Embed Size (px)
Citation preview

Olivier Henri-Rousseau I Application of the Frontier Molecular and Fernand Texier
lnstitut de Chimie Universite d'0ran I Orbital Theory to the Interpretation
Algerie of the hammett Correlations The Hammett correlations (eqn. (1)) are generally ex-
plained using the concept of partial charge stabilization ( I ) . 1'
log kj = log ho + pcj (1)
However, this concept does not allow an explanation of the chemical reactivity; it is for instance of no use in the expla- nation of regioselectivity and reactivity observed in concerted cycloadditions (2); furthermore, it is not fundamental in physics. Repartition of charges is a consequence of variations in energy, through the variation principle.
Our purpose is to show how a physical explanation of the Hammett relationship may be proposed in the framework of a frontier orbital method (3) which has provided a basis for explaining several aspects of chemical reactivity (4).
General Concepts Used In Molecular Orbital Theory When two atomic orbitals (AO's) of different atoms overlap
they lead to molecular orhitals (MO's). By using the super- position principle, the MO's may be written as linear combi- nations of AO's 9 For two overlapping AO's we have
In agreement with the variational principle, coeffir~entso and b me those which lead m a minimum energ). k.' for the MO. The use of the variational principle leads to the set of simul- taneous equations (3) when the overlap integral is neglected (5)
o(E, - E) + bHij = 0 (3)
oH;; + b(E, - E l = 0
E; and E j are the and rp, energies before interaction; Hij is the interaction Hamiltonian between q; and q,.
The interaction leads to one stabilized bonding MO and one destabilized antibonding MO (see Fig. 1). The stabilization energy AE in this degree of approximation is equal to the destabilization AE*.
a2 and b2 are the probabilities of finding the electrons de- scribed by J. either on the A 0 w or on qj; of course the nor- malization condition leads to a2 + b2 = 1.
By rearranging both eqns. (3) we obtain for the bonding MO
lnteractlng MO's and Second-Order Perlurbatlon Energy When two mulecules are nppr<gaching one ancdher, at the
onset of the reaction. t h e ~ r MO's h e ~ i n to interact. When the difference (Ei - E,) between the interacting
MO's rpj and rp, is greater than 4 H$ the interaction energy AE may he approximated by eqn. (5) (3)
m - A (Ei - E,)
( 5 )
In the general treatment of two interacting molecules R and S, all the interactions between one MO of R and any other of S may he a priori considered.
But as the difference (Ei - Ei) is in the denominator, it is
role in molecular interactions, which is analogous to that of valence AO's in molecules.
In the FMO approximation, only the nucleophile HOMO and the electrophile LUMO which are closest in energy are taken into account (3).
Relations Between Chemical Reactivity and FMO Energies Let us consider chemical transformations in which the ki-
netic step is a himolecular reaction between entities R and S; a t the beeinnine of the reaction. when R and S are aooroach- ing, t h e i r - ~ 0 ~ 3 ' s and LUMO'S interact; this lead; fo a sta- bilization enerw. which mav be evaluated hv eon. (5) even for .." . . . . . charged species.
Charged s~ec ies have hieher HOMO's and LUMO's. when - . they hear negative charges,and lower HOMO's and LUMO's, when thev hear ~osit ive ones: the stabilization enerev is therefweireater when ~harged nucleophile and rlrctro;;hilc are involved (I?!. This stah~liziiti~n incrraw may hc rimsidered as the quantum reflection of the classic couiombic interac- tion.
If we compare, for instance, the reaction of two different electrophiles SI and S2 with the same nucleophile R, it is then possible to assume a relation between the stabilization ener- gies and the reaction rates; the difference in the stabilization energies AE may he considered as reflecting the future dif- ference in the activation energies, as illustrated in Figure 2.
In such a consideration, there is the implicit assumption that the activation energy is a result of a difference between a constant repulsive term and a variable attractive one whose perturbation energy (eqn. (5)) is the prefiguration a t the be- ginning of the reaction. The hypothesis is therefore that the stronger the perturhation energy AE, the smaller the activa- tion enerev.
