Embed Size (px)
Citation preview

Applied Chemistry

Chapter- 1
STRUCTURE OF ATOM (MH)
Chemistry as important branch of science (H):
Chemistry has a reputation for being a complicated and boring science, but for the most part, that reputation is undeserved. Fireworks and explosions are based on chemistry, so it's definitely not a boring science. If you take classes in chemistry, you'll apply math and logic, which can make studying chemistry a challenge if you are weak in those areas. However, anyone can understand the basics of how things work and that's the study of chemistry. In a nutshell, the importance of chemistry is that it explains the world around you.
Chemistry Explains... (SH)
• Cooking Chemistry explains how food changes as you cook it, how it rots, how to preserve food, how your body uses the food you eat, and how ingredients interact to make food.
• Cleaning Part of the importance of chemistry is it explains how cleaning works. You use chemistry to help decide what cleaner is best for dishes, laundry, yourself, and your home. You use chemistry when you use bleaches and disinfectants and even ordinary soap and water. How do they work? That's chemistry!
• Medicine You need to understand basic chemistry so you can understand how vitamins, supplements, and drugs can help or harm you. Part of the importance of chemistry lies in developing and testing new medical treatments and medicines.
• Environmental Issues Chemistry is at the heart of environmental issues. What makes one chemical a nutrient and another chemical a pollutant? How can you clean up the environment? What processes can produce the things you need without harming the environment?
We're all chemists. We use chemicals every day and perform chemical reactions without thinking much about them. Chemistry is important because everything you do is chemistry! Even your body is made of chemicals. Chemical reactions occur when you breathe, eat, or just sit there reading. All matter is made of chemicals, so the importance of chemistry is that it's the study of everything.
Importance of Taking Chemistry (SH)
Everyone can and should understand basic chemistry, but it may be important to take a course in chemistry or even make a career out of it. It's important to understand chemistry if you are studying any of the sciences because all of the sciences involve matter and the interactions between types of matter. Students wanting to become doctors, nurses, physicists, nutritionists, geologists, pharmacists, and (of course) chemists all study chemistry. You might want to

make a career of chemistry because chemistry-related jobs are plentiful and high-paying. The importance of chemistry won't be diminished over time, so it will remain a promising career path.
Basic concept of Elements Mixture and compound (H)
Elements (SH) –
A chemical element is a pure chemical substance consisting of one type of atom distinguished by its atomic number, which is the number ofprotons in its nucleus. Elements are divided into metals, metalloids, and non-metals. Familiar examples of elements include carbon, oxygen (non-metals), silicon, arsenic (metalloids), aluminium, iron, copper,
gold, mercury, and lead (metals).
The lightest chemical elements, including hydrogen, helium (and smaller amounts of lithium, beryllium and boron), are thought to have been produced by various cosmic processes during the Big Bang and cosmic-ray spallation. Production of heavier elements, from carbon to the very heaviest elements, proceeded by stellar nucleosynthesis, and these were made available for later solar system and planetary formation byplanetary nebulae and supernovae, which blast these elements into space. The high abundance of oxygen, silicon, and iron on Earth reflects their common production in such stars, after the lighter gaseous elements and their compounds have been subtracted. While most elements are generally viewed as stable, a small amount of natural transformation of one element to another also occurs at the present time through decay of radioactive elements as well as other natural nuclear processes.
Mixture (SH)
Two or more substances which have been combined such that each substance retains its own chemical identity.
In chemistry, a mixture is a material system made up of two or more different substances which are mixed but are not combined chemically. A mixture refers to the physical combination of two or more substances on which the identities are retained and are mixed in the form of alloys, solutions, suspensions, and colloids.
Mixtures are the product of a mechanical blending or mixing of chemical substances like elements and compounds, without chemical bonding or other chemical change, so that each ingredient substance retains its own chemical properties and makeup. Despite that there are no chemical changes to its constituents, the physical properties of a mixture, such as its melting point, may differ from those of the components. Some mixtures can be separated into their components by physical (mechanical or thermal) means. Azeotropes can be considered as a kind of mixture which usually pose considerable difficulties regarding the separation processes required to obtain their constituents (physical or chemical processes or, even a blend of them).

Mixturesmixture propertieseen, as homogenother suhomogena solute a
Compoun
A chemicchemicalreactionsa fixed rbonds. Cbonds, sabonds, ornot consmultiple molecule
ChemicaA chemicthe reactaside.
A chemsubstancereaction)individua
As an edenoted:
2H
Balance form sod
1. Get yohelp yousulfate, a
s can be eithin which th
es. A heterogthere are tw
neous mixturubstances. Sneous mixand solvent p
nd (SH) –
cal compoul elements s. Chemical cratio of atomChemical calts held toger complexesidered chematoms of
es or polyato
al Equation, ical equatioant entities a
mical equatioes) and the . The two aral substance'
example, th
HCl + 2Na →
the equationdium sulfate
ourself an unu out. If youand water are
her homogenhe compositigeneous mixwo or more re of the ga
Salt, sugar, xtures. A present is als
und is a purthat can
compounds ms that are hcompounds ether by ion held togeth
mical compoa single el
omic molecu
its balancingon is the are given on
on consists chemical fo
re separated 's chemical f
e equation
→ 2NaCl +
n that takesand water."
nbalanced equ know whae, you'd be a
neous or heteion is uniforxture is a typhases pres
aseous substand many homogene
so a solution
re chemical be separ
have a uniquheld togethecan be mol
nic bonds, inher by coordiounds, even lement (suc
ules.
g , implicatiosymbolic
n the left han
of the cheormula of thby an arrow
formula is se
for the rea
H2
place when
quation. Hereat the formu
able to write
erogeneous. rm and everype of mixtusent. One exances nitrog
other subseous mixtun.
substance cated into ue and definer in a definlecular comp
ntermetallic cinate covalen
if they conh as H2, S
ons and limitrepresentatio
nd side and
emical formhe products w symbol (eparated from
action of hy
n sodium hy
e's where youla of sodiuthe followin
A homogenry part of thure in whichxample of a
gen, oxygen,stances dissure in w
onsisting ofsimpler
ned chemicalned spatial apounds heldcompounds hnt bonds. Punsist of molS8, etc.), wh
tations (H) on of a cthe product
mulas of the(substances, usually r
m others by
ydrochloric
ydroxide rea
ou use your kum hydroxidng unbalance
neous mixturhe solution h the compoa mixture is, and smallesolve in wawhich ther
f two or msubstances l structure; tharrangementd together held togetheure chemicallecules that
hich are cal
chemical reentities on t
e reactants s formed in read as "yiela plus sign.
acid with so
acts with su
knowledge ode, sulfuric ed equation:
re is a type ohas the sam
onents can bs air. Air is er amounts oater to formre is bot
more differenby chemic
hey consist ot by chemic
by covalener by metalll elements arcontain onl
lled diatom
eaction wherthe right han
(the startinthe chemic
ds") and eac
odium can b
ulfuric acid t
of formulas tacid, sodium
of me be
a of m th
nt al of al nt ic re ly
mic
re nd
ng al ch
be
to
to m

2. Draw boxes around all the chemical formulas. Draw the boxes:
3. Make an element inventory. In this inventory, your job is to figure out how many atoms of each element you have on the left and right sides of the equation. Now, if you look at the equation, you should be able to see that on the left side of the equation there is one sodium atom, five oxygen atoms (one from the sodium hydroxide, four from the sulfuric acid), three hydrogen atoms (one from the sodium hydroxide, two from the sulfuric acid), and one sulfur atom. On the right side of the equation, there are two atoms of sodium, one atom of sulfur, five atoms of oxygen (four from the sodium sulfate and one from the water), and two atoms of hydrogen. Thus, your element inventory should look like this:
4. Write numbers in front of each of the boxes until the inventory for each element is the same both before and after the reaction.Now, what happens when we put a number in front of a formula? Basically, anything in that box is multiplied by that number, because we're saying that we have that many of that kind of molecule. So, looking at the inventory, what should we do?
Well, we can see that on the left side of the inventory, there is one atom of sodium and on the right there are two. The solution: Stick a "2" in front of the sodium hydroxide on the left side of the equation so that the numbers of sodium atoms are the same on both sides of the equation. When we do this, the new atom inventory should look like this: (I'll let you figure out how this is done)

Now what? Well, looking at the new inventory, we can see that we now have two sodium atoms on both the left and the right sides, but the others still don't match up. What to do?
You can see from the inventory that on the right side of the equation, there are two hydrogen atoms and on the left there are four. Using your amazing powers of mathematics (and hopefully not needing to use a calculator), you can see that two multiplied by the number two becomes four. That's what you need to do. How? Put a "2" in front of the water on the right side of the equation to make the hydrogens balance out. Now that this is done, you should make a new inventory that looks something like this:
Since both sides of the inventory match, the equation is now balanced! All other equations will balance in exactly the same way.

Types of Reactions (mh)
I. Formulas show chemistry at a standstill. Equations show chemistry in action.
A. Equations show:
1. the reactants which enter into a reaction. 2. the products which are formed by the reaction. 3. the amounts of each substance used and each substance produced.
B. Two important principles to remember:
1. Every chemical compound has a formula which cannot be altered. 2. A chemical reaction must account for every atom that is used. This is an application
of the Law of Conservation of Matter which states that in a chemical reaction atoms are neither created nor destroyed.
C. Some things to remember about writing equations:
1. The diatomic elements when they stand alone are always written H2, N2, O2, F2, Cl2, Br2, I2
2. The sign, → , means "yields" and shows the direction of the action. 3. A small delta, (D), above the arrow shows that heat has been added. 4. A double arrow, ↔ , shows that the reaction is reversible and can go in both
directions. 5. Before beginning to balance an equation, check each formula to see that it is
correct. NEVER change a formula during the balancing of an equation. 6. Balancing is done by placing coefficients in front of the formulas to insure the same
number of atoms of each element on both sides of the arrow.
Practice Balancing Equations
7. Always consult the Activity Series of metals and nonmetals before attempting to write equations for replacement reactions.
8. If a reactant or product is a solid, (s) is placed after the formula. 9. If a reactant or product is a gas, (g) is placed after it. 10. If a reactant or product is in water solution, (aq) is placed after it. 11. Some products are unstable and break down (decompose) as they are produced during
the reaction. You need to be able to recognize these products when they occur and write the decomposition products in their places.
Examples:
• H2CO3(aq) → H2O(l) + CO2(g)

Carbonic acid, as in soft drinks, decomposes when it is formed.
• H2SO3(aq) → H2O(l) + SO2(g)
Sulfurous acid also decomposes as it is formed.
• NH4OH(aq) → NH3(g) + H2O(l)
You can definitely smell the odor of ammonia gas because whenever "ammonium hydroxide" is formed it decomposes into ammonia and water.
D. Rules for writing equations.
1. Write down the formula(s) for any substance entering into the reaction. Place a plus (+) sign between the formulas as needed and put the yield arrow after the last one.
2. Examine the formulas carefully and decide which of the four types of equations applies to the reaction you are considering. On the basis of your decision, write down the correct formulas for all products formed, placing them to the right of the arrow.
Four basic types of chemical reactions (mh)
A. Synthesis (composition):
• two or more elements or compounds may combine to form a more complex compound.
• Basic form: A + X → AX
Examples of synthesis reactions:
1. Metal + oxygen → metal oxide
EX. 2Mg(s) + O2(g) → 2MgO(s)
2. Nonmetal + oxygen → nonmetallic oxide
EX. C(s) + O2(g) → CO2(g)
3. Metal oxide + water → metallic hydroxide
EX. MgO(s) + H2O(l) → Mg(OH)2(s)
4. Nonmetallic oxide + water → acid

EX. CO2(g) + H2O(l) → ; H2CO3(aq)
5. Metal + nonmetal → salt
EX. 2 Na(s) + Cl2(g) → 2NaCl(s)
6. A few nonmetals combine with each other.
EX. 2P(s) + 3Cl2(g) → 2PCl3(g)
These two reactions must be remembered:
1. N2(g) + 3H2(g) → 2NH3(g) 2. NH3(g) + H2O(l) → NH4OH(aq)
B. Decomposition:
• A single compound breaks down into its component parts or simpler compounds. • Basic form: AX → A + X
Examples of decomposition reactions:
1. Metallic carbonates, when heated, form metallic oxides and CO2(g).
EX. CaCO3(s) → CaO(s) + CO2(g)
2. Most metallic hydroxides, when heated, decompose into metallic oxides and water.
EX. Ca(OH)2(s) → CaO(s) + H2O(g)
3. Metallic chlorates, when heated, decompose into metallic chlorides and oxygen.
EX. 2KClO3(s) → 2KCl(s) + 3O2(g)
4. Some acids, when heated, decompose into nonmetallic oxides and water.
EX. H2SO4 → H2O(l) + SO3(g)
5. Some oxides, when heated, decompose.
EX. 2HgO(s) → 2Hg(l) + O2(g)
6. Some decomposition reactions are produced by electricity.

EX. 2H2O(l) → 2H2(g) + O2(g)
EX. 2NaCl(l) → 2Na(s) + Cl2(g)
C. Replacement:
• a more active element takes the place of another element in a compound and sets the less active one free.
• Basic form: A + BX → AX + B or AX + Y → AY + X
Examples of replacement reactions:
1. Replacement of a metal in a compound by a more active metal.
EX. Fe(s) + CuSO4(aq) → FeSO4(aq) + Cu(s)
2. Replacement of hydrogen in water by an active metal.
EX. 2Na(s) + 2H2O(l) → 2NaOH(aq) + H2(g)
EX. Mg(s) + H2O(g) → MgO(s) + H2(g)
3. Replacement of hydrogen in acids by active metals.
EX. Zn(s) + 2HCl(aq) → ZnCl2(aq) + H2(g)
4. Replacement of nonmetals by more active nonmetals.
EX. Cl2(g) + 2NaBr(aq) → 2NaCl(aq) + Br2(l)
D. Ionic:
• occurrs between ions in aqueous solution. A reaction will occurr when a pair of ions come together to produce at least one of the following:
1. a precipitate 2. a gas 3. water or some other non-ionized substance.
• Basic form: AX + BY → AY + BX
Examples of ionic reactions:
1. Formation of precipitate.

EX. NaCl (aq) + AgNO3(aq) → NaNO3(aq) + AgCl(s)
EX. BaCl2(aq) + Na2 SO4(aq) → 2NaCl(aq) + BaSO4(s)
2. Formation of a gas.
EX. HCl(aq) + FeS(s) → FeCl2(aq) + H2S(g)
3. Formation of water. (If the reaction is between an acid and a base it is called a neutralization reaction.)
EX. HCl(aq) + NaOH(aq) → NaCl(aq) + H2O(l)
4. Formation of a product which decomposes.
EX. CaCO3(s) + HCl(aq) → CaCl2(aq) + CO2(g) + H2O(g)
Combustion of Hydrocarbons (mh)
Another important type of reaction, in addition to the four types above, is that of the combustion of a hydrocarbon. When a hydrocarbon is burned with sufficient oxygen supply, the products are always carbon dioxide and water vapor. If the supply of oxygen is low or restricted, then carbon monoxide will be produced. This is why it is so dangerous to have an automobile engine running inside a closed garage or to use a charcoal grill indoors.
• Hydrocarbon (CxHy) + O2(g) → CO2(g) + H2O(g) • EX. CH4(g) + 2O2(g) → CO2(g) + 2H2O(g) • EX. � 2C4H10(g) + 13O2(g) → 8CO2(g) + 10H2O(g)
Implications and limitations. (H)
• It does not mention the state of the substances. So (s) for solid, (l) for liquid, (g) for gas and (vap) for vapor may be added.
• The reaction may or may not be complete. Equation does not reveal it.
• It does not give any information regarding the speed of the reaction.
• It does not give the concentration of the substances. In some cases, terms like diluted and concentrated may be added.
• It does not give the conditions of temperature, pressure, catalyst, etc. This is overcome by mentioning these above or below the arrow e.g.,

• It does not give any idea about color changes, which has to be mentioned separately.
• It does not give any indication regarding the production or absorption of heat. This is mentioned separately.
• Some reactions are reversible. They are represented by
or
.
Significances of chemical equation(H) - coefficients of a chemical reaction indicate relative, not absolute, amounts of reactants and products o usually shows the smallest numbers of atoms, molecules or ions that will satisfy the law of conservation of mass - relative masses of the reactants and products of a chemical reaction can be determined from the reaction’s coefficients o can convert moles to mass in grams by multiplying by the molar mass - reverse reaction for a chemical equation has the same relative amounts of substances as the forward reaction - chemical equations give useful quantitative information but it does NOT give an indication of whether or not the reaction will ever take place. Recapitulation of Fundamental Particles of atom(H)
The atom is a basic unit of matter that consists of a dense central nucleus surrounded by a cloud of negatively charged electrons. Theatomic nucleus contains a mix of positively charged protons and electrically neutral neutrons (except in the case of hydrogen-1, which is the only stable nuclide with no neutrons). The electrons of an atom are bound to the nucleus by the electromagnetic force. Likewise, a group of atoms can remain bound to each other by chemical bonds based on the same force, forming a molecule. An atom containing an equal number of protons and electrons is electrically neutral, otherwise it is positively or

negatively charged and is known as an ion. An atom is classified according to the number of protons and neutrons in its nucleus: the number of protons determines the chemical element, and the number of neutrons determines the isotope of the element.[1]
Protons, Neutrons, and Electrons (SH)
The electron is a subatomic particle with a negative elementary electric charge.
An electron has no known components or substructure. It is generally thought to be an elementary particle.[2] An electron has a mass that is approximately 1/1836 that of the proton.[9] The intrinsic angular momentum (spin) of the electron is a half-integer value in units of ħ, which means that it is a fermion. The antiparticle of the electron is called the positron; it is identical to the electron except that it carries electrical and other charges of the opposite sign. When an electron collides with a positron, both particles may be totally annihilated, producing gamma ray photons.
The proton is a subatomic particle with the symbol p or p+ and a positive electric charge of 1 elementary charge. One or more protons are present in the nucleus of each atom. The number of protons in each atom is its atomic number. The name proton was given to the hydrogen nucleus by Ernest Rutherford in 1920, because in previous years he had discovered that the hydrogen nucleus (known to be the lightest nucleus) could be extracted from the nuclei of nitrogen by collision, and was thus a candidate to be a fundamental particle and building block of nitrogen, and all other heavier atomic nuclei.
The free proton (a proton not bound to nucleons or electrons) is a stable particle that has not been observed to break down spontaneously to other particles. Free protons are found naturally in a number of situations in which energies or temperatures are high enough to separate them from electrons,
The neutron is a subatomic hadron particle which has the symbol n or n0, no net electric charge and a mass slightly larger than that of a proton. With the exception of hydrogen-1, nuclei of atoms consist of protons and neutrons, which are therefore collectively referred to asnucleons. The number of protons in a nucleus is the atomic number and defines the type of element the atom forms. Neutrons are necessary within an atomic nucleus as they bind with protons via the nuclear force; protons are unable to bind with each other (see diproton) because their mutual electromagnetic repulsion is stronger than the attraction of the nuclear force.[4] The number of neutrons is the neutron number and determines the isotope of an element. For example, the abundant carbon-12 isotope has 6 protons and 6 neutrons, while the very rare radioactive carbon-14 isotope has 6 protons and 8 neutrons.
Modern atomic structure(H)
Near the end of the 18th century, two laws about chemical reactions emerged without referring to the notion of an atomic theory. The first was the law of conservation of mass, formulated byAntoine Lavoisier in 1789, which states that the total mass in a chemical reaction remains constant (that is, the reactants have the same mass as the products). The second was the law of definite proportions. First proven by the French chemist Joseph Louis

Proust inelementsregardles
John Dalproportioof the mwill be rtheir maswere tin(100g of tDalton fochemistrytwo oxyg
Dalton adifferent than it abcomplexiheavier a
Dalton pand thougmore comof the atoresults in
.
In 1803 substanceobtained Thomson
Dalton ewith the elements
n 1799, this , then the
ss of the qua
lton studied aons: if two e
masses of theratios of smsses were eit(II) oxide antin will combfound an atoy - in the cagen atoms.
also believedproportions
bsorbednitroity of the g
and larger tha
roposed thatgh they cannmplex structom, since Dan an empirica
Dalton orales. This papthese figure
n.
stimated thehydrogen a atoms exis
law states masses of
antity or sour
and expandelements cam
e second elemmall integers.
ther 88.1% tnd tin dioxidbine either w
omic theory ase of Prous
d atomic ths: for exampogen. Daltongases' respecan nitrogen m
t each chemnot be alteretures (chemialton reacheal fashion.
lly presentedper was publes. The meth
e atomic weatom taken ast in molecu
that if a cthe constitu
rce of the ori
ed upon this me together tment whichFor instanc
tin and 11.9%de respectivewith 13.5g o
of matter cst's tin oxide
eory could ple, he founn hypothesizctive particlemolecules (N
mical elemend or destroycal compound his conclu
d his first llished in 18hod was firs
eights accordas unity. Houles — e.g.
compound iuents will aiginal substa
previous woto form mor
h combine wce, Proust h% oxygen orely). Dalton
or 27g of oxycould eleganes, one tin a
explain whnd that watezed this wases. Indeed, cN2).
nt is composeyed by cheminds). This m
usions by exp
list of relati805, but he dst revealed i
ding to the mowever, Dalt
pure oxyge
s broken dalways haveance.
ork and devere than one c
with a fixed mhad studied tr 78.7% tin
n noted fromygen; 13.5 antly explain
atom will co
y water abser absorbeds due to the carbon diox
ed of atomsical means, t
marked the fiperimentatio
ive atomic wdid not discuin 1807 by
mass ratios ton did not en exists as
down into ite the same
eloped the lacompound, tmass of the tin oxides anand 21.3% o
m these perand 27 form n this comm
mbine with
sorbed diffecarbon dioxdifferences
xide molecul
of a single,they can com
first truly scion and exam
weights for uss there exhis acquaint
in which thconceive th O2. He als
ts constituene proportion
aw of multipthen the ratio
first elemennd found thoxygen (thesrcentages tha ratio of 1:2
mon pattern ieither one o
erent gases ixide far bettes in mass anles (CO2) ar
, unique typmbine to formientific theor
mination of th
a number oxactly how htance Thoma
ey combinehat with somso mistakenl
nt ns,
le os nt at se at 2. in or
in er nd re
e, m ry he
of he as
d, me ly

believed (so he thflawed hheavier t1 gram o1806 he he retainthe oxygefor the w
The flawAvogadrpressure,does not of the mnumerouof hydrogconstant to form tthe atommoleculeviewed Adoubtingcommuni
In 1827, floating Einstein
Werner H
In quantuinequalitipropertieas positiopreciselyknown, agiven byprinciplestandard
where ħ i
Historicain physic
that the simhought waterhis results. Fthan hydrogeof hydrogen concluded thed this weigen atom mus
water molecu
w in Daltono had propo contain equaffect the ga
molecule itsus gases by sgen will reapressure and
two particlesic mass of o
es and atomAvogadro's the truth oity only afte
the British in water
theorized th
Heisenbers’s
um mechanies asserting
es of on x and momy the positionand vice very Werner He. A more fdeviation of
is the reduce
ally, the unccs, called th
mplest compor was HO, nFor instanceen atoms, beand believe
hat the atomght for the rest weigh 8 rele (HO), or
n's theory wosed that equal numbersas volume thself). Avogadstudying the act with just d temperaturs of water. Toxygen and v
ms. Howeverideas as e
f Avogadroer about 1860
botanist Robconstantly at this Brow
s Uncertainty
nics, the uncg a fundamena particlmentum p, n of some parsa. The origeisenberg in formal inequf momentum
ed Planck co
certainty prihe observer
ound betweenot H2O). Te, in 1803 hecause in wed the formu
mic weight oest of his lifelative to hy16 if one ass
was correctequal volumes of moleculhat it occupidro's law alvolumes at one liter ofre), it meant
Thus, Avogavarious other, in the firsexcessively 's ideas. Th0.
bert Brownjiggled abo
wnian motion
y principle
certainty prntal limit to e knowncan be kn
article is detginal heuris
1927, afteruality relatin
m σp was deri
onstant.
inciple has effect, whic
en any two ehis, in addithe believed ater he meaula for wate
of oxygen mfe. Others at ydrogen equasumes the m
ed in principes of any twles (in otheries, which inllowed him which they r
f oxygen to pt a single ox
adro was abler elements, st half of thhypothetica
ese ideas w
observed thout for no n was caused
rinciple is athe precision
n as cnown simulttermined, thetic argumenr whom it ing the standived by Earl
been confuch notes tha
elements is tion to the c
that oxygeasured 5.5 grer was HO.
must actually this time ha
als 1, if one amodern water
ple in 1811wo gases, ar words, the n turn is muc
to deduce reacted. Forproduce twoxygen molecle to offer mand made a
he nineteenthal, and had
were widely
hat dust partapparent
d by the wate
any of a vn with which
complementataneously. e less precis
nt that such is sometime
dard deviatioe Hesse Ken
used with a at measurem
always one crudity of hen atoms werams of oxy
Adopting bbe 7 rather
ad already cassumes Dal
r formula.
1 by Amedeat equal tem
mass of a gch larger tha
the diatomr instance: sio liters of wcule splits in
more accurateclear distinch century m
d theoreticalaccepted in
ticles inside reason. In er molecules
variety of h certain paiary variaFor instanc
sely its moma limit shou
es named thon of positionnard.
somewhat sments of cer
atom of eacis equipmenere 5.5 timegen for ever
better data, ithan 5.5, anoncluded thlton's formu
eo Avogadrmperature angas's particlean the volum
mic nature oince two literater vapor (
n two in ordee estimates oction betweemost chemisl reasons fothe chemic
pollen grain1905, Albe
s.
mathematicirs of physicables, succe, the mor
mentum can buld exist wahe Heisenberon σx and th
similar effertain system
ch nt, es ry in nd at la
o. nd es
me of rs at er of en sts or al
ns ert
al al ch re be as rg he
ct ms

cannot be made without affecting the systems. Heisenberg offered such an observer effect at the quantum level as a physical "explanation" of quantum uncertainty. It has since become clear, however, that the uncertainty principle is inherent in the properties of all wave-like systems, and that it arises in quantum mechanics simply due to the matter wave nature of all quantum objects. Thus, the uncertainty principle actually states a fundamental property of quantum systems, and is not a statement about the observational success of current technology. It must be emphasized that measurement does not mean only a process in which a physicist-observer takes part, but rather any interaction between classical and quantum objects regardless of any observer.
Since the uncertainty principle is such a basic result in quantum mechanics, typical experiments in quantum mechanics routinely observe aspects of it. Certain experiments, however, may deliberately test a particular form of the uncertainty principle as part of their main research program. These include, for example, tests of number-phase uncertainty relations in quantum optics systems. Applications are for developing extremely low noise technology such as that required in gravitational-wave interferometers.
Atomic Orbital (sh)
An atomic orbital is a mathematical function that describes the wave-like behavior of either one electron or a pair of electrons in an atom.[1] This function can be used to calculate the probability of finding any electron of an atom in any specific region around the atom's nucleus. The term may also refer to the physical region or space where the electron can be calculated to be present, as defined by the particular mathematical form of the orbital.
Each orbital in an atom is characterized by a unique set of values of the three quantum numbers n, ℓ, and m, which correspond to the electron's energy, angular momentum, and an angular momentum vector component, respectively. Any orbital can be occupied by a maximum of two electrons, each with its own spin quantum number. The simple names s orbital, p orbital, d orbital and f orbital refer to orbitals with angular momentum quantum number ℓ = 0, 1, 2 and 3 respectively. These names, together with the value of n, are used to describe the electron configurations. They are derived from the description by early spectroscopists of certain series of alkali metal spectroscopic lines as sharp, principal, diffuse, and fundamental. Orbitals for ℓ > 3 are named in alphabetical order.
Atomic orbitals are the basic building blocks of the atomic orbital model (alternatively known as the electron cloud or wave mechanics model), a modern framework for visualizing the submicroscopic behavior of electrons in matter. In this model the electron cloud of a multi-electron atom may be seen as being built up (in approximation) in an electron configuration that is a product of simpler hydrogen-like atomic orbitals. The repeating periodicity of the blocks of 2, 6, 10, and 14 elements within sections of the periodic table arises naturally from the total number of electrons which occupy a complete set of s, p, d and f atomic orbitals, respectively.

