinra in treating the signs and
symptoms of rheumatoid arthritis, with a significant improvement in various clinical and
biochemical markers of disease activity, as well as in the Larsen radiographic scores,
in comparison with placebo 228. In fact, after one year of treatment with anakinra,
rheumatoid arthritis patients experienced a reduction in disease activity 233 and also a
clear decrease in joint structural damage progression 233-235. Anakinra was also shown
to be generally well tolerated and safe 228. Nonetheless, this therapy has several
disadvantages, such as the need for a subcutaneous daily injection, because it has a
very short half-life of 4-6 hours. There is also a need for a large (10 to 1000 fold)
excess of IL-1Ra to effectively block the effect of IL-1β, which often results in painful
injection-site reactions 236, 237.
Surprisingly, the efficacy of anakinra for rheumatoid arthritis treatment was lower
than the one observed with TNF antagonists, in contrast to what was anticipated given
the critical role of IL-1β in rheumatoid arthritis physiopathology 238, 228, 239. Therefore,
rheumatologists currently rarely use anakinra for treating rheumatoid arthritis.
The disappointing results of anakinra have raised the question of whether anakinra
failed due to drug design problems or whether IL-1β was the wrong cytokine to target in
rheumatoid arthritis. Of note, all studies that investigated the efficacy of anakinra were
conducted in patients with established rheumatoid arthritis that had already failed other
therapies 240. Unlike other biologic treatments, an early intervention trial was not
performed. Furthermore, the modest clinical benefits of anakinra could reflect the
functional redundancy in TLR and IL-1R signalling pathways. Importantly, a subset of
patients did respond very well to anakinra treatment, supporting the idea that IL-1β is a
crucial cytokine that drives inflammation and joint destruction in rheumatoid arthritis, at
least in certain phases of the disease or in the context of specific patient’s
characteristics. The difference of anakinra efficacy in autoinflammatory versus
autoimmune diseases may in part reflect the fact that autoimmune disorders develop
as a result of a more complex combination of hereditary and environmental factors,
which lead to the regulatory failure of adaptive immune responses. It is therefore

Introduction
- 28 -
crucial to elucidate where IL-1β is positioned in the cascade of inflammatory responses
and to understand whether it directly mediates inflammation or acts instead upstream
of T cells as an initiator. The IL-1β activation pathway, which is regulated by many
steps, clearly has the potential to provide several putative targets for therapy
intervention in rheumatoid arthritis.

- 29 -
The pathological activation of the immune system seems to occur years before the
clinical onset of rheumatoid arthritis. However, the majority of the studies have been
focused in the established phase of rheumatoid arthritis, while the knowledge about the
immune mechanisms involved in disease onset is still very scarce. Importantly, it is
during the early phase of this disease that the immune system might still be malleable
enough to allow some immunologic intervention. A “therapeutic window of opportunity”
has been described in early rheumatoid arthritis in which it is still possible to prevent
permanent damage and to achieve remission in a large proportion of the patients. As
previously reviewed, the efficacy of anakinra for rheumatoid arthritis treatment was
lower than it was anticipated given the evident IL-1β critical role in disease
physiopathology. However, this treatment strategy was not fully explored, as it was not
tested in early rheumatoid arthritis patients and the subset of the patients that
responded very well was not properly characterized. Therefore, further studies are
clearly needed to fully understand the contribution of IL-1β for early rheumatoid arthritis
physiopathology and to test a potential dual target of IL-1β and TNF to be introduced in
the early phase of disease development when there is a unique “therapeutic window of
opportunity” and the possibility to change the outcome of the disease.

- 30 -

Chapter II
Aims

- 32 -

Aims
- 33 -
The main goal of the present work was to study the role of inflammasome
activation and IL-1 secretion in rheumatoid arthritis.
The specific aims of this study were:
I. To quantify cytokine and chemokine levels in sera and synovial fluid
samples from patients with early and established rheumatoid arthritis;
II. To study inflammasome activation by the quantification of caspase-1
activation in blood samples from patients with early and established
rheumatoid arthritis;
III. To identify drugs which down-regulate the production of IL-1β and TNF;
IV. To study the in vivo early and late anti-inflammatory effects of drugs that
simultaneously down-regulate IL-1β and TNF levels using a rat model of
arthritis;
V. To study the early and late anti-inflammatory effects of a drug that
specifically inhibits RORγT in a rat model of arthritis.

- 34 -

Chapter III
Results

- 36 -

Results
- 37 -
In agreement with the Decreto-Lei 388/70, art. 8º, parágrafo 2, the results
presented and discussed in this thesis were published or submitted for publication in
the following scientific peer-reviewed journals:
I. Rita A. Moura*, Rita Cascão*, Inês Perpétuo, Helena Canhão, Elsa Vieira-
Sousa, Ana F. Mourão, Ana M. Rodrigues, Joaquim Polido-Pereira, Mário
V. Queiroz, Henrique S. Rosário, Maria M. Souto-Carneiro, Luis Graca,
João E. Fonseca. Cytokine pattern in very early rheumatoid arthritis
favours B cell activation and survival. Rheumatology (Oxford) 2011;
50(2):278-282
* Rita A. Moura and Rita Cascão contributed equally to this work.
II. Rita Cascão*, Rita A. Moura*, Inês Perpétuo, Helena Canhão, Elsa Vieira-
Sousa, Ana F. Mourão, Ana M. Rodrigues, Joaquim Polido-Pereira, Mário
V. Queiroz, Henrique S. Rosário, Maria M. Souto-Carneiro, Luis Graca,
João E. Fonseca. Identification of a cytokine network sustaining neutrophil
and Th17 activation in untreated early rheumatoid arthritis. Arthritis
Research and Therapy 2010; 12(5):R196
* Rita Cascão and Rita A. Moura contributed equally to this work.
III. Rita Cascão, Joaquim Polido-Pereira, Helena Canhão, Ana M. Rodrigues,
Márcio Navalho, Helena Raquel, Ana F. Mourão, Catarina Resende, João
E. Fonseca, Luis F. Moita. Caspase-1 is active since the early phase of
Rheumatoid Arthritis. Clinical and Experimental Rheumatology 2012;
30(1):144
IV. Rita Cascão, Bruno Vidal, Helena Raquel, Ana Neves-Costa, Nuno
Figueiredo, Vineet Gupta, João E. Fonseca, Luis F. Moita. Effective
treatment of rat adjuvant-induced arthritis by celastrol. Autoimmunity
Reviews 2012 Mar 3. [Epub ahead of print]

Results
- 38 -
V. Rita Cascão, Bruno Vidal, Helena Raquel, Ana Neves-Costa, Nuno
Figueiredo, Vineet Gupta, João E. Fonseca, Luis F. Moita. Gambogic acid
is a potent anti-inflammatory and anti-proliferative drug in a rat model of
adjuvant-induced arthritis. Under revision

Results
- 39 -
Cytokine pattern in very early rheumatoid
arthritis favours B cell activation and survival

Results
- 40 -

Concise report
Cytokine pattern in very early rheumatoid arthritisfavours B-cell activation and survival
Rita A. Moura1,*, Rita Cascao1,*, Ines Perpetuo1, Helena Canhao1,2,Elsa Vieira-Sousa1,2, Ana F. Mourao1,3, Ana M. Rodrigues1,2,Joaquim Polido-Pereira1,2, Mario V. Queiroz2, Henrique S. Rosario4,Maria M. Souto-Carneiro5, Luis Graca6,7 and Joao E. Fonseca1,2
Abstract
Objectives. B cells play an important role in the perpetuation of RA, particularly as autoantibody-
producing cells. The ICs that further develop deposit in the joints and aggravate the inflammatory process.
However, B-cell contribution in the very early stage of the disease remains unknown. The main goal of this
work was to determine the concentration of cytokines potentially relevant for B-cell activation in serum
from very early polyarthritis patients, with <6 weeks of disease duration, who latter on evolved into very
early RA (VERA).
Methods. A proliferation-inducing ligand (APRIL), B-cell activating factor (BAFF) and IL-21 levels were
measured by ELISA in the serum of VERA, other very early arthritis (VEA), established RA patients and
controls. SF samples of established RA were also analysed.
Results. VERA patients have higher levels of APRIL and BAFF as compared with VEA, established RA and
controls. Furthermore, APRIL and BAFF levels are also significantly elevated in RA-SF when compared
with serum.
Conclusions. The increased levels of APRIL and BAFF in VERA patients suggests that B-cell activation
and the development of autoreactive B-cell responses might be crucial in early phases of RA. Therefore,
APRIL and BAFF could be promising targets for therapy in the early phase of RA.
Key words: B cells, VERA, Synovial fluid, APRIL, BAFF.
Introduction
B cells play critical roles in RA pathogenesis. They are the
source of RFs and anti-CCP autoantibodies, which
contribute to IC formation in the joints. These cells are
also efficient antigen-presenting cells and contribute to
T-cell activation through expression of co-stimulatory
molecules. B cells simultaneously respond and produce
chemokines and cytokines that promote leucocyte infiltra-
tion into the joints, formation of ectopic lymphoid struc-
tures, angiogenesis and synovial hyperplasia. Moreover,
B-cell depletion therapy with rituximab, as well as the
promising results obtained with atacicept in a Phase Ib
trial [1], confirmed the importance of these cells in estab-
lished RA [2, 3]. However, B-cell participation in the early
phase of the disease is not yet completely understood.
Interestingly, our previous studies showed a significant
decrease of pre-switch memory B cells (IgD+CD27+) in
peripheral blood of very early RA (VERA) patients, with
<6 weeks of disease duration, when compared with
controls [4]. In accordance with the results of another
group, it is highly likely that this B-cell subset migrates
towards the SM, contributing to the onset of the synovitis
1Rheumatology Research Unit, Instituto de Medicina Molecular,Faculdade de Medicina da Universidade de Lisboa, 2RheumatologyDepartment, Centro Hospitalar de Lisboa Norte, EPE, Hospital deSanta Maria, 3Rheumatology Department, Centro Hospitalar de LisboaOcidental, EPE, Hospital Egas Moniz, 4Microvascular Biology andInflammation Unit, Instituto de Medicina Molecular, Faculdade deMedicina da Universidade de Lisboa, Lisbon, 5Center forNeurosciences and Cell Biology, Autoimmunity Group, Coimbra,6Cellular Immunology Unit, Instituto de Medicina Molecular,Faculdade de Medicina da Universidade de Lisboa, Lisbon and7Instituto Gulbenkian de Ciencia, Oeiras, Portugal.
Correspondence to: Joao E. Fonseca, Rheumatology Research Unit,Instituto de Medicina Molecular, Edifıcio Egas Moniz, Faculdade deMedicina da Universidade de Lisboa, Av. Professor Egas Moniz,1649-028 Lisbon, Portugal. E-mail: [email protected]
*Rita A. Moura and Rita Cascao contributed equally to this study.
Submitted 18 June 2010; revised version accepted 3 September 2010.
! The Author 2010. Published by Oxford University Press on behalf of the British Society for Rheumatology. All rights reserved. For Permissions, please email: [email protected]
RHEUMATOLOGY
Rheumatology 2011;50:278–282
doi:10.1093/rheumatology/keq338
Advance Access publication 2 November 2010
BA
SIC
SC
IEN
CE
at Harvard U
niversity on January 25, 2012http://rheum
atology.oxfordjournals.org/D
ownloaded from
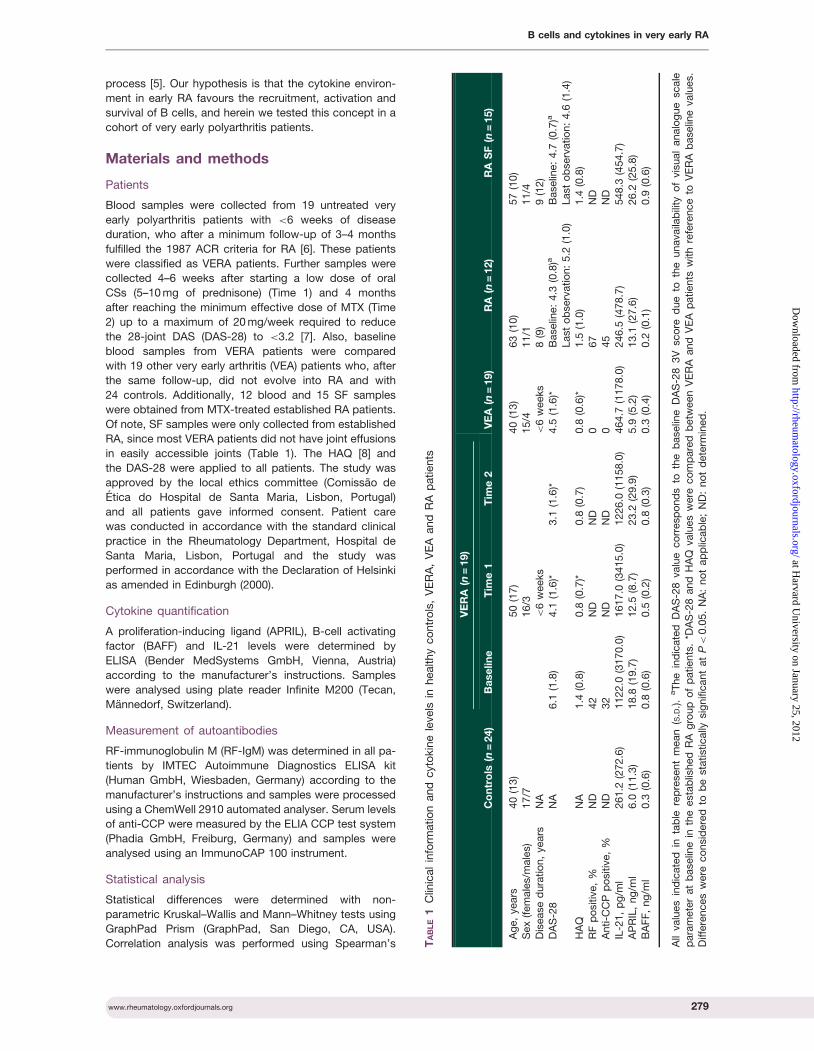
process [5]. Our hypothesis is that the cytokine environ-
ment in early RA favours the recruitment, activation and
survival of B cells, and herein we tested this concept in a
cohort of very early polyarthritis patients.
Materials and methods
Patients
Blood samples were collected from 19 untreated very
early polyarthritis patients with <6 weeks of disease
duration, who after a minimum follow-up of 3–4 months
fulfilled the 1987 ACR criteria for RA [6]. These patients
were classified as VERA patients. Further samples were
collected 4–6 weeks after starting a low dose of oral
CSs (5–10 mg of prednisone) (Time 1) and 4 months
after reaching the minimum effective dose of MTX (Time
2) up to a maximum of 20 mg/week required to reduce
the 28-joint DAS (DAS-28) to <3.2 [7]. Also, baseline
blood samples from VERA patients were compared
with 19 other very early arthritis (VEA) patients who, after
the same follow-up, did not evolve into RA and with
24 controls. Additionally, 12 blood and 15 SF samples
were obtained from MTX-treated established RA patients.
Of note, SF samples were only collected from established
RA, since most VERA patients did not have joint effusions
in easily accessible joints (Table 1). The HAQ [8] and
the DAS-28 were applied to all patients. The study was
approved by the local ethics committee (Comissao de
Etica do Hospital de Santa Maria, Lisbon, Portugal)
and all patients gave informed consent. Patient care
was conducted in accordance with the standard clinical
practice in the Rheumatology Department, Hospital de
Santa Maria, Lisbon, Portugal and the study was
performed in accordance with the Declaration of Helsinki
as amended in Edinburgh (2000).
Cytokine quantification
A proliferation-inducing ligand (APRIL), B-cell activating
factor (BAFF) and IL-21 levels were determined by
ELISA (Bender MedSystems GmbH, Vienna, Austria)
according to the manufacturer’s instructions. Samples
were analysed using plate reader Infinite M200 (Tecan,
Mannedorf, Switzerland).
Measurement of autoantibodies
RF-immunoglobulin M (RF-IgM) was determined in all pa-
tients by IMTEC Autoimmune Diagnostics ELISA kit
(Human GmbH, Wiesbaden, Germany) according to the
manufacturer’s instructions and samples were processed
using a ChemWell 2910 automated analyser. Serum levels
of anti-CCP were measured by the ELIA CCP test system
(Phadia GmbH, Freiburg, Germany) and samples were
analysed using an ImmunoCAP 100 instrument.
Statistical analysis
Statistical differences were determined with non-
parametric Kruskal–Wallis and Mann–Whitney tests using
GraphPad Prism (GraphPad, San Diego, CA, USA).
Correlation analysis was performed using Spearman’s TA
BL
E1
Clin
ical
info
rmatio
nand
cyto
kin
ele
vels
inhealthy
co
ntr
ols
,V
ER
A,
VE
Aand
RA
patients
Co
ntr
ols
(n=
24)
VE
RA
(n=
19)
VE
A(n
=19)
RA
(n=
12)
RA
SF
(n=
15)
Ba
se
lin
eT
ime
1T
ime
2
Ag
e,
years
40
(13)
50
(17)
40
(13)
63
(10)
57
(10)
Sex
(fem
ale
s/m
ale
s)
17/7
16/3
15/4
11/1
11/4
Dis
ease
dura
tio
n,
years
NA
<6
weeks
<6
weeks
8(9
)9
(12)
DA
S-2
8N
A6.1
(1.8
)4.1
(1.6
)*3.1
(1.6
)*4.5
(1.6
)*B
aselin
e:
4.3
(0.8
)aB
aselin
e:
4.7
(0.7
)a
Last
ob
serv
atio
n:
5.2
(1.0
)Last
ob
serv
atio
n:
4.6
(1.4
)
HA
QN
A1.4
(0.8
)0.8
(0.7
)*0.8
(0.7
)0.8
(0.6
)*1.5
(1.0
)1.4
(0.8
)
RF
po
sitiv
e,
%N
D42
ND
ND
067
ND
Anti-C
CP
po
sitiv
e,
%N
D32
ND
ND
045
ND
IL-2
1,
pg
/ml
261.2
(272.6
)1122.0
(3170.0
)1617.0
(3415.0
)1226.0
(1158.0
)464.7
(1178.0
)246.5
(478.7
)548.3
(454.7
)
AP
RIL
,ng
/ml
6.0
(11.3
)18.8
(19.7
)12.5
(8.7
)23.2
(29.9
)5.9
(5.2
)13.1
(27.6
)26.2
(25.8
)B
AF
F,
ng
/ml
0.3
(0.6
)0.8
(0.6
)0.5
(0.2
)0.8
(0.3
)0.3
(0.4
)0.2
(0.1
)0.9
(0.6
)
All
valu
es
ind
icate
din
tab
lere
pre
sent
mean
(S.D
.).
aT
he
ind
icate
dD
AS
-28
valu
eco
rresp
ond
sto
the
baselin
eD
AS
-28
3V
sco
red
ue
toth
eunavaila
bili
tyo
fvis
ual
analo
gue
scale
para
mete
rat
baselin
ein
the
esta
blis
hed
RA
gro
up
of
patients
.*D
AS
-28
and
HA
Qvalu
es
were
co
mp
are
db
etw
een
VE
RA
and
VE
Ap
atients
with
refe
rence
toV
ER
Ab
aselin
evalu
es.
Diffe
rences
were
co
nsid
ere
dto
be
sta
tistically
sig
nific
ant
at
P<
0.0
5.
NA
:no
tap
plic
ab
le;
ND
:no
td
ete
rmin
ed
.
www.rheumatology.oxfordjournals.org 279
B cells and cytokines in very early RA
at Harvard U
niversity on January 25, 2012http://rheum
atology.oxfordjournals.org/D
ownloaded from

test. Differences were considered to be statistically
significant for P< 0.05.
Results
Characterization of patients and disease evaluation
A total of 38 polyarthritis patients with <6 weeks of
disease duration were consecutively included. VERA
patients had a mean (S.D.) age of 59 (17) years; 84.2%
were female, 42% were RF positive and 32% anti-CCP
positive. The baseline DAS-28 and HAQ were 6.1 (1.8) and
1.4 (0.8), respectively. After treatment with CSs and MTX
there was a significant reduction of both DAS-28 and HAQ
values (Table 1). VEA patients were classified as having
SpA (five cases), SLE (four cases), crystal-induced arthritis
(two cases), SS (one case), paraneoplastic polyarthritis
related to multiple myeloma (one case) and arthritis asso-
ciated with HIV infection (one case), and five patients
entered spontaneous remission before 3 months of
follow-up, remaining without a specific diagnosis and
were thus classified as presenting a self-limited polyarthri-
tis. These early polyarthritis patients represent a subset of
a larger cohort previously described by our group [4].
Furthermore, unpaired blood and SF samples were col-
lected from established RA patients who had similar
DAS-28 and HAQ values to those of VERA patients at
baseline (Table 1).
APRIL and BAFF levels are increased in VERApatients at baseline
At baseline, APRIL and BAFF levels were significantly
higher in VERA patients as compared with both VEA and
controls (Fig. 1A). No differences were observed between
treated or untreated VERA patients, or between VEA and
controls (Table 1). Moreover, no significant differences in
IL-21 levels could be observed in VERA when compared
with either VEA patients and controls, or after therapy with
CSs and MTX (data not shown). Furthermore, there was
no correlation between DAS-28, anti-CCP or RF autoanti-
bodies and APRIL and BAFF serum concentrations (data
not shown).
VERA patients have higher APRIL and BAFF levelsin comparison with established RA patients
In order to compare early RA with the chronic phase,
we also analysed established RA serum samples.
Importantly, VERA patients, in baseline and even after
MTX treatment, had higher circulating levels of both
APRIL (P< 0.05) and BAFF (P< 0.001) in comparison
with established RA. However, regarding IL-21, no differ-
ences could be found in VERA when compared with
established RA (data not shown). Furthermore, no signifi-
cant differences could be observed in established RA in
comparison with controls (data not shown).
Established RA-SF has increased levels of APRIL,BAFF and IL-21
To verify whether in established RA patients a B-cell
activation environment could be present in the joint fluid
despite lower APRIL and BAFF serum levels, we tested
the same cytokine panel in established RA-SF. APRIL,
BAFF and IL-21 levels were in fact increased locally in
the joints of established RA patients in comparison with
RA serum (Fig. 1B).
Discussion
In the present work, a cytokine pattern favouring B-cell
activation is observed in RA patients with <6 weeks of
disease duration, when compared with other causes of
VEA and established RA. Our previous results demon-
strated a significant decrease of pre-switch memory
B cells (IgD+CD27+) in peripheral blood of VERA patients
[4]. This is in agreement with the report from another
group referring to a migration of this B-cell subset towards
the SM [5]. In fact, in established RA, ectopic germinal
centre-like structures develop in the inflamed synovial
tissue that support survival of B cells and autoantibody
production [9]. Therefore, we also analysed established
RA-SF samples and depicted the presence of a
cytokine-based B-cell survival environment that could
explain the maintenance of potentially autoreactive B
cells in the synovium. Our results demonstrated that
VERA patients have increased APRIL and BAFF levels
when compared with VEA and controls. Interestingly,
APRIL was also increased in VERA patients’ serum as
compared with established RA and its levels were even
higher in RA-SF suggesting a local up-regulation in the
synovium. APRIL affects not only the class-switch recom-
bination process [10–12], but also plasma cell differenti-
ation and survival [13, 14], which could thus explain the
maintenance of autoreactive B cells in the joints [15].
Actually, a highly positive association between the infiltra-
tion of plasma cells and SF levels of APRIL has been
demonstrated in RA patients [15, 16]. BAFF, similar to
APRIL, is a fundamental B-cell survival factor and we
have also detected increased serum levels in VERA
patients when compared with established RA. Moreover,
BAFF was also significantly elevated in RA-SF in compari-
son with RA serum. Previous studies have demonstrated
that BAFF increases the chemokine (C-X-C motif) ligand
13 (CXCL13)-dependent chemotaxis of memory B cells
through BAFF receptor (BAFF-R) triggering [17].
Therefore, increased BAFF levels in RA could support
the migration of pre-switch memory B cells towards RA
synovium, thus justifying the decrease of this B-cell sub-
population in circulation. Furthermore, increased IL-21
levels in RA-SF support local plasma cell differentiation
and autoantibody production [18]. Additionally, therapy
with neither CSs nor MTX affected cytokine production
in VERA patients. However, the patients with established
RA that we have studied were on chronic treatment with
MTX and low-dose CSs and this might have influenced the
serum levels of APRIL and BAFF. The effect of low-dose
CSs and MTX on cytokine production in RA patients is
still controversial [19, 20]. So, an absence of effect of
short-term therapy with CSs and MTX on the cytokines
analysed was not entirely unexpected in VERA, but an
effect on chronic-treated patients might in fact occur.
280 www.rheumatology.oxfordjournals.org
Rita A. Moura et al.
at Harvard U
niversity on January 25, 2012http://rheum
atology.oxfordjournals.org/D
ownloaded from
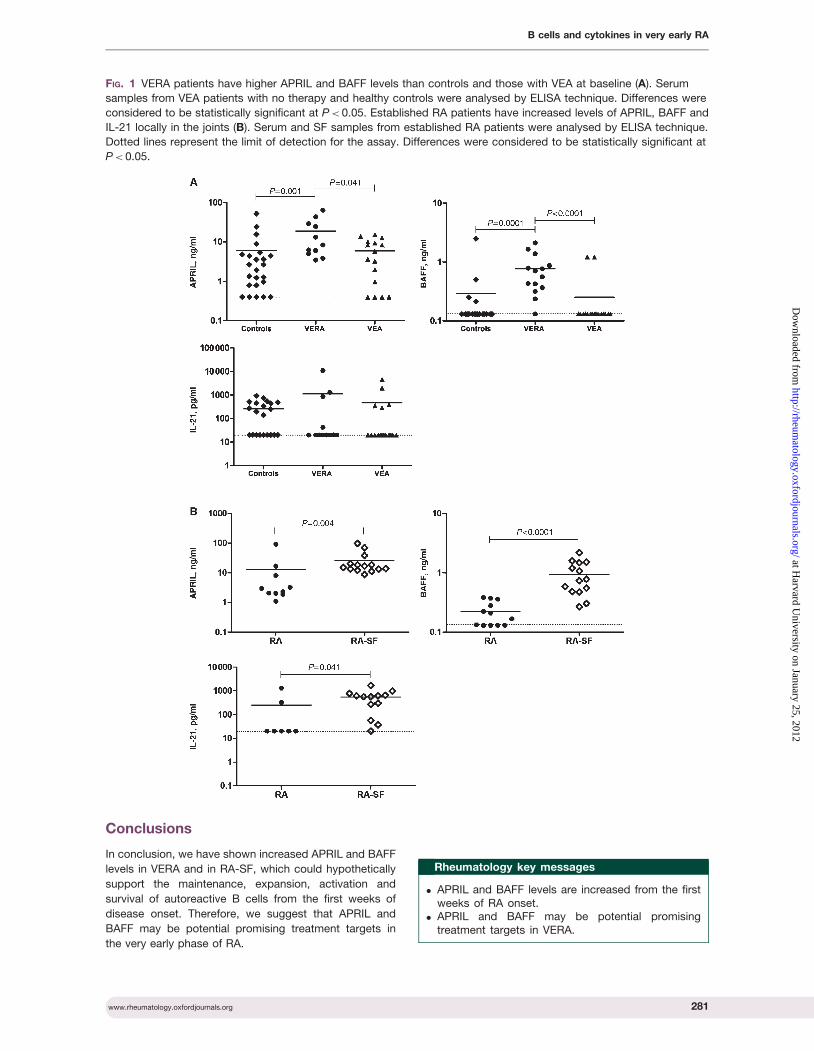
Conclusions
In conclusion, we have shown increased APRIL and BAFF
levels in VERA and in RA-SF, which could hypothetically
support the maintenance, expansion, activation and
survival of autoreactive B cells from the first weeks of
disease onset. Therefore, we suggest that APRIL and
BAFF may be potential promising treatment targets in
the very early phase of RA.
Rheumatology key messages
. APRIL and BAFF levels are increased from the firstweeks of RA onset.
. APRIL and BAFF may be potential promisingtreatment targets in VERA.
FIG. 1 VERA patients have higher APRIL and BAFF levels than controls and those with VEA at baseline (A). Serum
samples from VEA patients with no therapy and healthy controls were analysed by ELISA technique. Differences were
considered to be statistically significant at P< 0.05. Established RA patients have increased levels of APRIL, BAFF and
IL-21 locally in the joints (B). Serum and SF samples from established RA patients were analysed by ELISA technique.
Dotted lines represent the limit of detection for the assay. Differences were considered to be statistically significant at
P< 0.05.
www.rheumatology.oxfordjournals.org 281
B cells and cytokines in very early RA
at Harvard U
niversity on January 25, 2012http://rheum
atology.oxfordjournals.org/D
ownloaded from

Acknowledgements
The authors would like to acknowledge Bruno Vidal for
technical assistance.
Funding: This work was supported by a grant from
Sociedade Portuguesa de Reumatologia/Schering-
Plough 2005. R.A.M. and R.C. were funded by
Fundacao para a Ciencia e a Tecnologia (FCT) SFRH/
BD/30247/2006 and SFRH/BD/40513/2007, respectively.
M.M.S.-C. was funded by Marie Curie Intra-European
Fellowship PERG-2008-239422 and an EULAR Young
Investigator Award.
Disclosure statement: The authors have declared no
conflicts of interest.
References
1 Tak PP, Thurlings RM, Rossier C et al. Atacicept in
patients with rheumatoid arthritis: results of a multicenter,phase Ib, double-blind, placebo-controlled,
dose-escalating, single-, repeated-dose study. Arthritis
Rheum 2008;58:61–72.
2 Edwards JC, Cambridge G. Sustained improvement inrheumatoid arthritis following a protocol designed to
deplete B lymphocytes. Rheumatology 2001;40:205–11.
3 Leandro MJ, Edwards JC, Cambridge G. Clinical outcome
in 22 patients with rheumatoid arthritis treated with
B lymphocyte depletion. Ann Rheum Dis 2002;61:883–8.
4 Moura RA, Weinmann P, Pereira PA et al. Alterations on
peripheral blood B-cell subpopulations in very early
arthritis patients. Rheumatology 2010;49:1082–92.
5 Souto-Carneiro MM, Mahadevan V, Takada K et al.Alterations in peripheral blood memory B cells in patients
with active rheumatoid arthritis are dependent on the
action of tumour necrosis factor. Arthritis Res Ther 2009;
11:R84.
6 Arnett FC, Edworthy SM, Bloch DA et al. The AmericanRheumatism Association 1987 revised criteria for the
classification of rheumatoid arthritis. Arthritis Rheum 1988;
31:315–24.
7 Prevoo ML, van’t Hof MA, Kuper HH, van Leeuwen MA,van de Putte LB, van Riel PL. Modified disease activity
scores that include twenty-eight-joint counts.
Development and validation in a prospective longitudinal
study of patients with rheumatoid arthritis. Arthritis Rheum1995;38:44–8.
8 Fries JF, Spitz P, Kraines RG, Holman HR. Measurement
of patient outcome in arthritis. Arthritis Rheum 1980;23:
137–45.
9 Humby F, Bombardieri M, Manzo A et al. Ectopic
lymphoid structures support ongoing production of
class-switched autoantibodies in rheumatoid synovium.
PLoS Med 2009;6:e1.
10 Litinskiy MB, Nardelli B, Hilbert DM et al. DCs
induce CD40-independent immunoglobulin class
switching through BLyS and APRIL. Nat Immunol 2002;3:
822–9.
11 Castigli E, Scott S, Dedeoglu F et al. Impaired IgA class
switching in APRIL-deficient mice. Proc Natl Acad Sci
USA 2004;101:3903–8.
12 Castigli E, Wilson SA, Scott S et al. TACI and BAFF-R
mediate isotype switching in B cells. J Exp Med 2005;201:
35–9.
13 Belnoue E, Pihlgren M, McGaha TL et al. APRIL is critical
for plasmablast survival in the bone marrow and poorly
expressed by early-life bone marrow stromal cells. Blood
2008;111:2755–64.
14 Bossen C, Cachero TG, Tardivel A et al. TACI, unlike
BAFF-R, is solely activated by oligomeric BAFF and APRIL
to support survival of activated B cells and plasmablasts.
Blood 2008;111:1004–12.
15 Dong W, Li X, Liu H, Zhu P. Infiltrations of plasma cells in
synovium are highly associated with synovial fluid levels of
APRIL in inflamed peripheral joints of rheumatoid arthritis.
Rheumatol Int 2009;29:801–6.
16 Gabay C, Krenn V, Bosshard C, Seemayer CA,
Chizzolini C, Huard B. Synovial tissues concentrate
secreted APRIL. Arthritis Res Ther 2009;11:R144.
17 Badr G, Borhis G, Lefevre EA et al. BAFF enhances
chemotaxis of primary human B cells: a particular synergy
between BAFF and CXCL13 on memory B cells. Blood
2008;111:2744–54.
18 Ettinger R, Sims GP, Fairhurst AM et al. IL-21 induces
differentiation of human naive and memory B cells into
antibody-secreting plasma cells. J Immunol 2005;175:
7867–79.
19 Barrera P, Boerbooms AM, Demacker PN, van de
Putte LB, Gallati H, van der Meer JW. Circulating
concentrations and production of cytokines and soluble
receptors in rheumatoid arthritis patients: effects of a
single dose methotrexate. Br J Rheumatol 1994;33:
1017–24.
20 Chikanza IC, Kozaci D, Chernajovsky Y. The molecular
and cellular basis of corticosteroid resistance.
J Endocrinol 2003;179:301–10.
282 www.rheumatology.oxfordjournals.org
Rita A. Moura et al.
at Harvard U
niversity on January 25, 2012http://rheum
atology.oxfordjournals.org/D
ownloaded from


