Embed Size (px)
Citation preview

An Epigenetic Antimalarial Resistance Mechanism InvolvingParasite Genes Linked to Nutrient Uptake*
Received for publication, March 10, 2013, and in revised form, May 7, 2013 Published, JBC Papers in Press, May 28, 2013, DOI 10.1074/jbc.M113.468371
Paresh Sharma‡, Kurt Wollenberg§, Morgan Sellers‡, Kayvan Zainabadi‡, Kevin Galinsky¶, Eli Moss¶,Wang Nguitragool‡, Daniel Neafsey¶, and Sanjay A. Desai‡1
From the ‡Laboratory of Malaria and Vector Research and §Bioinformatics and Computational Biosciences Branch, Office of CyberInfrastructure and Computational Biology, NIAID, National Institutes of Health, Bethesda, Maryland 20852 and the ¶Broad Instituteof Harvard and MIT, Cambridge, Massachusetts 02142
Background:Malaria parasites acquire antimalarial resistance through incompletely understood mechanisms.Results:Resistance to blasticidin S results from reversible silencing of parasite clag genes through histonemodificationswithoutDNA level changes.Conclusion: Sophisticated epigenetic control of clag genes permits regulated control of nutrient and antimalarial transport atthe host membrane.Significance:This resistancemechanism allows rapid parasite adaptation to environmental pressures and isworrisome for drugdiscovery efforts.
Acquired antimalarial drug resistance produces treatmentfailures and has led to periods of global disease resurgence. InPlasmodium falciparum, resistance is known to arise throughgenome-level changes such asmutations and gene duplications.We now report an epigenetic resistance mechanism involvinggenes responsible for the plasmodial surface anion channel, anutrient channel that also transports ions and antimalarial com-pounds at the host erythrocytemembrane. Twoblasticidin S-re-sistant lines exhibited markedly reduced expression of claggenes linked to channel activity, but had no genome-levelchanges. Silencing aborted production of the channel proteinand was directly responsible for reduced uptake. Silencingaffected clag paralogs on two chromosomes and was mediatedby specific histone modifications, allowing a rapidly reversibledrug resistance phenotype advantageous to the parasite. Thesefindings implicate a novel epigenetic resistancemechanism thatinvolves reduced host cell uptake and is a worrisome liability forwater-soluble antimalarial drugs.
Lost efficacy of chloroquine, an inexpensive and safe drugthat once cured malaria with a single oral dose, contributedsignificantly to failedmalaria eradication programs in the 1960s(1). Resistance to subsequently deployed drugs has aggravatedmalaria control programs, led to use of combination therapiesthat increase both cost and side effects, and forced increasedinvestment in drug discovery and development. Reports ofdelayed Plasmodium falciparum clearance with artemisinins(2), the current mainstay of treatment, raise fears that recentgains in malaria control may also be lost. Understanding theresistance mechanisms of the parasite is critical to optimal
usage of existing drugs and the development of new, less sus-ceptible therapies.Antimalarial resistance is known to arise through DNA-level
changes in parasite genes that encode the drug target or effluxpumps. For example, specific mutations in the targets of atova-quone and sulfadoxine-pyrimethamine are known to reduceinhibition by these drugs (3, 4). pfcrt and pfmdr1, genes fortransporters that mediate efflux of chloroquine, mefloquine,and possibly other antimalarials (5–7), also incur specific causalmutations. In addition to mutations, copy number variationthrough genomic recombination events have also been impli-cated in drug resistance (8, 9).Reduced drug uptake by infected erythrocytes represents a
distinct and recently proposed mechanism of acquired resis-tance. Although not yet described with approved drugs, it hasbeen reported for blasticidin S and leupeptin (10–12). Theseagents inhibit parasite ribosomes and intracellular proteases,respectively. In both cases, resistant parasites exhibit changes inthe plasmodial surface anion channel (PSAC),2 an unusualbroad selectivity ion channel present only on infected erythro-cytes. Nevertheless, the precise mechanism of drug resistanceand its molecular basis remain unknown, partly due to possiblecontributions from putative host transporters.Wenow report a novel epigeneticmechanism responsible for
blasticidin S resistance. Our studies reveal histone modifica-tions that suppress transcription of parasite clag3 (cytoadher-ence linked antigen), genes recently linked to PSAC activity(13). Expression of clag2, an uncharacterized paralog, is alsocurtailed in resistant parasites, suggesting a role for this paralogin transport and revealing sophisticated regulation of this genefamily. The ability to acquire and revert from drug-resistant
* This work was supported, in whole or in part, by the National Institutes ofHealth Intramural Research Program, NIAID, and by a Global Health Pro-gram grant from the Bill and Melinda Gates Foundation.
1 To whom correspondence should be addressed: 12735 Twinbrook Parkway,Rockville, MD 20842. Tel.: 301-435-7552; Fax: 301-435-2201; E-mail:[email protected].
2 The abbreviations used are: PSAC, plasmodial surface anion channel; clagand CLAG, cytoadherence linked antigen genes and proteins, respectively;PhTMA�, phenyl-trimethyl ammonium; hDHFR, human dihydrofolatereductase; ISPA, isolate specific PSAC antagonist; ANOVA, analysis of vari-ance; qPCR, quantitative PCR; msp2, merozoite surface protein-2; BisTris,2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 288, NO. 27, pp. 19429 –19440, July 5, 2013Published in the U.S.A.
JULY 5, 2013 • VOLUME 288 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 19429
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from

phenotypes via a reversible epigenetic mechanism confers sur-vival advantage under diverse challenges and is worrisome formalaria control programs.
EXPERIMENTAL PROCEDURES
Parasite Culture and in Vitro Selection of Drug-resistantParasites—P. falciparum laboratory lines were cultivated bystandard methods and maintained under 5% O2, 5% CO2, 90%N2 at 37 °C. Mutant parasite lines resistant to either blasticidinS alone (FCB-br1) or to both blasticidin S and leupeptin (FCB-2mut) were previously generated through in vitro selectionsapplied to the wild-type FCB line (10, 11); both are limitingdilution clones. The 2C3 clone was obtained by limiting dilu-tion cloning of a blasticidin S-sensitive line resulting from cul-tivation of FCB-br1 without drug pressure. 2C3-R1, 2C3-S1,2C3-R2, and 2C3-S2 were then generated by sequential appli-cation and removal of 2.5�g/ml of blasticidin S to continuouslypropagated cultures; with each cycle, steady-state phenotypeswere observedwithin 6–28 days after changes in drug pressure.Computational Analyses—CLAG protein sequences were
aligned using MUSCLE (14); the alignment was inspected andimprovedmanually. A Bayesian phylogenetic tree was then cal-culated using MrBayes version 3.1.2 (15). Convergence wasachieved after 2 million generations; the resulting phylogenywas the consensus of 3002 trees. A maximum parsimony phy-logeny, calculated using MEGA5 (16), produced the sametopology. Ancestral sequences were reconstructed using thePAML version 4.5 maximum likelihood algorithm (17) appliedto a calculated Bayesian tree topology with the Jones-Taylor-Thorton substitution model (18). Mean pairwise sequencedistance within each paralog clade was calculated using theJones-Taylor-Thorton substitution model with � distributedrate heterogeneity among sites as implemented in MEGA5.DNA Sequencing—Candidate genes and their upstream pro-
moter regions were sequenced by standard methods afteramplification with specific primers and high fidelity Speed-STARHS DNA polymerase (Takara Bio, Shiga, Japan). Whole-genome sequencing used 200-bp fragment libraries andHiSeq2000 hardware (Illumina). 101-bp paired reads weremapped to the P. falciparum 3D7 version 8.0 referenceassembly.Quantitative RT-PCR—Gene expression was measured with
real-time PCR using total RNA harvested 28 h after sorbitolsynchronization of cultures. Contaminating genomicDNAwasremoved by DNase I treatment (TURBO DNA-free kit,Ambion). RNAwas adjusted to 5 �g/reaction and reverse tran-scribed at 50 °C using oligo(dT) priming and SuperScriptIII(Invitrogen). The resulting cDNA was used for quantitativereal-time PCR to determine expression levels of the indicatedgenes. PF07_0073, a constitutively expressed gene, was used asa normalization control to allow comparisons between sensi-tive and resistant parasite lines.Real-time PCR was performed in single-plex using primers
listed in Table 1 and theQuantiTect SYBRGreen PCR kit (Qia-gen). Primers were selected based on specificity for the desiredgene, an amplicon of �200 bp, and matched melting tempera-tures. Amplification was quantified by the iCycler iQ multi-color real-time PCR system (Bio-Rad) using a three-stage ther-
mal profile. After heating cDNA to 95 °C for 15 min, annealingand extension were performed for 40 cycles consisting of 94 °Cfor 30 s followed by 52 °C for 30 s. The final stage used gradualheating from 55 to 95 °C, incrementing by 0.5 °C/30 s; this dis-sociation protocol was used to confirm the specificity of primerbinding and product formation. Two negative controls, omis-sion of reverse transcriptase or cDNA template, were per-formed with each experiment to exclude genomic DNA con-tamination. All reactions were done in triplicate; the averagethreshold cycle (CT) was used for data analysis. Each experi-ment was performed on at least three separate RNA harvests.Gene expression was then quantified using the 2���CT
