Embed Size (px)
Citation preview

Analysis of the SINE S1 Pol III promoter from Brassica;impact of methylation and in¯uence of external sequences
Philippe Arnaud1², Yasushi Yukawa2², Laurence Lavie1, Thierry Pe lissier1, Masahiro Sugiura2 and Jean-Marc Deragon1,*
1CNRS UMR6547 and GDR2157, Biomove, Universite Blaise Pascal Clermont-Ferrand II, 63177 AubieÁre Cedex,
France, and2Center for Gene Research, Nagoya University, Nagoya 464±8602, Japan
Received 17 November 2000; revised 16 February 2001; accepted 23 February 2001.*For correspondence (fax +33 473407777; e-mail [email protected]).²The authors wish it to be known that, in their opinion, the ®rst two authors should be regarded as joint First Authors
Summary
Transcription is an important control point in the transposable element mobilization process. To better
understand the regulation of the plant SINE (Short Interspersed Elements) S1, its promoter sequence
was studied using an in vitro pol III transcription system derived from tobacco cells. We show that the
internal S1 promoter can be functional although upstream external sequences were found to enhance
this basal level of transcription. For one putative `master' locus (na7), three CAA triplets (in positions
±12, ±7 and ±2) and two overlapping TATA motifs (in positions ±54 to ±43) were important to stimulate
transcription. For this locus, two transcription initiation regions were characterized, one centered on
position + 1 (®rst nucleotide of the S1 element) and one centered on position ± 19 independently of the
internal motifs. The CAA triplets only in¯uence transcription in + 1 and work in association with the
internal motifs. We show that methylation can inhibit transcription at the na7 locus. We also observe
that S1 RNA is cleaved in a smaller Poly (A) minus product by a process analogous to the maturation of
mammalian SINEs.
Keywords: RNA polymerase III, RNA processing, retroposable element, Alu, DNA methylation.
Introduction
RNA polymerase III (RNA pol III) is responsible for the
synthesis of most small cytoplasmic RNAs, some small
nuclear RNAs, SINE retroposon RNAs and several viral
RNAs (reviewed in Paule and White, 2000; Schmid, 1998;
Willis, 1993). The genes transcribed by RNA pol III can be
separated into four types. Type-1 is restricted to 5S
ribosomal RNA genes and is characterized by a tripartite
intragenic promoter consisting of an A and C block
separated by an intermediate I element. Type-2 genes are
characterized by a bipartite intragenic promoter consisting
of an A and B block usually separated by 30±60 bp. Most
genes encoding tRNA, the adenovirus VA genes and SINE
retroposons are members of this group. Type-3 genes rely
solely on promoter elements upstream of the initiation
site. This group includes small RNA-like U6 and 7SK in all
higher eukaryotes, as well as U3 and 7SL in plants (Heard
et al., 1995). Finally, type-4 gene transcription is dependent
on intragenic and external elements. This group include
the Xenopus selenocysteine tRNA gene, the EBER genes of
Epstein-Barr virus, the human 7SL gene and the vault RNA
gene (Vilalta et al., 1994).
The type-2 genes require, in addition to RNA pol III, two
fractions named TFIIIB and TFIIIC to initiate transcription.
For mammalians, TFIIIC can be separated into two
components called TFIIIC1 and TFIIIC2. TFIIIC2 binds to
the B block and then serves to recruit TFIIIC1 that makes
contact with the A block region. The TFIIIC factors can then
recruit the TBP (TATA-binding protein) containing TFIIIB
fraction through protein±protein interactions. The TFIIIB-
TFIIIC-DNA complex serves to recruit RNA pol III. The
5¢-¯anking region can also participate in the initiation
process in cooperation with the internal element. A
pyrimidine-purine dinucleotide at or near the initiation
site and a TATA element in position ± 32 to ± 24 are
important for optimal type-2 gene expression (Choisne
et al., 1997; Ulmanov and Folk, 1995; Zecherle et al., 1996).
The presence of the TATA motif enables the TBP portion of
TFIIIB to interact directly with DNA so that TFIIIC is not
The Plant Journal (2001) 26(3), 295±305
ã 2001 Blackwell Science Ltd 295

solely responsible for its recruitment. Type-3 and type-4
genes also possess TATA motifs in their 5¢-¯anking region.
For type-4 genes, the TATA motif compensates for less
than optimal intragenic elements. For example, in the case
of the type-4 vault gene, TFIIIC can not interact with the
internal promoter unless it is recruited by a direct inter-
action with TFIIIB bound to the external TATA motifs
(Vilalta et al., 1994). The TATA motif in type-3 genes works
in association with other upstream sequences to recruit
the RNA pol III, independently of intragenic motif.
Mammalians type-3 promoters utilize TFIIIC1 but not
TFIIIC2 (Oettel et al., 1997) and the TFIIIB fraction employed
by these promoters is distinct from the one used by the
other types of genes (Teichmann et al., 1997). Also, the
TFIIIB that assembles on a TATA containing promoter is
different from the ones that is implicated in the initiation of
TATA-less promoter (Park et al., 1997). The organization of
the factors that nucleate the RNA pol III can therefore vary
among type-2, type-3 and type-4 genes.
SINEs are an abundant class of transposable elements
found in a wide variety of eukaryotes (Deininger, 1989;
Okada and Ohshima, 1995). The presence of an internal pol
III promoter in mammalian SINEs was initially suggested
as being directly responsible for their high copy number
since new elements would keep their capacity to be
transcribed and to engage in new retroposition cycles
(Roger, 1985). However, evolutionary and molecular
studies have shown that only a few `master' loci are active
in retroposition and that, in most cases, the SINE type-2
internal promoter is silent (Deininger and Batzer, 1995).
Several mechanisms appear to be involved in this silenc-
ing, including DNA methylation, the formation of a
repressive chromatin structure and the binding of positive
and negative trans-acting factors (Schmid, 1998). SINE loci
that escape this negative regulation are usually associated
with external enhancers (Chesnokov and Schmid, 1996;
Deininger et al., 1996). A productive combination between
internal signals (provided by the SINE element) and
external signals (provided by the integration site) can
therefore result in ef®cient transcription for a limited
number of SINE loci. These transcriptionally active loci
are candidates for `master sequences' responsible for the
ampli®cation of SINE subfamilies (Deininger et al., 1996).
SINE retroposition is also controlled at the post-tran-
scriptional level. SINE transcriptional termination usually
occurs at the ®rst run of Ts (at least four) ¯anking the
insertion site. On average the primary SINE transcript
contains 200 nucleotides of unique sequence from the
insertion site. The nature of this 3¢ sequence will in¯uence
the lifetime and processing of SINE RNA (Maraia et al.,
1992). Mammalian SINE RNAs (Alu, B1, ID) are post-
transcriptionally processed in smaller, Poly (A) minus,
products called sc (small cytoplasmic) SINE RNAs
(Deininger et al., 1996; Maraia et al., 1993; Maraia, 1991).
In vivo, an uncharacterized 3¢-end processing activity
removes the terminal Poly (A) region and, for Alu, also
cleaves the A-rich regions between the two monomers.
Since the terminal Poly (A) region is likely to be important
for reverse transcription and integration (Boeke, 1997;
Deininger et al., 1996), this processing should limit SINE
retroposition. For Alu and B1, this 3¢-end processing was
proposed to be regulated in vitro by the RNA terminus-
binding protein La (Goodier and Maraia, 1998). In addition
to this post-transcriptional processing, the appropriate
folding of SINE RNA in conserved secondary structure
domains derived from ancestral functional RNAs (7SL or
tRNA) is probably also important for ef®cient retroposition
(Sinnett et al., 1992).