Types of orbitals (sh)
Atomic orbitals can be the hydrogen-like "orbitals" which are exact solutions to the Schrödinger equation for a hydrogen-like "atom" (i.e., an atom with one electron). Alternatively, atomic orbitals refer to functions that depend on the coordinates of one electron (i.e. orbitals) but are used as starting points for approximating wave functions that depend on the simultaneous coordinates of all the electrons in an atom or molecule. The coordinate systems chosen for atomic orbitals are usually spherical coordinates (r,�θ,�φ) in atoms and cartesians (x,�y,�z) in polyatomic molecules. The advantage of spherical coordinates (for atoms) is that an orbital wave function is a product of three factors each dependent on a single coordinate: ψ(r,�θ,�φ) = R(r)�Θ(θ)�Φ(φ).
The angular factors of atomic orbitals Θ(θ) Φ(φ) generate s, p, d, etc. functions as real combinations of spherical harmonics Yℓm(θ,�φ) (where ℓ and m are quantum numbers). There are typically three mathematical forms for the radial functions R(r) which can be chosen as a starting point for the calculation of the properties of atoms and molecules with many electrons.
1. the hydrogen-like atomic orbitals are derived from the exact solution of the Schrödinger Equation for one electron and a nucleus, for a hydrogen-like atom. The part of the function that depends on the distance from the nucleus has nodes (radial nodes) and decays as e−(constant × distance).
2. The Slater-type orbital (STO) is a form without radial nodes but decays from the nucleus as does the hydrogen-like orbital.
3. The form of the Gaussian type orbital (Gaussians) has no radial nodes and decays as e(−distance squared).
Although hydrogen-like orbitals are still used as pedagogical tools, the advent of computers has made STOs preferable for atoms and diatomic molecules since combinations of STOs can replace the nodes in hydrogen-like atomic orbital. Gaussians are typically used in molecules with three or more atoms. Although not as accurate by themselves as STOs, combinations of many Gaussians can attain the accuracy of hydrogen-like orbitals.
The order of filling orbitals - the Aufbau Principle

Aufbau is a German word meaning building up or construction. We imagine that as you go from one atom to the next in the Periodic Table, you can work out the electronic structure of the next atom by fitting an extra electron into the next available orbital.
Electrons fill low energy orbitals (closer to the nucleus) before they fill higher energy ones. Where there is a choice between orbitals of equal energy, they fill the orbitals singly as far as possible.
This filling of orbitals singly where possible is known as Hund's rule. It only applies where the orbitals have exactly the same energies (as with p orbitals, for example), and helps to minimise the repulsions between electrons and so makes the atom more stable.
The diagram (not to scale) summarises the energies of the orbitals up to the 4p level that you will need to know when you are using the Aufbau Principle.
Notice that the s orbital always has a slightly lower energy than the p orbitals at the same energy level, so the s orbital always fills with electrons before the corresponding p orbitals.
The real oddity is the position of the 3d orbitals. They are at a slightly higher level than the 4s - and so it is the 4s orbital which you fill first, followed by all the 3d orbitals and then the 4p orbitals.
Similar confusion occurs at higher levels, with so much overlap between the energy levels that you don't fill the 4f orbitals until after the 6s.
Bohr’s model of atom (H)
Bohr model, introduced by Niels Bohr in 1913, depicts the atom as small, positively charged nucleussurrounded by electrons that travel in circular orbits around the nucleus—similar in structure to the solar system, but with attraction provided by electrostatic forces rather than gravity. After the cubic model (1902), the plum-pudding model (1904), the Saturnian model(1904), and the Rutherford model (1911) came the Rutherford–Bohr model or just Bohr model for short (1913). The improvement to the Rutherford model is

mostly a quantum physical interpretation of it. The Bohr model has been superseded, but the quantum theory remains sound.
The modern model of the atom is based on quantum mechanics. The Bohr Model contains some errors, but it is important because it describes most of the accepted features of atomic theory without all of the high-level math of the modern version. Unlike earlier models, the Bohr Model explains the Rydberg formula for the spectral emission lines of atomic hydrogen.
The Bohr Model is a planetary model in which the negatively-charged electrons orbit a small, positively-charged nucleus similar to the planets orbiting the Sun (except that the orbits are not planar). The gravitational force of the solar system is mathematically akin to the Coulomb (electrical) force between the positively-charged nucleus and the negatively-charged electrons.
Main Points of the Bohr Model (SH)
• Electrons orbit the nucleus in orbits that have a set size and energy. • The energy of the orbit is related to its size. The lowest energy is found in the smallest
orbit. • Radiation is absorbed or emitted when an electron moves from one orbit to another.
Bohr Model of Hydrogen (SH)
The simplest example of the Bohr Model is for the hydrogen atom (Z = 1) or for a hydrogen-like ion (Z > 1), in which a negatively-charged electron orbits a small positively-charged nucleus. Electromagnetic energy will be absorbed or emitted if an electron moves from one orbit to another. Only certain electron orbits are permitted. The radius of the possible orbits increases as n2, where n is the principal quantum number. The 3 → 2 transition produces the first line of the Balmer series. For hydrogen (Z = 1) this produces a photon having wavelength 656 nm (red light).

Problems
• It viola know
• The Bmome
• It mak• It does• The B• It does
Line Spe
The emiswavelengmoving bdetectingas spectro
the speclevels. Telectron j
s with the Bo
ates the Heiwn radius anBohr Modeentum. kes poor preds not predict
Bohr Model ds not explain
ectrum of Hy
ssion spectrugths given bybetween eneg the presencoscopy techn
tral lines of The simplestjumps from
ohr Model (S
senberg Uncnd orbit. l provides
dictions regat the relativedoes not expn the Zeeman
ydrogen (H)
um of atomiy the Rydbergy levels ince of hydrogniques devel
hydrogen cot model of a higher ene
SH)
certainty Prin
an incorrec
arding the spe intensities olain fine strun Effect.
ic hydrogenrg formula.
n the atom. Tgen and calcloped.
orrespond tothe hydroge
ergy to a low
nciple becau
ct value fo
pectra of largof spectral liucture and h
is divided inThese obser
The spectral culating red s
o particular jen atom is g
wer, a photon
use it consid
or the groun
ger atoms. ines. yperfine stru
nto a numberved spectralseries are im
shifts. Furth
jumps of thegiven by th
n of a specifi
ers electrons
nd state or
ucture in spe
er of spectral lines are dumportant in aher series we
e electron behe Bohr modic wavelengt
s to have bot
rbital angula
ectral lines.
al series, witue to electronastronomy foere discovere
etween energdel. When ath is emitted
th
ar
th ns or ed
gy an d.

The specstarting fwithin ea→ 3 lineseries, sas hyperfor more observe tand addior nitrogeobservatiwaveleng
Rydberg
The eneremitted/a
wherconstone o
Modern c
An atomione elect
ctral lines afrom the loach series. Fe is called "uch as thefine transitioclosely grouthese series itional linesen in the aiions of sugths.
formula(SH
rgy differenabsorbed pho
re n is the intant. Meaninover infinity
concept of a
ic orbital is tron or a pa
are grouped ongest waveFor example,Paschen-del
e 21 cm linons. Thefine uped thinnerfrom pure h
s can be cair). Lines ounlight, as
H)
nces betweenotons, is give
nitial energngful values
is taken to b
tomfour (H)
a mathematir of electro
into series elength/lowe, the 2 → 1lta" (Pa-δ). ne; these costructure als
r lines, due thydrogen sa
aused by othutside of ththe atmosp
n levels in ten by the Ry
gy level, n′ iare returne
be zero.
)
tical functionons in an at
according est frequencyline is calleSome hydroorrespond tso results in to relativisticamples in a her element
he visible spphere absor
the Bohr mydberg formu
s the final d only when
n that descritom. This fu
to n′. Linesy of the sed "Lyman-aogen spectrato much rasingle spectc correctionlab. Many ots (such as hpectrum typrbs most in
model, and hula:
energy levn n is greate
ibes the wavunction can
s are namedries, using
alpha" (Ly-αal lines fall arer atomic tral lines apps. Typically of the lines ahelium if usically cannonfra-red an
hence the wa
el, and R is
er than n′ and
ve-like behabe used to
d sequentiallGreek letter
α), while theoutside thesevents suc
pearing as twone can onl
are very fainsing sunlighot be seen ind ultraviol
avelengths o
the Rydberd the limit o
avior of eithecalculate th
ly rs 7
se ch
wo ly nt
ht, in et
of
rg of
er he

probability of finding any electron of an atom in any specific region around the atom's nucleus. The term may also refer to the physical region where the electron can be calculated to be, as defined by the particular mathematical form of the orbital.
Each orbital in an atom is characterized by a unique set of values of the three quantum numbers n, ℓ, and m, which correspond to the electron's energy, angular momentum, and an angular momentum vector component, respectively. Any orbital can be occupied by a maximum of two electrons, each with its own spin quantum number. The simple names s orbital, p orbital, d orbital and f orbital refer to orbitals with angular momentum quantum number ℓ = 0, 1, 2 and 3 respectively. These names, together with the value of n, are used to describe the electron configurations. They are derived from the description by early spectroscopists of certain series of alkali metal spectroscopic lines as sharp, principal, diffuse, and fundamental. Orbitals for l > 3 are named in alphabetical order (omitting j).
Atomic orbitals are the basic building blocks of the atomic orbital model (alternatively known as the electron cloud or wave mechanics model), a modern framework for visualizing the submicroscopic behavior of electrons in matter. In this model the electron cloud of a multi-electron atom may be seen as being built up (in approximation) in an electron configuration that is a product of simpler hydrogen-like atomic orbitals. The repeating periodicity of the blocks of 2, 6, 10, and 14 elements within sections of the periodic table arises naturally from the total number of electrons which occupy a complete set of s, p, d and f atomic orbitals, respectively.
Quantum numbers (H)
Quantum numbers describe values of conserved quantities in the dynamics of the quantum system. Perhaps the most peculiar aspect of quantum mechanics is the quantization of observable quantities, since quantum numbers are discrete sets of integers or half-integers. This is distinguished from classical mechanics where the values can range continuously. Quantum numbers often describe specifically the energies of electrons in atoms, but other possibilities include angular momentum, spin, etc. Any quantum system can have one or more quantum numbers; it is thus difficult to list all possible quantum numbers.
shells, subshells, orbital (H)
An electron shell, also called a principle energy level may be thought of as an orbit followed by electrons around an atom's nucleus. The closest shell to the nucleus is called the "1 shell" (also called "K shell"), followed by the "2 shell" (or "L shell"), then the "3 shell" (or "M shell"), and so on farther and farther from the nucleus. The shells correspond with the principal quantum numbers (1, 2, 3, 4...) or are labeled alphabetically with letters used in the X-ray notation (K, L, M, …).
Each shell can contain only a fixed number of electrons: The 1st shell can hold up to two electrons, the 2nd shell can hold up to eight electrons, the 3rd shell can hold up to 18, and 4th shell can hold up to 32 and so on. Since electrons are electrically attracted to the nucleus, an

atom's elbeen commay havexist in th
The electthe atom
Each shmore ato
Pauli’s ex
The Paultwo identstate simuidentical was form
For examif n, ℓ, anand so on
The Pauinteger spinclude eneutrons)fermionsdifferent
For examhas spin everyday
"Half-int
is theory oparticles the samefor super
Aufbau E
The Aufbused to postulateelectronsrespect to
lectrons willmpletely filleve two or evhese shells s
trons in the o; it is called
ell consistsmic orbitals
xclusion prin
li exclusitical fermionultaneously.fermions is
mulated by A
mple, no twond mℓ are then.
uli exclusionpin"), whileelementary ), electrons a, and are theoverall "spin
mple helium0 and is a bo
y matter, from
teger spin"
(reduf quantum mwith integer
e quantum strconductivity
Energy ranki
bau principldetermine
es a hypothes. As they aro the nucleus
l generally oed by other ven three incsee electron c
outermost octhe valence
s of one o.
nciple (H)
on princins (particles . A more rs anti-symme
Austrian phys
o electrons ie same, ms m
n principle ge bosons (par
particles sucand neutrinoerefore subjn", which de
m-3 has spin oson. As sucm its large-s
means th
uced Planck'smechanics fer spin havetates. Bosony, and the W
ing rule (H)
le (from thethe electron
etical procese added, thes and those e
occupy outerelectrons. H
complete ouconfiguratio
ccupied shelshell.
r more subs
iple is thewith half
rigorous statetric with resicist Wolfga
in a single amust be diffe
governs therticles with ch as quark
os. In addect to the Paetermines wh
1/2 and is thch, the Pauli cale stability
hat the in
s constant) ermions are e symmetric s include th
W and Z boso
e German Aun configuratiss in which
ey assume thelectrons alr
r shells only However, thuter shells. Fon.
ll (or shells)
shells, and
e quantum f-integer spintement is thspect to excang Pauli in
atom can haferent such th
e behavior o"integer sp
ks (the condition, protonauli exclusiohether they a
herefore a feexclusion pr
y, to the chem
ntrinsic angul
times a halfdescribed b
wave functie photon, th
ons.
ufbau meaniion of an atan atom is
heir most stabready there.
if the more is is not a sFor an expla
determine t
each subsh
mechanicn) may ochat the tota
change of th1925.
ave the samehat the elect
of all fermioin") are not
nstituent pans and neutron principle are fermions
ermion, in corinciple undmical behav
lar momen
f-integer (1/2by antisymmions; unlike
he Cooper pa
ng "buildintom, molecus "built up" ble condition
inner shellsstrict requireanation of w
the chemical
hell consist
al principle cupy the sal wave fun
he particles.
e four quanttrons have op
ons (particlet subject to rticles of ons and soas well. Ato
s or bosons
ontrast to heerpins many
vior of atoms
ntum value
2, 3/2, 5/2, metric states
fermions thairs which ar
ng up, constule or ion. Tby progress
ns (electron
s have alreadement: Atomwhy electron
l properties o
ts of one o
that nsamequantum
nction for twThe princip
tum numberpposite spin
s with "halit. Fermionprotons an
ome atoms aroms can hav
elium-4 whicy properties os.
of fermion
etc.). In ths. In contrashey may sharre responsib
truction": The principsively addinorbitals) wit
dy ms ns
of
or
no m
wo le
rs; ns,
lf-ns nd re ve
ch of
ns
he st, re le
is le
ng th

According to the principle, electrons fill orbitals starting at the lowest available energy levels before filling higher levels (e.g. 1s before 2s). The number of electrons that can occupy each orbital is limited by the Pauli exclusion principle. If multiple orbitals of the same energy are available.
Orbital concept types of bonds covalency (H) The valence bond theory was proposed by Heitler and London to explain the formation of covalent bond quantitatively using quantum mechanics.
The main postulates of this theory are as follows:
* A covalent bond is formed by the overlapping of two half filled valence atomic orbitals of two different atoms.
* The electrons in the overlapping orbitals get paired and confined between the nuclei of two atoms.
* The electron density between two bonded atoms increases due to overlapping. This confers stability to the molecule.
* Greater the extent of overlapping, stronger is the bond formed.
* The direction of the covalent bond is along the region of overlapping of the atomic orbitals i.e., covalent bond is directional.
Comparison of valence bond and molecular orbital theory (SH)
In some respects valence bond theory is superior to molecular orbital theory. When applied to the simplest two-electron molecule, H2, valence bond theory, even at the simplest Heitler-London approach, gives a much closer approximation to the bond energy, and it provides a much more accurate representation of the behavior of the electrons as chemical bonds are formed and broken. In contrast simple molecular orbital theory predicts that the hydrogen molecule dissociates into a linear superposition of hydrogen atoms and positive and negative hydrogen ions, a completely unphysical result. This explains in part why the curve of total energy against interatomic distance for the valence bond method lies below the curve for the molecular orbital method at all distances and most particularly so for large distances. This situation arises for all homonuclear diatomic molecules and is particularly a problem for F2, where the minimum energy of the curve with molecular orbital theory is still higher in energy than the energy of two F atoms.
The concepts of hybridization are so versatile, and the variability in bonding in most organic compounds is so modest, that valence bond theory remains an integral part of the vocabulary of organic chemistry. However, the work of Friedrich Hund, Robert Mulliken, and Gerhard Herzberg showed that molecular orbital theory provided a more appropriate description of the spectroscopic, ionization and magnetic properties of molecules. The deficiencies of valence bond theory became apparent when hypervalent molecules (e.g. PF5) were explained without the use of d orbitals that were crucial to the bonding hybridisation scheme proposed for such

molecules by Pauling. Metal complexes and electron deficient compounds (e.g. diborane) also appeared to be well described by molecular orbital theory, although valence bond descriptions have been made.
In the 1930s the two methods strongly competed until it was realised that they are both approximations to a better theory. If we take the simple valence bond structure and mix in all possible covalent and ionic structures arising from a particular set of atomic orbitals, we reach what is called the full configuration interaction wave function. If we take the simple molecular orbital description of the ground state and combine that function with the functions describing all possible excited states using unoccupied orbitals arising from the same set of atomic orbitals, we also reach the full configuration interaction wavefunction. It can be then seen that the simple molecular orbital approach gives too much weight to the ionic structures, while the simple valence bond approach gives too little. This can also be described as saying that the molecular orbital approach is too delocalised, while the valence bond approach is too localised.
The two approaches are now regarded as complementary, each providing its own insights into the problem of chemical bonding. Modern calculations in quantum chemistry usually start from (but ultimately go far beyond) a molecular orbital rather than a valence bond approach, not because of any intrinsic superiority in the former but rather because the MO approach is more readily adapted to numerical computations. However better valence bond programs are now available.
The formation of a covalent bond involves the overlapping of half-filled atomic orbitals. The covalent bonds can be classified into two different categories depending upon the type of overlapping. These are: (a) Sigma covalent bond (b) Pi covalent bond. (a) Sigma ( σ) bond. This type of covalent bond is formed by the axial overlapping of half-filled atomic orbitals. The atomic orbitals overlap along the inter-nuclear axis and involve end to end or head on overlap. The electron cloud formed as a result of axial overlap is cylindrically symmetrical about inter-nuclear axis. The electrons constituting sigma bond are called sigma electrons. There can be three types of axial overlap among s and p-orbitals as discussed below:
(z) s-s overlap. It involves mutual overlap of half-filled s-orbitals of the atoms approaching to form a bond. The bond formed is called s-s σ bond •
(ii) s-p overlap. It involves mutual overlap of half-filled s-orbital of the one atom with half-filled p-orbital of the other. The bond so formed is called s-p σ bond.
(iii) p-p overlap. It involves mutual overlap of half-filled p-orbitals of the two atoms. The bond so formed is called p-p σ bond.

(b) Pi (1t) Bond. This type of covalent bond is formed by the lateral or sidewise overlap of the atomic orbitals. The orbital overlap takes place in such a way that their axes are parallel to each other but perpendicular to the internuclear axis. The pi bond consists of two charge clouds above and below the plane of the atoms involved in the bond formation. The electrons involved in the 1t-bond formation are called n-electrons. SOME CHARACTERISTIC FEATURES OF π – BONDS (SH) A pi (π) bond is constituted by side ways overlap of orbital perpendicular to the internuclear axis, some characteristic features are:
(i) All the atoms directly attached to the carbon atoms of double bond lie in the same plane. For example, in CH2 = CH2 all the six atoms (2 carbon atoms and 4 hydrogen atoms) lie in the same plane. (ii) Only the unhybridised p-orbitals perpendicular to the plane of the molecule from pi bonds. (iii) Rotation of one C~ fragment with respect to other interferes with maximum overlap of p-orbitals and, therefore, such rotation about carbon-carbon double bond (C = C) is restricted. (iv) The electron charge cloud of the 1t-bond is placed above and below the plane of bonding atoms. This results in the electrons being easily available to the attacking reagents. In general, n-bonds provide the most reactive centres in the molecules containing multiple bonds. It may be noted that:
(i) Sigma bond is stronger than pi bond. It is because of the fact that overlapping of atomic orbitals can take place to a greater extent during the formation of sigma bond whereas overlapping of orbitals occurs to a smaller extent during the formation of pi bond. (ii) Pi bond between the two atoms is formed only in addition to a sigma bond. It is because of the fact that the atoms constituting a single bond prefer to form a strong sigma bond rather than a weak pi bond. Thus, pi bond is always present in molecules having multiple bonds, i.e., double or triple bond. In other words, a single bond cannot be a pi bond. (iii) The shape of molecule is controlled by the sigma framework (orientations of sigma bonds) around the central atom. Pi bonds are superimposed on sigma bonds hence they simply modify the dimensions of the molecule.

Atom Nucleus
The nucleus is the very dense region consisting of protons and neutrons at the center of an atom. It was discovered in 1911 as a result of Ernest Rutherford's interpretation of the 1909 Geiger–Marsden gold foil experiment performed by Hans Geiger and Ernest Marsden under Rutherford's direction. The proton–neutron model of nucleus was proposed by Dmitry Ivanenko in 1932. Almost all of the mass of an atom is located in the nucleus, witha very small contribution from the electron cloud.
The diameter of the nucleus is in the range of 1.75 fm (1.75×10−15 m) for hydrogen to about 15 fm for the heaviest atoms, such as uranium. These dimensions are much smaller than the diameter of the atom itself (nucleus + electron cloud), by a factor of about 23,000(uranium) to about 145,000 (hydrogen).
The branch of physics concerned with studying and understanding the atomic nucleus,including its composition and the forces which bind it together, is called nuclear physics.
The nucleus was discovered in 1911, as a result of Ernest Rutherford's efforts to test Thomson's "plum pudding model" of the atom. The electron had already been discovered earlier by J.J. Thomson himself, and knowing that atoms are neutral, Thomson postulated thatthere must be a positive charge as well. In his plum pudding model, Thomson stated that anatom consisted of negative electrons randomly scattered within a sphere of positive charge. Ernest Rutherford later devised an experiment that involved the deflection of alpha particlesat a thin sheet of metal foil. He reasoned that if Thomson's model were correct, the immensealpha particles would easily pass through the foil with very little deviation in their paths. Tohis surprise, many of the particles were deflected at very large angles. Because the mass ofalpha particles is about 8000 times that of an electron, it became apparent that a very strong force was present that allowed the particles to be deflected. He realized that the plum puddingmodel could not be accurate and that the deflections of the alpha particles could only becaused by a center of concentrated charge that contained most of the atom's mass. Thus, the idea of a nuclear atom—an atom with a dense center of positive charge—became justified.

Atomic number(SH)
In chemistry and physics, the atomic number (also known as the proton number) is the number of protons found in the nucleus of an atom and therefore identical to the charge number of the nucleus. It is conventionally represented by the symbol Z. The atomic number uniquely identifies a chemical element. In an atom of neutral charge, the atomic number is also equal to the number of electrons.
The atomic number, Z, should not be confused with the mass number, A, which is the number of nucleons, the total number of protons and neutrons in the nucleus of an atom. The number of neutrons, N, is known as the neutron number of the atom; thus, A = Z + N (these quantities are always whole numbers). Since protons and neutrons have approximately thesame mass (and the mass of the electrons is negligible for many purposes), and the mass defect of nucleon binding is always small compared to the nucleon mass, the atomic mass of any atom, when expressed in unified atomic mass units (making a quantity called the "relative isotopic mass,") is roughly (to within 1%) equal to the whole number A.
Atoms having the same atomic number Z but different neutron number N, and hence different atomic masses, are known as isotopes. A little more than three-quarters of naturally occurring elements exist as a mixture of isotopes (see monoisotopic elements), and the average isotopic mass of an isotopic mixture for an element (called the relative atomic mass) in a definedenvironment on Earth, determines the element's standard atomic weight. Historically, it was these atomic weights of elements (in comparison to hydrogen) that were the quantitiesmeasurable by chemists in the 19th century.
Mass number(SH)
The mass number (A), also called atomic mass number or nucleon number, is the total number of protons and neutrons (together known as nucleons) in an atomic nucleus. Because protons and neutrons both are baryons, the mass number A is identical with the baryon number B as of the nucleus as of the whole atom or ion. The mass number is different for each different isotope of a chemical element. This is not the same as the atomic number (Z) which denotes the number of protons in a nucleus, and thus uniquely identifies an element.Hence, the difference between the mass number and the atomic number gives the number of neutrons (N) in a given nucleus: N=A−Z.
The mass number is written either after the element name or as a superscript to the left of anelement's symbol. For example, the most common isotope of carbon is carbon-12, or 12C, which has 6 protons and 6 neutrons. The full isotope symbol would also have the atomicnumber (Z) as a subscript to the left of the element symbol directly below the mass number: 12 6C. This is technically redundant, as each element is defined by its atomic number, so it isoften omitted.

These are the elements having same atomic number but different mass number. They havethe same atomic number because the number of protons inside their nuclei remains the same.The difference in their mass number is due to the difference in their number of neutrons.
Since they are neutral isotopes are elements having same number of electrons, which makethem to possess identical chemical properties. Let us see some examples 1H1, 1H2, 1H3 are all isotopes of hydrogen. They all have their atomic number to be unity but the number ofneutrons are 0, 1, 2 and z respectively. 17Cl37, 17Cl35 are isotopes of chlorine. They have 17 protons in the nucleus but have number of neutrons equal to 20 and 18 respectively.Practically every element consists of a mixture of several isotopes. The relative abundance ofdifferent isotopes differs from element to element. For example chlorine is composed of two isotopes of masses 34.98U and 36.98U, which are nearly integral multiples of the mass ofhydrogen atom. Their relative abundances are 75.4 and 24.6 percent respectively. Mass ofnatural chlorine atom can be found as
= 35.47.
The isotope can occur either naturally or can be produced artificially in the laboratory.
Isobars Isotopes are chemically same and physically different. But the converse is true in isobars.That is isobars are elements, which are chemically different but physically same. So, isobars are atoms of different elements having the same atomic mass but different atomic number.Since their number of electrons is different, their chemical properties are different. The lightnuclei have unstable isobars. Heavy nuclei have stable isobars and these occur in pairs.Suppose the number of protons of one isobar matches with that of another they are called asmirror-nuclides of each other.
Examples of isobars are
Since isobars are different elements they appear in different places in the periodic table.
Isotones Isotones are elements having the same number of neutrons. Examples of isotones areChlorine - 37 and Potassium - 39. Both have 20 neutrons in their nuclei.
Characteristics of isotopes

(i) Isotopes of an element have the same number of proton inside their nuclei .as a result; allthe isotopes of an element contain the same number of electrons.
(ii) Different isotopes of an element have different mass numbers. So isotopes show thefollowing characteristics.
(a) Since, isotopes of an element have the same number of protons and electrons; hence allthe isotopes of an element show the same chemical properties, same electronicconfigurations, and the same number of valence electrons. For example , for the threeisotopes of oxygen,
16
8O 178O 18
8O
No. of protons 8 8 8
No. of electrons 8 8 8
Electronic configuration 2.6 2.6 2.6
No. of Valence electrons 6 6 6
Isotopes of an element have different masses. So, the properties which depend upon theatomic mass should be different for different isotopes. Many physical properties e.g., meltingpoint, boiling point, density, etc., depend upon the atomic mass. So different isotopes of anelement show different physical properties. For example, physical properties of the twoisotopes of hydrogen are different.
Characteristics of Isobars:
a. Their atomic masses are nearly equal. b. They possess different chemical and physical properties. c. They take different places in the periodic table. d. They possess different radioactive properties.
Application:
1. - Isotopes are used in the kinetic study of reaction mechanism 2. - C-14 isotopes are used in finding the age of fossils. 3. - It is used widely in spectroscopy for the study of metal ions 4. - Isotopes are also used to study the plant metabolic functions.