Results
- 41 -
Identification of a cytokine network sustaining
neutrophil and Th17 activation in untreated
early rheumatoid arthritis

Results
- 42 -

RESEARCH ARTICLE Open Access
Identification of a cytokine network sustainingneutrophil and Th17 activation in untreatedearly rheumatoid arthritisRita Cascão1†, Rita A Moura1†, Inês Perpétuo1, Helena Canhão1,2, Elsa Vieira-Sousa1,2, Ana F Mourão1,3,Ana M Rodrigues1,2, Joaquim Polido-Pereira1,2, Mário V Queiroz2, Henrique S Rosário4, Maria M Souto-Carneiro5,Luis Graca6†, João E Fonseca1,2*†
Abstract
Introduction: Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease characterized by sustainedsynovitis. Recently, several studies have proposed neutrophils and Th17 cells as key players in the onset andperpetuation of this disease. The main goal of this work was to determine whether cytokines driving neutrophiland Th17 activation are dysregulated in very early rheumatoid arthritis patients with less than 6 weeks of diseaseduration and before treatment (VERA).
Methods: Cytokines related to neutrophil and Th17 activation were quantified in the serum of VERA andestablished RA patients and compared with other very early arthritis (VEA) and healthy controls. Synovial fluid (SF)from RA and osteoarthritis (OA) patients was also analyzed.
Results: VERA patients had increased serum levels of cytokines promoting Th17 polarization (IL-1b and IL-6), aswell as IL-8 and Th17-derived cytokines (IL-17A and IL-22) known to induce neutrophil-mediated inflammation. Inestablished RA this pattern is more evident within the SF. Early treatment with methotrexate or corticosteroids ledto clinical improvement but without an impact on the cytokine pattern.
Conclusions: VERA patients already display increased levels of cytokines related with Th17 polarization andneutrophil recruitment and activation, a dysregulation also found in SF of established RA. 0 Thus, our data suggestthat a cytokine-milieu favoring Th17 and neutrophil activity is an early event in RA pathogenesis.
IntroductionRheumatoid arthritis (RA), the most common chronicautoimmune disease, affects approximately 1% of thepopulation worldwide. This disease comprises a syn-drome of pain, stiffness, and symmetrical synovitiswhich leads to joint destruction, functional disability,and substantial comorbidity due to the involvement ofmultiple organs and systems. The migration of leuko-cytes toward the synovium is crucial for the establish-ment of a chronic inflammatory process in RA [1-3].This multi-regulated mechanism involves interactions
with endothelial cells through cell adhesion moleculesand complex cytokine and chemokine pathways.Neutrophils specifically play an important role in the
onset and perpetuation of RA, not only as interleukin(IL)-producing cells but also as cells responsible for therelease of high amounts of reactive oxygen species anddestructive enzymes, such as metalloproteases, contri-buting to joint erosions [4]. Neutrophils are among thefirst leukocytes to arrive at sites of inflammation. Infact, these cells are the most abundant in the synovialfluid (SF) of patients with active RA, and previousresults from our group showed that the synovial tissueis heavily infiltrated by neutrophils in the first weeks ofRA onset [5]. Interestingly, in animal models of arthritis,neutrophil depletion prevented joint inflammation ifneutrophil-depleting antibodies were given before the
* Correspondence: [email protected]† Contributed equally1Rheumatology Research Unit, Instituto de Medicina Molecular, Edifício EgasMoniz, Faculdade de Medicina da Universidade de Lisboa, Av. Professor EgasMoniz, Lisboa, 1649-028, PortugalFull list of author information is available at the end of the article
Cascão et al. Arthritis Research & Therapy 2010, 12:R196http://arthritis-research.com/content/12/5/R196
© 2010 Cascão et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative CommonsAttribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction inany medium, provided the original work is properly cited.

induction of arthritis. Moreover, when the depletingantibody was given very early after the induction ofarthritis, complete abrogation of the inflammatorysymptoms was achieved [6].T helper 17 (Th17) cells have also been proposed to
have a relevant role in the early phase of RA throughthe production of IL-17 [7,8]. This cytokine promotesthe recruitment and survival of neutrophils, induces thesecretion of proinflammatory cytokines and the upregu-lation of RANKL (receptor activator of nuclear factor-kappa B ligand), and stimulates the activity of matrixmetalloproteases, leading to cartilage catabolism andbone resorption [9,10]. The recruitment, activation, andeffector function of Th17 cells and neutrophils are dri-ven by a network of cytokines and chemokines secretedby multiple cellular sources. In established RA, it hasbeen reported that IL-1b, IL-6, IL-8, IL-17, and tumornecrosis factor are elevated in the serum and this corre-lates with a higher disease activity [11-13]. Nevertheless,our knowledge of the influence of the cytokine networkon RA onset remains limited. The characterization ofthe cytokine profile at this stage, where the transitionfrom an acute to a chronic inflammatory phase occurs,may lead to the identification of early key players, withpotential implications for early treatment strategies.Thus, the main goal of our work was to determine
whether cytokines driving neutrophil and Th17 cell acti-vation and proinflammatory function were already pre-sent in very early RA (with less than 6 weeks of diseaseduration) and how this early cytokine environment dif-fers from established RA. We also evaluated whether theintroduction of low-dose corticosteroids and methotrex-ate (MTX) therapy had any influence on the cytokineprofile observed at that early stage of the disease. Wefound that cytokines related to Th17 polarization andneutrophil recruitment and activation were elevated inearly RA and that the conventional therapeutic options,though able to control clinical manifestations of the dis-ease, were ineffective in reversing this underlying proin-flammatory drive.
Materials and methodsPatientsBlood samples were obtained from 38 consecutiveuntreated polyarthritis patients with less than 6 weeks ofdisease duration. Some of these patients (19), after aminimum follow-up of 3 months, fulfilled the 1987American College of Rheumatology (ACR) criteria forRA [14]. These patients were classified as very early rheu-matoid arthritis (VERA) patients, and further sampleswere collected 4 to 6 weeks after starting a low dose oforal corticosteroids (5 to 10 mg of prednisone) (time 1)and 4 months after reaching the minimum effective doseof MTX (time 2) (up to a maximum of 20 mg/week) that
was required to reduce the disease activity score using 28joint counts (DAS28) to less than 3.2 [15]. The remainingearly arthritis patients (19), who did not develop RA,were classified as very early arthritis (VEA). Baselineblood samples from VERA and VEA patients were com-pared with 27 healthy donors used as controls. Addition-ally, 12 blood and 15 SF samples were obtained frompatients with established RA. SF samples were also col-lected from 10 patients with osteoarthritis (OA) (Rheu-matology Department, Hospital de Santa Maria, Lisbon,Portugal) (Table 1). Owing to the clinical characteristicsof the VEA patients, SF in easily accessible joints was notavailable in VERA and VEA patients and thus SF was notanalyzed in these groups of patients. The health assess-ment questionnaire (HAQ) [16] and DAS28 were appliedto all patients. The study was approved by the local ethicscommittee, and all patients signed an informed consentform. Patient care was conducted in accordance withstandard clinical practice, and the study was performedin accordance with the Declaration of Helsinki asamended in Edinburgh (2000).
Cytokine quantificationIL-1b, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12(p70), IL-17A,IL-22, IL-23, and interferon-gamma levels were mea-sured in the serum and SF by FlowCytomix assay kit(Bender MedSystems, Vienna, Austria) in accordancewith the instructions of the manufacturer. Standardcurves for each cytokine were generated by using refer-ence cytokine concentrations supplied by the manufac-turer. Samples were acquired with a FACS Calibur flowcytometer (BD Biosciences, San Jose, CA, USA). Rawdata of the flow cytometry bead assay were analyzed byFlowCytomix Pro 2.2 software (Bender MedSystems).
Measurement of autoantibodiesRheumatoid factor (RF)-IgM was determined in allpatients by means of an IMTEC Autoimmune Diagnos-tics ELISA [enzyme-linked immunosorbent assay] kit(Human GmbH, Wiesbaden, Germany) in accordancewith instructions of the manufacturer, and samples wereprocessed using a ChemWell 2910 automated analyzer(GMI, Ramsey, Minnesota, USA). Serum levels of anti-cyclic citrullinated peptide (anti-CCP) were measured byELIA™ CCP test system (Phadia GmbH, Freiburg, Ger-many), and samples were analyzed with an ImmunoCAP100 instrument (Phadia GmbH).
Statistical analysisStatistical differences were determined with non-parametric Kruskal-Wallis, Mann-Whitney, andWilcoxon signed-rank tests and GraphPad Prism (Graph-Pad Software, Inc., San Diego, CA, USA). Correlationanalysis was performed with the Spearman test.
Cascão et al. Arthritis Research & Therapy 2010, 12:R196http://arthritis-research.com/content/12/5/R196
Page 2 of 8
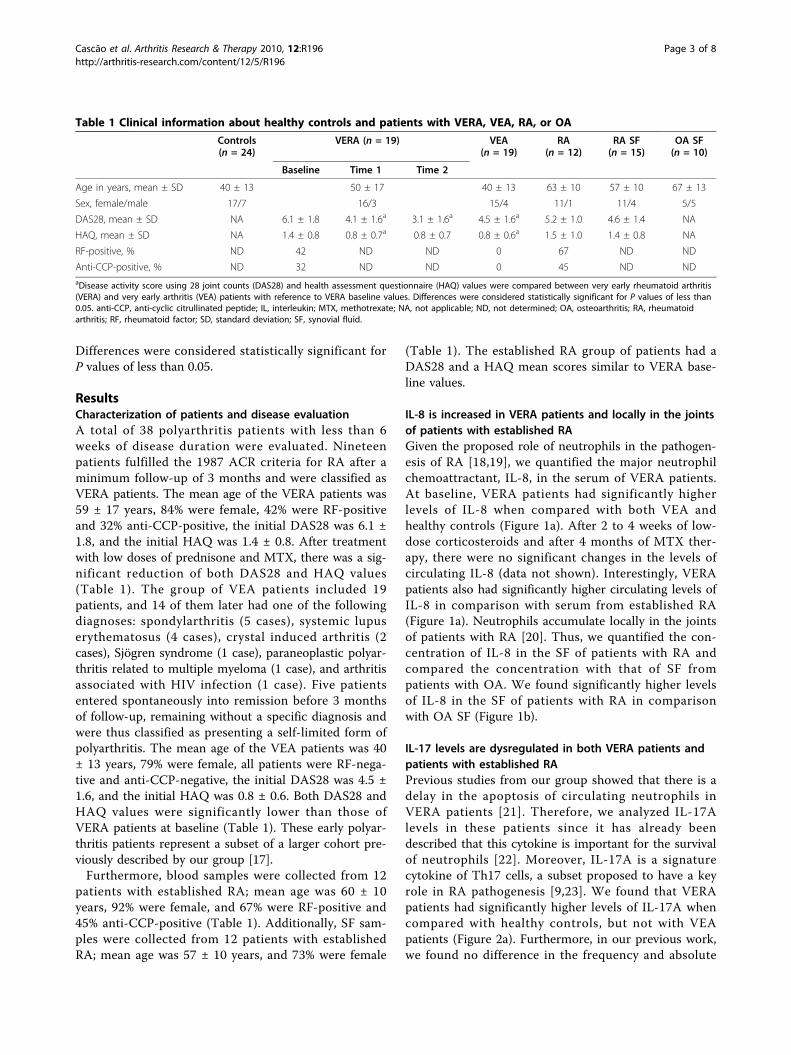
Differences were considered statistically significant forP values of less than 0.05.
ResultsCharacterization of patients and disease evaluationA total of 38 polyarthritis patients with less than 6weeks of disease duration were evaluated. Nineteenpatients fulfilled the 1987 ACR criteria for RA after aminimum follow-up of 3 months and were classified asVERA patients. The mean age of the VERA patients was59 ± 17 years, 84% were female, 42% were RF-positiveand 32% anti-CCP-positive, the initial DAS28 was 6.1 ±1.8, and the initial HAQ was 1.4 ± 0.8. After treatmentwith low doses of prednisone and MTX, there was a sig-nificant reduction of both DAS28 and HAQ values(Table 1). The group of VEA patients included 19patients, and 14 of them later had one of the followingdiagnoses: spondylarthritis (5 cases), systemic lupuserythematosus (4 cases), crystal induced arthritis (2cases), Sjögren syndrome (1 case), paraneoplastic polyar-thritis related to multiple myeloma (1 case), and arthritisassociated with HIV infection (1 case). Five patientsentered spontaneously into remission before 3 monthsof follow-up, remaining without a specific diagnosis andwere thus classified as presenting a self-limited form ofpolyarthritis. The mean age of the VEA patients was 40± 13 years, 79% were female, all patients were RF-nega-tive and anti-CCP-negative, the initial DAS28 was 4.5 ±1.6, and the initial HAQ was 0.8 ± 0.6. Both DAS28 andHAQ values were significantly lower than those ofVERA patients at baseline (Table 1). These early polyar-thritis patients represent a subset of a larger cohort pre-viously described by our group [17].Furthermore, blood samples were collected from 12
patients with established RA; mean age was 60 ± 10years, 92% were female, and 67% were RF-positive and45% anti-CCP-positive (Table 1). Additionally, SF sam-ples were collected from 12 patients with establishedRA; mean age was 57 ± 10 years, and 73% were female
(Table 1). The established RA group of patients had aDAS28 and a HAQ mean scores similar to VERA base-line values.
IL-8 is increased in VERA patients and locally in the jointsof patients with established RAGiven the proposed role of neutrophils in the pathogen-esis of RA [18,19], we quantified the major neutrophilchemoattractant, IL-8, in the serum of VERA patients.At baseline, VERA patients had significantly higherlevels of IL-8 when compared with both VEA andhealthy controls (Figure 1a). After 2 to 4 weeks of low-dose corticosteroids and after 4 months of MTX ther-apy, there were no significant changes in the levels ofcirculating IL-8 (data not shown). Interestingly, VERApatients also had significantly higher circulating levels ofIL-8 in comparison with serum from established RA(Figure 1a). Neutrophils accumulate locally in the jointsof patients with RA [20]. Thus, we quantified the con-centration of IL-8 in the SF of patients with RA andcompared the concentration with that of SF frompatients with OA. We found significantly higher levelsof IL-8 in the SF of patients with RA in comparisonwith OA SF (Figure 1b).
IL-17 levels are dysregulated in both VERA patients andpatients with established RAPrevious studies from our group showed that there is adelay in the apoptosis of circulating neutrophils inVERA patients [21]. Therefore, we analyzed IL-17Alevels in these patients since it has already beendescribed that this cytokine is important for the survivalof neutrophils [22]. Moreover, IL-17A is a signaturecytokine of Th17 cells, a subset proposed to have a keyrole in RA pathogenesis [9,23]. We found that VERApatients had significantly higher levels of IL-17A whencompared with healthy controls, but not with VEApatients (Figure 2a). Furthermore, in our previous work,we found no difference in the frequency and absolute
Table 1 Clinical information about healthy controls and patients with VERA, VEA, RA, or OA
Controls(n = 24)
VERA (n = 19) VEA(n = 19)
RA(n = 12)
RA SF(n = 15)
OA SF(n = 10)
Baseline Time 1 Time 2
Age in years, mean ± SD 40 ± 13 50 ± 17 40 ± 13 63 ± 10 57 ± 10 67 ± 13
Sex, female/male 17/7 16/3 15/4 11/1 11/4 5/5
DAS28, mean ± SD NA 6.1 ± 1.8 4.1 ± 1.6a 3.1 ± 1.6a 4.5 ± 1.6a 5.2 ± 1.0 4.6 ± 1.4 NA
HAQ, mean ± SD NA 1.4 ± 0.8 0.8 ± 0.7a 0.8 ± 0.7 0.8 ± 0.6a 1.5 ± 1.0 1.4 ± 0.8 NA
RF-positive, % ND 42 ND ND 0 67 ND ND
Anti-CCP-positive, % ND 32 ND ND 0 45 ND NDaDisease activity score using 28 joint counts (DAS28) and health assessment questionnaire (HAQ) values were compared between very early rheumatoid arthritis(VERA) and very early arthritis (VEA) patients with reference to VERA baseline values. Differences were considered statistically significant for P values of less than0.05. anti-CCP, anti-cyclic citrullinated peptide; IL, interleukin; MTX, methotrexate; NA, not applicable; ND, not determined; OA, osteoarthritis; RA, rheumatoidarthritis; RF, rheumatoid factor; SD, standard deviation; SF, synovial fluid.
Cascão et al. Arthritis Research & Therapy 2010, 12:R196http://arthritis-research.com/content/12/5/R196
Page 3 of 8
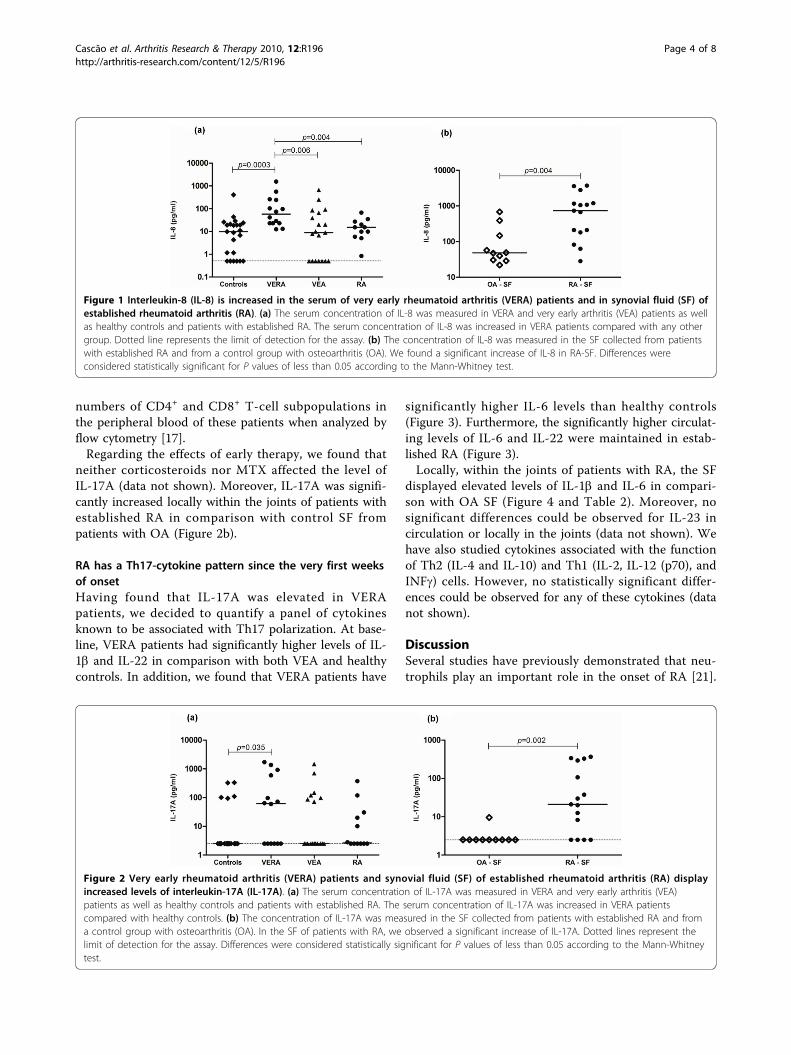
numbers of CD4+ and CD8+ T-cell subpopulations inthe peripheral blood of these patients when analyzed byflow cytometry [17].Regarding the effects of early therapy, we found that
neither corticosteroids nor MTX affected the level ofIL-17A (data not shown). Moreover, IL-17A was signifi-cantly increased locally within the joints of patients withestablished RA in comparison with control SF frompatients with OA (Figure 2b).
RA has a Th17-cytokine pattern since the very first weeksof onsetHaving found that IL-17A was elevated in VERApatients, we decided to quantify a panel of cytokinesknown to be associated with Th17 polarization. At base-line, VERA patients had significantly higher levels of IL-1b and IL-22 in comparison with both VEA and healthycontrols. In addition, we found that VERA patients have
significantly higher IL-6 levels than healthy controls(Figure 3). Furthermore, the significantly higher circulat-ing levels of IL-6 and IL-22 were maintained in estab-lished RA (Figure 3).Locally, within the joints of patients with RA, the SF
displayed elevated levels of IL-1b and IL-6 in compari-son with OA SF (Figure 4 and Table 2). Moreover, nosignificant differences could be observed for IL-23 incirculation or locally in the joints (data not shown). Wehave also studied cytokines associated with the functionof Th2 (IL-4 and IL-10) and Th1 (IL-2, IL-12 (p70), andINFg) cells. However, no statistically significant differ-ences could be observed for any of these cytokines (datanot shown).
DiscussionSeveral studies have previously demonstrated that neu-trophils play an important role in the onset of RA [21].
Figure 1 Interleukin-8 (IL-8) is increased in the serum of very early rheumatoid arthritis (VERA) patients and in synovial fluid (SF) ofestablished rheumatoid arthritis (RA). (a) The serum concentration of IL-8 was measured in VERA and very early arthritis (VEA) patients as wellas healthy controls and patients with established RA. The serum concentration of IL-8 was increased in VERA patients compared with any othergroup. Dotted line represents the limit of detection for the assay. (b) The concentration of IL-8 was measured in the SF collected from patientswith established RA and from a control group with osteoarthritis (OA). We found a significant increase of IL-8 in RA-SF. Differences wereconsidered statistically significant for P values of less than 0.05 according to the Mann-Whitney test.
Figure 2 Very early rheumatoid arthritis (VERA) patients and synovial fluid (SF) of established rheumatoid arthritis (RA) displayincreased levels of interleukin-17A (IL-17A). (a) The serum concentration of IL-17A was measured in VERA and very early arthritis (VEA)patients as well as healthy controls and patients with established RA. The serum concentration of IL-17A was increased in VERA patientscompared with healthy controls. (b) The concentration of IL-17A was measured in the SF collected from patients with established RA and froma control group with osteoarthritis (OA). In the SF of patients with RA, we observed a significant increase of IL-17A. Dotted lines represent thelimit of detection for the assay. Differences were considered statistically significant for P values of less than 0.05 according to the Mann-Whitneytest.
Cascão et al. Arthritis Research & Therapy 2010, 12:R196http://arthritis-research.com/content/12/5/R196
Page 4 of 8
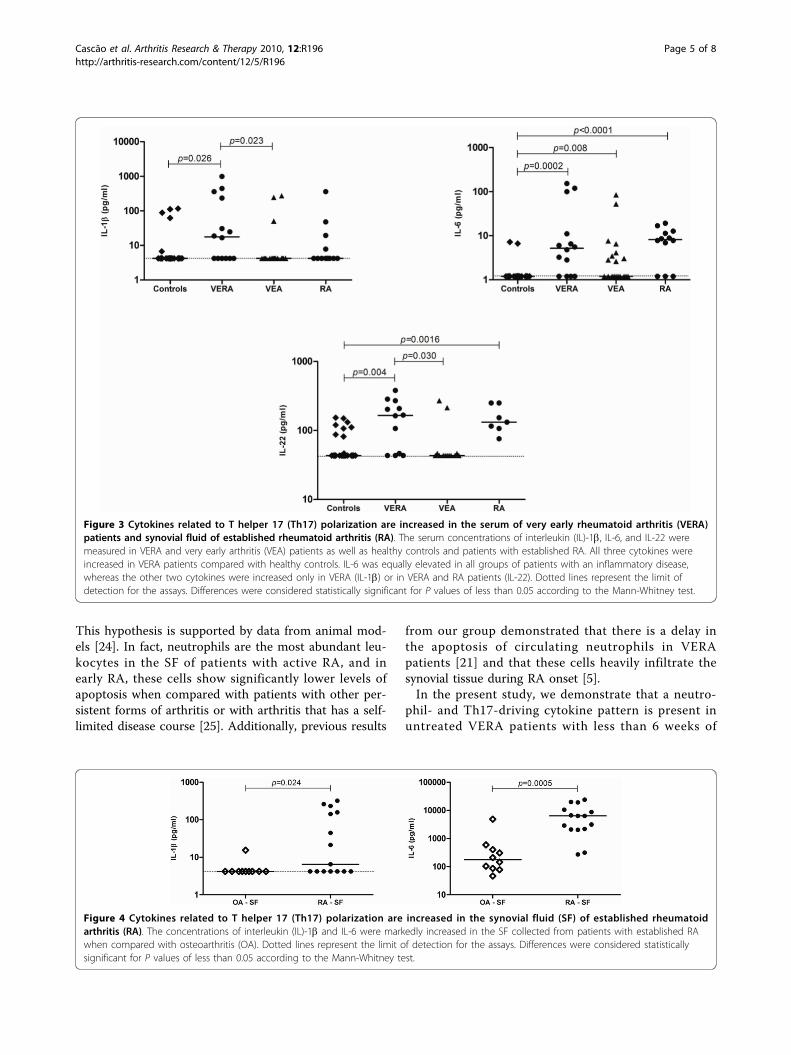
This hypothesis is supported by data from animal mod-els [24]. In fact, neutrophils are the most abundant leu-kocytes in the SF of patients with active RA, and inearly RA, these cells show significantly lower levels ofapoptosis when compared with patients with other per-sistent forms of arthritis or with arthritis that has a self-limited disease course [25]. Additionally, previous results
from our group demonstrated that there is a delay inthe apoptosis of circulating neutrophils in VERApatients [21] and that these cells heavily infiltrate thesynovial tissue during RA onset [5].In the present study, we demonstrate that a neutro-
phil- and Th17-driving cytokine pattern is present inuntreated VERA patients with less than 6 weeks of
Figure 3 Cytokines related to T helper 17 (Th17) polarization are increased in the serum of very early rheumatoid arthritis (VERA)patients and synovial fluid of established rheumatoid arthritis (RA). The serum concentrations of interleukin (IL)-1b, IL-6, and IL-22 weremeasured in VERA and very early arthritis (VEA) patients as well as healthy controls and patients with established RA. All three cytokines wereincreased in VERA patients compared with healthy controls. IL-6 was equally elevated in all groups of patients with an inflammatory disease,whereas the other two cytokines were increased only in VERA (IL-1b) or in VERA and RA patients (IL-22). Dotted lines represent the limit ofdetection for the assays. Differences were considered statistically significant for P values of less than 0.05 according to the Mann-Whitney test.
Figure 4 Cytokines related to T helper 17 (Th17) polarization are increased in the synovial fluid (SF) of established rheumatoidarthritis (RA). The concentrations of interleukin (IL)-1b and IL-6 were markedly increased in the SF collected from patients with established RAwhen compared with osteoarthritis (OA). Dotted lines represent the limit of detection for the assays. Differences were considered statisticallysignificant for P values of less than 0.05 according to the Mann-Whitney test.
Cascão et al. Arthritis Research & Therapy 2010, 12:R196http://arthritis-research.com/content/12/5/R196
Page 5 of 8
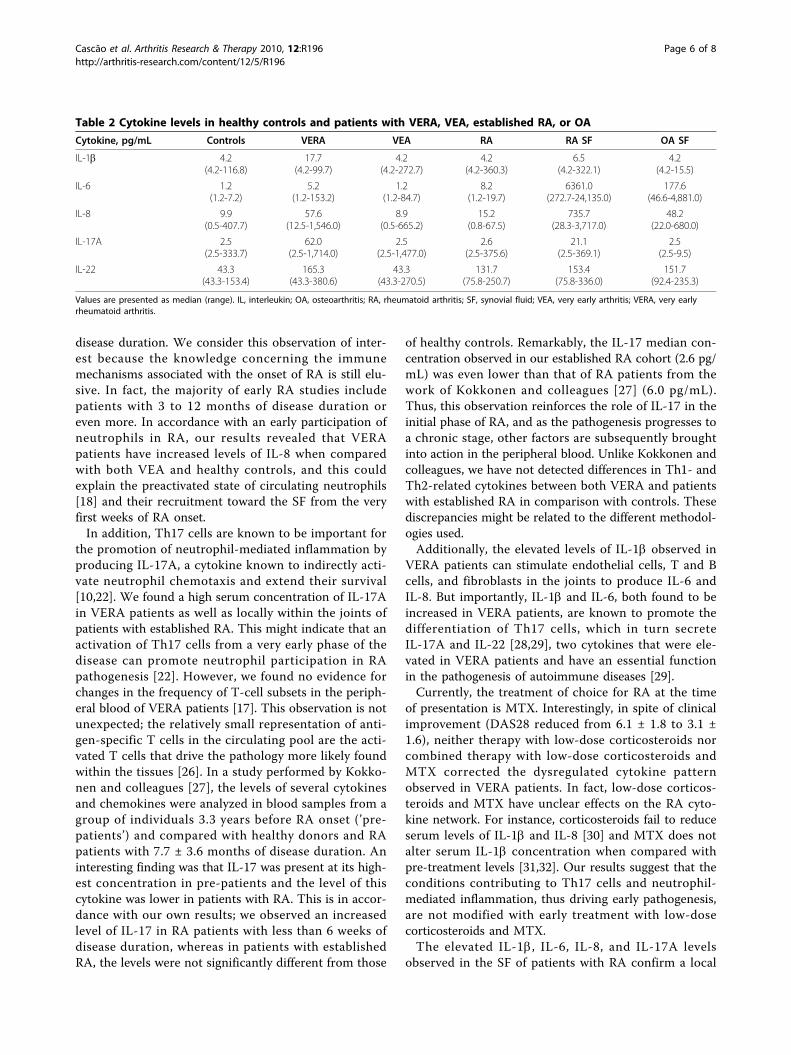
disease duration. We consider this observation of inter-est because the knowledge concerning the immunemechanisms associated with the onset of RA is still elu-sive. In fact, the majority of early RA studies includepatients with 3 to 12 months of disease duration oreven more. In accordance with an early participation ofneutrophils in RA, our results revealed that VERApatients have increased levels of IL-8 when comparedwith both VEA and healthy controls, and this couldexplain the preactivated state of circulating neutrophils[18] and their recruitment toward the SF from the veryfirst weeks of RA onset.In addition, Th17 cells are known to be important for
the promotion of neutrophil-mediated inflammation byproducing IL-17A, a cytokine known to indirectly acti-vate neutrophil chemotaxis and extend their survival[10,22]. We found a high serum concentration of IL-17Ain VERA patients as well as locally within the joints ofpatients with established RA. This might indicate that anactivation of Th17 cells from a very early phase of thedisease can promote neutrophil participation in RApathogenesis [22]. However, we found no evidence forchanges in the frequency of T-cell subsets in the periph-eral blood of VERA patients [17]. This observation is notunexpected; the relatively small representation of anti-gen-specific T cells in the circulating pool are the acti-vated T cells that drive the pathology more likely foundwithin the tissues [26]. In a study performed by Kokko-nen and colleagues [27], the levels of several cytokinesand chemokines were analyzed in blood samples from agroup of individuals 3.3 years before RA onset (’pre-patients’) and compared with healthy donors and RApatients with 7.7 ± 3.6 months of disease duration. Aninteresting finding was that IL-17 was present at its high-est concentration in pre-patients and the level of thiscytokine was lower in patients with RA. This is in accor-dance with our own results; we observed an increasedlevel of IL-17 in RA patients with less than 6 weeks ofdisease duration, whereas in patients with establishedRA, the levels were not significantly different from those
of healthy controls. Remarkably, the IL-17 median con-centration observed in our established RA cohort (2.6 pg/mL) was even lower than that of RA patients from thework of Kokkonen and colleagues [27] (6.0 pg/mL).Thus, this observation reinforces the role of IL-17 in theinitial phase of RA, and as the pathogenesis progresses toa chronic stage, other factors are subsequently broughtinto action in the peripheral blood. Unlike Kokkonen andcolleagues, we have not detected differences in Th1- andTh2-related cytokines between both VERA and patientswith established RA in comparison with controls. Thesediscrepancies might be related to the different methodol-ogies used.Additionally, the elevated levels of IL-1b observed in
VERA patients can stimulate endothelial cells, T and Bcells, and fibroblasts in the joints to produce IL-6 andIL-8. But importantly, IL-1b and IL-6, both found to beincreased in VERA patients, are known to promote thedifferentiation of Th17 cells, which in turn secreteIL-17A and IL-22 [28,29], two cytokines that were ele-vated in VERA patients and have an essential functionin the pathogenesis of autoimmune diseases [29].Currently, the treatment of choice for RA at the time
of presentation is MTX. Interestingly, in spite of clinicalimprovement (DAS28 reduced from 6.1 ± 1.8 to 3.1 ±1.6), neither therapy with low-dose corticosteroids norcombined therapy with low-dose corticosteroids andMTX corrected the dysregulated cytokine patternobserved in VERA patients. In fact, low-dose corticos-teroids and MTX have unclear effects on the RA cyto-kine network. For instance, corticosteroids fail to reduceserum levels of IL-1b and IL-8 [30] and MTX does notalter serum IL-1b concentration when compared withpre-treatment levels [31,32]. Our results suggest that theconditions contributing to Th17 cells and neutrophil-mediated inflammation, thus driving early pathogenesis,are not modified with early treatment with low-dosecorticosteroids and MTX.The elevated IL-1b, IL-6, IL-8, and IL-17A levels
observed in the SF of patients with RA confirm a local
Table 2 Cytokine levels in healthy controls and patients with VERA, VEA, established RA, or OA
Cytokine, pg/mL Controls VERA VEA RA RA SF OA SF
IL-1b 4.2(4.2-116.8)
17.7(4.2-99.7)
4.2(4.2-272.7)
4.2(4.2-360.3)
6.5(4.2-322.1)
4.2(4.2-15.5)
IL-6 1.2(1.2-7.2)
5.2(1.2-153.2)
1.2(1.2-84.7)
8.2(1.2-19.7)
6361.0(272.7-24,135.0)
177.6(46.6-4,881.0)
IL-8 9.9(0.5-407.7)
57.6(12.5-1,546.0)
8.9(0.5-665.2)
15.2(0.8-67.5)
735.7(28.3-3,717.0)
48.2(22.0-680.0)
IL-17A 2.5(2.5-333.7)
62.0(2.5-1,714.0)
2.5(2.5-1,477.0)
2.6(2.5-375.6)
21.1(2.5-369.1)
2.5(2.5-9.5)
IL-22 43.3(43.3-153.4)
165.3(43.3-380.6)
43.3(43.3-270.5)
131.7(75.8-250.7)
153.4(75.8-336.0)
151.7(92.4-235.3)
Values are presented as median (range). IL, interleukin; OA, osteoarthritis; RA, rheumatoid arthritis; SF, synovial fluid; VEA, very early arthritis; VERA, very earlyrheumatoid arthritis.
Cascão et al. Arthritis Research & Therapy 2010, 12:R196http://arthritis-research.com/content/12/5/R196
Page 6 of 8