When ice properties of the substrate are slightly changed in suhstituents. the ereatest Dart of the variation in the oer- . turbation energy, following from interaction with a same re- agent, depends on the variations in the FMO energies. H3 may he considered as constant toward (E, - Ej) in eqn. (5).
If the gap between the HOMO of the nucleophile R and the LUMO of the electrophile S decreases, when the properties of S are modified, the perturhation energy AE increases and the reaction rate K2 is enhanced.
Some recent papers show that there are good linear rela- tionships between the logarithm of the reaction rate Ka and the inverse of the FMO gap (26, 7,8)
obvious that thv interactions he'lween the frontier molecular log K z = - A T X urhitals (FMO's), i.e. the highest orrupied (HO\fO) anrl the E - E , lowest unoccupied (1.111\101 MO'sof R and S, play aprivileged where A and R are experimmrul constanti.
Volume 55. Number 7, J L ! ~ 1978 1 437
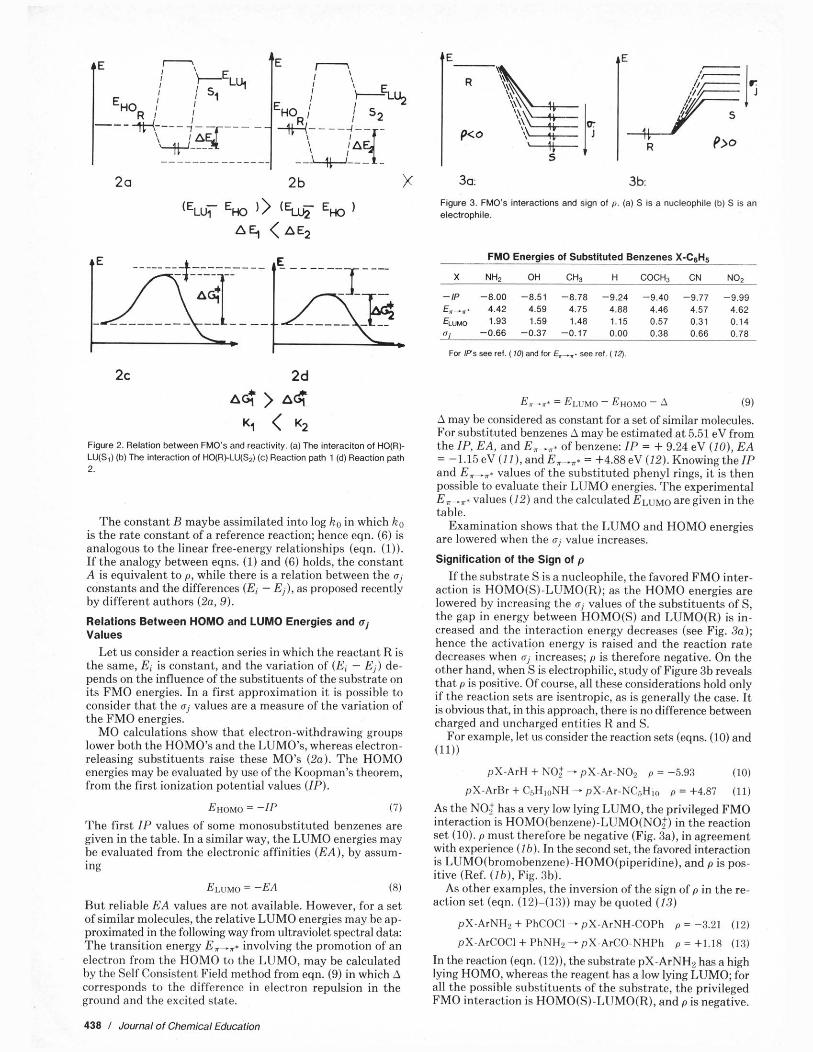
Figure 2. Relation between FMO's and reactivity. (a) The interacitan of HO(R)- LU(S,) (b) The interaction of HO(RI.LU(S2) (c) Reaction path 1 (d) Reaction path 2.