Nuclear binding energy is the energy required to split a nucleus of an atom into its component parts. The component parts are neutrons and protons, which are collectively called nucleons. The binding energy of nuclei is always a positive number, since all nuclei require net energy to separate them into individual protons and neutrons. Thus, the mass of an atom's nucleus is always less than the sum of the individual masses of the constituent protons and neutrons when separated. This notable difference is a measure of the nuclear bindingenergy, which is a result of forces that hold the nucleus together. Because these forces result in the removal of energy when the nucleus is formed, and this energy has mass, mass isremoved from the total mass of the original particles, and the mass is missing in the resultingnucleus. This missing mass is known as the mass defect, and represents the energy released when the nucleus is formed.
The term nuclear binding energy may also refer to the energy balance in processes in whichthe nucleus splits into fragments composed of more than one nucleon, and in this case thebinding energies for the fragments, as compared to the whole, may be either positive ornegative, depending on where the parent nucleus and the daughter fragments fall on thenuclear binding energy curve. If new binding energy is available when light nuclei fuse, or when heavy nuclei split, either of these processes result in releases of the binding energy.This energy, available as nuclear energy, can be used to produce electricity (nuclear power) or as a nuclear weapon. When a large nucleus splits into pieces, excess energy is emitted as photons (gamma rays) and as kinetic energy of a number of different ejected particles (nuclear fission products).
Total mass is conserved throughout all such processes, so long as the system is isolated.During each nuclear transmutation, the "mass defect" mass is relocated to, or carried awayby, other particles that are no longer a part of the original nucleus.
The nuclear binding energies and forces are on the order of a million times greater than the electron binding energies of light atoms like hydrogen.
The mass defect of a nucleus represents the mass of the energy of binding of the nucleus, and is the difference between the mass of a nucleus and the sum of the masses of the nucleons of which it is composed. Determining the relevant nuclear binding energy encompasses threesteps of calculation, which involves the creation of mass defect by removing the mass asreleased energy.
Mass defect
The fundamental reason for the "mass defect" is Albert Einstein's formula E = m c2, expressing the equivalence of energy and mass. By this formula, adding energy also increases mass (both weight and inertia), whereas removing energy decreases mass.
If a combination of particles contains extra energy—for instance, in a molecule of the explosive TNT—weighing it reveals some extra mass, compared to its end products after anexplosion. (The weighing must be done after the products have been stopped and cooled, however, as the extra mass must escape from the system as heat before its loss can benoticed, in theory.) On the other hand, if one must inject energy to separate a system ofparticles into its components, then the initial weight is less than that of the components after

they are separated. In the latter case, the energy injected is "stored" as potential energy, which shows as the increased mass of the components that store it. This is an example of the fact that energy of all types is seen in systems as mass, since mass and energy are equivalent, andeach is a "property" of the other.
The latter scenario is the case with nuclei such as helium: to break them up into protons and neutrons, one must inject energy. On the other hand, if a process existed going in the oppositedirection, by which hydrogen atoms could be combined to form helium, then energy wouldbe released. The energy can be computed using E = Δm c2 for each nucleus, where Δm is the difference between the mass of the helium nucleus and the mass of four protons (plus twoelectrons, absorbed to create the neutrons of helium).
For elements heavier than oxygen, the energy that can be released by assembling them from lighter elements decreases, up to iron. For nuclei heavier than iron, one actually releasesenergy by breaking them up into 2 fragments. That is how energy is extracted by breaking up uranium nuclei in nuclear power reactors.
The reason the trend reverses after iron is the growing positive charge of the nuclei. Theelectric force may be weaker than the nuclear force, but its range is greater: in an ironnucleus, each proton repels the other 25 protons, while the nuclear force only binds closeneighbors.
As nuclei grow bigger still, this disruptive effect becomes steadily more significant. By thetime polonium is reached (84 protons), nuclei can no longer accommodate their large positivecharge, but emit their excess protons quite rapidly in the process of alpha radioactivity—the emission of helium nuclei, each containing two protons and two neutrons. (Helium nuclei are an especially stable combination.) This process becomes so rapid that nuclei with more than92 protons are not found naturally on Earth.
Nuclear binding energy curve[edit source | editbeta]
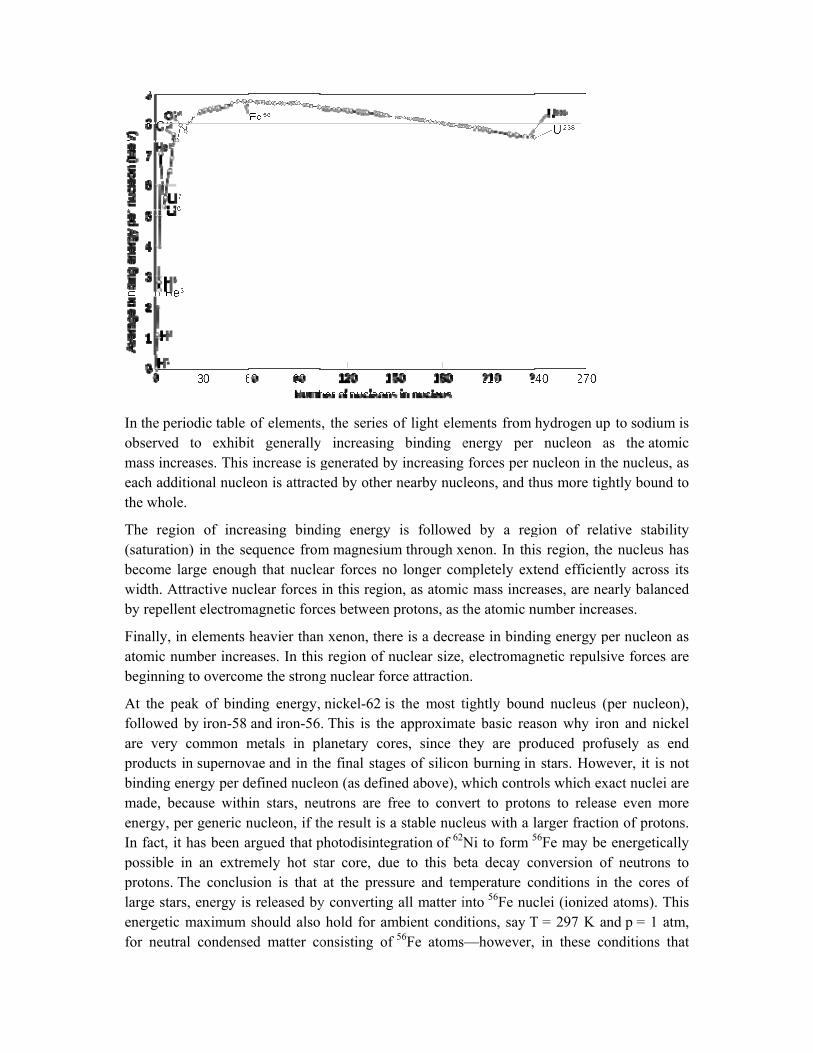
In the perobservedmass increach addthe whole
The regi(saturatiobecome lwidth. Aby repell
Finally, iatomic nbeginning
At the pfollowedare very products binding emade, beenergy, pIn fact, itpossible protons. Tlarge starenergeticfor neutr
riodic table d to exhibireases. This
ditional nuclee.
ion of increon) in the selarge enoug
Attractive nucent electrom
in elements umber increg to overcom
eak of bindd by iron-58 common min supernov
energy per decause withiper generic nt has been ain an extreThe conclusrs, energy isc maximum ral condense
of elementsit generallyincrease is g
eon is attract
easing bindequence fromh that nucleclear forces
magnetic forc
heavier thaneases. In thisme the strong
ding energy, and iron-56
metals in plvae and in thdefined nuclein stars, neunucleon, if thargued that pemely hot stsion is that s released byshould also
ed matter co
, the series y increasinggenerated byted by other
ding energy m magnesiumear forces noin this regio
ces between
n xenon, thers region of ng nuclear for
nickel-62 is. This is thelanetary corhe final stageon (as definutrons are fhe result is aphotodisintegtar core, duat the press
y convertinghold for am
onsisting of
of light elemg binding ey increasing nearby nucl
is followem through xo longer comon, as atomicprotons, as t
re is a decrenuclear size,rce attraction
s the most te approximares, since th
ges of siliconned above), wfree to conva stable nuclgration of 62N
ue to this besure and tem
g all matter imbient condif 56Fe atoms
ments from henergy per forces per n
leons, and th
d by a regenon. In thimpletely extc mass increthe atomic n
ease in bindi electromagn.
tightly bounate basic reahey are pron burning inwhich contro
vert to protoleus with a lNi to form 5
eta decay comperature cointo 56Fe nucitions, say T—however,
hydrogen upnucleon a
nucleon in thhus more tig
gion of relas region, thetend efficieneases, are nenumber incre
ing energy pnetic repulsi
nd nucleus (ason why irooduced profn stars. Howols which exons to releaslarger fractio56Fe may be onversion oonditions inclei (ionizedT = 297 K an
in these co
p to sodiumas the atomhe nucleus, aghtly bound t
ative stabilite nucleus hantly across iearly balanceeases.
per nucleon aive forces ar
(per nucleonon and nickfusely as enever, it is no
xact nuclei arse even moron of proton
energeticallf neutrons t
n the cores od atoms). Thnd p = 1 atmonditions th
is ic as to
tyas its ed
asre
n), el
ndot re re
ns.ly to of
his m, at

nuclei of
It is genemechanisbuild-up being relexplodesradioisotodecay-posupernovalpha-addnuclide in
Measurin
The fact of the traThe attrato each odistance. nuclei apnuclei larprotons mresult of bound asmore thastable, an
Semiemp
Nuclear bound elsplitting elementsnuclei are
As seen they mayand energwhen a c
Nuclear bsum of th
f atoms are in
erally believstic reasons,of 14 helium
leased into t. However,ope finally
owered lightvae, such asdition procen the univer
ng the bindin
that the maxade-off in theactive nucleaother, has aHowever, t
part, falls offrger than abomore than off additional s their size inan 209 nuclend are subjec
pirical formu
fusion produlements (suthe heavies (such as bare the most ti
above in they be easily mgy. The atomollection of
binding enehe masses of
nhibited from
ved that iron-, because itsm nuclei insthe interstell nickel-56 decays to irt curve of SN 1987A
esses, or elsse.
ng energy
ximum bindie effects of tar force (stroa limited ranthe repellingf with distanout four nucffsets any binstrong forcencreases, thoeons (larger ct to spontan
ula for nucle
uces energy uch as hydrost elements rium and kryightly bound
e example omeasured as fmic binding free nucleon
rgy can be cf the number
m fusing into
-56 is more s unstable pside supernolar medium then decay
ron-56 with such a proc. In a star, se there wo
ing energy itwo opposin
ong nuclear fnge due to g electromagnce much mocleons in diamnding energye interactionough most or than about neous decay
ar binding e
by combinogen into he(such as ur
ypton). Bothd of all.
of deuteriumfractional menergy is si
ns are joined
computed frr of free neut
o the most st
common tharogenitor nicvas, where iin a matter
ys to cobalt-a half life
cess has bethere are no
ould presum
s found in mng forces thaforce), whica rapid exp
gnetic force, ore slowly (ameter, the ady that results
ns. Such nucof them are s
6 nucleonsto smaller n
energy
ing the veryelium), andranium and ph processes p
m, nuclear biass deficits, imply the amd together to
rom the difftrons and pr
table and low
an nickel isockel-56 is coit has no timr of a few m-56 within aof about 77
een observedo good way
mably be mo
medium-sizedat have differch binds protponential dec
which acts as the inversdditional reps between fuclei become still stable. F in diameter
nuclei.
y lightest elnuclear fiss
plutonium) produce ener
nding energaccording to
mount of eneform a nucl
ference in motons that m
w energy sta
otopes in theopiously ma
me to decay tminutes, as ta few week7.3 days. Thd to happenys to create ore of this
d nuclei is arent range chtons and neucrease in thbetween proe square of d
pelling forceurther added increasinglFinally, nuclr) are all to
ements intosion produceinto more trgy, because
gies are largeo the equivaergy (and meus.
mass of a nucmake up the n
ate of matter
e universe foade by stageto iron beforthe supernovks, then thhe radioactivn in type IInickel-62 bhighly stab
a consequencharacteristicutrons equallhis force witotons to forcdistance). Fo
e of additionnucleons as
ly less tightllei containin
oo large to b
more tightles energy btightly boune middle-size
e enough thalence of mas
mass) released
cleus, and thnucleus. Onc
.
or ed re vahis ve Iaby le
ce cs.ly thce oral a lyngbe
lyby nd ed
atss d,
hece

this mass difference, called the mass defect or mass deficiency, is known, Einstein's mass-energy equivalence formula E = mc² can be used to compute the binding energy of any nucleus. Early nuclear physicists used to refer to computing this value as a "packing fraction"calculation.
For example, the atomic mass unit (1 u) is defined as 1/12 of the mass of a 12C atom—but the atomic mass of a 1H atom (which is a proton plus electron) is 1.007825 u, so each nucleon in 12C has lost, on average, about 0.8% of its mass in the form of binding energy.
Forces(SH)
Nuclei are bound together by the residual strong force (nuclear force). The residual strong force is a minor residuum of the strong interaction which binds quarks together to form protons and neutrons. This force is much weaker between neutrons and protons because it is mostly neutralized within them, in the same way that electromagnetic forces between neutral atoms (such as van der Waals forces that act between two inert gas atoms) are much weakerthan the electromagnetic forces that hold the parts of the atoms internally together (for example, the forces that hold the electrons in an inert gas atom bound to its nucleus).
The nuclear force is highly attractive at the distance of typical nucleon separation, and thisoverwhelms the repulsion between protons which is due to the electromagnetic force, thus allowing nuclei to exist. However, because the residual strong force has a limited rangebecause it decays quickly with distance (see Yukawa potential), only nuclei smaller than a certain size can be completely stable. The largest known completely stable (e.g., stable toalpha, beta, and gamma decay) nucleus is lead-208 which contains a total of 208 nucleons (126 neutrons and 82 protons). Nuclei larger than this maximal size of 208 particles areunstable and (as a trend) become increasingly short-lived with larger size, as the number of neutrons and protons which compose them increases beyond this number. However, bismuth-209 is also stable to beta decay and has the longest half-life to alpha decay of any known isotope, estimated at a billion times longer than the age of the universe.
The residual strong force is effective over a very short range (usually only a few fermis; roughly one or two nucleon diameters) and causes an attraction between any pair of nucleons. For example, between protons and neutrons to form [NP] deuteron, and also between protons and protons, and neutrons and neutrons.
Nuclear models (SH)
Although the standard model of physics is widely believed to completely describe the composition and behavior of the nucleus, theoretically generating predictions from it is muchmore difficult than for most other areas of particle physics. This is essentially because perturbation theory, a widely used mathematical tool, is not applicable to quantum chromodynamics (the theory of thestrong force) at the energy scales relevant to the nucleus. As a result, experiments have historically been compared to relatively crude models whichare necessarily imperfect. None of these models completely explain experimental data onnuclear structure.

The nuclpredict. Fis roughparticularconfigura
The stabcan be ap
wherneutr0.2 fmconst
Valency
Valence, atom hasof bonds atoms thapossible
The valenin the forof one anand can fa single a
Over thedescribintheory (1and all th
ear radius (RFor stable nuhly proportiorly in nuclations:
le nucleus hpproximated
re A = Atomrons N) and fm, dependintant.
(H)
also knowns formed, or can vary. Th
at may combfor the given
nce of an elrming of valnd thus can fform two sigatom. Alkyl
e last centung the c927), molec
he advanced
In cyclohevalence of
R) is considuclei (not halonal to the lei containi
has approximd by the follo
mic mass nr0 = 1.25 fm
ng on the nu
n as valence can form, whe IUPAC dbine with thn element.
ement depenlence bondsform one covgma bonds tand hydroxy
ury, the conchemical bcular orbitalsmethods of
exanonoximef three.
dered to be olo nuclei or
cube root ing many n
mately a conowing formu
number (the m = 1.25 × 10ucleus in qu
or valence nwith one or mdefinition lime atom that
nds on the n. A univalenvalent bond.to two differyl ions are un
ncept of valbond, inclus(1928), valequantum ch
e , the nitrog
one of the bother unstabof the mas
nucleons, a
nstant densitula,
number o0−15 m. In thuestion, but
number, is tmore other amits valence is the maxim
number of vant (monovale A divalent
rent atoms onivalent exa
lence evolvuding Lewisence shell e
hemistry.
gen atom has
basic quantitble distorted ss number (as they arra
ty and theref
f protons Zhis equation,this is less t
the number atoms. For mto the maxim
mum numbe
alence electrent) atom, iomolecular enr one sigma
amples; oxo
ved into a rs structureelectron pair
s three valen
ties that anynuclei) the n
(A) of the ange in mo
fore the nuc
Z, plus the, the constanthan 20% ch
of valence bmost elementmum number of valence
rons that mayon or group ntity has a vbond plus o
legends are
range of apes (1916), var repulsion
nce bonds an
y model munuclear radiunucleus, anore spheric
clear radius
e number ont r0 varies bhange from
bonds a givets the numbe
er of univalene bonds that
y be involvehas a valenc
valence of twone pi bond tdivalent.
pproaches foalence bontheory (1958
nd therefore
ustusnd al
R
of by
a
en ernt is
ed ce
wo to
ornd 8)
a

Electronic theory of valency (H)
Basic assumption of electronic theory of valency The outermost shell of an atom is known as the valence shell. The electrons present in the valence shell are called valence electrons.
K L M Na - 11 - 2 8 1
Valence shell - M Valence electrons - 1
Core electrons - 2 + 8 = 10 Question The electronic configurations of some of the elements are detailed below. How many valence and core electrons do the following have?
• Al: 2, 8, 3
• Mg: 2, 8, 2
• Cl: 2, 8, 7
• Ca: 2, 8, 8, 2. Solution
• Valence electrons = 3; core electrons = 10
• Valence electrons = 2; core electrons = 10
• Valence electrons = 7; core electrons = 10
• Valence electrons = 2; core electrons = 18
Significance of valence electrons
• Valence electrons of an atom are responsible for chemical reactions as they take part in them.
• Elements having same number of valence electrons in their atoms possess similar chemical properties. All alkali metals have one valence electron in their atom. Thus, their chemical properties are similar.

• Elements having 1, 2 or 3 electrons in the valence shell are metals. Exception is H and He. Elements having 4 to 7 electrons in their valence shell are non-metals.
Lets try out: Classify the following elements as metals or non-metals:
A - 2,8,2 - It is a metal as it has 2 valence electrons B - 2,8,6 - It is a non-metal as it has 6 valence electrons
C - 2,6 - It is a non-metal as it has 6 valence electrons
• The number of the valence shell in an atom determines the period number to which the elements belong in the Periodic Table.
Example Na - 11 : 2, 8, 1 Number of the valence shell - 3
Therefore, sodium is placed in period 3 of the periodic table. Example Ca - 20 : 2, 8, 8, 2 Number of the valence shell - 4
Therefore, sodium belongs to period 4 of the periodic table.
Ionic Bond(SH)
An ionic bond is a type of chemical bond formed through an electrostatic attraction between two oppositely charged ions. Ionic bonds are formed due to the attraction between an atomthat has lost one or more electron (known as a cation) and an atom that has gained one or more electrons (known as an anion). Usually, the cation is a metal atom and the anion is a nonmetal atom.
It is important to recognize that pure ionic bonding - in which one atom "steals" an electron from another - cannot exist: all ionic compounds have some degree of covalent bonding, or electron sharing. Thus, the term "ionic bond" is given to a bond in which the ionic character

is grealarge elecpolar (ionBonds wNeverthe
Ionic comgenerally
Formatio
A formawhose ioconfiguraof anotheelectron(atom becgases forconfiguratwo entit
For examcombinedatoms eain a 1:1 r
N
ReprLithifluordonais isointera
The energthe aaccepother
ater than ctronegativitnic) than oth
with partiallyeless, ionic b
mpounds coy have a high
on(SH)
ation of an onization eneation and after element (s), again to comes an ar elements ations for d-bies forms the
mple, commod, the sodiu
ach gain an eratio to form
Na + Cl → N
resentation oum has a lo
rine atom, wated by oelectronic waction betwe
removal of gy. There maddition of mpting the catr releases ene
the covalty differenceher forms of
y ionic and pbonding is co
onduct electrh melting po
ionic bond ergy is low, ter releasing(usually nonattain a stab
anion. Typicin the s
block and f-e ionic bond
on table salt m atoms eaelectron to fo
m sodium chlo
Na+ + Cl− →
of ionic bonw ionization
which has a the lithi
withhelium aeen the two a
electrons fray also be e
more than ontion's valencergy and thu
lent charae exists betwf covalent bopartially covonsidered to
ricity when moint and tend
proceeds wreleases som some of its
n metal), whble electron cally, the stas-block and -block elemed. It is forme
is sodium cch lose an e
form anions oride (NaCl)
NaCl
nding betwen energy andpositive ele
ium atomand fluorinatoms forms
rom the catienergy changne electron tce electrons us lowers the
cter - thween the twoonding whervalent charabe a form of
molten or ind to be solubl
when the atme of its eleelectron(s) t
hose electronconfiguratioable electro
thep-blockents. The eled to attain st
chloride. Whelectron, form(Cl−). These).
eenlithium ad readily givectron affinit
m. The ne is isoes an ionic bo
ion is endotges associateto form anioand the sub
e overall ene
hat is, a o atoms, caure electrons
acter are callf non covale
n solution, ble in water.
tom of an ectron(s) to the atom bec
n affinity is pon and after on configurak, and palectrostatic table octate
hen sodium (ming catione ions are th
and fluorine tves up its lonty and accepend resu
electronic wnd.
thermic, raised with breaons. Howevesequent attra
ergy of the sy
bond inusing the bon
are shared mled polar coent bonding.
but not as a
element (usachieve a stcomes a catipositive, theaccepting e
ation is one articular stabattraction bstructure.
(Na) and chlns (Na+), anden attracted
to form lithne valence epts the elec
ult is thwith neon.
sing the sysaking of exiser, the actionaction of theystem.
n which nd to be mormore equallyvalent bond
a solid. The
sually metaltable electroion. The atomen accepts thelectron(s) th
of the nobble electro
between thes
lorine (Cl) ard the chlorinto each othe
ium fluoridelectron to thtron that wahat lithium
Electrostat
stem's overasting bonds on of the anioe ions to eac
a rey.
ds.
ey
l),on mhe hele
on se
re ne er
de. he asmic
alloronch

Ionic bonding will occur only if the overall energy change for the reaction is favorable –when the reaction is exothermic. The larger the resulting energy change, the stronger thebond. The low electronegativity of metals and high electronegativity of non-metals means that the reaction is most favorable between a metal and a non-metal.
Structure(SH)
Ionic compounds in the solid state form lattice structures. The two principal factors indetermining the form of the lattice are the relative charges of the ions and their relativesizes. Some structures are adopted by a number of compounds; for example, the structureof the rock salt sodium chloride is also adopted by many alkali halides, and binary oxides such as MgO.
Bond strength (SH)
For a solid crystalline ionic compound the enthalpy change in forming the solid from gaseous ions is termed the lattice energy. The experimental value for the lattice energy can be determined using the Born-Haber cycle. It can also be calculated using the Born-Landé equation as the sum of the electrostatic potential energy, calculated by summing interactions between cations and anions, and a short range repulsive potential energy term. The electrostatic potential can be expressed in terms of the inter-ionic separation and a constant (Madelung constant) that takes account of the geometry of the crystal. The Born-Landé equation gives a reasonable fit to the lattice energy of e.g. sodium chloride where thecalculated value is −756 kJ/mol which compares to −787 kJ/mol using the Born-Haber cycle.
Polarization effects(SH)
Ions in crystal lattices of purely ionic compounds are spherical; however, if the positive ion is small and/or highly charged, it will distort the electron cloud of the negative ion, an effect summarised in Fajans' rules. This polarization of the negative ion leads to a build-up of extra charge density between the two nuclei, i.e., to partial covalency. Larger negative ions are more easily polarized, but the effect is usually only important when positive ions with charges of 3+ (e.g., Al3+) are involved. However, 2+ ions (Be2+) or even 1+ (Li+) show some polarizing power because their sizes are so small (e.g., LiI is ionic but has some covalent bonding present). Note that this is not the ionic polarization effect which refers to displacement of ions in the lattice due to the application of an electric field.
A covalent bond is the chemical bond that involves the sharing of electron pairs between atoms. The stable balance of attractive and repulsive forces between atomswhen they share electrons is known as covalent bonding.[1] For many molecules, the sharing of electrons allows each atom to attain the equivalent of a full outer shell, corresponding to astable electronic configuration. Covalent bonding includes many kinds of interactions, including σ-bonding, π-bonding, metal-to-metal bonding, agostic interactions, and three-center two-electron bonds. The term covalent bond dates from 1939. The prefix co- meansjointly, associated in action, partnered to a lesser degree, etc.; thus a "co-valent bond",

in essence, means that the atoms share "valence", such as is discussed in valence bond theory. In the molecule H2, the hydrogen atoms share the two electrons via covalent bonding. Covalency is greatest between atoms of similar electronegativities. Thus, covalent bonding does not necessarily require the two atoms be of the same elements, only that they be of comparableelectronegativity. Covalent bonding which entails sharing of electrons over more than twoatoms is said to be delocalized.
Comparison with covalent bonds(SH)
In an ionic bond, the atoms are bound by attraction of opposite ions, whereas, ina covalent bond, atoms are bound by sharing electrons to attain stable electronconfigurations. In covalent bonding, the molecular geometry around each atom is determined by Valence shell electron pair repulsion VSEPR rules, whereas, in ionic materials, the geometry follows maximum packing rules.
Purely ionic bonds cannot exist, as the proximity of the entities involved in the bondallows some degree of sharing electron density between them. Therefore, all ionic bonds have some covalent character.
Thus, an ionic bond is considered a bond where the ionic character is greater than the covalent character. The larger the difference in electronegativity between the two atoms involved in the bond, the more ionic (polar) the bond is. Bonds with partially ionic and partially covalent character are called polar covalent bonds.
For example, Na–Cl and Mg–O bonds have a few percent covalency, while Si–O bonds are usually ~50% ionic and ~50% covalent.
Electrical conductivity (SH) Ionic compounds, if molten or dissolved, can conduct electricity because the ions in these conditions are freeto move and carry electrons between the anode and the cathode. In the solid form,however, they cannot conduct because the electrons are held together too tightly for themto move. However, some ionic compounds can conduct electricity when solid. This is dueto migration of the ions themselves under the influence of an electric field. Thesecompounds are known as fast ion conductors.
Electrovalency (H)
This is the type of chemical bonding, that is established by the actual transference of one or more valence electrons, from a metallic atom to a non-metallic atom, so that each of the 2 elements can attain the stable electronic configuration of their respective nearest inertelements in the periodic table. This results in the formation of oppositely charged ions(cationand anion) which are held by electrostatic force of attraction and they in turn form anelectrovalent compound. For example, Na and Cl will form an electrovalent compound NaCl.So first Na(11), being a metal, will give away 1 electron to attain the stable state of Neon(10),

its nearest inert element. Then Na will become (Na+). That 1 electron will be accepted byCl(17) so that Cl can attain the stable state of Argon(18), its nearest inert element. Cl will thus become (Cl-). So Na+ + Cl- = NaCl
Electrovalency is a measurement of the net electric charge of an ion and is used when balancing chemical reactions. Electrovalency is related to the concepts of electronegativity and valence electrons, and indicates the number of electrons necessary for an ion to have a balanced electric charge.
Atoms that have an almost full or almost empty valence shells tend to be very reactive. Atoms that are strongly electronegative (as is the case with halogens) often only have one or two missing electrons in their valence shell, and frequently bond with other molecules or gain electrons to form anions. Atoms that are weakly electronegative (such as alkali metals) have relatively few valence electrons that can easily be lost to atoms that are stronglyelectronegative. As a result, weakly electronegative atoms tend to lose their electrons and form cations.
The electrovalency of an element or compound is expressed as a charge. Atoms or moleculesthat have lost electrons have an electrovalency greater than zero and are known as cations. When an atom or molecule gains electrons, it is called an anion. When an atom or moleculehas an electrovalency of zero, it has no net electric charge. When writing about an ion, theconvention is to write the chemical formula followed by the electrovalency as a superscript, illustrated below:
Ag+, Co2+, Fe3+, CN−, CO32−, PO4
3−. When an ion only contains a single atom it is called a monatomic ion, and when it contains more than one atom, it is called a polyatomic ion. On the above listed Ag+ would be a monatomic cation and PO4
3− would be a polyatomic anion.
Covalency(SH)
This is the number of electron pairs an atom can share with other atoms. The total number oforbitals available in the valence shell is known as covalency, whether the orbitals arecompletely filled or empty . For example, the electronic configuration of Boron (AtomicNumber 5) is 1s2 2s2 2p1. So, there are only two shells. The second shell contains one 2sorbitals and three 2p orbitals resulting total four orbitals in second shell. Therefore, Boron isrestricted to a maximum covalency of 4 since only four(one s and three p) orbitals areavailable for bonding.
Co ordinate bond(SH)
A co-ordinate bond (also called a dative covalent bond) is a covalent bond (a shared pair ofelectrons) in which both electrons come from the same atom.