method (19). EachCT of the target gene was first normalized tothe CT of the Pf07_0073 control to obtain a �CT value for eachparasite line. The difference between this �CT value and thecorrespondingmeasurement for the blasticidin S-sensitive par-ent yielded the ��CT. Because exponential cDNA amplifica-tion is expected to double the product with each cycle, the value2���CT then estimates the fold-change in gene expression inthe mutant parasite.Genome-wide Expression Microarrays—Whole genome
expression profiling was performed using an available 70-meroligonucleotide microarray that represents �6000 open read-ing frames of the P. falciparum genome (20). Total RNA wasisolated from tightly synchronized trophozoite-stage culturesof FCB and FCB-br1, labeled with Cy3 or Cy5-dUTP, and usedfor two-color hybridization with microarray probes based onthe parasite genome sequence 3D7 reference. Dye-swap exper-iments were performed to exclude possible artifacts. Mean sig-nals from 7 independent hybridization experiments were cal-culated without filtering. Probes that do not map to thePlasmoDB transcript set were removed. Hits were defined asgenes producing a hybridization signal �100 absorbance unitsand a 10-fold change in expression.Chromatin Immunoprecipitation and qPCR—qCHIP exper-
iments were carried out as described previously (21). Synchro-nous trophozoite-infected erythrocytes were lysed with 0.05%saponin, washed, and fixed with 1% formaldehyde for 5 min at
TABLE 1qPCR primers
Gene (parasiteline) Forward and reverse primer sequences
PF07_0073control
5�-AAGTAGCAGGTCATCGTGGTT-3�5�-CATAAAAAATGGAGGATATACAGGTAT-3�
clag2 5�-CTCTTACTACTTATTATCTATCTCTCA-3�5�-CCAGGCGTAGGTCCTTTAC-3�
clag3.1 (FCB andderivatives)
5�-ACCCATAACTACATATTTTCTAGTAATG-3�5�-TCTGAACTAGGAGGCCAACC-3�
clag3.1 (3D7Atransgene)
5�-ACCCATAACTACATATTTTCTAGTAATG-3�5�-CAGGGGATTTATAACCACTAGCATTAC-3�
clag3.2 5�-ACCCATAACTACATATTTTCTAGTAATG-3�5�-TTCAGCAGCAAGTCCGTGA-3�
clag8 5�-GTTACTACAACATTCCTGATTCAG-3�5�-AATGAAAATATAAAAATGCTGGGGGAT-3�
clag9 5�-TACCATTAGTGTTTTATACACTTAAGG-3�5�-CCAAAATATGGCCAAGTACTTGC-3�
rhoph2 5�-GACATGATATCCAAAAAGGTAATATCA-3�5�-ACTAGAAAAATCATATACTGGTTTGTG-3�
rhoph3 5�-GTAGATGAAGATGCTCACCATG-3�5�-GTATAATTCTATTTCTAAATCTTGATCCTT-3�
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
19430 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 27 • JULY 5, 2013
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from

room temperature. After quenching with 1.25 M glycine, nucleiwere isolated (Wizard DNA purification kit, Promega), lysed in1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.0, with Complete,EDTA-free Protease Inhibitor Mixture (Roche Applied Sci-ence) at 4 °C, and sonicated to obtain DNA fragments of 500 bpaverage size. After centrifugation (16,000 � g, 20 min), thelysate was pre-cleared with protein G-agarose/salmon spermDNA (Millipore) in 167mMNaCl, 0.01% SDS, 1%TritonX-100,1 mM EDTA, 20mMTris, pH 8.0, for 1 h at 4 °C. After reservinga fraction for input qPCR, equal fractions were incubated over-night with 1:100 dilutions of anti-H3, anti-H3K9me3, anti-H3K9ac, anti-H3K4me3, or no antibody (Millipore andAbcam). The immunoprecipitate was recovered on proteinG-agarose/salmon sperm DNA after a 4-h incubation, andwashed with increasing stringency: the above buffer with 150and 500mMNaCl followed by 0.25 M LiCl, 1% Triton X-100, 1%sodium deoxycholate, 1 mM EDTA, 20 mM Tris, pH 8.0, andfinally with TE buffer. DNA was eluted with 1% SDS, 0.1 M
NaHCO3; cross-links were reversed by overnight incubation at65 °C. DNA was purified and precipitated prior to qPCR withgene-specific primers (Table 2). Enrichment of histone modifi-cations is presented as the log2 ratio of eachmutant or revertantline relative to the wild-type FCB parent after normalizationagainst the control H3 antibody in each experiment.DNA Transfection to Prevent clag3 Silencing—The full-
length clag3.1 open reading frame along with the native 599-bp3� UTR was cloned from the 3D7A parasite line into thepiggyBac integration plasmid pXL-BACII-DHFR (22). DNAsequence encoding the FLAG epitope tag was inserted imme-diately before the stop codon of the gene. A 1105-bp 5� UTRupstream of the msp2 start ATG, previously shown to driveexpression of a reporter gene (23), was amplified using 5�-CCAACCGTCGACGAATTCTTATTCTTGCCATCC-3� and5�-CCGGAACTCGAGTTTGACTAATATAATATGTTA-3�,digested with SalI and XhoI, and cloned into the plasmidupstream of the clag3.1 start ATG. Plasmid construction wasconfirmed by DNA sequencing.Uninfected erythrocytes were loaded with this plasmid and
the pHTH helper vector carrying the piggyBac gene. 2C3 para-
sites were cultivated in these erythrocytes for 4 days prior toaddition of 2.5 nM WR99210 to select for integration. Trans-fected parasites were detected after 7 weeks of continuous cul-ture. All experiments used limiting dilution clones obtainedwith the C-SNARF method (24).Southern Blot Hybridization—Genomic DNA was digested
with the XhoI restriction enzyme, separated on a 0.8% agarosegel at 40 volts for 16–20 h, and transferred to Hybond-N�
membrane (GE Healthcare). A DNA probe specific for hdhfrwas amplified with primers (5�-ATTTCCAGAGAATGACCA-CAAC-3� and 5�-TTAAGATGGCCTGGGTGATTC-3�) andlabeled with digoxigenin-dUTP according to the manufactur-er’s instructions (DIG DNA Labeling and Detection kit, RocheApplied Science). Hybridization was performed overnight at39 °C in DIG-Easy Hyb prior to washing twice in 2� SSC with0.1% SDS for 10 min at RT and twice in 1� SSC with 0.5% SDSfor 15 min at 52 °C. The membrane was then incubated withanti-digoxigenin-AP Fab fragments at a dilution of 1:10,000 fol-lowed by visualization on HyBlot CL x-ray film using CDP-Starsubstrate (Roche).Immunoblots—Total cell lysates were prepared fromPercoll-
enriched trophozoite-infected cells by lysis in 7.5 mM
Na2HPO4, 1 mM EDTA, pH 7.5, with HALT protease inhibitormixture (Thermo Scientific). This lysate was denatured andreduced in LDS Sample Buffer with 100 mMDTT prior to elec-trophoresis on NuPAGE� Novex 4–12% BisTris gels in MESSDS buffer (Invitrogen) and transfer to nitrocellulose mem-branes. The nitrocellulose membranes were blocked (3% skimmilk in PBS with 0.1% Tween 20) and probed with either anti-CLAG3 or anti-FLAG antibody (Cell Signaling Technology)before detection with HRP-conjugated secondary antibody(Pierce) at a 1:3000 dilution in blocking buffer. The blotswere developed using enhanced chemiluminescent substrates(Immobilon, Millipore, or SuperSignal West Pico, Pierce) andvisualized on HyBlot CL x-ray film. All blots used matchedloading of samples, as confirmed by Coomassie Blue staining ofgels or Ponceau S staining of blots for hemoglobin.Solute Permeability Measurements—Infected erythrocyte
permeability to organic solutes was quantified with a light scat-
TABLE 2qCHIP primers
Gene(qChIP fragment, Fig. 3) Forward and reverse primer sequences
Position fromstart codon
clag2 (1) 5�-CTAGAAGGAATATAAGAACAG-3� � 1054 to � 9345�-ATATGCGTATGGTGTGTATTC-3�
clag2 (2) 5�-CAACTACGCAATGTGCAGTAA-3� � 392 to � 2185�-GGTCTTATAACGTCTTTATCC-3�
clag2 (3) 5�-CATCCGTTAAATCATCATTGT-3� � 8 to � 1345�-TTTCCAAATTCATATTCATC-3�
clag2 (4) 5�-GTGAACCAGATACAAAAGAAT-3� � 5081 to � 52735�-TATTGTAATCTTGTAATCATC-3�
clag3.1 (1) 5�-GATTTTATAATGCACTCATTAATAA-3� � 1026 to � 8965�-AATAAATGTATACGTAATTAGAACA-3�
clag3.1 (2) 5�-TTTCCATTGCACCGATATTAAAA-3� � 562 to � 4685�-GAAGTGAGATAAAAATACATATTC-3�
clag3.1 (3) 5�-TCAAAATGAAAATGATACCATTAGT-3� � 87 to � 1865�-CATGGATTTTAATTGTTCAATATTG-3�
clag3.1 (4) 5�-TAGTAATGAGAATTAGTTGGACA-3� � 4273 to � 44435�-ATAAATATTTGGATGCTTCAGCA-3�
clag9 (1) 5�-CATATGCGTCTTGCCTTGCAC-3� � 1015 to � 8675�-CATGATTATATGTATTGCTAAA-3�
clag9 (2) 5�-GATATATGGATTCAAAATTTG-3� � 195 to � 675�-AATATTCTTTTTCTTAGTGAC-3�
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
JULY 5, 2013 • VOLUME 288 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 19431
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from

tering assay (25). Trophozoite stage-infected RBCs wereenriched to �95% parasitemia by Percoll-sorbitol density gra-dient centrifugation, washed in 150 mM NaCl, 20 mM HEPES,0.1 mg/ml of BSA, pH 7.4, and resuspended at 5% hematocrit.Osmotic lysis due to PSAC-mediated solute uptake was initi-ated by addition of 40 volumes of buffered lysis solution (280mM sorbitol or 145 mM PhTMA-Cl with 20 mMNa-HEPES, 0.1mg/ml BSA, pH 7.4) with inhibitor as indicated at 37 °C. Con-tinuous tracking of 700 nm light transmittance through the cellsuspension was used to quantify lysis kinetics and estimatesolute permeability.GrowthAssays—Parasite growth uponblasticidin S challenge
was assessed with microscopic examination of Giemsa-stainedsmears or SYBRGreen I detection of parasiteDNAas describedpreviously (26); these methods produced similar results.Statistical Analyses—One-way analysis of variance (ANOVA)
was used to evaluate the hypothesis of no difference betweenthree or more groups. If significance was reached (p � 0.05),post hoc pairwise comparisons used Student’s t tests.