role for these cytokines in the maintenance of synovitis.Moreover, IL-6 can support a continuous recruitment ofautoreactive B cells toward the synovium [33,34], contri-buting to an exacerbation of the inflammatory processbecause of the production of autoantibodies andimmune complexes.
ConclusionsTaken together, our data reinforce the potential rele-vance of therapies targeting IL-1b [35,36] and IL-6[37,38] in early RA. In addition, the data establish IL-8and IL-17A as other potential therapeutic targets at anearly stage of the disease. Finally, we found that MTXand corticosteroids, though effective in reducing diseaseactivity in VERA patients, do not appear to correctunderlying cytokine dysregulation driving the Th17/neu-trophil-mediated inflammation.
AbbreviationsACR: American College of Rheumatology; anti-CCP: anti-cyclic citrullinatedpeptide; DAS28: disease activity score using 28 joint counts; HAQ: healthassessment questionnaire; IL: interleukin; MTX: methotrexate; OA:osteoarthritis; RA: rheumatoid arthritis; RF: rheumatoid factor; SF: synovialfluid; Th17: T helper 17; VEA: very early arthritis; VERA: very early rheumatoidarthritis.
AcknowledgementsThe authors would like to acknowledge the technical assistance of BrunoVidal. This work was supported by a grant from Sociedade Portuguesa deReumatologia/Schering-Plough 2005. RAM and RC were funded byFundação para a Ciência e a Tecnologia (FCT) SFRH/BD/30247/2006 andSFRH/BD/40513/2007, respectively. MMS-C was funded by Marie Curie Intra-European Fellowship PERG-2008-239422 and a EULAR Young InvestigatorAward.
Author details1Rheumatology Research Unit, Instituto de Medicina Molecular, Edifício EgasMoniz, Faculdade de Medicina da Universidade de Lisboa, Av. Professor EgasMoniz, Lisboa, 1649-028, Portugal. 2Rheumatology Department, CentroHospitalar de Lisboa Norte, EPE, Hospital de Santa Maria, Av. Professor EgasMoniz, Lisboa, 1649-028, Portugal. 3Rheumatology Department, CentroHospitalar de Lisboa Ocidental, EPE, Hospital Egas Moniz, Rua da Junqueira,126, Lisboa,1300, Portugal. 4Microvascular Biology and Inflammation Unit,Instituto de Medicina Molecular, Edifício Egas Moniz, Faculdade de Medicinada Universidade de Lisboa, Av. Professor Egas Moniz, Lisboa, 1649-028,Portugal. 5Chronic Inflammation Group, Center for Neurosciences and CellBiology, Rua Larga 6, Coimbra, 3030, Portugal. 6Cellular Immunology Unit,Instituto de Medicina Molecular, Edifício Egas Moniz, Faculdade de Medicinada Universidade de Lisboa, Av. Professor Egas Moniz, Lisboa, 1649-028,Portugal.
Authors’ contributionsRC and RAM equally performed all of the laboratorial work, data collection,and statistical analysis and wrote the paper. IP contributed to some of thelaboratory experiments. HC, ES, AFM, AMR, and JP-P were responsible for theselection, follow-up, and medical care of patients enrolled in this study andhelped review the paper. MVQ participated as the head of theRheumatology Department of Hospital de Santa Maria, which approved thestudy and patients’ management. HSR and MMS-C made a substantialintellectual contribution to the present work and revised it critically. LG andJEF, as senior authors, conceived of the study, participated in its design andcoordination, and contributed important intellectual input to the draft of themanuscript. All authors read and approved the final manuscript.
Competing interestsThe authors declare that they have no competing interests.
Received: 7 June 2010 Revised: 9 September 2010Accepted: 20 October 2010 Published: 20 October 2010
References1. Kim HJ, Krenn V, Steinhauser G, Berek C: Plasma cell development in
synovial germinal centers in patients with rheumatoid and reactivearthritis. J Immunol 1999, 162:3053-3062.
2. Fonseca JE, Cortez-Dias N, Francisco A, Sobral M, Canhao H, Resende C,Castelao W, Macieira C, Sequeira G, Saraiva F, da Silva JA, Carmo-Fonseca M,Viana Queiroz M: Inflammatory cell infiltrate and RANKL/OPG expressionin rheumatoid synovium: comparison with other inflammatoryarthropathies and correlation with outcome. Clin Exp Rheumatol 2005,23:185-192.
3. Fonseca JE, Edwards JC, Blades S, Goulding NJ: Macrophagesubpopulations in rheumatoid synovium: reduced CD163 expression inCD4+ T lymphocyte-rich microenvironments. Arthritis Rheum 2002,46:1210-1216.
4. Cascao R, Rosario HS, Souto-Carneiro MM, Fonseca JE: Neutrophils inrheumatoid arthritis: More than simple final effectors. Autoimmun Rev2010, 9:531-535.
5. Mourao AF, Canhao H, Sousa E, Cascao R, da Costa JB, de Almeida LS,Oliveira ME, Gomes MM, Queiroz MV, Fonseca JE: From a neutrophilicsynovial tissue infiltrate to a challenging case of rheumatoid arthritis.Acta Reumatol Port 2010, 35:228-231.
6. Cascao R, Rosario HS, Fonseca JE: Neutrophils: warriors and commandersin immune mediated inflammatory diseases. Acta Reumatol Port 2009,34:313-326.
7. Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P:Human interleukin-17: A T cell-derived proinflammatory cytokineproduced by the rheumatoid synovium. Arthritis Rheum 1999, 42:963-970.
8. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S,Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T: IL-17 in synovialfluids from patients with rheumatoid arthritis is a potent stimulator ofosteoclastogenesis. J Clin Invest 1999, 103:1345-1352.
9. Stamp LK, James MJ, Cleland LG: Interleukin-17: the missing link betweenT-cell accumulation and effector cell actions in rheumatoid arthritis?Immunol Cell Biol 2004, 82:1-9.
10. Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C,Cosmi L, Lunardi C, Annunziato F, Romagnani S, Cassatella MA: Evidencefor a cross-talk between human neutrophils and Th17 cells. Blood 2010,115:335-343.
11. Nagatani K, Itoh K, Nakajima K, Kuroki H, Katsuragawa Y, Mochizuki M,Aotsuka S, Mimori A: Rheumatoid arthritis fibroblast-like synoviocytesexpress BCMA and are stimulated by APRIL. Arthritis Rheum 2007,56:3554-3563.
12. Chen DY, Hsieh TY, Chen YM, Hsieh CW, Lan JL, Lin FJ: Proinflammatorycytokine profiles of patients with elderly-onset rheumatoid arthritis: acomparison with younger-onset disease. Gerontology 2009, 55:250-258.
13. Petrovic-Rackov L, Pejnovic N: Clinical significance of IL-18, IL-15, IL-12and TNF-alpha measurement in rheumatoid arthritis. Clin Rheumatol2006, 25:448-452.
14. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS,Healey LA, Kaplan SR, Liang MH, Luthra HS, et al: The AmericanRheumatism Association 1987 revised criteria for the classification ofrheumatoid arthritis. Arthritis Rheum 1988, 31:315-324.
15. Prevoo ML, van’t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB,van Riel PL: Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinalstudy of patients with rheumatoid arthritis. Arthritis Rheum 1995, 38:44-48.
16. Fries JF, Spitz P, Kraines RG, Holman HR: Measurement of patient outcomein arthritis. Arthritis Rheum 1980, 23:137-145.
17. Moura RA, Weinmann P, Pereira PA, Caetano-Lopes J, Canhao H, Sousa E,Mourao AF, Rodrigues AM, Queiroz MV, Souto-Carneiro MM, Graca L,Fonseca JE: Alterations on peripheral blood B-cell subpopulations in veryearly arthritis patients. Rheumatology (Oxford) 2010, 49:1082-1092.
18. Cedergren J, Forslund T, Sundqvist T, Skogh T: Intracellular oxidativeactivation in synovial fluid neutrophils from patients with rheumatoid
Cascão et al. Arthritis Research & Therapy 2010, 12:R196http://arthritis-research.com/content/12/5/R196
Page 7 of 8

arthritis but not from other arthritis patients. J Rheumatol 2007,34:2162-2170.
19. Poubelle PE, Chakravarti A, Fernandes MJ, Doiron K, Marceau AA:Differential expression of RANK, RANK-L, and osteoprotegerin bysynovial fluid neutrophils from patients with rheumatoid arthritis and byhealthy human blood neutrophils. Arthritis Res Ther 2007, 9:R25.
20. Assi LK, Wong SH, Ludwig A, Raza K, Gordon C, Salmon M, Lord JM, Scheel-Toellner D: Tumor necrosis factor alpha activates release of Blymphocyte stimulator by neutrophils infiltrating the rheumatoid joint.Arthritis Rheum 2007, 56:1776-1786.
21. Weinmann P, Moura RA, Caetano-Lopes JR, Pereira PA, Canhao H,Queiroz MV, Fonseca JE: Delayed neutrophil apoptosis in very earlyrheumatoid arthritis patients is abrogated by methotrexate therapy. ClinExp Rheumatol 2007, 25:885-887.
22. Parsonage G, Filer A, Bik M, Hardie D, Lax S, Howlett K, Church LD, Raza K,Wong SH, Trebilcock E, Scheel-Toellner D, Salmon M, Lord JM, Buckley CD:Prolonged, granulocyte-macrophage colony-stimulating factor-dependent, neutrophil survival following rheumatoid synovial fibroblastactivation by IL-17 and TNFalpha. Arthritis Res Ther 2008, 10:R47.
23. Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS, Shnier R,Portek IJ: Synovial membrane cytokine expression is predictive of jointdamage progression in rheumatoid arthritis: a two-year prospectivestudy (the DAMAGE study cohort). Arthritis Rheum 2006, 54:1122-1131.
24. Vossenaar ER, Nijenhuis S, Helsen MM, van der Heijden A, Senshu T, vanden Berg WB, van Venrooij WJ, Joosten LA: Citrullination of synovialproteins in murine models of rheumatoid arthritis. Arthritis Rheum 2003,48:2489-2500.
25. Andersson AK, Li C, Brennan FM: Recent developments in theimmunobiology of rheumatoid arthritis. Arthritis Res Ther 2008, 10:204.
26. Duarte J, Agua-Doce A, Oliveira VG, Fonseca JE, Graca L: Modulation of IL-17 and Foxp3 expression in the prevention of autoimmune arthritis inmice. PLoS One 2010, 5:e10558.
27. Kokkonen H, Soderstrom I, Rocklov J, Hallmans G, Lejon K, RantapaaDahlqvist S: Up-regulation of cytokines and chemokines predates theonset of rheumatoid arthritis. Arthritis Rheum 2010, 62:383-391.
28. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K,Collins M, Fouser LA: Interleukin (IL)-22 and IL-17 are coexpressed byTh17 cells and cooperatively enhance expression of antimicrobialpeptides. J Exp Med 2006, 203:2271-2279.
29. Pene J, Chevalier S, Preisser L, Venereau E, Guilleux MH, Ghannam S,Moles JP, Danger Y, Ravon E, Lesaux S, Yssel H, Gascan H: Chronicallyinflamed human tissues are infiltrated by highly differentiated Th17lymphocytes. J Immunol 2008, 180:7423-7430.
30. Chikanza IC, Kozaci D, Chernajovsky Y: The molecular and cellular basis ofcorticosteroid resistance. J Endocrinol 2003, 179:301-310.
31. Barrera P, Haagsma CJ, Boerbooms AM, Van Riel PL, Borm GF, Van dePutte LB, Van der Meer JW: Effect of methotrexate alone or incombination with sulphasalazine on the production and circulatingconcentrations of cytokines and their antagonists. Longitudinalevaluation in patients with rheumatoid arthritis. Br J Rheumatol 1995,34:747-755.
32. Segal R, Mozes E, Yaron M, Tartakovsky B: The effects of methotrexate onthe production and activity of interleukin-1. Arthritis Rheum 1989,32:370-377.
33. Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaisnes CN, VanSnick J: Interleukin-6 in synovial fluid and serum of patients withrheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum1988, 31:784-788.
34. Gabay C: Interleukin-6 and chronic inflammation. Arthritis Res Ther 2006,8(Suppl 2):S3.
35. Smeets TJ, Barg EC, Kraan MC, Smith MD, Breedveld FC, Tak PP: Analysis ofthe cell infiltrate and expression of proinflammatory cytokines andmatrix metalloproteinases in arthroscopic synovial biopsies: comparisonwith synovial samples from patients with end stage, destructiverheumatoid arthritis. Ann Rheum Dis 2003, 62:635-638.
36. Joosten LA, Abdollahi-Roodsaz S, Heuvelmans-Jacobs M, Helsen MM, vanden Bersselaar LA, Oppers-Walgreen B, Koenders MI, van den Berg WB: Tcell dependence of chronic destructive murine arthritis induced byrepeated local activation of Toll-like receptor-driven pathways: crucialrole of both interleukin-1beta and interleukin-17. Arthritis Rheum 2008,58:98-108.
37. al-Janadi M, al-Balla S, al-Dalaan A, Raziuddin S: Cytokine production byhelper T cell populations from the synovial fluid and blood in patientswith rheumatoid arthritis. J Rheumatol 1993, 20:1647-1653.
38. Youinou P, Jamin C: The weight of interleukin-6 in B cell-relatedautoimmune disorders. J Autoimmun 2009, 32:206-210.
doi:10.1186/ar3168Cite this article as: Cascão et al.: Identification of a cytokine networksustaining neutrophil and Th17 activation in untreatedearly rheumatoid arthritis. Arthritis Research & Therapy 2010 12:R196.
Submit your next manuscript to BioMed Centraland take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit
Cascão et al. Arthritis Research & Therapy 2010, 12:R196http://arthritis-research.com/content/12/5/R196
Page 8 of 8

Results
- 43 -
Caspase-1 is active since the early phase of
rheumatoid arthritis

Results
- 44 -

144
Letters to the EditorsCaspase-1 is active since the early phase of rheumatoid arthritis
Sirs,We have recently reported increased interleu-kin (IL)-1β levels in very recent onset rheu-matoid arthritis (RA) (1). Together with IL-6, which we also found to be increased in these patients, IL-1β can promote the polarization of Th17 cells, which have an essential role in the pathogenesis of autoimmune diseases (2), such as RA. It has also been reported that RA synovial tissue contains higher concen-trations of cytokines including IL-1β and shows activation of the nuclear transcription factor NF-κB (3), suggesting that IL-1β/IL1 receptor signalling may contribute to arthritis onset and progression (4). Caspase-1 is activated by multiprotein com-plexes known as inflammasomes. One of the best characterised examples is the NLRP3 inflammasome (5), which is able to process immature proinflammatory cytokines, such as pro-IL-1β and pro-IL-18 (6). Interestingly, it has been reported that polymorphisms in the nlrp3 gene are associated with increased susceptibility and a worse prognosis in RA (7, 8). Therefore, we hypothesised that the inflammasome could be an active contributor to the inflammatory response in RA and set out to evaluate the activation of caspase-1 in early and established RA patients.To study the early phase of RA, we collected blood samples from 10 untreated early RA (ERA) and 9 untreated early arthritis patients who have not evolved to RA (EA) with less than 12 months of disease duration. ERA patients had a mean age of 48±9 years and DAS28 of 5.0±1.8. Among ERA patients, 90% were female, 60% were rheumatoid fac-tor (RF) positive and 40% were anti-cyclic citrullinated peptide (anti-CCP) positive. EA patients had mean age of 60±17 years and DAS28 of 5.5±2.3. In the EA group, 67% were female, 22% were anti-CCP positive and all patients were RF negative. To study the chronic phase of RA, we collected blood samples from 11 established RA patients. Established RA patients had a mean age of 54±10 years and DAS28 of 4.5±1.3, 70% were RF positive and 36% were anti-CCP positive, and 80% of them were females. This group of patients had a mean disease dura-tion of 13±8 years and all of them were under methotrexate (MTX) treatment with a mean dose of 14±5 mg/week and a low dose of prednisolone (less than 10 mg). We also col-lected blood samples from 10 age- and sex-matched healthy donors for comparison.We measured caspase-1 activity using the Carboxyfluorescein FLICA Detection kit (Immunochemistry Technologies, LLC, USA) following the reagent instructions. Samples were acquired using a FACS Calibur (BD biosciences, USA) and analysed using FlowJo software (Tree Star Inc). Statistical differenc-es were determined with parametric unpaired t-test and Bonferroni’s multiple comparison
test using GraphPad Prism (GraphPad, San Diego, CA), and differences were considered statistically significant for p<0.05.We found that ERA patients have signifi-cantly higher levels of basal active caspase-1 than in healthy controls (Fig. 1, p=0.0222). Also established RA patients have higher basal levels of active caspase-1 as compared to healthy controls (Fig. 1, p=0.0301). In ad-dition, no differences were found when com-paring EA with healthy controls or between ERA and EA patients, neither between ERA and EA with established RA patients. Of note, we previously did not find any significant differences in the percentages of leukocyte populations between patients and healthy controls (9, 10).These results show that the level of active caspase-1 is increased in circulating leuko-cytes of early and established RA patients, supporting our hypothesis that the caspase-1 pathway is already activated since the early phase of RA, plays a role in this disease and may be a promising treatment target in early RA. Given the relative inefficacy of blocking IL-1β in established RA we thus suggest that this treatment strategy should be re-evalu-ated in early RA patients.The authors would like to acknowledge Ana Lopes and Bruno Vidal for their technical as-sistance. This work was supported by a grant (SFRH/BD/40513/2007) from Fundação para a Ciência e a Tecnologia (FCT) and by an un-restricted research grant from Pfizer. Work in Luis Moita’s laboratory is supported by FCT (PIC/IC/82991/2007 and PTDC/SAU-MII/100780/2008) and FLAD.
R. CASCÃO1, MScJ. POLIDO-PEREIRA1,2, MDH. CANHÃO1,2, MD, PhDA.M. RODRIGUES1,2, MDM. NAVALHO1,3, MDH. RAQUEL1, PhDA. NEVES-COSTA1, PhDA.F. MOURÃO1,4, MDC. RESENDE2, MDJ.A.P. DA SILVA2, MDJ.E. FONSECA1,2*, MD, PhDL.F. MOITA1*, MD, PhD 1Instituto de Medicina Molecular, Faculdade de Medicina da Universidade de Lisboa, Lisbon;
2Rheumatology Department, Centro Hospitalar de Lisboa Norte, EPE, Hospital de Santa Maria, Lisbon; 3Radiology Department, Hospital da Luz, Lisbon; 4Rheumatology Department, Cen-tro Hospitalar de Lisboa Ocidental, EPE, Hospital Egas Moniz, Lisbon, Portugal.*Joint senior authors.Address correspondence to: Dr Luis Ferreira Moita, Cell Biology of the Immune System Unit, Instituto de Medicina Molecular, Edifício Egas Moniz, Faculdade de Medicina da Universidade de Lisboa, Av. Professor Egas Moniz, 1649-028 Lisbon, Portugal.E-mail: [email protected] interests: none declared.
References 1. CASCAO R, MOURA RA, PERPETUO I et al.: Identi-
fication of a cytokine network sustaining neutrophil and Th17 activation in untreated early rheumatoid arthritis. Arthritis Res Ther 2010; 12: R196.
2. PENE J, CHEVALIER S, PREISSER L et al.: Chroni-cally inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol 2008; 180: 7423-30.
3. AUPPERLE K, BENNETT B, HAN Z et al.: NF-kappa B regulation by I kappa B kinase-2 in rheumatoid ar-thritis synoviocytes. J Immunol 2001; 166: 2705-11.
4. SIDIROPOULOS PI, GOULIELMOS G, VOLOU-DAKIS GK, PETRAKI E, BOUMPAS DT: Inflamma-somes and rheumatic diseases: evolving concepts. Ann Rheum Dis 2008; 67: 1382-9.
5. HOFFMAN HM, MUELLER JL, BROIDE DH, WAN-DERER AA, KOLODNER RD: Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muck-le-Wells syndrome. Nat Genet 2001; 29: 301-5.
6. DOWDS TA, MASUMOTO J, ZHU L, INOHARA N, NUNEZ G: Cryopyrin-induced interleukin 1beta se-cretion in monocytic cells: enhanced activity of dis-ease-associated mutants and requirement for ASC. J Biol Chem 2004; 279: 21924-8.
7. KASTBOM A, VERMA D, ERIKSSON P et al.: Ge-netic variation in proteins of the cryopyrin inflam-masome influences susceptibility and severity of rheumatoid arthritis (the Swedish TIRA project). Rheumatology (Oxford) 2008; 47: 415-7.
8. KASTBOM A, JOHANSSON M, VERMA D, SODERKVIST P, RANTAPAA-DAHLQVIST S: CARD8 p.C10X polymorphism is associated with inflammatory activity in early rheumatoid arthritis. Ann Rheum Dis 2010; 69: 723-6.
9. WEINMANN P, MOURA RA, CAETANO-LOPES JR et al.: Delayed neutrophil apoptosis in very early rheu-matoid arthritis patients is abrogated by methotrexate therapy. Clin Exp Rheumatol 2007; 25: 885-7.
10. MOURA RA, WEINMANN P, PEREIRA PA et al.: Alterations on peripheral blood B-cell subpopula-tions in very early arthritis patients. Rheumatology (Oxford) 2010; 49: 1082-92.
Fig. 1. Early and established RA patients have a persistent activation of caspase-1. No-tice that, in contrast to EA patients, both in early and established RA patients there are higher active caspase-1 levels. (Unpaired t-test and Bonferroni’s multiple com-parison test).
© Clinical and Experimental Rheumatology 2012


Results
- 45 -
Effective treatment of rat adjuvant-induced
arthritis by celastrol

Results
- 46 -

Autoimmunity Reviews xxx (2012) xxx–xxx
AUTREV-01244; No of Pages 7
Contents lists available at SciVerse ScienceDirect
Autoimmunity Reviews
j ourna l homepage: www.e lsev ie r .com/ locate /aut rev
Review
Effective treatment of rat adjuvant-induced arthritis by celastrol
R. Cascão a, B. Vidal a, H. Raquel a, A. Neves-Costa a, N. Figueiredo a,b, V. Gupta c,J.E. Fonseca a,d,1,⁎, L.F. Moita a,1,⁎a Instituto de Medicina Molecular, Faculdade de Medicina da Universidade de Lisboa, Lisbon, Portugalb Gulbenkian Programme for Advanced Medical Education, Lisbon, Portugalc Division of Nephrology and Hypertension, Department of Medicine, University of Miami, Miami, FL 33136, USAd Rheumatology Department, Centro Hospitalar de Lisboa Norte, EPE, Hospital de Santa Maria, Lisbon, Portugal
⁎ Corresponding authors at: Instituto de Medicina MoPortugal. Tel.: +351 217999544; fax: +351 217999459
E-mail addresses: [email protected] (R. Cascão)[email protected] (N. Figueiredo), vgupta2@
1 Joint senior authors.
1568-9972/$ – see front matter © 2012 Elsevier B.V. Alldoi:10.1016/j.autrev.2012.02.022
Please cite this article as: Cascão R, et adoi:10.1016/j.autrev.2012.02.022
a b s t r a c t
a r t i c l e i n f oArticle history:Received 7 February 2012Accepted 29 February 2012Available online xxxx
Keywords:CelastrolDigoxinWistar AIA rat modelIL-1βCaspase-1Th17 cells
We have previously reported an increase in interleukin (IL)-1β and IL-17 levels, and a continuous activationof caspase-1 in early rheumatoid arthritis (RA) patients. These results suggest that drugs targeting IL-1βregulatory pathways, in addition to tumor necrosis factor (TNF), may constitute promising therapeutic agentsin early RA. We have recently used a THP-1 macrophage-like cell line to screen 2320 compounds for thosethat down-regulate both IL-1β and TNF secretion. Celastrol was one of the most promising therapeutic can-didates identified in that study. Our main goal in the present work was to investigate whether administrationof celastrol is able to attenuate inflammation in a rat model of adjuvant-induced arthritis (AIA). Moreover,since IL-1β is known to play a role in the polarization of Th17 cells, we also investigate whether administra-tion of digoxin, a specific inhibitor of Th17 cells polarization, is able to attenuate inflammation in the same ratmodel. We found that celastrol administration significantly suppressed joint inflammation. The histologicaland immunohistochemical evaluation revealed that celastrol-treated rats had a normal joint structure withcomplete abrogation of the inflammatory infiltrate and cellular proliferation. In contrast, we observed thatdigoxin administration significantly ameliorated inflammation but only if administrated in the early phaseof disease course (after 4 days of disease induction), and it was not efficient at inhibiting the infiltration ofimmune cells within the joint and in preventing damage. Thus, our results suggest that celastrol has signifi-cant anti-inflammatory and anti-proliferative properties and can constitute a potential anti-inflammatorydrug with therapeutic efficacy in the treatment of immune-mediated inflammatory diseases such as RA.Furthermore, we find that early inhibition of Th17 cells polarization ameliorates arthritis but it is not as effec-tive as celastrol.
© 2012 Elsevier B.V. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 02. Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0
2.1. Compounds . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 02.2. IL-1β and TNF secretion assay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 02.3. Cell culture . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 02.4. Animal experimental design . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 02.5. Histological and immunohistochemical evaluation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 02.6. Intracellular IL-17 staining . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 02.7. Statistical analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0
3. Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 03.1. Celastrol decreases IL-1β and TNF secretion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 03.2. Celastrol inhibits the activation of NF-kB and caspase-1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0
lecular, Edifício Egas Moniz, Faculdade de Medicina da Universidade de Lisboa, Av. Professor Egas Moniz, 1649-028 Lisbon,., [email protected] (B. Vidal), [email protected] (H. Raquel), [email protected] (A. Neves-Costa),med.miami.edu (V. Gupta), [email protected] (J.E. Fonseca), [email protected] (L.F. Moita).
rights reserved.
l, Effective treatment of rat adjuvant-induced arthritis by celastrol, Autoimmun Rev (2012),

2 R. Cascão et al. / Autoimmunity Reviews xxx (2012) xxx–xxx
3.3. Celastrol is able to suppress inflammation in Wistar rat adjuvant-induced arthritis . . . . . . . . . . . . . . . . . . . . . . . . . . 03.4. Celastrol prevents joint immune cells infiltration and proliferation as well as cartilage and bone erosions . . . . . . . . . . . . . . . . 03.5. Digoxin delayed the course and reduced the severity of arthritis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 03.6. Digoxin prevents proliferation but not infiltration of immune cells within joints . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0
4. Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 05. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0List of abbreviations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0Conflict of interests . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0Funding . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0Take-home messages . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0
1. Introduction
Rheumatoid arthritis (RA) is a chronic systemic immune-mediated inflammatory disease characterized by synovial hyperplasiacaused by a large proliferative cellular infiltrate of immune cells, highexpression of proinflammatory cytokines and consequent erosion andremodelling of joint cartilage and bone. RA management strategy hasbeen revolutionized in the last decade with the discovery of specifictreatments targeted against cytokines, such as tumor necrosis factor(TNF), and immune (B and T) cells. The current treatment goal forRA is to achieve a state of disease remission but, despite all availabletherapeutic approaches, RA remains an incurable, progressive, debili-tating and destructive disease with only 20% of patients reachingremission [1]. Moreover, these novel treatment strategies are onlyeffective in around 70% of the patients, with many of them eventuallyloosing response to the drugs or being forced to interrupt drug ad-ministration due to adverse effects. Anti-TNF treatment has beenshown to be more effective when introduced early in the diseasecourse [2,3]. However, still a relatively large proportion of patientsfail to respond in these optimal conditions.
We have previously reported increased levels of interleukin (IL)-1β in very recent onset arthritis and in the synovial fluid of estab-lished RA patients [4]. This observation may be explained by activa-tion of caspase-1, the pro-inflammatory enzyme which is activatedby the inflammasome and which is responsible for the processing ofpro-IL-1β, which we have also observed to be increased both inearly and established RA patients [4]. Interestingly, the role of theinflammasomes has been recently addressed in the context of RA. Infact, it has been reported that polymorphisms in NLRP3 and CARD8inflammasome genes are associated with anti-citrullinated proteinantibodies (ACPA)-positive RA, an increased susceptibility for the dis-ease and a worse prognosis for these patients [5–7]. Inflammasomesare activated by several different foreign and self-antigens and recentevidences suggest that these multiprotein complexes may participatein the development of the new syndrome termed ASIA, ‘Autoimmune(Auto-inflammatory) Syndrome [8], which is induced by adjuvantsand assembles a spectrum of immune-mediated diseases triggeredby an adjuvant stimulus [9,10].
The importance of IL-1β in the early phase of RA is furtherhighlighted by reports of its ability to promote the differentiation ofTh17 cells [11,12] through the induction of the transcription factorsIFR4 and RORγt expression [11]. These cells are characterized by theproduction of IL-17, a cytokine that is also up-regulated in the earlyphase of RA [13]. Interestingly, IL-17 serum levels and Th17 frequencyare decreased in Cryopyrin-associated periodic syndromes (CAPS)patients following in vivo IL-1β blockage [14]. Therefore, it is possiblethat IL-1β plays an important role in early rather than late stages ofthe disease and that pathways regulating this cytokine and TNF,such as the inflammasome/caspase-1 and NF-kB, can potentiallyconstitute promising combined therapeutic targets. Based on thisbackground and on the results of a recent drug screen performed inour laboratory for compounds that simultaneously inhibit IL-1β and
Please cite this article as: Cascão R, et al, Effective treatment of ratdoi:10.1016/j.autrev.2012.02.022
TNF secretion (Figueiredo et al., unpublished), we have identifiedcelastrol as a promising therapeutic candidate for arthritis. Celastrol,a pentacyclic-triterpene extract from Trypterigium wilfordii Hook, isused in traditional Chinese medicine and was recently shown topossess anti-tumor [15,16] and anti-inflammatory [17] effects. Ouraim in this study was to investigate whether celastrol administrationis able to attenuate inflammation in a rat model of adjuvant-inducedarthritis (AIA) and which mechanisms might be important for itsprotective effect. Moreover, since IL-1β is known to play a role inthe polarization of Th17 cells, we have also analyzed the anti-inflammatory and anti-proliferative properties of digoxin, a specificinhibitor of RORγt transcriptional activity and consequently inhibitorof Th17 cells polarization [18], in the same rat model. We found thatcelastrol, in contrast to digoxin, has significant anti-inflammatory andanti-proliferative properties and can putatively constitute an anti-inflammatory drug with therapeutic efficacy in the treatment ofimmune-mediated inflammatory diseases such as RA.
2. Methods
2.1. Compounds
Celastrol and digoxin were purchased from Sigma (Missouri, USA).
2.2. IL-1β and TNF secretion assay
THP-1 cells were stimulated with 4% PFA-fixed DH5 Escherichia coli(E. coli) at a Multiplicity of Infection (MOI) of 20 bacterial cells perTHP-1 cell, 1 hour after incubation with celastrol. Cell supernatantswere collected and IL-1β and TNF cytokines quantified by enzymelinked immunosorbent assay (ELISA) technique (R&D systems,Minnesota, USA) according to the provider's instructions.
2.3. Cell culture
THP-1 (ATCC TIB-202) macrophage-like cell line and THP-1/NF-kBreporter cell line were cultured in R10 - RPMImedia 1640 supplemen-ted with 10% (v/v) fetal bovine serum, 1% (v/v) penicillin-streptomycin, 1% (v/v) pyruvate, 1% (v/v) L-glutamine, 1% (v/v) non-essential aminoacids, 1% (v/v) hepes buffer and 2-mercaptoethanolto a final concentration of 0.05 M, as recommended by the AmericanTissue Culture Collection (ATCC). Cells were cultured at 250.000cells/mL, incubated with 10 μM of celastrol for 1 h at 37 °C 5% CO2,and then stimulated with PFA-fixed E. coli (20 E. coli per cell) for 8 hand 24 h at 37 °C 5% CO2. Simultaneously, non-stimulated negativecontrol cells were also cultured at the same density as the stimulatedpopulation for comparison. Caspase-1 activity wasmeasured in THP-1macrophage-like cell line using the Carboxyfluorescein FLICA Detec-tion kit for Caspase Assay (Immunochemistry Technologies, LLC,Minnesota, USA) following the reagent instructions. Briefly, cellsfrom the different assays were protected from light exposure whileincubated for 1 hour at 37 °C with 30X FLICA solution at a 1:30 ratio.
adjuvant-induced arthritis by celastrol, Autoimmun Rev (2012),