Formal c
formal that electrelative e
The form
Wherstate)and Bmole
Whenmoleatom
FormThis
Exam
• C• N• d• si
An electrcounthe n
.
charge conce
charge (FCtrons in a celectronegati
mal charge of
re V is the ); N is the nB is the totaecule. There
n determininecule, the stru
ms is minimiz
mal charge isis significan
mples:
Carbon in meNitrogen in N
ouble bondeingle bonded
alternative ronegativity
nts the numbnumber of ato
ept(H)
) is the chemical boivity.
f any atom in
number of
number of nal number ofare two elec
ng the correcucture is chozed.
s a test to dent when draw
ethane: FC =NO2
-: FC = 5ed oxygen ind oxygen in N
method foy is by oxidber of electrooms bonded
charge asond are sha
n a molecule
valence elecnon-bonding f electrons s
ctrons shared
ct Lewis struosen such th
etermine the wing structur
= 4 - 0 - (8÷25 - 2 - (6÷2) =n NO2
-: FC =NO2
-: FC =
or assigningdation numbons that an ato the atom
signed to ared equall
e can be calc
ctrons of thevalence ele
shared in covd per single c
ucture (or prat the forma
efficiency ores.
2) = 0 = 0
= 6 - 4 - (4÷26 - 6 - (2÷2)
g charge ber. Other ratom uses inof interest.
an atom in ly between
culated by th
e atom in isectrons on thvalent bondcovalent bon
redominant ral charge (wi
of electron d
2) = 0 ) = -1
to an atomrelated concn bonding, a
a moleculn atoms, r
he following
solation (atohis atom in ts with other
nd.
resonance stithout sign) o
distribution o
m taking cepts are vaand coordina
le, assuminregardless o
equation:
om in grounthe molecul
r atoms in th
tructure) for on each of th
of a molecul
into accounalence, whication numbe
ng of
nd e; he
a he
e.
ntch er,

Hybridization sp, sp2, sp3, (consider BeF2, BF3, CH4) molecules.(H)
Hybridization is a theoretical process involving the combination of atomic orbitals to create anew set of orbitals that take part in covalent bonding.SP Hybridization involves the hybridization of one s- orbital and one p orbital to produce two sp-hybrid orbitals.BeF2 molecule is an example of SP hybridization. Bereyllium has no unpaired electrons toform bonds with fluorine atoms.So one electrons jumps from 2s orbital to the first sub shell in2p orbital.The two orbitals with unpaired electrons fuse together to make two hybrid orbitals with forma both two fluorine atoms.
The unpaired electrons in hybrid orbitals form covalent bonds with unpaired electrons in theside atoms.The bond angle between two sp hybridized orbitals is 180o - See more at: http://biochemhelp.com/BeF2-hybridization.html#sthash.qEJxcawz.dpuf
Hybridization of B in BF3 molecule is sp2.(SH) One 2s orbital and two 2p orbital of B undergo hybridization to produce three sp2 hybrid orbitals. Each of the 3 hybrid orbital is used for the formation of sigma bonds with F atoms.

sp3 hybri
Hybridistetrahedrthe corre
Carbon's
The carbpz are mtwo covasimplest excitationthere are
As the afour C-H
Quantumrequires equivalenvalence-sfunctions
ids (SH)
ation descrirally coordinct symmetry
ground state
on atom canmeaningless alent bondsw
of the carbn of an electfour singly
additional boH bonds is en
m mechanicathat they b
nce, theangushell (Core s[3] to form f
ibes the bonnated carbony to bond to t
e configurat
n utilize its tat this poin
with two hydenes. The ctron from thoccupied orb
ond energy mnergetically f
lly, the lowebe formed ular distribu
orbitals afour sp3 hybr
nding atomsn (e.g., methathe 4 hydrog
ion is 1s2 2s
C↑↓ ↑↓
1s 2s
two singly ont, as they drogen atomcarbon atomhe doubly ocbitals.
C*↑↓ ↑
1s 2
more than cfavoured.
est energy isfrom equivtions of the
are almost rids.
C* ↑↓ ↑
s from an aane CH4), thgen atoms. 2 2px
1 2py1 o
↓ ↑ ↑
s 2px 2py 2pz
occupied p-tydo not fill
ms, yielding tm can also bccupied 2s o
↑ ↑ ↑
s 2px 2py 2p
compensates
s obtained ifvalent orbita
orbitals chanever invo
↑ ↑ ↑
atom's pointhe carbon sh
or more easil
z
ype orbitals l in any pathe "free radbond to fourrbital to the
pz
s for the exc
f the four boals on the cange via a lolved in bo
t of view. Thould have 4
ly read:
(the designaarticular orddical" methyr hydrogen empty 2p o
citation, the
onds are equcarbon. To linear combionding) s
That is, for 4 orbitals wit
ations px py oder), to formylene CH2, th
atoms by aorbital, so th
formation o
ivalent whicachieve th
ination of thand p wav
ath
or mhe anat
of
ch hishe ve

In CH4, (sigma) b
sp2 hybri
Ethene st
Other camethane.
For this the doublIn sp2 hy
forming acarbon atcovalent carbon ahydrogendata.
sp hybrid
four sp3 hybonds (that i
ids[edit sour
tructure
rbon based . Take, for ex
molecule, le bond betw
ybridisation t
a total of thrtoms form a bonds with
atoms perpen–carbon bo
ds
ybrid orbitals, four singl
tran
ce | editbeta
compounds xample, ethe
carbon wilween the carthe 2s orbital
ree sp2 orbitaσ bond by ohydrogen by
endicular tonds are all o
1s sp
ls are overlae covalent b
slates into
]
and other mene (C2H4). E
ll sp2 hybridrbons, and ol is mixed w
C*↑↓ ↑
1s sp
als with oneoverlapping y s–sp2 over
o the molecof equal stre
p3 sp3 sp3 sp
apped by hybonds) of the
molecules mEthene has a
dise, becauonly three σ
with only two
↑ ↑ ↑ ↑
p2 sp2 sp2 2p
p orbital remtwo sp2 orbi
rlap all with cular plane ngth and len
3
ydrogen's 1se same length
may be explaa double bon
se one π (pσ bonds are o of the three
p
maining. In itals and eac120° anglesis formed
ngth, which
s orbital, yih and strengt
ained in a sind between t
pi) bond is formed per e available 2
ethylene (ethch carbon ato. The π bondby 2p–2p agrees with
ielding fourth.
imilar way athe carbons.
required focarbon atom
2p orbitals:
hene) the twom forms twd between thoverlap. Thexperiment
σ
as
or m.
wo wo he he al

A schem
The chemhybridisa
In this msp orbitalinacetylea σ bondhydrogen
Brief con
The perioof theiratare preseof the tabelementsblock to that.
The rowshaving nincorporapropertiesynthesizother varare widel
LONG F
GR
s-bl
atic presenta
mical bondination.
model, the 2ls and tw
ene (ethyne) d and two an in a σ s–sp
ncept of mod
odic table is tomic numbeented in ordeble comprise below. Ththe left, the
s of the tablnames suchates recurrines of the elzed, elementriant—providly used in ch
FORM OF M
ROUPS
lock d-block
ation of hybr
ng in comp
2s orbital miwo remain(C2H2) cons
additional π b overlap at 1
dern periodic
a tabular arers, electroner of increases an 18-col
he table cane p-block to
le are calledh as halogenng trends, anlements andts. As a resudes a useful
hemistry and
MODERN PE
k
rid orbitals s
ounds such
C*↑↓
1s
ixes with onning unchsists of sp–spbonds forme
180° angles.
c table of ele
rrangement on configuratising atomic lumn-by-7-r also be dethe right, t
d periods; thens or noble ny such tabled predict thult, a period framework
d other scien
ERIODIC T
sp
as alkynes w
↑ ↑ ↑ ↑
sp sp 2p 2p
nly one of hanged p op overlap beed by p–p o
ements.(H)
of the chemions, and recnumber (nu
row main grieconstructedthe d-block i
e columns agases. Sinc
e can be usehe propertiesdic table—w
for analyzince.
TABLE
with triple b
the three porbitals. Thetween the twoverlap. Eac
ical elementcurring chemumber of proid of elemen
d into four rn the middl
are called groce, by defid to derive rs of new, y
whether in thngchemical b
p-bloc
bonds is exp
orbitals resuhe chemicwo carbon ach carbon a
ts, organizedmical propertotons). The snts, with a drectangular ble, and the f
oups, with snition, a prelationshipsyet to be d
he standard fbehavior, an
ck
plained by s
ulting in twcal bondinatoms forminalso bonds t
d on the basties. Elemenstandard formdouble row oblocks: the f-block below
some of thesperiodic tabs between thdiscovered oform or somnd such table
sp
wo ng ng to
is nts m of s-w
se le he or
me es

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
IA
IIA
IIIB
IVB
VB
VIB
VIIB
VIII IBIIB
IIIA
IVA
VAVIA
VIIA
0
ns1
ns2np6
P E R I O D
1
1H ns2
ns2
np1ns2
np2ns2
np3ns2
np4 ns2
np5 2He
2
3Li 4Be
5B 6C 7N 8O 9F 10Ne
3
11
Na
12
Mg
(n-1)d1-10 ns1-2 13Al 14Si 15P 16S 17C
l 18Ar
4
19
K 20Ca
21Sc
22
Ti 23
V 24
Cr
25
Mn
26Fe
27
Co28Ni
29
Cu30Zn
31Ga
32Ge
33As
34Se
35Br 36Kr
5
37Rb
38Sr 39Y 40
Zr 41
Nb
42
Mo
43Tc
44
Ru45
Rh46Pd
47
Ag48
Cd 49In50Sn
51Sb
52Te 53I 54Xe
6
55Cs
56Ba
57-71
72
Hf 73Ta
74
W 75
Re76
Os77Ir 78Pt 79
Au80
Hg 81Tl 82Pb
83Bi
84Po
85At 86Rn
7
87Fr
88Ra
89-103
104
Rf 105
Db 106
Sg 107
Bh108
Hs109
Mt110
Ds 111
Rg112
Cn
f-block
Lanthanoids
57La
58
Ce 59Pr
60
Nd 61Pm
62Sm
63Eu
64Gd
65Tb
66
Dy67Ho
68Er
69Tm
70Yb
71Lu
Actinoids
89Ac
90
Th 91Pa
92
U 93
Np94Pu
95
Am
96Cm
97
Bk98
Cf99Es
100Fm
101
Md102
No 103
Lr
MODERN PERIODIC LAW
The modern periodic law was proposed by Moseley. He found the relation between

atomic numbers (Z) and the frequencies (ν) of X-rays produced when the atoms of different elements are bombarded with cathode rays. The relation between the square root of frequency (√ν) of highest energy emission line, called Kα line, with the atomic number, Z was found to be linear.
The mathematical relation can be presented as:
√ν = a(Z-b)
Where a & b are constants, characteristic of elements.
Later on it was clearly established that an element can be characterized by its atomicnumber, Z and not by the atomic weight. It was also found that there is a relation betweenelectronic configuration and properties of elements. The number of electrons in an atom andits electronic configuration are in turn are related to the atomic number.
Thus the modern periodic law can be stated as:
"The chemical and physical properties of elements are the periodic functions of theiratomic numbers and electronic configurations."
The modern long form of periodic table was constructed based on above law. Thefollowing points are considered while constructing the periodic table.
* The elements in the periodic table are arranged in the increasing order of the atomicnumber.
* Every row, also called as period, in the periodic table starts with the filling up ofdifferentiating electron into a new quantum shell.
* The elements in a vertical column called as group should get similar outer electronicconfiguration since it is observed that the elements with similar outer electronic configuration show similar chemical properties.
SALIENT FEATURES OF LONG FORM OF MODERN PERIODIC TABLE
The long form of modern periodic table consists of seven rows called periods and eighteen columns called groups.
Periods:
* Each period starts with an alkali metal and ends with an inert gas element.
* The first period is a very short period with only two elements i.e., Hydrogen (H) &Helium (He). In this period, the 1s orbital is being filled up.
1
1H
2He
* The second period starts with Lithium (Li) and ends with Neon (Ne) and contains 8elements. It is called first short period. In this group, the 2s & 2p orbitals are being filled up.

2
3Li 4Be
5B 6C 7N 8O 9F 10Ne
* The third period also contain 8 elements i.e., from Sodium (Na) to Argon (Ar). It iscalled second short period. The 3s & 3p orbitals are being filled up in this period.
3
11Na 12Mg
13Al 14Si 15P 16S 17Cl 18Ar
* The fourth period is the first long period with 18 elements, it starts with Potassium (K) and ends with Krypton (Kr). It also includes 10 elements belonging to 3d series i.e., fromScandium (Sc) to Zinc (Zn). In this period, not only 4s & 4p and also the 3d orbitals are beingfilled up by electrons.
4
19
K 20Ca
21Sc
22Ti
23
V 24Cr
25Mn
26Fe
27Co
28Ni
29Cu
30Zn
31Ga
32Ge
33As
34Se
35Br
36Kr
* The fifth period is the second long period with 18 elements, it starts with Rubidium(Rb) and ends with Xenon (Xe). It also includes 10 elements belonging to 4d series i.e. from Yttrium (Y) to Cadmium (Cd). The 5s & 5p along with 4d orbitals are filled up by electrons.
5
37Rb
38Sr
39
Y 40Zr
41Nb
42Mo
43Tc
44Ru
45Rh
46Pd
47Ag
48Cd
49In
50Sn
51Sb
52Te
53
I 54Xe
* The sixth period is the longest period with 32 elements. It not only includes 10 elements belonging to 5d series i.e., Lanthanum (La), Hafnium (Hf) to Mercury (Hg) but also contains14 elements belonging the 4f series called lanthanides (Cerium (Ce) to Lutetium (Lu)).
In this period, the 6s & 6p along with 4f & 5d orbitals are being filled up.
6
55Cs
56Ba
57-71
72Hf
73Ta
74
W 75Re
76Os
77Ir
78Pt
79Au
80Hg
81Tl
82Pb
83Bi
84Po
85At
86Rn
Lanthanoids
57La
58Ce
59Pr
60Nd
61Pm
62Sm
63Eu
64Gd
65Tb
66Dy
67Ho
68Er
69Tm
70Yb
71Lu
Note: Lanthanum is also considered to be the part of f-block since its properties resemble more to lanthanoids.
* The seventh period is an incomplete period. It starts with Fr. It also includes the 14

elements belonging to 5f series called actinides (Thorium (Th) to Lawrencium (Lr)).
In this period, the 7s & 5f orbitals are filled up. Since this is an incomplete period, thefilling up of 6d is not yet completed.
7
87Fr 88Ra 89-103
104Rf 105Db 106Sg 107Bh 108Hs 109Mt 110Ds 111Rg 112Cn
Actinoids
89Ac
90Th
91Pa
92
U 93Np
94Pu
95Am
96Cm
97Bk
98Cf
99Es
100Fm
101Md
102No
103Lr
Note: Actinium is also considered to be the part of f-block since its properties resemble more to actinoids.
* The Lanthanides and actinides are placed below the periodic table separately.
Groups:
* The 18 groups in the periodic table are numbered from 1 to 18 according to IUPACconvention.
However, according to American convention, these are also denoted by IA , IIA, IIIB,IVB, VB, VIB, VIIB, VIII (which actually includes 3 groups), IB ,IIB, IIIA, IVA, VA, VIA,VIIA and 0 (zero).
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
IA IIA IIIB IVB VB VIB VIIB VIII IB IIB IIIA IVA VA VIA VIIA 0
* The elements of IA to VII A groups i.e., 1, 2, 13, 14, 15, 16 & 17 groups are calledas representative elements.
* The zero group (or 18th group) elements are called as inert gases or noble gases. This group includes He, Ne, Ar, Kr, Xe and Rn.
* The elements of IIIB to IIB i.e., from groups 3 to 12 are called as transition elements.
* However the Lanthanoids and Actinoids are also considered to be the part of IIIB group (i.e., group 3). These are usually called as inner transition elements.
* The elements present in a group show similar physical & chemical properties since theyhave similar outer electronic configurations.

Review Questions(H) 1. Define the significance of chemistry as the branch of science? 2. Define atomic orbital. 3. What is Quantum numbers.Explain with examples. 4. What is law of Afbau. Define its use with two examples. 5. Define Bohr’s model. Also give their limitations. 6” Pauli’s exclusion principle”. Explain. 7. Give the introduction of modern periodic table. What are the limitations of it. 8. What is hybridization? Explain with two examples. 9. There are17 proton and 18 neutrons in a atomic nucleus of element. Determine it’s valency and atomic weight. 10. What is isotopes and isobars? Give the difference between them. Also describe itscharacteristics. 11. Explain Binding energy of nucleus. Also explain binding energy per nucleon. 12. Explain nuclear forces. 13. What do you mean by atom four concepts? 14. Define the concept of Line Spectrum of Hydrogen. 15. Give the formation of ss,pp and sp bonding with example. 16. Write short notes on-: Electron, proton, atomic number, isotopes, . 17. Explain-: electrovalence, covalence, covalent bond. Coordinate bond. 18. What is the mechanism of formation of covalent bond? Explain.
19. Explain Werner Heisenberg’s Uncertainty principle.
20. Define atomic orbital. Also explain its types.
.


Chapter-2
CHEMICAL EQUATION, OXIDATION & REDUCTION (MH)
Oxidation & Reduction reactions is defined as adding oxygen to form an oxide (oxidation) or removing oxygen (reduction). They always occur together. For example, in the burning of hydrogen
2H2 + O2 -> 2H2O
the hydrogen is oxidized and the oxygen is reduced. The combination of nitrogen and oxygen which occurs at high temperatures follows the same pattern.
N2 + O2 -> 2NO
This formation of nitric oxide oxidizes the nitrogen and reduces the oxygen. In some reactions, the oxidation is most prominent. For example in the burning of methane,
CH4 + 2O2 -> CO2 + 2H2O
both carbon and hydrogen are oxidized (gain oxygen). The accompanying reduction of oxygen is perhaps easier to see when you describe reduction as thegaining of hydrogen.
On the other hand, the reaction of lead dioxide at high temperatures appears to be just reduction.
2PbO2 -> 2PbO + O2 The reduction of the lead dioxide is clear, but the associated oxidation of oxygen is easier to see when you describe oxidation as the losing of electrons.
Oxidation and reduction in terms of oxygen transfer
Definitions

• Oxidation is gain of oxygen. • Reduction is loss of oxygen.
For example, in the extraction of iron from its ore:
Because both reduction and oxidation are going on side-by-side, this is known as a redox reaction.
Oxidising and reducing agents
An oxidising agent is substance which oxidises something else. In the above example, the iron(III) oxide is the oxidising agent.
A reducing agent reduces something else. In the equation, the carbon monoxide is the reducing agent.
Oxidising agents give oxygen to another substance.
• Reducing agents remove oxygen from another substance.
Oxidation and reduction in terms of hydrogen transfer
These are old definitions which aren't used very much nowadays. The most likely place you will come across them is in organic chemistry.
Definitions
• Oxidation is loss of hydrogen. • Reduction is gain of hydrogen.
Notice that these are exactly the opposite of the oxygen definitions.
For example, ethanol can be oxidised to ethanal:

The Role of Oxidation Numbers in Oxidation-Reduction Reactions(H)
Consider the following reaction.
CO(g) + H2O(g) CO2(g) + H2(g)
As can be seen in the figure below, the total number of electrons in the valence shell of each atom remains constant in this reaction.
What changes in this reaction is the oxidation state of these atoms. The oxidation state of carbon increases from +2 to +4, while the oxidation state of the hydrogen decreases from +1 to 0.
Oxidation and reduction are therefore best defined as follows. Oxidation occurs when the oxidation number of an atom becomes larger. Reduction occurs when the oxidation number of an atom becomes smaller.
Electronic Concept of Oxidation and Reduction (Modern Concept) (H)
The process in which any substance (atom, ion or molecule) loses one or more electrons is called oxidation. Thus, it is a deelectronation process. The substance which loses electrons is said to be oxidised.
E. g.
The above reactions are known as oxidation half-reactions. In these reactions are oxidised to, , respectively.

The process in which any substance (atom, ion or molecule) gains one or more electrons is called reduction. Thus it is an electronation process. The substance which gains electrons is said to be reduced.
E. g.
The above reactions are known as reduction: half-reactions. In these reactions and are reduced to , respectively.
In brief, oxidation involves following things.
(i) Addition of oxygen or other electro negative element.
[Mg is oxidised here]
(ii) Removal of or other electro positive element.
(iii) Increase of oxidation no.
(iv) Loss of electrons.
• During oxidation acidic nature increases. • Burning of coal, rusting of iron and combustion reactions involve oxidation. • Just reverse process occurs during reduction.
Oxidising and Reducing agents
A substance which oxidises others but itself undergoes reduction is known as oxidising agent or oxidant. Thus we can say that oxidising agent is an electron acceptor. A substance which undergoes oxidation but reduces others is known as reducing agent or reductant. In other words a reducing agent is an electron loser.
Some important oxidising agents are etc.

Some important reducing agents are etc.
Redox reactions (direct and indirect) (H) (i) An overall reaction in which oxidation and reduction takes place simultaneously is called redox or oxidation-reduction reaction. These reactions involve transfer of electrons from one atom to another. Thus every redox reaction is made up of two half reactions; One half reaction represents the oxidation and the other half reaction represents the reduction. (ii) Types of redox reaction (a) Direct redox reaction: The reactions in which oxidation and reduction takes place in the same vessel are called direct redox reactions. (b) Indirect redox reaction: The reactions in which oxidation and reduction takes place in different vessels are called indirect redox reactions. Indirect redox reactions are the basis of electro-chemical cells. (c) Intermolecular redox reactions: In which one substance is oxidised while the other is reduced. For example, Here, Al is oxidised to while is reduced to Fe. (d) Intramolecular redox reactions: In which one element of a
compound is oxidised while the other is reduced.
For example,
Here, in is reduced to in KCl while in is oxidised to . Methods of balancing redox reaction (H) There are two methods of balancing redox reaction. The first one is the oxidation number method which is based on the change in the oxidation number of the reducing and the oxidizing agents. The other method is called as Half reaction method, which involves the splitting of the redox reaction into two half reaction and then balancing both the oxidization half reaction and the reduction half reaction. The steps involved in the balancing of the redox reaction by half reaction method is Step 1: First write down the unbalanced net ionic equation for the redox reaction. Step 2: Separate the equation into half reaction. That is, write down the redox reaction separately into reduction half reaction and oxidation half reaction.

Step 3: Balance the atoms, other than oxygen and hydrogen, in each of the half reaction. Step 4: If the reaction takes place in an acid medium use H+ and H2O to balance the oxygen and hydrogen ion. Step 5: Balance the charges on each half reaction by adding appropriate amount of electrons on the reactant side in case of the reduction and on the product side in case of oxidation. Step 6: Balance the electrons in the two half reactions. That is, make the electrons in the oxidation half reaction equal to that of the reduction half reaction. Step 7: Add the two half reactions, that is the balanced reduction half reaction and cancel the common term in the two reactions. That is the electron will be cancelled and then the common ions would also be cancelled. Redox Reaction Examples Reduction oxidation can be explained in terms of the electronic concept also. Reduction is considered as a process in which an electron is added and oxidation is when the electron is removed from the element. With change in oxidation state also the redox reaction can be said to have taken place. Reduction is said to have taken place when the oxidation number of the element decreases. For example consider the reaction Fe3+ accepting the electron and forming Fe2+, since the oxidation number of the Iron decrease from 3 to 2 it is called as reduction reaction. When the oxidation number increases then it is the oxidation reaction. As we have already seen, reduction and oxidation occurs in the same reaction. The reduction and oxidation can be separated into two half reaction.
Consider for example the reaction of
2Na(s) + Cl2 (g → 2Na+ Cl– (s) or 2NaCl (s) The reaction can be separated into two parts
2Na(s) → 2Na+(g) + 2e-
Cl2(g) + 2e- → 2Cl-(g)
The first one that is the conversion of the sodium to sodium cation is oxidation half reaction. The second process is reduction half reaction in which chlorine is converted into chloride ion.

An oxidiabove reait reduces Applicati 1. Metall Metal oxreduced reduction 2. Photos Photosyncarbohyd
CO2 is rerequired
3. Combu Combustthat meetproduce hin the prdoing ph
C6H12O6( 4. Energy Many eleexample,and oxyg Redox QuestionFerrous Solution
zing agent isaction, chlors the other g
ions of Redo
lurgical Proc
xides are redto iron in a
n in an electr
synthesis(SH
nthesis is thedrates in pres
educed to cfor this reac
ustion of Fu
tion of fuelsts our daily heat and lighresence of oysical and m
(aq) + 6O2(g
y Generation
ectrochemica, electrical engen used in f
n: Balance thi
s one which rine is the ox
group and ge
ox Reactions
cesses(SH)
duced to ma blast furnarolytic cell.
H)
e process bysence of ligh
arbohydratection is provi
els and Food
is an oxidatneeds. On
ht energies.Ioxygen and mental work.
g) 6C
n(SH)
al cells basenergy for sp
fuel cells, wi
Reac
he chemicaion
gets reducedxidizing age
ets itself oxid
s
metals using ace using co
y which greht.
and water ided by sunl
ds(SH)
tion reactioncombustion
In living cellenergy is re
CO2(g) + 6H2
ed on redox pace applicatith hydrogen
ction
al reaction in
d and oxidizent and sodiudized.
suitable redoke. Al2O3 is
en plants co
is oxidizedight.
n. Fuels are (oxidation a
ls, glucose, Celeased. Thi
2O(I) + Ener
reactions artions is met n and oxygen
occurring bthe
zes the other um is called
ducing agents reduced to
onvert carbo
d to oxygen
the most imaccompanieC6H12O6 is ois energy is
rgy
re used for gout of the re
n electrodes.
Problems
between poac
atom. For ethe reducing
ts. For examo aluminium
on dioxide an
n. The activ
mportant soud by heat an
oxidized to Cutilized by
generating eleaction betw
otassium diccidic
example in thg agent, sinc
mple Fe2O3
m by cathod
nd water int
vation energ
urce of energnd light) the
CO2 and watethe body fo
lectricity. Foween hydroge
(SH
chromate anmedium
he ce
is ic
to
gy
gy ey er or
or en
H)
nd m.