RESULTS
Blasticidin S Resistance Is Not Linked to DNA LevelChanges—A clonal PSAC mutant termed FCB-br1 was previ-ously generated by in vitro selection with blasticidin S (10);subsequent selection for leupeptin resistance followed by clon-ing yielded a distinct double mutant referred to as FCB-2mut(11). Both mutants exhibit decreased organic solute perme-abilities and altered PSAC activity in patch clamp studies. Toexplore whether genome-level changes account for thesemutant channel phenotypes, we first considered the two clag3genes of these mutants. These genes have recently been impli-cated in infected erythrocyte permeability (13); how theencoded proteins contribute to PSAC is unclear at present.DNA sequencing of both genes in FCB-br1 and FCB-2mut didnot reveal either mutations or indels relative to the blasticidinS-sensitive FCB parental line (not shown).Although most plasmodial species have only two or three
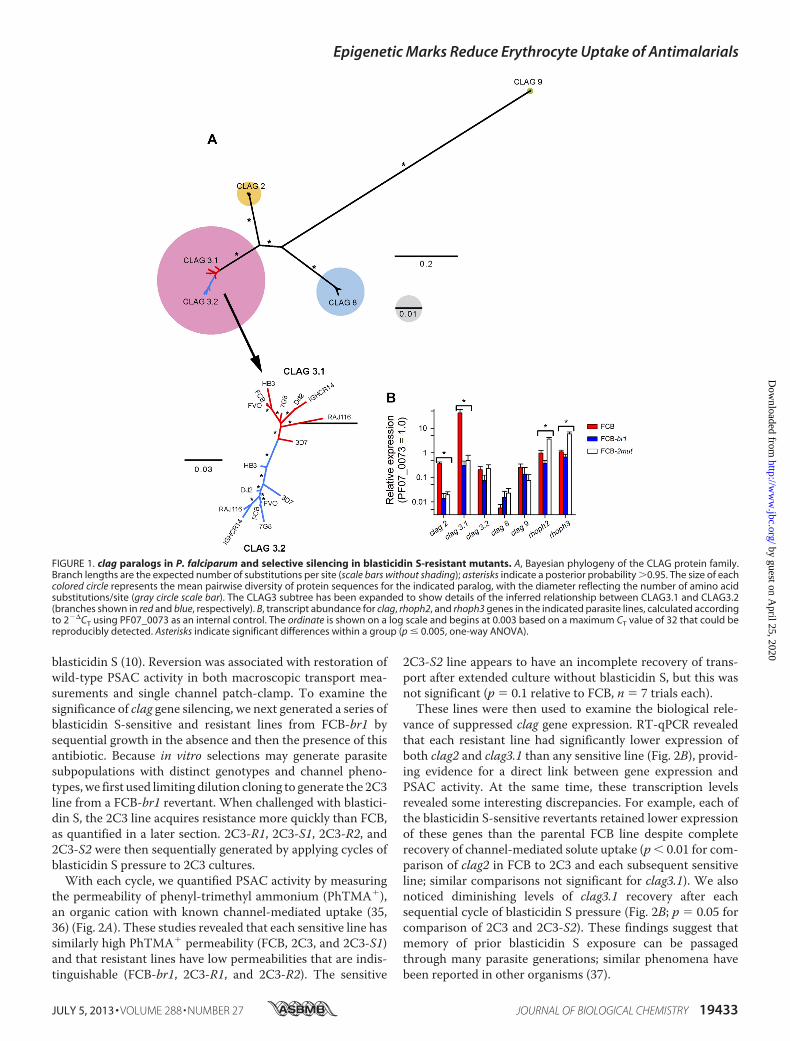
clag genes in their genomes (27, 28), P. falciparum has under-gone significant expansion to have five members in this genefamily. To explore whether the other paralogs contribute toPSAC, we performed phylogenetic analysis of clag genes fromlaboratory lines representing Africa, South America, andSoutheast Asia and found that each paralog has primarysequence elements that distinguish it from all other clag prod-ucts, with the clag9 group being the most divergent (Fig. 1A).Notably, even though clag3.1 and clag3.2 undergo mutuallyexclusive expression and should therefore serve fully overlap-ping roles, these paralogs also segregate into discrete cladeswith high confidence (posterior probability 1.0). These find-ings suggest relatively ancient expansion and ongoing evolutionof the clag gene family in P. falciparum.CLAG3.1 and CLAG3.2 both carry a �30 residue variable
domain located some 1100 residues from the N terminus of theprotein (13, 29), accounting for the substantial pairwise diver-sity within this clade (pink circle, Fig. 1A). This domain isexposed at the host cell surface, consistent with selection forvariability by host immunity. Our analysis of CLAG2 andCLAG8 sequences also revealed pairwise diversity, although at
somewhat lower levels (smaller colored circles, Fig. 1A). Inter-estingly, the diversity in these proteins was also largely attrib-utable to a variable domain at a similar position in the primarysequence, suggesting that these paralogs may have transmem-brane topologies similar to that of CLAG3. In contrast, CLAG9sequences lack a variable domain and have accrued few poly-morphisms in P. falciparum.Chain termination sequencing of the five clag paralogs did
not reveal any changes in either FCB-br1 or FCB-2mut relativeto the sensitive FCB parental line. Because unrelated genesmayalso contribute to PSAC activity (30), we then used next gener-ation sequencing to search for responsible mutations or indels.We analyzed coverage and determined that 95% of codingregions achieved a minimum of 15� coverage; multigene fam-ilies such as the clag genes were also well covered (data notshown). Only six genes had variants that met our SNP callingthreshold in one or both resistant parasites, but these did notconfirm with chain termination sequencing. Thus, deepsequencing suggests that blasticidin S resistance does not resultfrom DNA level changes.Suppressed clag2 and clag3 Expression in BothMutants—We
next used real-time quantitative reverse transcriptase PCR(RT-qPCR) to evaluate possible changes in transcription withprimers specific for each open reading frame of the clag genes(Table 1). rhoph2 and rhoph3, genes whose products make sta-ble interactions with CLAG proteins (31, 32), were also exam-ined. In synchronous trophozoite-stage cultures, the wild-typeFCB parent expressed clag3.1 at levels that were 42 16-foldhigher than the constitutively expressed control gene,PF07_0073 (Fig. 1B). Its nearby paralog, clag3.2, was expressedat much lower levels (0.2 0.07 relative to PF07_0073), con-sistent with monoallelic expression of these two family mem-bers (33). Preferential expression of one or the other clag3 genehas been documented for various lines (13, 34), but it is unclearwhether this reflects differences between the proteins that con-fer an in vitro survival advantage to parasites.We found marked reductions in expression of both clag3.1
and clag2 in FCB-br1 cultures grown in the continuous pres-ence of 2.5 �g/ml of blasticidin S (140- and 26-fold, respec-tively; p � 10�4 each, 8 independent trials). FCB-2mut, whencultivated with blasticidin S and leupeptin, exhibited compara-bly reduced expression of the same paralogs (87- and 18-foldlower than FCB, respectively; p � 10�4). Both mutants showedsmall changes in expression of clag3.2, clag8, and clag9, butthese did not reach statistical significance. Although expressionof rhoph2 was decreased in FCB-br1, both rhoph2 and rhoph3exhibited increased expression in FCB-2mut (Fig. 1B, p �0.005). Because expression changed in opposite directions forthese two mutants and the changes were relatively modest, wedid not examine rhoph2 and rhoph3 further in this study.Whole genome expression microarray experiments excludedglobal changes in parasite gene expression and revealed a smallnumber of candidates for further study (data not shown, GEOSeries accession number GSE47579, www.ncbi.nlm.nih.gov/geo/query/acc.cgi?accGSE47579).clag2 and clag3.1 Silencing Is Reversible in FCB-br1, but Not
in FCB-2mut—We previously reported that the FCB-br1mutant loses its resistance phenotype when cultivated without
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
19432 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 27 • JULY 5, 2013
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from
blasticidin S (10). Reversion was associated with restoration ofwild-type PSAC activity in both macroscopic transport mea-surements and single channel patch-clamp. To examine thesignificance of clag gene silencing, we next generated a series ofblasticidin S-sensitive and resistant lines from FCB-br1 bysequential growth in the absence and then the presence of thisantibiotic. Because in vitro selections may generate parasitesubpopulations with distinct genotypes and channel pheno-types, we first used limiting dilution cloning to generate the 2C3line from a FCB-br1 revertant. When challenged with blastici-din S, the 2C3 line acquires resistance more quickly than FCB,as quantified in a later section. 2C3-R1, 2C3-S1, 2C3-R2, and2C3-S2 were then sequentially generated by applying cycles ofblasticidin S pressure to 2C3 cultures.With each cycle, we quantified PSAC activity by measuring
the permeability of phenyl-trimethyl ammonium (PhTMA�),an organic cation with known channel-mediated uptake (35,36) (Fig. 2A). These studies revealed that each sensitive line hassimilarly high PhTMA� permeability (FCB, 2C3, and 2C3-S1)and that resistant lines have low permeabilities that are indis-tinguishable (FCB-br1, 2C3-R1, and 2C3-R2). The sensitive
2C3-S2 line appears to have an incomplete recovery of trans-port after extended culture without blasticidin S, but this wasnot significant (p 0.1 relative to FCB, n 7 trials each).