The S1 element is a small (180 bp) plant SINE that was
®rst described and studied in Brassica napus and is widely
distributed among Cruciferae (Deragon et al., 1994; Gilbert
et al., 1997; Lenoir et al., 1997; Tatout et al., 1999). S1
present all structural characteristic features found in
SINEs, including a primary and secondary sequence
homology to tRNA, a 3¢ Poly (A) region and internal
conserved pol III motifs (an A and B block) (Deragon et al.,
1994). Surprisingly, S1 seems to share many characteris-
tics with mammalian SINEs, like a similar subfamily
distribution (Deragon et al., 1994), a similar target site
selection (Tatout et al., 1998), a preferential colocalization
with MAR elements (Tikonov et al. 2001), and the capacity
to act as nucleation center for de novo methylation
(Arnaud et al., 2000). Also, S1 elements are highly
methylated in vivo and their transcription is severely
repressed in B. napus (Deragon et al., 1996; Goubely
et al., 1999). The objective of this work was to test the
functionality of the internal S1 promoter and the putative
impact of external sequences. We were also interested to
determine if methylation could repress S1 transcription.
Results
Two different S1 loci were selected for the in vitro study.
The ®rst locus, na7, contains an S1 element that is identical
to the consensus sequence of the young `Ea' subfamily
(Deragon et al., 1996; Lenoir et al., 1997). This element was
shown by a sequence-speci®c primer-extension to be
transcribed in vivo (P. Arnaud, C. Goubely and
J.M. Deragon, unpublished). The na7 sequence is also
identical to the sequence of the major S1 transcript
described in a previous study (Ea1 in Deragon et al.,
1996). It is therefore likely that the S1 element at the na7
locus is a major contributor of S1 RNA in vivo. The second
locus, na2, contains an S1 element that is identical to the
consensus sequence of the youngest (and Brassica napus-
speci®c) `A' subfamily (Deragon et al., 1996; Lenoir et al.,
1997). However, a transcript from the `A' subfamily was
296 Philippe Arnaud et al.
ã Blackwell Science Ltd, The Plant Journal, (2001), 26, 295±305
not detected in a previous study and it is not known
whether this element is transcribed in vivo.
S1 transcription is highly in¯uenced by upstream
external sequences.
Several lines of evidence indicate that there may be
differences in the initiation of pol III transcription between
animals compared with plants and fungi (Choisne et al.,
1997; Connelly et al., 1994). Although most in vitro studies
on plant pol III promoters have been carried out in extracts
from animal or S. cerevisiae nuclei, we decided to use a
plant in vitro pol III transcription system recently
developed from BY-2 tobacco cultured cells (Fan and
Sugiura, 1996). For na7, three constructions were tested
(see Figure 1a). Na7±1 contains the S1 element without its
Poly (A) tail, na7±2 the S1 element with the Poly (A) and
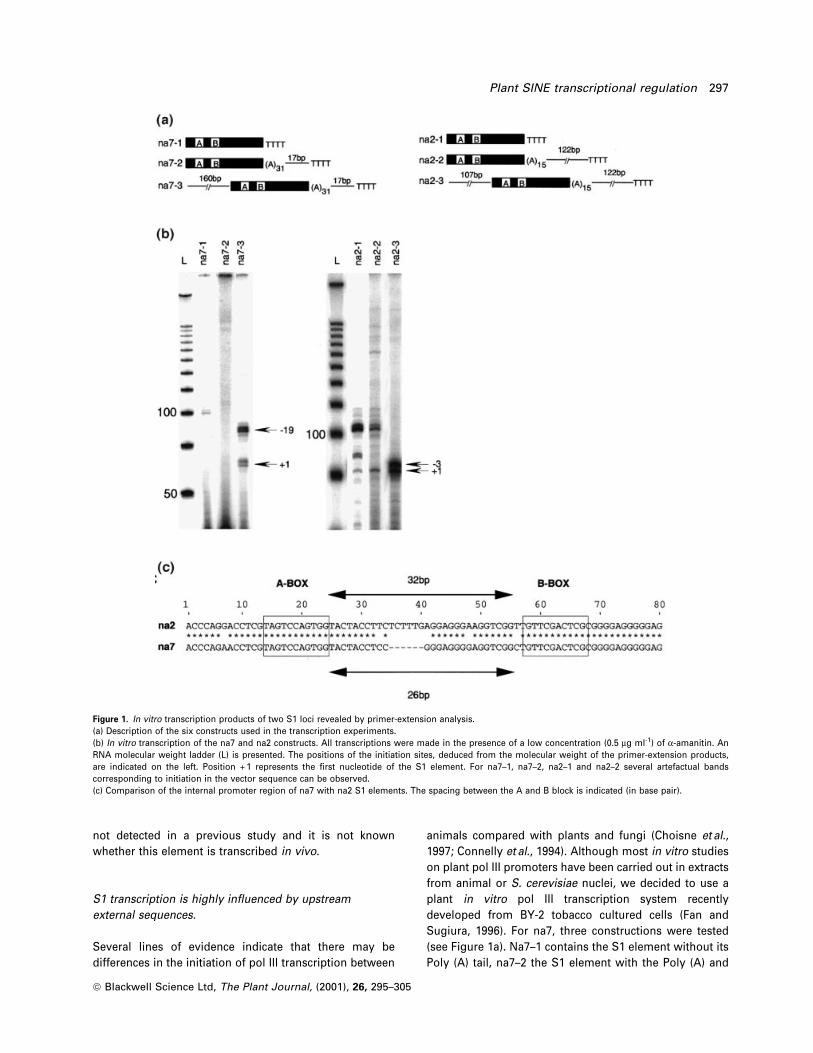
Figure 1. In vitro transcription products of two S1 loci revealed by primer-extension analysis.(a) Description of the six constructs used in the transcription experiments.(b) In vitro transcription of the na7 and na2 constructs. All transcriptions were made in the presence of a low concentration (0.5 mg ml-1) of a-amanitin. AnRNA molecular weight ladder (L) is presented. The positions of the initiation sites, deduced from the molecular weight of the primer-extension products,are indicated on the left. Position + 1 represents the ®rst nucleotide of the S1 element. For na7±1, na7±2, na2±1 and na2±2 several artefactual bandscorresponding to initiation in the vector sequence can be observed.(c) Comparison of the internal promoter region of na7 with na2 S1 elements. The spacing between the A and B block is indicated (in base pair).
Plant SINE transcriptional regulation 297
ã Blackwell Science Ltd, The Plant Journal, (2001), 26, 295±305

17 bp of 3¢ ¯anking genomic sequences (up to a TTTT
terminator) and na7±3 is composed of na7±2 plus 160 bp of
5¢ ¯anking genomic sequences. These three constructs
were tested in the in vitro assay and transcription products
were analyzed by speci®c primer-extensions (see
Experimental procedures section). Using, the na7±1 and
na7±2 constructs, no speci®c transcription product was
detected (Figure 1b). However, using the na7±3 construct,
we have detected transcriptional products resistant to low
concentration of a-amanitin (0.5 mg ml-1) which inhibit pol
II activity completely in vitro (Yukawa et al., 1997). The
precise sizing of these primer-extension products revealed
two transcriptional initiation regions: one centered in ± 19
and another centered in + 1 (+ 1 being the ®rst nucleotide
of the S1 element). The ± 19 region (referred to as the
upstream region) is composed of products initiated in ± 23,
± 19, ± 17 and ± 14, while the + 1 region is composed of
products initiated in ± 1, + 1, + 2 and + 3 (see Figure 2c).