3R. Cascão et al. / Autoimmunity Reviews xxx (2012) xxx–xxx
NF-kB activity was measured in THP-1/NF-kB reporter cell line. Lenti-viral particles carrying a NF-kB-responsive GFP-expressing reportergene (Cignal Lenti Reporters, SABiosciences, Maryland, USA) wereused to infect THP-1 cells and to establish a stable cell line. All sampleswere analyzed by flow cytometry using a FACS Calibur (BD biosci-ences, New Jersey, USA). The data collected were further analyzedusing FlowJo software (Tree Star Inc, Oregon, USA).
2.4. Animal experimental design
Wistar AIA rats were purchased from Charles River LaboratoriesInternational (Massachusetts, USA). Female Wistar AIA rats weighing125–150 g were maintained under specific pathogen free (SPF) con-ditions and all experiments were approved by the Animal User andEthical Committees at the Instituto de Medicina Molecular, accordingto the Portuguese law and the European recommendations. Celastroland digoxin were administrated at a dose of 1 μg/g and 2 μg/g bodyweight every day, respectively [18,19]. Drugs and vehicle controlwere dissolved in normal saline solution and injected intraperitone-ally to AIA rats (N=5–10 animals per group) after 4 days (earlytreatment group) and after 11 days (late treatment group) of diseaseinduction, when arthritis was already present. The inflammatoryscore, ankle perimeter and body weight were measured during theperiod of treatment. Inflammatory signs were evaluated by countingthe score of each joint in a scale of 0–3 (0 — absence; 1 — erythema;2 — erythema and swelling; 3 — deformities and functional impair-ment) [20]. The total score of each animal was defined as the sumof the partial scores of each affected joint. Rats were sacrificedafter 19 days of disease evolution and paw samples were collectedfor histological and immunohistochemical evaluation.
2.5. Histological and immunohistochemical evaluation
For histopathological observation, paw, lung, liver, kidney and pan-creas samples were collected at the time of sacrifice. Samples werefixed immediately in 10% neutral buffered formalin solution and thendehydrated with increasing ethanol concentrations (70%, 96% and100%). Paw samples, after being fixed, were also decalcified in 10%formic acid. Samples were next embedded in paraffin, sectioned andstained with hematoxylin and eosin for morphological examination.Paws were also used for immunohistological staining with Ki67 anti-body, a cellular proliferation marker. Tissue sections were incubatedwith primary antibody against rat polyclonal Ki67 (Abcam, Cambridge,UK) andwith EnVision+ (Dako, Glostrup, Denmark). Colourwas devel-oped in solution containing diaminobenzadine-tetrahydrochloride(Sigma, Missouri, USA), 0.5% H2O2 in phosphate-buffered saline buffer(pH 7.6). Slides were counterstained with hematoxylin and mounted.All images were acquired using a Leica DM 2500 (Leica microsystems,Wetzlar, Germany) microscope equipped with a colour camera. Dataregarding the degree of proliferation of synovial cells was scored from0–3 (0 — fewer than three layers; 1 — three to four layers; 2 — five tosix layers; 3 — more than six layers). Lymphoid cell infiltration wasscored from 0–3 (0 — none to diffuse infiltration; 1 — lymphoid cellaggregate; 2— lymphoid follicles; 3— lymphoid follicles with germinalcenter formation) [21].
2.6. Intracellular IL-17 staining
Spleen cells were cultured in complete cell culture media at 37 °C5% CO2. Cells were stimulated for 3 h with brefeldin A (0.01 mg/ml)(Epicenter Technologies, Nebraska, USA), phorbol 12-myristate 13-acetate (PMA) (50 ng/ml) (Sigma, Missouri, USA) and ionomycin(500 ng/ml) (Calbiochem, Darmstadt, Germany). Next, cells werepermeabilised using saponin (Sigma, Missouri, USA) and stainedwith anti-IL17 FITC (Biolegend, California, USA) to detect intracellularIL-17. Lastly, cells were acquired with a FACS LSR Fortessa (BD
Please cite this article as: Cascão R, et al, Effective treatment of ratdoi:10.1016/j.autrev.2012.02.022
biosciences, New Jersey, USA) and data collected were further ana-lyzed using FlowJo software (Tree Star Inc, Oregon, USA).
2.7. Statistical analysis
Statistical differences were determined with non-parametricKruskal-Wallis and Mann–Whitney tests using GraphPad Prism(GraphPad, California, USA). Differences were considered statisticallysignificant for pb0.05.
3. Results
3.1. Celastrol decreases IL-1β and TNF secretion
Based on the hypothesis that drugs which block the secretion ofboth IL-1β and TNF might be particularly effective at decreasingearly disease activity in RA, we have recently used the humanTHP-1 macrophage-like cell line to screen for drugs that can simulta-neously down-regulate the secretion of both cytokines. Among the2320 tested drugs included in the Spectrum collection (MicrosourceDiscovery Systems, Connecticut, USA), we found 45 that significantlydecrease the levels of both IL-1β and TNF secretion (Figueiredo et al.,unpublished). We further narrowed the selection by taking intoaccount the possible human tolerability to chronic exposure andother biological properties that could be of interest in the contextof RA. Celastrol was thus selected for testing in a rat model ofadjuvant-induced arthritis (AIA), due to its prior human use in tradi-tional Chinese medicine and due to its concomitant alleged anti-proliferative effects [15,16]. We started by validating the effect ofcelastrol in the inhibition of IL-1β and TNF secretion in a humanTHP-1 macrophage-like cell line, by using increasing concentrationsof celastrol for 1 hour before challenging them with PFA-fixed E. colifor 6 hours. The conditioned media was then probed for the secretionof either IL-1β or TNF using ELISA technique. Celastrol was very effec-tive at inhibiting the secretion of both cytokines over a wide range oftested concentrations (Fig. 1A).
3.2. Celastrol inhibits the activation of NF-kB and caspase-1
The sequence of events culminating in IL-1β secretion is complex,but it can be summarized in two steps: induction of pro-IL-1β and itsprocessing by activated caspase-1 (reviewed in [22]). Both pro-IL-1βand TNF depend on NF-kB activation for the transcription of theirrespective mRNAs. We therefore tested the effect of celastrol onthese key pathways. To investigate its effect in the activation of NF-kB, we used an NF-kB reporter cell line made by stably infectingTHP-1 cells with a commercial lentiviral GFP reporter under the con-trol of a minimal CMV promoter and tandem repeats of the NF-kBtranscriptional response element (TRE). We found that celastrol wasable to suppress NF-kB reporter activation upon E. coli stimulationin comparison with cells that were also stimulated but did not receivetreatment (Fig. 1B). To test the effect of this drug in caspase-1 proces-sing and activation we used a caspase-1 fluorescent substrate andmeasured the relative active caspase-1 levels using FACS. Also inthis case, celastrol administration decreased the activation ofcaspase-1 (Fig. 1B). We can thus conclude that celastrol inhibits NF-kB and caspase-1 activation.
3.3. Celastrol is able to suppress inflammation in Wistar ratadjuvant-induced arthritis
To study the anti-inflammatory properties of celastrol in vivo, AIArats were treated daily with this drug after the disease had alreadybecome symptomatic. We started the treatment after 4 days of dis-ease induction (early treatment group) and after 11 days of diseaseinduction (late treatment group). The inflammatory score and ankle
adjuvant-induced arthritis by celastrol, Autoimmun Rev (2012),
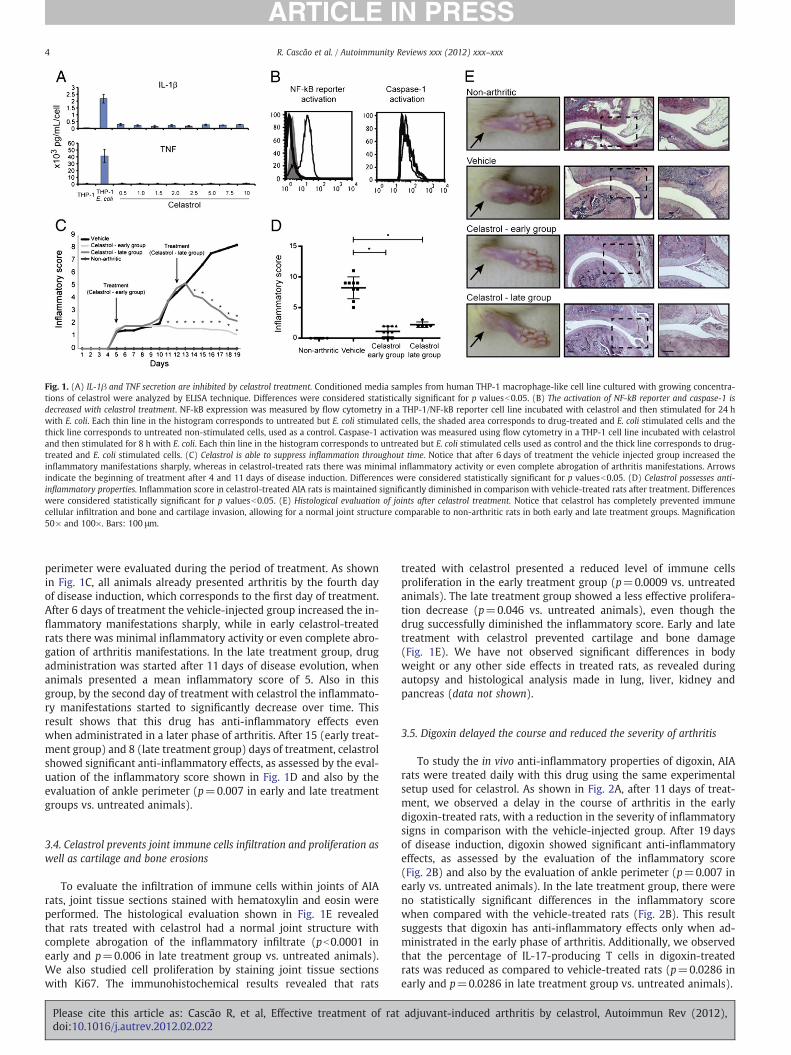
Fig. 1. (A) IL-1β and TNF secretion are inhibited by celastrol treatment. Conditioned media samples from human THP-1 macrophage-like cell line cultured with growing concentra-tions of celastrol were analyzed by ELISA technique. Differences were considered statistically significant for p valuesb0.05. (B) The activation of NF-kB reporter and caspase-1 isdecreased with celastrol treatment. NF-kB expression was measured by flow cytometry in a THP-1/NF-kB reporter cell line incubated with celastrol and then stimulated for 24 hwith E. coli. Each thin line in the histogram corresponds to untreated but E. coli stimulated cells, the shaded area corresponds to drug-treated and E. coli stimulated cells and thethick line corresponds to untreated non-stimulated cells, used as a control. Caspase-1 activation was measured using flow cytometry in a THP-1 cell line incubated with celastroland then stimulated for 8 h with E. coli. Each thin line in the histogram corresponds to untreated but E. coli stimulated cells used as control and the thick line corresponds to drug-treated and E. coli stimulated cells. (C) Celastrol is able to suppress inflammation throughout time. Notice that after 6 days of treatment the vehicle injected group increased theinflammatory manifestations sharply, whereas in celastrol-treated rats there was minimal inflammatory activity or even complete abrogation of arthritis manifestations. Arrowsindicate the beginning of treatment after 4 and 11 days of disease induction. Differences were considered statistically significant for p valuesb0.05. (D) Celastrol possesses anti-inflammatory properties. Inflammation score in celastrol-treated AIA rats is maintained significantly diminished in comparison with vehicle-treated rats after treatment. Differenceswere considered statistically significant for p valuesb0.05. (E) Histological evaluation of joints after celastrol treatment. Notice that celastrol has completely prevented immunecellular infiltration and bone and cartilage invasion, allowing for a normal joint structure comparable to non-arthritic rats in both early and late treatment groups. Magnification50× and 100×. Bars: 100 μm.
4 R. Cascão et al. / Autoimmunity Reviews xxx (2012) xxx–xxx
perimeter were evaluated during the period of treatment. As shownin Fig. 1C, all animals already presented arthritis by the fourth dayof disease induction, which corresponds to the first day of treatment.After 6 days of treatment the vehicle-injected group increased the in-flammatory manifestations sharply, while in early celastrol-treatedrats there was minimal inflammatory activity or even complete abro-gation of arthritis manifestations. In the late treatment group, drugadministration was started after 11 days of disease evolution, whenanimals presented a mean inflammatory score of 5. Also in thisgroup, by the second day of treatment with celastrol the inflammato-ry manifestations started to significantly decrease over time. Thisresult shows that this drug has anti-inflammatory effects evenwhen administrated in a later phase of arthritis. After 15 (early treat-ment group) and 8 (late treatment group) days of treatment, celastrolshowed significant anti-inflammatory effects, as assessed by the eval-uation of the inflammatory score shown in Fig. 1D and also by theevaluation of ankle perimeter (p=0.007 in early and late treatmentgroups vs. untreated animals).
3.4. Celastrol prevents joint immune cells infiltration and proliferation aswell as cartilage and bone erosions
To evaluate the infiltration of immune cells within joints of AIArats, joint tissue sections stained with hematoxylin and eosin wereperformed. The histological evaluation shown in Fig. 1E revealedthat rats treated with celastrol had a normal joint structure withcomplete abrogation of the inflammatory infiltrate (pb0.0001 inearly and p=0.006 in late treatment group vs. untreated animals).We also studied cell proliferation by staining joint tissue sectionswith Ki67. The immunohistochemical results revealed that rats
Please cite this article as: Cascão R, et al, Effective treatment of ratdoi:10.1016/j.autrev.2012.02.022
treated with celastrol presented a reduced level of immune cellsproliferation in the early treatment group (p=0.0009 vs. untreatedanimals). The late treatment group showed a less effective prolifera-tion decrease (p=0.046 vs. untreated animals), even though thedrug successfully diminished the inflammatory score. Early and latetreatment with celastrol prevented cartilage and bone damage(Fig. 1E). We have not observed significant differences in bodyweight or any other side effects in treated rats, as revealed duringautopsy and histological analysis made in lung, liver, kidney andpancreas (data not shown).
3.5. Digoxin delayed the course and reduced the severity of arthritis
To study the in vivo anti-inflammatory properties of digoxin, AIArats were treated daily with this drug using the same experimentalsetup used for celastrol. As shown in Fig. 2A, after 11 days of treat-ment, we observed a delay in the course of arthritis in the earlydigoxin-treated rats, with a reduction in the severity of inflammatorysigns in comparison with the vehicle-injected group. After 19 daysof disease induction, digoxin showed significant anti-inflammatoryeffects, as assessed by the evaluation of the inflammatory score(Fig. 2B) and also by the evaluation of ankle perimeter (p=0.007 inearly vs. untreated animals). In the late treatment group, there wereno statistically significant differences in the inflammatory scorewhen compared with the vehicle-treated rats (Fig. 2B). This resultsuggests that digoxin has anti-inflammatory effects only when ad-ministrated in the early phase of arthritis. Additionally, we observedthat the percentage of IL-17-producing T cells in digoxin-treatedrats was reduced as compared to vehicle-treated rats (p=0.0286 inearly and p=0.0286 in late treatment group vs. untreated animals).
adjuvant-induced arthritis by celastrol, Autoimmun Rev (2012),
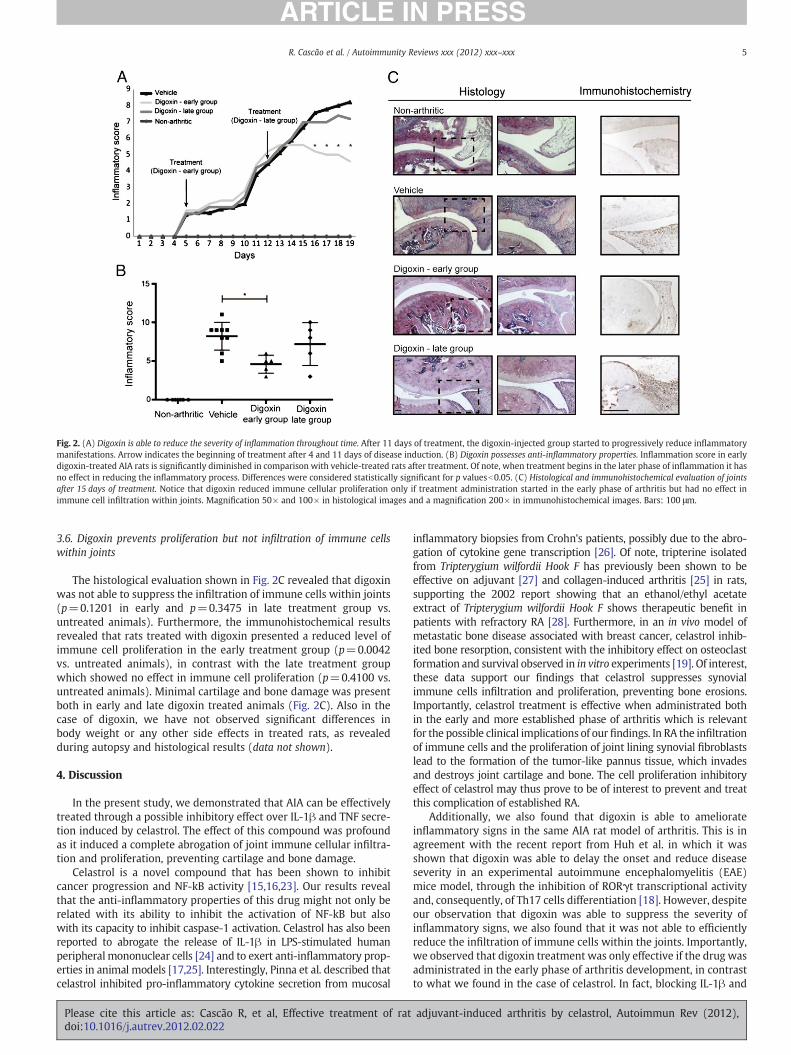
Fig. 2. (A) Digoxin is able to reduce the severity of inflammation throughout time. After 11 days of treatment, the digoxin-injected group started to progressively reduce inflammatorymanifestations. Arrow indicates the beginning of treatment after 4 and 11 days of disease induction. (B) Digoxin possesses anti-inflammatory properties. Inflammation score in earlydigoxin-treated AIA rats is significantly diminished in comparison with vehicle-treated rats after treatment. Of note, when treatment begins in the later phase of inflammation it hasno effect in reducing the inflammatory process. Differences were considered statistically significant for p valuesb0.05. (C) Histological and immunohistochemical evaluation of jointsafter 15 days of treatment. Notice that digoxin reduced immune cellular proliferation only if treatment administration started in the early phase of arthritis but had no effect inimmune cell infiltration within joints. Magnification 50× and 100× in histological images and a magnification 200× in immunohistochemical images. Bars: 100 μm.
5R. Cascão et al. / Autoimmunity Reviews xxx (2012) xxx–xxx
3.6. Digoxin prevents proliferation but not infiltration of immune cellswithin joints
The histological evaluation shown in Fig. 2C revealed that digoxinwas not able to suppress the infiltration of immune cells within joints(p=0.1201 in early and p=0.3475 in late treatment group vs.untreated animals). Furthermore, the immunohistochemical resultsrevealed that rats treated with digoxin presented a reduced level ofimmune cell proliferation in the early treatment group (p=0.0042vs. untreated animals), in contrast with the late treatment groupwhich showed no effect in immune cell proliferation (p=0.4100 vs.untreated animals). Minimal cartilage and bone damage was presentboth in early and late digoxin treated animals (Fig. 2C). Also in thecase of digoxin, we have not observed significant differences inbody weight or any other side effects in treated rats, as revealedduring autopsy and histological results (data not shown).
4. Discussion
In the present study, we demonstrated that AIA can be effectivelytreated through a possible inhibitory effect over IL-1β and TNF secre-tion induced by celastrol. The effect of this compound was profoundas it induced a complete abrogation of joint immune cellular infiltra-tion and proliferation, preventing cartilage and bone damage.
Celastrol is a novel compound that has been shown to inhibitcancer progression and NF-kB activity [15,16,23]. Our results revealthat the anti-inflammatory properties of this drug might not only berelated with its ability to inhibit the activation of NF-kB but alsowith its capacity to inhibit caspase-1 activation. Celastrol has also beenreported to abrogate the release of IL-1β in LPS-stimulated humanperipheral mononuclear cells [24] and to exert anti-inflammatory prop-erties in animal models [17,25]. Interestingly, Pinna et al. described thatcelastrol inhibited pro-inflammatory cytokine secretion from mucosal
Please cite this article as: Cascão R, et al, Effective treatment of ratdoi:10.1016/j.autrev.2012.02.022
inflammatory biopsies from Crohn's patients, possibly due to the abro-gation of cytokine gene transcription [26]. Of note, tripterine isolatedfrom Tripterygium wilfordii Hook F has previously been shown to beeffective on adjuvant [27] and collagen-induced arthritis [25] in rats,supporting the 2002 report showing that an ethanol/ethyl acetateextract of Tripterygium wilfordii Hook F shows therapeutic benefit inpatients with refractory RA [28]. Furthermore, in an in vivo model ofmetastatic bone disease associated with breast cancer, celastrol inhib-ited bone resorption, consistent with the inhibitory effect on osteoclastformation and survival observed in in vitro experiments [19]. Of interest,these data support our findings that celastrol suppresses synovialimmune cells infiltration and proliferation, preventing bone erosions.Importantly, celastrol treatment is effective when administrated bothin the early and more established phase of arthritis which is relevantfor the possible clinical implications of our findings. In RA the infiltrationof immune cells and the proliferation of joint lining synovial fibroblastslead to the formation of the tumor-like pannus tissue, which invadesand destroys joint cartilage and bone. The cell proliferation inhibitoryeffect of celastrol may thus prove to be of interest to prevent and treatthis complication of established RA.
Additionally, we also found that digoxin is able to ameliorateinflammatory signs in the same AIA rat model of arthritis. This is inagreement with the recent report from Huh et al. in which it wasshown that digoxin was able to delay the onset and reduce diseaseseverity in an experimental autoimmune encephalomyelitis (EAE)mice model, through the inhibition of RORγt transcriptional activityand, consequently, of Th17 cells differentiation [18]. However, despiteour observation that digoxin was able to suppress the severity ofinflammatory signs, we also found that it was not able to efficientlyreduce the infiltration of immune cells within the joints. Importantly,we observed that digoxin treatment was only effective if the drug wasadministrated in the early phase of arthritis development, in contrastto what we found in the case of celastrol. In fact, blocking IL-1β and
adjuvant-induced arthritis by celastrol, Autoimmun Rev (2012),

6 R. Cascão et al. / Autoimmunity Reviews xxx (2012) xxx–xxx
TNF simultaneously results in a significant inhibitory effect in arthritisprogression and severity, even when administrated in a later phaseof disease course, with a complete abrogation of the inflammatoryscore, infiltration and proliferation of immune cells within jointsand prevention of structural damage. In contrast, we observed thatdigoxin had a slower and less efficient effect on disease progression.These data indicate that IL-1β, in the context of arthritis, might playa role independent from Th17/IL-17. Besides inducing Th17 cell polar-ization, IL-1β also directly stimulates the influx of neutrophils andmacrophages into the damaged site. These cells in turn can destroythe tissue by the release of proteases and reactive oxygen species[29], and also by the formation of osteoclasts [30] leading to tissuedamage and consequent functional disability characteristic of RA pa-tients. IL-1β, together with IL-6 and TNF, also has a potent capacityto induce the receptor activator of nuclear factor kappa-B ligand(RANKL) expression on synovial fibroblasts/osteoblasts and to facili-tate RANK signaling, thus directly contributing to the bone destruc-tion process. In contrast, IL-17 seems to have a more limited effecton inflammatory cell influx and consequent inflammatory symptoms,as we observed in this study. Moreover, in agreement with this fact, arecent phase II study testing an anti-IL-17A drug in RA did not achieveits primary end point [31]. This does not preclude the important in-volvement of Th17 cells in driving the innate immune inflammationtowards the adaptive (auto)immune chronic inflammation in RA,involving several other cytokines apart from IL-17A [32].
Despite this apparent crucial role of IL-1β signaling in RA, clinicalbenefits after IL-1β inhibition have been modest compared to anti-TNF drugs, at least in moderate to severe long established RA. Further,in 2004 a study which tested the efficacy of combination therapy
Fig. 3. Proposed mechanism for celastrol anti-inflammatory effects. Celastrol showed anti-inof arthritis development and abrogation of joint immune cellular infiltration and proliferatiof caspase-1 and NF-kB activation and, consequently, lead to a decrease in IL-1β and TNF scaspase recruitment domain, CARD — caspase recruitment domain, DAMP — danger associkappa-light-chain-enhancer of activated B cells, NLRP — NOD-like receptor protein, PAMP —
TNF — tumor necrosis factor.
Please cite this article as: Cascão R, et al, Effective treatment of ratdoi:10.1016/j.autrev.2012.02.022
using anakinra and etanercept in 244 long-standing and very activeRA patients who have been treated unsuccessfully with MTX showedthat concomitant IL-1β and TNF inhibition provides no added benefitand increased infections as compared to etanercept alone [33]. Possi-bly, in the context of RA inhibiting the IL-1β pathway at the receptorlevel is not an effective strategy but an upstream inhibition mightwork better, at least, in animal models. On top of that, downregulat-ing TNF and IL-1β production might be safer than inhibitingcompletely TNF and IL-1β.
5. Conclusions
In conclusion, celastrol can putatively constitute an anti-inflammatoryand anti-proliferative drug with therapeutic efficacy in the treatmentof immune-mediated inflammatory diseases such as RA, possibly throughthe down-regulation of caspase-1 and inhibition of NF-kB activation(Fig. 3). Its anti-proliferative effects might be of additional value in RAto counteract the formation of the characteristic pannus leading to thedestruction of cartilage and bone. In addition, we have shown thatdespite the ability to decrease the severity of early inflammatory signsin AIA, digoxin is not effective in reducing the infiltration of immunecells within the joints. We thus suggest that the isolated inhibition ofTh17 polarization might be a strategy with limited efficacy, at least inestablished RA. Further animal experimentation is required to deter-mine the real efficacy and safety of celastrol for arthritis treatment butthese results suggest that it might be worth of entering into phase Iclinical trials. Simultaneously, this study highlights the need for moreresearch on the role of the inflammasome/caspase-1 and NF-kB path-ways in the etiopathology of RA.
flammatory and anti-proliferative properties in vivo, promoting a complete suppressionon, preventing cartilage and bone damage. This compound induced a down-regulationecretion. ASC — adaptor molecule apoptosis associated speck-like protein containing aated molecular pattern, IL — interleukin, IL-1R — IL-1 receptor, NF-kB — nuclear factorpathogen associated molecular pattern, PYR — pyrin domain, TLR — toll-like receptor,
adjuvant-induced arthritis by celastrol, Autoimmun Rev (2012),

7R. Cascão et al. / Autoimmunity Reviews xxx (2012) xxx–xxx
List of abbreviations
RA rheumatoid arthritisTNF tumor necrosis factorIL interleukinACPA anti-citrullinated protein antibodiesAIA adjuvant-induced arthritisCAPS cryopyrin-associated periodic syndromesMOI multiplicity of infectionELISA enzyme linked immunosorbent assaySPF specific pathogen freeTRE transcriptional response elementEAE experimental autoimmune encephalomyelitisRANKL receptor activator of nuclear factor kappa-B ligand
Conflict of interests
The authors declare that they have no competing interests.
Funding
This work was supported by a grant (SFRH/BD/40513/2007)from Fundação para a Ciência e a Tecnologia (FCT). Work in LuisMoita's laboratory is supported by FCT (PIC/IC/82991/2007 andPTDC/SAU-MII/100780/2008) and Fundação Luso-Americana para oDesenvolvimento (FLAD). The Gulbenkian Programme for AdvancedMedical Education was sponsored by Fundação Calouste Gulbenkian,Fundação Champalimaud, Ministério da Saúde e FCT.
Take-home messages
• Celastrol has significant anti-inflammatory and anti-proliferativeproperties.
• Digoxin is not as effective as celastrol treatment for AIA.• Blocking IL-1β and TNF (upstream to their receptors) is effective inarthritis.
• Therapies targeting IL-1β pathway should be re-evaluated in RApatients.
Acknowledgements
The authors would like to acknowledge Ana Lopes for technicalassistance and Ana Luisa Caetano for technical support with FACS.
References
[1] Lindqvist E, Saxne T, Geborek P, Eberhardt K. Ten year outcome in a cohort ofpatients with early rheumatoid arthritis: health status, disease process, anddamage. Ann Rheum Dis 2002;61:1055–9.
[2] Emery P, Breedveld FC, Hall S, Durez P, Chang DJ, Robertson D, et al. Comparison ofmethotrexate monotherapy with a combination of methotrexate and etanerceptin active, early, moderate to severe rheumatoid arthritis (COMET): a randomised,double-blind, parallel treatment trial. Lancet 2008;372:375–82.
[3] Klarenbeek NB, Guler-Yuksel M, van der Kooij SM, Han KH, Ronday HK, KerstensPJ, et al. The impact of four dynamic, goal-steered treatment strategies on the 5-year outcomes of rheumatoid arthritis patients in the BeSt study. Ann RheumDis 2011;70:1039–46.
[4] Cascao R, Polido-Pereira J, Canhao H, Rodrigues AM, Navalho M, Raquel H, et al.Caspase-1 is active since the early phase of rheumatoid arthritis. Clin Exp Rheumatol2012;30(1):0144.
[5] Kastbom A, Johansson M, Verma D, Soderkvist P, Rantapaa-Dahlqvist S. CARD8p.C10X polymorphism is associated with inflammatory activity in early rheuma-toid arthritis. Ann Rheum Dis 2010;69:723–6.
[6] Fontalba A, Martinez-Taboada V, Gutierrez O, Pipaon C, Benito N, Balsa A, et al.Deficiency of the NF-kappaB inhibitor caspase activating and recruitment domain
Please cite this article as: Cascão R, et al, Effective treatment of ratdoi:10.1016/j.autrev.2012.02.022
8 in patients with rheumatoid arthritis is associated with disease severity. JImmunol 2007;179:4867–73.
[7] Kastbom A, Verma D, Eriksson P, Skogh T, Wingren G, Soderkvist P. Geneticvariation in proteins of the cryopyrin inflammasome influences susceptibilityand severity of rheumatoid arthritis (the Swedish TIRA project). Rheumatology(Oxford) 2008;47:415–7.
[8] Shoenfeld Y, Agmon-Levin N. ‘ASIA’ — autoimmune/inflammatory syndromeinduced by adjuvants. J Autoimmun 2011;36:4–8.
[9] Agmon-Levin N, Paz Z, Israeli E, Shoenfeld Y. Vaccines and autoimmunity. Nat RevRheumatol 2009;5:648–52.
[10] Balofsky A, Agmon-Levin N, Shoenfeld Y. The new H1N1 and HPV vaccines and oldfears. Curr Opin Rheumatol 2010;22:431–6.
[11] Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Criticalregulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity2009;30:576–87.
[12] Kryczek I, Wei S, Vatan L, Escara-Wilke J, Szeliga W, Keller ET, et al. Cutting edge:opposite effects of IL-1 and IL-2 on the regulation of IL-17+ T cell pool IL-1subverts IL-2-mediated suppression. J Immunol 2007;179:1423–6.
[13] Cascao R, Moura RA, Perpetuo I, Canhao H, Vieira-Sousa E, Mourao AF, et al.Identification of a cytokine network sustaining neutrophil and Th17 activationin untreated early rheumatoid arthritis. Arthritis Res Ther 2010;12:R196.
[14] Lasiglie D, Traggiai E, Federici S, Alessio M, Buoncompagni A, Accogli A, et al. Roleof IL-1 beta in the development of human T(H)17 cells: lesson from NLPR3 mutatedpatients. PLoS One 2011;6:e20014.
[15] He D, Xu Q, Yan M, Zhang P, Zhou X, Zhang Z, et al. The NF-kappa B inhibitor,celastrol, could enhance the anti-cancer effect of gambogic acid on oral squamouscell carcinoma. BMC Cancer 2009;9:343.
[16] Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, et al. Inhibition of NF-kappa Bactivation through targeting I kappa B kinase by celastrol, a quinone methidetriterpenoid. Biochem Pharmacol 2006;72:1311–21.
[17] Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potentantioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer'sdisease. Prog Neuropsychopharmacol Biol Psychiatry 2001;25:1341–57.
[18] Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, et al. Digoxin andits derivatives suppress TH17 cell differentiation by antagonizing RORgammatactivity. Nature 2011;472:486–90.
[19] Idris AI, Libouban H, Nyangoga H, Landao-Bassonga E, Chappard D, Ralston SH.Pharmacologic inhibitors of IkappaB kinase suppress growth and migration ofmammary carcinosarcoma cells in vitro and prevent osteolytic bone metastasisin vivo. Mol Cancer Ther 2009;8:2339–47.
[20] da Silva JA, Fonseca JE, Graca L, Moita L, Carmo-Fonseca M. Reinnervation of post-arthritic joints in the rat. Clin Exp Rheumatol 1996;14:43–51.
[21] Tsubaki T, Arita N, Kawakami T, Shiratsuchi T, Yamamoto H, Takubo N, et al.Characterization of histopathology and gene-expression profiles of synovitis inearly rheumatoid arthritis using targeted biopsy specimens. Arthritis Res Ther2005;7:R825–36.
[22] Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis.Nat Immunol 2009;10:241–7.
[23] Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from theChinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresseshuman prostate cancer growth in nude mice. Cancer Res 2006;66:4758–65.
[24] Ngassapa O, Soejarto DD, Pezzuto JM, Farnsworth NR. Quinone-methide triter-penes and salaspermic acid from Kokoona ochracea. J Nat Prod 1994;57:1–8.
[25] Li H, Jia YF, Pan Y, Pan DJ, Li D, Zhang LX. Effect of tripterine on collagen-inducedarthritis in rats. Zhongguo Yao Li Xue Bao 1997;18:270–3.
[26] Pinna GF, Fiorucci M, Reimund JM, Taquet N, Arondel Y, Muller CD. Celastrolinhibits pro-inflammatory cytokine secretion in Crohn's disease biopsies. BiochemBiophys Res Commun 2004;322:778–86.
[27] Li H, Zhang YY, Tan HW, Jia YF, Li D. Therapeutic effect of tripterine on adjuvantarthritis in rats. J Ethnopharmacol 2008;118:479–84.
[28] Tao X, Younger J, Fan FZ, Wang B, Lipsky PE. Benefit of an extract of TripterygiumWilfordii Hook F in patients with rheumatoid arthritis: a double-blind, placebo-controlled study. Arthritis Rheum 2002;46:1735–43.
[29] Cascao R, Rosario HS, Souto-Carneiro MM, Fonseca JE. Neutrophils in rheumatoidarthritis: more than simple final effectors. Autoimmun Rev 2010;9:531–5.
[30] Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between theimmune and bone systems. Nat Rev Immunol 2007;7:292–304.
[31] Genovese M, Durez P, Richards H, Hugot S, Thangavelu K, Mpofu S. Secukinumab(AIN457), a novel monoclonal antibody targeting IL-17A demonstrates efficacyin active rheumatoid arthritis patients despite stable methotrexate treatment:results of a phase IIb study. ACR; 2010. abstr #L9:2010.
[32] Ferraccioli G, Zizzo G. The potential role of Th17 in mediating the transitionfrom acute to chronic autoimmune inflammation: rheumatoid arthritis as amodel. Discov Med 2011;11:413–24.
[33] Genovese MC, Cohen S, Moreland L, Lium D, Robbins S, Newmark R, et al. Combi-nation therapy with etanercept and anakinra in the treatment of patients withrheumatoid arthritis who have been treated unsuccessfully with methotrexate.Arthritis Rheum 2004;50:1412–9.
adjuvant-induced arthritis by celastrol, Autoimmun Rev (2012),