Step 1 Fe2+
(aq) + Cr2O7 2– (aq) → Fe3+ (aq) + Cr3+(aq)
Step 2 Oxidation half : Fe2+ (aq) → Fe3+ (aq) Reduction half : Cr2O7 2– (aq) → Cr3+
(aq) Step 3 Cr2O7 2– (aq) →2 Cr3+
(aq) Step 4 Cr2O7 2– (aq) + 14H+ (aq) → 2 Cr3+
(aq) + 7H2O (l) Fe2+ (aq) → Fe3+ (aq) + e– Step 5 Cr2O7 2– (aq) + 14H (aq) + 6e → Cr3+
(aq) + 7H2O (l) Step 6 6 Fe2+ (aq) → Fe3+ (aq) + 6e Step 7 6 Fe2+ (aq) + Cr2O7 2– (aq) + 14H+(aq) → 6 Fe3+ (aq) + Cr3+
(aq)+ 7H2O(l)
Balancing redox reactions by oxidation number method (H) There is important method for balancing oxidation-reduction reactions. This is :
Oxidation number method- Rules for Assigning Oxidation Numbers
• The oxidation number of an atom is zero in a neutral substance that contains atoms of only one element. Thus, the atoms in O2, O3, P4, S8, and aluminum metal all have an oxidation number of 0.
• The oxidation number of monatomic ions is equal to the charge on the ion. The oxidation number of sodium in the Na+ ion is +1, for example, and the oxidation number of chlorine in the Cl- ion is -1.
• The oxidation number of hydrogen is +1 when it is combined with a nonmetal. Hydrogen is therefore in the +1 oxidation state in CH4, NH3, H2O, and HCl.
• The oxidation number of hydrogen is -1 when it is combined with a metal. Hydrogen is therefore in the -1 oxidation state in LiH, NaH, CaH2, and LiAlH4.

• The metals in Group IA form compounds (such as Li3N and Na2S) in which the metal atom is in the +1 oxidation state.
• The elements in Group IIA form compounds (such as Mg3N2 and CaCO3) in which the metal atom is in the +2 oxidation state.
• Oxygen usually has an oxidation number of -2. Exceptions include molecules and polyatomic ions that contain O-O bonds, such as O2, O3, H2O2, and the O2
2- ion. • The nonmetals in Group VIIA often form compounds (such as AlF3, HCl, and
ZnBr2) in which the nonmetal is in the -1 oxidation state. • The sum of the oxidation numbers of the atoms in a molecule is equal to the charge
on the molecule. • The most electronegative element in a compound has a negative oxidation number.
Oxidation Number Method (SH) During a redox reaction, the total increase in oxidation number must be equal to total decrease in oxidation number. This is the basic principle for balancing chemical equations. In addition, the number of atoms of each kind on one side of the equation must be equal to the number of atoms of the corresponding elements on the other side (the law of conservation of mass should not be violated). The following steps should be followed: Steps for balancing redox equations by oxidation number method (SH)
• Write the skeleton redox reaction.
• Indicate the oxidation number of atoms in each compound above the symbol of the element.
• Identify the element or elements, which undergo a change in oxidation number, one whose oxidation number increases (reducing agent) and the other whose oxidation number decreases (oxidizing agent).
• Calculate the increase or decrease in oxidation numbers per atom. Multiply this number of increase/decrease of oxidation number, with the number of atoms, which are undergoing change.
• Equate the increase in oxidation number with decrease in oxidation number on the reactant side by multiplying the formulae of the oxidizing and reducing agents.
• Balance the equation with respect to all other atoms except hydrogen and oxygen.
• Finally, balance hydrogen and oxygen.
• For reactions taking place in acidic solutions, add H+ ions to the side deficient in hydrogen atoms.
• For reactions taking place in basic solutions, add H2O molecules to the side deficient in hydrogen atoms and simultaneously add equal number to OH- ions on the other side of the equation.

Let us discuss the above method stepwise with the help of reaction between zinc and hydrochloric acid.
Step 1 The skeleton equation is:
Step 2 Oxidation number of various atoms involved in the reaction
Step 3 The oxidation number of zinc has increased from 0 to +2 while that of
The oxidation number of zinc has increased from 0 to +2 while that of hydrogen has decreased from +1 to 0. However, the oxidation number of chlorine remains same on both sides of the equation. Therefore, zinc is reducing agent while HCl is oxidizing agent in reaction and the changes are shown as:
Step 4 The increase and decrease in oxidation number per atom can be indicated as: O.N. increases by 2 per atom
Step 5 The increase in oxidation number of 2 per atom can be balanced with decrease in oxidation number of 1 per atom if Zn atoms are multiplied by 1 and HCl by 2. The equation will be:
Problem

12. Copper reacts with nitric acid. A brown gas is formed and the solution turns blue. The equation may be written as:
Balance the equation by oxidation number method. Solution Step 1 Skeleton equation
Step 2 Writing oxidation numbers of each atom
Step 3:
The oxidation number of copper has increased from 0 to +2 while that of nitrogen has decreased from +5 to +4. Step 4 Show, the increase/decrease of oxidation number
Step 5 Balance the increase/decrease in oxidation number by multiplying NO3- by 2 and Cu by 0.
Step 6 Balance other atoms except H and O as

Step 7 Reaction takes place in acidic medium, so add H+ ions to the side deficient in H+ and balance H and O atoms:
Another way to understand this method-(SH)
Balancing Redox Equations Using the Oxidation Number Method In most situations that call for balancing an equation, you are not told whether the reaction is redox or not. In these circumstances, you can use a procedure called the oxidation number method, which is outlined below.
Sample Study Sheet: Balancing Redox Equations Using the Oxidation Number Technique Tip-off – If you are asked to balance an equation and if you are not told whether the reaction is a redox reaction or not, you can use the following procedure. General Steps
Step 1: Try to balance the atoms in the equation by inspection, that is, by the standard technique for balancing non-redox equations. (Many equations for redox reactions can be easily balanced by inspection.) If you successfully balance the atoms, go to Step 2. If you are unable to balance the atoms, go to Step 3. Step 2: Check to be sure that the net charge is the same on both sides of the equation. If it is, you can assume that the equation is correctly balanced. If the charge is not balanced, go to Step 3. Step 3: If you have trouble balancing the atoms and the charge by inspection, determine the oxidation numbers for the atoms in the formula, and use them to decide whether the reaction is a redox reaction. If it is not redox, return to Step 1 and try again. If it is redox, go to Step 4. Step 4: Determine the net increase in oxidation number for the element that is oxidized and the net decrease in oxidation number for the element that is reduced. Step 5: Determine a ratio of oxidized to reduced atoms that would yield a net increase in oxidation number equal to the net decrease in oxidation number (a ratio that makes the number of electrons lost equal to the number of electrons gained). Step 6: Add coefficients to the formulas so as to obtain the correct ratio of the atoms whose oxidation numbers are changing. (These coefficients are usually placed in front of the formulas on the reactant side of the arrow.) Step 7: Balance the rest of the equation by inspection.
EXAMPLE 1 – Balancing Redox Reactions Using the Oxidation Number Method

Balance the following redox equation using either the “inspection” technique or the “oxidation number” method. Be sure to check that the atoms and the charge are balanced.
HNO3(aq) + H3AsO3(aq) → NO(g) + H3AsO4(aq) + H2O(l)
Solution: Step 1: Try to balance the atoms by inspection. Step 2: The H and O atoms are difficult to balance in this equation. You might arrive at the correct balanced equation using a “trial and error” technique, but if you do not discover the correct coefficients fairly quickly, proceed to Step 3. Step 3: Is the reaction redox?
The N atoms change from +5 to +2, so they are reduced. This information is enough to tell us that the reaction is redox. (The As atoms, which change from +3 to +5, are oxidized.)
Step 4: Determine the net increase in oxidation number for the element that is oxidized and the net decrease in oxidation number for the element that is reduced.
As +3 to +5 Net Change = +2
N +5 to +2 Net Change = −3
Step 5: Determine a ratio of oxidized to reduced atoms that would yield a net increase in oxidation number equal to the net decrease in oxidation number.
As atoms would yield a net increase in oxidation number of +6. (Six electrons would be lost by three arsenic atoms.) 2 N atoms would yield a net decrease of −6. (Two nitrogen atoms would gain six electrons.) Thus the ratio of As atoms to N atoms is 3:2.
Step 6: To get the ratio identified in Step 5, add coefficients to the formulas which contain the elements whose oxidation number is changing.
2HNO3(aq) + 3H3AsO3(aq) → NO(g) + H3AsO4(aq) + H2O(l)
Step 7: Balance the rest of the equation by inspection. 2HNO3(aq) +3H3AsO3(aq) → 2NO(g) + 3H3AsO4(aq) + H2O(l).
Conclusion (H) -:
(i) Oxidation is a process in which one or more electrons are lost or valency of the element increases.

(ii) Reduction is a process in which one or more electrons are gained or valency of the element decreases.
(iii) Oxidizing agent is a material which can gain one or more electrons, i.e., valency decreases.
(iv) Reducing agent is a material which can lose one or more electrons, i.e., valency increases.
(v) Redox reaction involves two half reactions, one involving loss of electron or electrons (oxidation) and the other involving gain of electrons or electrons (reduction).
Solved Example –
2AgCl(s) + H2(g) → 2 H+(aq) + 2 Ag(s) + 2 Cl- identify the atoms that undergo oxidation or reduction and list the oxidizing and reducing agents.
Solution: The first step is to assign oxidation states to each atom in the reaction.
• AgCl: Ag has a +1 oxidation state Cl has a -1 oxidation state
• H2 has an oxidation state of zero • H+ has a +1 oxidation state • Ag has an oxidation state of zero. • Cl- has a -1 oxidation state. The next step is to check what happened to each element in the reaction. • Ag went from +1 in AgCl(s) to 0 in Ag(s). The silver atom gained an electron. • H went from 0 in H2(g) to +1 in H+(aq). The hydrogen atom lost an electron. • Cl kept its oxidation state constant at -1 throughout the reaction. Oxidation involves the loss of electrons and reduction involves the gain of electrons.
For this reaction, hydrogen gas was oxidized with the oxidizing agent being silver chloride. Silver was reduced with the reducing agent being H2 gas.
Example 1
Balance the equation from the following two half-reactions:

Cr2O72- + 14 H+ + 6 e- --> 2 Cr3+ + 7 H2O
H2C2O4 --> 2 CO2 + 2 H+ + 2 e-
Solution Multiply the second equation by 3 and then add them algebraically so that the electrons in the two half-reaction equations cancel completely.
Cr2O72- + 14 H+ + 6 e- --> 2 Cr3+ + 7 H2O
3 H2C2O4 --> 6 CO2 + 6 H+ + 6 e- add the two equations and cancel the electrons to give the overvall equation Cr2O7
2- + 8 H+ + 3 H2C2O4 --> 2 Cr3+ + 7 H2O + 6 CO2
Discussion This example illustrate balancing redox equations in acid solutions.
Example 2
Balance the equation from the two half-reactions: Cd --> Cd2+ + 2 e-
4 H+ + NO3- + 3 e- --> NO + 2 H2O
Solution The first half-reaction has 2 electrons, whereas the second one has 3. The lowest common multiple of 2 and 3 is 6. Thus, you multiply the first equation by 3 and the second one by 2. The half-reaction equations become
3 Cd --> 3 Cd2+ + 6 e- 8 H+ + 2 NO3
- + 6 e- --> 2 NO + 4 H2O add the two equations and cancel the electrons to give the overvall equation 3 Cd + 8 H+ + 2 NO3
- --> 3 Cd2+ + 2 NO + 4 H2O
Discussion The last step in balancing oxidation and reduction reactions is simple.
Example 3
In a basic solution, Fe(OH)2 and Fe(OH)3 are solids. The former may be oxidized by H2O2.
Fe(OH)2 + H2O2 --> Fe(OH)3 + H2O. Balance this equation.
Solution The balanced half-reactions are:

Fe(OH)2 + OH- --> Fe(OH)3 (s) + e- H2O2 + 2 e- --> 2 OH-
Thus, the balanced equation is 2 Fe(OH)2 + H2O2 --> 2 Fe(OH)3
Discussion Note that the balanced equation does not have an H2O in it.
Example 4
Balance the following reaction, which is carried out in an acidic solution: I- + IO3
- --> I2
Solution The half-reactions are:
2 I- --> I2 + 2 e- (oxidized) 2 IO3
- + 10 e- --> I2 (reduced) The balanced equation is
6 H+ + 5 I- + IO3- ® 3 I2 + 3H2O.
EXAMPLE – Balancing Redox Equations for Reactions Run in Acidic Conditions: Balance the following redox equation using the “half-reaction” method.
Cr2O72−(aq) + HNO2(aq)
→ Cr3+(aq) + NO3−(aq) (acidic)
Solution: Step 1: Write the skeletons of the oxidation and reduction half-reactions.
You will usually be given formulas for two reactants and two products. In such cases, one of the reactant formulas is used in writing one half-reaction, and the other reactant formula is used in writing the other half-reaction. (In most cases, you do not need to know which reactant is oxidized and which is reduced.) The product formula in each half-reaction must include all of the elements in the reactant formula except hydrogen and oxygen. There are circumstances that make this step more complicated, but we will stick to simpler examples at this stage.
Cr2O72− → Cr3+
HNO2 → NO3−
Step 2: Balance all elements other than H and O. To balance the chromium atoms in our first half-reaction, we need a two in front of Cr3+.
Cr2O72− → 2Cr3+
HNO2 → NO3−
Step 3: Balance the oxygen atoms by adding H2O molecules on the side of the arrow where O atoms are needed.

The first half-reaction needs seven oxygen atoms on the right, so we add seven H2O molecules.
Cr2O72− → 2Cr3+ + 7H2O
The second half-reaction needs one more oxygen atom on the left, so we add one H2O molecule.
HNO2 + H2O → NO3−
Step 4: Balance the hydrogen atoms by adding H+ ions on the side of the arrow where H atoms are needed.
The first half-reaction needs 14 hydrogen atoms on the left to balance the 14 hydrogen atoms in the 7 H2O molecules, so we add 14 H+ ions to the left.
Cr2O72− + 14H+ → 2Cr3+ + 7H2O
The second half-reaction needs three hydrogen atoms on the right to balance the three hydrogen atoms on the left, so we add 3 H+ ions to the right.
HNO2 + H2O → NO3− + 3H+
Step 5: Balance the charge by adding electrons, e-.
The electrons go on the side of the equation with the highest charge (most positive or least negative). We add enough electrons make the charge on that side of the equation equal to the charge on the other side of the equation.
The sum of the charges on the left side of the chromium half-reaction is +12 (-2 for the Cr2O7
2− plus +14 for the 14 H+). The sum of the charges on the right side of the chromium half-reaction is +6 (for the 2 Cr3+). If we add six electrons to the left side, the sum of the charges on each side of the equation becomes +6.
6e− + Cr2O72− + 14H+ → 2Cr3+ + 7H2O
The sum of the charges on the left side of the nitrogen half-reaction is zero. The sum of the charges on the right side of the nitrogen half-reaction is +2 (−1 for the nitrate plus +3 for the 3 H+). If we add two electrons to the right side, the sum of the charges on each side of the equation becomes zero.
HNO2 + H2O → NO3− + 3H+ + 2e−
(Although it is not necessary, you can check that you have added the correct number of electrons by looking to see whether the net change in oxidation number for each half-reaction is equal to the number of electrons gained or lost. Because the two Cr atoms in Cr2O7
2− are changing from +6 to +3, the net change in oxidation number is 2(−3) or −6. This would require six electrons, so we have added the correct number of electrons to the first half-reaction. The N atom in HNO2 changes from +3 to +5, so the net change is +2. Two electrons would be lost in this change, so we have added the correct number of electrons to the second half-reaction.)
Step 6: If the number of electrons lost in the oxidation half-reaction is not equal to the number of electrons gained in the reduction half-reaction, multiply one or both of the half- reactions by a number that will make the number of electrons gained equal to the number lost.

For the chromium half-reaction to gain six electrons, the nitrogen half-reaction must lose six electrons. Thus we multiply the coefficients in the nitrogen half-reaction by 3.
6e− + Cr2O72− + 14H+ → 2Cr3+ + 7H2O
3(HNO2 + H2O → NO3− + 3H+ + 2e−)
or
6e− + Cr2O72− + 14H+ → 2Cr3+ + 7H2O
3HNO2 + 3H2O → 3NO3− + 9H+ + 6e−
Step 7: Add the 2 half-reactions as if they were mathematical equations. The 3 H2O in the second half-reaction cancel three of the 7 H2O in the first half-reaction to yield 4 H2O on the right of the final equation. The 9 H+ on the right of the second half-reaction cancel nine of the 14 H+ on the left of the first half-reaction leaving 5 H+on the left of the final equation.
Cr2O72− + 3HNO2 + 5H+
→ 2Cr3+ + 3NO3− + 4H2O
Step 8: Check to make sure that the atoms and the charge balance.
The atoms in our example balance and the sum of the charges is +3 on each side, so our equation is correctly balanced.
Cr2O72−(aq) + 3HNO2(aq) + 5H+(aq)
→ 2Cr3+(aq) + 3NO3− (aq) + 4H2O(l)
EXAMPLE – Balancing Redox Reactions Using the Half-Reaction Method: Balance the following redox equation using the “half-reaction” method.
Cr(OH)3(s) + ClO3−(aq)
→ CrO42−(aq) + Cl−(aq) (basic)
Solution: Step 1:
Cr(OH)3 → CrO42−
ClO3− → Cl−
Step 2: (Not necessary for this example) Cr(OH)3 → CrO4
2− ClO3
− → Cl− Step 3:
Cr(OH)3 + H2O → CrO42−
ClO3− → Cl− + 3H2O
Step 4: Cr(OH)3 + H2O → CrO4
2− + 5H+ ClO3
− + 6H+ → Cl− + 3H2O Step 5:

Cr(OH)3 + H2O → CrO42− + 5H+ + 3e−
ClO3− + 6H+ + 6e− → Cl− + 3H2O
Step 6: 2(Cr(OH)3 + H2O → CrO4
2− + 5H+ + 3e− ) ClO3
− + 6H+ + 6e− → Cl− + 3H2O
or
2Cr(OH)3 + 2H2O → 2CrO42− + 10H+ + 6e−
ClO3− + 6H+ + 6e− → Cl− + 3H2O
Step 7: 2Cr(OH)3(s) + ClO3
-(aq) → 2CrO4
2−(aq) + Cl−(aq) + H2O(l) + 4H+(aq) Step 8: Because there are 4 H+ on the right side of our equation above, we add 4 OH- to each side of the equation.
2Cr(OH)3 + ClO3− + 4OH−
→ 2CrO42− + Cl− + H2O + 4H+ + 4OH−
Step 9: Combine the 4 H+ ions and the 4 OH- ions on the right of the equation to form 4 H2O.
2Cr(OH)3 + ClO3− + 4OH-
→ 2CrO42− + Cl− + H2O + 4H2O
Step 10: Cancel or combine the H2O molecules. 2Cr(OH)3(s) + ClO3
−(aq) + 4OH−(aq) → 2CrO4
2−(aq) + Cl−(aq) + 5H2O(l) Step 11: The atoms in our equation balance, and the sum of the charges in each side is −5. Our equation is balanced correctly. EXAMPLE Balancing Redox Reactions Using the Oxidation Number Method Balance the following redox equation using either the “inspection” technique or the “oxidation number” method. Be sure to check that the atoms and the charge are balanced.
NO2(g) + H2(g) → NH3(g) + H2O(l) Solution: The atoms in this equation can be balanced by inspection. (You might first place a 2 in front of the H2O to balance the O’s, then 7/2 in front of the H2 to balance the H’s, and then multiply all the coefficients by 2 to get rid of the fraction.)
2NO2(g) + 7H2(g) → 2NH3(g) + 4H2O(l) We therefore proceed to Step 2. For the reaction between NO2and H2, the net charge on both sides of the equation in Step 1 is zero. Because the charge and the atoms are balanced, the equation is correctly balanced.

Excercise
Balance the following reactions -:
(1) Mn2+(aq) + NaBiO3(s) → Bi3+(aq) + MnO4–(aq)
(2) KMnO4 + Na2SO3 + H2O → MnO2 + Na2SO4 + KO (3) C3H8 + O2 → CO2 + H2O (4) Cu(s) + HNO3(aq) → Cu2+(aq) + NO(g).
(5) CuSO4 + 2NaOH -> Cu(OH)2 + Na2SO4.
(6) Fe (metal) + Cu2+ -> Fe2+ + Cu (metal).
(7) Zn + 2HCl -> Zn2+ + H2 +2Cl-.
(8) CuS + HNO3 -> Cu SO4 + NO (g) + H2O.
(9) Au3+(aq) + I−(aq) → Au(s) + I2(s) (10) NO2(g) + H2(g) → NH3(g) + H2O(l)

Chapter-3
ELECTROLYSIS – (MH)
CONCEPT OF ELECTROLYSIS - In chemistry and manufacturing, electrolysis is a method of using a direct electric current (DC) to drive an otherwise non-spontaneous chemical reaction. Electrolysis is commercially highly important as a stage in the separation of elements from naturally occurring sources such as ores using an electrolytic cell.
Electrolysis is the passage of dielectric current through an ionic substance that is either molten or dissolved in a suitable solvent, resulting in chemical reactions at the electrodes and separation of materials.
The main components required to achieve electrolysis are :
• An electrolyte : a substance containing free ions which are the carriers of electric current in the electrolyte. If the ions are not mobile, as in a solid salt then electrolysis cannot occur.
• A direct current (DC) supply : provides the energy necessary to create or discharge the ions in the electrolyte. Electric current is carried by electrons in the external circuit.
• Two electrodes : an electrical conductor which provides the physical interface between the electrical circuit providing the energy and the electrolyte
Electrodes of metal, graphite and semiconductor material are widely used. Choice of suitable electrode depends on chemical reactivity between the electrode and electrolyte and the cost of manufacture.
PROCESS OF ELECTROLYSIS(SH)
The key process of electrolysis is the interchange of atoms and ions by the removal or addition of electrons from the external circuit. The desired products of electrolysis are often in a different physical state from the electrolyte and can be removed by some physical processes. For example, in the electrolysis of brine to produce hydrogen and chlorine, the products are gaseous. These gaseous products bubble from the electrolyte and are collected.
2 NaCl + 2 H2O → 2 NaOH + H2 + Cl2
Oxidation and reduction at the electrodes
Oxidation of ions or neutral molecules occurs at the anode, and the reduction of ions or neutral molecules occurs at the cathode. For example, it is possible to oxidize ferrous ions to ferric ions at the anode:
Fe2+ aq → Fe3+ aq + e–
It is also possible to reduce ferricyanide ions to ferrocyanide ions at the cathode:

F
Nc
IpEa
Tea
E
Teaththroah
R
T
•
Faraday’
First law
e(CN)3- 6 +
Neutral molecan be reduce
+ 2 e–
n the last exprovided by Electrolysis alkaline wate
The substancelectrodes. Ita Gas diffusi
Energy chang
The amount energy of thearbitrarily che enthalpy he electric in
released in of steam intoabsorbed frohigher than th
Related techn
The followin
• Electrochin Standawhich usand electdistinct. reverse".
s law of elec
of electroly
+ e– → Fe(CN
ecules can aed to hydroq
+ 2 H+ →
xample, H+ an acid in
reactions iner solutions,
ces oxidisedt is possibleon electrode
ges during e
of electricale reaction pclose to zchange divinput is largethe form o
o hydrogen aom the surrohe electric in
niques
ng technique
hemical celard electrodeseful power trodes, electA chemical
ctrolysis.(H)
ysis –
N)4-6
also react atquinone at th
ions (hydron the solutionvolving H+
reactions in
d or reducede to have elee)
electrolysis
l energy thaplus the losszero, so thded by the f
er than the enof heat. In and oxygen oundings, annput.
s are related
lls, includine potential ican be extratrolysis and cell should
)
t either electhe cathode:
gen ions) alon, or the s+ions are fainvolving OH
d can also bectrolysis in
at must be adses in the syhe maximufree energy cnthalpy chan
some caseat high temp
nd the heatin
d to electroly
ng the hydin order to gacted. Althou
the operatiod not be thou
trode. For e
lso take partsolvent itselfirly common- (hydroxide
be the solvenvolving gas
dded equals ystem. The lum thermodychange of thnge of the rees, for instaperature, the
ng value of t
ysis:
drogen fuel generate an ugh related von of electro
ught of as pe
example: p-B
t in the reacf (water, mn in acidic
e ions) are co
nt (usually ses. (Such a
the change losses can (
ynamic efficihe reaction. Ieaction, so soance, in thee opposite isthe produced
cell, utiliseelectrical p
via the interochemical cerforming "e
Benzoquinon
ction, and armethanol etc.
solutions. Iommon.
water) or ths when usin
in Gibbs fre(in theory) biency equaIn most caseome energy e electrolyss true. Heat d hydrogen
e differencepotential fromaction of ion
cells are quielectrolysis i
ne
re .). In
he ng
ee be als es, is
sis is is
es m ns te in

In 1832,electric ccharge pa
Where, m
q
k
Second la
Faraday electrolytdirectly p
Mathem
Faraday's
wher
• m• Q• F• M• z
Note
For Flarge
For of M
In the
a
Michael Facurrent throuassed throug
m = quantity
= electric c
= proporti
aw of electro
discovered tes connecteproportional
atical form
s laws can be
re:
m is the massQ is the total = 96485 C m
M is the molais the valenc
that M/z is
Faraday's firer m will be.
Faraday's sM/z (equivalen
e simple cas
and then to
where:
araday reporugh a moltengh the circuit
of element
charge
onality cons
olysis
that when ted in series, to their equ
e summarize
s of the substelectric charmol−1 is the ar mass of thcy number o
the same as
rst law, M, F
second law,nt weight) th
se of constan
rted that then or dissolvt. This becam
stant
the same amthe mass of ivalent weig
ed by
tance liberatrge passed thFaraday con
he substanceof ions of the
the equivale
F, and z are
, Q, F, andhe larger m w
nt-current ele
e quantity oved salt is prme the basis
mount of elf substance lights.
ted at an elechrough the snstant
e substance (
ent weight of
constants,
z are constawill be.
ectrolysis,
of elements roportional tof the first l
lectricity is iberated/dep
ctrode in graubstance
(electrons tra
f the substan
so that the
ants, so th
lead
separated bto the quantilaw of electr
passed throposited at the
ams
ansferred per
nce altered.
larger the v
at the larg
ding to
by passing aity of electrrolysis:
ough differene electrodes
r ion).
value of Q th
ger the valu
an ric
nt is
he
ue

Electroly
One impo
2
Hydror elsuggealmo
The eelectrcombenergtextsof heboth Somehowecatalyerron(becohenceThe tof thefficihydro
NREin enthe n
Abouused proce
• n is t• t is th
In the mothe electr
Hereis a f
ysis of wate
ortant use of
H2O(l) → 2
rogen can belectric motoested as on
ost complete
energy effirolyser is abustion withgy. Heat/enth, as 144 MJ/
eat/enthalpy,types of teche reports qever, much ytic technol
neous tendeoming obsole the efficietheoretical m
he hydrogen iency), theseogen's chem
EL estimated nergy terms)near term and
ut 4% of hydonsite. Hyd
ess, and conv
the amount ohe total time
ore complicaric current I(
e t is the totalfunction of ta
r (H)
f electrolysis
2 H2(g) + O2
e used as a fuors via hyde approach dependence
ciency of wa measure o oxygen, or halpy values/kg. Note tha, which has lhnology. In
quote efficiehigher prac
logy, such ncy to uselete) does noency appearsmaximum co
and oxygene values refe
mical energy;
that 1 kg of) could be pd $2.27 in th
drogen gas pdrogen is usverting heav
of substancee the constan
ated case of ( ) integrate
l electrolysisau.
s of water is
(g); E0 = -1.
fuel for powedrogen fuel to shift eco
e upon hydro
water electrof the enthsome other s for hydrogat fuel cells led to some the reaction
encies betwctical efficieas 95% eff the 'Loweot represent s lower than onsiders the tn from waterer only to th the energy l
f hydrogen (roduced by
he long term.
produced wosed for the cvy petroleum
("number ofnt current wa
a variable eed over time
s time. Pleas
to produce h
229 V
ering internacells (see
onomies of ocarbons for
rolysis varihalpy contailater reaction
gen are well (not electroconfusion w, some energeen 50% aencies are aficiency. In er Heating the total amwhen using
total amountr. Note that he efficiencylost in gener
(roughly equwind power
.
orldwide is creation of
m sources to l
f moles") libas applied.
electrical cur:
se note that t
hydrogen.
al combustioHydrogen vthe world fenergy (See
ies widely. ned in the n), comparepublished inlysers) cann
when calculagy is lost as and 70% foavailable withe US theValue' for
mount of eneg the more at of energy r(in more br
y of convertrating the ele
uivalent to 3red electroly
created by eammonia folighter fracti
berated: n = m
rrent, the tota
tau is used a
on engines bvehicle). Thfrom the cue hydrogen e
The effichydrogen
d with the inn science annot utilise thiating efficienheat, a usele
or alkaline ith the use ere is still a
efficienciesergy within taccurately derequired for roader conteting electricaectricity is no
3 kg, or 4 L,ysis for betw
electrolysis, or fertilizer vions via hyd
m/M
al charge Q
s the current
by combustiohis has bee
urrent state oeconomy.)
iency of a(to under gnput electricd engineerinis full amounncy values foess byproducelectrolyserof PEM an
an occasions. This valuthe hydrogenefined valuethe formatio
exts of energal energy intot included.
of petroleumween $5.55 i
and normallvia the Habe
drocracking.
is
t I
on en of
an go al
ng nt or ct. rs; nd nal ue n,
es. on gy to
m in
ly er

Electrocrystallization (H)
A specialized application of electrolysis involves the growth of conductive crystals on one of the electrodes from oxidized or reduced species that are generated in situ. The technique has been used to obtain single crystals of low-dimensional electrical conductors, such as charge-transfer salts.
Engineering applications –(H) There are following applications of electrolysis in engineering.
1. electrometallurgy 2. electroplating 3. electrorefining
Electrometallurgy(SH) Electrometallurgy is a term used for processes that refine or purify metals using electricity. It can also be a general term for electrical processes used to plate one metal with another for decorative or corrosion resistance purposes. Metal processing using electricity is generally not the first step in metals purification, but rather a later or final step used to create very pure metals for other industrial processes. Raw ore mined from the ground can contain a valuable metal such as gold, copper or aluminum with a large amount of impurities. Some processing can be done by melting the ores at high temperatures and separating the desirable metals. These processes give metals that still can contain an undesirable percentage of minerals or other metals. Electrometallurgy can be used in a number of ways to purify or separate the remaining products. Electrowinning(SH) is a process that uses electric current passing through a water-based bath, called a cell, to separate the metal molecules, or ions, to a rod or plate. An electrical circuit consists of a positive and negative charge and a way for electrical current to flow between them. When two charged electrodes, called the cathode and anode, are placed in the cell, the metal ions will collect on one of the electrodes. Careful control of the voltage and current flow can create very pure metal deposits. This is a common process for purifying copper from less pure copper mixtures Another electrometallurgy process is electrorefining, which uses greater electrical currents to heat and melt partially refined ores to extract metals. This is a common process for aluminum refining, and is called the Hall process. In this process, partially refined aluminum oxide is first made from aluminum ore called bauxite. The aluminum oxide is then mixed with cryolite, a mineral composed of sodium, aluminum, and fluorine, which melts at a much lower temperature than aluminum oxide. When the mixture is exposed to high electrical currents, it melts and produces pure aluminum metal.