We did a similar analysis with the na2 locus. Again, three
constructs were tested (na2±1, na2±2 and na2±3) that differ
in the presence or absence of ¯anking sequences (see
Figure 1a). For na2±1 and na2±2, a transcriptional product
resistant to a low concentration of a-amanitin and corres-
ponding to an initiation in position + 1 was detected
(Figure 1b). The higher molecular weight bands also
observed for na2±1 and na2±2 correspond to initiations
in the vector sequence. For na2±3, in the presence of
107 bp of 5¢ ¯anking sequences, the + 1 signal was
enhanced and a second signal, corresponding to an
initiation in ± 3 was detected (Figure 1b). These results
show that genomic region upstream of S1 elements can
in¯uence transcription by RNA pol III. For na7, the
upstream region seems to be required for transcription
while for na2 just the internal promoter allows transcrip-
tion in vitro with the 5¢-external sequence acting as an
enhancer. To understand this difference we have com-
pared the promoter region of na7 and na2 (Figure 1c).
While the A and B blocks are identical, the spacing
between them is not. The two motifs are separated by
26 bp for na7 and 32 bp for na2, the optimal spacing of the
A and B motifs in tRNA being between 30 and 60pb
(Sprague, 1995).
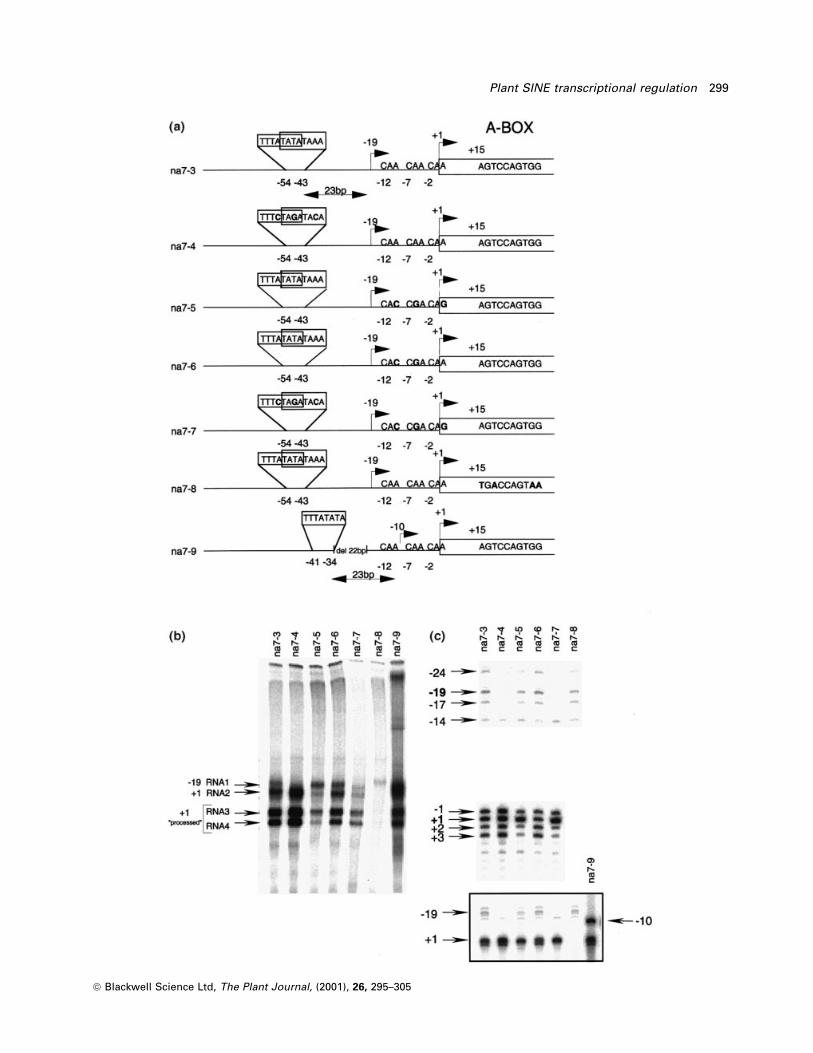
Analysis of the na7 5¢ genomic region
The genomic region upstream of S1 at the na7 locus
presents two putative cis-acting elements that could be
responsible for its transcriptional activating properties
(Figure 2a). First, three CAA trinucleotides are present in
positions ± 12, ± 7 and ± 2. A CAA triplet at or immediately
preceding the transcription start site (usually in positions
± 7 to ± 3), was found to be important for the transcription
ef®ciency of plant tRNA genes (Choisne et al., 1997).
Deletion of this triplet led to a signi®cant decrease in
transcription ef®ciency for the tRNALeu gene from
Phaseolus vulgaris (Choisne et al., 1997). Second, two
overlapping TATA motifs are present in positions ± 54 to
± 43 (Figure 2a). TATA elements are generally known to act
as enhancers for the pol II system but have also been
implicated as enhancers of pol III transcription (Willis,
1993). The importance of these two cis-acting elements
was tested by mutagenesis. The direct radioactive labeling
of RNAs during the in vitro transcription using normal or
mutagenized templates is presented in Figure 2(b). The
two largest RNAs (RNA1 approximately 230 bases and
RNA2 approximately 210 bases) correspond to an initiation
of transcription in the ± 19 or + 1 region and a termination
of transcription at the TTTT site of the construct. These
products were expected from the primer-extension results
(Figure 1b). The two other products (RNA3 and RNA4) are
smaller (approximately 175 and approximately 160 bases,
respectively) and result from the maturation of RNA2 (as
described later). The primer-extension products obtained
from the in vitro transcription of the different constructs
are presented in Figure 2(c).
Mutagenesis of the three CAA triplets (na7±5) resulted in
a reduction in transcription ef®ciency for the + 1 region
(evaluated by combining the intensity of RNA2, 3 and 4)
without affecting initiation in the upstream region (RNA1,
Figure 2b). Leaving one of the three CAA triplets overlap-
ping the initiation site (na7±6) resulted in a small reduction
in initiation ef®ciency in the + 1 region (RNA2, 3 4)
compared with wild type situation (na7±3, Figure 2b).
Mutation of the two TATA motifs (na7±4) completely
abolished initiation in the upstream region (except for
the ± 14 site, described later) (Figure 2b,c). This mutation
also enhanced transcription in the + 1 region (Figure 2b). A
construction where the CAA triplets and the TATA motifs
were mutated (na7±7) did not allow transcription initiation
in the upstream region (except for the ± 14 position) and
resulted in a reduction of the initiation ef®ciency in the + 1
region (Figure 2b,c). Finally, a mutation in the internal A
block (na7±8, Figure 2b,c) did not affect initiation in the
upstream region but completely abolished initiation in
Figure 2. Functional analysis of the 5¢ ¯anking region from the na7 locus.(a) Description of the constructs used in the transcription experiments. The position of the TATA and CAA motifs is indicated. Spacing (in base pair)between the TATA motifs and the upstream initiation site is also indicated.(b) Labeled in vitro transcription products after 120 min of transcription. RNA3 and RNA4 are processed products from RNA2 (see also Figure 3).(c) Primer-extension products obtained from the in vitro transcription of the different constructs. A different migration time, that includes na7±9, is shownin the box.