Results
- 47 -
Gambogic acid is a potent anti-inflammatory
and anti-proliferative drug in a rat model of
adjuvant-induced arthritis

Results
- 48 -

1
GAMBOGIC ACID IS A POTENT ANTI-INFLAMMATORY AND 1
ANTI-PROLIFERATIVE DRUG IN A RAT MODEL OF ADJUVANT-2
INDUCED ARTHRITIS 3
4
R. Cascãoa, B. Vidal
a, H. Raquel
a, A. Neves-Costa
a, N. Figueiredo
a,b, V. Gupta
c, J. E. 5
Fonsecaa,d*
, L. F. Moitaa*
6
7
aInstituto de Medicina Molecular, Faculdade de Medicina da Universidade de Lisboa, 8
Lisbon, Portugal. 9
bGulbenkian Programme for Advanced Medical Education, Lisbon, Portugal. 10
cDivision of Nephrology and Hypertension, Department of Medicine, University of 11
Miami, Miami, FL 33136, U.S.A. 12
dRheumatology Department, Centro Hospitalar de Lisboa Norte, EPE, Hospital de 13
Santa Maria, Lisbon, Portugal. 14
* Joint senior authors. 15
16
Corresponding author: 17
Luis Ferreira Moita 18
Cell Biology of the Immune System Unit 19
Instituto de Medicina Molecular 20
Edifício Egas Moniz 21
Faculdade de Medicina da Universidade de Lisboa 22
Av. Professor Egas Moniz 23
1649-028 Lisbon 24
Tel: (+351) 217999544 25

2
Fax: (+351) 217999459 26
Email: [email protected] 27
28
ABSTRACT 29
Background: In a previous study, we have reported a continuous activation of caspase-30
1 in rheumatoid arthritis (RA) patients in the early phase of the disease, as well as 31
increased levels of interleukin (IL)-1. These observations raised the hypothesis that 32
drugs targeting IL-1 regulatory pathways, as well as tumor necrosis factor (TNF), may 33
be particularly effective for the treatment of early RA. We have recently identified 34
gambogic acid as one of the most promising therapeutic candidates to simultaneously 35
block the secretion of IL-1 and TNF after screening ~2320 compounds, using a THP1 36
macrophage-like cell line challenged with E. coli. Our main goal here is to investigate 37
whether administration of gambogic acid is able to attenuate inflammation in a rat 38
model of adjuvant-induced arthritis (AIA). Methodology/Principle Findings: 39
Gambogic acid was administered to AIA rats in the early phase of arthritis (4 days after 40
disease induction) for a period of 15 days. The inflammatory score, ankle perimeter and 41
body weight were evaluated during the time of treatment. Rats were sacrificed after 19 42
days of disease progression and paw samples were collected for histological and 43
immunohistochemical evaluation. We found that inflammation in joints was 44
significantly suppressed with administration of gambogic acid. Histological and 45
immunohistochemical evaluation of treated rats revealed normal joint structures with 46
complete abrogation of the inflammatory infiltrate and cellular proliferation. 47
Conclusions/Significance: Our results suggest that gambogic acid has significant anti-48
inflammatory properties and can putatively constitute an anti-inflammatory drug with 49

3
therapeutic efficacy in the treatment of inflammatory diseases such as RA, possibly 50
through the down-regulation of IL-1β and TNF secretion. 51
52
INTRODUCTION 53
Rheumatoid arthritis (RA), which afflicts about 1% of the world population, is the most 54
common of the inflammatory joint diseases. This disease can have a very aggressive 55
course and poor outcome as inferred by the analysis of its social impact (after 10 years, 56
more than 50% of the RA patients are too impaired to perform professional activities) 57
[1] and life expectancy diminishes 10 years due to disease activity and associated 58
comorbidities [2]. RA is a chronic systemic inflammatory disease characterized by 59
synovial hyperplasia caused by a large proliferative cellular infiltrate of leukocytes, high 60
expression levels of proinflammatory cytokines and consequent erosion of joint 61
cartilage and bone. The therapeutic approach of RA has been revolutionized in the last 62
decade with the discovery of specific targeted treatments. However, despite all available 63
therapeutic options, RA remains a progressive, destructive and debilitating disease with 64
only 20% of patients reaching remission [3]. Anakinra, an antagonist of interleukin 65
(IL)-1, was approved for RA treatment in the last decade. However, the real impact on 66
disease activity has been shown in practice to be lower than what was anticipated from 67
clinical trial results, casting doubts on the role of IL-1β as a therapeutic target [4]. 68
Nonetheless, we have previously reported increased levels of IL-1β in very recent onset 69
arthritis and in the synovial fluid of established RA patients [5]. This observation could 70
be explained by the activation of caspase-1 that we also have observed both in early and 71
established RA patients [6]. Therefore, it is possible that IL-1β plays an important role 72
in early rather than late stages of the disease and that pathways regulating this cytokine, 73
such as caspase-1 and NF-kB activation, can potentially constitute promising 74

4
therapeutic targets for specific drugs. The effect might be further boosted if an 75
inhibitory effect on tumor necrosis factor (TNF) can also be achieved. Based on the 76
results of a recent drug screen for compounds that simultaneously inhibit IL-1β and 77
TNF secretion (Figueiredo et al., unpublished), we chose gambogic acid as a promising 78
therapeutic candidate for the treatment of arthritis. Gambogic acid is a polyprenylated 79
xanthone abundant in resin derived from Garcinia hanburyi and G. Morella and is used 80
in Southeast Asia complementary and alternative medicine [7]. Recent studies showed 81
that gambogic acid could inhibit the growth of a wide range of tumor cells [8]. Our aim 82
in this study is to investigate whether gambogic acid administration is able to attenuate 83
inflammation in a rat model of adjuvant-induced arthritis (AIA). 84
85
METHODS 86
Ethics statement 87
All experiments were approved by the Animal User and Ethical Committees at the 88
Instituto de Medicina Molecular, according to the Portuguese law and the European 89
recommendations. 90
91
Compound 92
Gambogic acid was purchased from Santa Cruz Biotechnology (Santa Cruz, USA). 93
94
IL-1 and TNF quantification 95
THP-1 cells were stimulated with 4% PFA-fixed DH5 Escherichia coli (E. coli) at a 96
Multiplicity of Infection (MOI) of 20 bacterial cells per THP-1 cell, 1 hour after 97
incubation with gambogic acid. Cell supernatants were collected and IL-1β and TNF 98

5
cytokines quantified by enzyme linked immunosorbent assay (ELISA) (R&D systems, 99
USA) according to the provider's instructions. 100
101
AIA rat model and assessment of arthritis 102
Female Wistar AIA rats were purchased from Charles River Laboratories International 103
(Massachusetts, USA) and maintained under specific pathogen free (SPF) conditions. 104
Gambogic acid was administrated at a dose of 4µg/g body weight every day. Drug and 105
vehicle control were dissolved in normal saline solution and injected intraperitoneally to 106
AIA rats (N=5-10 animals per group) after 4 days of disease induction, when arthritis 107
was already present, and then for a period of 15 days. The inflammatory score, ankle 108
perimeter and body weight were measured during the time of treatment. Inflammatory 109
signs were evaluated trough the counting of the score of each joint in a scale of 0‐3 (0 – 110
absence; 1 – erythema; 2 – erythema and swelling; 3 – deformities and functional 111
impairment). The total score of each animal was defined as the sum of the partial scores 112
of each affected joint [9]. Rats were sacrificed after 19 days of disease evolution and 113
paw samples were collected for histological and immunohistochemical evaluation. 114
115
Histology and Immunohistochemistry 116
For histopathological observation, paws, lungs, livers, kidneys and pancreas samples 117
were collected at the time of sacrifice. Samples were fixed immediately in 10% neutral 118
buffered formalin solution and then dehydrated using increased ethanol concentrations 119
(70%, 96% and 100%). Paw samples, after being fixed, were also decalcified in 10% 120
formic acid. Samples were next embedded in paraffin, sectioned and stained with 121
hematoxylin and eosin for morphological examination. Paws were also used for 122
immunohistochemical staining with Ki67 antibody, a cellular proliferation marker. 123

6
Tissue sections were incubated with primary antibody against rat polyclonal Ki67 124
(Abcam, UK) and with EnVision+ (Dako, Denmark). Colour was developed in solution 125
containing diaminobenzadine-tetrahydrochloride (Sigma, USA), 0.5% H2O2 in 126
phosphate-buffered saline buffer (pH 7.6). Slides were counterstained with hematoxylin 127
and mounted. All images were acquired using a Leica DM 2500 (Leica microsystems, 128
Germany) microscope equipped with a colour camera. Data regarding the degree of 129
proliferation of synovial cells was scored from 0-3 (0 – fewer than three layers; 1 – 130
three to four layers; 2 – five to six layers; 3 – more than six layers). Lymphoid cell 131
infiltration was scored from 0-3 (0 – none to diffuse infiltration; 1 – lymphoid cell 132
aggregate; 2 – lymphoid follicles; 3 – lymphoid follicles with germinal center 133
formation) [10]. 134
135
Caspase-1 and NF-kB assay 136
THP1 (ATCC TIB-202) macrophage-like cell line and THP1/NF-kB reporter cell line 137
were cultured in R10 - RPMI media 1640 supplemented with 10% (v/v) fetal bovine 138
serum, 1% (v/v) penicillin-streptomycin, 1% (v/v) pyruvate, 1% (v/v) L-glutamine, 1% 139
(v/v) non-essential aminoacids, 1% (v/v) hepes buffer and 2-mercaptoethanol to a final 140
concentration of 0.05M, as recommended by the American Tissue Culture Collection 141
(ATCC). Cells were cultured at 250.000 cells/mL, incubated with 10µM of gambogic 142
acid for 1h at 37ºC 5% CO2, and then stimulated with PFA-fixed E. coli (20 E. coli per 143
cell) for 8h and 24h at 37ºC 5% CO2. Simultaneously, non-stimulated negative control 144
cells were also cultured at the same density as the stimulated population for comparison. 145
Caspase-1 activity was measured in THP1 macrophage-like cell line using the 146
Carboxyfluorescein FLICA Detection kit for Caspase Assay (Immunochemistry 147
Technologies, LLC) following the reagent instructions. Briefly, cells from the different 148

7
assays were protected from light exposure while incubated for 1 hour at 37ºC with 30X 149
FLICA solution at a 1:30 ratio. NF-kB activity was measured in THP1/NF-kB reporter 150
cell line. Lentiviral particles carrying a NF-kB-responsive GFP-expressing reporter gene 151
(Cignal Lenti Reporters, SABiosciences, USA) were used to infect THP-1 cells and to 152
establish a stable cell line. All samples were analyzed by flow cytometry using a FACS 153
Calibur (BD biosciences, USA). The data collected were further analyzed using FlowJo 154
software (Tree Star Inc, USA). 155
156
Statistical analysis 157
Statistical differences were determined with non-parametric Kruskal-Wallis and Mann-158
Whitney tests using GraphPad Prism (GraphPad, USA). Differences were considered 159
statistically significant for p < 0.05. 160
161
RESULTS 162
Gambogic acid reduces IL-1β and TNF production 163
To study the effect of this drug in the inhibition of IL-1β and TNF secretion we treated 164
the human THP-1 macrophage-like cell line with growing concentrations of gambogic 165
acid for 1 hour before challenging them with PFA-fixed E. coli for 6 hours. The 166
conditioned media was then probed for the secretion of either IL-1β or TNF using 167
ELISA. Gambogic acid significantly inhibited the secretion of both cytokines over a 168
wide range of concentrations (Fig. 1), confirming the previously reported effect of 169
gambogic acid in blocking the secretion of these cytokines [11] and validating our 170
earlier finding (Figueiredo et al., unpublished). 171
172

8
173
Figure 1. IL-1β and TNF secretion are inhibited by gambogic acid treatment. Media 174
samples from human THP-1 macrophage-like cell line cultured with growing 175
concentrations of gambogic acid were analyzed by ELISA technique. Differences were 176
considered statistically significant for p values < 0.05. 177
178
Gambogic acid inhibits the activation of NF-kB and Caspase-1 179
Pro-IL-1 and TNF both depend on NF-kB activation for the transcription of their 180
respective mRNAs. Pro-IL-1 processing is further dependent on the activation of 181
caspase-1. We therefore tested the effect of gambogic acid on these key pathways. To 182
investigate the effect of this drug in the activation of NF-kB, we used an NF-kB reporter 183
cell line created by stably infecting THP-1 cells with a commercial lentiviral GFP 184
reporter under the control of a minimal CMV promoter and tandem repeats of the NF-185
kB transcriptional response element (TRE). Gambogic acid was able to suppress NF-kB 186
reporter activation upon E. coli stimulation in comparison with cells that were also 187
stimulated but did not receive treatment (Fig. 2A). To test the effect of this drug in 188
caspase-1 processing and activation we used a caspase-1 fluorescent substrate, and 189
measured relative active caspase-1 levels using FACS. Also in this case, gambogic acid 190
significantly decreased the activation of caspase-1 (Fig. 2B). 191
192

9
193
Figure 2. (A) NF-kB reporter activation is suppressed by gambogic acid treatment. NF-194
kB expression was measured by flow cytometry in a THP-1/NF-kB reporter cell line 195
incubated with gambogic acid and then stimulated for 24h with E. coli. Each thin line in 196
the histogram corresponds to untreated but E. coli stimulated cells, the shaded area 197
corresponds to drug-treated and E. coli stimulated cells and the thick line corresponds to 198
untreated non-stimulated cells as a control. Caspase-1 activation is decreased with 199
gambogic acid treatment. (B) Caspase-1 activation was measured using flow cytometry 200
in a THP-1 cell line incubated with gambogic acid and then stimulated for 8h with E. 201
coli. Each thin line in the histogram corresponds to untreated but E. coli stimulated cells 202
used as control and the thick line corresponds to drug-treated and E. coli stimulated 203
cells. 204
205
Gambogic acid is able to suppress inflammation in Wistar rat adjuvant-induced 206
arthritis 207
To study the in vivo anti-inflammatory properties of gambogic acid, AIA rats were 208
treated daily with this drug after disease had already started to be symptomatic and for a 209
period of 15 days. The inflammatory score and ankle perimeter were evaluated during 210
the period of treatment. As shown in Fig. 3, by the 4th
day all induced animals already 211
presented arthritis. All induced animals received either vehicle or gambogic acid at that 212
time point. After 6 days of treatment the vehicle injected group increased sharply the 213
inflammatory manifestations, whereas in gambogic acid-treated rats there was minimal 214
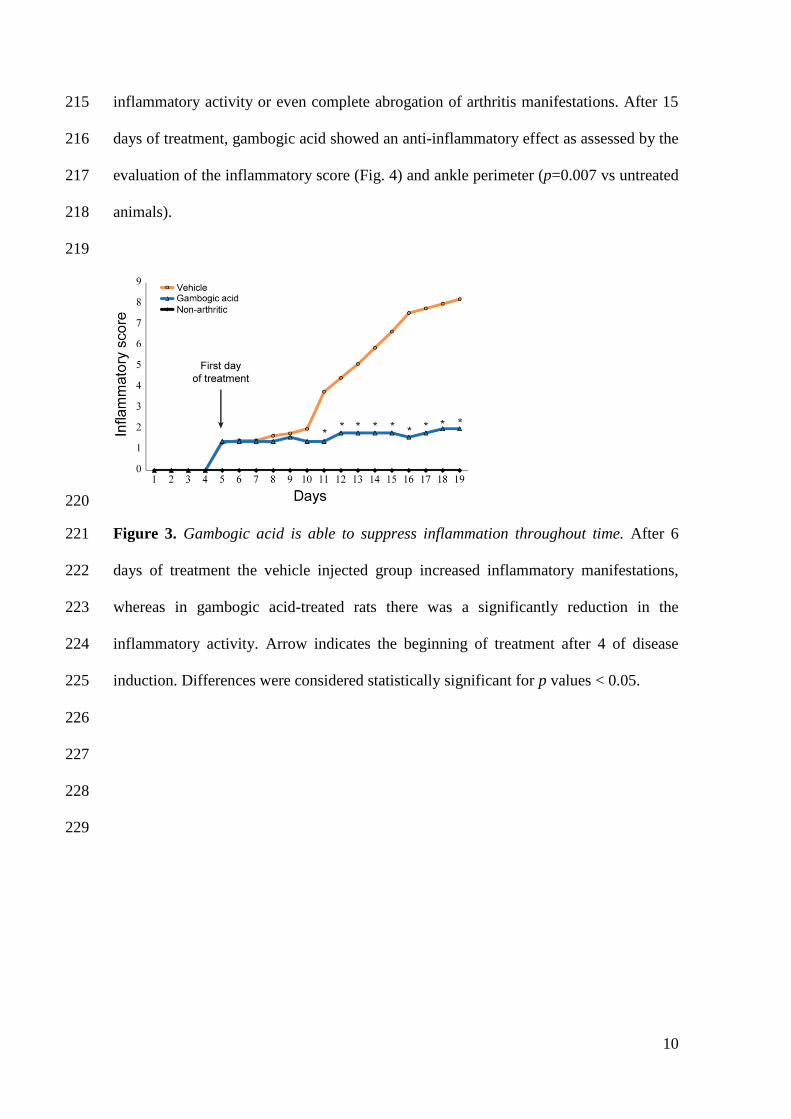
10
inflammatory activity or even complete abrogation of arthritis manifestations. After 15 215
days of treatment, gambogic acid showed an anti-inflammatory effect as assessed by the 216
evaluation of the inflammatory score (Fig. 4) and ankle perimeter (p=0.007 vs untreated 217
animals). 218
219
220
Figure 3. Gambogic acid is able to suppress inflammation throughout time. After 6 221
days of treatment the vehicle injected group increased inflammatory manifestations, 222
whereas in gambogic acid-treated rats there was a significantly reduction in the 223
inflammatory activity. Arrow indicates the beginning of treatment after 4 of disease 224
induction. Differences were considered statistically significant for p values < 0.05. 225
226
227
228
229

11
230
Figure 4. Gambogic acid possess anti-inflammatory properties. Inflammation score in 231
gambogic acid-treated AIA rats is significantly diminished in comparison with vehicle-232
treated rats after treatment. Differences were considered statistically significant for p 233
values < 0.05. 234
235
Gambogic acid prevents joint inflammatory infiltration and proliferation 236
To evaluate the infiltration of immune cells within joints in AIA rats, joint tissue 237
sections were stained with hematoxylin and eosin. The histological evaluation shown in 238
Fig. 5 revealed that rats treated with gambogic acid had a normal joint structure with 239
complete abrogation of the inflammatory infiltrate (p<0.0001 vs untreated animals). In 240
contrast, vehicle-treated rats exhibited infiltration of inflammatory cells, bone invasion 241
and erosions (Fig. 5). We have not observed significant differences in body weight or 242
any other side effects in treated rats, as revealed during autopsy and histological 243
analysis made in lung, liver, kidney and pancreas (data not shown). We also studied the 244
levels of proliferation of immune cells, by staining joint tissue sections with Ki67. The 245
immunohistochemical results revealed that rats treated with gambogic acid presented 246
reduced proliferation of immune cells within joints (p=0.0098 vs untreated animals). 247
248

12
249
Figure 5. Histological and immunohistochemical evaluation of joints after 15 days of 250
treatment. Non-arthritic control (A), vehicle-treated (B) and gambogic acid-treated (C) 251
AIA rats (magnification 50x and 100x in histological images and a magnification 200x 252
in immunohistochemical images). Notice that gambogic acid has completely prevented 253
immune cellular infiltration, proliferation and bone invasion. 254
255
DISCUSSION 256
Our results demonstrated that treatment with gambogic acid protected Wistar AIA rats 257
from arthritis development with a complete abrogation of joint immune cellular 258
infiltration and proliferation, preventing cartilage and bone damage. 259
Previous reports have already demonstrated that gambogic acid can inhibit the growth 260
of a wide variety of tumor cell lines, possibly due to its ability to induce apoptosis [12] 261
via the transferrin receptor (TfR1) [13]. Additionally, recent data have shown that this 262
drug can inhibit NF-kB signalling pathway in human leukemia cancer cells [8] and in a 263
non-cancerigenous macrophagic cell line [14] also via TfR1. Therefore, the anti-264
inflammatory effects of gambogic acid appear to be mediated by the inhibition of NF-265

13
kB activation pathway which in turn leads to the silencing of most of the inflammatory 266
genes. In our study we demonstrated that the anti-inflammatory properties of this drug 267
in AIA rats might not only be related with its ability to suppress the activation of NF-kB 268
but also to its effect on inhibiting caspase-1 activation. Importantly, in RA the inflamed 269
synovium expands into and destroys the underlying cartilage and bone, resulting in 270
irreversible erosion of the bone and in the loss of normal joint architecture leading to 271
disability [15]. The inhibitory effect of gambogic acid in cellular proliferation can thus 272
prove relevant for the management of RA course. 273
In conclusion, gambogic acid constitutes an effective drug in a rat model of RA, 274
possibly by its ability to down-regulate caspase-1 and NF-kB activation and by blocking 275
synovial hyperplasia due to its anti-proliferative properties. Further animal 276
experimentation is required to explore the safety of this compound for the treatment of 277
inflammatory diseases, such as RA, but these results hold the promise that gambogic 278
acid could be worth of entering into phase I clinical trials. 279
280
Acknowledgements 281
The authors would like to acknowledge Ana Lopes for technical assistance and Ana 282
Luisa Caetano for technical support with FACS. 283
284
Financial Disclosure 285
This work was supported by a grant (SFRH/BD/40513/2007) from Fundação para a 286
Ciência e a Tecnologia (FCT) and by an unrestricted research grant from Pfizer. Work 287
in Luis Moita’s laboratory is supported by FCT (PIC/IC/82991/2007 and PTDC/SAU-288
MII/100780/2008) and Fundação Luso-Americana para o Desenvolvimento (FLAD). 289

14
The funders had no role in study design, data collection and analysis, decision to 290
publish, or preparation of the manuscript. 291
292
Authors’ contributions 293
RC performed all the laboratorial work, data collection, statistical analysis and writing 294
of the paper. BV contributed in animal manipulation, histological and 295
immunohistochemical experiments. HR, ANC and NF participated in some laboratory 296
experiments. VG contributed with the drugs used in the primary screening. LFM and 297
JEF as senior authors conceived the study, participated in its design and coordination, 298
and contributed in the draft of the manuscript with important intellectual input. All 299
authors read and approved the final manuscript. 300
301
REFERENCES 302
1. Yelin E, Callahan LF (1995) The economic cost and social and psychological impact 303
of musculoskeletal conditions. National Arthritis Data Work Groups. Arthritis 304
Rheum 38: 1351-1362. 305
2. Radner H, Smolen JS, Aletaha D (2010) Impact of comorbidity on physical function 306
in patients with rheumatoid arthritis. Ann Rheum Dis 69: 536-541. 307
3. Lindqvist E, Saxne T, Geborek P, Eberhardt K (2002) Ten year outcome in a cohort 308
of patients with early rheumatoid arthritis: health status, disease process, and 309
damage. Ann Rheum Dis 61: 1055-1059. 310
4. Goldbach-Mansky R (2009) Blocking interleukin-1 in rheumatic diseases. Ann N Y 311
Acad Sci 1182: 111-123. 312

15
5. Cascao R, Moura RA, Perpetuo I, Canhao H, Vieira-Sousa E, et al. (2010) 313
Identification of a cytokine network sustaining neutrophil and Th17 activation in 314
untreated early rheumatoid arthritis. Arthritis Res Ther 12: R196. 315
6. Rita Cascão, Joaquim Polido-Pereira, Helena Canhão, Ana M. Rodrigues, Márcio 316
Navalho, et al. (2011) Caspase-1 is active since the early phase of Rheumatoid 317
arthritis. Clin Exp Rheumatol in press. 318
7. Han QB, Cheung S, Tai J, Qiao CF, Song JZ, et al. (2005) Stability and cytotoxicity 319
of gambogic acid and its derivative, gambogoic acid. Biol Pharm Bull 28: 2335-320
2337. 321
8. Pandey MK, Sung B, Ahn KS, Kunnumakkara AB, Chaturvedi MM, et al. (2007) 322
Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF-induced 323
apoptosis through modulation of the nuclear factor-kappaB signaling pathway. 324
Blood 110: 3517-3525. 325
9. da Silva JA, Fonseca JE, Graca L, Moita L, Carmo-Fonseca M (1996) Reinnervation 326
of post-arthritic joints in the rat. Clin Exp Rheumatol 14: 43-51. 327
10. Tsubaki T, Arita N, Kawakami T, Shiratsuchi T, Yamamoto H, et al. (2005) 328
Characterization of histopathology and gene-expression profiles of synovitis in 329
early rheumatoid arthritis using targeted biopsy specimens. Arthritis Res Ther 7: 330
R825-836. 331
11. Zhang L, Yi Y, Chen J, Sun Y, Guo Q, et al. Gambogic acid inhibits Hsp90 and 332
deregulates TNF-alpha/NF-kappaB in HeLa cells. Biochem Biophys Res 333
Commun 403: 282-287. 334
12. Zhang HZ, Kasibhatla S, Wang Y, Herich J, Guastella J, et al. (2004) Discovery, 335
characterization and SAR of gambogic acid as a potent apoptosis inducer by a 336
HTS assay. Bioorg Med Chem 12: 309-317. 337

16
13. Kasibhatla S, Jessen KA, Maliartchouk S, Wang JY, English NM, et al. (2005) A 338
role for transferrin receptor in triggering apoptosis when targeted with gambogic 339
acid. Proc Natl Acad Sci U S A 102: 12095-12100. 340
14. Palempalli UD, Gandhi U, Kalantari P, Vunta H, Arner RJ, et al. (2009) Gambogic 341
acid covalently modifies IkappaB kinase-beta subunit to mediate suppression of 342
lipopolysaccharide-induced activation of NF-kappaB in macrophages. Biochem 343
J 419: 401-409. 344
15. Tak PP, Bresnihan B (2000) The pathogenesis and prevention of joint damage in 345
rheumatoid arthritis: advances from synovial biopsy and tissue analysis. 346
Arthritis Rheum 43: 2619-2633. 347
348
349
350

Chapter IV
Discussion

- 50 -

Discussion
- 51 -
Rheumatoid arthritis is a chronic immune-mediated inflammatory disease that is
mainly characterized by symmetrical synovitis of the small joints of hands and feet. The
migration and infiltration of leukocytes towards the synovium is crucial for the
establishment of the chronic inflammatory process of the disease 241-243. These multi-
regulated mechanisms involve interactions with endothelial cells through cell adhesion
molecules and complex cytokine networks.
Significant efforts have been made during the last 20 years to identify the cytokine
profile of rheumatoid arthritis. In established rheumatoid arthritis, it has already been
reported that IL-1β, IL-6, IL-8, IL-17A and TNF levels are elevated in the serum of
patients and that these levels are correlated with disease activity 244-246. Nevertheless,
the knowledge about the relative influence of different cytokine pathways on disease
onset remains scarce. The characterization of the cytokine profile at the early stage of
rheumatoid arthritis may lead to the identification of early key players, thus extending
our knowledge in the disease physiopathology and highlighting new potential treatment
targets for early disease.
Accordingly, the first goal and one of the major novelties of the present study was
the analysis of the early cytokine and chemokine environment in the first few weeks of
rheumatoid arthritis onset. For this purpose, a cohort of untreated polyarthritis patients
with less than six weeks of disease duration was followed up, allowing the identification
of a subset of patients that later on evolved into rheumatoid arthritis, defined as very
early rheumatoid arthritis (VERA) patients. The remaining patients either have self-
limited forms of arthritis or evolved into other chronic inflammatory joint diseases, being
classified as very early arthritis (VEA) patients. Clinical and laboratorial evaluations of
VERA patients were performed at baseline without therapy, after short-term treatment
with low-dose corticosteroids and after 4 months of MTX. The panel of cytokines
selected to be studied in VERA patients was selected to include all the major known
rheumatoid arthritis pathogenic players, which encompassed complex cytokine-driven
interactions between resident synovial and infiltrating inflammatory cells, as illustrated
in Figure 1.
Importantly, it was found that VERA patients have increased circulating levels of
cytokines related with the recruitment (IL-6), maturation and survival (a proliferation-
inducing ligand, APRIL and BLyS) of B cells, as well as a cytokine profile that supports
neutrophil recruitment (IL-8) and Th17 cells polarization (IL-1β and IL-6) and activation
(IL-17A and IL-22) when compared with VEA and established rheumatoid arthritis
patients under MTX treatment (Figure 5). Furthermore, the analysis of synovial fluid
samples from established rheumatoid arthritis patients, representative of the

Discussion
- 52 -
inflammatory process that occurs locally in the joints, also revealed a similar cytokine
pattern. Considering the cytokine profile observed in the very early phase of the
disease, it is tempting to consider that after an initial unknown triggering event,
macrophages and neutrophils are attracted to the joints and are activated by monocyte
chemotactic protein (MCP)-1 and IL-8 chemotactic gradients, respectively 247, 248. In
fact, previous results demonstrated that there is a delay in the apoptosis of circulating
neutrophils in VERA patients 34 and that these cells heavily infiltrate the synovial tissue
during disease onset 23. Hence, neutrophils together with macrophages are the most
abundant cells infiltrated into the joints of rheumatoid arthritis patients. Notably,
activated macrophages and fibroblasts are able to produce APRIL, which affects the
class-switch recombination process 249, 250 and induces plasma cell differentiation 249-253,
possibly explaining the maintenance of autoreactive B cells in joints 254. Subsequent
steps in rheumatoid arthritis progression could include the production by autoreactive B
cells of autoantibodies and immune complexes that deposit in joints and activate Fcγ-
receptors expressed by macrophages 255-257, further increasing the inflammatory
process through the production of proinflammatory cytokines, such as TNF and IL-1β
by macrophages 255, 258, 259. In addition, IL-1β can stimulate endothelial cells, T and B
cells, and fibroblasts to produce IL-6 and IL-8. Furthermore, IL-1β together with IL-6 is
able to drive Th17 cells polarization 260, 261. Activated Th17 cells could thus produce
high levels of IL-17A which is able to extend neutrophil survival 262, 263 and stimulate
osteoclastogenesis leading to bone destruction 264-266, and IL-22 which helps B cells in
autoantibody production 267-269. On the other hand, once activated, neutrophils can also
release high amounts of BLyS 35 that in turn can stimulate the function and survival of
autoreactive B cells 270-272. Moreover, increased IL-6 levels support a scenario of
continuous recruitment of autoreactive B cells toward the synovium during rheumatoid
arthritis progression 65, 95, thus contributing to the chronic inflammatory process.
Moreover, in the established phase of the disease, the increased levels of IL-21, mainly
produced by CD4+ T cells, including Th17 cells 273-275, can not only induce plasma cell
differentiation and autoantibody production 276, but also significantly enhance Th17
cells proliferation 277. Interestingly, cytokines associated with the function of Th1 and
Th2 cells were not significantly elevated in the early phase of the disease. This result
suggests that Th17-cell activation is the most relevant T-cell related occurrence in very
early rheumatoid arthritis, reinforcing the notion that this might be a Th17-driven
disease.
Currently, MTX is the gold standard treatment for rheumatoid arthritis at the time of
presentation. Of note, neither initial therapy with low dose corticosteroids nor MTX