The constant R maybe assimilated into log ko in which ko is the rate constant of a reference reaction; hence eqn. (6) is analogous to the linear free-energy relationships (eqn. (1)). If the analogy between eqns. (1) and ( 6 ) holds, the constant A is equivalent top , while there is a relation hetween the aj constants and the differences (Ej - Ej), as proposed recently by different authors (2a, 9).
Relations Between HOMO and LUMO Energies and a! Values
Let us consider a reaction series in which the reactant R is the same, Ej is constant, and the variation of (Ej - Ej) de- pends on the influence of the substituents of the substrate on its FMO energies. In a first approximation it is possihle to consider that the cj values are a measure of the variation of the FMO energies.
MO calculations show that electron-withdrawing groups lower both the HOMO'S and the LUMO's, whereas electron- releasing substitnents raise these MO's (20). The HOMO energies may be evaluated by use of the Koopman's theorem, from the first ionization potential values (IP).
E H O M ~ = -1P (7)
The first IP values of some monosubstituted benzenes are given in the table. In a similar way, the LUMO energies may be evaluated from the electronic affinities (EA), by assum- ing
ELUMO = -EA (8)
But reliable EA values are not available. However, for a set of similar molecules, the relative LUMO energies may be ap- proximated in the following way from ultraviolet spectral data: The transition energy E,+ involving the promotion of an electron from the HOMO to the LUMO, may he calculated by the Self Consistent Field method from eqn. (9) in which A corresponds to the difference in electron repulsion in the ground and the excited state.
Figure 3. FMO's interactions and sign of p. (a) S is a nucieophiie (b) S is an slectrophile.
FMO Energies of Substituted Benzenes X-C.H, -- -.
X NH, OH CHS H COCH* CN NOz
-IP -8.00 -8.51 -8.78 -9.24 -9.40 -9.77 -9.99 E,-,. 4.42 4.59 4.75 4.88 4.46 4.57 4.62 E L ~ O 1.93 1.59 1.48 1.15 0.57 0.31 0.14
-0.66 -0.37 -0.17 0.00 0.38 0.66 0.78
For IPS see ref. ( 1 4 and for E,,. see ref. (12).
E,-+ = EI.UMO - EHOMO - A (9)
A may be considered as constant for a set of similar molecules. For substituted benzenes A may be estimated a t 5.51 eV from the IP, EA, and E, .,* of henzene: I P = + 9.24 eV (lo), EA = -1.15eV ( I I ) , and Ex-,* = +4.88eV (12). Knowing t h e I P and E,-.,. values of the substituted phenyl rings, it is then possible to evaluate their LUMO energies. The experimental E,-.,a values (12) and the calculated E L U M ~ are given in the table.
Examination shows that the LUMO and HOMO energies are lowered when the oJ value increases.
Signilication of the Sign of p If the substrate S is a nucleophile, the favored FMO inter-
action is HOMO(S)-LUMO(R); as the HOMO energies are lowered by increasing the oJ values of the substituents of S, the gap in energy hetween HOMO(S) and LUMO(R) is in- creased and the interaction energy decreases (see Fig. 3a); hence the activation energy is raised and the reaction rate decreases when o, increases; p is therefore negative. On the other hand, when S is electrophilic, study of Figure 3b reveals that p is positive. Of course, all these considerations hold only if the reaction sets are isentroplc, as is generally the case. I t is ohvious that, in this approach, there is nodifference between charged and uncharged entities R and S.
For example, let us consider the reaction sets (eqns. (10) and (11))
pX-ArH + NO: - pX-Ar-NO2 p = -5.93 (10)
pX-ArRr + CSHIONH - pX-Ar-NC5Hlo P = f4.87 (11)
As the NO: has a very low lying LUMO, the privileged FMO interaction is HOMO(henzene)-LUMO(N0:) in the reaction set (10). p must therefore be negative (Fig. 3a), in agreement with experience ( I h). In the second set, the favored interaction is LUMO(bromohenzene)-HOMO(piperidine), and p is pos- itive (Ref. ( l b ) , Fig. 3h).