298 Philippe Arnaud et al.
ã Blackwell Science Ltd, The Plant Journal, (2001), 26, 295±305
Plant SINE transcriptional regulation 299
ã Blackwell Science Ltd, The Plant Journal, (2001), 26, 295±305

the + 1 region. These results suggest that the CAA motifs
work in association with the internal A and B blocks to
stimulate transcription in the + 1 region while the TATA
motifs work independently of the internal blocks to stimu-
late transcription in the upstream region.
In our case, the TATA motifs are not in consensus
positions (± 34 to ± 29) to stimulate initiation in the + 1
region. To determine if the TATA motifs could stimulate
transcription in the + 1 region if correctly positioned, a
22-bp deletion was made deleting one of the TATA
motives and placing the remaining one in position ± 34
(construction na7±9, Figure 2a). No stimulation of tran-
scription was observed in the + 1 region but initiation at
the upstream site was displaced to position ± 10 (Figure
2b,c). In the na7±9 construction, the ± 10 site is located
23 bp downstream of the TATA motif (Figure 2a).
Similarly, in the na7±3 construction, the upstream site is
centered on position ± 19, 23 bp downstream of the TATA
motifs. These results agree with the mutation experiments
and suggest that the TATA motifs of na7±3 and the
remaining TATA motif of na7±9 are directing the transcrip-
tional initiation in the upstream region. Since all transcrip-
tion initiation events are resistant to low concentrations of
a-amanitin, the TATA motifs are controlling the initiation
site of RNA polymerase III, independently of the internal
motifs. Initiation in position ± 14 is not dependent on the
presence of the TATA, CAA or internal A block and is of
unknown origin.
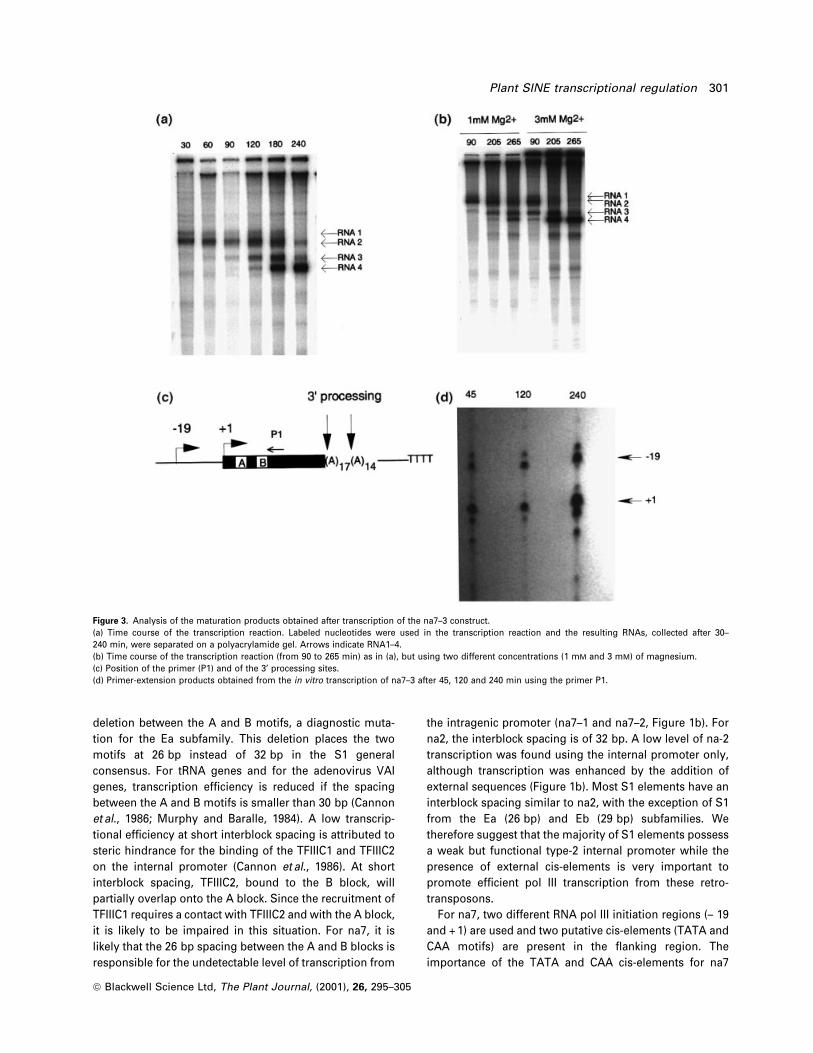
3¢-end maturation of S1 RNAs.
The RNA3 and RNA4 products observed after direct
labeling of RNAs during the in vitro transcription (Figure
2b), could result from a different termination of transcrip-
tion and/or from a processing event. To distinguish
between these two possibilities, a time course of the
transcription reaction was done (Figure 3a). After 30 min of
transcription, only the two largest RNA products are
visible. After 60 min, RNA3 is also visible and accumulates
up to 180 min before decreasing in intensity at 240 min.
RNA4 is visible at 90 min and accumulates to a maximum
at 240 min. This smallest RNA is the major product after
240 min of transcription. The production of RNA3 and 4 is
stimulated by an increase in the concentration of magne-
sium (Figure 3b), an essential factor for RNA processing
(Yukawa et al., 1997). These results suggest that RNA3 and
4 result from RNA processing and not from the use of
different transcription termination sites. To determine if
this processing concerns the 5¢ region, we analyzed by
primer-extension the RNAs produced after 45 min,
120 min and 240 min using primer P1 (Figure 3c).
Additional (smaller) primer-extension products expected
if the RNA is 5¢ end processed, are not detected at 120 and
240 min compared with 45 min (Figure 3d). Also, the
relative intensity between the products was unchanged
between 45 and 240 min (Figure 3d). These results suggest
that the processing did not implicate the 5¢ end of the S1
RNA. Mutation analysis revealed that the amount of the
two maturation products (RNA3 and 4) is correlated with
the production of RNA 2 (initiated in the + 1 region) and
not with the production of RNA1 (initiated the ± 19 region)
(Figure 2b compare na7±4 and na7±8). The sizes of RNA3
and 4 are compatible with a 3¢-end processing of RNA 2
that would cleave the Poly (A) region of S1 to generate
Poly (A) minus S1 RNA (Figure 3a,c). RNA3 would be an
intermediate, with a ®rst cut in the Poly (A) region (Figure
3c), and RNA4 would be the ®nal Poly (A) minus product.
This is compatible with the observation that RNA3 appears
sooner (60 min) compared with RNA 4 (90 min) in the time
course experiment and that the decrease in the amount of
RNA3 is correlated with an increase in the amount of
RNA4. RNA1 is also degraded in the time course experi-
ment (Figures 3a, 240 min). This degradation does not
generate detectable processing products (Figure 2b, na7±8
and Figure 3a) and since the 5¢ primer-extension (± 19)
product is not reduced at 240 min (Figure 3d), it must be
limited to the 3¢-end of the RNA.
Impact of methylation on S1 transcription.
The impact of DNA methylation on the in vitro transcrip-
tion of na7±3 was tested. The na7±3 construct was
methylated using the CpG-speci®c SssI methylase. Three
different treatments were done and the level of methyl-
ation was estimated by bisul®te (genomic sequencing)
analysis (see Experimental procedures section). Thirteen
to 16 clones were sequenced for each treatment (T1 to T3).