Discussion
- 53 -
affected cytokine production in VERA patients. Thus, despite being able to control
clinical manifestations of the disease, these conventional therapies were ineffective in
reversing the proinflammatory cytokine production that underlies inflammation in VERA
patients. Such an effect on cytokine production can presumably occur for long-term
treatments, as is the case of established rheumatoid arthritis patients who do not show
increased levels of circulating cytokines.
These results support the pathogenic role of, not only adaptive immune cells such
as B and Th17 cells, but also of innate immune cells, namely macrophages and
neutrophils in rheumatoid arthritis onset and chronicity. Particularly, these observations
also suggest that B cells and Th17-related cytokines targeted therapies could be
beneficial for rheumatoid arthritis patients in the first weeks of disease onset.
The results of this thesis are additionally supported by recently published data. It
has been reported that in pre-arthritis patients (3.3 years before disease onset), there
are increased levels of several circulating cytokines, such as IL-1β, IL-2, IL-4, IL-6, IL-
7, IL-10, IL-12, IL-17, interferon (IFN)γ and TNF along with MCP-1, all of them
associated with the development of inflammation and possibly predictive of the onset of
rheumatoid arthritis. An interesting finding was that IL-17 concentration was present at
its highest level in pre-arthritis patients and decreased afterwards in rheumatoid
arthritis patients after 7.7 months of disease duration 278. This is in accordance with our
observations that the concentration of IL-17A was increased in VERA patients but
similar to healthy controls in the established phase of the disease. Accordingly, it is
conceivable that in individuals who will develop rheumatoid arthritis, there is a trend
towards Th1/Th2/Th17 lineages and macrophage responses 278-280. Other study using
synovial fluid samples also demonstrated that the cytokines which better distinguished
patients with early synovitis (rheumatoid arthritis and crystal synovitis) from controls
with no inflammation (osteoarthritis) were IL-1β, IL-6 and IL-2. In contrast, the cytokines
that better distinguished persistent synovitis of rheumatoid arthritis from other
synovitides were IL-1β, IL-2 and IL-13 281. Interestingly, Meyer et al recently
demonstrated that in DMARD-naïve rheumatoid arthritis patients (with less than 2
years of disease duration) there are increased circulating levels of Th1 and
monocyte/macrophage-derived cytokines, possibly counter-regulated by cytokines
produced by Th2 cells. In contrast, IL-17 was barely detectable in this cohort of
patients, which is in agreement with the notion that the early phase of rheumatoid
arthritis is primarily driven by a Th17 profile, which is gradually replaced by a Th1
pattern during the progression of the disease. Surprisingly, several studies have
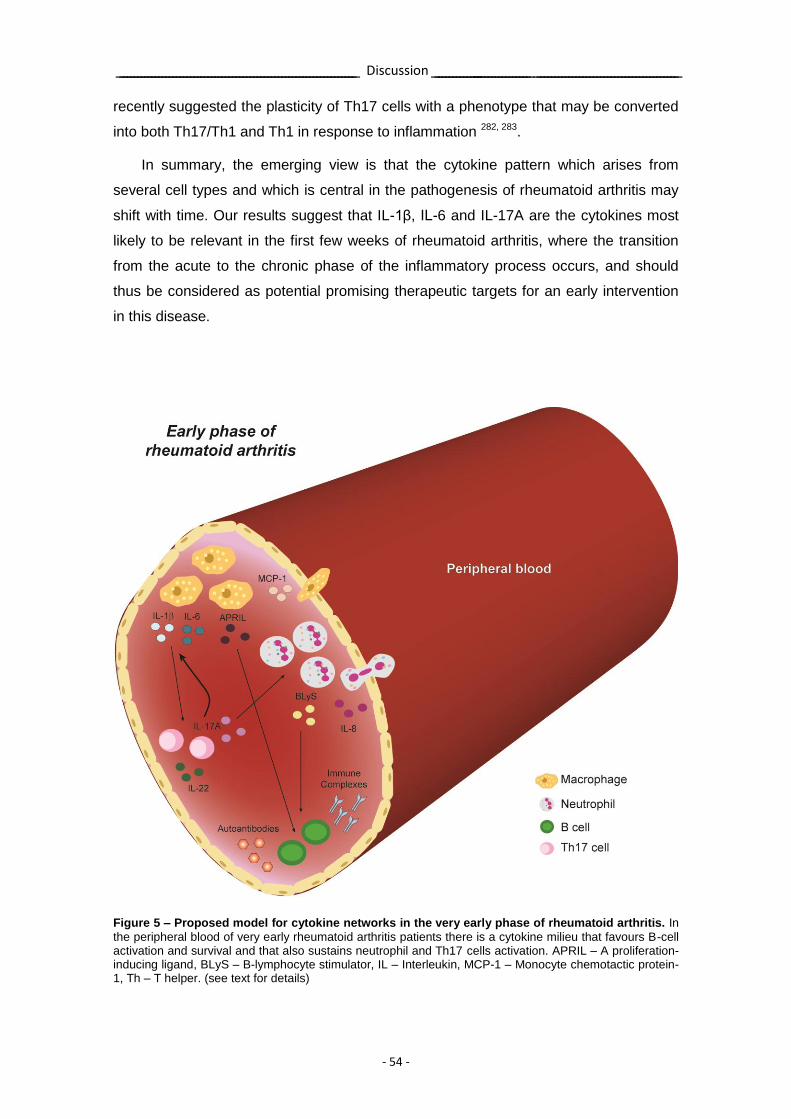
Discussion
- 54 -
recently suggested the plasticity of Th17 cells with a phenotype that may be converted
into both Th17/Th1 and Th1 in response to inflammation 282, 283.
In summary, the emerging view is that the cytokine pattern which arises from
several cell types and which is central in the pathogenesis of rheumatoid arthritis may
shift with time. Our results suggest that IL-1β, IL-6 and IL-17A are the cytokines most
likely to be relevant in the first few weeks of rheumatoid arthritis, where the transition
from the acute to the chronic phase of the inflammatory process occurs, and should
thus be considered as potential promising therapeutic targets for an early intervention
in this disease.
Figure 5 – Proposed model for cytokine networks in the very early phase of rheumatoid arthritis. In
the peripheral blood of very early rheumatoid arthritis patients there is a cytokine milieu that favours B-cell activation and survival and that also sustains neutrophil and Th17 cells activation. APRIL – A proliferation-inducing ligand, BLyS – B-lymphocyte stimulator, IL – Interleukin, MCP-1 – Monocyte chemotactic protein-1, Th – T helper. (see text for details)

Discussion
- 55 -
Several studies have documented the relevance of IL-1β in rheumatoid arthritis
physiopathology through diverse mechanisms. IL-1β, which is mainly produced by
macrophages 146, 147, is able to increase the secretion of several cytokines and
chemokines, to elevate the production of MMPs that are responsible for cartilage
damage 59-61, to enhance the expression and survival of T cells and, consequently,
enhance the help for B cell production of autoantibodies 62, to promote the
differentiation of Th17 cells 63, 64, to activate endothelial cells and chondrocytes 45, 267
and also to induce the differentiation of osteoclasts 65, 66, thus triggering the destructive
inflammatory process characteristic of rheumatoid arthritis, as illustrated in Figure 2.
IL-1 family cytokines, such as IL-1β, are abundantly expressed in rheumatoid
arthritis. In fact, in established rheumatoid arthritis there are augmented levels of IL-1β,
which are associated with a higher disease activity 223, 224, 226, 245, and there is also
activation of NF-kB 284 in the synovial fluid and tissue. Additionally, it has been
suggested that synovial membrane mRNA levels of this cytokine are also predictive of
damage progression 67. An interesting finding of the present study was the observation
that IL-1β levels were increased not only in the synovial fluid of established rheumatoid
arthritis patients, but also in the peripheral blood of patients in the early phase of the
disease.
Furthermore, the NLRP3 inflammasome is able to activate caspase-1 285, 286 which,
in turn activates IL-1β enabling the secretion of this cytokine 287-289. Recently, it has
been reported that polymorphisms in NLRP3 and CARD8 genes are associated with
ACPA-positive rheumatoid arthritis, increased susceptibility for the disease and a
worse prognosis for these patients 218-220.
Therefore, these observations together with the above-mentioned results,
prompted us to hypothesize that pathways which regulate IL-1β secretion, such as
caspase-1 and inflammasome activation, could be actively contributing to the
inflammatory response in rheumatoid arthritis. To address this question, the activation
of caspase-1 was evaluated in untreated early rheumatoid arthritis (ERA) patients with
less than one year of disease duration and established rheumatoid arthritis patients
under treatment with MTX. Likewise, other untreated early arthritis (EA) patients who
did not evolve to rheumatoid arthritis and had less than one year of disease duration
were also analyzed.
Strikingly, it was found for the first time that the levels of active caspase-1 are
increased in circulating leukocytes in rheumatoid arthritis patients since the early phase
of the disease, which could explain the increased levels of IL-1β after rheumatoid

Discussion
- 56 -
arthritis onset (Figure 6). In contrast, there were no differences when comparing EA
patients with healthy controls, corroborating the previous IL-1β results in which IL-1β
levels were increased in the early phase of rheumatoid arthritis but not in early non-
rheumatoid arthritis patients in comparison with healthy controls. Of interest, it has
been already reported in Sjögren’s syndrome-like prone mice that inflammatory
caspases are essential in promoting a proinflammatory microenvironment in the target
tissues of Sjögren’s syndrome before the onset of this autoimmune disease 290. In this
study, it was demonstrated that caspase-11, which activates caspase-1 via a non-
canonic pathway 291, plays a critical role in the susceptibility of mice to Sjögren’s
syndrome-like disease, by up-regulating caspase-1 mediated pathways, such as IL-1β
production. The authors suggest the existence of an abnormal regulation of the
inflammasome in these mice in the early pathogenesis of Sjögren’s syndrome 290. It
may be possible that individuals bearing variants in genes coding for inflammasome
components, which confer susceptibility for rheumatoid arthritis, might produce higher
levels of inflammasome components, thus implying that upon an inflammasome
activating event, more inflammasome complexes may be formed, ultimately producing
more inflammatory cytokines that might trigger the activity of the disease, as already
hypothesized based on a very recent study performed by Sui et al 292.
In conclusion, the results of this thesis support our initial hypothesis that the
caspase-1 pathway is already activated since the early phase of rheumatoid arthritis
and that IL-1β, caspase-1 and the proteins that regulate the activation of
inflammasomes may be potentially promising treatment targets in the early phase of
this disease.
Despite this apparent crucial role of IL-1β signaling in rheumatoid arthritis, clinical
benefits after IL-1β inhibition in these patients have been modest. Several trials have
shown the efficacy of anakinra in treating symptoms of a subset of rheumatoid arthritis,
with significant improvement in many different clinical, biochemical and radiographic
parameters 228. Unexpectedly, the efficacy of this IL-1β blocking therapy was lower
than TNF inhibitors in treating rheumatoid arthritis 228, 238, 239.
Notably, even though the disappointing observations of anakinra have raised some
controversy regarding the importance of IL-1β in rheumatoid arthritis, our results do
support the relevant role of IL-1β signaling in the pathogenesis of this disease since its
onset, suggesting that this treatment strategy should be re-evaluated in early
rheumatoid arthritis patients.
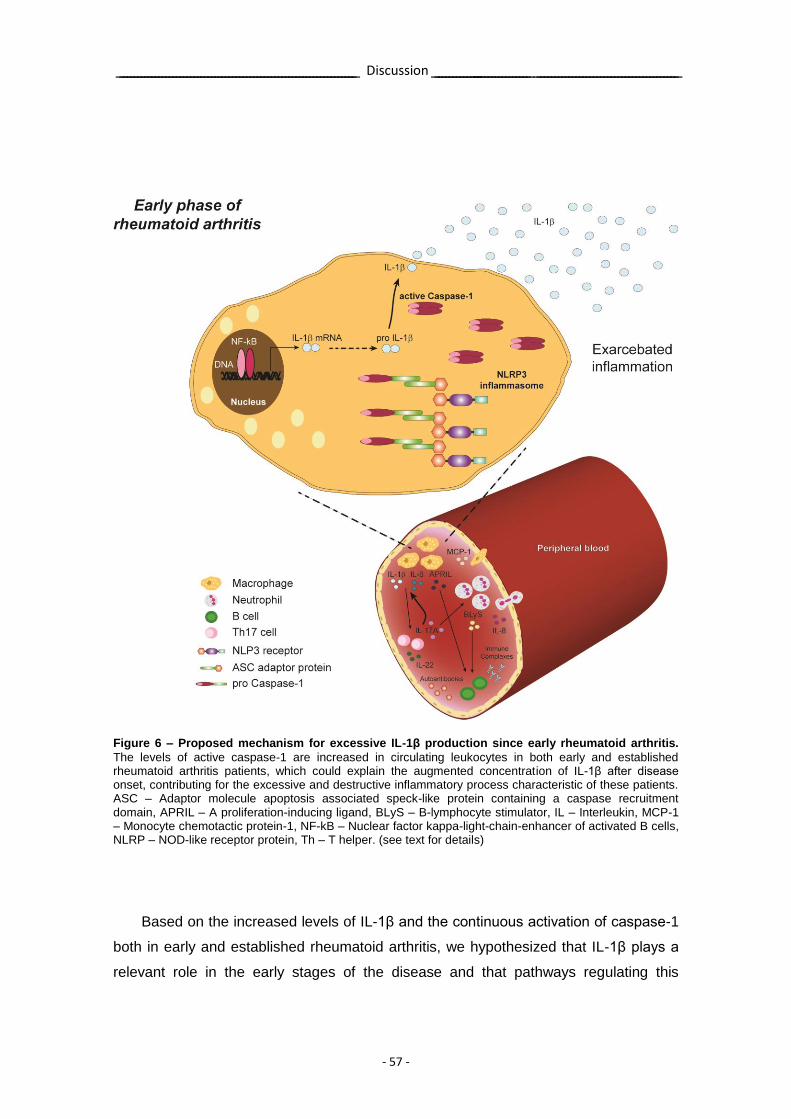
Discussion
- 57 -
Figure 6 – Proposed mechanism for excessive IL-1β production since early rheumatoid arthritis.
The levels of active caspase-1 are increased in circulating leukocytes in both early and established rheumatoid arthritis patients, which could explain the augmented concentration of IL-1β after disease onset, contributing for the excessive and destructive inflammatory process characteristic of these patients. ASC – Adaptor molecule apoptosis associated speck-like protein containing a caspase recruitment domain, APRIL – A proliferation-inducing ligand, BLyS – B-lymphocyte stimulator, IL – Interleukin, MCP-1 – Monocyte chemotactic protein-1, NF-kB – Nuclear factor kappa-light-chain-enhancer of activated B cells, NLRP – NOD-like receptor protein, Th – T helper. (see text for details)
Based on the increased levels of IL-1β and the continuous activation of caspase-1
both in early and established rheumatoid arthritis, we hypothesized that IL-1β plays a
relevant role in the early stages of the disease and that pathways regulating this

Discussion
- 58 -
cytokine and TNF, such as caspase-1 and NF-kB activation, could constitute promising
combined therapeutic targets.
Our laboratory used a THP-1 macrophage-like cell line to screen 2320 drugs for
those that simultaneously inhibit IL-1β and TNF secretion. We have selected celastrol
and gambogic acid as the two most effective drugs in inhibiting both IL-1β and TNF
secretion due to the reduction in caspase-1 and NF-kB activation. Celastrol is a novel
pentacyclic-triterpene compound extracted from Trypterigium wilfordii Hook F, which is
used in traditional Chinese medicine and that was recently shown to be able to
suppress cancer progression 293, 294, to inhibit NF-kB activity 295, to abrogate the
secretion of IL-1β in lipopolysaccharide-stimulated human peripheral mononuclear cells
296 and to inhibit proinflammatory cytokine secretion in mucosal inflammatory biopsies
from Crohn’s patients, possibly due to the blockade of cytokine gene transcription 297.
Gambogic acid is a polyprenylated xanthone abundant in resin derived from Garcinia
hanburyi and Garcinia Morella, which is used in Southeast Asia traditional medicine 298,
and that has recently been shown to inhibit the growth of a wide variety of tumor cell
lines 299, possible due to its ability to induce apoptosis 300, 301, to abrogate NF-kB
signalling pathway in human leukemia cancer cells 299 and in a non-cancerigenous
macrophagic cell line 302, both via the transferrin receptor (TfR1). Celastrol and
gambogic acid were further tested in vivo in a rat model of adjuvant-induced arthritis
(AIA) in order to evaluate their anti-inflammatory properties. These drugs were
administrated to AIA rats in the early phase of arthritis development. Administration
was through a therapeutic approach, since it started in the early phase of arthritis, after
4 days of disease induction, with all animals already displaying signs of inflammation.
Additionally, celastrol was also administrated in the established phase of arthritis, after
11 days of disease induction when the animals already displayed a marked
inflammatory score.
Animal models of rheumatoid arthritis have been extensively used for many years
in the evaluation of anti-arthritic agents 303, 304. The most widely used model is the AIA
in rats 305, 306, which shares key features related to human rheumatoid arthritis making
them a critical tool in drug development. There are some similarities in disease
characteristics when comparing AIA rat model and human rheumatoid arthritis, namely
the presence of acute phase proteins, polyarthritis, synovitis and bursitis/tendonitis,
erosion of cartilage and bone, the influx of neutrophils, the homing of macrophages to
soft tissues, the increased levels of synovial cytokines such as IL-1β, IL-6 and TNF,
and the occurrence of edema. The AIA rat model, when compared with other models of
arthritis such as collagen-induced arthritis, clearly exhibits the greatest magnitude of

Discussion
- 59 -
disease, as measured by edema, cellular influx, cytokine levels and markers of
cartilage and bone destruction. The prolonged time frame and the recurrent nature of
disease activity of human rheumatoid arthritis are not replicated in this model, as is
also the case of some other features of this disease, including RF and synovial
lymphoid follicles that are absent in the AIA rat model. This animal model was used to
test arthritis therapies such as steroids and NSAIDs 303, 304, as well as to assess
immunomodulatory drugs such as MTX, cyclosporine A and therapies designed to
block cyclooxygenase 2, TNF or IL-1β 307-315. Overall, the AIA model has been the most
extensively used arthritic rat model by the pharmaceutical industry because it has an
excellent track record for predicting both activity and toxicity 313. Paw swelling in AIA rat
model is robust, with increases up to 3.5-fold times control volume and, in general,
maximal swelling occurring by day 19 and then plateaus. Thus, studies are generally
completed at this time point.
We found, for the first time, that wistar AIA rats can be effectively treated by both
celastrol and gambogic acid possibly through an inhibitory effect over TNF and IL-1β
secretion, induced by the down-regulation of caspase-1 and NF-kB activation. These
drugs showed anti-inflammatory and anti-proliferative properties, promoting a complete
suppression of arthritis development and abrogation of joint immune cellular infiltration
and proliferation, preventing cartilage and bone damage (Figure 7). Interestingly,
celastrol treatment was effective when administrated both in the early and later phase
of arthritis, which is relevant for the possible clinical implications of our findings. Our
results demonstrated that the anti-inflammatory properties of these drugs might not
only be related to their ability to suppress the activation of NF-kB but also to their ability
to inhibit caspase-1 activation, as observed in the in vitro experiments performed in
THP-1 macrophage-like cell line.
The role of NF-kB in the pathogenesis of rheumatoid arthritis has been already
described. In fact, the inhibition of NF-kB in animal models has shown the ability to
inhibit inflammatory arthritis 316-319, demonstrating that NF-kB may be an important
therapeutic target in this disease. Indeed, NF-kB participates in the transcription of the
genes encoding many proinflammatory cytokines and chemokines, in the regulation of
the different immune cells and in the regulation of the expression of adhesion
molecules and matrix MMPs 320-323. As recently reviewed, the Ikβ kinase IKKβ is
essential for the inflammatory cytokine-induced activation of NF-kB 324. Blocking IKKβ
could therefore be part of a therapeutic strategy for the treatment of inflammation. In
this context, it was demonstrated by Wen and co-workers that the inhibition of IKKβ
inhibits NF-kB signaling in human synovial fibroblasts, chondrocytes and mast cells 325,

Discussion
- 60 -
and that it reduces NF-kB activity in rats with induced polyarthritis 326. Importantly, it
has already been reported that both celastrol and gambogic acid are able to inhibit
IKKβ activity 295, 302.
Our results are also supported by a study from van den Berg et al, in which the
authors suggest potential uncoupled activities of TNF and IL-1β in joint swelling and
ongoing cartilage destruction 327. The authors also point out that the claim of TNF being
a driving force for IL-1 production in rheumatoid arthritis should be viewed with care 327.
Their results in animal models of arthritis suggest that anti-TNF treatment is not
sufficient and that a combination therapy with anti-IL-1 provided the best protection
against disease progression, thus suggesting that this could be a promising strategy for
rheumatoid arthritis treatment 327. Synovial biopsy immunostainings of rheumatoid
arthritis patients have shown distinct TNF and IL-1β localisation patterns 328 and no
significant IL-1β reduction in synovial cells after treatment with TNF antagonists 327.
Additionally, systemic treatment with TNF antagonists does not reduce IL-1β mRNA in
circulating leukocytes of most patients 327. In fact, it has already been reported that
there are different time dependent roles for IL-1β and TNF in the various stages of
collagen-induced arthritis, since anti-TNF therapy showed efficacy only shortly after
disease onset while anti-IL-1 therapy ameliorated both early and established arthritis 83.
TNF is in fact sufficient to drive inflammation but IL-1 is a crucial mediator for
inflammatory cartilage and bone degradation 70. Bendele et al have demonstrated that
a combination of treatment with agents blocking both IL-1 and TNF receptors provides
substantially more clinical benefits than either agent alone in animal models of arthritis
313. This result could be explained by a synergistic effect between IL-1 and TNF. This
synergism has been already reported by others 327, 329-333 and may be explained by the
ability of TNF to induce IL-1 and vice-versa 334. Consequently, a double blocking of IL-1
and TNF strategy could be beneficial for the treatment of inflammatory diseases such
as rheumatoid arthritis. Taking into consideration the apparent advantage of a
combination therapy blocking both IL-1 and TNF, in 2004 a trial tested the efficacy of
combination therapy using anakinra and etanercept in 244 long-standing and very
active rheumatoid arthritis patients who have been treated unsuccessfully with MTX 335.
Results from this study revealed that combination therapy with anakinra and etanercept
provided no added benefit for these patients comparing with etanercept alone and
increased the frequency of infection related adverse effects 335. The reasons for the
discrepancies between preclinical and clinical results are unclear but several theories
are discussed by the authors of this study 335 such as negative interaction between
compounds and the development of anti-anakinra antibodies. Moreover, these

Discussion
- 61 -
discrepancies might be additionally explained by the fact that anakinra has some drug
concept limitations that can limit its effectiveness in blocking IL-1. Finally, the very
advanced disease stage of the patients could have also limited a sufficient evaluation
of this combination treatment. Our results thus suggest that a combination therapy
inhibiting both IL-1β and TNF production is effective not only in the early but also in the
later phases of arthritis and is safe, since these drugs diminish the levels of both
cytokines, instead of silencing their receptors as is the case of etanercept and
anakinra.
Our findings are also in agreement with data already published, showing that a
tripterine isolated from Trypterigium wilfordii Hook F was effective on adjuvant 336 and
collagen-induced arthritis 337 in rats, supporting the 2002 report from Tao et al revealing
that an ethanol/ethyl acetate extract of Trypterigium wilfordii Hook F possess
therapeutic benefits in patients with refractory rheumatoid arthritis 338. Interestingly, our
results are also supported by data from a study performed in an in vivo model of
metastatic bone disease associated with breast cancer, in which celastrol inhibited
bone resorption, consistent with the inhibitory effect on osteoclast formation and
survival that was already observed in in vitro experiments 339. In rheumatoid arthritis the
infiltration of immune cells and the proliferation of joint lining synovial fibroblasts lead to
the formation of the tumor-like pannus tissue, which invades and destroys the
underlying joint cartilage and bone, resulting in irreversible erosion of the bone and in
the loss of normal joint structure, causing functional disability 225. The cell proliferation
inhibitory properties of celastrol and gambogic acid may thus prove to be of interest to
prevent and treat this complication of established rheumatoid arthritis. This is exactly
compatible with our experiments that have shown an inhibition of the inflammatory
activity, cell proliferation and bone erosions. Although further animal experimentation is
required to fully assess the safety of these drugs for human arthritis treatment, these
results point out that both celastrol and gambogic acid might be worth entering into
phase I clinical trials.
To sum up, these data support our hypothesis that IL-1β dependent inflammatory
effects, preceded by caspase-1 and NF-kB activation, have a role in the very early
pathogenesis of rheumatoid arthritis.
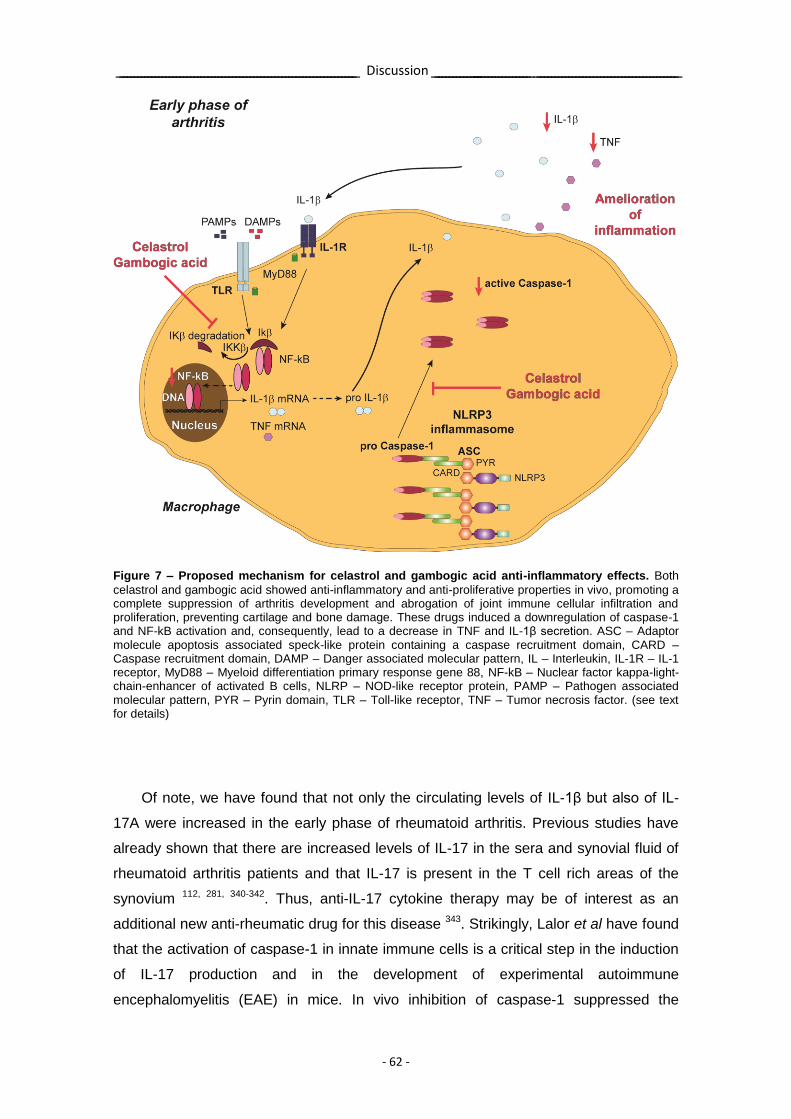
Discussion
- 62 -
Figure 7 – Proposed mechanism for celastrol and gambogic acid anti-inflammatory effects. Both
celastrol and gambogic acid showed anti-inflammatory and anti-proliferative properties in vivo, promoting a complete suppression of arthritis development and abrogation of joint immune cellular infiltration and proliferation, preventing cartilage and bone damage. These drugs induced a downregulation of caspase-1 and NF-kB activation and, consequently, lead to a decrease in TNF and IL-1β secretion. ASC – Adaptor molecule apoptosis associated speck-like protein containing a caspase recruitment domain, CARD – Caspase recruitment domain, DAMP – Danger associated molecular pattern, IL – Interleukin, IL-1R – IL-1 receptor, MyD88 – Myeloid differentiation primary response gene 88, NF-kB – Nuclear factor kappa-light-chain-enhancer of activated B cells, NLRP – NOD-like receptor protein, PAMP – Pathogen associated molecular pattern, PYR – Pyrin domain, TLR – Toll-like receptor, TNF – Tumor necrosis factor. (see text for details)
Of note, we have found that not only the circulating levels of IL-1β but also of IL-
17A were increased in the early phase of rheumatoid arthritis. Previous studies have
already shown that there are increased levels of IL-17 in the sera and synovial fluid of
rheumatoid arthritis patients and that IL-17 is present in the T cell rich areas of the
synovium 112, 281, 340-342. Thus, anti-IL-17 cytokine therapy may be of interest as an
additional new anti-rheumatic drug for this disease 343. Strikingly, Lalor et al have found
that the activation of caspase-1 in innate immune cells is a critical step in the induction
of IL-17 production and in the development of experimental autoimmune
encephalomyelitis (EAE) in mice. In vivo inhibition of caspase-1 suppressed the

Discussion
- 63 -
development of Th17 cells and the induction of EAE. These new evidences suggest
that caspase-1 might also be an efficient drug target in immune mediated inflammatory
diseases 344. An interesting report dissecting CAPS inflammatory environment have
demonstrated the central role of IL-1β in Th17 cells differentiation, since these patients
showed significantly increased IL-17 serum levels as well as a higher frequency of
Th17 cells, which were decreased following in vivo IL-1β blockade 345. This expansion
of Th17 phenotype in CAPS patients may be a simple epiphenomenon secondary to
the over-secretion of IL-1β due to NLRP3 mutations or it may be contributing to the
maintenance of chronic inflammation.
To help distinguish between a direct role for IL-1β as an effector in the
inflammatory process or as an upstream initiator of inflammation by driving the
differentiation of Th17 cells, we analyzed the anti-inflammatory effects of digoxin in the
same rat model of arthritis, using the same experimental design as described above. A
very recent paper from Huh et al has shown that digoxin is a specific inhibitor of RORγt
transcriptional activity and consequently an inhibitor of Th17 cells polarization 346.
Digoxin is currently recommended for patients with heart failure who are symptomatic
despite standard therapy and for controlling the ventricular rate in atrial fibrillation, with
an important role in optimizing care in certain acute and chronic cardiac conditions 347.
We found for the first time that digoxin was able to ameliorate the inflammatory
signs of the AIA rat model of arthritis, which is in accordance with the reported effect of
digoxin in delaying the onset and reducing disease severity in an EAE mice model, by
the inhibition of Th17 cells differentiation 346. We observed that digoxin treatment was
effective if administrated in the early phase of arthritis development but not if given in
the later phase of the disease course. Although digoxin was able to efficiently suppress
the severity of inflammatory signs, it was not able to significantly reduce the infiltration
of immune cells within the joints. Contrarily, treatment with the previously mentioned
drugs that block IL-1β and TNF simultaneously, without inhibiting IL-17, have a
significant inhibitory effect in arthritis progression and severity, even when
administrated in a later phase of the disease course, with a complete abrogation of the
inflammatory score, infiltration and proliferation of immune cells. Moreover, digoxin had
a slower and less efficient effect on disease progression comparing with these drugs.
In conclusion, these data suggest that despite the role of IL-1β as a driver of Th17
cells polarization, the inhibition of this pathway is not effective in the treatment of
arthritis. Indeed, IL-1β contributes to rheumatoid arthritis physiopathology not only by
the induction of Th17 cells differentiation but also by promoting the migration of
macrophages and neutrophils to the damaged joints, thus driving the release of

Discussion
- 64 -
proteases and ROS 348 and promoting the differentiation of osteoclasts 349. Likewise, IL-
1β, together with TNF and IL-6, has also a significant capacity to induce RANKL
expression on synovial fibroblasts/osteoblasts and to facilitate RANK signaling, thus
directly contributing to the bone resorption and destructive process. In contrast, IL-17
seems to have a more limited effect on inflammatory cell influx and subsequent
inflammatory signs, as suggested by the results of digoxin treatment. This does not
preclude however the important role of Th17 cells in driving the innate immune
inflammation towards the adaptive autoimmune inflammation in rheumatoid arthritis,
involving several other cytokines besides IL-17. Nevertheless, the results regarding
anti-IL-17 therapy in rheumatoid arthritis are still contradictory. Two different clones of
anti-IL-17 antibody are currently undergoing clinical trials in patients with rheumatoid
arthritis. A study performed by Genovese et al has shown that one of these clones, a
humanized anti–IL-17 monoclonal antibody (LY2439821), added to oral DMARDs
improved signs and symptoms of rheumatoid arthritis with no significant adverse safety
signal noted 350. More recently, the same author reported a phase II placebo-controlled
trial with 237 patients testing the second clone, an anti-IL-17A drug (secukinumab, also
called AIN457) in rheumatoid arthritis where the primary end point was not met 351, 352.
This drug was significantly more effective after 12 weeks, but at week 16 - the pre-
specified point for evaluating the primary outcome (percentage of patients achieving an
ACR20 response) was not met - a rise in placebo response did not allow statistically
significant difference between the treatment and placebo groups 351. Interestingly,
combination therapy with soluble IL-1R and IL-17R is more effective for controlling
synovial inflammation and bone resorption than each therapy alone in an ex vivo model
of rheumatoid arthritis synovium and bone explants 353. These results suggest that the
isolated inhibition of Th17 cells polarization might be a strategy with limited efficacy in
rheumatoid arthritis, at least in the established phase of the disease.
In summary, our results, as compared with other published studies, have shown
that caspase-1 inhibitors are efficient in different experimental models of immune
mediated inflammatory diseases 354-358. The study discussed here provides new and
original evidence for the relevance of IL-1β, including caspase-1/NF-kB/inflammasome
activation, as possible drug targets for immune mediated inflammatory diseases,
especially if introduced in the early phase of the disease.

Discussion
- 65 -
Overall, the results of this thesis suggest that an earlier intervention on the IL-1β
pathway might be of beneficial clinical use to induce early remission in rheumatoid
arthritis patients. It cannot be claimed that IL-1β is the only factor, but these results
imply that interfering with IL-1β in the context of a network of cooperating mediators is
sufficient to down-regulate inflammation and progressive articular destruction.
Therefore, it is worthwhile pursuing the possibility that IL-1β is at least an adjuvant
therapeutic target in the early phase of arthritis.

