Embed Size (px)
Citation preview

at SciVerse ScienceDirect
Tetrahedron 68 (2012) 6781e6802
Contents lists available
Tetrahedron
journal homepage: www.elsevier .com/locate/ tet
Tetrahedron report number 980
Applications of enzymatic and non-enzymatic methods to accessenantiomerically pure compounds using kinetic resolution and racemisation
Marwa Ahmed a, Tamsin Kelly b, Ashraf Ghanema,*
aChirality Program, Faculty of Applied Science, University of Canberra, Australian Capital Territory (ACT), AustraliabNational Centre for Forensic Studies, Faculty of Applied Science, University of Canberra, ACT, Australia
a r t i c l e i n f o
Article history:Received 9 May 2012Available online 23 May 2012
* Corresponding author. E-mail address: ash(A. Ghanem).
0040-4020/$ e see front matter Crown Copyright �doi:10.1016/j.tet.2012.05.049
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67812. Kinetic resolution: general . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6782
2.1. Kinetic resolution using lipases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67822.1.1. Determination of absolute configuration of secondary alcohols using lipase-catalysed KR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67832.1.2. Factors affecting activity and selectivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67842.1.3. Strategies for controlling and enhancing enantioselectivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67842.1.4. Examples of lipase-catalysed reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6785
3. Deracemisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67873.1. Stepwise racemisation (cyclic deracemisation) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6787
3.1.1. Deracemisation reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67883.2. In situ racemisation (dynamic kinetic resolution) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6790
3.2.1. Enzyme-catalysed racemisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67913.2.2. Non-enzyme-catalysed racemisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67913.2.3. Organocatalysts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6796
4. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6798References and notes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6798Biographical sketch . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6801
1. Introduction
The augmented awareness of the importance of chirality and itsstrong associationwith biological activity created an immense needfor development of enantiomerically pure compounds at a reducedcost. The global sales of chiral technology products in 2008were 4.3billion dollars, which increased to 4.5 billion dollars in 2009(representing 3.3% growth). The market is expected to expand ata compound annual growth rate (CAGR) of 2.8% to reach 5.1 billion
2012 Published by Elsevier Ltd. All
dollars by 2014.1 Various methods have been reported to accessenantiomerically pure compounds including synthesis from chiralpool, asymmetric synthesis from prochiral substrates and resolu-tion of racemic mixtures2 (Fig. 1).
Among these methods, the enzyme-catalysed transformationsto obtain enantiomerically pure compounds have become in-creasingly considered in the manufacture of a wide range of in-termediates in the pharmaceutical, agrochemical, fine-chemicaland food industry. Nowadays, it is replacing chemical catalysisto overcome the disadvantages of undesired by-products, toxiceffluents and poor substrate selectivity.
For many years, commercial enzyme suppliers, such as Novo-zyme, Amano and Roche have supplied the market with an
rights reserved.

Fig. 1. Different methods to access enantiomerically pure compounds.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026782
economical source of hydrolases, and the new generation of en-zyme suppliers, such as Diversa, Biocatalytics, Cambrex, Julich En-zyme Products, Combinature and others have expanded thespectrum of enzyme platforms to include nitrilases, epoxide hy-drolases, transaminases, p450 enzymes, halogenases, ketor-eductases and aldolases, among others. As more and more genomicDNA sequences become available, alongwith the use of non-natural
Fig. 2. Classical kinetic resolution.
enzyme libraries generated from protein evolution using ePCR,gene-shuffling and gene reassembly, it is expected that this spec-trum will continue to grow rapidly.2
As a result of improved understanding of biochemistry, fer-mentation techniques and recovery methods as well as the versa-tile transformations that can be catalysed by enzymes, the numberof enzymes utilised in industry has been growing progressively.Among the most popular utilised enzymes, lipases are considereda milestone in the resolution of racemic mixtures. Lipases andracemases have been widely used to synthesise enantiomericallypure compounds via different approaches, including kinetic reso-lution, dynamic kinetic resolution and deracemisation. This reviewsummarises literature over the past five years on the use of en-zymes in kinetic resolution, dynamic kinetic resolution and dera-cemisation of racemic chiral compounds.
2. Kinetic resolution: general
The search for new and efficient methods for the synthesis ofenantiomerically pure organic compounds has been an active area
of research in organic chemistry. To date, resolution of racemates isthe most common industrial approach for the synthesis of enan-tiomerically pure compounds. Kinetic resolution (KR) is defined asa process where the two enantiomers of a racemate are trans-formed into products at different rates. If the KR is efficient, one ofthe enantiomers will be transformed into the desired product whilethe other is recovered unchanged (Fig. 2).3
This procedure has the limitation of having a maximum theo-retical yield of 50%, as only one of the two enantiomers will react.Many efforts have been devoted to circumvent this limitation andto afford compounds with the same high enantiomeric purity, butwith much improved yield. Therefore, classical kinetic resolutionevolved into dynamic kinetic resolution (DKR) where two steps arecombined, a resolution step of the KR with an in situ racemisationof the chirally labile substrate. Hence, all of the substrate can beconverted into a single enantiomer with 100% theoretical yield(Fig. 3).3,4
2.1. Kinetic resolution using lipases
Lipases (triacylglycerol ester hydrolases) are ubiquitous en-zymes ,which belong to the serine hydrolase enzyme family. Theycatalyse hydrolysis of fats and oils to free fatty acids and glycerol.Since the reaction is reversible, they can also catalyse the formationof acylglycerols from free fatty acids and glycerol. Therefore, lipasesare the most commonly used enzymes in synthetic organic chem-istry for hydrolysis of carboxylic acid esters in aqueous solvents or

Fig. 3. Dynamic kinetic resolution.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e6802 6783
the transesterification in organic solvents.5 Lipases do not requirecofactors, can be used either in the free or immobilised form, areinexpensive, readily available and accept a wide range of substrateswhile retaining their high chemo-, regio- and stereo-selectivities.All of these properties render them a useful tool in organic syn-thesis.6 Moreover, lipases work at the lipidewater interface andtherefore they do not require a water-soluble substrate and canwork efficiently in the hydrophobic medium of the organic sol-vents. They are also able to catalyse the reactions under mildconditions (ambient temperature, around neutral pH).
Lipases have been widely used to catalyse three main types ofasymmetric transformation, KR of racemic alcohols and carboxylicacids, enantiotopic group differentiation of meso-diols and meso-dicarboxylic acids and, finally enantiotopic group differentiation ofprochiral dicarboxylic acids and diols (Fig. 4).6
1. Hydrolysis (aqueous medium)
R1 OR2
O
lipase
waterR1 O
O
2. Esterification (organic medium)
R1 OH
O
lip
org.sR2 R3
OH
3. Transesterification a) alcoholysis
R1 OR2
O
lip
org.soR3 R4
OH
b) acidolysis
R1 OR2
O
lip
org.soR3 OH
O
4. Interesterification
R1 OR2
O
lip
org.sR3 OR4
O
Fig. 4. Some reactions catalysed by lipases, including hydrolysis (aqueous medium
To achieve high enantioselectivity in a KR process, the reactionmust be rapid and irreversible. Since the enantiomeric purity of theproduct and the remaining unreacted substrate diminishes as thereverse reaction proceeds, several strategies have been developedto suppress reversibility of the reaction to ensure high enantio-meric purity. The use of certain acyl donors that have the ability tobe converted into volatile products and shift the reaction in onedirection is one of the successful tools. For example, the use of vinylesters or isopropenyl esters produces vinyl or isopropenyl alcohols,respectively, which in turn are tautomerised to the volatile acet-aldehyde or acetone to shift the (trans)esterification reaction for-ward. Moreover, the amount of water in the reaction mixture willdetermine the direction of lipase-catalysed reaction, i.e., underanhydrous conditions, only (trans)esterification is favoured, whilehydrolysis is favoured in an aqueous medium.7
2.1.1. Determination of absolute configuration of secondary alcoholsusing lipase-catalysed KR. Jing and Kazlauskas postulated an em-pirical rule to predict, which enantiomer reacts faster in a lipase-catalysed esterification reaction of a racemic secondary alcohol,based on the relative sizes of the two substituents in the secondaryalcohol.8 X-ray structures of lipases showed that the alcohol bind-ing pocket contains a large hydrophobic pocket accessible to sol-vent and another smaller pocket. Thus, the enantiopreference oflipases allows the determination of absolute configuration of sec-ondary alcohols. The reliability of this method depends on thesimilarity of the unknown molecule to a molecule with an estab-lished absolute configuration in KR. One disadvantage of this ap-proach is that the empirical rule to date only applies to secondary
H
R2OH
ase
olventR1 O
O
H2O
R3
R2
ase
lventR1 O
O
R2OH
R4
R2
ase
lventR3 OR2
O
OH
O
R1
ase
olventR3 OR4
O
OR2
O
R3
), esterification (organic medium), transesterification and interesterification.