Although the protocol for each treatment was similar, the
level of methylation was found to vary greatly. For the T1
treatment, only three clones out of 16 were found
completely methylated and the average CpG methylation
was 52%. A second treatment (T2) was more ef®cient with
®ve clones out of 13 completely methylated and an
average CpG methylation of 70%. The third treatment
(T3) was the most ef®cient with 11 out of 13 clones
completely methylated and an average CpG methylation of
95%. We used the product of these three different
treatments in the in vitro assay. We observed an inhibition
of transcription that is proportional to the methylation
level of the templates with a complete inhibition using the
highly methylated template (not shown).
Discussion
We show here that the transcription of a putative S1
`master' locus from the Ea subfamily, the na7 locus,
depends largely on upstream ¯anking sequences. The
reason for this dependence is probably linked to a 6-bp
300 Philippe Arnaud et al.
ã Blackwell Science Ltd, The Plant Journal, (2001), 26, 295±305
deletion between the A and B motifs, a diagnostic muta-
tion for the Ea subfamily. This deletion places the two
motifs at 26 bp instead of 32 bp in the S1 general
consensus. For tRNA genes and for the adenovirus VAI
genes, transcription ef®ciency is reduced if the spacing
between the A and B motifs is smaller than 30 bp (Cannon
et al., 1986; Murphy and Baralle, 1984). A low transcrip-
tional ef®ciency at short interblock spacing is attributed to
steric hindrance for the binding of the TFIIIC1 and TFIIIC2
on the internal promoter (Cannon et al., 1986). At short
interblock spacing, TFIIIC2, bound to the B block, will
partially overlap onto the A block. Since the recruitment of
TFIIIC1 requires a contact with TFIIIC2 and with the A block,
it is likely to be impaired in this situation. For na7, it is
likely that the 26 bp spacing between the A and B blocks is
responsible for the undetectable level of transcription from
the intragenic promoter (na7±1 and na7±2, Figure 1b). For
na2, the interblock spacing is of 32 bp. A low level of na-2
transcription was found using the internal promoter only,
although transcription was enhanced by the addition of
external sequences (Figure 1b). Most S1 elements have an
interblock spacing similar to na2, with the exception of S1
from the Ea (26 bp) and Eb (29 bp) subfamilies. We
therefore suggest that the majority of S1 elements possess
a weak but functional type-2 internal promoter while the
presence of external cis-elements is very important to
promote ef®cient pol III transcription from these retro-
transposons.
For na7, two different RNA pol III initiation regions (± 19
and + 1) are used and two putative cis-elements (TATA and
CAA motifs) are present in the ¯anking region. The
importance of the TATA and CAA cis-elements for na7
Figure 3. Analysis of the maturation products obtained after transcription of the na7±3 construct.(a) Time course of the transcription reaction. Labeled nucleotides were used in the transcription reaction and the resulting RNAs, collected after 30±240 min, were separated on a polyacrylamide gel. Arrows indicate RNA1±4.(b) Time course of the transcription reaction (from 90 to 265 min) as in (a), but using two different concentrations (1 mM and 3 mM) of magnesium.(c) Position of the primer (P1) and of the 3¢ processing sites.(d) Primer-extension products obtained from the in vitro transcription of na7±3 after 45, 120 and 240 min using the primer P1.
Plant SINE transcriptional regulation 301
ã Blackwell Science Ltd, The Plant Journal, (2001), 26, 295±305

transcription was tested. First, we observed that mutations
in the two TATA motifs abolish initiation in the upstream
region (except for position ± 14, Figure 2, na7±4) and led to
an augmentation of the initiation in the + 1 region (Figure
2, na7±4). The deletion of one TATA motif and the
displacement of the remaining one resulted in an identical
displacement of the upstream (± 19) site without enhanc-
ing transcription in the + 1 region (Figure 2, na7±9). Also,
the mutation of the internal A block did not abolish the
upstream site (Figure 2, na7±8). These results suggest that
initiation of transcription at the upstream site requires at
least one of the two overlapping TATA motifs and is
completely independent of the intragenic motifs, a situ-
ation usually encountered for type 3 promoter. In the
upstream region, position ± 14 is behaving differently from
the other sites. Initiation in position ± 14 is independent of
the TATA, CAA and A motif and may be directed by an
uncharacterized upstream cis-element or, more likely,
represent an artifact of in vitro transcription.
Mutation of the three CAA motifs reduces the initiation
ef®ciency in the + 1 region without affecting the upstream
region (Figure 2b, na7±5, na7±7). Leaving only one of the
three CAA (the one overlapping the initiation site) resulted
in a small reduction of initiation ef®ciency in the + 1 region
(RNA2, 3 4) compared with wild type situation (na7±3 and
na7±6, Figure 2b). The CAA motifs work in association with
the internal promoter since mutations in the A block
completely abolish initiation in the + 1 region (Figure 2b,c,
na7±8). These results suggest that transcription from
the + 1 region is enhanced by the presence of at least
one CAA motif at the initiation site and is dependent on the
internal promoter, a situation characteristic of type 2
promoter. To our knowledge, the na7 locus represents
the ®rst example of a mixed type-2/type-3 RNA pol III
promoter. The transcription ef®ciency in the + 1 region is
higher in construction na7±5 (CAA mutated in CAG at the
initiation site) than for construction na7±2 or na7±1 (CAA
changed for TTA after cloning in pGEM-T) (Figures 1b and
2b). This can be explained if the CAG triplet at the initiation
site conserved a transcription stimulating activity com-
pared with the TTA triplet.
Based on these results, we suggest that the composition
and organization of the factors that nucleate the RNA pol III
are different for the ± 19 and + 1 region. For the + 1 region,
The ®rst round of transcription would be slowly initiated
by the weak (type 2) S1 internal promoter. The CAA motifs
would then participate in the reinitiation as this was
recently shown for tRNA in plants (Yukawa et al., 2000).
In the presence of at least one CAA motif, multiple-round
of transcription would take place leading to the accumu-
lation of S1 RNA initiated in the + 1 region. For the
initiation in the upstream region, the complex ®xed on
the TATA motif could be different and organized as
suggested for type-3 promoter (Yoon et al., 1995). In that
case the complex would consist in a TBP fraction loosely
associated to certain TFIIIB TAFs and bound by a TFIIIC1-
like fraction. This complex could recruit RNA pol III,
possibly in association with uncharacterized upstream
cis-elements, and initiate transcription independently of
the intragenic motifs. In vitro initiations in the ± 19 and + 1
region are likely to be in competition and the augment-
ation of transcription in the + 1 region following mutation
of the TATA motif (Figure 2b, na7±3, na7±4) can be
explained by the abolition of this competition in favor of
the + 1 region.
We have shown previously that S1 elements, including
the na7 locus, are highly methylated in vivo (Goublely
et al., 1999; Arnaud et al., 2000). The impact of DNA
methylation on pol III transcription is not clear and relies
on a small number of reports, all using animal systems
(Jutterman et al., 1991; Minarovits et al., 1992) and con-
cludes in a more or less severe inhibition following
methylation. While the general tendency for methylation
is to inhibit pol III transcription, exceptions exist (Besser
et al., 1990). Here we present for the ®rst time the impact of
methylation on a plant pol III promoter. In our plant in vitro
assay, we detect a transcriptional inhibition that is directly
correlated to the level of template methylation. The
inhibition is complete at high methylation levels. From
these observations, we suggest that the very low level of
general S1 transcription in vivo (Deragon et al., 1996)
results from S1 internal promoter methylation. An import-
ant question then is how, despite its high level of
methylation, the na7 locus can be ef®ciently transcribed
in vivo? We suggest that the presence of external motifs at
this locus can by-pass the down-regulation imposed on S1
internal promoters. In that scenario, initiation in the ±19
region would be necessary to maintain the internal
promoter accessible for transcription factors initiating in
the + 1 region. The combination of TATA motifs (promot-
ing initiation in the ±19 region) and CAA motifs (promoting
initiation in the + 1 region) could give a particular tran-
scriptional advantage to this locus over other S1 loci. This
could explain why na7 is most likely responsible for the
major S1 transcript and may well represent the `master'
locus of the Ea subfamily. However, at this point, we can
not exclude the possibility that the ±19 site is not used
in vivo.