- 66 -

Chapter V
Conclusions

- 68 -

Conclusions
- 69 -
The study of IL-1β since the early phase of rheumatoid arthritis supports an early
influence of this cytokine in disease progression and has revealed that:
1) In very early rheumatoid arthritis and in the synovial fluid of established
rheumatoid arthritis patients there is a cytokine milieu, which supports the activation,
expansion and survival of autoreactive Th17 and B cells as well as macrophages and
neutrophils since the first weeks of disease onset;
2) IL-1β is one of the cytokines with an increased concentration detected since the
early phase of rheumatoid arthritis until the established phase of the disease;
3) Early short-term treatment with corticosteroids or MTX leads to clinical
improvement, but does not seem to have an impact in the cytokine pattern of very early
rheumatoid arthritis patients;
4) The caspase-1 pathway, which is responsible for IL-1β activation, is activated
since the early phase of rheumatoid arthritis, reinforcing the potential relevance of
therapies targeting IL-1β in the early course of this disease;
5) Drugs targeting IL-1β and TNF simultaneously, upstream to their receptors,
namely celastrol and gambogic acid, have anti-inflammatory and anti-proliferative
properties in the treatment of arthritis and thus might constitute therapeutic approaches
with significant efficacy in the treatment of immune-mediated inflammatory diseases
such as rheumatoid arthritis;
6) Digoxin, which inhibits Th17 cells differentiation, despite being able to decrease
the severity of early inflammatory signs in AIA rat model, is not effective in reducing the
infiltration of immune cells within the joints, suggesting that the isolated inhibition of
Th17 cells polarization might be a strategy with limited efficacy, at least in established
rheumatoid arthritis.
The understanding of the pathogenesis of rheumatoid arthritis remains, however,
far from complete. Therapeutic targets and biological windows of opportunity identified
from advances made in studies as the one presented herein have contributed to the
expansion of treatment options for rheumatoid arthritis, in a series of successful
transitions from bench to bedside. This growing knowledge may ultimately make
complete remission a real goal for rheumatoid arthritis patients.

- 70 -

References

- 72 -

References
- 73 -
1. Michaud K, Wolfe F. Comorbidities in rheumatoid arthritis. Best Pract Res Clin
Rheumatol 2007;21:885-906
2. Meune C, Touze E, Trinquart L, et al. Trends in cardiovascular mortality in patients with
rheumatoid arthritis over 50 years: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford) 2009;48:1309-13
3. Sokka T, Kautiainen H, Mottonen T, et al. Work disability in rheumatoid arthritis 10
years after the diagnosis. J Rheumatol 1999;26:1681-5
4. Alamanos Y, Drosos AA. Epidemiology of adult rheumatoid arthritis. Autoimmun Rev
2005;4:130-6
5. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet 2010;376:1094-108
6. McQueen FM, Stewart N, Crabbe J, et al. Magnetic resonance imaging of the wrist in
early rheumatoid arthritis reveals a high prevalence of erosions at four months after symptom onset. Ann Rheum Dis 1998;57:350-6
7. McGonagle D, Conaghan PG, O'Connor P, et al. The relationship between synovitis
and bone changes in early untreated rheumatoid arthritis: a controlled magnetic resonance imaging study. Arthritis Rheum 1999;42:1706-11
8. Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an
American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569-81
9. Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987
revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315-24
10. Lanchbury JS. The HLA association with rheumatoid arthritis. Clin Exp Rheumatol
1992;10:301-4
11. Ligeiro D, Fonseca JE, Abade O, et al. Influence of human leucocyte antigen-DRB1 on
the susceptibility to rheumatoid arthritis and on the production of anti-cyclic citrullinated peptide antibodies in a Portuguese population. Ann Rheum Dis 2007;66:246-8
12. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to
understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 1987;30:1205-13
13. Blass S, Engel JM, Burmester GR. The immunologic homunculus in rheumatoid
arthritis. Arthritis Rheum 1999;42:2499-506
14. Harrison BJ. Influence of cigarette smoking on disease outcome in rheumatoid arthritis.
Curr Opin Rheumatol 2002;14:93-7
15. Morgan AW, Thomson W, Martin SG, et al. Reevaluation of the interaction between
HLA-DRB1 shared epitope alleles, PTPN22, and smoking in determining susceptibility to autoantibody-positive and autoantibody-negative rheumatoid arthritis in a large UK Caucasian population. Arthritis Rheum 2009;60:2565-76
16. Symmons DP, Bankhead CR, Harrison BJ, et al. Blood transfusion, smoking, and
obesity as risk factors for the development of rheumatoid arthritis: results from a primary care-based incident case-control study in Norfolk, England. Arthritis Rheum 1997;40:1955-61
17. Klareskog L, Stolt P, Lundberg K, et al. A new model for an etiology of rheumatoid
arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 2006;54:38-46
18. Shiozawa S, Shiozawa K, Fujita T. Morphologic observations in the early phase of the
cartilage-pannus junction. Light and electron microscopic studies of active cellular pannus. Arthritis Rheum 1983;26:472-8
19. McCachren SS, Haynes BF, Niedel JE. Localization of collagenase mRNA in
rheumatoid arthritis synovium by in situ hybridization histochemistry. J Clin Immunol 1990;10:19-27
20. Gravallese EM, Darling JM, Ladd AL, et al. In situ hybridization studies of stromelysin
and collagenase messenger RNA expression in rheumatoid synovium. Arthritis Rheum 1991;34:1076-84
21. Kobayashi I, Ziff M. Electron microscopic studies of the cartilage-pannus junction in
rheumatoid arthritis. Arthritis Rheum 1975;18:475-83

References
- 74 -
22. Karouzakis E, Neidhart M, Gay RE, et al. Molecular and cellular basis of rheumatoid
joint destruction. Immunol Lett 2006;106:8-13
23. Mourao AF, Canhao H, Sousa E, et al. From a neutrophilic synovial tissue infiltrate to a
challenging case of rheumatoid arthritis. Acta Reumatol Port 2010;35:228-31
24. Girard JP, Springer TA. High endothelial venules (HEVs): specialized endothelium for
lymphocyte migration. Immunol Today 1995;16:449-57
25. Paleolog EM. Angiogenesis in rheumatoid arthritis. Arthritis Res 2002;4 Suppl 3:S81-90
26. Sidiropoulos PI, Goulielmos G, Voloudakis GK, et al. Inflammasomes and rheumatic
diseases: evolving concepts. Ann Rheum Dis 2008;67:1382-9
27. Huang QQ, Pope RM. The role of toll-like receptors in rheumatoid arthritis. Curr
Rheumatol Rep 2009;11:357-64
28. Ma Y, Pope RM. The role of macrophages in rheumatoid arthritis. Curr Pharm Des
2005;11:569-80
29. Beech JT, Andreakos E, Ciesielski CJ, et al. T-cell contact-dependent regulation of CC
and CXC chemokine production in monocytes through differential involvement of NFkappaB: implications for rheumatoid arthritis. Arthritis Res Ther 2006;8:R168
30. Toubi E, Kessel A, Slobodin G, et al. Changes in macrophage function after rituximab
treatment in patients with rheumatoid arthritis. Ann Rheum Dis 2007;66:818-20
31. Abrahams VM, Cambridge G, Lydyard PM, et al. Induction of tumor necrosis factor
alpha production by adhered human monocytes: a key role for Fcgamma receptor type IIIa in rheumatoid arthritis. Arthritis Rheum 2000;43:608-16
32. Seibl R, Birchler T, Loeliger S, et al. Expression and regulation of Toll-like receptor 2 in
rheumatoid arthritis synovium. Am J Pathol 2003;162:1221-7
33. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol
2006;6:173-82
34. Weinmann P, Moura RA, Caetano-Lopes JR, et al. Delayed neutrophil apoptosis in very
early rheumatoid arthritis patients is abrogated by methotrexate therapy. Clin Exp Rheumatol 2007;25:885-7
35. Scapini P, Carletto A, Nardelli B, et al. Proinflammatory mediators elicit secretion of the
intracellular B-lymphocyte stimulator pool (BLyS) that is stored in activated neutrophils: implications for inflammatory diseases. Blood 2005;105:830-7
36. Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci 2008;13:2400-7
37. Scapini P, Lapinet-Vera JA, Gasperini S, et al. The neutrophil as a cellular source of
chemokines. Immunol Rev 2000;177:195-203
38. Rooney M, Whelan A, Feighery C, et al. The immunohistologic features of synovitis,
disease activity and in vitro IgM rheumatoid factor synthesis by blood mononuclear cells in rheumatoid arthritis. J Rheumatol 1989;16:459-67
39. Jenkins RN, Nikaein A, Zimmermann A, et al. T cell receptor V beta gene bias in
rheumatoid arthritis. J Clin Invest 1993;92:2688-701
40. Uematsu Y, Wege H, Straus A, et al. The T-cell-receptor repertoire in the synovial fluid
of a patient with rheumatoid arthritis is polyclonal. Proc Natl Acad Sci U S A 1991;88:8534-8
41. Boissier MC, Assier E, Falgarone G, et al. Shifting the imbalance from Th1/Th2 to
Th17/treg: the changing rheumatoid arthritis paradigm. Joint Bone Spine 2008;75:373-5
42. Leipe J, Skapenko A, Lipsky PE, et al. Regulatory T cells in rheumatoid arthritis.
Arthritis Res Ther 2005;7:93
43. Skapenko A, Leipe J, Lipsky PE, et al. The role of the T cell in autoimmune
inflammation. Arthritis Res Ther 2005;7 Suppl 2:S4-14
44. Nykanen P, Bergroth V, Raunio P, et al. Phenotypic characterization of 3H-thymidine
incorporating cells in rheumatoid arthritis synovial membrane. Rheumatol Int 1986;6:269-71
45. Kohem CL, Brezinschek RI, Wisbey H, et al. Enrichment of differentiated
CD45RBdim,CD27- memory T cells in the peripheral blood, synovial fluid, and synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum 1996;39:844-54

References
- 75 -
46. Koch AE, Robinson PG, Radosevich JA, et al. Distribution of CD45RA and CD45RO T-
lymphocyte subsets in rheumatoid arthritis synovial tissue. J Clin Immunol 1990;10:192-9
47. Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu
Rev Immunol 1996;14:397-440
48. Boissier MC, Chiocchia G, Bessis N, et al. Biphasic effect of interferon-gamma in
murine collagen-induced arthritis. Eur J Immunol 1995;25:1184-90
49. Hirota K, Hashimoto M, Yoshitomi H, et al. T cell self-reactivity forms a cytokine milieu
for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med 2007;204:41-7
50. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev
Immunol 2007;7:429-42
51. Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis.
N Engl J Med 2001;344:907-16
52. Leslie D, Lipsky P, Notkins AL. Autoantibodies as predictors of disease. J Clin Invest
2001;108:1417-22
53. Samuels J, Ng YS, Coupillaud C, et al. Impaired early B cell tolerance in patients with
rheumatoid arthritis. J Exp Med 2005;201:1659-67
54. Keystone E. B cell targeted therapies. Arthritis Res Ther 2005;7 Suppl 3:S13-8
55. Looney RJ. B cell-targeted therapy for rheumatoid arthritis: an update on the evidence.
Drugs 2006;66:625-39
56. Stohl W. Therapeutic targeting of B lymphocyte stimulator (BLyS) in the rheumatic
diseases. Endocr Metab Immune Disord Drug Targets 2006;6:351-8
57. Moura RA, Weinmann P, Pereira PA, et al. Alterations on peripheral blood B-cell
subpopulations in very early arthritis patients. Rheumatology (Oxford) 2010;49:1082-92
58. Catalan D, Aravena O, Sabugo F, et al. B cells from rheumatoid arthritis patients show
important alterations in the expression of CD86 and FcgammaRIIb, which are modulated by anti-tumor necrosis factor therapy. Arthritis Res Ther 2010;12:R68
59. Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases.
Blood 2011;117:3720-32
60. Jacques C, Gosset M, Berenbaum F, et al. The role of IL-1 and IL-1Ra in joint
inflammation and cartilage degradation. Vitam Horm 2006;74:371-403
61. Dinarello CA. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J
Immunol 2011;41:1203-17
62. Ben-Sasson SZ, Hu-Li J, Quiel J, et al. IL-1 acts directly on CD4 T cells to enhance
their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A 2009;106:7119-24
63. Chung Y, Chang SH, Martinez GJ, et al. Critical regulation of early Th17 cell
differentiation by interleukin-1 signaling. Immunity 2009;30:576-87
64. Kryczek I, Wei S, Vatan L, et al. Cutting edge: opposite effects of IL-1 and IL-2 on the
regulation of IL-17+ T cell pool IL-1 subverts IL-2-mediated suppression. J Immunol 2007;179:1423-6
65. Fonseca JE, Santos MJ, Canhao H, et al. Interleukin-6 as a key player in systemic
inflammation and joint destruction. Autoimmun Rev 2009;8:538-42
66. Yamamoto Y, Udagawa N, Matsuura S, et al. Osteoblasts provide a suitable
microenvironment for the action of receptor activator of nuclear factor-kappaB ligand. Endocrinology 2006;147:3366-74
67. Kirkham BW, Lassere MN, Edmonds JP, et al. Synovial membrane cytokine expression
is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort). Arthritis Rheum 2006;54:1122-31
68. Joosten LA, Helsen MM, Saxne T, et al. IL-1 alpha beta blockade prevents cartilage
and bone destruction in murine type II collagen-induced arthritis, whereas TNF-alpha blockade only ameliorates joint inflammation. J Immunol 1999;163:5049-55
69. van den Berg WB, Joosten LA, Helsen M, et al. Amelioration of established murine
collagen-induced arthritis with anti-IL-1 treatment. Clin Exp Immunol 1994;95:237-43

References
- 76 -
70. Zwerina J, Redlich K, Polzer K, et al. TNF-induced structural joint damage is mediated
by IL-1. Proc Natl Acad Sci U S A 2007;104:11742-7
71. Keffer J, Probert L, Cazlaris H, et al. Transgenic mice expressing human tumour
necrosis factor: a predictive genetic model of arthritis. EMBO J 1991;10:4025-31
72. Probert L, Plows D, Kontogeorgos G, et al. The type I interleukin-1 receptor acts in
series with tumor necrosis factor (TNF) to induce arthritis in TNF-transgenic mice. Eur J Immunol 1995;25:1794-7
73. Boissier MC. Cell and cytokine imbalances in rheumatoid synovitis. Joint Bone Spine
2011;78:230-4
74. Nawroth PP, Bank I, Handley D, et al. Tumor necrosis factor/cachectin interacts with
endothelial cell receptors to induce release of interleukin 1. J Exp Med 1986;163:1363-75
75. Butler DM, Maini RN, Feldmann M, et al. Modulation of proinflammatory cytokine
release in rheumatoid synovial membrane cell cultures. Comparison of monoclonal anti TNF-alpha antibody with the interleukin-1 receptor antagonist. Eur Cytokine Netw 1995;6:225-30
76. Haworth C, Brennan FM, Chantry D, et al. Expression of granulocyte-macrophage
colony-stimulating factor in rheumatoid arthritis: regulation by tumor necrosis factor-alpha. Eur J Immunol 1991;21:2575-9
77. Chin JE, Winterrowd GE, Krzesicki RF, et al. Role of cytokines in inflammatory
synovitis. The coordinate regulation of intercellular adhesion molecule 1 and HLA class I and class II antigens in rheumatoid synovial fibroblasts. Arthritis Rheum 1990;33:1776-86
78. Dayer JM, Beutler B, Cerami A. Cachectin/tumor necrosis factor stimulates collagenase
and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J Exp Med 1985;162:2163-8
79. Bertolini DR, Nedwin GE, Bringman TS, et al. Stimulation of bone resorption and
inhibition of bone formation in vitro by human tumour necrosis factors. Nature 1986;319:516-8
80. Saxne T, Palladino MA, Jr., Heinegard D, et al. Detection of tumor necrosis factor alpha
but not tumor necrosis factor beta in rheumatoid arthritis synovial fluid and serum. Arthritis Rheum 1988;31:1041-5
81. Brennan FM, Chantry D, Jackson A, et al. Inhibitory effect of TNF alpha antibodies on
synovial cell interleukin-1 production in rheumatoid arthritis. Lancet 1989;2:244-7
82. Brennan FM, Chantry D, Jackson AM, et al. Cytokine production in culture by cells
isolated from the synovial membrane. J Autoimmun 1989;2 Suppl:177-86
83. Joosten LA, Helsen MM, van de Loo FA, et al. Anticytokine treatment of established
type II collagen-induced arthritis in DBA/1 mice. A comparative study using anti-TNF alpha, anti-IL-1 alpha/beta, and IL-1Ra. Arthritis Rheum 1996;39:797-809
84. Van Snick J. Interleukin-6: an overview. Annu Rev Immunol 1990;8:253-78
85. Yap SH, Moshage HJ, Hazenberg BP, et al. Tumor necrosis factor (TNF) inhibits
interleukin (IL)-1 and/or IL-6 stimulated synthesis of C-reactive protein (CRP) and serum amyloid A (SAA) in primary cultures of human hepatocytes. Biochim Biophys Acta 1991;1091:405-8
86. Suwa T, Hogg JC, English D, et al. Interleukin-6 induces demargination of intravascular
neutrophils and shortens their transit in marrow. Am J Physiol Heart Circ Physiol 2000;279:H2954-60
87. Muraguchi A, Hirano T, Tang B, et al. The essential role of B cell stimulatory factor 2
(BSF-2/IL-6) for the terminal differentiation of B cells. J Exp Med 1988;167:332-44
88. Jego G, Bataille R, Pellat-Deceunynck C. Interleukin-6 is a growth factor for
nonmalignant human plasmablasts. Blood 2001;97:1817-22
89. Romano M, Sironi M, Toniatti C, et al. Role of IL-6 and its soluble receptor in induction
of chemokines and leukocyte recruitment. Immunity 1997;6:315-25
90. Maruotti N, Cantatore FP, Crivellato E, et al. Angiogenesis in rheumatoid arthritis. Histol
Histopathol 2006;21:557-66

References
- 77 -
91. Hashizume M, Hayakawa N, Mihara M. IL-6 trans-signalling directly induces RANKL on
fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology (Oxford) 2008;47:1635-40
92. Hashizume M, Mihara M. Desirable effect of combination therapy with high molecular
weight hyaluronate and NSAIDs on MMP production. Osteoarthritis Cartilage 2009;17:1513-8
93. Suzuki M, Hashizume M, Yoshida H, et al. IL-6 and IL-1 synergistically enhanced the
production of MMPs from synovial cells by up-regulating IL-6 production and IL-1 receptor I expression. Cytokine 2010;51:178-83
94. Silacci P, Dayer JM, Desgeorges A, et al. Interleukin (IL)-6 and its soluble receptor
induce TIMP-1 expression in synoviocytes and chondrocytes, and block IL-1-induced collagenolytic activity. J Biol Chem 1998;273:13625-9
95. Houssiau FA, Devogelaer JP, Van Damme J, et al. Interleukin-6 in synovial fluid and
serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum 1988;31:784-8
96. Madhok R, Crilly A, Watson J, et al. Serum interleukin 6 levels in rheumatoid arthritis:
correlations with clinical and laboratory indices of disease activity. Ann Rheum Dis 1993;52:232-4
97. Mihara M, Moriya Y, Kishimoto T, et al. Interleukin-6 (IL-6) induces the proliferation of
synovial fibroblastic cells in the presence of soluble IL-6 receptor. Br J Rheumatol 1995;34:321-5
98. Kremer JM, Blanco R, Brzosko M, et al. Tocilizumab inhibits structural joint damage in
rheumatoid arthritis patients with inadequate responses to methotrexate: results from the double-blind treatment phase of a randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum 2011;63:609-21
99. Garnero P, Thompson E, Woodworth T, et al. Rapid and sustained improvement in
bone and cartilage turnover markers with the anti-interleukin-6 receptor inhibitor tocilizumab plus methotrexate in rheumatoid arthritis patients with an inadequate response to methotrexate: results from a substudy of the multicenter double-blind, placebo-controlled trial of tocilizumab in inadequate responders to methotrexate alone. Arthritis Rheum 2010;62:33-43
100. Mihara M, Kasutani K, Okazaki M, et al. Tocilizumab inhibits signal transduction
mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int Immunopharmacol 2005;5:1731-40
101. Nishimoto N, Hashimoto J, Miyasaka N, et al. Study of active controlled monotherapy
used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an x ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis 2007;66:1162-7
102. Smolen JS, Beaulieu A, Rubbert-Roth A, et al. Effect of interleukin-6 receptor inhibition
with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet 2008;371:987-97
103. Li X, Yuan FL, Lu WG, et al. The role of interleukin-17 in mediating joint destruction in
rheumatoid arthritis. Biochem Biophys Res Commun 2010;397:131-5
104. Metawi SA, Abbas D, Kamal MM, et al. Serum and synovial fluid levels of interleukin-17
in correlation with disease activity in patients with RA. Clin Rheumatol 2011;30:1201-7
105. Fossiez F, Djossou O, Chomarat P, et al. T cell interleukin-17 induces stromal cells to
produce proinflammatory and hematopoietic cytokines. J Exp Med 1996;183:2593-603
106. Yao Z, Fanslow WC, Seldin MF, et al. Herpesvirus Saimiri encodes a new cytokine, IL-
17, which binds to a novel cytokine receptor. Immunity 1995;3:811-21
107. Yao Z, Painter SL, Fanslow WC, et al. Human IL-17: a novel cytokine derived from T
cells. J Immunol 1995;155:5483-6
108. Lundy SK, Sarkar S, Tesmer LA, et al. Cells of the synovium in rheumatoid arthritis. T
lymphocytes. Arthritis Res Ther 2007;9:202

References
- 78 -
109. Paradowska A, Masliniski W, Grzybowska-Kowalczyk A, et al. The function of
interleukin 17 in the pathogenesis of rheumatoid arthritis. Arch Immunol Ther Exp (Warsz) 2007;55:329-34
110. Sylvester J, Liacini A, Li WQ, et al. Interleukin-17 signal transduction pathways
implicated in inducing matrix metalloproteinase-3, -13 and aggrecanase-1 genes in articular chondrocytes. Cell Signal 2004;16:469-76
111. Chabaud M, Lubberts E, Joosten L, et al. IL-17 derived from juxta-articular bone and
synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res 2001;3:168-77
112. Chabaud M, Durand JM, Buchs N, et al. Human interleukin-17: A T cell-derived
proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum 1999;42:963-70
113. Koenders MI, Lubberts E, Oppers-Walgreen B, et al. Blocking of interleukin-17 during
reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol 2005;167:141-9
114. Lubberts E, Koenders MI, van den Berg WB. The role of T-cell interleukin-17 in
conducting destructive arthritis: lessons from animal models. Arthritis Res Ther 2005;7:29-37
115. Guidelines for the management of rheumatoid arthritis: 2002 Update. Arthritis Rheum
2002;46:328-46
116. Goekoop-Ruiterman YP, de Vries-Bouwstra JK, Allaart CF, et al. Clinical and
radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): a randomized, controlled trial. Arthritis Rheum 2005;52:3381-90
117. Quinn MA, Conaghan PG, O'Connor PJ, et al. Very early treatment with infliximab in
addition to methotrexate in early, poor-prognosis rheumatoid arthritis reduces magnetic resonance imaging evidence of synovitis and damage, with sustained benefit after infliximab withdrawal: results from a twelve-month randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2005;52:27-35
118. Gaffo A, Saag KG, Curtis JR. Treatment of rheumatoid arthritis. Am J Health Syst
Pharm 2006;63:2451-65
119. Polido-Pereira J, Vieira-Sousa E, Fonseca JE. Rheumatoid arthritis: what is refractory
disease and how to manage it? Autoimmun Rev 2011;10:707-13
120. Sizova L. Approaches to the treatment of early rheumatoid arthritis with disease-
modifying antirheumatic drugs. Br J Clin Pharmacol 2008;66:173-8
121. Wolfe F, Hawley DJ, Cathey MA. Termination of slow acting antirheumatic therapy in
rheumatoid arthritis: a 14-year prospective evaluation of 1017 consecutive starts. J Rheumatol 1990;17:994-1002
122. Pincus T, Marcum SB, Callahan LF. Longterm drug therapy for rheumatoid arthritis in
seven rheumatology private practices: II. Second line drugs and prednisone. J Rheumatol 1992;19:1885-94
123. Ortendahl M, Holmes T, Schettler JD, et al. The methotrexate therapeutic response in
rheumatoid arthritis. J Rheumatol 2002;29:2084-91
124. Cronstein BN. Low-dose methotrexate: a mainstay in the treatment of rheumatoid
arthritis. Pharmacol Rev 2005;57:163-72
125. Cash JM, Klippel JH. Second-line drug therapy for rheumatoid arthritis. N Engl J Med
1994;330:1368-75
126. Nagashima M, Matsuoka T, Saitoh K, et al. Treatment continuation rate in relation to
efficacy and toxicity in long-term therapy with low-dose methotrexate, sulfasalazine, and bucillamine in 1,358 Japanese patients with rheumatoid arthritis. Clin Exp Rheumatol 2006;24:260-7
127. Keystone EC. The role of tumor necrosis factor antagonism in clinical practice. J
Rheumatol Suppl 1999;57:22-8
128. Saag KG, Teng GG, Patkar NM, et al. American College of Rheumatology 2008
recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum 2008;59:762-84

References
- 79 -
129. Emery P, Breedveld FC, Hall S, et al. Comparison of methotrexate monotherapy with a
combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet 2008;372:375-82
130. Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: A multicenter,
randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 2006;54:26-37
131. Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory
to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum 2006;54:2793-806
132. Genovese MC, Becker JC, Schiff M, et al. Abatacept for rheumatoid arthritis refractory
to tumor necrosis factor alpha inhibition. N Engl J Med 2005;353:1114-23
133. Tak PP, Rigby WF, Rubbert-Roth A, et al. Inhibition of joint damage and improved
clinical outcomes with rituximab plus methotrexate in early active rheumatoid arthritis: the IMAGE trial. Ann Rheum Dis 2011;70:39-46
134. Keystone E, Emery P, Peterfy CG, et al. Rituximab inhibits structural joint damage in
patients with rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitor therapies. Ann Rheum Dis 2009;68:216-21
135. Genant HK, Peterfy CG, Westhovens R, et al. Abatacept inhibits progression of
structural damage in rheumatoid arthritis: results from the long-term extension of the AIM trial. Ann Rheum Dis 2008;67:1084-9
136. Molto A, Olive A. Anti-IL-1 molecules: new comers and new indications. Joint Bone
Spine 2010;77:102-7
137. Cohen S, Hurd E, Cush J, et al. Treatment of rheumatoid arthritis with anakinra, a
recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2002;46:614-24
138. Bresnihan B, Alvaro-Gracia JM, Cobby M, et al. Treatment of rheumatoid arthritis with
recombinant human interleukin-1 receptor antagonist. Arthritis Rheum 1998;41:2196-204
139. Alten R, Gomez-Reino J, Durez P, et al. Efficacy and safety of the human anti-IL-1beta
monoclonal antibody canakinumab in rheumatoid arthritis: results of a 12-week, phase II, dose-finding study. BMC Musculoskelet Disord 2011;12:153
140. Cohen SB, Dore RK, Lane NE, et al. Denosumab treatment effects on structural
damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum 2008;58:1299-309
141. Vander Cruyssen B, Van Looy S, Wyns B, et al. Four-year follow-up of infliximab
therapy in rheumatoid arthritis patients with long-standing refractory disease: attrition and long-term evolution of disease activity. Arthritis Res Ther 2006;8:R112
142. Karlsson JA, Kristensen LE, Kapetanovic MC, et al. Treatment response to a second or
third TNF-inhibitor in RA: results from the South Swedish Arthritis Treatment Group Register. Rheumatology (Oxford) 2008;47:507-13
143. Calabrese LH. Molecular differences in anticytokine therapies. Clin Exp Rheumatol
2003;21:241-8
144. Sims JE, Nicklin MJ, Bazan JF, et al. A new nomenclature for IL-1-family genes. Trends
Immunol 2001;22:536-7
145. Dinarello CA. Biologic basis for interleukin-1 in disease. Blood 1996;87:2095-147
146. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family.
Annu Rev Immunol 2009;27:519-50
147. Martinon F, Petrilli V, Mayor A, et al. Gout-associated uric acid crystals activate the
NALP3 inflammasome. Nature 2006;440:237-41

References
- 80 -
148. Lamkanfi M, Kanneganti TD. Nlrp3: an immune sensor of cellular stress and infection.
Int J Biochem Cell Biol 2010;42:792-5
149. Eriksson U, Kurrer MO, Sonderegger I, et al. Activation of dendritic cells through the
interleukin 1 receptor 1 is critical for the induction of autoimmune myocarditis. J Exp Med 2003;197:323-31
150. Schindler R, Clark BD, Dinarello CA. Dissociation between interleukin-1 beta mRNA
and protein synthesis in human peripheral blood mononuclear cells. J Biol Chem 1990;265:10232-7
151. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell
2010;140:805-20
152. Burns K, Martinon F, Tschopp J. New insights into the mechanism of IL-1beta
maturation. Curr Opin Immunol 2003;15:26-30
153. Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is
required for interleukin-1 beta processing in monocytes. Nature 1992;356:768-74
154. Romanowski MJ, Scheer JM, O'Brien T, et al. Crystal structures of a ligand-free and
malonate-bound human caspase-1: implications for the mechanism of substrate binding. Structure 2004;12:1361-71
155. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering
activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002;10:417-26
156. Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821-32
157. Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates sensitization to
ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 2009;183:792-6
158. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern
recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009;183:787-91
159. O'Connor W, Jr., Harton JA, Zhu X, et al. Cutting edge:
CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF-kappa B suppressive properties. J Immunol 2003;171:6329-33
160. Cassel SL, Eisenbarth SC, Iyer SS, et al. The Nalp3 inflammasome is essential for the
development of silicosis. Proc Natl Acad Sci U S A 2008;105:9035-40
161. Dostert C, Petrilli V, Van Bruggen R, et al. Innate immune activation through Nalp3
inflammasome sensing of asbestos and silica. Science 2008;320:674-7
162. Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in
response to toxins and ATP. Nature 2006;440:228-32
163. Meissner F, Molawi K, Zychlinsky A. Superoxide dismutase 1 regulates caspase-1 and
endotoxic shock. Nat Immunol 2008;9:866-72
164. Zhou R, Tardivel A, Thorens B, et al. Thioredoxin-interacting protein links oxidative
stress to inflammasome activation. Nat Immunol 2010;11:136-40
165. Petrilli V, Papin S, Dostert C, et al. Activation of the NALP3 inflammasome is triggered
by low intracellular potassium concentration. Cell Death Differ 2007;14:1583-9
166. Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the
NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008;9:847-56
167. Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the
innate immune response to amyloid-beta. Nat Immunol 2008;9:857-65
168. Bryan NB, Dorfleutner A, Kramer SJ, et al. Differential splicing of the apoptosis-
associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J Inflamm (Lond) 2010;7:23
169. Chae JJ, Wood G, Masters SL, et al. The B30.2 domain of pyrin, the familial
Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A 2006;103:9982-7
170. Joosten LA, Netea MG, Fantuzzi G, et al. Inflammatory arthritis in caspase 1 gene-
deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum 2009;60:3651-62

References
- 81 -
171. Guma M, Ronacher L, Liu-Bryan R, et al. Caspase 1-independent activation of
interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum 2009;60:3642-50
172. Brough D, Rothwell NJ. Caspase-1-dependent processing of pro-interleukin-1beta is
cytosolic and precedes cell death. J Cell Sci 2007;120:772-81
173. Rubartelli A, Cozzolino F, Talio M, et al. A novel secretory pathway for interleukin-1
beta, a protein lacking a signal sequence. EMBO J 1990;9:1503-10
174. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1beta secretion.
Cytokine Growth Factor Rev 2011;22:189-95
175. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal 2010;3:cm1
176. Liu S, Chen ZJ. Expanding role of ubiquitination in NF-kappaB signaling. Cell Res
2011;21:6-21
177. Horai R, Saijo S, Tanioka H, et al. Development of chronic inflammatory arthropathy
resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med 2000;191:313-20
178. Colotta F, Re F, Muzio M, et al. Interleukin-1 type II receptor: a decoy target for IL-1 that
is regulated by IL-4. Science 1993;261:472-5
179. Smeets RL, Joosten LA, Arntz OJ, et al. Soluble interleukin-1 receptor accessory
protein ameliorates collagen-induced arthritis by a different mode of action from that of interleukin-1 receptor antagonist. Arthritis Rheum 2005;52:2202-11
180. Vambutas A, DeVoti J, Goldofsky E, et al. Alternate splicing of interleukin-1 receptor
type II (IL1R2) in vitro correlates with clinical glucocorticoid responsiveness in patients with AIED. PLoS One 2009;4:e5293
181. Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune
system to autoinflammatory diseases. Cell 2004;117:561-74
182. Hoffman HM, Rosengren S, Boyle DL, et al. Prevention of cold-associated acute
inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet 2004;364:1779-85
183. Hawkins PN, Lachmann HJ, Aganna E, et al. Spectrum of clinical features in Muckle-
Wells syndrome and response to anakinra. Arthritis Rheum 2004;50:607-12
184. Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem
inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med 2006;355:581-92
185. Lachmann HJ, Lowe P, Felix SD, et al. In vivo regulation of interleukin 1beta in patients
with cryopyrin-associated periodic syndromes. J Exp Med 2009;206:1029-36
186. Pascual V, Allantaz F, Arce E, et al. Role of interleukin-1 (IL-1) in the pathogenesis of
systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med 2005;201:1479-86
187. Gattorno M, Tassi S, Carta S, et al. Pattern of interleukin-1beta secretion in response to
lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum 2007;56:3138-48
188. Dinarello CA, Ikejima T, Warner SJ, et al. Interleukin 1 induces interleukin 1. I. Induction
of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol 1987;139:1902-10
189. Hoffman HM, Mueller JL, Broide DH, et al. Mutation of a new gene encoding a putative
pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001;29:301-5
190. Masters SL, Simon A, Aksentijevich I, et al. Horror autoinflammaticus: the molecular
pathophysiology of autoinflammatory disease (*). Annu Rev Immunol 2009;27:621-68
191. Meng G, Zhang F, Fuss I, et al. A mutation in the Nlrp3 gene causing inflammasome
hyperactivation potentiates Th17 cell-dominant immune responses. Immunity 2009;30:860-74
192. Brydges SD, Mueller JL, McGeough MD, et al. Inflammasome-mediated disease animal
models reveal roles for innate but not adaptive immunity. Immunity 2009;30:875-87