These lines were then used to examine the biological rele-vance of suppressed clag gene expression. RT-qPCR revealedthat each resistant line had significantly lower expression ofboth clag2 and clag3.1 than any sensitive line (Fig. 2B), provid-ing evidence for a direct link between gene expression andPSAC activity. At the same time, these transcription levelsrevealed some interesting discrepancies. For example, each ofthe blasticidin S-sensitive revertants retained lower expressionof these genes than the parental FCB line despite completerecovery of channel-mediated solute uptake (p � 0.01 for com-parison of clag2 in FCB to 2C3 and each subsequent sensitiveline; similar comparisons not significant for clag3.1). We alsonoticed diminishing levels of clag3.1 recovery after eachsequential cycle of blasticidin S pressure (Fig. 2B; p 0.05 forcomparison of 2C3 and 2C3-S2). These findings suggest thatmemory of prior blasticidin S exposure can be passagedthrough many parasite generations; similar phenomena havebeen reported in other organisms (37).
FIGURE 1. clag paralogs in P. falciparum and selective silencing in blasticidin S-resistant mutants. A, Bayesian phylogeny of the CLAG protein family.Branch lengths are the expected number of substitutions per site (scale bars without shading); asterisks indicate a posterior probability �0.95. The size of eachcolored circle represents the mean pairwise diversity of protein sequences for the indicated paralog, with the diameter reflecting the number of amino acidsubstitutions/site (gray circle scale bar). The CLAG3 subtree has been expanded to show details of the inferred relationship between CLAG3.1 and CLAG3.2(branches shown in red and blue, respectively). B, transcript abundance for clag, rhoph2, and rhoph3 genes in the indicated parasite lines, calculated accordingto 2��CT using PF07_0073 as an internal control. The ordinate is shown on a log scale and begins at 0.003 based on a maximum CT value of 32 that could bereproducibly detected. Asterisks indicate significant differences within a group (p � 0.005, one-way ANOVA).
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
JULY 5, 2013 • VOLUME 288 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 19433
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from
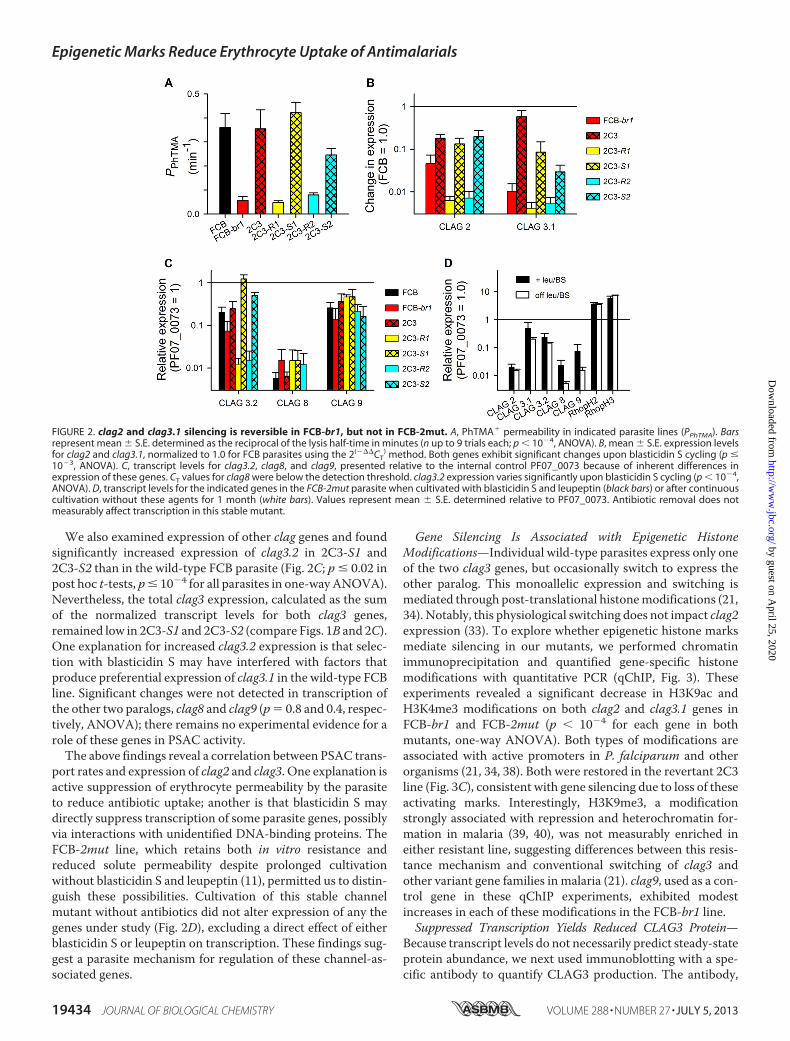
We also examined expression of other clag genes and foundsignificantly increased expression of clag3.2 in 2C3-S1 and2C3-S2 than in the wild-type FCB parasite (Fig. 2C; p � 0.02 inpost hoc t-tests, p� 10�4 for all parasites in one-wayANOVA).Nevertheless, the total clag3 expression, calculated as the sumof the normalized transcript levels for both clag3 genes,remained low in 2C3-S1 and 2C3-S2 (compare Figs. 1B and 2C).One explanation for increased clag3.2 expression is that selec-tion with blasticidin S may have interfered with factors thatproduce preferential expression of clag3.1 in the wild-type FCBline. Significant changes were not detected in transcription ofthe other two paralogs, clag8 and clag9 (p 0.8 and 0.4, respec-tively, ANOVA); there remains no experimental evidence for arole of these genes in PSAC activity.The above findings reveal a correlation between PSAC trans-
port rates and expression of clag2 and clag3. One explanation isactive suppression of erythrocyte permeability by the parasiteto reduce antibiotic uptake; another is that blasticidin S maydirectly suppress transcription of some parasite genes, possiblyvia interactions with unidentified DNA-binding proteins. TheFCB-2mut line, which retains both in vitro resistance andreduced solute permeability despite prolonged cultivationwithout blasticidin S and leupeptin (11), permitted us to distin-guish these possibilities. Cultivation of this stable channelmutant without antibiotics did not alter expression of any thegenes under study (Fig. 2D), excluding a direct effect of eitherblasticidin S or leupeptin on transcription. These findings sug-gest a parasite mechanism for regulation of these channel-as-sociated genes.
Gene Silencing Is Associated with Epigenetic HistoneModifications—Individual wild-type parasites express only oneof the two clag3 genes, but occasionally switch to express theother paralog. This monoallelic expression and switching ismediated through post-translational histonemodifications (21,34). Notably, this physiological switching does not impact clag2expression (33). To explore whether epigenetic histone marksmediate silencing in our mutants, we performed chromatinimmunoprecipitation and quantified gene-specific histonemodifications with quantitative PCR (qChIP, Fig. 3). Theseexperiments revealed a significant decrease in H3K9ac andH3K4me3 modifications on both clag2 and clag3.1 genes inFCB-br1 and FCB-2mut (p � 10�4 for each gene in bothmutants, one-way ANOVA). Both types of modifications areassociated with active promoters in P. falciparum and otherorganisms (21, 34, 38). Both were restored in the revertant 2C3line (Fig. 3C), consistent with gene silencing due to loss of theseactivating marks. Interestingly, H3K9me3, a modificationstrongly associated with repression and heterochromatin for-mation in malaria (39, 40), was not measurably enriched ineither resistant line, suggesting differences between this resis-tance mechanism and conventional switching of clag3 andother variant gene families in malaria (21). clag9, used as a con-trol gene in these qChIP experiments, exhibited modestincreases in each of these modifications in the FCB-br1 line.Suppressed Transcription Yields Reduced CLAG3 Protein—
Because transcript levels do not necessarily predict steady-stateprotein abundance, we next used immunoblotting with a spe-cific antibody to quantify CLAG3 production. The antibody,
FIGURE 2. clag2 and clag3.1 silencing is reversible in FCB-br1, but not in FCB-2mut. A, PhTMA� permeability in indicated parasite lines (PPhTMA). Barsrepresent mean S.E. determined as the reciprocal of the lysis half-time in minutes (n up to 9 trials each; p � 10�4, ANOVA). B, mean S.E. expression levelsfor clag2 and clag3.1, normalized to 1.0 for FCB parasites using the 2(���CT
) method. Both genes exhibit significant changes upon blasticidin S cycling (p �10�3, ANOVA). C, transcript levels for clag3.2, clag8, and clag9, presented relative to the internal control PF07_0073 because of inherent differences inexpression of these genes. CT values for clag8 were below the detection threshold. clag3.2 expression varies significantly upon blasticidin S cycling (p � 10�4,ANOVA). D, transcript levels for the indicated genes in the FCB-2mut parasite when cultivated with blasticidin S and leupeptin (black bars) or after continuouscultivation without these agents for 1 month (white bars). Values represent mean S.E. determined relative to PF07_0073. Antibiotic removal does notmeasurably affect transcription in this stable mutant.