When the na7±3 transcription products were directly
labeled in our assay, four RNA were detected instead of the
two expected ones (initiation in ± 19 and + 1 regions and
termination at the TTTT site). The time course analysis
shows that the two (unexpected) smaller products are
gradually generated in the assay and that their appearance
can be speeded up by an increase (from 1 mM to 3 mM) in
magnesium (Figure 3b). Since RNA processing in the
extract used was shown to be minimal at 1 mM and
optimal over 3 mM of magnesium (Yukawa et al., 1997), we
302 Philippe Arnaud et al.
ã Blackwell Science Ltd, The Plant Journal, (2001), 26, 295±305

suggest that the smaller products resulted from the
processing of the largest ones. Size determination of
RNA products, primer-extension analysis (Figure 3) and
transcription from the mutant templates (Figure 2b)
suggest that the two smallest RNAs (RNA3 and 4) are
3¢-processed Poly (A) minus forms of the RNA initiated in
the + 1 region (RNA2). RNA3 is probably an intermediate
resulting of a ®rst cleavage in the Poly (A) tail while RNA4
is the ®nal Poly (A) minus product. This S1 Poly (A) minus
RNA is similar to the small cytoplasmic mammalian SINE
RNA obtained after the in vitro incubation of a full length
SINE RNA with nuclear extract or after SINEs were
expressed in vivo in Xenopus oocytes (Deininger et al.,
1996; Maraia et al., 1993; Maraia, 1991). Since retroposition
appears to be limited to unprocessed transcripts, the
formation of S1 Poly (A) minus RNA should lower the
retropositional potential of S1 transcripts. By analogy to
mammalian system, the 3¢-end processing of S1 RNA
would constitute, after transcription, a second barrier to
retroposition.
Experimental procedures
Constructions
Na2±3 (GeneBank AF101144) and na7±3 (GeneBank AF101149)loci were ampli®ed by PCR using Brassica napus genomic DNAand cloned in pGem-T easy vector (Promega, Charbonniere,France). The PCR primers used were na2±3.1: TCAACTCGGCCGC-TAAC, na2±3.2: GAATATTTACGCACTATCACG, na7±3.1: GAGG-TAGACAACGATTAGTGG, na7±3.2: CCAAAACAAATGTTTTGTCT-TGA. Using different internal PCR primer combinations and na2±3or na7±3 as template we were able to generate (and clone) PCRproducts corresponding to na2±1, na2±2, na7±1 and na7±2 loci.The na7±4 to na7±9 constructs were generated using themegaprimer PCR method (Barik, 1998) and na7±3 as template.Brie¯y, mutagenesis by the megaprimer method requires threeprimers, one in 5¢ (na7±3.1), one in 3¢ (na7±3.2) of na7±3 and aninternal primer encoding the sequence changes needed. Theinternal and the 5¢-end (na7±3.1) primers were used in a ®rstround of PCR ampli®cation using the Vent DNA polymerase at60°C for 35 cycles under conditions recommended by themanufacturer (New England Biolabs, St. Quentin, Yvelines,France). This PCR product was gel-puri®ed and used as a`megaprimer' (10 mM) with the 3¢-end (na7±3.2) primer in a secondround of PCR ampli®cation using the HotStart Taq DNApolymerase at 60°C for 35 cycles as recommended by themanufacturer (Qiagen). The ®nal PCR product was gel-puri®edand cloned in the pGemT-easy vector. The deletion mutant na7±9was generated from na7±3 by the PCR inverse method (Yukawaet al., 2000). All PCR products were puri®ed by Qiaquick gelextraction Kit (Qiagen, Courtaboeuf, France). Cloning in pGem-Teasy vector was performed as previously described (Arnaud et al.,2000). Plasmid DNA constructs were prepared using the Quia®lterplasmid Kit (Qiagen).
Preparation of tobacco nuclear extract
Tobacco nuclear extracts were prepared by a modi®cation of theprocedure of Fan and Sugiura (1995) and Yukawa et al. (1997).
Tobacco cultured cells (BY-2 cell line) (Nagata et al., 1992) wereharvested at middle log phase (approximately 85 h after inocula-tion) with Miracloth (Calbiochem, USA) from a 1.5 litter culture.The cells are digested in 500 ml of enzyme solution [2% Cellulase`Onozuka' RS (Yakult Pharmachemical, Tokyo, Japan) and 0.2%Pectolyase Y-23 (Kikkoman, Tokyo, Japan) in the LS mediumcontaining 0.38 M mannitol and 3% sucrose, pH 5.5] at 30°C for50 min. Protoplasts were collected by centrifugation at 250 g for2 min at 2°C, washed twice with ice cold 0.38 M mannitol (pH 5.5).The pellet was suspended in 300 ml nuclear isolation buffer (NIB)[15 mM HEPES-KOH (pH 7.9), 18% (w/v) Ficoll 400, 4 mM MgSO4,1 mM NaF, 1 mM EGTA, 0.5 mM EDTA, 3 mM DTT, 0.5 mM PMSF,0.5 mM benzamidine hydrochloride, 1.5 mg/ml pepstatin A, 1 mgml±1 leupeptin] and for braking cell membrane, vacuum-®ltratedtwice through one layer of 20 mm nylon mesh (Schweiz.Seidengazefabrik AG Thal, Switzerland). The ®ltrate was centri-fuged at 2500 g for 12 min at 2°C and the nuclear pellet wassuspended with 250 ml NIB and centrifuged at 2500 g for 10 minat 2°C. Washed nuclei were suspended in 3 volumes of nuclearextraction buffer (NEB) [25 mM HEPES-KOH (pH 7.9), 20% (v/v)glycerol, 4 mM MgSO4, 0.4 mM EGTA, 1 mM NaF, 5 mM DTT, 3 mgml±1 pepstatin A, 2 mg ml±1 leupeptin]. Ammonium sulfate wasadded to 0.42 M, and then rotated at 2°C for 30 min. Nuclear lysatewas centrifuged at 200 000 g for 1 h and the supernatant wassubjected to precipitation of 60% saturated ammonium sulfate indialysis buffer (DB) [20 mM HEPES-KOH (pH 7.9), 20% (v/v)glycerol, 4 mM MgSO4, 0.2 mM EGTA, 0.1 mM EDTA, 2 mM DTT,0.5 mM PMSF, 0.5 mM benzamidine hydrochloride]. The precipi-tate was dissolved with 2 ml of DB and dialyzed twice with MWCO12 000 cellulose membrane (Wako Chemical USA, Richemond,VA, USA) at 4°C for 1.5 h each against 500 ml DB. The resultingnuclear extract was aliquoted and frozen in a deep freezer.
In vitro transcription and primer-extension.