M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026784
alcohols. Lipases also catalyse esterification of primary alcohols andcarboxylic acids and the hydrolysis of lactones, although no reliablerules have been reported for these other classes, because appar-ently minor changes in substituents can reverse the enantiopre-ference.9,10 The reliability of the method depends on: (1) a largedifference in the size of the two substituents, so that the rule inFig. 5 can be applied unambiguously, (2) high enantioselectivity inthe KR, showing that the lipase strongly favours one enantiomer,and (3) known enantiopreference of the chosen lipase with similarsecondary alcohols. In acylation reactions, the enantiomer shownin Fig. 5 reacts faster, while, in hydrolysis reactions, the ester of thisenantiomer reacts faster.8
OH
LM
Fig. 5. Empirical rule to predict, which enantiomer of secondary alcohol reacts fasterin lipase-catalysed reactions. M: medium-sized substituent, e.g., methyl; L: largesubstituent, e.g., phenyl.
2.1.2. Factors affecting activity and selectivity. Interfacial activationof lipases occurs at the lipidewater interface, a phenomenon thatcan be traced to the unique structural characteristics of this class ofenzymes.11e13 Lipases contain a helical oligopeptide unit thatshields the active site (called the lid). When the lid interacts witha hydrophobic interface, it undergoes conformational changes insuch away that exposes the active site, providing free access for thesubstrate. The active site is a triad composed of serine, histidine andaspartate. Acyl enzyme complexes are the crucial intermediates inall lipase-catalysed reactions.14,15 There are some factors that affectactivity and selectivity of lipase-catalysed reactions, including thenature of the acylating agent, temperature, pH and solventselection.
As previously mentioned, the use of vinyl acetate as an acyldonor yields the volatile acetaldehyde and renders the reactionirreversible. However, acetaldehyde is known to deactivate certainlipases, e.g., those from Candida rugosa and Geotrichum candidum.Immobilisation of the lipase (Section 2.1.3) may solve the problem,but the use of ethoxyvinyl esters, such as 1-ethoxyvinyl acetate orfuroate constitutes an alternative approach, because they irre-versibly release volatile and non-harmful ethyl acetate, as in thedesymmetrisation of meso-diols (Fig. 6).16
OH
OH EtO O R1
O
lipase
O
OH
R1
O
MeCOOEt
Fig. 6. Use of ethoxyvinyl esters as acyl donors.
When applying lipases in a non-aqueous medium, it is impor-tant to consider the degree of residual water content, because itaffects the activity and stereoselectivity. The removal of tightlybound water can cause thermodenaturation of the enzyme and,therefore, it is not desired. It is reported that enantioselectivity isenhanced upon lowering the temperature at the expense of longerreaction times.17e19 Lipases are active under a broad range of pH
values, with optimum activity around pH 9. Substrate hydrolysis inan acidic medium is poor and totally absent at pH 2.20
Since most organic compounds are hydrophobic in nature, it isnecessary to conduct biocatalytic transformations in non-aqueousmedia. Furthermore, conducting biocatalytic transformations inthe presence of water leads to side reactions, such as hydrolysis,decomposition, racemisation and polymerisation and makes itdifficult to remove water after completion of the reaction. Theselimitations created the need to develop procedures to performbiocatalytic transformations in organic solvents.21This also pro-vides the advantages of better product yield, higher substrate sol-ubility and ease of enzyme recovery after reaction completion.Nevertheless, the use of enzymes in organic solvents sometimesleads to decreased catalytic activities (due to the heterogeneoussystem), reaction rate and enantioselectivity. To overcome the re-duced catalytic activity of lipase in polar organic solvents like di-methyl sulfoxide (DMSO) or dimethylformamide (DMF), changes inthe enzyme surface by chemical methods successfully yieldedbiocatalysts that worked better in nearly anhydrous media.22 Re-cently, the use of lipase in organic solvents has been extended tosupercritical fluids and ionic liquids with tuneable solvent prop-erties, employing an environmentally friendly approach.6,23
2.1.3. Strategies for controlling and enhancing enantioselectivi-ty. Subtle modification of the substrate leads to a change in theenantioselectivity of the reaction. This approach is one of thesimplest methods for obtaining changes in optical purity. In a studyconducted by Stampfer et al., oxidative KR of sec-alcohols withwhole lyophilised cells of Rhodococcus ruber DSM 44541 wasemployed. The enantioselectivity was significantly improved byintroducing a C]C bond adjacent to the alcohol moiety. Moreover,high stereodifferentiation between the E- and Z-configured doublebonds occurred as the Z-isomers were not converted. Similarselectivity-enhancing effects were observed with acetylenicanalogues.24
2.1.3.1. Immobilisation and chemical modification. Despite theinteresting properties of lipases, they may be unstable and mayshow neither optimum activity nor optimum enantioselectivityunder certain conditions. Some of them are relatively expensive.Therefore, the recycling of lipases via their immobilisation on dif-ferent supports could be a possible solution, due to the reduced costalong with the enhanced stability and reuse of the biocatalyst.
Different strategies have been used to immobilise crude lipase,suchas adsorption, linkage, or bondingof the enzyme to an insolublesupport or entrapment of the enzyme in polymeric gels or encap-
sulation.25 Several parameters of the immobilisation support shouldbe considered:mechanical strength, chemical and physical stability,hydrophilicity/lipophilicity and enzyme- loading capacity.26
Another strategy to enhance lipase enantioselectivity andmodify its catalytic properties is via physical or chemical modifi-cations. Coating Candida antarctica lipase B (CaLB) with polymers,such as dextran sulfate enhanced its enantioselectivity in the

M. Ahmed et al. / Tetrahedron 68 (2012) 6781e6802 6785
hydrolysis of mandelic acid methyl ester.27 Similarly, succinylation,hydroxyethylamidation or aminoethylamidation were applied toCaLB with different results.27 Due to the simplicity and short time-consumption of these modifications, these strategies seem to bevery powerful and an easy-to-perform tool to enhance CaLBenantioselectivity. The same strategy was applied through site-directed chemical modification of a lipase from Geobacillus ther-mocatenulatus (BTL2) using tailor-made polymers. Godoy et al. in-corporated 2-pyridyldisulfide-activated polymers in the Cys64 ofBLT2, previously immobilised on CNBr-agarose or glyoxyl-agaroseby a thiol-disulfide exchange. Cys64 is located close to the mov-ing area of the protein, that is, associated with the opening/closingchanges of BTL2 and the introduced polymers seems to modify themobility of these groups, greatly altering BTL2 properties. Thechemically modified BTL2 immobilised preparations were irre-versibly inhibitedmore rapidly than non-modified ones, suggestingthat the modification can favour a higher exposition of the catalyticserine to the medium.28 Another study demonstrated that CaLB canbe stabilised up to 60-fold by treatment with ethylene glycol bis(-succinimidyl succinate) and subsequent immobilisation on CNS-type mesoporous silicate.29 These mesoporous silicates are able toaccommodate the enzyme within their long channel pores, pro-tecting it from external-shear forces or from extreme temperatureor pH. Such conditions are often encountered in industrial pro-cesses and can lead to enzyme destruction.
Interestingly, Pseudomonas cepacia lipase amino groups weremodifiedwith pyromellitic dianhydride, and then transesterification
R
OH MeCOCl
Et3N, DMAPCH2Cl2
R
OAc
solvent
MeOHlipase
R
OAc
R
OH
(S) (R)
OAclipase
solventR
OH
R
OAc
(S) (R)
R =S
N
racemic racemic
Scheme 1. Biotransformation of racemic phenothiazinyl ethanol derivatives and their acetates.
reactions were conducted in n-octane and DMF and compared tounmodified pH-tuned lipase. High initial reaction rates in DMFwere observedwith themodified lipasewhile unmodified pH-tunedlipases showed no activity at all in DMF.22
2.1.3.2. Directed evolution and enzyme libraries. Microbiallipases are widely used in industrial applications, due to their highstability, substrate specificity and lower production costs whencompared to other sources. Moreover, the broad diversity of mi-croorganisms improves their biotechnological importance andjustifies the search for new lipases.30 Filamentous fungi are con-sidered the major lipase producers and are currently the preferredsources, since they produce extracellular lipases, facilitating theextraction from fermentation media.31,32 Furthermore, the use ofdirected evolution can be very helpful to optimise existing lipaseswith respect to desired properties. An interesting example of theinfluence of binding-site engineering and enhanced or reversedenzyme enantioselectivity was reported by Lafaqui�ere et al.33 Thegroup targeted three amino acids (L17, V266 and L287) in Bur-kholderis cepacia lipase (BCL) for mutagenesis aimed at tailoringenzyme enantioselectivity. The three single mutants showed sig-nificantly improved enantioselectivity. After analysis of substratedocking and access trajectories, construction of a further 13 doublemutants was performed. Among them, an outstanding improved
mutant of BCL showed an E value of 178 and 15-fold enhancedactivity, compared to the wild-type BCL.
Engineering of CaLB was also performed to broaden its appli-cations to cover bulky substrates. An in-silico library of 2400 CaLBvariants was built and screened in silico by substrate-imprinteddocking. Eleven enzyme variants were then expressed in Escher-ichia coli BL21 and the hydrolysis of two branched fatty acids wasperformed. Five variants showed an initial increase in activity; themost active variant was purified and further characterised.34
2.1.4. Examples of lipase-catalysed reactions. Hundreds of in-vestigations have been reported on the use of lipases in KR of ra-cemic alcohols, esters and amines. The reliability of KR data ishighly dependent on the accurate determination of the enantio-meric excess (ee) of both substrate and product. This is based uponthe development of a reliable analytical procedure for the simul-taneous baseline separation of both substrate and product in onerun by chromatography.35 The following section discusses litera-ture over the past five years on lipase-catalysed KR reactions.
2.1.4.1. Lipase-catalysed KR of secondary alcohols. Synthesis ofenantiomers of phenothiazinyl ethanol derivatives and their ace-tates was reported by Brem et al. C. antarctica lipase A (CaLA) andCaLB were utilised in acylation of the racemic alcohols and/orenantioselective methanolysis of the corresponding racemic esters(Scheme 1).36 Both (R)- and (S)-enantiomers were obtained at highenantioselectivity (E 200) and with high ee (>99%).
The same group also studied CaLA and CaLB enantiopreferenceover a wide variety of racemic ethyl 3-hydroxy-3-(furyl/thienyl)propanoates. High enantioselectivity (E >200) allowed the sepa-ration of enantiomer at the maximum theoretical yield. Surpris-ingly, they reported the inverted enantiopreference of CaLA,despite its low enantioselectivity (E 14e54). They concluded thatthe behaviour of CaLA and CaLB depends on the substratestructure.37
The use of supercritical fluids instead of organic solvents wasalso studied. Utcz�as et al. utilised supercritical carbon dioxide asa solvent in immobilised CaLB-catalysed KR of trans-2-hydroxycyclohexanecarbonitrile (Scheme 2). The procedure is en-vironmentally friendly and it was successful to achieve the maxi-mum theoretical conversion with high enantioselectivity (E 100)and high ee (99.8% for (1S,2R) and 98% for (1R,2S)).38
OAc
CNOH
**CaLB
trans-
CNOH
trans-
CNOAc
trans-
SS RR
Scheme 2. Enzymatic resolution of trans-2-hydroxycyclohexanecarbonitrile.