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
19434 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 27 • JULY 5, 2013
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from

raised by immunizingmice with a 141-residue C-terminal frag-ment of CLAG3 (13), may also recognize CLAG2 based onsequence similarity, but low identity with CLAG8 or CLAG9
suggests it will not recognize these paralogs. Immunoblottingwith this antibody revealed a markedly reduced signal in eachblasticidin S-resistant parasite (Fig. 4A), consistent withreduced CLAG3 abundance due to suppressed transcription.Peripheral membrane protein extraction with Na2CO3
revealed that CLAG3 is detected as an integral membrane pro-tein in FCB parasites, but is below the detection threshold inFCB-br1 (Fig. 4B). It is possible that CLAG3 delivery to the hostmembrane is also compromised in blasticidin S-resistant para-sites, as reported for ion channel proteins in other systems (41).We were unable to examine such post-translational regulationbecause the low CLAG3 levels in our mutants prohibited pro-tein trafficking studies.Resistance Requires Suppressed clag3 Expression—To deter-
mine whether clag gene silencing is sufficient to produce blas-ticidin S resistance and altered PSAC activity, we sought aDNAtransfection approach that would prevent silencing under blas-ticidin S pressure. We reasoned that use of an unrelated pro-moter to drive expression may allow escape from the aboveepigenetic mechanisms. At the same time, it is important topreserve the stage specificity of clag gene expression becausetranscripts made on an incorrect schedule in the parasite lifecyclemay yield protein that either fails to interact with essentialligands or does not traffic properly (42). We selected the wellcharacterized promoter of merozoite surface protein-2 (msp2,PFB0300c) because it, like that of all clag genes, directsmaximaltranscription at the schizont stage (23, 43). Finally, to avoidsilencing mechanisms that depend on the location of the genewithin the parasite genome and local heterochromatin forma-tion, we selected the piggyBac transposase system. Stable com-plementation of genes using this transposase allows randomintegration at one of numerous sites throughout the parasitegenome (44), a potential advantage under our circumstances.Fig. 5A shows the pXL-M2–120w vector, which carries a
1105-bp msp2 promoter followed by the full-length clag3.1cDNAcloned from3D7Aparasites, a C-terminal FLAGepitopetag, and a 599-bp 3� UTR native to clag3.1. This gene and ahuman dihydrofolate reductase (hDHFR) selectable markerwere inserted in tandem between two inverted terminal repeat
FIGURE 3. qChIP analysis of histone modifications associated with resis-tance. A, gene structures and positions of fragments amplified by qPCR; blackboxes indicate exons. Notice the similar size and exon structures of these genes;scale bar, 1000 bp. B–D, mean S.E. enrichment of gene fragment-specific his-tone modifications in FCB-br1, the 2C3 revertant, and FCB-2mut lines (panels B–D,respectively), presented relative to the parental FCB line. Positive and negativevalues indicate enrichment and depletion in daughter parasites, respectively.
FIGURE 4. Consequent reductions in CLAG3 abundance. A, immunoblotusing whole-cell lysates from the indicated parasites and anti-CLAG3 anti-body. The primary band at �160 kDa corresponds to CLAG3 and is markedlyreduced in each blasticidin S-resistant parasite. B, immunoblot using Na2CO3-extracted membranes, showing that incorporation of CLAG3 into the hostmembrane is below detectable levels in FCB-br1.
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
JULY 5, 2013 • VOLUME 288 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 19435
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from
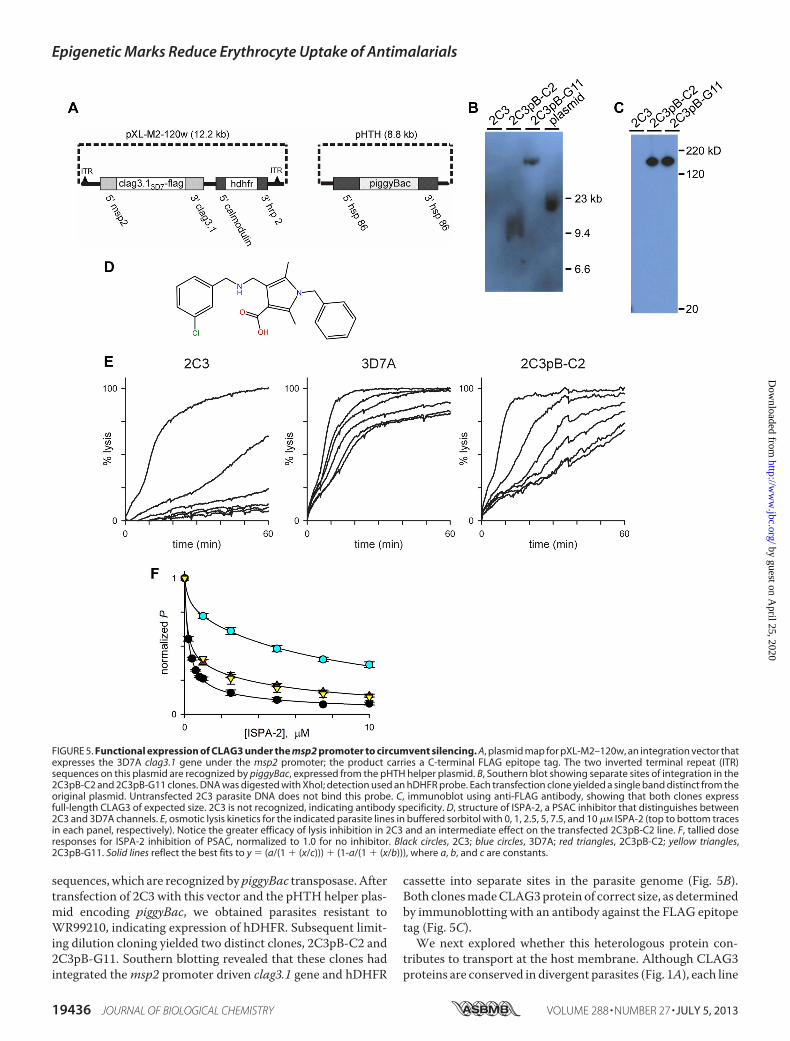
sequences, which are recognized by piggyBac transposase. Aftertransfection of 2C3 with this vector and the pHTH helper plas-mid encoding piggyBac, we obtained parasites resistant toWR99210, indicating expression of hDHFR. Subsequent limit-ing dilution cloning yielded two distinct clones, 2C3pB-C2 and2C3pB-G11. Southern blotting revealed that these clones hadintegrated themsp2 promoter driven clag3.1 gene and hDHFR
cassette into separate sites in the parasite genome (Fig. 5B).Both clonesmadeCLAG3protein of correct size, as determinedby immunoblotting with an antibody against the FLAG epitopetag (Fig. 5C).We next explored whether this heterologous protein con-
tributes to transport at the host membrane. Although CLAG3proteins are conserved in divergent parasites (Fig. 1A), each line
FIGURE 5. Functional expression of CLAG3 under the msp2 promoter to circumvent silencing. A, plasmid map for pXL-M2–120w, an integration vector thatexpresses the 3D7A clag3.1 gene under the msp2 promoter; the product carries a C-terminal FLAG epitope tag. The two inverted terminal repeat (ITR)sequences on this plasmid are recognized by piggyBac, expressed from the pHTH helper plasmid. B, Southern blot showing separate sites of integration in the2C3pB-C2 and 2C3pB-G11 clones. DNA was digested with XhoI; detection used an hDHFR probe. Each transfection clone yielded a single band distinct from theoriginal plasmid. Untransfected 2C3 parasite DNA does not bind this probe. C, immunoblot using anti-FLAG antibody, showing that both clones expressfull-length CLAG3 of expected size. 2C3 is not recognized, indicating antibody specificity. D, structure of ISPA-2, a PSAC inhibitor that distinguishes between2C3 and 3D7A channels. E, osmotic lysis kinetics for the indicated parasite lines in buffered sorbitol with 0, 1, 2.5, 5, 7.5, and 10 �M ISPA-2 (top to bottom tracesin each panel, respectively). Notice the greater efficacy of lysis inhibition in 2C3 and an intermediate effect on the transfected 2C3pB-C2 line. F, tallied doseresponses for ISPA-2 inhibition of PSAC, normalized to 1.0 for no inhibitor. Black circles, 2C3; blue circles, 3D7A; red triangles, 2C3pB-C2; yellow triangles,2C3pB-G11. Solid lines reflect the best fits to y (a/(1 � (x/c))) � (1-a/(1 � (x/b))), where a, b, and c are constants.