In vitro transcription reactions from S1 elements using tobacconuclear extracts were done as previously described (Yukawa et al.,1997) with minor modi®cations. Brie¯y, reaction was performedin a 20-ml volume containing 30 mM HEPES-KOH (pH 7.9), 3 mM
MgSO4, 80 mM KOAc, 0.1 mM EGTA, 2 mM DTT, 10% glycerol,0.5 mM each of ATP, CTP, UTP, 25 mM GTP, 37 kBq [a-32P]GTP,0.2 pmol circular plasmids, 0.5 mg ml±1 a-amanitin and 15 mgtobacco nuclear extract. After incubation at 28°C for the indicatedtime, the 32P-labeled RNA was extracted by phenolation or TotalRNA SafeKit (BIO101, La Jolla, CA, USA). The extracted RNA wasseparated by 5±8% polyacrylamide gel containing 7 M urea andTBE. Radioactivity was detected by Bio-Imaging Analyzer BAS-2000 II (Fuji Photo Film, Tokyo, Japan).
For primer-extension assay, in vitro transcription was per-formed in the same reaction mix except for 1 mM each of 4NTPsas substrates. The primers used were P1: CCGCGAGTCGA-ACAGCC, corresponding to position 48±64 of the S1 element atthe na7 locus and pBN2: CCCCCTCCCCGCCAGTCGAACAACCG,corresponding to position 53±78 of the S1 element at the na2locus. Total nucleic acid was extracted twice with phenol/chloro-form and once with chloroform. After ethanol precipitation with100 fmol [5¢ 32P]primers, the resulting pellet was dissolved in 10 mlof reverse transcription buffer [50 mM Tris±HCl (pH 8.3), 75 mM
KCl, 3 mM MgCl2 and 10 mM DTT], and subjected to denaturationat 70°C for 5 min and annealing at 60°C for 10 min. After adding10 ml of RTase cocktail [1 mM each of 4dNTPs, 100 mM actinomy-cin D, 10 U RNase inhibitor, 100 U ReverTra Ace (Toyobo, Japan)in 1x RTase buffer], the mixture was incubated at 50°C for 1 h.Reaction was stopped by phenol/chloroform treatment. After
Plant SINE transcriptional regulation 303
ã Blackwell Science Ltd, The Plant Journal, (2001), 26, 295±305

ethanol precipitation, the pellet was dissolved in loading buffer[95% formamide, 5 mM EDTA, 0.1% bromophenol blue, 0.1%xylene cyanol], and then separated with 8% polyacrylamide gelcontaining 7 M urea and TBE. The sequencing ladder wasprepared from the same combinations of templates and primersusing Thermosequenase Cycle Sequencing Kit (AmershamPharmacia, Saclay, France). Detection was the same as above.
Methylation and bisul®te treatments.
The na7±3 construct was methylated by the SssI CpG methylase(New England Biolabs, St. Quentin, Yvelines, France) for 4 h at37°C using 2.5 units m±1g of DNA. The methylation ef®ciency wastested using the bisul®te DNA modi®cation method performed aspreviously described (Arnaud et al., 2000).
Acknowledgements
We thank Alain Lenoir for technical help and Christophe Tatoutand Charles White for helpful discussions. This work wassupported by the CNRS (as part of the Genome Project), by theARC foundation (1996±98) and by Universite Blaise Pascal. Y. Y. isa Research Fellow of the Japan Society for the Promotion ofScience.
References
Arnaud, P., Goubely, C., Pe lissier, T. and Deragon, J.M. (2000).SINE retroposons can be used in vivo as nucleation centers forde novo methylation. Mol. Cell. Biol. 20, 3434±3441.
Barik, S. (1998) Mutagenesis by megaprimer PCR. In GeneticEngineering with PCR (Horton, R.M. and Tait, R.C., eds),Wymondham, UK: Horizon Scienti®c Press, pp. 25±37.
Besser, D., Gotz, F., Schulze-Foster, K., Wagner, H., Kroger, H. andSimon, D. (1990) DNA methylation inhibits transcription byRNA polymerase III of tRNA gene, but not of a 5S rRNA gene.FEBS Lett. 269, 358±362.
Boeke, J.D. (1997) LINEs and Alus-the polyA connection. NatureGenet. 16, 6±7.
Cannon, R.E., Wu, G.J. and Railey, J.F. (1986) Function of andinteractions between the A and B blocks in adenovirus type2-speci®c VARNA1 gene. Proc. Natl Acad. Sci. USA, 83, 1285±1289.
Chesnokov, I. and Schmid, C.W. (1996) Flanking sequences of anAlu source stimulate transcription in vitro by interacting withsequence-speci®c transcription factors. J. Mol. Evol. 42, 30±36.
Choisne, N., Carneiro, V.T.C., Pelletier, G. and Small, I. (1997)Implication of 5¢-¯anking sequence elements in expression of aplant tRNAleu gene. Plant Mol. Biol. 36, 113±123.
Connelly, S., Mashallsay, C., Leader, D., Brown, J.W.S. andFilipowicz, W. (1994) Small nuclear RNA genes transcribed byeither RNA polymerase II or RNA polymerase III in monocotplants share three promoter elements and use a strategy toregulate gene expression different from that used by their dicotplant counterparts. Mol. Cell. Biol. 14, 5910±5919.
Deininger, P.L. (1989) SINEs: short interspersed repeated DNAelements in higher eukaryotes. In Mobile DNA (Berg, D.E. andHowe, M.M., eds), Washington, DC, USA: American Society forMicrobiology, pp. 619±636.
Deininger, P.L. and Batzer, M.A. (1995) SINE master genes andpopulation biology. in the Impact of Short InterspersedElements (Sines) on the Host Genome (Maraia, R.J., ed.),Austin, Texas, USA: R. G. Landes Company, Springer, pp. 43±60.
Deininger, P.L., Tiedge, H., Kim, J. and Brosius, J. (1996)Evolution, expression and possible function of a master genefor ampli®cation of an interspersed repeated DNA family inrodents. Prog. Nucl Acid Res. Mol. Biol. 52, 67±88.
Deragon, J.M., Landry, B.S., Pe lissier, T., Tutois, S. and Picard, G.(1994). Analysis of retroposition in plants based on a family ofSINEs from Brassica napus. J. Mol. Evol. 39, 78±386.
Deragon, J.M., Gilbert, N., Rouquet, L., Lenoir, A., Arnaud, P. andPicard, G. (1996) A transcriptional analysis of the S1Bn(Brassica napus) family of SINE retroposons. Plant Mol. Biol.32, 869±878.
Fan, H. and Sugiura, M. (1995) A plant basal in vitro systemsupporting accurate transcription of both RNA polymerase II-and III-dependent genes (Suppl.)of green leaf component (s)drives accurate transcription of a light-responsive rbcS gene.EMBO J. 14, 1024±1031.
Fan, H. and Sugiura, M. (1996) Basal and activated in vitrotranscription in plants by RNA polymerase II and III. Meth.Enzymol. 273, 268±277.
Gilbert, N., Arnaud, P., Lenoir, A., Warwick, S.I., Picard, G. andDeragon, J.M. (1997) Plant S1 SINEs as a model to studyretroposition. Genetica, 10, 155±160.
Goodier, J.L. and Maraia, R.J. (1998) Terminator-speci®c recyclingof a B1-Alu transcription complex by RNA polymerase III ismediated by the RNA terminus-binding protein La. J. Biol.Chem. 273, 26110±26116.