M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026786
Additionally, theeffectofdifferent ionic liquidson lipases-catalysedKR of secondary alcohols was studied by another group. Zhou et al.
reported a remarkably enhanced enantioselectivity (E >200) com-bined with nearly complete KR of racemic 1-phenylethanol. Thereported catalyst/ionic liquid system could be recycled several timeswithout reducing the activity and enantioselectivity.39A series of aliphatic and aromatic cyanohydrins were also re-solved in an organic medium using different lipases (Scheme 3).The effect of molecular sieves, different acyl donors, temperatureand variable organic solvents was also studied. Optimised resolu-tion conditions were achieved using lipase PS-30 in the presence of4�A molecular sieves using diethyl ether as solvent, vinyl acetate asacyl donor and at 15 �C with high enantioselectivities (E >200).19
R CN
OH
R CN
OAc
R CN
OH
(S) (R)
OAclipase PS-30
solventracemic
R= X ,,X
Scheme 3. Kinetic resolution of cyanohydrins using lipase PS-30.
The b-blocking drug, betaxolol, was resolved via an efficientchemoenzymatic procedure in 2011.40 A new lipase strain (Rhodo-torula mucilaginosaDQ832198) was used as biocatalyst for the KR ofthe key acetylated intermediates (Scheme 4). Excellent ee (>99%)was obtained under very mild conditions. The biocatalyst is quitestable and could be used several times with little decrease of theresolving ability.
AcO
O NOAc Ac
lipase DQ832198
AcO
O NOH Ac
AcO
O NOAc Ac
HO
O NOH H
O
O NOH H
NaOH
(S)-betaxolol
Br
Scheme 4. Synthesis of (S)-betaxolol via Rhodotorula mucilaginosa DQ832198 lipase.
Similarly, racemic atenolol was resolved by immobilised CaLB inan organic medium. Barbosa et al. immobilised CaLB on Eupergit Cunder different conditions and compared the performance of dif-ferent CaLB biocatalysts. The (R)-enantiomer was preferred in allcases but the performances of the biocatalysts were substantially
different; with high differences in reaction rates, yields and enan-tioselectivities. The results confirmed that the use of differentimmobilisation protocols may be a powerful tool for altering en-zyme properties when used in organic medium.41
2.1.4.2. Lipase catalysedKRofamines andaminoacids. Enantiopureamines are important in the organic synthesis of chiral buildingblocks for pharmaceuticals and agrochemicals.
Piliss~ao et al. studied the influence of pure organic solvents ororganic solvent/ionic liquid mixtures on the CaLB-catalysed KR ofracemic phenylethylamine using different acyl donors.42 The re-sults showed that the choice of medium and acyl donor is crucial inorder to obtain enantiopure products with high enantioselectivity.
Similarly,1-(heteroaryl)ethanamines were resolved via differentlipases, resulting in (R)-acetamides and (S)-amines (Scheme 5).43
This study compared the effect of different lipases and organicsolvents on KR of different heteroaryl amines. CaLB showed the bestresults in the presence of tetrahydrofuran (THF) or tert-butylmethyl ether (TBME), depending on the solubility properties of thestarting materials.
Trans-cycloalkane-1,2-diamines and boron-containing aminesand amides were also resolved in a similar way using CaLB withhigh enantioselectivity (E >200).44,45
Zhao et al. reported the KR of amino acids using the ionic liquid,1-ethyl-3-methylimidazolium acetate. Different DL-amino acidsesters were used in this study, with DL-phenylalanine methyl estershowing the highest ee (>98%). Interestingly, high ee (98.2%) wasalso found in deuterium oxide (D2O) rather than ordinary water.The isotopic effect was explained in terms of protein stabilisationby D2O.46
2.1.4.3. Lipase-catalysed KR of acids. A wide range of racemicnon-steroidal anti-inflammatory drugs (NSAIDs) have been re-
solved through lipase-catalysed KR35,47e52 (Scheme 6). Variouslipases were screened in organic solvents, aqueous media50 or us-ing an alcohol alone,48 to conduct either esterification or hydrolysisreactions. Similarly, a series of 3-arylalkanoic acids was resolvedwith excellent enantiopurity.53

N
X NH2EtOAc
lipase
N
X NH2
(S)
N
X NHAc
(R)X = NH, S,O
Scheme 5. Enzymatic kinetic resolution of racemic amines.
lipase
OH
R COOH
CH3
R COOH
CH3
R
CH3
O
(S)-acid (R)-ester
O
racemic
Scheme 6. General scheme for lipase-catalysed KR of racemic acidic drugs.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e6802 6787
3. Deracemisation
Deracemisation covers reactions in which two enantiomers areinterconverted by a stereoinversion process where a racemate canbe converted into non-racemate with 100% theoretical yield,without a change in the composition of the molecule and withoutintermediate separation of materials. Deracemisation reactionstend to involve redox processes, e.g., interconversion of a chiralsecondary alcohol via the ketone or interconversion of amines viathe corresponding imine (Fig. 7).54
D-amino acid oxidase [H]
R COOH
NH2
R COOH
NH
R COOH
NH2Oxidation Reduction
Fig. 7. Deracemisation of amino acids via amino acid oxidases.
SRfast
I SSslow
SR, SS: substrate enantiomersI: intermediate
back reactionnon selective
Fig. 8. Cyclic deracemisation.
In contrast to KR, where the maximum theoretical yield inachieving an enantiomerically pure product is 50% and the otherenantiomer is considered waste, deracemisation allows the syn-thesis of a single enantiomer from a racemic starting material.
Chemical racemisation techniques depend largely on drasticconditions, such as high temperature, strong acid or base catalysisor via chirally labile intermediates. Consequently, process controlover chemical racemisation is very limited and the development ofside reactions, such as elimination, rearrangement and condensa-tion has set a low ceiling on the commercial utility of this process.55
In contrast, enzymatic racemisation takes place at intrinsicallymilder conditions e.g., ambient temperature, atmospheric pressureand neutral pH, and therefore the tendency for formation of by-products is greatly eliminated.56
Racemases are a small group of enzymes, classified underisomerases, having a great potential to catalyse several reactionsknown to be impossible by the conventional methods. Bio(-catalytic) racemisation becomes very useful when combined withenantioselective transformation to furnish (what is called)deracemisation.
Three main concepts have to be highlighted:
� Stepwise racemisation: after separation of the unwantedunreacted enanatiomer in KR, it is subjected to racemisationand then the racemate is subjected again to KR in a subsequentcycle. Typically, five cycles are required to produce the desiredenantiomer with high yield (95%).57
� In situ racemisation: or dynamic kinetic resolution (DKR)where the undesired unreacted enantiomer is racemised in situ
in the presence of the enantioselective reaction. KR will resultin 50% of the desired enantiomer while the other undesiredenantiomer (50%) will be racemised in situ and subsequentlyresolved. After a while, the slow-reacting substrate enantiomerwill be converted into the desired enantiomerically pureproduct in 100% theoretical and optical yields.58e60
� Auxiliary racemisation where both enantiomers of a racemicstarting material are converted into an achiral product.This delivers the non-chiral product in 100% theoreticalyield.61,62
The next sections will provide more details of the first twoconcepts.
3.1. Stepwise racemisation (cyclic deracemisation)
This process involves an oxidation-reduction sequence whereone of the enantiomers from the racemic starting material isenantioselectively oxidised to a non-chiral intermediate (mostlikely a ketone or imine) and then this intermediate is non-selectively reduced to racemic starting material. After many cy-cles, the starting material should be mainly the unreacted enan-tiomer in 100% theoretical yield and 100% ee.54 The feasibility ofthis process depends on the compatibility of the two reactions(oxidation-reduction) in one pot (Figs. 7 and 8). A combination ofmetal catalysts and biocatalysts has been used.