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
19436 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 27 • JULY 5, 2013
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from
carries a unique polymorphic region near the C terminus. Thispolymorphic region is exposed at the host erythrocyte surfaceand interacts with a subset of PSAC inhibitors (13), which arereferred to as ISPA compounds (for isolate-specific PSACantagonists) to highlight their ability to discriminate betweenchannels linked to specific genotypes. Because piggyBac com-plementation should yield parasites that express clag3.1 prod-ucts from both 3D7A and 2C3 lineages, we surveyed severalISPA hits from recent high-throughput PSAC inhibitor screens(13, 26). This revealed ISPA-2 as a compound with greaterpotency for inhibition of channels from the 2C3 parasite thanfrom3D7A (Fig. 5,D–F; p� 10�9, n 7–10 trials each). Trans-port studies revealed that the two transfection clones haveidentical ISPA-2 inhibition dose responses that are intermedi-ate between those of 2C3 and 3D7A. Although inhibition moreclosely resembled that of 2C3, the change from this untrans-fected parent was significant (p � 10�4 for comparisons of 2C3to either 2C3pB-C2 or 2C3pB-G11 at 1 �M ISPA-2). Becausethe two clones carry the 3D7A transgene at separate sites intheir genomes, their similar inhibition phenotypes suggest thatexpression level, rather than genomic environment, determineschannel block by ISPA-2. Importantly, the intermediate inhibi-tion dose response is consistent with contributions from clag3products encoded by both 2C3 and 3D7A parasites. One possi-bility is that these pseudodiploid parasites assemble channelsconsisting of the two CLAG3 isoforms and therefore exhibitinhibition that is a weighted average of the individual isoforms.The absolute change in ISPA-2 affinity cannot, however, beused to estimate the relative copy number of the two channelisoforms because the precise role and stoichiometry of CLAG3contribution to PSAC is unknown.We then challenged both transfected and untransfected lines
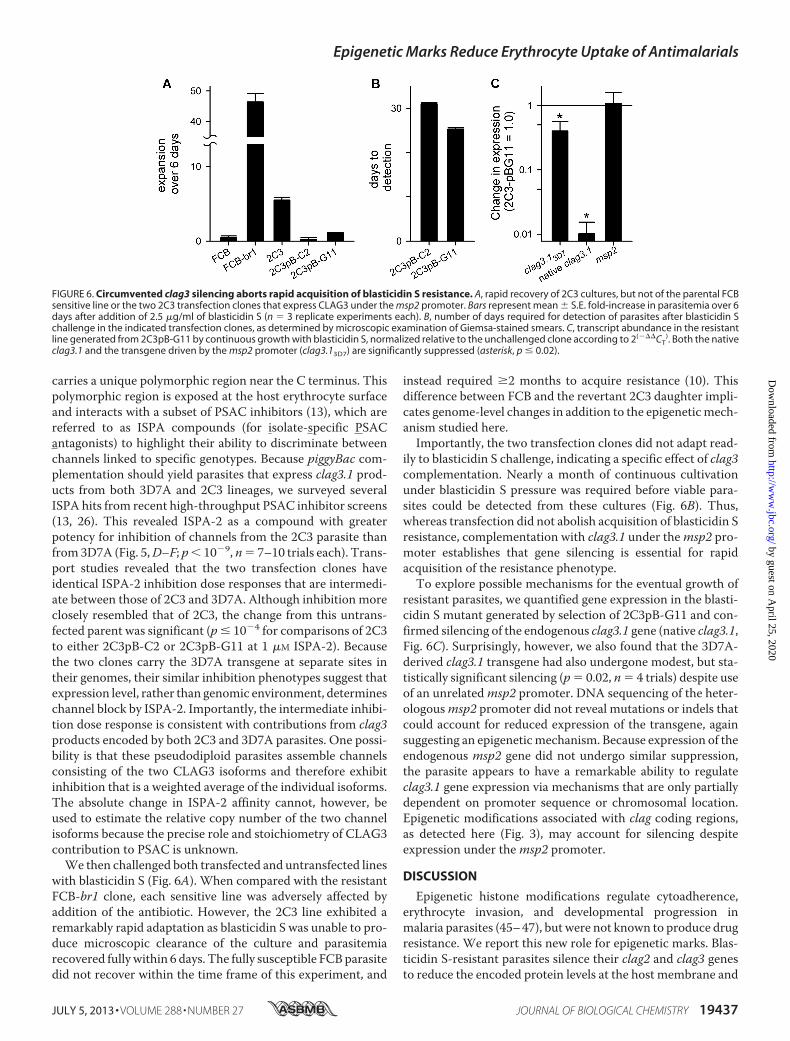
with blasticidin S (Fig. 6A). When compared with the resistantFCB-br1 clone, each sensitive line was adversely affected byaddition of the antibiotic. However, the 2C3 line exhibited aremarkably rapid adaptation as blasticidin S was unable to pro-duce microscopic clearance of the culture and parasitemiarecovered fullywithin 6 days. The fully susceptible FCBparasitedid not recover within the time frame of this experiment, and
instead required �2 months to acquire resistance (10). Thisdifference between FCB and the revertant 2C3 daughter impli-cates genome-level changes in addition to the epigeneticmech-anism studied here.Importantly, the two transfection clones did not adapt read-
ily to blasticidin S challenge, indicating a specific effect of clag3complementation. Nearly a month of continuous cultivationunder blasticidin S pressure was required before viable para-sites could be detected from these cultures (Fig. 6B). Thus,whereas transfection did not abolish acquisition of blasticidin Sresistance, complementation with clag3.1 under themsp2 pro-moter establishes that gene silencing is essential for rapidacquisition of the resistance phenotype.To explore possible mechanisms for the eventual growth of
resistant parasites, we quantified gene expression in the blasti-cidin S mutant generated by selection of 2C3pB-G11 and con-firmed silencing of the endogenous clag3.1 gene (native clag3.1,Fig. 6C). Surprisingly, however, we also found that the 3D7A-derived clag3.1 transgene had also undergone modest, but sta-tistically significant silencing (p 0.02, n 4 trials) despite useof an unrelatedmsp2 promoter. DNA sequencing of the heter-ologousmsp2 promoter did not reveal mutations or indels thatcould account for reduced expression of the transgene, againsuggesting an epigeneticmechanism. Because expression of theendogenous msp2 gene did not undergo similar suppression,the parasite appears to have a remarkable ability to regulateclag3.1 gene expression via mechanisms that are only partiallydependent on promoter sequence or chromosomal location.Epigenetic modifications associated with clag coding regions,as detected here (Fig. 3), may account for silencing despiteexpression under themsp2 promoter.
DISCUSSION
Epigenetic histone modifications regulate cytoadherence,erythrocyte invasion, and developmental progression inmalaria parasites (45–47), but were not known to produce drugresistance. We report this new role for epigenetic marks. Blas-ticidin S-resistant parasites silence their clag2 and clag3 genesto reduce the encoded protein levels at the host membrane and
FIGURE 6. Circumvented clag3 silencing aborts rapid acquisition of blasticidin S resistance. A, rapid recovery of 2C3 cultures, but not of the parental FCBsensitive line or the two 2C3 transfection clones that express CLAG3 under the msp2 promoter. Bars represent mean S.E. fold-increase in parasitemia over 6days after addition of 2.5 �g/ml of blasticidin S (n 3 replicate experiments each). B, number of days required for detection of parasites after blasticidin Schallenge in the indicated transfection clones, as determined by microscopic examination of Giemsa-stained smears. C, transcript abundance in the resistantline generated from 2C3pB-G11 by continuous growth with blasticidin S, normalized relative to the unchallenged clone according to 2(���CT
). Both the nativeclag3.1 and the transgene driven by the msp2 promoter (clag3.13D7) are significantly suppressed (asterisk, p � 0.02).
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
JULY 5, 2013 • VOLUME 288 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 19437
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from

curtail channel-mediated uptake of this antibiotic. We con-firmed a direct contribution to resistance by expressing clag3under a heterologous promoter to circumvent epigeneticsilencing. Although blasticidin S is not used to treat humanmalaria, our findings suggest antimalarial drugs that requirechannel-mediated uptake to reach their intracellular targetsmay also be prone to resistance and clinical failure by thismechanism. Although chloroquine and artemisinin are lipo-philic and do not appear to require channel-mediated uptake(48, 49), drug discovery programs increasingly prefer highwater solubility to improve bioavailability (50);many such com-pounds may depend on uptake via PSAC to reach their intra-cellular parasite targets. Thus, future antimalarial drugs mayencounter the resistance mechanism described here.Our findings implicate a new level of regulated expression for
clag genes. Although epigenetic switching between the twoclag3 genes is established (13, 33, 34), the silencing reportedhere has some distinctive features. First, blasticidin S-inducedsilencing affects clag2 and both clag3 genes, whereas monoal-lelic expression suppresses individual clag3 genes withoutreducing total transcript levels. Second, this resistance mecha-nism uses a distinct pattern of epigenetic marks as it does notinvolve H3K9me3, a modification linked to silencing andmonoallelic expression in parasite gene families (Fig. 3)(39).Third, this new layer of regulation appears to use epigeneticmodifications that can be more rapidly applied or removed.Although switching between the two clag3 genes occurs with ahalf-time of �2 months after perturbation (13), we found thatsensitive 2C3 parasites recover and expand within only 6 daysafter blasticidin S challenge (Fig. 6A). Reversion to the sensitivephenotype also occurs rapidly as transport was restored in lessthan 1 month upon removal of blasticidin S (Fig. 2A). Finally,whereas conventional silencing and switching between the twoclag3 genes has only modest effects on channel properties (13),the epigenetic changes described here yield marked reductionsin solute transport (Fig. 2). We propose that silencing of indi-vidual clag3 genes in sensitive parasites serves to protect thesilenced paralog from host immunity (51, 52), but that themechanism reported here functions to regulate host cell per-meability in response to nutrient availability and drug pressure(53). For example, in vivo suppression of PSACmay contributeto development of dormant bloodstream parasites, as observedafter drug exposure (54). Regulated host cell permeability mayalso be important for fine-tuning changes in erythrocyte cationconcentrations (55).Gene silencing in resistant parasites represents independent
evidence for a role of clag genes in nutrient uptake at the hostmembrane. Nevertheless, it is not safe to conclude that CLAGproteins form ion channels on their own. They lack sequencehomology to known channels and have fewer predicted trans-membrane domains than most pore-forming proteins. CLAGproteins also have proposed roles in cytoadherence and proteintrafficking (56–58), raising additional questions about how theprotein may alter host cell permeability. Conclusive answerswill require multiple experimental approaches including heter-ologous expression andDNA transfections inmalaria parasites.Our new evidence linking the uncharacterized CLAG2 para-
log to PSAC activity provides a framework for examining the
elusive composition of these channels. If future studies revealthat CLAG proteins form aqueous pores directly, individualparalogs may define distinct parts of the functional channel.This model is reminiscent of the acetylcholine receptor, whichis composed of four related subunits that undergo controlledassembly into a gated ion channel (59). If these parasite proteinsfunction as transport activators rather than as stable channelsubunits, then our findings predict that CLAG2will also exhibitsimilar enzymatic activity.A recently generated clag3 knockdown parasite may also
help determine the precise role of CLAG proteins (34). There,transfection was used to replace the clag3.2 gene with anhDHFR selectable marker cassette. Subsequent selection forhDHFR expression reportedly silenced clag3.1 and yieldedundetectable expression of both clag3 genes. The resultingknockdown parasite may therefore resemble our blasticidinS-resistant lines, which exhibit �100-fold reduced expressionof clag3. The transport properties and drug sensitivities of theknockdown parasite were not studied there, but should be pur-sued. One possibility is that the newly implicated CLAG2 playsa compensatory role to sustain transport.Although silencing of clag2 and clag3 are tightly associated
with blasticidin S resistance, several findings suggest thatsilencing alone is not sufficient to account for the observedtransport phenotypes. 1) Whole cell patch clamp measure-ments reveal that FCB-br1 has a 7-fold reduction in PSAC-specific Cl� currents (10), which may be less than expected bythe measured reductions in transcript and CLAG3 protein(Figs. 2 and 4). 2) Altered single channel gating and solute selec-tivity in these mutants is also inconsistent with the conserv-ative prediction that transcriptional down-regulation yieldsa reduced number of otherwise unchanged channels (10, 11). 3)Although FCB-br1 and FCB-2mut exhibit similar levels of claggene silencing, thesemutants have very different channel prop-erties and responses to removal of selective pressure. 4) FCBand the revertant 2C3 lines switch to the resistant phenotype atdifferent rates (Fig. 6A). One explanation for these findings isthat other proteins are also required to form functional chan-nels at the host membrane. Then, the slow initial acquisition ofblasticidin S resistance in FCB may reflect one or more DNAlevel changes in an unidentified channel component. Suchchanges may then be permissive for clag2 and clag3 silencing,which appears to be the final step in reducing host cell perme-ability and antibiotic sensitivity. Another possibility is thatPSAC is formed exclusively by CLAG paralogs assembled as aheteromeric complex. In this scenario, differential silencing ofspecific subunits may lead to assembly of altered channels. Theavailability of mutants with altered channel phenotypes, robusttransport methodologies, and new molecular approachesdescribed here should help reveal the structure and function ofthis unusual ion channel.