ElectroplThis is anacid bathbath and plate or items suElectroplcorrosionAnother electroplavaporizedcharge, wair is remdepositioelectricalelectronicProcess
Electropl
The anoddirect cupositive negative is oxidizeassociatethe metaanode tosolution
lating(H) n electromet
h containing given an el
deposit as auch as chrolated metals n-resistant coprocess useating, becaud to give it
which causesmoved is typon is used wl circuits. Itcs and some
lating of a m
de and cathourrent — a bterminal of terminal. W
ed from the e with the anallic, zero vao Cu2+ by loto form cop
tallurgy techa dissolved
lectrical chaa thin layer ome parts u
not only havovering to exed in electrouse a metal an electrica
s a very fine pically used where watert also has te industrial p
metal (Me) w
ode in the elbattery or, mthe supply,
When the exzero valenc
nions in the salence state. osing two epper sulfate.
hnique wheremixture of mrge can attraover the obj
used for auve a shiny dxtend the lifeometallurgy i
is depositeal charge, an
layer of metto remove o
r-acid solutithe ability tprocesses.
with copper in
lectroplatingmore comm, and the caternal powee state to fosolution. The
For exampelectrons. Th. At the cath
e an electricmetal and othact the meta
bject. This isutomobiles, decorative apfe of the partis physical
ed on anothend the surfactal to be depoxygen that ions might to form ver
n a copper su
g cell are botonly, a rect
athode (articer supply is orm cations we cations are
ple, in an ache Cu2+ asshode, the C
cal current isher minerals
al ions to itss a commonmotorcycles
ppeal, but the. vapor depoer surface. ce to be coaposited. A va
can contamdamage the
ry thin laye
ulfate bath
th connectedifier. The an
cle to be plaswitched on
with a positie reduced at id solution,ociates withu2+ is reduc
s passed thros. An object s surface, whn process to s and homee chrome pla
sition. This The metal i
ated is givenacuum cham
minate the pre parts, suchrs that can
d to an externode is conated) is conn, the metal ive charge. Tthe cathodecopper is ox
h the anioned to metal
ough a wateplaced in th
hich will thecreate plate
e applianceating acts as
is similar tis electricall
n the opposimber where th
roduct. Vapoh as in som
be useful i
rnal supply onnected to thnnected to th
at the anodThese catione to deposit ixidized at th
n SO42- in th
lic copper b
er-his en ed es. a
to ly te he or
me in
of he he de ns in he he by

gaining two electrons. The result is the effective transfer of copper from the anode source to a plate covering the cathode.
The plating is most commonly a single metallic element, not an alloy. However, some alloys can be electrodeposited, notably brass and solder.
Many plating baths include cyanides of other metals (e.g., potassium cyanide) in addition to cyanides of the metal to be deposited. These free cyanides facilitate anode corrosion, help to maintain a constant metal ion level and contribute to conductivity. Additionally, non-metal chemicals such ascarbonates and phosphates may be added to increase conductivity.
When plating is not desired on certain areas of the substrate, stop-offs are applied to prevent the bath from coming in contact with the substrate. Typical stop-offs include tape, foil, lacquers, and waxes.
Strike
Initially, a special plating deposit called a "strike" or "flash" may be used to form a very thin (typically less than 0.1 micrometer thick) plating with high quality and good adherence to the substrate. This serves as a foundation for subsequent plating processes. A strike uses a high current density and a bath with a low ion concentration. The process is slow, so more efficient plating processes are used once the desired strike thickness is obtained.
The striking method is also used in combination with the plating of different metals. If it is desirable to plate one type of deposit onto a metal to improve corrosion resistance but this metal has inherently poor adhesion to the substrate, a strike can be first deposited that is compatible with both. One example of this situation is the poor adhesion of electrolytic nickel on zinc alloys, in which case a copper strike is used, which has good adherence to both.
Brush electroplating
A closely related process is brush electroplating, in which localized areas or entire items are plated using a brush saturated with plating solution. The brush, typically a stainless steel body wrapped with a cloth material that both holds the plating solution and prevents direct contact with the item being plated, is connected to the positive side of a low voltage direct-current power source, and the item to be plated connected to the negative. The operator dips the brush in plating solution then applies it to the item, moving the brush continually to get an even distribution of the plating material. Brush electroplating has several advantages over tank plating, including portability, ability to plate items that for some reason cannot be tank plated (one application was the plating of portions of very large decorative support columns in a building restoration), low or no masking requirements, and comparatively low plating solution volume requirements. Disadvantages compared to tank plating can include greater operator involvement (tank plating can frequently be done with minimal attention), and inability to achieve as great a plate thickness.
Electroless deposition

Usually acurrent) ione elecelectrolesform:
In prreducchemother
A mabathsdiverslowechara
Clean
Cleanprevepriordetertest fheld up, awill descrelectrSurfaoff.
Effec
Electworkresistexamis a r
Uses
Electropla thin laywhich meas car par
an electrolytis used for e
ctrode and nss process n
rinciple anycer half-cell
mistry. Electrr metals like
ajor benefit s are not nerse shapes aner and canacteristics, el
nliness
nliness is esent adhesionr to electrorgent cleaninfor cleanlinevertical. Hy
allowing the retain an unribes a versioroplating practants such
cts
troplating ckpiece. An etance. An ex
mple of a merequired attri
lating is a usyer of a difetal of the obrts, bath taps
tic cell (conselectrodeposno external needs to con
water-basemust be hig
roless nickelsilver, gold
of this appeeded, reducnd types of not create lectroless de
ssential to sn of the coaoplating. Clng, electro-cless is the waydrophobic cwater to dra
nbroken sheon of this tesrocess can as soap red
changes thexample of axample of a chanical chaibute in tooli
seful processfferent metabject lacks. s, kitchen ga
sisting of twition. In consource of e
ntain a reduc
d reducer cgh enough tol plating useand copper
proach over cing the cosurface. Thsuch thick
eposition is q
successful elating. ASTMeaning procleaning, andaterbreak tescontaminantain rapidly. Pet of water st. This test displace the
duce the sens
e chemical, a chemical ch
physical change is a chaing industry
s. It is widell. The layerFor example
as burners, w
wo electrodentrast, an eleelectric currcing agent s
can be used o overcome es hypophosptypically us
electroplatinst of produ
he downside plates of
quite commo
lectroplatingM B322 is acesses inclu
d acid treatmst, in which s such as oiPerfectly clethat does ndoes not detese easily ssitivity of th
physical, hange is wh
hange is a change in tensi.
ly used in inr of metal de chromium
wheel rims an
es, electrolytectroless deprent. Howevso that the e
although ththe energy bphite as the e low molec
ng is that pouction. The t
is that the pmetal. As on in the dec
g, since moa standard gude solvent
ment etc. Thethe surface
ils cause thean metal surot bead up tect hydrophsince the sohe test and m
and mechahen nickel plhange in theile strength o
ndustry for cdeposited ha
plating is dond many oth
te, and exterposition procver, the solelectrode rea
he redox pobarriers inhereducer wh
cular weight
ower sourcetechnique cplating proca conseque
corative arts.
lecular layeguide for clet cleaning, e most comm
is thoroughe water to berfaces are hyor drain off
hilic contamiolutions are must be thoro
anical propelating impro
e outward apor surface ha
coating metaas some desione on manyhers.
rnal source ocess uses onllution for thaction has th
otential of therent in liquihile plating oaldehydes.
es and platincan also plaess is usuallnce of thes.
ers of oil caeaning meta
hot alkalinmon industrihly rinsed anead and breaydrophilic anf. ASTM F2inants, but th
water-basedoughly rinse
erties of thoves corrosioppearance. Aardness whic
al objects witired propertyy objects suc
of ly he he
he id of
ng te ly se
an als ne al
nd ak nd 22 he d. ed
he on An ch
th y, ch

ELECTROREFINIG (SH) In the Electrorefining process, the impure metallic anode is dissolved to produce purified cathode. Like the Electrowinning process, this too removes metal impurities and used on an economic scale for mass production of purified metals. To explain this further, the process consists of anodes which contain unrefined impure metal and when current is passed during the electrolytic process, the anodes corrode into the solution so that the pure metal is extracted and deposited on the cathodes. Earlier instances of Electrorefining use can be dated back to Maximilian when he successfully experimented with copper Electrowinning in 1847 and in 1870; it was patented by James Elkington for commercial use where the first Electrowinning plant was opened in Wales. The first electrometallurgy plan in the United States was opened in Newark, New Jersey in 1883 under the name of Balbach and Sons Refining and Smelting Company. Applications (SH) Some of the most common metals used with electrometallurgy process include the alkali metals, cobalt, zinc, gold, silver, lead, copper and chromium. Aluminum is the metal which can be produced through this method only. Commercially, the process has also been used to utilize spent nuclear fuel but no strategic outcome has been derived, In future, electrometallurgy can be used to separate heavy metals like strontium and plutonium from less toxic uranium.
ELECTROPLATING (SH)
Electroplating has, over recent decades, evolved from an art to an exact science. This development is seen as responsible for the ever-increasing number and widening types of applications of this branch of practical science and engineering. Some of the technological areas in which means and methods of electroplating constitute an essential component are all aspects of electronics: macro and micro, optics, opto-electronics, and sensors of most types, to name only a few. In addition a number of key industries such as the automobile industry (that uses for example chrome plating to enhance the corrosion resistance of metal parts) adopt the methods even where other methods, such as evaporation, sputtering, chemical vapor deposition (CVD) and the like are an option. That is so for reasons of economy and convenience. By way of illustration it should be noted that that modern electroplating equips the practitioner with the ability to predesign the properties of surfaces and in the case of electroforming those of the whole part. Furthermore, the ability to deposit very thin

multilayers (less than a millionth of a cm) via electroplating represents yet a new avenue of producing new materials.
Electroplating is often also called "electrodeposition", and the two terms are used interchangeably. As a matter of fact, "electroplating" can be considered to occur by the process of electrodeposition. Electrodeposition is the process of producing a coating, usually metallic, on a surface by the action of electric current. The deposition of a metallic coating onto an object is achieved by putting a negative charge on the object to be coated and immersing it into a solution which contains a salt of the metal to be deposited (in other words, the object to be plated is made the cathode of an electrolytic cell). The metallic ions of the salt carry a positive charge and are thus attracted to the object. When they reach the negatively charged object (that is to be electroplated), it provides electrons to reduce the positively charged ions to metallic form. Figure 1 is a schematic presentation of an electrolytic cell for electroplating a metal "M" from an aqueous (water) solution of metal salt "MA".
To further illustrate the foregoing, let us assume that one has an object made of one of the common metals, like copper, and that it has been properly pre-cleaned. We should want to plate it with, say, nickel. A wire will have to be attached to the object while the other end of the wire should be attached to the negative pole of abattery (or a power supply). To the positive pole of the battery (or power supply) we connect another wire with its other end connected to a rod made of nickel. Next we fill the cell with a solution of the metal salt to be plated. It is possible to use a molten salt and in some not so common cases, such as the deposition of tungsten, that is what is done. In most, more common, cases though the salt is simply dissolved in water. In our present example the nickel chloride salt dissociates in water to positively charged nickel cations and negatively charged chloride anions. As the object to be plated is negatively charged it attracts the positively charged nickel cations, and electrons flow from the object to the cations to neutralize them (to reduce them) to metallic form. Meanwhile the negatively charged chloride anions are attracted to the positively charged nickel rod (known as the anode of the electrolytic cell). At the anode electrons are removed from the nickel metal,oxidizing it to the nickel cations. Thus we see that the nickel dissolves as ions into the solution. That is how replacement nickel is supplied to the solution for that which has been plated out and one retains a solution of nickel chloride in the cell.
Nickel chloride is used here to exemplify the process of electroplating for a number of reasons. First among those is simplicity. It is not recommended, however, that nickel be used for, say, school science demonstrations because some individuals are quite allergic to it. We further do not recommend that chloride salts be used because those are amenable to release chlorine gas. For school or amateur type demonstration we recommend plating copper coins with zinc or nickel coins with copper.

ELECTROPLATING

Industria
Hall-Her
• Produ• Coul
durinelectr
• Produ• Produ• Produ• Produ
an an
Electroly
• Electobtaimolteimpo
• Anodto cothis p
• Produ• Elect
indus• Produ• Elect
al uses
roult process
uction of aluometric tech
ng electrolyrolysis uction of chluction of soduction of peruction of el
node.
ysis has many
trometallurgin the pure fen form is s
ortant chemicdization is arrosion. For
process. Theuction of oxtroplating is stries for funuction of hytrolytic Etch
s for producin
uminium, lithhniques can sis by mea
lorine and sodium chloratrfluorinated ectrolytic co
y other uses
gy is the proform of metseparated bycal uses. (Wan electrolyexample, sh
e process is aygen for spaused in lay
nctional or dydrogen for fhing of metal
ngaluminium
hium, sodiumbe used to
asuring the
odium hydrote and potassorganic com
opper as a c
:
ocess of redtal using eley electrolysi
Water is produytic procesships are savealso used to dacecraft and nering metalsecorative pu
fuel, using a l surfaces lik
m
m, potassiumo determineamount of
oxide sium chlorat
mpounds succathode, from
duction of mectrolysis. Fis into sodiuuced at the ss that makeed from beindecorate surnuclear subms to fortify turposes, as incheap sourc
ke tools or kn
m, magnesium the amounelectricity
te ch as trifluorom refined co
metals fromor example,um and oxyame time.) es the surf
ng corroded brfaces. marines. them. Electrn vehicle bodce of electricnives with a
m, calcium nt of matterrequired to
oacetic acid opper of low
m metallic c sodium hy
ygen, both o
face of meby oxygen in
roplating is udies and nick
cal energy. permanent m
r transformeperform th
wer purity a
compounds tdroxide in if which hav
etals resistann the water b
used in mankel coins.
mark or logo
ed he
as
to its ve
nt by
ny
o.

HYDROLYSIS (H)
Hydrolysis is a chemical reaction during which molecules of water (H2O) are split into hydrogen cations (H+, conventionally referred to as protons) and hydroxideanions (OH−) in the process of a chemical mechanism.[1][2] It is the type of reaction that is used to break down certain polymers, especially those made by step-growth polymerization. Such polymer degradation is usually catalysed by either acid, e.g., concentrated sulfuric acid (H2SO4), or alkali, e.g., sodium hydroxide (NaOH).
Hydrolysis is a chemical process in which a certain molecule is split into two parts by the addition of a molecule of water. One fragment of the parent molecule gains ahydrogen ion (H+) from the additional water molecule. The other group collects the remaining hydroxyl group (OH−).
A characteristic feature of the hydrolysis of esters and of most other organic compounds is that a third substance, ordinarily an acid or a base, increases the rate at which the chemical change takes place. In the biochemical process of digestion, enzymes secreted by the digestive tract catalyze the hydrolysis of complex molecules into forms that the body organisms can assimilate. Proteins are decomposed to amino acids, fats to fatty acids and glycerol, and starches and complex sugars to glucose and other simple sugars; enzymes such as lipases, amylases, and proteinases catalyze the hydrolysis of fats, carbohydrates, and proteins, respectively.
Hydrolysis involving ionic compounds may be illustrated by the chemical changes occurring in an aqueous solution of the salt sodium acetate. In solution, the ionic constituents of the salt (the acetate ion and the sodium ion) separate; water molecules combine with the acetate ions to form acetic acid and hydroxide ions. Acetic acid dissociates reversibly into acetate ions and hydrogen ions, but only to a very small extent, so that the ionic content of the solution is largely sodium and hydroxide ions. Hence, the solution exhibits basic properties (i.e., turns red litmus paper blue).

The most common hydrolysis occurs when a salt or weak base (or both) is dissolved in water. Water autoionizes into negative hydroxyl ions and positive hydrogen ions. The salt breaks down into positive and negative ions. For example, sodium acetate dissociates in water into sodium and acetate ions. Sodium ions react very little with hydroxyl ions whereas acetate ions combine with hydrogen ions to produce neutral acetic acid, and the net result is a relative excess of hydroxyl ions, causing a basic solution.
Acid–base-catalyzed hydrolyses are very common; one example is the hydrolysis ofamides or esters. Their hydrolysis occurs when the nucleophile (a nucleus-seeking agent, e.g., water or hydroxyl ion) attacks the carbon of the carbonyl group of theester or amide. In an aqueous base, hydroxyl ions are better nucleophiles than dipoles such as water. In acid, the carbonyl group becomes protonated, and this leads to a much easier nucleophilic attack. The products for both hydrolyses are compounds with carboxylic acid groups.
Hydrolysis of Acidic Salts
A salt formed between a strong acid and a weak base is an acid salt. Ammonia is a weak base, and its salt with any strong acid gives a solution with a pH lower than 7. For example, let us consider the reaction:
HCl + NH4OH = NH4+ + Cl- + H2O
In the solution, the NH4+ ion reacts with water (called hydrolysis) according to the equation:
NH4+ + H2O = NH3 + H3O+.
The acidity constant can be derived from Kw and Kb. [H3O+] [NH3] [OH-] Ka = ---------------- ------ [NH4
+] [OH-] = Kw / Kb = 1.00e-14 / 1.75e-5 = 5.7e-10.
Example 1
What is the concentration of NH4+, NH3, and H+ in a 0.100 M NH4NO3 solution?
Solution Assume that [NH3] = x, then [H3O+] = x, and you write the concentration below the formula in the reaction:
NH4+ + H2O = NH3 + H3O+
0.100-x x x Ka = 5.7E-10. x2

= ------- 0.100-x Since the concentration has a value much greater than Ka, you may use x = (0.100*5.7E(-10))1/2
= 7.5E-6
[NH3
] = [H+] = x = 7.5E-6 M
pH = -log7.5e-6 = 5.12
[NH4
+] = 0.100 M
Discussion Since pH = 5.12, the contribution of [H+] due to self ionization of water may therefore be neglected.
Hydrolysis and Basic Salts
A basic salt is formed between a weak acid and a strong base. The basicity is due to the hydrolysis of the conjugate base of the (weak) acid used in the neutralization reaction. For example, sodium acetate formed between the weak acetic acid and the strong base NaOH is a basic salt. When the salt is dissolved, ionization takes place:
NaAc = Na+ + Ac- In the presence of water, Ac- undergo hydrolysis:
H2O + Ac- = HAc + OH- And the equilibrium constant for this reaction is Kb of the conjugate base Ac- of the acid HAc. Note the following equilibrium constants: [HAc] [OH-] Kb = ----------- [Ac-] [HAc] [OH-] [H+] Kb = ----------- --- [Ac-] [H+] [HAc] [OH-][H+] Kb = ---------- --------- [Ac-] [H+] = Kw / Ka = 1.00e-14 / 1.75e-5 = 5.7e-10. Thus,
Ka Kb = Kw or
pKa + pKb = 14
N o t e Acetic acid Ka=1.75e-5Ammonia Kb=1.75e-5

for a conjugate acid-base pair. Let us look at a numerical problem of this type.
Example 2
Calculate the [Na+], [Ac-], [H+] and [OH-] of a solution of 0.100 M NaAc (at 298 K). (Ka = 1.8E-5)
Solution Let x represent [H+], then
H2O + Ac- = HAc + OH- 0.100-x x x x2 --------- = (1E-14)/(1.8E-5) = 5.6E-10 0.100-x Solving for x results in x = sqrt( 0.100*5.6E-10) = 7.5E-6 [OH-] = [HAc] = 7.5E-6 [Na+] = 0.100 F
Discussion This corresponds to a pH of 8.9 or [H+] = 1.3E-9.
Note that Kw / Ka = Kb of Ac-, so that Kb rather than Ka may be given as data in this question.
Salts of weak acids and weak bases
A salt formed between a weak acid and a weak base can be neutral, acidic, or basic depending on the relative strengths of the acid and base.
If Ka(cation) > Kb(anion) the solution of the salt is acidic.
If Ka(cation) = Kb(anion) the solution of the salt is neutral.
If Ka(cation) < Kb(anion) the solution of the salt is basic.
Example 3
Arrange the three salts according to their acidity. NH4CH3COO (ammonium acetate), NH4CN (ammonium cyanide), and NH4HC2O4 (ammonium oxalate).

S
BUFFER
A buffer base or aof strongsolution. wide varrange so solution f
PRINCIP
Buffer sequilibriu
H
Ka(aceKa(hyKa(oxKb(NH
olution
ammoammoammo
R SOLUTIO
is an aqueoa weak base g acid or bas
Buffer soluriety of chem
they utilizefound in nat
PLE OF BUF
olutions achum between
HA H+ + A
etic ydrogen xalic H3) = 1.8E-5
onium onium onium cyanid
N(H)
ous solutionand its conje is added t
utions are usmical applicae a buffer soture is blood
FFERING(S
hieve their the acid HA
A-
accac
5.
oxalate acet
de -- basic K
n consisting jugate acid. to it and thued as a meaations. Manyolution to m.
SH)
resistance
A and its con
cid) cyanide) cid)
--tate
Ka(c) < Kb(N
of a mixtuIts pH chan
us it is usedans of keepiny life forms
maintain a co
to pH channjugate base
= =
=
acidic,--
NH3)
ure of a weanges very littd to preventng pH at a ns thrive onlyonstant pH.
nge becauseA-.
=
Ka(o) ne
ak acid and tle when a st changes innearly constay in a relativOne exampl
e of the pre
1.85E-6.2E-15.6E-2
> Kb(NHeutral Ka = K
its conjugatsmall amoun
n the pH of ant value in vely small ple of a buffe
esence of a
5, 0, 2,
H3) Kb
te nt a a
H fer
an

When some strong acid is added to an equilibrium mixture of the weak acid and its conjugate base, the equilibrium is shifted to the left, in accordance with Le Chatelier's principle. Because of this, the hydrogen ion concentration increases by less than the amount expected for the quantity of strong acid added.
Similarly, if strong alkali is added to the mixture the hydrogen ion concentration decreases by less than the amount expected for the quantity of alkali added. The effect is illustrated by the simulated titration of a weak acid with pKa = 4.7. The relative concentration of undissociated acid is shown in blue and of its conjugate base in red. The pH changes relatively slowly in the buffer region, pH = pKa ± 1, centered at pH = 4.7 where [HA] = [A-]. The hydrogen ion concentration decreases by less than the amount expected because most of the added hydroxide ion is consumed in the reaction
OH- + HA → H2O + A-
and only a little is consumed in the neutralization reaction which results in an increase in pH.
OH- + H+ → H2
Acidic buffer solutions
An acidic buffer solution is simply one which has a pH less than 7. Acidic buffer solutions are commonly made from a weak acid and one of its salts - often a sodium salt.
A common example would be a mixture of ethanoic acid and sodium ethanoate in solution. In this case, if the solution contained equal molar concentrations of both the acid and the salt, it would have a pH of 4.76. It wouldn't matter what the concentrations were, as long as they were the same.
You can change the pH of the buffer solution by changing the ratio of acid to salt, or by choosing a different acid and one of its salts.
Alkaline buffer solutions
An alkaline buffer solution has a pH greater than 7. Alkaline buffer solutions are commonly made from a weak base and one of its salts.

A frequently used example is a mixture of ammonia solution and ammonium chloride solution. If these were mixed in equal molar proportions, the solution would have a pH of 9.25. Again, it doesn't matter what concentrations you choose as long as they are the same.
How do buffer solutions work?
A buffer solution has to contain things which will remove any hydrogen ions or hydroxide ions that you might add to it - otherwise the pH will change. Acidic and alkaline buffer solutions achieve this in different ways.
Acidic buffer solutions
We'll take a mixture of ethanoic acid and sodium ethanoate as typical.
Ethanoic acid is a weak acid, and the position of this equilibrium will be well to the left:
Adding sodium ethanoate to this adds lots of extra ethanoate ions. According to Le Chatelier's Principle, that will tip the position of the equilibrium even further to the left.
The solution will therefore contain these important things:
• lots of un-ionised ethanoic acid; • lots of ethanoate ions from the sodium ethanoate; • enough hydrogen ions to make the solution acidic.
Other things (like water and sodium ions) which are present aren't important to the argument.
Adding an acid to this buffer solution
The buffer solution must remove most of the new hydrogen ions otherwise the pH would drop markedly.
Hydrogen ions combine with the ethanoate ions to make ethanoic acid. Although the reaction is reversible, since the ethanoic acid is a weak acid, most of the new hydrogen ions are removed in this way.

Since most of the new hydrogen ions are removed, the pH won't change very much - but because of the equilibria involved, it will fall a little bit.
Adding an alkali to this buffer solution
Alkaline solutions contain hydroxide ions and the buffer solution removes most of these.
This time the situation is a bit more complicated because there are two processes which can remove hydroxide ions.
Removal by reacting with ethanoic acid
The most likely acidic substance which a hydroxide ion is going to collide with is an ethanoic acid molecule. They will react to form ethanoate ions and water.