M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026788
Soda et al. reported the deracemisation of a series of a-hydroxyacids and a-amino acids in good yields and high enantioselectiv-ities (>98%)by combining a-amino or a-hydroxy acid oxidase andsodium borohydride (NaBH4) as a reducing agent.63 However, so-dium borohydride is sensitive to water and easily decomposed atneutral and low pH, requiring a large excess to be added, whichusually leads to rapid inactivation of the oxidase enzyme. Alter-natively, sodium cyanoborohdride is more stable at low-to-neutralpH. Nevertheless, its industrial application is limited because of itstoxicity and its reduced activity, compared to sodium borohydride(Scheme 7).64
NH
HN porcine kidney D-amino acid
oxidase
NaCNBH3(3 equiv)
86% yield99% ee
NH
HN
COOH COOH
Scheme 7. Use of sodium cyanoborohydride as reducing agent.
Moreover, Alexandre et al. utilised an alternative reducing sys-tem, Catalytic transfer hydrogenation (CTH), where the imine isreduced by hydrogen released from a metal or metal complex anda hydrogen source as the reaction proceeds. Different amine bo-ranes were utilised as reducing agents, which possess betterproperties, compared to sodium borohydride and sodium cyano-borohydride (Scheme 8).65
COOH
NH2L-amino acid oxidase
COOH
NH
amine borane
Scheme 8. Deracemisation of DL-leucine with L-amino acid oxidase and amine borane.
3.1.1. Deracemisation reactions.
3.1.1.1. Deracemisation of secondary alcohols. Various microor-ganisms are able to racemise racemic secondary alcohols via twodifferent alcohol dehydrogenases with complementary enantio-specificity. In this regard, Demir et al. reported the deracemisationof racemic benzoin using Rhizopus oryzae ATCC 9363 (Scheme 9).Interestingly, by controlling the pH of the medium, they were ableto control the absolute configuration of the produced enantiomer.Thus, at pH 7.5e8.5, the (R)-enantiomer was the major product(with 75% yield and 97% ee), while at pH 4e5, the (S)-enantiomerwas the predominant product (with 71% yield and 85% ee).66
O
O
R. oryzae
pH 7.5-8.5
O
OH pH 7.5-8.5
R. oryzae
Scheme 9. Deracemisation of be
Similarly, Titu and Chadha were successful in synthesisinga series of enantiomerically pure (S)-alkyl 3-(hetero-2-yl)-3-hydroxypropanoates in yields of up to 75% and high ee (upto 99%) using whole cells of Candida parapsilosis ATCC 7330(Scheme 10).67 Thangavel also reported the synthesis of pure (S)-enantiomers of (3E,5E)-alkyl-2-hydroxy-6-arylhexa-3,5-dienoates(Scheme 11) and alkyl (3E)-4-(hetero-2-yl)-2-hydroxybut-3-enoates using the same Candida species (Scheme 12) in yields ofup to 80% and 99% ee.68,69
Moreover, enantiomerically pure allylic alcohols were success-fully prepared by using whole cells of C. parapsilosis ATCC 7330(Scheme 13). The (R)-enantiomers were prepared in yields of up to79% and high ee (99%).70
Surprisingly, Voss et al. reported simultaneous biocatalytic oxi-dation and reduction through combining molecular oxygen(required for the oxidation step) with a stereoselective reducingagent (alcohol dehydrogenase) and cofactor recycling system. Thismethodwas successful inproducing the (S)-enantiomer in 91% yieldand 99% ee. It was also demonstrated that the opposite enantiomerwas accessible through a related system (Scheme 14).71e73
Another one-pot oxidation/reduction sequence using the samesingle catalyst was reported by Voss et al.,74 where the (R)-alcoholwas inverted to the corresponding (S)-enantiomer using lyophi-lised cells of various Rhodococci spp. They utilised non-selectiveoxidation of the racemic secondary alcohol and stereoselectiveasymmetric reduction of the corresponding ketone. The processresulted in 82% yield and 92% ee (Scheme 15).
Another group reported the use of a ruthenium catalyst for cleanoxidation of secondary alcohol into ketone then pressurisation of
the reaction mixture with hydrogen to reduce the ketone back toalcohol with a good level of enantioselectivity for most of thealcohols examined (Scheme 16).75
3.1.1.2. Deracemisation of carboxylic acids. Deracemisation ofmandelic acid to yield optically pure L-phenylglycine was reportedby Resch et al. The process involved three sequential steps: race-misation, enantioselective oxidation and stereoselctive reductiveamination. Racemisation takes place by mandelate racemase thenoxidation and reduction reactions take place simultaneously asso-ciated with cofactor recycling to give the product at 94% yield and97% ee (Fig. 9).76
H
R. oryzae
pH 4-5
O
OH
nzoin using Rhizopus oryzae.
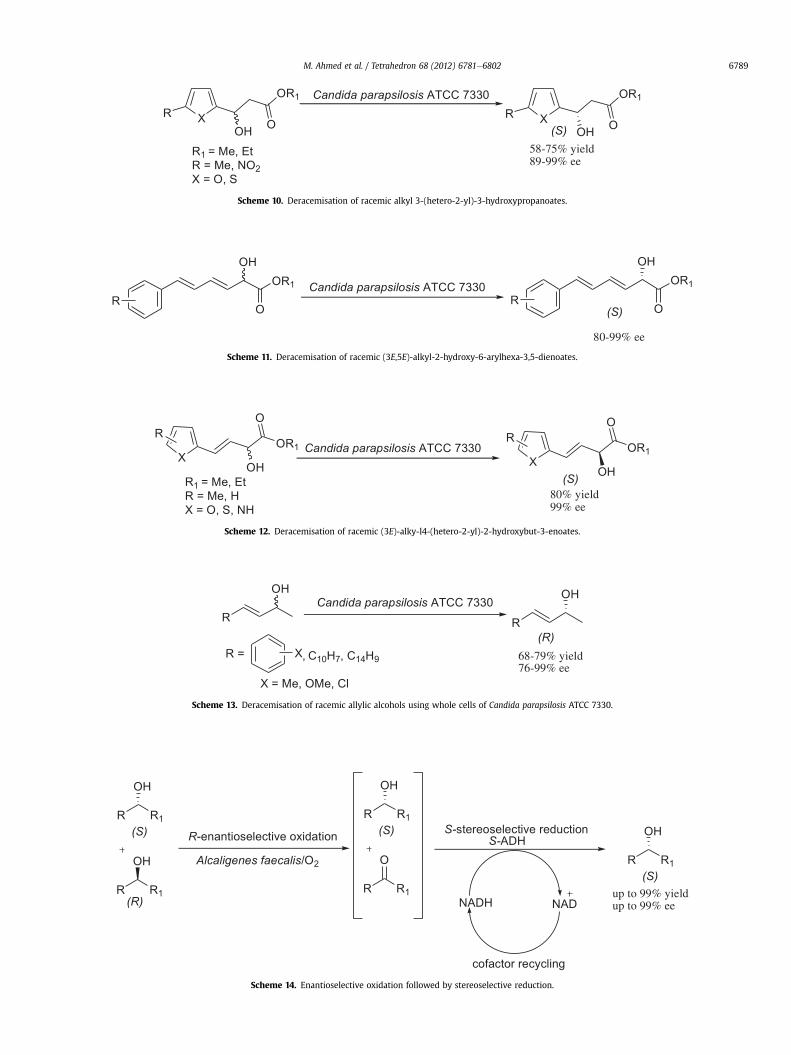
58-75% yield89-99% ee
X
Candida parapsilosis ATCC 7330
(S)
R1 = Me, EtR = Me, NO2X = O, S
ROR1
OH O XROR1
OH O
Scheme 10. Deracemisation of racemic alkyl 3-(hetero-2-yl)-3-hydroxypropanoates.
Candida parapsilosis ATCC 7330
(S)
R
80-99% ee
OR1
OH
OR
OR1
OH
O
Scheme 11. Deracemisation of racemic (3E,5E)-alkyl-2-hydroxy-6-arylhexa-3,5-dienoates.
80% yield99% ee
Candida parapsilosis ATCC 7330
(S)R1 = Me, EtR = Me, HX = O, S, NH
X
ROR1
O
OH X
ROR1
O
OH
Scheme 12. Deracemisation of racemic (3E)-alky-l4-(hetero-2-yl)-2-hydroxybut-3-enoates.
R
OH
68-79% yield76-99% ee
Candida parapsilosis ATCC 7330
(R)
R
OH
R = X
X = Me, OMe, Cl
C10H7, C14H9,
Scheme 13. Deracemisation of racemic allylic alcohols using whole cells of Candida parapsilosis ATCC 7330.
R R1
OH
R R1
OH
(S)
(R)
R-enantioselective oxidation
Alcaligenes faecalis/O2
R R1
OH
R R1
O
(S) S-stereoselective reductionS-ADH
NADH
cofactor recycling
R R1
OH
(S)
up to 99% yieldup to 99% eeNAD
Scheme 14. Enantioselective oxidation followed by stereoselective reduction.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e6802 6789

n-C8H17
OH non-enaniospecific oxidation
(S)
Step 1
n-C8H17
O
n-C8H17
OHstereoselective reduction
Step 2
Scheme 15. Oxidation/reduction deracemisation in one-pot, two-step fashion using single biocatalyst.
(S)
Ar R
OH[RuCl2(benzene)]2then H2 (10 bar)
Ar R
OH
up to 97% yieldup to 99% ee
Scheme 16. Oxidation of secondary alcohols into ketones then reduction withhydrogen.
94% yield> 97% ee
Ph COOH
OH
Ph COOH
OH
mandelate racemase
alcohol dehydrogenase
Ph COOH
Oamino acid dehydrogenase
Ph COOH
NH2
L-enantiomer
Fig. 9. Deracemisation of mandelic acid to L-phenylglycine.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026790
Additionally, Kato et al. developed a new method for the prep-aration of optically active a-substituted carboxylic acids. Using thegrowing cell system of Nocardia diaphanozonaria JCM3208, race-mates of 2-aryl- and 2-aryloxy-propanoic acid are deracemisedsmoothly and the (R)-enantiomer is recovered in high yield (>50%)and 69% ee (Scheme 17). They also discovered a new enzyme sys-tem for the biotransformation and it proceeds by the same mech-anism as in rat liver.77
>50% yield69% ee
(R)
COOH
Me
XNocardia diaphanozonaria JCM3208 COOH
Me
X
Scheme 17. Microbial deracemisation of 2-arylpropanoic acid.
Another interesting examplewas reported by Larissegger-Schnellet al. for the deracemisation of 3-phenyllactic acid and 2-hydroxy-4-phenylbutanoic acid. It was achieved through lipase-catalysed KR(using porous ceramic-immobilised lipase: lipase PSeCII) followedby racemisation of the non-reacting enantiomer using Lactobacillusparacasei DSM 20008.78 Due to the necessity to switch betweenaqueous-organic solvent systems, it was not possible to achieveracemisation in a dynamic process. Attempts at in situ racemisationin organic solvents were not successful (Scheme 18).
3.1.1.3. Deracemisation of amino acids and amines. Deracemisa-tion of DL-a-amino acids was reported in many publications using
either D- or L-amino acid oxidases and then reducing the in-termediate imine by a non-selective reducing agent, e.g., hydridereducing agents (such as sodium borohydride or sodium cyano-borohydride) or amine boranes.64,65
Racemic a-chiral primary amines were deracemised to enan-tiomerically pure amines in up to 99% yield and 99% ee by Kosze-lewski et al. The process involved a one-pot, two-step procedurewhere KR of a racemic mixture was first performed usingu-transaminase to yield an intermediate ketone, which was thenstereoselectively transformed into an amine by reductive amina-tion (Scheme 19).79
On the other hand, cyclic secondary amines80 were successfullyderacemised by performing direct evolution of the monoamineoxidase from Aspergillus niger. The new variant displayed highcatalytic activity and enantioselectivity towards chiral secondaryamines; the secondary amines are then subjected to reductionusing ammonia borane to give the enantiomerically pure isomer(Scheme 20).
3.2. In situ racemisation (dynamic kinetic resolution)
Since the early 1990s, several approaches have been developedto combine both KR along with in situ racemisation to overcomethe 50% yield barrier associated with KR. The process starts with KRwhere one of the enantiomers is transformed quickly into theproduct retaining the other unreacted enantiomer. Then, as thefaster-reacting enantiomer is depleted during the enantioselectivereaction, the equilibrium of (R)-/(S)- is constantly readjusted byracemisation of the slow-reacting counterpart (S)- (Fig. 3). To em-phasise the ‘non-static character’ of the process, the term DKR wasapplied. In contrast to KR, DKR was successful in achieving the

COOH
OHPh lipase PS-CII
OAc
COOH2
OHPh
COOH
OAcPh
Lactobacillus paracasei DSM 20008 56% yield>99% ee
(R) (S)
COOH
OAcPh porcine pancreatic lipase
COOH2
OHPh
COOH
OAcPh
(S) (R)
Ac2O
Lactobacillus paracasei DSM 20008
Ac2O
40% yield98% ee
Scheme 18. Deracemisation of 3-phenyllactic acid.
R1 R2
NH2
R1 R2
NH2
(R)-selective transaminase
(R)
(S) R1 R2
NH2
R1 R2
O
(S) (S)-selective transaminaseR1 R2
NH2
(S)
pyruvate D-Ala L-Ala pyruvate
LDH
lactate
GDH
NADH
NAD+
glucose
gluconolactone
Scheme 19. One-pot, two-step deracemisation of a-chiral primary amines.
(R)
95% yield99% ee
NH
Me
(S)-amine oxidase
ammonia boraneNH
Me
Scheme 20. Deracemisation of cyclic secondary amines.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e6802 6791
enantiomerically pure isomer in 100% theoretical yield.58 As a ruleof thumb, the rate of racemisation should be equal to or higherthan, the rate of enantioselective transformation.81 Racemisation ofthe substrate can be performed either thermally, chemically, bio-catalytically or even spontaneously. Racemisationmethods that canbe performed in a single step under mild conditions are suitable forDKR. Conditions must be adjusted to avoid racemisation of theproduct.
There are three basic requirements for an efficient DKR: (a) anefficient KR has to be identified (i.e., kR>>kS); (b) an efficient rac-emisation method has to be chosen (i.e., krac >10 kS); and (c) the KRand racemisation procedures should be compatible with eachother.82 Since the most commonly used enzymes work under mildconditions of temperature and pressure, catalysts used for race-misation should be carefully selected in order not to destroy theenzymes. An ideal catalyst for racemisation should have a broad
substrate scope, be effective under mild conditions of temperatureand pressure and tolerate enzyme working conditions.
3.2.1. Enzyme-catalysed racemisation. Since the reactions catalysedby enzymes are performed under mild conditions, enzymatic rac-emisation is considered an attractive option in DKR.
Choi et al. reported the synthesis of (R)-mandelic acid ethyl esterfrom racemic mandelic acid by recombinant mandelate racemase.The DKR process was achieved via an aqueous/organic two- phasesystem and two enzymes, where KR first took place with lipase inan organic solvent followed by an in situ racemisation of theunreacted isomer, (S)-mandelic acid, in an aqueous medium byrecombinant mandelate racemase. The procedure was carried outin a hollow-fibre membrane bioreactor, with (R)-mandelic acidethyl ester being obtained in 65% yield and 98% ee (Scheme 21).83
Other racemases, e.g., lactate racemase and different amino acidracemases, could be useful for in situ racemisation of differenta-amino acids.
3.2.2. Non-enzyme-catalysed racemisation. Several mechanismswere adopted for the in situ racemisation reaction, including viaprotonation/deprotonation, via addition/elimination, via oxidation/reduction and via nucleophilic substitution.
3.2.2.1. In situ racemisation via protonation/deprotonation. Asenzymes are usually stable at pH values close to 7, base-catalysedracemisation is considered the easiest and most common mecha-nism of racemisation. However, it is limited to substrates