Acknowledgments—We thankGodfrey Lisk, LanxuanDoan, andTet-suya Furuya for technical help as well as Dyann Wirth, Sarah Volk-man, and members of the Desai laboratory for helpful discussions.
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
19438 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 27 • JULY 5, 2013
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from

REFERENCES1. Greenwood, B. (2009) Can malaria be eliminated? Trans. R. Soc. Trop.
Med. Hyg. 103, S2–S52. Anderson, T. J., Nair, S., Nkhoma, S., Williams, J. T., Imwong, M., Yi, P.,
Socheat, D., Das, D., Chotivanich, K., Day, N. P., White, N. J., and Don-dorp, A. M. (2010) High heritability of malaria parasite clearance rateindicates a genetic basis for artemisinin resistance in western Cambodia.J. Infect. Dis. 201, 1326–1330
3. Korsinczky,M., Chen,N., Kotecka, B., Saul, A., Rieckmann, K., andCheng,Q. (2000) Mutations in Plasmodium falciparum cytochrome b that areassociated with atovaquone resistance are located at a putative drug-bind-ing site. Antimicrob. Agents Chemother. 44, 2100–2108
4. Gregson, A., and Plowe, C. V. (2005) Mechanisms of resistance of malariaparasites to antifolates. Pharmacol. Rev. 57, 117–145
5. Foote, S. J., Kyle, D. E., Martin, R. K., Oduola, A. M., Forsyth, K., Kemp,D. J., and Cowman, A. F. (1990) Several alleles of the multidrug-resistancegene are closely linked to chloroquine resistance in Plasmodium falcipa-rum. Nature 345, 255–258
6. Sidhu, A. B., Verdier-Pinard, D., and Fidock, D. A. (2002) Chloroquineresistance in Plasmodium falciparummalaria parasites conferred by pfcrtmutations. Science 298, 210–213
7. Duraisingh, M. T., and Cowman, A. F. (2005) Contribution of the pfmdr1gene to antimalarial drug-resistance. Acta Tropica 94, 181–190
8. Price, R. N., Uhlemann, A. C., Brockman, A., McGready, R., Ashley, E.,Phaipun, L., Patel, R., Laing, K., Looareesuwan, S., White, N. J., Nosten, F.,and Krishna, S. (2004) Mefloquine resistance in Plasmodium falciparumand increased pfmdr1 gene copy number. Lancet 364, 438–447
9. Nair, S., Miller, B., Barends, M., Jaidee, A., Patel, J., Mayxay, M., Newton,P., Nosten, F., Ferdig, M. T., and Anderson, T. J. (2008) Adaptive copynumber evolution in malaria parasites. PLoS Genet. 4, e1000243
10. Hill, D. A., Pillai, A. D., Nawaz, F., Hayton, K., Doan, L., Lisk, G., andDesai,S. A. (2007) A blasticidin S-resistant Plasmodium falciparummutant witha defective plasmodial surface anion channel. Proc. Natl. Acad. Sci. U.S.A.104, 1063–1068
11. Lisk, G., Pain, M., Sellers, M., Gurnev, P. A., Pillai, A. D., Bezrukov, S. M.,and Desai, S. A. (2010) Altered plasmodial surface anion channel activityand in vitro resistance to permeating antimalarial compounds. Biochim.Biophys. Acta 1798, 1679–1688
12. Lisk, G., Pain, M., Gluzman, I. Y., Kambhampati, S., Furuya, T., Su, X. Z.,Fay, M. P., Goldberg, D. E., and Desai, S. A. (2008) Changes in the plasmo-dial surface anion channel reduce leupeptin uptake and can confer drugresistance in P. falciparum-infected erythrocytes. Antimicrob. AgentsChemother. 52, 2346–2354
13. Nguitragool, W., Bokhari, A. A., Pillai, A. D., Rayavara, K., Sharma, P.,Turpin, B., Aravind, L., andDesai, S. A. (2011)Malaria parasite clag3 genesdetermine channel-mediated nutrient uptake by infected red blood cells.Cell 145, 665–677
14. Edgar, R. C. (2004) MUSCLE. Multiple sequence alignment with highaccuracy and high throughput. Nucleic Acids Res. 32, 1792–1797
15. Ronquist, F., and Huelsenbeck, J. P. (2003) MrBayes 3. Bayesian phyloge-netic inference under mixed models. Bioinformatics 19, 1572–1574
16. Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S.(2011)MEGA5.Molecular evolutionary genetics analysis usingmaximumlikelihood, evolutionary distance, and maximum parsimony methods.Mol. Biol. Evol. 28, 2731–2739
17. Yang, Z. (2007) PAML 4. Phylogenetic analysis by maximum likelihood.Mol. Biol. Evol. 24, 1586–1591
18. Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992) The rapid genera-tion of mutation data matrices from protein sequences. Comput. Appl.Biosci. 8, 275–282
19. Livak, K. J., and Schmittgen, T. D. (2001) Analysis of relative gene expres-sion data using real-time quantitative PCR and the 2(�[/Delta][/Del-ta]CT) method.Methods 25, 402–408
20. Llinás, M., Bozdech, Z., Wong, E. D., Adai, A. T., and DeRisi, J. L. (2006)Comparative whole genome transcriptome analysis of three Plasmodiumfalciparum strains. Nucleic Acids Res. 34, 1166–1173