References
- 82 -
193. Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1
Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 2008;58:2443-52
194. Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the
cryopyrin-associated periodic syndrome. N Engl J Med 2009;360:2416-25
195. Martinon F. Update on biology: uric acid and the activation of immune and inflammatory
cells. Curr Rheumatol Rep 2010;12:135-41
196. Terkeltaub RA. Clinical practice. Gout. N Engl J Med 2003;349:1647-55
197. Joosten LA, Netea MG, Mylona E, et al. Engagement of fatty acids with Toll-like
receptor 2 drives interleukin-1beta production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum 2010;62:3237-48
198. Dinarello CA. How interleukin-1beta induces gouty arthritis. Arthritis Rheum
2010;62:3140-4
199. So A, De Smedt T, Revaz S, et al. A pilot study of IL-1 inhibition by anakinra in acute
gout. Arthritis Res Ther 2007;9:R28
200. Terkeltaub R. Update on gout: new therapeutic strategies and options. Nat Rev
Rheumatol 2010;6:30-8
201. So A, De Meulemeester M, Pikhlak A, et al. Canakinumab for the treatment of acute
flares in difficult-to-treat gouty arthritis: Results of a multicenter, phase II, dose-ranging study. Arthritis Rheum 2010;62:3064-76
202. Terkeltaub R, Sundy JS, Schumacher HR, et al. The interleukin 1 inhibitor rilonacept in
treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis 2009;68:1613-7
203. Scott P, Ma H, Viriyakosol S, et al. Engagement of CD14 mediates the inflammatory
potential of monosodium urate crystals. J Immunol 2006;177:6370-8
204. Tramontini N, Huber C, Liu-Bryan R, et al. Central role of complement membrane attack
complex in monosodium urate crystal-induced neutrophilic rabbit knee synovitis. Arthritis Rheum 2004;50:2633-9
205. Announ N, Palmer G, Guerne PA, et al. Anakinra is a possible alternative in the
treatment and prevention of acute attacks of pseudogout in end-stage renal failure. Joint Bone Spine 2009;76:424-6
206. McGonagle D, Tan AL, Madden J, et al. Successful treatment of resistant pseudogout
with anakinra. Arthritis Rheum 2008;58:631-3
207. Joosten LA. Excessive interleukin-1 signaling determines the development of Th1 and
Th17 responses in chronic inflammation. Arthritis Rheum 2010;62:320-2
208. Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of
human interleukin 17-producing helper T cells. Nat Immunol 2007;8:950-7
209. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, et al. Interleukins 1beta and 6 but
not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol 2007;8:942-9
210. Koenders MI, Devesa I, Marijnissen RJ, et al. Interleukin-1 drives pathogenic Th17 cells
during spontaneous arthritis in interleukin-1 receptor antagonist-deficient mice. Arthritis Rheum 2008;58:3461-70
211. Nakae S, Saijo S, Horai R, et al. IL-17 production from activated T cells is required for
the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci U S A 2003;100:5986-90
212. van de Veerdonk FL, Lauwerys B, Marijnissen RJ, et al. The anti-CD20 antibody
rituximab reduces the Th17 cell response. Arthritis Rheum 2011;63:1507-16
213. Nakae S, Asano M, Horai R, et al. IL-1 enhances T cell-dependent antibody production
through induction of CD40 ligand and OX40 on T cells. J Immunol 2001;167:90-7
214. O'Sullivan BJ, Thomas HE, Pai S, et al. IL-1 beta breaks tolerance through expansion
of CD25+ effector T cells. J Immunol 2006;176:7278-87
215. Leadbetter EA, Rifkin IR, Hohlbaum AM, et al. Chromatin-IgG complexes activate B
cells by dual engagement of IgM and Toll-like receptors. Nature 2002;416:603-7

References
- 83 -
216. Joosten LA, Koenders MI, Smeets RL, et al. Toll-like receptor 2 pathway drives
streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J Immunol 2003;171:6145-53
217. Kyburz D, Rethage J, Seibl R, et al. Bacterial peptidoglycans but not CpG
oligodeoxynucleotides activate synovial fibroblasts by toll-like receptor signaling. Arthritis Rheum 2003;48:642-50
218. Kastbom A, Verma D, Eriksson P, et al. Genetic variation in proteins of the cryopyrin
inflammasome influences susceptibility and severity of rheumatoid arthritis (the Swedish TIRA project). Rheumatology (Oxford) 2008;47:415-7
219. Kastbom A, Johansson M, Verma D, et al. CARD8 p.C10X polymorphism is associated
with inflammatory activity in early rheumatoid arthritis. Ann Rheum Dis 2010;69:723-6
220. Fontalba A, Martinez-Taboada V, Gutierrez O, et al. Deficiency of the NF-kappaB
inhibitor caspase activating and recruitment domain 8 in patients with rheumatoid arthritis is associated with disease severity. J Immunol 2007;179:4867-73
221. Rosengren S, Hoffman HM, Bugbee W, et al. Expression and regulation of cryopyrin
and related proteins in rheumatoid arthritis synovium. Ann Rheum Dis 2005;64:708-14
222. Kolly L, Busso N, Palmer G, et al. Expression and function of the NALP3 inflammasome
in rheumatoid synovium. Immunology 2010;129:178-85
223. Shankar S, Handa R. Biological agents in rheumatoid arthritis. J Postgrad Med
2004;50:293-9
224. Gracie JA, Forsey RJ, Chan WL, et al. A proinflammatory role for IL-18 in rheumatoid
arthritis. J Clin Invest 1999;104:1393-401
225. Tak PP, Bresnihan B. The pathogenesis and prevention of joint damage in rheumatoid
arthritis: advances from synovial biopsy and tissue analysis. Arthritis Rheum 2000;43:2619-33
226. Dayer JM. The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid
arthritis. Rheumatology (Oxford) 2003;42 Suppl 2:ii3-10
227. Lettesjo H, Nordstrom E, Strom H, et al. Synovial fluid cytokines in patients with
rheumatoid arthritis or other arthritic lesions. Scand J Immunol 1998;48:286-92
228. Mertens M, Singh JA. Anakinra for rheumatoid arthritis: a systematic review. J
Rheumatol 2009;36:1118-25
229. Fleischmann RM, Schechtman J, Bennett R, et al. Anakinra, a recombinant human
interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis: A large, international, multicenter, placebo-controlled trial. Arthritis Rheum 2003;48:927-34
230. Tesser J, Fleischmann R, Dore R, et al. Concomitant medication use in a large,
international, multicenter, placebo controlled trial of anakinra, a recombinant interleukin 1 receptor antagonist, in patients with rheumatoid arthritis. J Rheumatol 2004;31:649-54
231. Schiff MH, DiVittorio G, Tesser J, et al. The safety of anakinra in high-risk patients with
active rheumatoid arthritis: six-month observations of patients with comorbid conditions. Arthritis Rheum 2004;50:1752-60
232. Nuki G, Bresnihan B, Bear MB, et al. Long-term safety and maintenance of clinical
improvement following treatment with anakinra (recombinant human interleukin-1 receptor antagonist) in patients with rheumatoid arthritis: extension phase of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2002;46:2838-46
233. Bresnihan B, Newmark R, Robbins S, et al. Effects of anakinra monotherapy on joint
damage in patients with rheumatoid arthritis. Extension of a 24-week randomized, placebo-controlled trial. J Rheumatol 2004;31:1103-11
234. Abramson SB, Amin A. Blocking the effects of IL-1 in rheumatoid arthritis protects bone
and cartilage. Rheumatology (Oxford) 2002;41:972-80
235. Bresnihan B, Cobby M. Clinical and radiological effects of anakinra in patients with
rheumatoid arthritis. Rheumatology (Oxford) 2003;42 Suppl 2:ii22-8
236. Campion GV, Lebsack ME, Lookabaugh J, et al. Dose-range and dose-frequency study
of recombinant human interleukin-1 receptor antagonist in patients with rheumatoid arthritis. The IL-1Ra Arthritis Study Group. Arthritis Rheum 1996;39:1092-101

References
- 84 -
237. Dinarello CA, Thompson RC. Blocking IL-1: interleukin 1 receptor antagonist in vivo and
in vitro. Immunol Today 1991;12:404-10
238. Burger D, Dayer JM, Palmer G, et al. Is IL-1 a good therapeutic target in the treatment
of arthritis? Best Pract Res Clin Rheumatol 2006;20:879-96
239. Nixon R, Bansback N, Brennan A. The efficacy of inhibiting tumour necrosis factor
alpha and interleukin 1 in patients with rheumatoid arthritis: a meta-analysis and adjusted indirect comparisons. Rheumatology (Oxford) 2007;46:1140-7
240. Thaler K, Chandiramani DV, Hansen RA, et al. Efficacy and safety of anakinra for the
treatment of rheumatoid arthritis: an update of the Oregon Drug Effectiveness Review Project. Biologics 2009;3:485-98
241. Kim HJ, Krenn V, Steinhauser G, et al. Plasma cell development in synovial germinal
centers in patients with rheumatoid and reactive arthritis. J Immunol 1999;162:3053-62
242. Fonseca JE, Cortez-Dias N, Francisco A, et al. Inflammatory cell infiltrate and
RANKL/OPG expression in rheumatoid synovium: comparison with other inflammatory arthropathies and correlation with outcome. Clin Exp Rheumatol 2005;23:185-92
243. Fonseca JE, Edwards JC, Blades S, et al. Macrophage subpopulations in rheumatoid
synovium: reduced CD163 expression in CD4+ T lymphocyte-rich microenvironments. Arthritis Rheum 2002;46:1210-6
244. Nagatani K, Itoh K, Nakajima K, et al. Rheumatoid arthritis fibroblast-like synoviocytes
express BCMA and are stimulated by APRIL. Arthritis Rheum 2007;56:3554-63
245. Chen DY, Hsieh TY, Chen YM, et al. Proinflammatory cytokine profiles of patients with
elderly-onset rheumatoid arthritis: a comparison with younger-onset disease. Gerontology 2009;55:250-8
246. Petrovic-Rackov L, Pejnovic N. Clinical significance of IL-18, IL-15, IL-12 and TNF-
alpha measurement in rheumatoid arthritis. Clin Rheumatol 2006;25:448-52
247. Dominical VM, Bertolo MB, Almeida CB, et al. Neutrophils of rheumatoid arthritis
patients on anti-TNF-alpha therapy and in disease remission present reduced adhesive functions in association with decreased circulating neutrophil-attractant chemokine levels. Scand J Immunol 2011;73:309-18
248. Koch AE, Kunkel SL, Harlow LA, et al. Enhanced production of monocyte
chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest 1992;90:772-9
249. Litinskiy MB, Nardelli B, Hilbert DM, et al. DCs induce CD40-independent
immunoglobulin class switching through BLyS and APRIL. Nat Immunol 2002;3:822-9
250. Castigli E, Wilson SA, Scott S, et al. TACI and BAFF-R mediate isotype switching in B
cells. J Exp Med 2005;201:35-9
251. Castigli E, Scott S, Dedeoglu F, et al. Impaired IgA class switching in APRIL-deficient
mice. Proc Natl Acad Sci U S A 2004;101:3903-8
252. Belnoue E, Pihlgren M, McGaha TL, et al. APRIL is critical for plasmablast survival in
the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood 2008;111:2755-64
253. Bossen C, Cachero TG, Tardivel A, et al. TACI, unlike BAFF-R, is solely activated by
oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood 2008;111:1004-12
254. Dong W, Li X, Liu H, et al. Infiltrations of plasma cells in synovium are highly associated
with synovial fluid levels of APRIL in inflamed peripheral joints of rheumatoid arthritis. Rheumatol Int 2009;29:801-6
255. Clavel C, Nogueira L, Laurent L, et al. Induction of macrophage secretion of tumor
necrosis factor alpha through Fcgamma receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum 2008;58:678-88
256. Blom AB, Radstake TR, Holthuysen AE, et al. Increased expression of Fcgamma
receptors II and III on macrophages of rheumatoid arthritis patients results in higher production of tumor necrosis factor alpha and matrix metalloproteinase. Arthritis Rheum 2003;48:1002-14

References
- 85 -
257. Iwahashi M, Yamamura M, Aita T, et al. Expression of Toll-like receptor 2 on CD16+
blood monocytes and synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum 2004;50:1457-67
258. Jovanovic DV, Di Battista JA, Martel-Pelletier J, et al. IL-17 stimulates the production
and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol 1998;160:3513-21
259. Sokolove J, Zhao X, Chandra PE, et al. Immune complexes containing citrullinated
fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum 2011;63:53-62
260. Liang SC, Tan XY, Luxenberg DP, et al. Interleukin (IL)-22 and IL-17 are coexpressed
by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 2006;203:2271-9
261. Pene J, Chevalier S, Preisser L, et al. Chronically inflamed human tissues are infiltrated
by highly differentiated Th17 lymphocytes. J Immunol 2008;180:7423-30
262. Pelletier M, Maggi L, Micheletti A, et al. Evidence for a cross-talk between human
neutrophils and Th17 cells. Blood 2010;115:335-43
263. Parsonage G, Filer A, Bik M, et al. Prolonged, granulocyte-macrophage colony-
stimulating factor-dependent, neutrophil survival following rheumatoid synovial fibroblast activation by IL-17 and TNFalpha. Arthritis Res Ther 2008;10:R47
264. Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with
rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 1999;103:1345-52
265. Yago T, Nanke Y, Ichikawa N, et al. IL-17 induces osteoclastogenesis from human
monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-alpha antibody: a novel mechanism of osteoclastogenesis by IL-17. J Cell Biochem 2009;108:947-55
266. Pollinger B, Junt T, Metzler B, et al. Th17 cells, not IL-17+ gammadelta T cells, drive
arthritic bone destruction in mice and humans. J Immunol 2011;186:2602-12
267. Vinuesa CG, Tangye SG, Moser B, et al. Follicular B helper T cells in antibody
responses and autoimmunity. Nat Rev Immunol 2005;5:853-65
268. Salzer U, Grimbacher B. Common variable immunodeficiency: The power of co-
stimulation. Semin Immunol 2006;18:337-46
269. Hickman-Brecks CL, Racz JL, Meyer DM, et al. Th17 cells can provide B cell help in
autoantibody induced arthritis. J Autoimmun 2011;36:65-75
270. Schneider P, MacKay F, Steiner V, et al. BAFF, a novel ligand of the tumor necrosis
factor family, stimulates B cell growth. J Exp Med 1999;189:1747-56
271. Cheema GS, Roschke V, Hilbert DM, et al. Elevated serum B lymphocyte stimulator
levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum 2001;44:1313-9
272. Bosello S, Youinou P, Daridon C, et al. Concentrations of BAFF correlate with
autoantibody levels, clinical disease activity, and response to treatment in early rheumatoid arthritis. J Rheumatol 2008;35:1256-64
273. Leonard WJ, Spolski R. Interleukin-21: a modulator of lymphoid proliferation, apoptosis
and differentiation. Nat Rev Immunol 2005;5:688-98
274. Korn T, Bettelli E, Gao W, et al. IL-21 initiates an alternative pathway to induce
proinflammatory T(H)17 cells. Nature 2007;448:484-7
275. Nurieva R, Yang XO, Martinez G, et al. Essential autocrine regulation by IL-21 in the
generation of inflammatory T cells. Nature 2007;448:480-3
276. Ettinger R, Sims GP, Fairhurst AM, et al. IL-21 induces differentiation of human naive
and memory B cells into antibody-secreting plasma cells. J Immunol 2005;175:7867-79
277. Niu X, He D, Zhang X, et al. IL-21 regulates Th17 cells in rheumatoid arthritis. Hum
Immunol 2010;71:334-41
278. Kokkonen H, Soderstrom I, Rocklov J, et al. Up-regulation of cytokines and chemokines
predates the onset of rheumatoid arthritis. Arthritis Rheum 2010;62:383-91

References
- 86 -
279. Deane KD, O'Donnell CI, Hueber W, et al. The number of elevated cytokines and
chemokines in preclinical seropositive rheumatoid arthritis predicts time to diagnosis in an age-dependent manner. Arthritis Rheum 2010;62:3161-72
280. Milman N, Karsh J, Booth RA. Correlation of a multi-cytokine panel with clinical disease
activity in patients with rheumatoid arthritis. Clin Biochem 2010;43:1309-14
281. Raza K, Falciani F, Curnow SJ, et al. Early rheumatoid arthritis is characterized by a
distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res Ther 2005;7:R784-95
282. Hueber AJ, Asquith DL, Miller AM, et al. Mast cells express IL-17A in rheumatoid
arthritis synovium. J Immunol 2010;184:3336-40
283. Nistala K, Adams S, Cambrook H, et al. Th17 plasticity in human autoimmune arthritis is
driven by the inflammatory environment. Proc Natl Acad Sci U S A 2010;107:14751-6
284. Aupperle K, Bennett B, Han Z, et al. NF-kappa B regulation by I kappa B kinase-2 in
rheumatoid arthritis synoviocytes. J Immunol 2001;166:2705-11
285. Stehlik C, Lee SH, Dorfleutner A, et al. Apoptosis-associated speck-like protein
containing a caspase recruitment domain is a regulator of procaspase-1 activation. J Immunol 2003;171:6154-63
286. Agostini L, Martinon F, Burns K, et al. NALP3 forms an IL-1beta-processing
inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004;20:319-25
287. Manji GA, Wang L, Geddes BJ, et al. PYPAF1, a PYRIN-containing Apaf1-like protein
that assembles with ASC and regulates activation of NF-kappa B. J Biol Chem 2002;277:11570-5
288. Dowds TA, Masumoto J, Zhu L, et al. Cryopyrin-induced interleukin 1beta secretion in
monocytic cells: enhanced activity of disease-associated mutants and requirement for ASC. J Biol Chem 2004;279:21924-8
289. Stehlik C, Fiorentino L, Dorfleutner A, et al. The PAAD/PYRIN-family protein ASC is a
dual regulator of a conserved step in nuclear factor kappaB activation pathways. J Exp Med 2002;196:1605-15
290. Bulosan M, Pauley KM, Yo K, et al. Inflammatory caspases are critical for enhanced cell
death in the target tissue of Sjogren's syndrome before disease onset. Immunol Cell Biol 2009;87:81-90
291. Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation
targets caspase-11. Nature 2011;479:117-21
292. Sui J, Li H, Fang Y, et al. NLRP1 gene polymorphism influences gene transcription and
is a risk factor for rheumatoid arthritis in Han Chinese. Arthritis Rheum 2011;
293. He D, Xu Q, Yan M, et al. The NF-kappa B inhibitor, celastrol, could enhance the anti-
cancer effect of gambogic acid on oral squamous cell carcinoma. BMC Cancer 2009;9:343
294. Yang H, Chen D, Cui QC, et al. Celastrol, a triterpene extracted from the Chinese
"Thunder of God Vine," is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res 2006;66:4758-65
295. Lee JH, Koo TH, Yoon H, et al. Inhibition of NF-kappa B activation through targeting I
kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol 2006;72:1311-21
296. Ngassapa O, Soejarto DD, Pezzuto JM, et al. Quinone-methide triterpenes and
salaspermic acid from Kokoona ochracea. J Nat Prod 1994;57:1-8
297. Pinna GF, Fiorucci M, Reimund JM, et al. Celastrol inhibits pro-inflammatory cytokine
secretion in Crohn's disease biopsies. Biochem Biophys Res Commun 2004;322:778-86
298. Han QB, Cheung S, Tai J, et al. Stability and cytotoxicity of gambogic acid and its
derivative, gambogoic acid. Biol Pharm Bull 2005;28:2335-7
299. Pandey MK, Sung B, Ahn KS, et al. Gambogic acid, a novel ligand for transferrin
receptor, potentiates TNF-induced apoptosis through modulation of the nuclear factor-kappaB signaling pathway. Blood 2007;110:3517-25

References
- 87 -
300. Zhang HZ, Kasibhatla S, Wang Y, et al. Discovery, characterization and SAR of
gambogic acid as a potent apoptosis inducer by a HTS assay. Bioorg Med Chem 2004;12:309-17
301. Kasibhatla S, Jessen KA, Maliartchouk S, et al. A role for transferrin receptor in
triggering apoptosis when targeted with gambogic acid. Proc Natl Acad Sci U S A 2005;102:12095-100
302. Palempalli UD, Gandhi U, Kalantari P, et al. Gambogic acid covalently modifies IkappaB
kinase-beta subunit to mediate suppression of lipopolysaccharide-induced activation of NF-kappaB in macrophages. Biochem J 2009;419:401-9
303. Bendele A. Animal models of rheumatoid arthritis. J Musculoskelet Neuronal Interact
2001;1:377-85
304. Kannan K, Ortmann RA, Kimpel D. Animal models of rheumatoid arthritis and their
relevance to human disease. Pathophysiology 2005;12:167-81
305. Philippe L, Gegout-Pottie P, Guingamp C, et al. Relations between functional,
inflammatory, and degenerative parameters during adjuvant arthritis in rats. Am J Physiol 1997;273:R1550-6
306. Van Eden W, Waksman BH. Immune regulation in adjuvant-induced arthritis: possible
implications for innovative therapeutic strategies in arthritis. Arthritis Rheum 2003;48:1788-96
307. Jaffee BD, Kerr JS, Jones EA, et al. The effect of immunomodulating drugs on
adjuvant-induced arthritis in Lewis rats. Agents Actions 1989;27:344-6
308. Rovensky J, Svik K, Stancikova M, et al. Effect of immunostimulatory ribomunyl on the
preventive treatment of rat adjuvant arthritis with cyclosporine and methotrexate. J Rheumatol 2003;30:2027-32
309. Rovensky J, Svik K, Matha V, et al. Combination treatment of rat adjuvant-induced
arthritis with methotrexate, probiotic bacteria Enterococcus faecium, and selenium. Ann N Y Acad Sci 2005;1051:570-81
310. Silva MA, Ishii-Iwamoto EL, Bracht A, et al. Efficiency of combined
methotrexate/chloroquine therapy in adjuvant-induced arthritis. Fundam Clin Pharmacol
2005;19:479-89
311. Noguchi M, Kimoto A, Kobayashi S, et al. Effect of celecoxib, a cyclooxygenase-2
inhibitor, on the pathophysiology of adjuvant arthritis in rat. Eur J Pharmacol 2005;513:229-35
312. Bendele A, McAbee T, Sennello G, et al. Efficacy of sustained blood levels of
interleukin-1 receptor antagonist in animal models of arthritis: comparison of efficacy in animal models with human clinical data. Arthritis Rheum 1999;42:498-506
313. Bendele AM, Chlipala ES, Scherrer J, et al. Combination benefit of treatment with the
cytokine inhibitors interleukin-1 receptor antagonist and PEGylated soluble tumor necrosis factor receptor type I in animal models of rheumatoid arthritis. Arthritis Rheum 2000;43:2648-59
314. Miwatashi S, Arikawa Y, Kotani E, et al. Novel inhibitor of p38 MAP kinase as an anti-
TNF-alpha drug: discovery of N-[4-[2-ethyl-4-(3-methylphenyl)-1,3-thiazol-5-yl]-2-pyridyl]benzamide (TAK-715) as a potent and orally active anti-rheumatoid arthritis agent. J Med Chem 2005;48:5966-79
315. Cole P, Rabasseda X. The soluble tumor necrosis factor receptor etanercept: a new
strategy for the treatment of autoimmune rheumatic disease. Drugs Today (Barc) 2004;40:281-324
316. Aya K, Alhawagri M, Hagen-Stapleton A, et al. NF-(kappa)B-inducing kinase controls
lymphocyte and osteoclast activities in inflammatory arthritis. J Clin Invest 2005;115:1848-54
317. Tas SW, Vervoordeldonk MJ, Hajji N, et al. Local treatment with the selective IkappaB
kinase beta inhibitor NEMO-binding domain peptide ameliorates synovial inflammation. Arthritis Res Ther 2006;8:R86
318. Okamoto H, Iwamoto T, Kotake S, et al. Inhibition of NF-kappaB signaling by
fenofibrate, a peroxisome proliferator-activated receptor-alpha ligand, presents a therapeutic strategy for rheumatoid arthritis. Clin Exp Rheumatol 2005;23:323-30

References
- 88 -
319. Okamoto H, Yoshio T, Kaneko H, et al. Inhibition of NF-kappaB signaling by fasudil as a
potential therapeutic strategy for rheumatoid arthritis. Arthritis Rheum 2010;62:82-92
320. Eck SL, Perkins ND, Carr DP, et al. Inhibition of phorbol ester-induced cellular adhesion
by competitive binding of NF-kappa B in vivo. Mol Cell Biol 1993;13:6530-6
321. Lai WC, Zhou M, Shankavaram U, et al. Differential regulation of lipopolysaccharide-
induced monocyte matrix metalloproteinase (MMP)-1 and MMP-9 by p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J Immunol 2003;170:6244-9
322. Vincenti MP, Coon CI, Brinckerhoff CE. Nuclear factor kappaB/p50 activates an
element in the distal matrix metalloproteinase 1 promoter in interleukin-1beta-stimulated synovial fibroblasts. Arthritis Rheum 1998;41:1987-94
323. Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-
13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res 2002;4:157-64
324. Ruocco MG, Karin M. IKK{beta} as a target for treatment of inflammation induced bone
loss. Ann Rheum Dis 2005;64 Suppl 4:iv81-5
325. Wen D, Nong Y, Morgan JG, et al. A selective small molecule IkappaB Kinase beta
inhibitor blocks nuclear factor kappaB-mediated inflammatory responses in human fibroblast-like synoviocytes, chondrocytes, and mast cells. J Pharmacol Exp Ther 2006;317:989-1001
326. Schopf L, Savinainen A, Anderson K, et al. IKKbeta inhibition protects against bone and
cartilage destruction in a rat model of rheumatoid arthritis. Arthritis Rheum 2006;54:3163-73
327. van den Berg WB, Joosten LA, Kollias G, et al. Role of tumour necrosis factor alpha in
experimental arthritis: separate activity of interleukin 1beta in chronicity and cartilage destruction. Ann Rheum Dis 1999;58 Suppl 1:I40-8
328. Kirkham B, Portek I, Lee CS, et al. Intraarticular variability of synovial membrane
histology, immunohistology, and cytokine mRNA expression in patients with rheumatoid arthritis. J Rheumatol 1999;26:777-84
329. van den Berg WB, Joosten LA, van de Loo FA. TNF alpha and IL-1 beta are separate
targets in chronic arthritis. Clin Exp Rheumatol 1999;17:S105-14
330. Kuiper S, Joosten LA, Bendele AM, et al. Different roles of tumour necrosis factor alpha
and interleukin 1 in murine streptococcal cell wall arthritis. Cytokine 1998;10:690-702
331. Okusawa S, Gelfand JA, Ikejima T, et al. Interleukin 1 induces a shock-like state in
rabbits. Synergism with tumor necrosis factor and the effect of cyclooxygenase inhibition. J Clin Invest 1988;81:1162-72
332. Movat HZ, Burrowes CE, Cybulsky MI, et al. Acute inflammation and a Shwartzman-like
reaction induced by interleukin-1 and tumor necrosis factor. Synergistic action of the cytokines in the induction of inflammation and microvascular injury. Am J Pathol 1987;129:463-76
333. Feige U, Hu YL, Gasser J, et al. Anti-interleukin-1 and anti-tumor necrosis factor-alpha
synergistically inhibit adjuvant arthritis in Lewis rats. Cell Mol Life Sci 2000;57:1457-70
334. Ikejima T, Okusawa S, Ghezzi P, et al. Interleukin-1 induces tumor necrosis factor
(TNF) in human peripheral blood mononuclear cells in vitro and a circulating TNF-like activity in rabbits. J Infect Dis 1990;162:215-23
335. Genovese MC, Cohen S, Moreland L, et al. Combination therapy with etanercept and
anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum 2004;50:1412-9
336. Li H, Zhang YY, Tan HW, et al. Therapeutic effect of tripterine on adjuvant arthritis in
rats. J Ethnopharmacol 2008;118:479-84
337. Li H, Jia YF, Pan Y, et al. Effect of tripterine on collagen-induced arthritis in rats.
Zhongguo Yao Li Xue Bao 1997;18:270-3
338. Tao X, Younger J, Fan FZ, et al. Benefit of an extract of Tripterygium Wilfordii Hook F in
patients with rheumatoid arthritis: a double-blind, placebo-controlled study. Arthritis Rheum 2002;46:1735-43

References
- 89 -
339. Idris AI, Libouban H, Nyangoga H, et al. Pharmacologic inhibitors of IkappaB kinase
suppress growth and migration of mammary carcinosarcoma cells in vitro and prevent osteolytic bone metastasis in vivo. Mol Cancer Ther 2009;8:2339-47
340. Hwang SY, Kim HY. Expression of IL-17 homologs and their receptors in the synovial
cells of rheumatoid arthritis patients. Mol Cells 2005;19:180-4
341. Ziolkowska M, Koc A, Luszczykiewicz G, et al. High levels of IL-17 in rheumatoid
arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol 2000;164:2832-8
342. Shahrara S, Huang Q, Mandelin AM, 2nd, et al. TH-17 cells in rheumatoid arthritis.
Arthritis Res Ther 2008;10:R93
343. Gaffen SL. Biology of recently discovered cytokines: interleukin-17--a unique
inflammatory cytokine with roles in bone biology and arthritis. Arthritis Res Ther 2004;6:240-7
344. Lalor SJ, Dungan LS, Sutton CE, et al. Caspase-1-processed cytokines IL-1beta and IL-
18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol 2011;186:5738-48
345. Lasiglie D, Traggiai E, Federici S, et al. Role of IL-1 beta in the development of human
T(H)17 cells: lesson from NLPR3 mutated patients. PLoS One 2011;6:e20014
346. Huh JR, Leung MW, Huang P, et al. Digoxin and its derivatives suppress TH17 cell
differentiation by antagonizing RORgammat activity. Nature 2011;472:486-90
347. Mittal MK, Chockalingam P, Chockalingam A. Contemporary indications and therapeutic
implications for digoxin use. Am J Ther 2011;18:280-7
348. Cascao R, Rosario HS, Souto-Carneiro MM, et al. Neutrophils in rheumatoid arthritis:
More than simple final effectors. Autoimmun Rev 2010;9:531-5
349. Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the
immune and bone systems. Nat Rev Immunol 2007;7:292-304
350. Genovese MC, Van den Bosch F, Roberson SA, et al. LY2439821, a humanized anti-
interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum 2010;62:929-39
351. Genovese M, Durez P, Richards H, et al. 2010. Secukinumab (AIN457), a novel
monoclonal antibody targeting IL-17A demonstrates efficacy in active rheumatoid arthritis patients despite stable methotrexate treatment: results of a phase IIb study. Presented at ACR
352. Buch MH, Emery P. New therapies in the management of rheumatoid arthritis. Curr
Opin Rheumatol 2011;23:245-51
353. Chabaud M, Miossec P. The combination of tumor necrosis factor alpha blockade with
interleukin-1 and interleukin-17 blockade is more effective for controlling synovial inflammation and bone resorption in an ex vivo model. Arthritis Rheum 2001;44:1293-303
354. Furlan R, Martino G, Galbiati F, et al. Caspase-1 regulates the inflammatory process
leading to autoimmune demyelination. J Immunol 1999;163:2403-9
355. Siegmund B. Interleukin-1beta converting enzyme (caspase-1) in intestinal
inflammation. Biochem Pharmacol 2002;64:1-8
356. Loher F, Bauer C, Landauer N, et al. The interleukin-1 beta-converting enzyme inhibitor
pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1 T-cell activation. J Pharmacol Exp Ther 2004;308:583-90
357. Paszkowski AS, Rau B, Mayer JM, et al. Therapeutic application of caspase
1/interleukin-1beta-converting enzyme inhibitor decreases the death rate in severe acute experimental pancreatitis. Ann Surg 2002;235:68-76
358. Ravizza T, Lucas SM, Balosso S, et al. Inactivation of caspase-1 in rodent brain: a
novel anticonvulsive strategy. Epilepsia 2006;47:1160-8

- 90 -

- 91 -
A presente dissertação foi realizada na Unidade de Investigação em Reumatologia e
na Unidade de Biologia Celular do Sistema Imunitário do Instituto de Medicina
Molecular, Faculdade de Medicina da Universidade de Lisboa, Lisboa, Portugal.
O trabalho aqui apresentado foi realizado no âmbito do projecto “The role of
inflammasome in the initiation and perpetuation of rheumatoid arthritis: contribution for
new targeted therapies.”, financiado pela Sociedade Portuguesa de Reumatologia e
por uma unrestricted research grant da empresa Pfizer.
Bolsa de Doutoramento da Fundação para a Ciência e a Tecnologia (referência:
SFRH/BD/40513/2007).




















![[HQ MS] Cascão Jovem - 01 - Monstro do Mar](https://img.pdfslide.net/doc/110x75/55cf9bb2550346d033a70d4a/hq-ms-cascao-jovem-01-monstro-do-mar.jpg)