(S)-mandelic acid
mandelate racemase
aqueous buffer (R)-mandelic acid ethyl ester(R)-mandelic acid
lipase/organic solvent
EtOH
65% yield98% ee
COOH
OH
COOH
OH
COOEt
OH
Scheme 21. DKR of (S)-mandelic acid.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026792
possessing a stereogenic centre with an acidic proton (e.g.,a-substituted carboxylic acid derivatives and a-substitutedketones).84e86
Schichl et al. reported the use of different salicylaldehyde de-rivatives in the racemisation of different a-amino acid ethyl esters.DKR procedures involved the use of alcalase enzyme (an endopro-teinase of the serine type) for the resolution step and different al-dehydes were reported for the in situ-racemisation step. Aftercondensation of the amino acid ester with the salicylaldehyde de-rivative, the acidity of the a-hydrogen atom at the chiral centre wasremarkably increased, facilitating rapid protonation/deprotonationand, hence, racemisation. Thereafter, hydrolysis of the intermediateSchiff base took place to liberate the racemised amino acid ester andthe aldehyde. The synthesised a-amino acids were obtained at highyield (up to94%)andopticalpurity (up to98%) (Schemes22and23).87
COOR
NH2 alcalase
different salicylaldehyde derivatives
Scheme 22. DKR of phenylalanine esters u
O
OHX
R COOR1
NH2
-H2O
+H2O
-H+
+H+
**
OH
N
COOR1R
X
Scheme 23. Proposed mechanism for aldehyde-ca
Similarly, an important class of non-steroidal anti-inflammatorydrugs (profens) was prepared through the DKR concept. (S)-nap-roxen, (S)-fenoprofen and (S)-suprofenwere synthesised from theirrespective thioesters using immobilised lipase and trioctylamine asthe racemising agent (Scheme 24).88
3.2.2.2. In situ racemisation via addition/elimination. Thismechanism will be suitable when addition/elimination reactionsare feasible, e.g., with cyanohydrins, hemiacetals, hemiaminals andhemithioacetals, where reversible addition/elimination reactionsare possible under mild conditions. In this context, silica-supportedBTAH (benzyltrimethylammonium hydroxide) was successfullyutilised as a racemising agent for different cyanohydrins togetherwith lipase PSeCII for efficient DKR of chiral cyanohydrins(Scheme 25).89
COOHCOOR
NH2 NH2
up to 87% yieldup to 98% ee
(S)
sing salicylaldehydes for racemisation.
+H+/ H2O-H+/ H2O
O
OHX R COOR1
NH2
intermediates
OH
N
COOR1R
XOH
N
COOR1R
X XOH
N
R COOR1
talysed racemisation of a-amino acid esters.

O
SCH2CF3
O
O
SCH2CF3
O
-H+ +H+
O
SCH2CF3
Olipase
O
OH
O
-H++H+
Scheme 24. Synthesis of (S)-fenoprofen via DKR using trioctylamine as racemising agent.
Ar H
O HO CN
base on silicaAr CN
OHlipase PS-CII
Ar CN
OH
Ar CN
OActransesterification
recrystallisation
90% yield93% ee
(S) >99% ee
OAc
Scheme 25. Lipase-catalysed DKR of cyanohydrins using silica-supported BTAH as racemising agent.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e6802 6793
3.2.2.3. In situ racemisation via oxidation/reduction. Reversibleoxidation/reduction is a common mechanism of racemisation.Oxidation removes hydrogen from the stereogenic carbon atomcreating a planar intermediate and then reduction or hydrogena-tion restores the tetrahedral configuration in a non-stereoselectivemanner and thus racemisation takes place. Oxidation and reductioncan take place either simultaneously or in two separate steps (withor without isolation of the oxidised intermediate) (Fig. 10).
oxidation reduction
oxidation reduction
R2 H
R1XH
R2
R1
X R2 H
R1XH
R2 H
R1XH
R2 H
R1XR3
R2
R1
XR3
R2 H
R1XR3
R2 H
R1XR3
Fig. 10. Racemisation via oxidation/reduction sequence.
3.2.2.4. In situ racemisation via nucleophilic substitution. Arather rare mechanism of racemisation, which occurs when re-versible nucleophilic substitution of a sec-halogen is displaced bythe same halide, is applicablewhen the halide is in an electronicallyactivated position. Therefore, this mechanism has been used forracemisation of a-halo esters (Fig. 11). Racemisation takes place byconsecutive inversion induced by additives, such as polar solvents,bases or halide salts.
3.2.2.5. Metal-catalysed racemisation. A wide range of metalcatalysts have been reported in the racemisation step for DKR.Several complexes of ruthenium, rhodium, iridium, palladium andother metals are known to catalyse rapid racemisation of differentsubstrates, but only a few complexes have proved to be compatiblewith the enzymatic reaction. An efficient catalyst must not interferewith the enzymatic transformation or be affected by the surfactants
used to support the enzyme. At the same time, the enzyme ortransformation by-products might slow down or inhibit the race-misation by the metal. Moreover, whereas racemisation is usuallyfaster at high temperature, the enzymes undergo denaturation athigh temperature. However, it has been recently shown that thethermostability of enzymes could be highly enhanced throughimmobilisation on certain supports, which also allows recovery andsubsequent reuse of the enzymes.90 Hence, the selection of the
appropriate metal catalyst is very crucial in DKR. Metal complexesbearing chiral ligands have played a great role in asymmetric syn-thesis and have been considered a breakthrough in organic syn-thesis over the past 35 years.3 To date, a significant number of chiralcatalysts have been reported in racemisation reactions. There aretwo general mechanisms by which racemisation occurs: either viahydrogen transfer or via p-allyl formation (Fig. 12).82
3.2.2.5.1. Ruthenium-based catalysts. Ruthenium-catalysed hy-drogenation has been widely reported in DKR of secondary alco-hols, a- and b-hydroxy acid esters. Ruthenium-based catalysts havethe advantage that they do not require an additional base as a co-catalyst (as one of the ligand oxygens act as a basic centre), and,therefore, eliminate the base-catalysed transesterification. In late1990s, B€ackvall et al. developed a robust ruthenium complex(Shvo’s catalyst) and a specially designed acyl donor (p-chlor-ophenyl acetate). It is activated by heat and therefore must be used

R
O
X
nucleophilic substitutionslow
R
O
Nu
epimerisation
R
O
X
nucleophilic substitutionfast
R
O
Numajor
Fig. 11. SN2 reactions on electronically activated chiral halides.
R2 H
R1XH
R2
R1
X R2 H
R1XHM
MH
H
M
R2R1
Nu
M R2R1
M
Nu R2R1
NuM: metalNu: nucleophile
Fig. 12. Metal-catalysed racemisation mechanisms via hydrogen transfer or p-allyl formation.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026794
in combination with thermostable enzymes (Fig. 13). It was suc-cessfully used in combinationwith CaLB for DKR ofmany secondaryalcohols and secondary diols (Scheme 26).82,91,92
O
Ph
Ph
Ph
Ph Ru
O
PhPh
Ph
Ph
H
Ru
CO OCOC CO
HOH
Ph Ph
Ph
Ru
Ph
PhPh
Ph
O
RuHOC
OCOC CO
(a)
Ph
Ph Ph
Ph
O
RuOC CO
(b)OH
R1 R2
HOH
Ph Ph
Ph
Ph
Ru HOC
OC
R1 R2
O Ph
Ph Ph
Ph
O
RuOC CO
OH
R1 HR2
1
1b1a
1a
1b
1b
Ph
Fig. 13. Racemisation mechanism using Shvo’s catalyst 1, dissociation of 1 into 1a and 1b, which is the active component for racemisation of an alcohol (step b).
Later on, another pre-catalyst was reported by Choi et al.,93,94
where the catalyst was able to racemise secondary alcohols atroom temperature (Fig. 14).
A highly efficient ruthenium catalyst was then described byB€ackvall et al.,95,96 which is the fastest reported hydrogen-transfercatalyst. This catalyst is widely used in DKR of a broad variety ofsecondary alcohols, b-hydroxy nitriles97 and other important chiralintermediates (Schemes 27 and 28).98e102
As naturally occurring lipases are (R)-selective for alcoholsaccording to Kazlauskas’ rule,8,103 DKR of racemic alcohols can onlybe used to produce the (R)-acetate. To produce the (S)-counterpart,
proteases rather than lipases are utilised in the resolution step.Bearing in mind that proteases are not thermostable, therefore onlymetal complexes that racemise the substrate at room temperatureare suitable for efficient (S)-selective DKR of secondaryalcohols.104,105
Since racemisation of amines is difficult and requires harshconditions, a powerful Shvo-type ruthenium catalyst wassynthesised by B€ackvall et al.. The catalyst was used in combination