21. Crowley, V. M., Rovira-Graells, N., Ribas de Pouplana, L., and Cortés, A.
(2011) Heterochromatin formation in bistable chromatin domains con-trols the epigenetic repression of clonally variant Plasmodium falciparumgenes linked to erythrocyte invasion.Mol. Microbiol. 80, 391–406
22. Balu, B., Shoue, D. A., Fraser, M. J., Jr., and Adams, J. H. (2005) High-efficiency transformation of Plasmodium falciparum by the lepidopterantransposable element piggyBac. Proc. Natl. Acad. Sci. U.S.A. 102,16391–16396
23. Wickham, M. E., Thompson, J. K., and Cowman, A. F. (2003) Character-isation of themerozoite surface protein-2 promoter using stable and tran-sient transfection in Plasmodium falciparum. Mol. Biochem. Parasitol.129, 147–156
24. Lyko, B., Hammershaimb, E. A., Nguitragool, W., Wellems, T. E., andDesai, S. A. (2012) A high-throughput method to detect Plasmodium fal-ciparum clones in limiting dilution microplates.Malar. J. 11, 124
25. Wagner, M. A., Andemariam, B., and Desai, S. A. (2003) A two-compart-ment model of osmotic lysis in Plasmodium falciparum-infected erythro-cytes. Biophys. J. 84, 116–123
26. Pillai, A. D., Pain, M., Solomon, T., Bokhari, A. A., and Desai, S. A. (2010)A cell-based high-throughput screen validates the plasmodial surface an-ion channel as an antimalarial target.Mol. Pharmacol. 77, 724–733
27. Kaneko, O. (2007) Erythrocyte invasion. Vocabulary and grammar of thePlasmodium rhoptry. Parasitol. Int. 56, 255–262
28. Iriko, H., Kaneko, O., Otsuki, H., Tsuboi, T., Su, X. Z., Tanabe, K., andTorii, M. (2008) Diversity and evolution of the rhoph1/clag multigenefamily of Plasmodium falciparum.Mol. Biochem. Parasitol. 158, 11–21
29. Alexandre, J. S., Kaewthamasorn, M., Yahata, K., Nakazawa, S., andKaneko, O. (2011) Positive selection on the Plasmodium falciparum clag2gene encoding a component of the erythrocyte-binding rhoptry proteincomplex. Trop. Med. Health 39, 77–82
30. Desai, S. A. (2012) Ion and nutrient uptake by malaria parasite-infectederythrocytes. Cell Microbiol. 14, 1003–1009
31. Kaneko, O., Tsuboi, T., Ling, I. T., Howell, S., Shirano, M., Tachibana, M.,Cao, Y. M., Holder, A. A., and Torii, M. (2001) The high molecular massrhoptry protein, RhopH1, is encoded by members of the clag multigenefamily in Plasmodium falciparum and Plasmodium yoelii. Mol. Biochem.Parasitol. 118, 223–231
32. Vincensini, L., Fall, G., Berry, L., Blisnick, T., and Braun Breton, C. (2008)TheRhopHcomplex is transferred to the host cell cytoplasm following redblood cell invasion by Plasmodium falciparum. Mol. Biochem. Parasitol.160, 81–89
33. Cortés, A., Carret, C., Kaneko, O., Yim Lim, B. Y., Ivens, A., and Holder,A. A. (2007) Epigenetic silencing of Plasmodium falciparum genes linkedto erythrocyte invasion. PLoS Pathog. 3, e107
34. Comeaux, C. A., Coleman, B. I., Bei, A. K.,Whitehurst, N., andDuraisingh,M. T. (2011) Functional analysis of epigenetic regulation of tandemRhopH1/clag genes reveals a role in Plasmodium falciparum growth.Mol.Microbiol. 80, 378–390
35. Staines, H. M., Rae, C., and Kirk, K. (2000) Increased permeability of themalaria-infected erythrocyte to organic cations. Biochim. Biophys. Acta1463, 88–98
36. Lisk, G., Scott, S., Solomon, T., Pillai, A. D., and Desai, S. A. (2007) Solute-inhibitor interactions in the plasmodial surface anion channel reveal com-plexities in the transport process.Mol. Pharmacol. 71, 1241–1250
37. Bhat, P. J., and Iyer, R. S. (2009) Epigenetics of the yeast galactose geneticswitch. J. Biosci. 34, 513–522
38. Pokholok, D. K., Harbison, C. T., Levine, S., Cole, M., Hannett, N.M., Lee,T. I., Bell, G. W., Walker, K., Rolfe, P. A., Herbolsheimer, E., Zeitlinger, J.,Lewitter, F., Gifford, D. K., and Young, R. A. (2005) Genome-wide map ofnucleosome acetylation and methylation in yeast. Cell 122, 517–527
39. Chookajorn, T., Dzikowski, R., Frank, M., Li, F., Jiwani, A. Z., Hartl, D. L.,and Deitsch, K. W. (2007) Epigenetic memory at malaria virulence genes.Proc. Natl. Acad. Sci. U.S.A. 104, 899–902
40. Lopez-Rubio, J. J., Gontijo, A. M., Nunes, M. C., Issar, N., HernandezRivas, R., and Scherf, A. (2007) 5� Flanking region of var genes nucleatehistone modification patterns linked to phenotypic inheritance of viru-lence traits in malaria parasites.Mol. Microbiol. 66, 1296–1305
41. Lukacs, G. L., and Verkman, A. S. (2012) CFTR. Folding, misfolding andcorrecting the �F508 conformational defect. TrendsMol. Med. 18, 81–91
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
JULY 5, 2013 • VOLUME 288 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 19439
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from

42. Rug, M., Wickham, M. E., Foley, M., Cowman, A. F., and Tilley, L. (2004)Correct promoter control is needed for trafficking of the ring-infectederythrocyte surface antigen to the host cytosol in transfected malaria par-asites. Infect. Immun. 72, 6095–6105
43. Kyes, S., Christodoulou, Z., Pinches, R., and Newbold, C. (2002) Stage-specific merozoite surface protein 2 antisense transcripts in Plasmodiumfalciparum.Mol. Biochem. Parasitol. 123, 79–83
44. Balu, B., Chauhan, C., Maher, S. P., Shoue, D. A., Kissinger, J. C., Fraser,M. J., Jr., andAdams, J. H. (2009) piggyBac is an effective tool for functionalanalysis of the Plasmodium falciparum genome. BMCMicrobiol. 9, 83
45. Freitas-Junior, L. H., Hernandez-Rivas, R., Ralph, S. A.,Montiel-Condado,D., Ruvalcaba-Salazar, O. K., Rojas-Meza, A. P., Mâncio-Silva, L., Leal-Silvestre, R. J., Gontijo, A. M., Shorte, S., and Scherf, A. (2005) Telomericheterochromatin propagation and histone acetylation control mutuallyexclusive expression of antigenic variation genes in malaria parasites. Cell121, 25–36
46. Jiang, L., López-Barragán, M. J., Jiang, H., Mu, J., Gaur, D., Zhao, K.,Felsenfeld, G., and Miller, L. H. (2010) Epigenetic control of the variableexpression of a Plasmodium falciparum receptor protein for erythrocyteinvasion. Proc. Natl. Acad. Sci. U.S.A. 107, 2224–2229
47. Malmquist, N. A., Moss, T. A., Mecheri, S., Scherf, A., and Fuchter, M. J.(2012) Small-molecule histone methyltransferase inhibitors display rapidantimalarial activity against all blood stage forms in Plasmodium falcipa-rum. Proc. Natl. Acad. Sci. U.S.A. 109, 16708–16713
48. Martínez, A., Rajapakse, C. S., Jalloh, D., Dautriche, C., and Sánchez-Del-gado, R. A. (2009) The antimalarial activity of Ru-chloroquine complexesagainst resistant Plasmodium falciparum is related to lipophilicity, basic-ity, and heme aggregation inhibition ability near water/n-octanol inter-faces. J. Biol. Inorg. Chem. 14, 863–871
49. Haynes, R. K. (2006) From artemisinin to new artemisinin antimalarials.Biosynthesis, extraction, old and new derivatives, stereochemistry andmedicinal chemistry requirements. Curr. Top. Med. Chem. 6, 509–537
50. Ritchie, T. J., Macdonald, S. J., Young, R. J., and Pickett, S. D. (2011) Theimpact of aromatic ring count on compound developability. Further in-sights by examining carbo- and hetero-aromatic and -aliphatic ring types.
Drug Discov. Today 16, 164–17151. Rasti, N.,Wahlgren,M., and Chen, Q. (2004)Molecular aspects ofmalaria
pathogenesis. FEMS Immunol. Med. Microbiol. 41, 9–2652. Ralph, S. A., and Scherf, A. (2005) The epigenetic control of antigenic
variation in Plasmodium falciparum. Curr. Opin. Microbiol. 8, 434–44053. Pillai, A. D., Nguitragool, W., Lyko, B., Dolinta, K., Butler, M.M., Nguyen,
S. T., Peet, N. P., Bowlin, T. L., and Desai, S. A. (2012) Solute restrictionreveals an essential role for clag3-associated channels in malaria parasitenutrient acquisition.Mol. Pharmacol. 82, 1104–1114
54. Teuscher, F., Gatton, M. L., Chen, N., Peters, J., Kyle, D. E., and Cheng, Q.(2010) Artemisinin-induced dormancy in Plasmodium falciparum.Dura-tion, recovery rates, and implications in treatment failure. J. Infect. Dis.202, 1362–1368
55. Pillai, A. D., Addo, R., Sharma, P., Nguitragool, W., Srinivasan, P., andDesai, S. A. (2013) Malaria parasites tolerate a broad range of ionic envi-ronments and do not require host cation remodeling.Mol. Microbiol. 88,20–34
56. Trenholme, K. R., Gardiner, D. L., Holt, D. C., Thomas, E. A., Cowman,A. F., and Kemp, D. J. (2000) clag9. A cytoadherence gene in Plasmodiumfalciparum essential for binding of parasitized erythrocytes to CD36. Proc.Natl. Acad. Sci. U.S.A. 97, 4029–4033
57. Ling, I. T., Florens, L., Dluzewski, A. R., Kaneko, O., Grainger, M., YimLim, B. Y., Tsuboi, T., Hopkins, J. M., Johnson, J. R., Torii, M., Bannister,L. H., Yates, J. R., 3rd, Holder, A. A., and Mattei, D. (2004) The Plasmo-dium falciparum clag9 gene encodes a rhoptry protein that is transferredto the host erythrocyte upon invasion.Mol. Microbiol. 52, 107–118
58. Goel, S., Valiyaveettil, M., Achur, R. N., Goyal, A., Mattei, D., Salanti, A.,Trenholme, K. R., Gardiner, D. L., and Gowda, D. C. (2010) Dual stagesynthesis and crucial role of cytoadherence-linked asexual gene 9 in thesurface expression of malaria parasite var proteins. Proc. Natl. Acad. Sci.U.S.A. 107, 16643–16648
59. Wanamaker, C. P., Christianson, J. C., andGreen,W.N. (2003) Regulationof nicotinic acetylcholine receptor assembly. Ann. N.Y. Acad. Sci. 998,66–80
Epigenetic Marks Reduce Erythrocyte Uptake of Antimalarials
19440 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 27 • JULY 5, 2013
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Eli Moss, Wang Nguitragool, Daniel Neafsey and Sanjay A. DesaiParesh Sharma, Kurt Wollenberg, Morgan Sellers, Kayvan Zainabadi, Kevin Galinsky,
Linked to Nutrient UptakeAn Epigenetic Antimalarial Resistance Mechanism Involving Parasite Genes
doi: 10.1074/jbc.M113.468371 originally published online May 28, 20132013, 288:19429-19440.J. Biol. Chem.
10.1074/jbc.M113.468371Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/288/27/19429.full.html#ref-list-1
This article cites 59 references, 15 of which can be accessed free at
by guest on April 25, 2020
http://ww
w.jbc.org/
Dow
nloaded from









![THE CONGRl:ENCE OF TWO PROGRAMMING LANGUAGE …This proof provides an introduction to the techniques developed loy Milne [8, 91, andused by bimandStracbey19] toJlrove th.: correctnessof](https://img.pdfslide.net/doc/110x75/60e42a9c7f183a545b3171d5/the-congrlence-of-two-programming-language-this-proof-provides-an-introduction.jpg)


