As other examples, the inversion of the sign of p in the re- action set (eqn. (12)-(13)) may be quoted (13)
pX-ArNH, + PhCOCI - pX-ArNH-COPh p = -3.21 (12)
pX-ArCOCI + PhNHz - pX-ArCO-NHPh p = tI.18 (13)
In the reaction (eqn. (1 2)), the substrate pX-ArNH2 has a high lying HOMO, whereas the reagent has a low lying LUMO; for all the possihle suhstit,uents of the substrate, the privileged FMO interaction is HOMO(S)-LUMO(R), and p is negative.
438 1 Journal of Chemical Education


PX- ArCOMe + RH, - p X- Ar-CH-Me P= t3 .06 (22)
I OH
pX- ArCOMe + NHiOH - pX-Ar-CMe-NHOH p= +0.32 (23)
I I OH
This leads to a greater p value for the first reaction type (14); the transition state is therefore nearer the product in reaction (22) than in (23). This MO consideration agrees with the classic conclusions (14).
Explanation of Solvent Effects
I t is ohvious that in the MO explanation of the Hammett correlations, the stabilization of partial charges in the tran- sition state does not nlav an important role. But in these tmditions the, solvent t:ffcrts may he explained.
I n thc cliis;ic explani~tinn, thr solvent plays a role hy sul- vating ionir charges involved in the reaction. Rut thestrungent rr i t i~ism of the usr uf thesolvcnt polaritv is that it does not raprcw the sprcific nature of interartions hetween solvent rnol tdec and 9011111~ (151. Fur Hammett rorrelatiuns a nar- rowing of the FMO's hy wlwnt effect u,ill Increase the mag- nitude of 0. while, an incrvase uf the mwn FMO gap will lead - . to smalle; p values (16).
Comparisons Between the Classic Approach and the MO Approach
In the cldssic and the nra. treatment of the reactivity there is a net tlvw of elwtronic chames frum the nucleo~hik to the electrophile. Nevertheless there are differences between the two explanations of the Hammett relationships about the significance of aj and p values, and the influence of substitu- ents on the charge separation in the transition state.
oj measures the polar effects of the substituents in the classic treatment and the lowering or the raising of the FMO energies in the MO approach. In the old explanation p is a measure of the susce~tibilitv of a reaction set to polar effects, whereas in the new it reflects thvsensitivity ;,f the charge transier. The inflocnr~:ofsuhstiturnts on the chargesepara- tion in the transition state is furthermore not o f t h e same nature in the two approaches.
Let us consider for example the reactions of benzoylchlo- rides and anilines
CI
6 - I Ar-NH, + Ar-C-CI -+ Ar-NH,---C-Ar -+
II Il
Classicallv the smallest charge separation occurs in the reac- .. . tion ofp-dimethylaminoaniline and I,-nitrobenzoylrhl~,ri~le while the crrntwt ic in\wlved in that o i p-nitmanilink ilnd - p-dimethylamino beuzoyl chloride.
In the MO treatment different results are obtained: the strongest charge transfer and thus the greatest charge sepa- ration is involved in the reaction of p-dimethylaminoaniline and p-nitrohenzoylchloride since the gap in the FMO's is here the smallest.
However there is no contradiction between the two expla- nations about the charge separation in the transition state because each approach takes into account one of the phe- nomena which act in the opposite way.
Another difference between the two exnlanations is that in the classic approach the discussions of the influence of suh- stituents in the transition state concern only the reacting sites,
Figure 5. Change of Ik favored FMO interaction with increasing c, values.
whereas, in the MO treatment, the substrate and the reagent are globally considered. In this last approach the driving force of the reaction is the charge transfer interaction (eqn. (5)) from one reactant to the other. But as this transfer is aided by electron-withdrawing groups on the electrophile and elec- tron-releasina substituents on the nucleonhile. there is no cont~adictio~hetween the conclusions of the classic and the new treatment concerning the reactivity.
Devlatlons from the Linear Hammett Relatlonshlps Deviations from linear Hammett relationships are generally
explained by change of mechanism (13). If downward curves cannot he explained by the FMO method, on the other hand, it is possible to understand upward curves in this framework without using a change of mechanism, when both the FMO interactions are not verv different.