R
OH CaLB, p-Cl-C6H4OAc
Shvo's catalyst R
OAc
up to 92% yield> 99% ee
(R)R = alkyl, aryl
Scheme 26. DKR of secondary alcohols using Shvo’s catalyst.
Ph Ph
Ph
Ru ClOC
OC
Ph Ph
87% yield98% ee
(R)
ArCN
OH OAcCaLB
ruthenium catalyst 3 ArCN
OAc
Ruthenium catalyst 3
Scheme 27. DKR of cyanohydrins using CaLB and ruthenium catalyst 3.
99% yield98% ee
(R)
OAc
ruthenium catalyst 3
ClOH
F
F
lipase PS-CIICl
OAcF
F
F
F
Scheme 28. DKR of secondary alcohols using lipase PSeCII and ruthenium catalyst 3.
Fig. 14. Ruthenium catalyst 2.
O
Ar
Ar
Ar
Ar Ru
O
ArAr
Ar
Ar
H
Ru
CO OCOC CO
H
Ar = p-MeO-C6H4
Ruthenium catalyst 4
up to 95% yieldup tp 99% ee
(R)
R1 R2
NH2OAc
CaLB
ruthenium catalyst 4 R1 R2
NHAc
Scheme 29. DKR of different secondary amines using CaLB and ruthenium catalyst 4.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e6802 6795
with CaLB for the DKR of variety of secondary amines with excellentyields and enantioselectivities (Scheme 29).106,107
Other ruthenium-based catalysts were also reported forDKR of different amines with different electronic properties. Acomprehensive study of the effect of different acyl donors aswell as a competitive racemisation study of the deuterated andnon-deuterated amine was carried out by the same group(Fig. 15).108
Another highly efficient ruthenium catalyst [RuCl2((S)-SDP)(R,R)-DPEN] was reported by Xie et al. for asymmetric hydro-genation of a-amino ketones. It displayed high enantio- and dia-stereoselectivities for preparing cis a-amino cycloalkanols109 and itwas used successfully for DKR of a-amino ketones with impressivediastereoselectivity (Scheme 30).110
Similarly, chiral b-hydroxy aryl sulfones were racemised ina similar manner in high yields (up to 95%) and high ee (up to 98%)by Ding et al. (Scheme 31).111
3.2.2.5.2. Non-ruthenium catalysts. Chiral complexes of metalsother than ruthenium have also been used in the context of DKR.However, their compatibility with the enzymatic reactions is themain prerequisite for efficient DKR. There were early successfulattempts to racemise secondary alcohols using rhodium, iridium,aluminium and palladium catalysts.112e114
Haak at al. developed a new DKR process for efficient synthesisof chiral epoxides from racemic haloalcohols. Haloalcohol de-hydrogenase was used for the KR of racemic b-haloalcohols to givethe corresponding (R)-epoxide and the unreacted (S)-enantiomerwas racemised with an iridium complex as a racemising catalyst. Inthis way, a series of epoxides were furnished in excellent yield andhigh ee (98%) (Scheme 32).115
On the other hand, Andrade et al. employed CaLB in combina-tion with Pd on barium sulfate for the DKR of organoselenium-containing 1-phenylethylamines to yield the correspondingamides in high yield (up to 87%) and high ee (99%) (Scheme 33).116
Similarly, Engstr€om described the use of a palladium nano-catalyst for DKR of b-amino esters in combination with CaLA.The products were obtained in high yield (up to 97%) and high ee(up to 99%) (Scheme 34).117

Fig. 15. Racemisation mechanism of secondary amines using ruthenium catalyst.
Ruthenium catalyst 5
P
P Ru
Cl
Cl
H2N
NH2
Ph
Ph
Ph
Ph
R1
NR3 R4
R2
Oruthenium catalyst 5
R1
R2
OHN R4R3
up tp 99.9% ee
Scheme 30. Asymmetric hydrogenation of a-amino ketones via DKR using rutheniumcatalyst 5.
Ruthenium catalyst 6
N
NH2
RuClPh
PhTs
ruthenium catalyst 6
O
SO2Ph
OH
SO2Ph
Scheme 31. DKR of chiral b-hydroxy aryl sulfones using ruthenium catalyst 6.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026796
A novel approach was developed by Akai et al. for DKR of allylicalcohols using a vanadate complex as a racemising agent in com-binationwith lipase for the KR step. An excellent yield and ee (99%)were obtained (Scheme 35).118
Interestingly, Berkessel et al. utilised inexpensive aluminium-based catalysts for racemisation of secondary alcohols at ambienttemperature. To increase the reactivity of the aluminium com-plexes, a bidentate ligand (such as binol) was used (Scheme 36).119
3.2.3. Organocatalysts. Simple organic molecules could be highlyefficient in enantioselective transformations and therefore could besuitable in the context of DKR. Organocatalysts have the advantages
of being robust, inexpensive, readily available and non-toxic. Thisincreased the interest towards their use in organic synthesis ex-cluding any environmental hazards associated with metal use.120
Berkessel et al. developed a diverse library of organocatalystsbearing a thiourea moiety and tertiary amine group (Fig. 16). Thesecatalysts were successful for alcoholytic DKR of azalactones with eeup to 95% (Scheme 37).121e123
Azalactones were also racemised by cinchona alkaloid-derived catalysts, which were highly efficient in providing thecorresponding chiral amino esters in excellent yields and highenantioselectivities (up to 88%).124,125 Additionally, Wakayamaand Ellman utilised quinidine to promote the reaction betweentert-butanesulfinyl chloride with different alcohols to producethe corresponding chiral alkyl tert-butanesulfinates viaa DKR process in high yield and optical purity (up to 89%)(Scheme 38).126

NIr
Me
H NCMe
PF6-
iridacycle racemisation catalyst
OHCl O
haloalcohol dehalogenase
90% yield98% ee
(R)
Scheme 32. DKR of haloalcohols using iridium catalyst as racemising agent.
NH2
EtSePd-BaSO4
CaLB- EtOAc
NHAc
EtSe
up to 87% yield 99% ee
(R)
Scheme 33. DKR of amines using Pd on BaSO4 as racemising agent.
up to 99% yield up to 99% ee
(R)
R OEt
ONH2 O CF3Pr
O
CaLA
Pd nanocatalystR OEt
ONHPr
O
Scheme 34. DKR of b-amino esters using palladium nanocatalyst.
91% yield99% ee
(R)
OH
OAcEtO
CaLB
VO(OSiPh3)3
, OAc
Scheme 35. DKR of allylic alcohols using vanadate complex as racemising agent.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e6802 6797
96% yield96% ee
(R)
OAcPh
CaLB
AlMe3
,
OH OAc
Scheme 36. DKR of secondary alcohols using AlMe3 as racemising agent.
NMe2
NH
NH
N
O
t-BuS( )
nR2
R1
Fig. 16. Thiourea and tertiary amine-based organocatalysts.
N O
O
Ph
urea/thiourea organocatalyst
N O
OR
Ph
HN
NH
X
OR3
H NR2
Bronsted base
Ph NH
OO HR1
OR3
H
R1 NH
NH
XR2
NR2
chiralspacer
X = O,S
Scheme 37. Alcoholytic DKR of azalactones using urea/thiourea organocatalysts.
N
OMe
N
OH
H
SO
t-Bu Cl ROH SO
t-Bu ORSO
t-Bu NH2
NaNH2
up to 99% yieldup to 89% ee
Scheme 38. Quinidine-catalysed DKR of tert-butanesulfinyl chloride.
M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026798
4. Conclusions
A considerable number of publications have been addressingbiocatalysis in the past five years. The use of lipase for KR of dif-ferent functional groups has been explored. Tailoring of the lipasebinding site to fit bulky substrates and to enhance enantiose-lectivity has been a promising tool to resolve challenging chiralsubstrates. However, KR has a maximum theoretical yield of 50%,and so the search for techniques that furnish highly enantiomeri-cally pure compounds at higher chemical yields is of great need.Hence, the deracemisation concept evolved where a continuous
process of converting the unreacted isomer into the racemicstarting material enables the synthesis of a single enantiomer froma racemic starting material in 100% theoretical yield and 100% ee.Different racemisation techniques are available, cyclic and in situ,both having the advantage of achieving high yields very close to100%. Deracemisation techniques are an attractive option in view ofthe simplicity of the system and compatibility of wide ranges ofenzymes/catalysts for a one-pot process. The increased availabilityof microbial genome sequences, coupled with advances in high-throughput screening technologies and directed evolution, willprovide enzymes with wide substrate specificity and selectivity aswell as compatible biocatalysts that are better able to operate underthe conditions of the reaction, especially when a metal catalyst isutilised.
Accurate chromatographic determination of the enantiomericexcess is the key factor for reliable kinetic resolution data.This can only be achieved via simultaneous baseline resolutionof the substrate and the product in one run withoutderivatisation.
References and notes
1. Dewan, S. S. BCC Research; BCC Research: 2010; http://bccresearch.com/report/chiral-technology-bio012e.html.
2. Ghanem,A.;Enantiomer Separation: Fundamentals andPracticalMethods.KluwerAcademic: 2005; 2336.
3. Pellissier, H. Tetrahedron 2011, 67, 3769e3802.4. Pellissier, H. Tetrahedron 2008, 64, 1563e1601.5. Schoffers, E.; Golebiowski, A.; Johnson, C. R. Tetrahedron 1996, 52, 3769e3826.6. Ghanem, A. Tetrahedron 2007, 63, 1721e1754.7. Klibanov, A. M. Trends Biotechnol. 1997, 15, 97e101.8. Jing, Q.; Kazlauskas, R. J. Chirality 2008, 20, 724e735.9. Hultin, P. G.; Mueseler, F. J.; Jones, J. B. J. Org. Chem. 1991, 56, 5375e5380.
10. Wimmer, Z. Tetrahedron 1992, 48, 8431e8436.