PH value
In chemia pH leare basic
The pH internatiocell witelectrodefor aqueo
pH measscience, eengineeri
In tec(solvatedconcentra
pH[edit s
pH is defa solution
This pH, whic
wcewIetose
R
e(H)
stry, pH is aess than 7or alkaline.
scale is traonal agreemth transferee and a standous solutions
surements aenvironmenting, nutrition
chnical ted) hydroniumation.
source | editb
fined as the n.[1]
definition wrespond to h, for the hy
where E is a constant, T iselectrons tranwhen pH is nternational
electromotiveo the hydro
solution. Thelectrode. Th
Reference ele
a measure of 7 are said
Pure water
aceable to ament.] Primarence, by mdard electrods can be don
are importantal scin, water trea
erms, pHm ion, more
beta]
decimal log
was adopted activity. Id
ydrogen ion c
measured ps the tempernsferred is odefined in te Standard ISe force (e.m
ogen ion acte reference
he hydrogen-
ectrode | con
f the acidity oto be acid
has a pH ver
a set of stary pH standameasuring tde such as th
ne with a glas
nt in mediciience, oceanatment & wa
H is th often expr
garithm of th
because iondeally, electcan be writte
potential, E0
rature in kelvone. It followerms of acti
SO 31-8 as fm.f.) between
tivity when electrode m
-ion selectiv
ncentrated so
or basicity odic and solury close to 7
andard soluard values athe potentiahe silver chlss electrode
ine, biology,ography, civ
ater purificati
he negative lressed as t
he reciprocal
n-selective eltrode potenten as
0 is the standvin, F is thews that elecivity. Precisefollows:[7] An a reference
they are bmay be a si
ve electrode i
olution of KC
of an aqueouutions with 7.
utions whosare determinal differencloride electrand a pH m
, chemistry, vil engion, and man
logarithm ofthe measure
l of the hydr
lectrodes, wtial, E, follo
dard electro Faraday con
ctrode potente measuremgalvanic cele electrode aboth immersilver chloridis a standard
Cl || test solu
us solution. Sa pH gre
e pH is esned using a ce betweenrode. Measur
meter or using
agriculture, gineering, ny other app
f the active of the hy
rogen ion ac
which are useows the Ner
ode potentialnstant. For Htial is propo
ment of pH isll is set up toand an electrsed in the sde electroded hydrogen e
ution | H2 | Pt
Solutions witeater than
stablished bconcentratio
n a hydrogerement of pg indicators.
forestry, foochemic
plications.
vity of thydronium io
ctivity, aH+, i
ed to measurrnst equation
l, R is the gaH+ number o
ortional to ps presented io measure throde sensitivsame aqueoue or a calomelectrode.
t
th 7
by on en H
od al
he on
in
re n,
as of H in he ve us el

Firstly, themf, ES, of unkno
The This
poten the "
To acumbreferhydrosolutorderTo imstandstandthen solutmoreobserComvalue
The p
p[H]
This of pHhydroconctitratknowconcalkalthat calibvalueNern
he cell is filis measured
own pH is me
difference bmethod of
ntial. The pr"Nerstian slo
apply this prbersome hydence electrogen ion ations of knor to accommmplement thdard solutiondard buffer's
adjusted, ution. Furthere than two brved pH val
mmercial stane at 25 °C an
pH scale is l
was the oriH in 1924. ogen ions dientrations. Oe a solution
wn concentrentration of li are knownthe measur
bration is usue for the stannst equation i
lled with a sd. Then the eeasured.
between the calibration
roportionalityope".
rocess in prdrogen electrrode. It is activity. IUPAown H+ activmodate the fahis approach n and the rea value. The
using the "sr details, arebuffer solutiolues to a strandard buffernd a correcti
logarithmic a
ginal definitHowever,
irectly, if theOne way to
n of known cation of strbackground
n, it is easy tored potentiaually carriedndard electroin the form
solution of kemf, EX, of
two measuravoids the n
y constant, 1
ractice, a glarode. A com
calibrated AC has pro
vity.[1] Two oact that the "to calibratio
ading on a preading from
slope" contre given in thons are usedaight line wr solutions uon factor to
and therefor
tion of Søreit is possibe electrode ido this, wh
concentratioong alkali i
d electrolyte.o calculate tal can be cd out using ode potentia
known hydrthe same ce
red emf valuneed to kno
1/z is ideally
ass electrodmbined glass
against bufoposed the or more bufslope" may
on, the electrpH meteris am a second srol, to be ehe IUPAC red the electrod
with respect tusually come
be applied f
e pH is a dim
ensen, whichble to measuis calibrated hich has been of a stronin the prese. Since the cthe concentracorrelated wa Gran plot]
al, E0, and a
rogen ion acell containing
ues is proporow the stand
y equal to
de is used ras electrode hffer solutiouse of a s
ffer solutiondiffer slight
rode is first idjusted to b
standard buffequal to theecommendatde is calibrato standard be with informfor other tem
mensionless
h was supersure the conin terms of
en used exteng acid with ence of a reconcentrationation of hyd
with concen] The calibraslope factor,
tivity and thg the solutio
rtional to pHdard electrod
ather than thhas an in-buins of knowset of buffe
ns are used itly from ideaimmersed ine equal to th
ffer solution e pH for thtions.[1] Wheated by fittinbuffer valuemation on th
mperatures.
quantity.
seded in favoncentration o
hydrogen ioensively, is t
a solution oelatively higns of acid androgen ions sntrations. Thation yields , f, so that th
he on
H. de
he ilt
wn fer in al. n a he is at en ng es. he
or of on to of gh nd so he
a he

Acid Bas
An acid–neutralizifor quantuse of thhow acid
Acid–bas
Alkalime
Alkalimeconcentraacidometsubstance
METHO
Before stthe reacti
cmAfthththee
Tcinec
Tpty
se titration(H
–base titratioing the acidtitative analy
he neutralizatds and bases
se titrations
etry and acid
etry is the ation of a batry, is the sae.
D
tarting the tiion, the poin
can be usedmeasuremenA slope facfunctioning hat the hydrhe titration. he standard
electrolyte. Tequal to the n
The glass ecalibrated innstance, if
electrode shochemical com
The differencpH = p[H] +ypes of mea
H)
n is the deted or base wysis of the ction reactionwill react if
can also be u
dimetry(SH)
specializedasic (synonyame concept
itration a suint at which e
d to derive hts of E. The
ctor of less correctly. Trogen ion a As it is co
state as beThus, the efnumerical va
electrode (an a mediumone wishes ould be calimposition, as
ce between p+ 0.04. It is asurement.
ermination owith an acidconcentrationn that occursf their formul
used to find
d analytic ymous to alkt of specializ
itable pH indequivalent am
hydrogen ioe slope facto
than 0.95 The presenceactivity coeffonstant, its veing the soffect of usinalue of conce
and other iom similar t
to measureibrated in a s detailed be
p[H] and pHcommon pr
of the concend or base on of an unks between aclas are know
percent puri
use of ackaline) substzed analytic
dicator mustmounts of th
on concentraor, f, is usual
indicates the of backgr
fficient is efvalue can bolution con
ng this proceentration.
on selectiveto the one e the pH of
solution reelow.
H is quite smractice to us
ntration of af known co
known acid ocids and bas
wn.
ity of chemic
id-base titrtance. Acidim acid-base t
t be chosen. he reactants
ations from lly slightly lhat the elecround electrffectively coe set to one
ntaining theedure is to m
e electrodesbeing inve
f a seawateresembling se
mall. It has bee the term "
an acid or baoncentration.or base solutses and the k
cals.
ration to dmetry, someitration, but
The equivalhave reacte
experimentless than onctrode is no
rolyte ensureonstant durine by definin
e backgrounmake activit
s) should bestigated. For sample, theawater in i
een stated th"pH" for bot
ase by exactl. This allowtion. It makeknowledge o
determine thetimes spelle
for an acid
lence point od, will have
tal ne. ot es ng ng nd ty
be or he its
at th
ly ws es of
he ed
dic
of a

pH dependent on the relative strengths of the acid and base used. The pH of the equivalence point can be estimated using the following rules:
• A strong acid will react with a strong base to form a neutral (pH = 7) solution. • A strong acid will react with a weak base to form an acidic (pH < 7) solution. • A weak acid will react with a strong base to form a basic (pH > 7) solution.
When a weak acid reacts with a weak base, the equivalence point solution will be basic if the base is stronger and acidic if the acid is stronger. If both are of equal strength, then the equivalence pH will be neutral. However, weak acids are not often titrated against weak bases because the colour change shown with the indicator is often quick, and therefore very difficult for the observer to see the change of colour.
The point at which the indicator changes colour is called the end point. A suitable indicator should be chosen, preferably one that will experience a change in colour (an end point) close to the equivalence point of the reaction.
First, the burette should be rinsed with the standard solution, the pipette with the unknown solution, and the conical flask with distilled water.
Secondly, a known volume of the unknown concentration solution should be taken with the pipette and placed into the conical flask, along with a small amount of the indicator chosen.
The known solution should then be allowed out of the burette, into the conical flask. At this stage we want a rough estimate of the amount of this solution it took to neutralize the unknown solution. The solution should be let out of the burette until the indicator changes colour and the value on the burette should be recorded. This is the first (or rough) titre and should be discluded from any calculations.
At least three more titrations should be performed, this time more accurately, taking into account roughly where the end point will occur. The initial and final readings on the burette (prior to starting the titration and at the end point, respectively) should be recorded. Subtracting the initial volume from the final volume will yield the amount of titrant used to reach the end point. The end point is reached when the indicator just changes colour permanently. This is best achieved by washing a hanging drop from the tip of the burette into the flask right at the end of the titration to achieve a drop that is smaller in volume than what can usually be achieved by just dripping solution off the burette.
Acid–base titration is performed with a phenolphthalein indicator, when it is a strong acid – strong base titration, a bromthymol blue indicator in weak acid – strong base reactions, and a methyl orange indicator for strong acid – weak base reactions. If the base is off the scale, i.e. a pH of >13.5, and the acid has a pH >5.5, then an Alizarin eyellow indicator may be used. On the other hand, if the acid is off the scale, i.e. a pH of <0.5, and the base has a pH <8.5, then a Thymol Blue indicator may be used.
.CHOICE OF INDICATOR(H)

A pH indicator is a halochromic chemical compound that is added in small amounts to a solution so that the pH (acidity or basicity) of the solution can be determined visually. Hence a pH indicator is a chemical detector for hydronium ions (H3O+) or hydrogen ions (H+) in the Arrhenius model. Normally, the indicator causes the colour of the solution to change depending on the pH. Indicators can also show change in other physical properties; for example,olfactory indicators show change in their odor.
In chemistry:
• pH indicator, a chemical detector for protons in acid-base titrations • Redox indicator, a chemical detector for redox titrations • Complexometric indicator, a chemical detector for metal ions in complexometric
titrations • Zeta potential, a property of interfaces in fluids for Zeta potential titration
In biology:
• Indicator (genus), a genus of birds in the honeyguide family • Environmental indicator of environmental health (pressures, conditions and responses)
• pH is a measure of the activity of hydrogen ions (H+) in a solution and, therefore, its acidity or alkalinity. The acidity of some common acids are shown in the table below.
Acid Normality pH
Acetic N 2.4
Acetic 0.1 N 2.9
Acetic 0.01 N 3.4
Alum 0.1 N 3.2
Arsenious saturated 5.0
Benzoic 0.1 N 3.0
Boric 0.1 N 5.2

Acid Normality pH
Carbonic saturated 3.8
Citric 0.1 N 2.2
Formic 0.1 N 2.3
Hydrochloric N 0.1
Hydrochloric 0.1 N 1.1
Hydrochloric 0.01 N 2.0
Hydrocyanic 0.1 N 5.1
Hydrogen sulfide 0.1 N 4.1
Lactic 0.1 N 2.4
Lemon Juice 2
Malic 0.1 N 2.2
Nitric 0.1N 1.0
Orthophosphoric 0.1 N 1.5
Oxalic 0.1 N 1.3
Salicylic saturated 2.4
Succinic 0.1N 2.7
Sulfuric N 0.3
Acid Normality pH
Sulfuric 0.1 N 1.2
Sulfuric 0.01 N 2.1
Sulfurous 0.1 N 1.5
Stomach Acid 1
Tartaric 0.1 N 2.2
Trichloracetic 0.1N 1.2
Vinegar 3
• Sponsored Links

ACID BASE THEORY(H)
An acid–base reaction is a chemical reaction that occurs between an acid and a base. Several concepts exist that provide alternative definitions for the reaction mechanisms involved and their application in solving related problems. Despite several differences in definitions, their importance becomes apparent as different methods of analysis when applied to acid–base reactions for gaseous or liquid species, or when acid or base character may be somewhat less apparent.
The term Lewis acid refers to a definition of acid published by Gilbert N. Lewis in 1923, specifically: An acid substance is one which can employ an electron lone pair from another molecule in completing the stable group of one of its own atoms. Thus, H3O+ is a Lewis acid, since it can accept a lone pair, completing its stable form, which requires two electrons.
The modern-day definition of Lewis acid, as given by IUPAC is a molecular entity (and the corresponding chemical species) that is an electron-pair acceptor and therefore able to react with a Lewis base to form a Lewis adduct, by sharing the electron pair furnished by the Lewis base. This definition is both more general and more specific—the electron pair need not be a lone pair (it could be the pair of electrons in a π bond, for example), but the reaction should give an adduct (and not just be a displacement reaction).
A Lewis base, then, is any species that donates a pair of electrons to a Lewis acid to form a Lewis adduct. For example, OH− and NH3 are Lewis bases, because they can donate a lone pair of electrons.
Some compounds, such as H2O, are both Lewis acids and Lewis bases, because they can either accept a pair of electrons or donate a pair of electrons, depending upon the reaction.
Usually the terms Lewis acid and Lewis base are defined within the context of a specific chemical reaction. For example, in the reaction of Me3B and NH3to give Me3BNH3, Me3B acts as a Lewis acid, and NH3 acts as a Lewis base. Me3BNH3 is the Lewis adduct.
Classically, the term "Lewis acid" is restricted to trigonal planar species with an empty p orbital, such as BR3 where R can be an organic substituent or a halide. For the purposes of discussion, even complex compounds such as Et3Al2Cl3 and AlCl3 are treated as trigonal planar Lewis acids. Metal ions such as Na+, Mg2+, and Ce3+, which are invariably complexed with additional ligands, are often sources of coordinatively unsaturated derivatives that form Lewis adducts upon reaction with a Lewis base. Other reactions might simply be referred to as "acid-catalyzed" reactions.
Reformulation of Lewis Theory
Lewis had suggested in 1916 that two atoms are held together in a chemical bond by sharing a pair of electrons. When each atom contributed one electron to the bond it was called a covalent bond. When both electrons come from one of the atoms it was called a dative covalent bond or coordinate bond. The distinction is not very clear-cut. For example, in the formation of an ammonium ion from ammonia and hydrogen the ammonia molecule donates a pair of electrons to the proton; the identity of the electrons is lost in the ammonium ion that

is formedand an el
A more mempty ato(LUMO)
Compari
A Lewis proton isLowry acused for very diffa very we
In anothebutylpyriexample role in dtiny proto
A Brønst
Lewis ac
Lewis dshowadd
Lewis accommon
• Examinclu• th• m• tr• p
d. Neverthellectron-pair a
modern definomic or mol) can accomm
son with Brø
base is often a Lewis acicid is also a the A—H b
ficult to protoeak Brønsted
er comparisoidine reacts tdemonstrate
determining ton.
ted–Lowry a
cids
diagrams sduct having a
cids are diveare those th
mples of Leude: he proton (H
metal cationsrigonal plan
pentahalides
less, Lewis sacceptor be
nition of a Llecular orbitamodate a pai
ønsted–Low
n a Brønstedid as it can aLewis base
bond as a lononate, yet std–Lowry ba
on of Lewis to form the hes that stericthe strength
acid is a prot
showing tha positive ch
erse. Simpleshat undergo a
ewis acids b
H+) and acidis, such as Li+
ar species, sof phosphor
suggested thclassified as
Lewis acid isal of low ir of electron
wry Theory
d–Lowry basaccept a pairas loss of H+
ne pair on thtill react withse but it form
and Brønstehydrochloridc factors, in
of the inter
ton donor, no
he formatiarge)
st are those ta reaction pr
based on th
ic compound+ and Mg2+, uch as BF3 arus, arsenic,
hat an electrs acid.
s an atomic energy. Thins.
se as it can dr of electron+ from the ac
he conjugateh a Lewis ams a strong a
ed–Lowry acde salt with Haddition to
raction betw
ot an electro
on of the
that react dirrior to formin
he general
ds onium ionoften as thei
and carbocatand antimon
ron-pair don
or moleculais lowest e
donate a pairns. The conjucid leaves th base. Howecid. For exaadduct with
cidity by BroHCl but doeelectron con
ween the bulk
on-pair accep
e ammonium
rectly with tng the adduc
definition o
ns, such as Nir aquo or ettions H3C+ ny
or be classif
ar species wienergy mole
r of electronugate base ohose electronever, a Lewiample,carbonBF3.
own and Kans not react w
nfiguration fky di-t-buty
ptor.
m ion (diag
the Lewis bact.
of electron
NH4+ and H3O
ther complex
fied as a bas
ith a localizeecular orbit
ns to H+;[5] thf a Brønsted
ns which weris base can bn monoxide
nner, 2,6-di-with BF3. Thfactors, play ylpyridine an
gram shoul
ase. But mor
pair accepto
O+ xes,
se
ed al
he d–re be is
-t-his
a nd
ld
re
or

• electron poor π-systems, such as enones and tetracyanoethylene
Again, the description of a Lewis acid is often used loosely. For example, in solution, bare protons do not exist.
Simple Lewis acids
The most studied examples of such Lewis acids are the boron trihalides and organoboranes, but other compounds exhibit this behavior:
BF3 + F− → BF4−
In this adduct, all four fluoride centres (or more accurately, ligands) are equivalent.
BF3 + OMe2 → BF3OMe2
Both BF4− and BF3OMe2 are Lewis base adducts of boron trifluoride.
In many cases, the adducts violate the octet rule, such as the triiodide anion:
I2 + I− → I3−
The variability of the colors of iodine solutions reflects the variable abilities of solvent to form adducts with the Lewis acid I2.
In some cases, the Lewis acids are capable of binding two Lewis bases, a famous example being the formation of hexafluorosilicate:
SiF4 + 2 F− → SiF62−
Complex Lewis acids
Most compounds considered to be Lewis acids require an activation step prior to formation of the adduct with the Lewis base. Well known cases are the aluminium trihalides, which are widely viewed as Lewis acids. Aluminium trihalides, unlike the boron trihalides, do not exist in the form AlX3, but as aggregates and polymers that must be degraded by the Lewis base. A simpler case is the formation of adducts of borane. Monomeric BH3 does not exist appreciably, so the adducts of borane are generated by degradation of diborane:
B2H6 + 2 H− → 2 BH4−
In this case, an intermediate B2H7− can be isolated.
Many metal complexes serve as Lewis acids, but usually only after dissociating a more weakly bound Lewis base, often water.
[Mg(H2O)6]2+ + 6 NH3 → [Mg(NH3)6]2+ + 6 H2O
H+ as Lewis acid

The proton (H+)� is one of the strongest but is also one of the most complicated Lewis acids. It is convention to ignore the fact that a proton is heavily solvated (bound to solvent). With this simplification in mind, acid-base reactions can be viewed as the formation of adducts:
• H+ + NH3 → NH4+
• H+ + OH− → H2O
Applications of Lewis acids
Typical example of a Lewis acid in action is in the Friedel–Crafts alkylation reaction.[3] The key step is the acceptance by AlCl3 of a chloride ion lone-pair, forming AlCl4− and creating the strongly acidic, that is, electrophilic, carbonium ion.
RCl +AlCl3 → R+ + AlCl4−
Lewis bases
A Lewis base is an atomic or molecular species where the HOMO is highly localized. Typical Lewis bases are conventional amines such as ammonia and alkyl amines. Other common Lewis bases include pyridine and its derivatives. Some of the main classes of Lewis bases are
• amines of the formula NH3−xRx where R = alkyl or aryl. Related to these are pyridine and its derivatives.
• phosphines of the formula PR3−xAx, where R = alkyl, A = aryl. • compounds of O, S, Se and Te in oxidation state 2, including
water, ethers, ketones
The most common Lewis bases are anions. The strength of Lewis basicity correlates with the pKa of the parent acid: acids with high pKa's give good Lewis bases. As usual, a weaker acid has a stronger conjugate base.
• Examples of Lewis bases based on the general definition of electron pair donor include: • simple anions, such as H– and F-. • other lone-pair-containing species, such as H2O, NH3, HO–,
and CH3–
• complex anions, such as sulfate • electron rich π-system Lewis bases, such as ethyne, ethene,
and benzene

The strength of Lewis bases have been evaluated for various Lewis acids, such as I2, SbCl5, and BF3.[8]
Historic acid–base theories
Lavoisier's oxygen theory of acids
The first scientific concept of acids and bases was provided by Lavoisier circa 1776. Since Lavoisier's knowledge of strong acids was mainly restricted tooxoacids, such as HNO3 (nitric acid) and Sulfuric acid. 4 (sulphuric acid), which tend to contain central atoms in high oxidation states surrounded by oxygen, and since he was not aware of the true composition of the hydrohalic acids (HF, HCl, HBr, and HI), he defined acids in terms of their containing oxygen which in fact he named from Greek words meaning "acid-former). The Lavoisier definition was held as absolute truth for over 30 years, until the 1810 article and subsequent lectures by Sir Humphry Davy in which he proved the lack of oxygen in H2S, H2Te, and the hydrohalic acids. However, Davy failed to develop a new theory, concluding that "acidity does not depend upon any particular elementary substance,
Liebig's hydrogen theory of acids
Circa 1838 Justus von Liebeg proposed that an acid is a hydrogen-containing substance in which the hydrogen could be replaced by a metal. This redefinition was based on his extensive work on the chemical composition of organic acids, finishing the doctrinal shift from oxygen-based acids to hydrogen-based acids started by Davy. Liebig's definition, while completely empirical, remained in use for almost 50 years until the adoption of the Arrhenius definition
Common acid–base theories
Arrhenius definition

Svante A
The first theory oestablishiPrize in C
As define
• an A(H+);
This causmodern tthat a bar
• an Athat i
The Arrhrefer to tdissolvedliquid am
The univformationthat in Aan acid aOH− are neutraliza
ac
Arrhenius
modern defof acids, it ing the preseChemistry in
ed by Arrhen
Arrhenius a;[6] that is, an
ses the prototimes, the usre proton do
Arrhenius basis, a base inc
henius definthe concentrd in toluenemmonia are n
versal aqueon of a water
Arrhenius aciand a base.[6]
neutralized,ation reactio
cid + base →
finition of acfollowed f
ence of ions n 1903.
nius:
acid is a sun acid increa
onation of wse of H+ is res not exist
se is a substacreases the c
nitions of acration of th
e are not acinot alkaline.
ous acid–basr molecule frid–base reac] This is ane, for they c
on can be put
→ salt + wate
cids and basefrom his 1in aqueous
ubstance thaases the conc
water, or the regarded as as a free spe
ance that disoncentration
cidity and alke solvent ioidic, and mo
se definitionfrom a protoctions, a salteutralization combine to t into a word
er
es was devis884 work solution and
at dissociatescentration of
creation of ta shorthand
ecies in aque
ssociates in n of OH− ion
kalinity are ons. Under tolten NaOH
n of the Arrn and hydroand water areaction - thform H2O,
d equation:
sed by Svantwith Friedri
d led to Arrh
s in water f H+ ions in a
the hydroniud for H3O+, eous solution
water to forns in an aque
restricted tothis definitio
H and solutio
rhenius conoxide ion. Thare formed frhe acid and b
the water
te Arrheniusich Wilhelm
henius receiv
to form hyan aqueous s
um (H3O+) iobecause it is
n.[10]
rm hydroxideous solution
o aqueous son, pure H2
ons of calci
ncept is deschis leads to
from the reacbase propertmolecule. T
s. A hydrogem Ostwald iving the Nob
ydrogen ionsolution.
on.[9] Thus, is now know
e (OH−) ionn.
solutions, an2SO4 and HCium amide i
cribed as ththe definitioction betweeties of H+ anThe acid-bas
en in el
ns
in wn
ns;
nd Cl in
he on en nd se

The positive ion from a base and the negative ion from an acid form a salt together - in other words, an acid-base neutralization reaction is a double-replacement reaction. For example, when a neutralization reaction takes place between hydrochloric acid (HCl) and sodium hydroxide (NaOH), the products are sodium chloride (common table salt) and water.
HCl(aq) + NaOH(aq) → NaCl + H2O
Notice how the cations and the anions merely switched places: the Na+ from the NaOH combined with the Cl- from the HCl to form NaCl, while the OH− from the NaOH combined with the H+ from the HCl to form H2O.
ACID AND BASE STRENGTH IN IONIZATION CONSTANT.(H)
Water has a very low concentration of ions that are detectable. Water undergoes self-ionization, where two water molecules interact to from a hydronium ion and a hydroxide ion.
H2O + H2O → H3O+ + OH-
To understand this process in further detail, we can take a closer look.
Even though water does not form a lot of ions, the existence of them is evident as is proved by the electrical conductivity of pure water. Water undergoes ionization because the powerfully electronegative oxygen atom takes the electron from a hydrogen atom, making the proton dissociate.
H-O-H → H+ + OH-
Here, two ions are formed:
1. Hydrogen ions H+ 2. Hydroxyl ions OH-
The hydrogen ions then react with water to form hydronium ions:
H+ + H2O → H3O+
Now, we since we have seen the break down of the process, this brings us back to our first analysis of self-ionization. Typically, hydrogen atoms are bonded with another water molecule resulting in a hydration that forms a hydronium and hydroxide ion.
H2O + H2O → H3O+ + OH-
In 1L of pure water at 25 degrees Celsius, there is 10-7 moles of hyrodronium ions or hydroxide ions at equilibrium. Let’s come back to the question of to what degree will a substance form ions in water? For water, we have learned that it will occur until 10-7 moles of either ion will ionize at the previous given conditions. Since this is during equilibrium, a constant can be formed.
H2O → H+ + OH-

Keq= [H+] [OH-]/[H2O]
Each molecule is expressed in concentrations of moles per Liter. You might be thinking… Hold on, what is this equation talking about? I will explain further on, once we understand the self-ionization of water.
Each H+, OH-, H2O are expressed in concentrations. The concentration of a substance is usually defined in moles per Liter. To find the concentration for water, we must calculate the moles per Liter then. Let’s think about it, this is relatively simple. Since we have 1 L of water, we have 1000 grams of water. To convert to moles, we simply divide by the molecular mass. Concentration therefore is calculated by: (1000/18) The answer is 55M of water. Now, we have (10-7)2/55.5 = 1.8 * 10-16 M
However, since there is not a lot of ionization, the change in water concentration is very small. It does not decrease very much. All of these concentrations are generally reliable and dependable. Experimentation has proved their accuracy as scientists use these standard values. The concentration of water is considered a constant. Kw = 55.5M But because water is a liquid and its molarity is invariable, we multiply the concentrations of the ions and use that answer for other empirical calculations. Keq = 10-14 M = [OH-] [H+] In chem. 2B, this standard equation is used
Kw = [H3O+] [OH-] = 10-14
Weak acids and bases, however do not behave the same way. Their amounts cannot be calculated as easily. This is because the ions do not fully dissociate in the solution. Weak acids have a higher pH than strong acids, because weak acids do not release all of its hydrogens.
The acid dissociation constant tell us the extent to which an acid dissociates
HCN(aq) + H2O?H3O+ + CN-
Ka= [H3O+] [CN-] / HCN
This equation is used fairly often when looking at equilibrium reactions. During equilibrium, the rates of the forward and backward reaction are the same. However, the concentrations tend to be varied. Since concentration is what gives us an idea of how much substance has dissociated, we can relate concentration ratios to give us a constant. K is found by first finding out the molarity of each substance
Example
Let’s calculate the ionization constant of a weak acid. Solve for Ka given 0.8M of hydrogen cyanide and 0.39 M for hyrodronium and cyanide ions
HCN(aq) + H2O → H3O+ + CN-
(HCN) is .8M (H3O+) is .0039M (CN-) is .0039M