For very nrgative n, valuei, the privileged FMO interaction is HOMO(S1-LIIMOtRj and o is negative. On theother hand for very positive uj values the reaction is governed hy the LUMO(S)-HOMO(R) interaction and p is positive (see Fig. 5). ~,
Such explanations have been used for particular 13-dipolar eycloadditions involving non-linear Hammett relations (17, 18); for instance the upward curve observed in the cycload- ditions of benzonitrile oxides to substituted aryl alkynes is related to an inversion in the charge transfer from the dipole to the dipolarophile. The henzonitrile oxide is electrophilic toward the arylacetylenes substituted by electron-releasina groups, and nucleophilic toward those hearing electron- withdrawing substituents; but for all the awlacetylenes the two bond makings may he considered as concerted in the same way (18).
Conclusion The rough MO treatment that we have exposed leads to a
physical explanation of the linear energy relationships without using the concept of charge stahilization. In this framework aj i s a constant measuring the lowering or the raising of the FMO energies, while p is a measure of the sensitivity to the substituents of the charge transfer involved in a reaction set.
When o is small the charee transfer is small and sliehtlv u ,
sensitive to the substituents; the trailsition state may Ilr thus considered as reactant-like. On the contrary if p is great the chargv transfer is great and vrry much influencrd hy the suhstituents; the transition state is product-like.
Nnn-linear upward frer rnrrgy relationships may he t x - plnined hy an inversion of the privileged FMO interaction without fundamental change in the mrchaniam.
These explanations fullou, from the variational and sur~er- position principles in a similar way to the explanation oi the chemical bonding. These analogies reveal the beauty of chemistry.
The authors are grateful to Professor G. Lamaty, Drs. M. Santelli and J. Bastide for helpful discussions and suaaes-
440 1 Journal of Chemical Educafion

Literature Cited
(1) (a) Hammett, L. P.. "Physical Orgsnie Chemistry: 2nd Ed., McCraw-Hill. New York. 1970 ,~ . I sndsq. (h) Jsffe, H. H..Chrm. Re", 53.191 (19531.
(2) (a) Sudmsnn, R.. Pure Appl Chrm., 46,669 (1974). (bl Hwk, K. N., Aerounts Chem. Ras., 8.361 11975). (c) Hcnri-Rousseau. 0.. and Baatide, J., in "The Chemistry of Aikynos," Pats! S., in press.
(31 Fukui. K.;'Theoryof Orientation and Stenoseleciion." Springer Verlag. Heidelben, 1970. p. 1 and $4.
(4) Fleming, I.. "Fmntie. OrhiW~ end OrgsnieChemical Reactions: John Wiley end Som, London. 1976, p. I and sq.
15) .lafie. H. H.. and Orchin. M.."Snnmetn.OrhiWs and S~eetra:'John Wiley sndSons.
(a) Desimoni, G., Gamba, A,. Monticdli, M.. Nieola. M., and Tecconi, G., J. Amer. Chsm.. Sm., 98,3947 (1976). (h) Loudon, G. M.. and Berke C., J. Amen Chsm. Sor., 96.1508 (1974).
Msier, J., and Turner, W., J. Chem. Soe. Trons. For 11,521 (19731. Burrow, P. P.. Miehejha, J. A , and Jordan. K. D.. J. Amer Chem. Soc., 98. 6392
(1976). Reo, C. N. R.."UV end Visible SpeNoscopy." Butterworth. London, 1975, p. 62. Shoner, J., "Corrslation snslysb in Organic Chemistry: Oxford. 1913, p. 20. (a) Genets, P.. and Lamsty, C.,Rull. Sac. Chim. F,., 669 (19681. (b) Genestp, P.. la-
rnsty,G.,andRoque, J. P.,RPc. TIOY. Chim.. 91,188 11972). Doughorty R. C., Tetrahedron Lett., 385 (1975). Blaise. P., and Henri-Rousseau. 0.. C R. Aeod Sci. FI, C285.125 (1977). SusfmannR.. TefrahedronL~tf.. 963U974). Bartida, J., Henri-Raussesu, 0.. and Steohsn, E., C R. Arod Sci. Fr.. C 278, 195
(1974).
Volume 55. Number 7, July 1978 1 441





