M. Ahmed et al. / Tetrahedron 68 (2012) 6781e6802 6799
11. Klibanov, A. M. Nature 2001, 409, 241e246.12. Koeller, K. M.; Wong, C.-H. Nature 2001, 409, 232e240.13. Schmid, A.; Dordick, J. S.; Hauer, B.; Kiener, A.; Wubbolts, M.; Witholt,
B. Nature 2001, 409, 258e268.14. Schmid, R. D.; Verger, R. Angew. Chem., Int. Ed. 1998, 37, 1608e1633.15. Villeneuve, P.; Muderhwa, J. M.; Graille, J.; Haas, M. J. J. Mol. Catal. B: Enzym.
2000, 9, 113e148.16. Akai, S.; Naka, T.; Fujita, T.; Takebe, Y.; Kita, Y. Chem. Commun. 2000,
1461e1462.17. L�opez-Serrano, P.; Wegman, M. A.; van Rantwijk, F.; Sheldon, R. A. Tetrahedron:
Asymmetry 2001, 12, 235e240.18. Cainelli, G.; De Matteis, V.; Galletti, P.; Giacomini, D.; Orioli, P. Chem. Commun.
2000, 2351e2352.19. Xu, Q.; Xie, Y.; Geng, X.; Chen, P. Tetrahedron 2010, 66, 624e630.20. Invernizzi, G.; Casiraghi, L.; Grandori, R.; Lotti, M. J. Biotechnol. 2009,141, 42e46.21. Kirchner, G.; Scollar, M. P.; Klibanov, A. M. J. Am. Chem. Soc. 1985, 107,
7072e7076.22. Solanki, K.; Gupta, M. N. Bioorg. Med. Chem. Lett. 2011, 21, 2934e2936.23. Matsuda, T.; Harada, T.; Nakamura, K.; Ikariya, T. Tetrahedron: Asymmetry
2005, 16, 909e915.24. Stampfer, W.; Kosjek, B.; Faber, K.; Kroutil, W. Tetrahedron: Asymmetry 2003,
14, 275e280.25. Mateo, C.; Palomo, J. M.; Fernandez-Lorente, G.; Guisan, J. M.; Fernandez-La-
fuente, R. Enzyme Microb. Technol. 2007, 40, 1451e1463.26. Silva, V. C. F.; Contesini, F. J.; Oliveira Carvalho, P. J. Ind. Microbiol. Biotechnol.
2009, 36, 949e954.27. Cabrera, Z.; Fernandez-Lorente, G.; Fernandez-Lafuente, R.; Palomo, J. M.;
Guisan, J. M. Process Biochem. 2009, 44, 226e231.28. Godoy, C. A.; de las Rivas, B.; Filice, M.; Fern�andez-Lorente, G.; Guisan, J. M.;
Palomo, J. M. Process Biochem. 2010, 45, 534e541.29. Forde, J.; Vakurov, A.; Gibson, T. D.; Millner, P.; Whelehan, M.; Marison, I. W.;
�O’F�ag�ain, C. J. Mol. Catal. B: Enzym. 2010, 66, 203e209.30. Contesini, F. J.; Lopes, D. B.; Macedo, G. A.; Nascimento, MdG.; Carvalho, PdO. J.
Mol. Catal. B: Enzym. 2010, 67, 163e171.31. Carvalho, PdO.; Contesini, F. J.; Bizaco, R.; Alves Macedo, G. Food Biotechnol.
(Philadelphia, PA, U. S.) 2005, 19, 183e192.32. Hita, E.; Robles, A.; Camacho, B.; Gonzalez, P. A.; Esteban, L.; Jimenez, M. J.;
Munio, M. M.; Molina, E. Biochem. Eng. J. 2009, 46, 257e264.33. Lafaqui�ere, V.; Barbe, S.; Puech-Guenot, S.; Guieysse, D.; Cort�es, J.; Monsan, P.;
Sim�eon, T.; Andr�e, I.; Remaud-Sim�eon, M. ChemBioChem 2009, 10, 2760e2771.34. Juhl, P. B.; Doderer, K.; Hollmann, F.; Thum, O.; Pleiss, J. J. Biotechnol. 2010, 150,
474e480.35. Ghanem, A.; Aboul-Enein, M. N.; El-Azzouny, A.; El-Behairy, M. F. J. Chroma-
togr., A 2010, 1217, 1063e1074.36. Brem, J.; Pilb�ak, S.; Paizs, C.; B�an�oczi, G.; Irimie, F.-D.; Tosa, M.-I.; Poppe, L.
Tetrahedron: Asymmetry 2011, 22, 916e923.37. Brem, J.; Liljeblad, A.; Paizs, C.; Tosa, M. I.; Irimie, F.-D.; Kanerva, L. T.
Tetrahedron: Asymmetry 2011, 22, 315e322.38. Utcz�as, M.; Sz�ekely, E.; Forr�o, E.; Szollosy, �A.; F€ul€op, F.; Sim�andi, B. Tetrahedron
Lett. 2011, 52, 3916e3918.39. Zhou, H.; Chen, J.; Ye, L.; Lin, H.; Yuan, Y. Bioresour. Technol. 2011, 102,
5562e5566.40. Li, Y.-H.; Huang, L.-H.; Liu, H.-M. Synth. Commun. 2011, 41, 2468e2474.41. Barbosa, O.; Ortiz, C.; Torres, R.; Fernandez-Lafuente, R. J. Mol. Catal. B: Enzym.
2011, 71, 124e132.42. Piliss~ao, C.; Carvalho, PdO.; Nascimento, MdG. Process Biochem. 2009, 44,
1352e1357.43. Alatorre-Santamaría, S.; Gotor-Fern�andez, V.; Gotor, V. Eur. J. Org. Chem. 2009,
2533e2538.44. Quijada, F. J.; Gonz�alez-Sabín, J.; Rebolledo, F.; Gotor, V. Tetrahedron 2009, 65,
8028e8034.45. Andrade, L. H.; Barcellos, T.; Santiago, C. G. Tetrahedron: Asymmetry 2010, 21,
2419e2424.46. Zhao, H.; Jackson, L.; Song, Z.; Olubajo, O. Tetrahedron: Asymmetry 2006, 17,
2491e2498.47. Ghanem, A. Chirality 2010, 22, 597e603.48. Foresti, M. L.; Galle, M.; Ferreira, M. L.; Briand, L. E. J. Chem. Technol. Biotechnol.
2009, 84, 1461e1473.49. Morrone, R.; D’Antona, N.; Lambusta, D.; Nicolosi, G. J. Mol. Catal. B: Enzym.
2010, 65, 49e51.50. Zhang, Y.-Y.; Liu, J.-H. Biochem. Eng. J. 2011, 54, 40e46.51. Yilmaz, E.; Sezgin, M.; Yilmaz, M. J. Mol. Catal. B: Enzym. 2010, 62, 162e168.52. Ch�avez-Flores, D.; Salvador, J. M. Biotechnol. J. 2009, 4, 1222e1224.53. Deasy, R. E.; Brossat, M.; Moody, T. S.; Maguire, A. R. Tetrahedron: Asymmetry
2011, 22, 47e61.54. Faber, K. Chem.dEur. J. 2001, 7, 5004e5010.55. Ebbers, E. J.; Ariaans, G. J. A.; Houbiers, J. P. M.; Bruggink, A.; Zwanenburg, B.
Tetrahedron 1997, 53, 9417e9476.56. Schnell, B.; Faber, K.; Kroutil, W. Adv. Synth. Catal. 2003, 345, 653e666.57. Strauss, U. T.; Faber, K. Tetrahedron: Asymmetry 1999, 10, 4079e4081.58. Huerta, F. F.; Minidis, A. B. E.; Baeckvall, J.-E. Chem. Soc. Rev. 2001, 30,
321e331.59. El Gihani, M. T.; Williams, J. M. J. Curr. Opin. Chem. Biol. 1999, 3, 11e15.60. Strauss, U. T.; Felfer, U.; Faber, K. Tetrahedron: Asymmetry 1999, 10, 107e117.61. Bae, H.-S.; Hong, S.-P.; Lee, S.-G.; Kwak, M.-S.; Esaki, N.; Sung, M.-H. J. Mol.
Catal. B: Enzym. 2002, 17, 223e233.
62. Bae, H.-S.; Lee, S.-G.; Hong, S.-P.; Kwak, M.-S.; Esaki, N.; Soda, K.; Sung, M.-H. J.Mol. Catal. B: Enzym. 1999, 6, 241e247.
63. Soda, K.; Oikawa, T.; Yokoigawa, K. J. Mol. Catal. B: Enzym. 2001, 11, 149e153.64. Beard, T. M.; Turner, N. J. Chem. Commun. (Cambridge) 2002, 246e247.65. Alexandre, F.-R.; Pantaleone, D. P.; Taylor, P. P.; Fotheringham, I. G.; Ager, D. J.;
Turner, N. J. Tetrahedron Lett. 2002, 43, 707e710.66. Demir, A. S.; Hamamci, H.; Sesenoglu, O.; Neslihanoglu, R.; Asikoglu, B.;
Capanoglu, D. Tetrahedron Lett. 2002, 43, 6447e6449.67. Titu, D.; Chadha, A. J. Mol. Catal. B: Enzym. 2008, 52-53, 168e172.68. Thangavel, V.; Chadha, A. Tetrahedron 2007, 63, 4126e4133.69. Vaijayanthi, T.; Chadha, A. Tetrahedron: Asymmetry 2007, 18, 1077e1084.70. Titu, D.; Chadha, A. Tetrahedron: Asymmetry 2008, 19, 1698e1701.71. Voss, C. V.; Gruber, C. C.; Kroutil, W. Angew. Chem., Int. Ed. 2008, 47,
741e745.72. Voss, C. V.; Gruber, C. C.; Faber, K.; Knaus, T.; Macheroux, P.; Kroutil, W. J. Am.
Chem. Soc. 2008, 130, 13969e13972.73. Voss, C. V.; Gruber, C. C.; Kroutil, W. Synlett 2010, 991e998.74. Voss, C. V.; Gruber, C. C.; Kroutil, W. Tetrahedron: Asymmetry 2007, 18,
276e281.75. Adair, G. R. A.; Williams, J. M. J. Chem. Commun. (Cambridge) 2007, 2608e2609.76. Resch, V.; Fabian, W. M. F.; Kroutil, W. Adv. Synth. Catal. 2010, 352, 993e997.77. Kato, D.-i.; Mitsuda, S.; Ohta, H. J. Org. Chem. 2003, 68, 7234e7242.78. Larissegger-Schnell, B.; Glueck, S. M.; Kroutil, W.; Faber, K. Tetrahedron 2006,
62, 2912e2916.79. Koszelewski, D.; Clay, D.; Rozzell, D.; Kroutil, W. Eur. J. Org. Chem. 2009,
2289e2292.80. Carr, R.; Alexeeva, M.; Dawson, M. J.; Gotor-Fernandez, V.; Humphrey, C. E.;
Turner, N. J. ChemBioChem 2005, 6, 637e639.81. Kitamura, M.; Tokunaga, M.; Noyori, R. Tetrahedron 1993, 49, 1853e1860.82. P�amies, O.; B€ackvall, J.-E. Curr. Opin. Biotechnol. 2003, 14, 407e413.83. Choi, W. J.; Lee, K. Y.; Kang, S. H.; Lee, S. B. Sep. Purif. Technol. 2007, 53, 178e182.84. Fogal, E.; Forzato, C.; Nitti, P.; Pitacco, G.; Valentin, E. Tetrahedron: Asymmetry
2000, 11, 2599e2614.85. Howarth, J.; James, P.; Dai, J. Tetrahedron Lett. 2001, 42, 7517e7519.86. Dehli, J. R.; Gotor, V. J. Org. Chem. 2002, 67, 6816e6819.87. Schichl, D. A.; Enthaler, S.; Holla, W.; Riermeier, T.; Kragl, U.; Beller, M. Eur.
J. Org. Chem. 2008, 3506e3512.88. Lin, H.-Y.; Tsai, S.-W. J. Mol. Catal. B: Enzym. 2003, 24e25, 111e120.89. Sakai, T.; Wang, K.; Ema, T. Tetrahedron 2008, 64, 2178e2183.90. Brady, D.; Jordaan, J. Biotechnol. Lett. 2009, 31, 1639e1650.91. Turner, N. J. Curr. Opin. Biotechnol. 2003, 14, 401e406.92. Martin-Matute, B.; B€ackvall, J.-E. Curr. Opin. Chem. Biol. 2007, 11, 226e232.93. Choi, J. H.; Kim, Y. H.; Nam, S. H.; Shin, S. T.; Kim, M.-J.; Park, J. Angew. Chem.,
Int. Ed. 2002, 41, 2373e2376.94. Choi, J. H.; Choi, Y. K.; Kim, Y. H.; Park, E. S.; Kim, E. J.; Kim, M.-J.; Park, J. J. Org.
Chem. 2004, 69, 1972e1977.95. Martin-Matute, B.; Edin, M.; Bogar, K.; Kaynak, F. B.; B€ackvall, J.-E. J. Am. Chem.
Soc. 2005, 127, 8817e8825.96. Martin-Matute, B.; Edin, M.; Bogar, K.; B€ackvall, J.-E. Abstracts of Papers, 229th
ACS National Meeting; United States: San Diego, CA, March 13e17, 2005;ORGN-947.
97. Traeff, A.; Lihammar, R.; B€ackvall, J.-E. J. Org. Chem. 2011, 76, 3917e3921.98. Thalen, L. K.; Sumic, A.; Bogar, K.; Norinder, J.; Persson, A. K. A.; Baeckvall, J.-E.
J. Org. Chem. 2010, 75, 6842e6847.99. Traff, A.; Bogar, K.; Warner, M.; B€ackvall, J.-E. Org. Lett. 2008, 10, 4807e4810.
100. Traff, A.; Bogar, K.; Warner, M.; B€ackvall, J.-E. Abstract of Papers, 238th ACSNational Meeting; United States: Washington, DC, August 16-20, 2009; ORGN-714.
101. Bogar, K.; Martin-Matute, B.; B€ackvall, J.-E. Beilstein J. Org. Chem. 2007, 3, 1e4.102. Bogar, K.; Vidal, P. H.; Leon, A. R. A.; B€ackvall, J.-E. Org. Lett. 2007, 9,
3401e3404.103. Kazlauskas, R. J.; Weissfloch, A. N. E.; Rappaport, A. T.; Cuccia, L. A. J. Org. Chem.
1991, 56, 2656e2665.104. Boren, L.; Martin-Matute, B.; Xu, Y.; Cordova, A.; B€ackvall, J.-E. Chem.dEur. J.
2005, 12, 225e232.105. Kim, M.-J.; Chung, Y. I.; Choi, Y. K.; Lee, H. K.; Kim, D.; Park, J. J. Am. Chem. Soc.
2003, 125, 11494e11495.106. P€atzold, J.; B€ackvall, J. E. J. Am. Chem. Soc. 2005, 127, 17620e17621.107. Thalen, L. K.; B€ackvall, J.-E. Beilstein J. Org. Chem. 2010, 6, 823e829.108. Thalen, L. K.; Zhao, D.; Sortais, J.-B.; Paetzold, J.; Hoben, C.; B€ackvall, J.-E.
Chem.dEur. J. 2009, 15, 3403e3410.109. Liu, S.; Xie, J.-H.; Wang, L.-X.; Zhou, Q.-L. Angew. Chem., Int. Ed. 2007, 46,
7506e7508.110. Xie, J.-H.; Liu, S.; Kong, W.-L.; Bai, W.-J.; Wang, X.-C.; Wang, L.-X.; Zhou, Q.-L. J.
Am. Chem. Soc. 2009, 131, 4222e4223.111. Ding, Z.; Yang, J.; Wang, T.; Shen, Z.; Zhang, Y. Chem. Commun. (Cambridge)
2009, 571e573.112. Kvintovics, P.; Bakos, J.; Heil, B. J. Mol. Catal. 1985, 32, 111e114.113. Allen, J. V.; Williams, J. M. J. Tetrahedron Lett. 1996, 37, 1859e1862.114. Dinh, P. M.; Howarth, J. A.; Hudnott, A. R.; Williams, J. M. J.; Harris, W. Tetra-
hedron Lett. 1996, 37, 7623e7626.115. Haak, R. M.; Berthiol, F.; Jerphagnon, T.; Gayet, A. J. A.; Tarabiono, C.; Postema,
C. P.; Ritleng, V.; Pfeffer, M.; Janssen, D. B.; Minnaard, A. J.; others. J. Am. Chem.Soc. 2008, 130, 13508e13509.
116. Andrade, L. H.; Silva, A. V.; Pedrozo, E. C. Tetrahedron Lett. 2009, 50,4331e4334.