Goubely, C., Arnaud, P., Tatout, C., Heslop-Harrison, J.S. andDeragon, J.M. (1999) S1 SINE retroposons are methylated atsymmetrical and non-symmetrical positions in Brassica napus:Identi®cation of a preferred target site for asymmetricalmethylation. Plant. Mol. Biol. 39, 243±255.
Heard, D.J., Filipowicz, W., Marques, J.P., Palme, K. andGualberto, J.M. (1995) An upstream U-snRNA gene-likepromoter is required for transcription of the Arabidopsisthaliana 7SL RNA gene. Nucl Acids Res. 23, 1970±1976.
Juttermann, R., Hosokawa, K., Kochanek, S. and Doer¯er, W.(1991) Adenovirus type 2 VAI RNA transcription by polymeraseIII is blocked by sequence-speci®c methylation. J. Virol. 65,1735±1742.
Lenoir, A., Cournoyer, B., Warwick, S.I., Picard, G. and Deragon,J.M. (1997). Evolution of SINE S1 retrotransposons inCruciferae plants pecies. Mol. Biol. Evol. 14, 934±941.
Maraia, R.J. (1991) The subset of mouse B1 (Alu-equivalent)sequences expressed as small processed cytoplasmictranscripts. Nucl Acids Res. 19, 5695±5702.
Maraia, R.J., Chang, D.Y., Wolffe, A.P., Vorce, R.L. and Hsu, K.(1992) The RNA polymerase III terminator used by a B1-Aluelement can modulate 3¢ processing of the intermediate RNAproduct. Mol. Cell. Biol. 12, 1500±1506.
Maraia, R.J., Driscoll, C.T., Bilyeu, T., Hsu, K. and Darlington, G.J.(1993) Multiple dispersed loci produce small cytoplasmic AluRNA. Mol. Cell. Biol. 13, 4233±4241.
Minarovits, J., Hu, L.F., Marcsek, Z., Minarovits-Kormuta, S.,Klein, G. and Ernberg, I. (1992) RNA polymerase III-transcribedEBER 1 and 2 transcription units are expressed andhypomethylated in the major Epstein-Barr virus-carrying celltypes. J. Gen. Virol. 73, 1687±1692.
Murphy, M.H. and Baralle, F.E. (1984) Construction and functionalanalysis of a series of synthetic RNA polymerase III promoters.J. Biol. Chem. 259, 10208±10211.
Nagata, T., Nemoto, Y. and Hasegawa, S. (1992) Tobacco BY-2cell line as the `HeLa' cell in the cell biology of higher plants. Int.Rev. Cytol. 132, 1±30.
Oettel, S., Hartel, F., Kober, I., Iben, S. and Seifart, K.H. (1997)
304 Philippe Arnaud et al.
ã Blackwell Science Ltd, The Plant Journal, (2001), 26, 295±305

Human transcription factors IIIC2 IIIC1 and novel componentIIIC0 ful®l different aspects of DNA binding to various pol IIIgenes. Nucl Acids Res. 25, 2440±2447.
Okada, N. and Ohshima, K. (1995) Evolution of tRNA-derivedSINEs. In The Impact of Short Interspersed Elements (Sines) onthe Host Genome (Maraia, R.J., ed.), Austin, Texas, USA: R. G.Landes Compagny, Springer, pp. 61±79
Park, J.M., Lee, J.Y., Hat®eld, D.L. and Lee, B.J. (1997) Differentialmode of TBP utilization in transcription of the tRNA (ser) sec geneand TATA-less class III genes. Gene, 196, 99±103.
Paule, M.R. and White, R.J. (2000) Transcription by RNApolymerase I and III. Nucl Acids Res. 28, 1283±1298.
Rogers, J.H. (1985) The origin and evolution of retroposons. Int.Rev. Cytol. 93, 187±279.
Schmid, C.W. (1998) Does SINE evolution preclude Alu function?Nucl Acids Res. 26, 4541±4550.
Sinnett, D., Richer, C., Deragon, J.M. and Labuda, D. (1992) AluRNA transcripts in human embryonal carcinoma cells. Model ofpost-transcriptional selection of master sequences. J. Mol. Biol.226, 689±706.
Sprague, K.U. (1995) Transcription of eukaryotic tRNA genes. In:Trna: Structure, Biosynthesis and Function (Soll, D. andRajBhandary, U., eds) Washington, DC, USA: AmericanSociety for Microbiology, pp. 31±50.
Tatout, C., Lavie, L. and Deragon, J.M. (1998). Similar target siteselection occurs in integration of plant and mammalianretroposons. J. Mol. Evol. 47, 463±470.
Tatout, C., Warwick, S.I., Lenoir, A. and Deragon, J.M. (1999) SINEinsertions as clade markers for wild crucifer species. Mol. Biol.Evol. 16, 1614±1621.
Teichmann, M., Dieci, G., Huet, J., Ruth, J., Sentenac, A. andSeifart, K.H. (1997) Functional interchangeability of TFIIIB
components from yeast and human cells in vitro. EMBO J. 16,4708±4716.
Tikhonov, A.P., Tatout, C., Lavie, L., Bennetzen, J.L., Avramova, Z.and Deragon, J.M. (2001) Matrix-Attachment Regions (MARs)as target sites for, SINE integration in Brassica genomes.Chrom. Res. (in press)
Ulmanov, B. and Folk, W. (1995) Analysis of the role of 5¢ and 3¢¯anking sequence elements upon in vivo expression of theplant tRNATrp genes. Plant Cell, 7, 1723±1734.
Vilalta, A., Kickhoerfer, V.A., Rome, L.H. and Johnson, D.L. (1994)The rat vault RNA gene contains a unique RNA polymerase IIIpromoter composed of both external and internal elements thatfunction synergistically. J. Biol. Chem. 269, 29752±29759.
Willis, I.M. (1993) RNA polymerase III. Genes, factors andtranscriptional speci®city. Eur. J. Biochem. 212, 1±11.
Yoon, J.B., Murphy, S., Bai, L., Wang, Z. and Roeder, R.G. (1995)Proximal sequence element-binding transcription factor (PTF)is a multisubunit complex required for transcription of bothRNA polymerase II and RNA polymerase III-dependent smallnuclear RNA genes. Mol. Cell. Biol. 15, 2019±2026.
Yukawa, Y., Sugita, M. and Sugiura, M. (1997) Ef®cient in vitrotranscription of plant nuclear tRNAser genes in a nuclearextract from tobacco cultured cells. Plant J. 12, 965±970.
Yukawa, Y., Sugita, M., Choisne, N., Small, I. and Sugiura, M.(2000) The TATA motif, the CAA motif and the Poly (T)transcription termination motif are all important fortranscription reinitiation on plant tRNA genes. Plant J. 22,439±447.
Zecherle, G.N., Whelen, S. and Hall, B.D. (1996) Purine arerequired at the 5¢ ends of newly initiated RNAs for optimalRNA polymerase III gene expression. Mol. Cell. Biol. 16, 5801±5810.
GeneBank accession numbers AF101144and AF101149.
Plant SINE transcriptional regulation 305
ã Blackwell Science Ltd, The Plant Journal, (2001), 26, 295±305












![Global analysis of DNA methylation in hepatocellular ......exome [14] as well as the promoter methylome [15]. In the present study, we initially performed promoter-targeted LHC-BS](https://img.pdfslide.net/doc/110x75/614a349a12c9616cbc6943eb/global-analysis-of-dna-methylation-in-hepatocellular-exome-14-as-well.jpg)
