M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026800
117. Engstr€oem, K.; Shakeri, M.; B€ackvall, J.-E. Eur. J. Org. Chem. 2011, 1827e1830.118. Akai, S.; Tanimoto, K.; Kanao, Y.; Egi, M.; Yamamoto, T.; Kita, Y. Angew. Chem.,
Int. Ed. 2006, 45, 2592e2595.119. Berkessel, A.; Sebastian-Ibarz, M. L.; M€uller, T. N. Angew. Chem., Int. Ed. 2006,
45, 6567e6570.120. Pellissier, H. Adv. Synth. Catal. 2011, 353, 659e676.121. Berkessel, A.; Mukherjee, S.; M€uller, T. N.; Cleemann, F.; Roland, K.; Bran-
denburg, M.; Neudoerfl, J.-M.; Lex, J. Org. Biomol. Chem. 2006, 4, 4319e4330.
122. Berkessel, A. Pure Appl. Chem. 2005, 77, 1277e1284.123. Berkessel, A.; Mukherjee, S.; Cleemann, F.; M€uller, T. N.; Lex, J. Chem. Commun.
(Cambridge) 2005, 1898e1900.124. Peschiulli, A.; Quigley, C.; Tallon, S.; Gun’ko, Y. K.; Connon, S. J. J. Org. Chem.
2008, 73, 6409e6412.125. Lee, J. W.; Ryu, T. H.; Oh, J. S.; Bae, H. Y.; Jang, H. B.; Song, C. E. Chem. Commun.
(Cambridge) 2009, 7224e7226.126. Wakayama, M.; Ellman, J. A. J. Org. Chem. 2009, 74, 2646e2650.

edron 68 (2012) 6781e6802 6801
M. Ahmed et al. / TetrahBiographical sketch
Dr. Ashraf Ghanem studied Chemistry at the University of Stuttgart, Germany wherehe conducted his master degree (MSc) with Prof. Franz Effenberger and Prof. G. H. El-Gemeie. In 1999, he joined the group of Prof. Rolf D. Schmid and Uwe Bornscheuer(now at the University of Greifswald, Germany) at the Institute for Technical Biochem-istry, University of Stuttgart, Germany. In 2000, he was awarded a Scholarship from theGerman Research Council (DFG) to conduct his PhD with Prof. Volker Schurig at theUniversity of Tuebingen, Germany. In 2002, he received his PhD and joined the groupof Prof. Paul Muller at the University of Geneva, Switzerland as post doctoral fellow. In2004, he was appointed Scientist and Head of the Biomedicinal Chemistry Unit,KFSHRC, Saudi Arabia. In 2008, he was appointed Pfizer research fellow/Lecturer atACROSS with Prof. Paul Haddad, School of Chemistry, University of Tasmania, Australia.In 2010, he was awarded the Australian Endeavour and the Japan Society for Promotionof Science (JSPS) Awards at the Kyoto Institute of Technology, Japan. In 2011 he re-turned back to Australia and was appointed Assistant Professor of Organic Chemistryat the University of Canberra. In 2012, He received the Discovery translation fundaward to develop high potential, but earlier stage commercial projects in Australia.Ashraf’s research interests lie in the area of asymmetric and enantioselective catalysisusing synthetic chiral metal or natural enzyme catalysts to access to enantiomericallypure compounds. He is also expert in the field of chiral separation using GC and HPLC.Ashraf is the Head of Biomedical Science Discipline and the Chirality group in Canberra(www.chiralitygroup.com). He teaches Organic Chemistry (>300 students) and Medic-inal Chemistry at the University of Canberra, Australia.
Marwa Ahmed studied pharmacy at Ain Shams University, Egypt. In 2002, she workedas a research assistant at the National Centre of Radiation Research and Technology,Egypt, where she started her research in synthetic pharmaceutical chemistry. In her re-search, she has studied the potential hazards of g-radiation on the biological systemand completed a masters thesis focused on the synthesis of new organic compoundswith potential antitumour and radioprotective activities. In late 2005, she joined theGerman University in Cairo where she taught Organic and Pharmaceutical Chemistryto pharmacy students. She was awarded her M.Sc. in Pharmaceutical Chemistry in2006. In 2008, she received of a short-term research scholarship from the German Ac-ademic Exchange Service (DAAD) to study the biological assay of some enzymes at theInstitute of Pharmacy at Eberhard Karls University in T€ubingen, Germany. Marwa wasalso awarded the Schlumberger Faculty for the Future Award Fellowship in 2011.Marwa is a recipient of the W.J, Weeden Postgraduate Research Scholarship, an award,which has led her to the University of Canberra, Australia to do her PhD in the chiralityprogram there under Dr. Ashraf Ghanem. Marwa’s doctoral research focuses on chiralsynthesis and analysis to develop fully integrated chiral synthesis/analysis systems forpharmaceutical industries.

M. Ahmed et al. / Tetrahedron 68 (2012) 6781e68026802
Dr. Tamsin Kelly completed a PhD in forensic toxicology at the University of Technol-ogy, Sydney (Australia) in 2004. She then undertook a post-doctoral fellowship at theNational Institute on Drug Abuse (NIDA) in Baltimore (USA). Her post-doctoral researchfocussed on the detection of in utero drug exposure of amphetamine related analoguesusing liquid chromatography-mass spectrometry (LCeMSeMS). She has extensive ex-perience in the development of analytical methods (such as LCeMSeMS and capillaryelectrophoresis (CE)) for the detection of achiral and chiral drugs in various biologicalspecimens, including plasma, urine, hair, and meconium, as well as the analysis ofclandestine drug seizures. She co-authored ‘Pharmacokinetics of Cocaine and Metabo-lites in Human Oral Fluid and Correlation with Plasma Concentrations following Con-trolled Administration’, which was awarded best paper published in Therapeutic DrugMonitoring during 2009e2011. Tamsin was a member of the Sydney 2000 Olympic andParalympic Drug Testing team. Her research interests include forensic toxicology, ap-plication of analytical chemistry to forensic and pharmaceutical related analyses, che-mometrics and chemical education. She is currently an Assistant Professor within theNational Centre for Forensic Studies, Faculty of Applied Science, University of Canberra(June 2009 to present).





