We can a
The equi
We can uconstant A smalle
Some com
C5H5N(a
We woulobvious. better.
Example
Using theKb= 1.5 *
Since weearlier (c
Kb= [OH
Because since we in molarican solve
To makeweak basis 4.7 * 1
Our appr5 percent
Accordinsolution,
H
The constoxonwritte
H
Brønste
assume here
librium cons
use the simiis a measure
er Kb corresp
mmon weak
aq) + H2O(l)
ld find Kb thLet’s do an
e previous e* 10-9
e already havconcentration
H-] [C5H5NH
pyridine is have 3M, le
ity due to die for them as
e the calculase. Now our 10-5 M
roximation wt the initial c
ng to Arrhenreleasing th
HA A− + H
equilibriumtant. The lib
nium) ion H3
en as an acid
HA + H2O
ed and Lowr
that [H3O+]
stant would b
ilar method e of the exteponds to a we
k bases: NH3
→ C5H5N+
he same wayn example th
equation, cal
ve our equan of products
H+]/ [C5H5N]
a weak baseet’s make 3Missociation. Ss being X. T
ation simple,equation be
was efficientconcentration
nius's originahe hydrogen
H+.
m constant fberated proto3O+, and sod–base react
A− + H3O+
ry generalise
= [CN-]
be Ka= .003
to find the Kent a base wieaker base, a
, NH4OH, H
+(aq) + OH-
y we did Ka
hat is a littl
lculate the c
ation, let’s ws divided by
= 1.5 * 10-9
e, we can asM-X our calSince our ioHUS: (X2/3-
, we can estecomes: (X2/
t because x tn for that est
al definition,ion H+ (a pr
for this dison combineso Arrhenius tion: +.
ed this furthe
92/.8 = 1.9E
Kb constant ill dissociateas a higher o
HS-, C5H5N
(aq)
. However, le more cha
oncentration
write the expy concentratio9
ssume that nculation. Su
ons are unkn-X) = 1.5 * 1
timate 3-X t/3) = 1.5 * 1
turned out totimation to b
an acid is a oton):
ssociation rs with a wat
later propo
er to a proton
E-5
for weak bae. Kb relates one a stronge
most problellenging to
n of [OH-] in
pression for on of reactan
not much ofubtracting X nown and the10-9
to be approx10-9 When w
o be pretty smbe justified.
substance th
eaction is kter moleculeosed that th
n exchange r
ases. Again, these molar
er base.
ems are not ahelp you un
n a 3M Pyri
the constantnts)
f it will disais going to bey are both
ximately 3 bwe solve for
mall. X mus
hat dissociat
known as e to give a he dissociatio
reaction:
an ionizatiority quantitie
as simple annderstand th
dine solution
t as discusse
ssociate. Anbe the changone mole, w
because of thX our answe
st be less tha
tes in aqueou
a dissociatiohydronium (oon should b
on es.
nd his
n.
ed
nd ge we
he er
an
us
on or be

ac
The acidcreating conjugatedefinitionaccepting
In solutioion, regarthan a pro
The desigBH+ of a
which is
H2O (aci
The hydrwater. Arespectiv
A broadeby the spwere a prhydrolys
BSimilarly
[AEquilibri
An acid specific e
H
the therm
where {Adimensioactivitiesderivatio
cid + base
d loses a pra conjugate
e base is An applies to g a proton an
on chemistryrdless of theoton.
gnation of ana base B diss
B
s the reverse
d) + B (base
roxide ion OAcids and bvely.
er definitionplitting of wroton donor,is equilibriu
B(OH)3 + 2 Hy, metal ion hAl(H2O)6]3+
ium constan
dissociationequilibrium b
HA + H2O
modynamic e
A} is the actionless. Activs of the rean of this exp
conjugate
roton, leavine acid. For
A− and the cother solven
nd forming t
y, it is comme solvent. In
n acid or basociates acco
BH+ + OH−
e of the equil
e) OH− (c
OH−, a well kbases are th
of acid dissater molecu, but it has bm:
H2O B(OHhydrolysis c+H2O [Ant
n constant ibetween a m
A− + H3O+
equilibrium c
ivity of the cvities of thactants are pression.
base + conju
ng a conjugaqueous so
conjugate acnts, such asthe conjugate
mon to use aqueous sol
se as "conjuording to
B + H2O
librium
conjugate ba
known basehus regarded
sociation incles. For exam
been confirm
H)4− + H3O+
auses ions suAl(H2O)5(OH
s a particulmonoprotic a+
constant, K
chemical spehe products
placed in
ugate acid.
gate base; tholutions of acid is the hydimethyl sue acid SH+.
H+ as an ablution H+ den
ugate" depen
O
se) + BH+ (c
, is here actd simply a
cludes hydromple, boric
med by Rama
+. uch as [Al(H
H)]2+ + H3O+
lar example acid, HA and
can be de
ecies A etc. Kof dissocia
the denomi
he proton isan acid HAydronium io
ulfoxide: the
bbreviation fnotes a solva
ds on the co
conjugate ac
ing as the cs donors an
olysis, in whacid (B(OH
an spectrosc
H2O)6]3+ to b+.
of an equild its conjugat
efined by
K is dimeation are pinator. See a
s transferredA, the base on. The Brø
solvent S a
for the solvaated hydroni
ontext. The c
cid).
onjugate basnd acceptor
hich protons )3) producesopy that this
behave as we
librium conste base A−, i
ensionless sinlaced in thactivity coe
d to the basis water; th
ønsted–Lowracts as a bas
ated hydrogeium ion rathe
conjugate aci
se of the acirs of proton
are produces H3O+ as if s is due to th
eak acids:
stant. For thin water,
nce activity he numeratoefficient for
e, he ry e,
en er
id
id ns
ed it
he
he
is or,
a

Since actalso be w
whercoeff
To aare dcondmighperchmostconst[H+]
ishmdppa
Adritq
M
tivity is the written as
re [HA] repficients.
avoid the determined, ditions in whht be hlorate (1 Mt concentratetant, approxifor the conc
s obtained. Thowever, thamedium usedifferent conpublished coparticular apand other the
Although Ka dimensionlesresult of omit is not unus
quoted with a
Monoprotic a
product of c
presents the
complicatiowhere possich Γ can bea soluti
M = 1 mol·dmed solutions imately 55 m
centration of
This is the dat all publised in their dnditions, asonstants refepplication, theories.
appears to ss or it wouitting the consual, particula dimension
acids
Variat
concentratio
e concentra
ons involvesible, in a e assumed toion of
m−3, a unit ofit can be as
mol·dm−3. Onf the hydroni
definition inshed dissociadeterminatios shown foer to an ionhey may be
have the duld not be pnstant term [larly in textsas, for exam
tion of pKa o
n and activit
ation of HA
ed in usingmedium of
o be always c0.1 M sod
f molar concssumed that n dividing Kium ion the e
n common usation constaon and that or acetic acinic strengthadjusted by
dimension ofossible to ta[H2O] from ts relating to mple, "Ka = 3
of acetic acid
ty coefficien
A and Γ is
g activities,f high ionic constant.[10] Fdium nitentration). Fthe concentr
K by the cexpression
se. pKa is deant values re
different vid in the il
h other thany means of
f concentratake its logarthe definingbiochemical300 M".
d with ionic
nt (γ) the de
s a quotien
, dissociatistrength, th
For exampletrate or 3Furthermore,ration of waonstant term
efined as −loefer to the values are ollustration an the one respecific ion
tion it musrithm. The i
g expression.l equilibria,
strength
finition coul
nt of activit
on constanhat is, undee, the medium3 Mpotassium, in all but thater, [H2O],
ms and writin
og10 Ka. Notspecific ionobtained witabove. Wheequired for
theory (SIT
st in fact billusion is th Neverthelesto see a valu
ld
ty
nts er m m he is
ng
te, ic th en
a T)
be he ss ue
S
Vw
Polyproti
% specie
See also: Aci
Variation of with the diffe
ic acid
es' formation
id#Monopro
the % formerence betwe
n as a functio
otic acids
mation of a meen the pH a
on of pH
monoprotic aand the pKa o
acid, AH, anof the acid.
nd its conjuggate base, AA−,

% specieacid. pKa
Polyprotithe first pas Ka2, etprotons.
eq
H
H
H
WhenexamH2POHPOdistri5.5 a
Whenbetwdiffercitric
In geexam
HH
es formation a1=3.13, pKa
ic acids are aproton may btc. Phosphor
quilibrium
H3PO4 H2
H2PO4− H
HPO42− PO
n the differmple, each spO4
− may be cO4
2− may be cibution diagr
and 10.
n the differeeen the pHrence, the m
c acid are bu
eneral, it ismple, for a di
H2A HA−
HA− A2− +
it can bespecies. Sit; that iillustrate to the ruthe case acid is tepKa1 > pK
calculated wa2 = 4.76, pK
acids that cabe denoted aric acid, H3P
PO4− + H+
HPO42− + H+
O43− + H+
rence betwepecies may crystallised crystallised ram shows
ence betweenH range of more the over
ffered over t
s true that iprotic acid,
+ H+ + H+
e seen that Since the pros the cause this rule, as
ule is found of VO2
+ (aqetrahedral, 4Ka2 for vana
with the proKa3=6.40.
an lose moreas Ka1 and thPO4, is an e
pKa value
pKa1 = 2.15
pKa2 = 7.20
pKa3 = 12.3
en successivbe considerefrom solutiofrom solutiothat the con
n successiveexistence o
rlap. The casthe whole ra
successive pH2A, the tw
the second oton carries of the tren
s do the valuit indicates
q), the vanad4-coordinateadium(V) ox
ogram HySS
e than one prhe constants example of a
7
ve pK valueed as an acion by adjuston by adjustncentrations
pK values iof the specise of citric aange of pH 2
pK values iwo equilibria
proton is ra positive ch
nd noted aboues for vanathat a major
dium is octahe. This is thoacids.
for a 10 mi
roton. The cofor dissociata polyprotic
es is about id in its owntment of pH tment of pH of the two i
is less than aies in equilacid is shown2.5 to 7.5.
ncrease (Paare
removed froharge extra wove. Phosphadic acid, H3
r change in hedral, 6-coohe basis for
illimolar solu
onstant for dtion of succeacid as it c
four or mon right;[15] Into about 5.5to about 10
ions are max
about four thibrium. Then at the right
auling's first
om a negatiwork is need
horic acid va3VO4. Whenstructure is
ordinate, whr an explana
ution of citr
dissociation oessive protoncan lose thre
re, as in thn fact salts o5 and salts o
0. The specieximum at p
here is overlae smaller tht; solutions o
t rule).[16] Fo
ively chargeded to removalues (above
n an exceptiooccurring. I
hereas vanadation of wh
ic
of ns ee
his of of es H
ap he of
or
ed ve e) on In ic
hy

Isoelectr
Main arti
For subsweightedweightedspecies othe exam
AA
S
[A
[H
IONIC E
ic point[edit
icle: isoelect
tances in sod by charge d sum of conof each type,
mple of glycin
AH2+ AH
AH A- + H
Substitute the
A-][H+]2 = K
At the isois equal t
H+]2 = K1K2
Ther
pae
W
We
EQUILIBIRI
t source | edi
tric point
olution the isvalue, of c
ncentrations , the isoelecne, defined a
+ H+; [AH][H+; [A-][H+]
e expression
K1K2[AH2+]
oelectric poito the concen
efore, taking
pI values acid#Chemicequilibrium w
Water self-io
Water posseequilibrium c
is given b
Wheassum
Ts
IUM (H)
itbeta]
soelectric poconcentration
of negativetric point caas AH. Ther
[H+] = K1[A= K2[AH]
n for [AH] in
int the concentration of th
g cologarithm
for aminocal propertiewith each oth
onization
sses both acconstant for
2 H2
by
en, as is usumed to be co
The self-ionispecial case
oint (pI) is dns of positively charged an be obtainee are two dis
AH2+]
nto the first e
entration of he negatively
ms, the pH is
o acids aes. When mher a full spe
cidic and bathe equilibri
2O OH− +
ually the casonstant, this
ization constof an acid di
defined as thvely chargedspecies. In ted directly fssociation eq
equation
the positively charged sp
s given by
are listed more than tw
eciation calc
asic propertiium
+ H3O+
e, the conceexpression m
tant of waissociation c
he pH at whd species isthe case thafrom the pKquilibria to c
ly charged species, A-, so
at Proteinogwo charged sculation may
ies. It is am
entration of may be repla
ater, Kw, is constant.
hich the sums equal to that there is on
K values. Takconsider.
species, AH2
o
genic aminspecies are iy be needed.
mphoteric. Th
water can baced by
thus just
m, he ne ke
2+,
no in
he
be
a

Ionization(SH)
Ionization is the process of converting an atom or molecule into an ion by adding or removing charged particles such aselectrons or ions. In the case of ionization of a gas, ion pairs are created consisting of a free electron and a positive ion.
Ionization is the gain or loss of electrons. The loss of electrons, which is the more common process in astrophysical environments, converts an atom into a positively charged ion, while the gain of electrons converts an atom into a negatively charged ion. In the subsequent discussion, we will use the terms ionization and ionize in the sense of losing electrons to form positive ions.
Types of ionization(SH)
The process of ionization works slightly differently depending on whether an ion with a positive or a negative electric charge is being produced. A positively charged ion is produced when an electron bonded to an atom (or molecule) absorbs the proper amount of energy to escape from the electric potential barrier that originally confined it, thus breaking the bond and freeing it to move. The amount of energy required is called the ionization energy. A negatively charged ion is produced when a free electron collides with an atom and is subsequently caught inside the electric potential barrier, releasing any excess energy.
In general, ionization can be broken down into two types: sequential ionization and non-sequential ionization. In classical physics, only sequential ionization can take place; refer to the Classical ionization section for more information. Non-sequential ionization violates several laws of classical physics; refer to the Quantum ionization section.
Classical ionization
Applying only classical physics and the Bohr model of the atom makes both atomic and molecular ionization entirely deterministic; that is, every problem will always have a definite and computable answer. According to classical physics, it is absolutely necessary that the energy of the electron exceeds the energy difference of the potential barrier it is trying to pass. In concept, this idea should make sense: The same way a person cannot jump over a one-meter wall without jumping at least one meter off the ground, an electron cannot get over a 13.6-eV potential barrier without at least 13.6 eV of energy.
Applying to positive ionization
According to these two principles, the energy required to release an electron is strictly greater than or equal to the potential difference between the current bound atomic or molecular orbital and the highest possible orbital. If the energy absorbed exceeds this potential, then the electron is emitted as a free electron. Otherwise, the electron briefly enters an excited state until the energy absorbed is radiated out and the electron re-enters the lowest available state.
Applying to negative ionization

Due to the shape of the potential barrier, according to these principles, a free electron must have an energy greater than or equal to that of the potential barrier in order to make it over. If a free electron has enough energy to do so, it will be bound to the lowest available energy state, and the remaining energy will be radiated away. If the electron does not have enough energy to surpass the potential barrier, then it is forced away by the electrostatic force, described by Coulombs Law, associated with the electric potential barrier.
Sequential ionization
Sequential ionization is a description of how the ionization of an atom or molecule takes place. For example, an ion with a +2 charge can be created only from an ion with a +1 charge or a +3 charge. That is, the numerical charge of an atom or molecule must change sequentially, always moving from one number to an adjacent, or sequential, number.
Quantum ionization
In quantum mechanics, ionization can still happen classically, whereby the electron has enough energy to make it over the potential barrier, but there is the additional possibility of tunnel ionization.
Tunnel ionization
Tunnel ionization is ionization due to quantum tunneling. In classical ionization, an electron must have enough energy to make it over the potential barrier, but quantum tunneling allows the electron simply to go through the potential barrier instead of going all the way over it because of the wave nature of the electron. The probability of an electron's tunneling through the barrier drops off exponentially with the width of the potential barrier. Therefore, an electron with a higher energy can make it further up the potential barrier, leaving a much thinner barrier to tunnel through and, thus, a greater chance to do so.
Non-sequential ionization
When the fact that the electric field of light is an alternating electric field is combined with tunnel ionization, the phenomenon of non-sequential ionization emerges. An electron that tunnels out from an atom or molecule may be sent right back in by the alternating field, at which point it can either recombine with the atom or molecule and release any excess energy or have the chance to further ionize the atom or molecule through high-energy collisions (electron re-scattering model]). This additional ionization is referred to as non-sequential ionization for two reasons: One, there is no order to how the second electron is removed, and, two, an atom or molecule with a +2 charge can be created straight from an atom or molecule with a neutral charge, so the integer charges are not sequential. Thus, the phenomenon of non-sequential ionization enormousely increase the probability of doubly charged ion formation at lower laser-field intensities. Moreover, more than two electrons can simultaneously ionize via non-sequential ionization.

The nonatoms whmomentuspace andtwo electan ionizadirectioncenter. Taxes x apotential direction
Atomic s
Accordinpotential
will e
Ithacuthpep
P
Ichwlaathrsb
-sequential hich until reum for both d may be trutrons as the ation of one
n on the two-The sequentiaand y when
channels fron.
stabilization
ng to a gene
experience th
t means thathe dipole ap
atom does ncounter-intuiultra-strong heory show
potential ofelectromagneprevents ioni
Population tr
n 1992, de could survivehave been exwith the fielaser pulse d
atoms. We sheoretical c
resonant excsuch as 6f ofbandwidth. T
ionization ccently were electrons re
ue for linear two dimens
e two-dimen-electron plaal ionization
n the two diom the hype
eral theory o
he time aver
t within this pproximationnot ionize atitive ionizatifield limit as that durin
f H2+ is fo
etic field isization.
rapping
Boer and Me in the highxcited by thed during theid not ionizehall refèr todculation th
citation into f Xe whidi cThese level
can be readithe only to
emains so lopolarization
sional atom wnsional electane emitted n on the otheimensionaler-nucleus an
of Cook et a
rage effectiv
.
approximatn whose timet all. In praion rates for and atomic ng the actionormed whics present in
Moller showehly excited ste dynamic Ste rising parte completely
o this phenomhat incomple
a common consists of 7ls along wit
ily understohandle numw that they
n but is not fwhere the sitron which from the muer hand is rehyper-electrnd then ioni
al., the electr
ve potential
tion the effece-dependent
actice, becaufields highestabilizationn of the lasch binds thn similarity
ed that Xe atates 4f, 5f, atark shift of t of the lasey these statemenon as "pete ionizatiolevel with
7 quasi-degrnth the cont
ood with onmerically. It h
effectively mfor the circulimultaneous results in jeultiply chargepresented bron is first ized by the h
ron in the ex
ctive potentit potential is use atoms ioer than for lon of ionizatier field, thehe electronto the hydr
atoms subjecand 6f . Thethe levels in
er pulse. Subs leaving bepopulation tron occurs wionization lnenite leveisinuum cons
e-dimensionhappens whemove in onelar. One mayionization o
ets of probaged nucleus by the emissguided by
hyper-electri
xternal rapid
ial for the ellinear is con
onize, it resower field vaion occurs. e time-averan even if rogen mole
cted to shorese states wento multiphotbsequent evoehind some hrapping".We
whenever theloss. We cons in the rangstitute a lam
nal models oen the angulae dimensiony then look of both is juability in 45or the squarions from ththe Coulomic field in 45
dly oscillatin
lectric field instant and thsults in lowealues, even iMore precis
aged effectivvery stron
ecule ion an
rt laser pulseere believed tton resonancolution of thhighly excitee mention there is parallnsider a stage of the lasembda system
of ar al at
ust 5° re he
mb 5°
ng
in he er in se ve ng nd
es to ce he ed he el te er m.

During the laser interaction, when the intensity is increased the levefs are shified and one of them is brought into muitiphoton resonance with the ground state and is popdated. Under subsequent action of the same pulse, due to interference in the transition amplitudes of the lambda system, the field cannot ionize the population completely and a fraction of the population will be trapped in a coherent superposition of the quasi degenerate levels. According to this explanation the states with higher angular rnomentum- with more sublevels- would have a higher probability of trapping the population. In general the strength of the trapping will be detennined by the strength of the two photon coupling between the quasi-degenerate levels via the continuum.In 1996, using the very stable laser and by minimizing the masking effects of the focal region expansion with increasing intensity, Talebpour et al observed structures on the curves of singly charged ions of Xe, Kr and Ar. These structures were attributed to electron trapping in the strong laser field.
Dissociation – distinction
A substance may dissociate without necessarily producing ions. As an example, the molecules of table sugar dissociate in water (sugar is dissolved) but exist as intact neutral entities. Another subtle event is the dissociation of sodium chloride (table salt) into sodium and chlorine ions. Although it may seem as a case of ionization, in reality the ions already exist within the crystal lattice. When salt is dissociated, its constituent ions are simply surrounded by water molecules and their effects are visible (e.g. the solution becomes electrolytic). However, no transfer or displacement of electrons occurs. Actually, the chemical synthesis of salt involves ionization. This is a chemical reaction.
Ionization energy (SH)
The ionization energy of an atom or molecule describes the amount of energy required to remove an electron from the atom or molecule in the gaseous state.
X → X+ + e-
The units for ionization energy vary from discipline to discipline. In physics, the ionization energy is typically specified in electron volts (eV) and refers to the energy required to remove a single electron from a single atom or molecule. In chemistry, the ionization energy is typically specified as a molarquantity (molar ionization energy or enthalpy) and is reported in it
.
degree of ionization (H)
The degree of ionization (also known as ionization yield in the literature) refers to the proportion of neutral particles, such as those in agas or aqueous solution, that are ionized into charged particles. A low degree of ionization is sometimes called partially ionized, and a very high degree of ionization as fully ionized.

It is defined as the ratio between the number of ionized molecules and the number of molecules dissolved in water. It can be represented as a decimal number or as a percentage.
Factors affecting ionization(H)
1) atomic radius: Smaller the atomic radius, the higher is the atomic energy. This is because the Coulombic attractive forces on the negatively charged valence electrons by the positively charged nucleus is much higher in the case of smaller atom-the force of attraction decreases with the increase in atomic radius, i.e separation between the electrons and the nucleus in accordance with the inverse square law. 2) Nuclear charge: the higher the positive charge of the nuceus, stronger is it's attraction for the electrons, and hence the harder it is to remove the electrons 3) Orbital penetration: It's easier to remove electrons from p orbitals than from s orbitals, because the s orbitals penetrate towards the nucleus more closely than the p orbitals, thus making the electrons in the s orbital feel greater nuclear attraction. 4) Electron pairing:within a subshell, paired electrons are easier to remove than unpaired ones. This is because repulsion between electrons in the same orbital is higher than repulsion between electrons in different orbitals 5) Shielding or screening effect of the inner orbitals: Due to the electron present in the inner orbitals, the electrons in the outermost orbitals feel lesser attraction for the nucleus than expected. In other words, the inner electron orbitals effectively screen or shield the outermost electrons from the nucleus, due to which the ionization energy decreases.
6) At normal dilution, value of is nearly 1 for strong electrolytes, while it is very less than 1 for weak electrolytes. 7) Higher the dielectric constant of a solvent more is its ionising power. Water is the most powerful ionising solvent as its dielectric constant is highest.
8) Dilution of solution Amount of solvent
9) Degree of ionisation of an electrolyte in solution increases with rise in temperature. 10) Presence of common ion: The degree of ionisation of an electrolyte decreases in the presence of a strong electrolyte having a common ion.

Ionization of Water(H) In the section on amphiprotic species, we saw that water can act as a very weak acidand a very weak base, donating protons to itself to a limited extent:
Applying the equilibrium law to this reaction, we obtain
However, as can be seen in the section on the law of chemical equilibrium, theconcentration of water has a constant value of 55.5 mol dm–3, and so its square can be multiplied by Kc to give a new constant Kw, called the ion- product constant of water:
(1) Measurements of the electrical conductivity of carefully purified water indicate that at 25°C [H3O+] = [OH–] = 1.00 × 10–7 mol dm–3, so that
(Since the equilibrium law is not obeyed exactly, even in dilute solutions, results of most equilibrium calculations are rounded to three significant figures. Hence the value of Kw = 1.00 × 10–14 mol2 dm–6 is sufficiently accurate for all such calculations.) The equilibrium constant Kw applies not only to pure water but to any aqueous solution at 25°C. Thus, for example, if we add 1.00 mol of the strong acid HNO3 to H2O to make a total volume of 1 dm3, essentially all the HNO3 molecules donate their protons to H2O:
and a solution in which [H3O+] = 1.00 mol dm–3 is obtained. Although this solution is very acidic, there are still hydroxide ions present. We can calculate their concentration by rearranging Eq. (1):

The addition of the HNO3 to H2O not only increases the hydronium-ion concentration but also reduces the hydroxide-ion concentration from an initially minute 10–7 mol dm–3to an even more minute 10–14 mol dm–3.
EXAMPLE Calculate the hydronium-ion concentration in a solution of 0.306 M Ba(OH)2.
Solution Since 1 mol Ba(OH)2 produces 2 mol OH– in solution, we have [OH–] = 2 × 0.306 mol dm–3 = 0.612 mol dm–3 Then
Ionization equilibrium in aqueous solutions (H)
We have already noted the importance of reactions in aqueous solutions in the chemical laboratory, in the natural environment, and in the human body. Many reactions in aqueous solutions involve weak acids or bases or slightly solublesubstances, and in such cases one or more equilibria are achieved in solution. Furthermore, the equilibrium state is usually reached almost instantaneously, and so we can use the equilibrium law to calculate the concentrations and amounts of substance of different species in solution. Such information enables us to understand, predict, and control what will happen in solution, and it has numerous practical applications. Equilibrium constants may be used to obtain information about reactions in solution, and in many cases the results of equilibrium calculations will be applied to practical problems. Acid-base reactions in aqueous solutions are intimately related to water’s ability to act as both a weak acid and a weak base, producing H3O+ and OH– by proton transfer. In any aqueous solution at 25°C:

and concentrations of H3O+ and OH– can vary from roughly 100 to 10–14 mol dm–3. This makes it convenient to define pH and pOH as:
Since molecules of a strong acid transfer their protons to water molecules completely, [H3O+] (and hence pH) can be obtained directly from the stoichiometric concentration of the solution. Similarly [OH–] and pOH may be obtained from the stoichiometric concentration of a strong base. In the case of weak acids and weak bases, proton-transfer reactions proceed to only a limited extent and a dynamic equilibrium is set up. In such cases an acid constant Ka or a base constant Kb as well as the stoichiometric concentration of weak acid or base are required to calculate [H3O+], [OH–], pH, or pOH.Ka and Kb for a conjugate acid-base pair are related, and their product is always Kw. Often it is necessary or desirable to restrict the pH of an aqueous solution to a narrow range. This can be accomplished by means of a buffer solution―one which contains a conjugate weak acid-weak base pair. If a small amount of strong base is added to abuffer, the OH– ions are consumed by the conjugate weak acid, so they have little influence on pH. Similarly, a small amount of strong acid can be consumed by the conjugate weak base in a buffer. To a good approximation the [H3O+] in a buffer solution depends only on Ka for the weak acid and the stoichiometric concentrations of the weak acid and weak base. Indicators for acid-base titrations are conjugate acid-base pairs, each member of which is a different color. An indicator changes from the color of the conjugate acid to the color of the conjugate base as pH increases from approximately pKIn – 1 to pKIn + 1. For titrations involving only strong acids and strong bases, several indicators are usually capable of signaling the endpoint because there is a large jump in within ± 0.05 cm3 of the exact stoichiometric volume of titrant. In the case of titrations which involve a weak acid or a weak base, a buffer solution is involved and the jump in pH is smaller. Consequently greater care is required in selection of an appropriate indicator. Common ion effect(H) The common ion effect is responsible for the reduction in solubility of an ionic precipitate when a soluble compound combining one of the ions of the precipitate is added to the solution in equilibrium with the precipitate. It states that if the concentration of any one of the ions is increased, then, according to Le Chatelier's principle, the ions in excess should combine with the oppositely charged ions. Some of the salt will be precipitated until the ionic product is equal to the solubility of the product. In simple words, common ion effect is

defined acommon
Solubility
The solucommon solution o
A prawater froreduce tcarbonateand finelused in th
The saltineffect. Ssolubilityand incre
Sea, bracnormal excess So
Buffering
A bufferacid.[3] Aexample,dissociatdissociatAccordinsuppress dissociatiionization
NC
Tw
Calculati
In order the amoueither in
as the supprion.
y Effects(SH
ubility of a with that sa
ofsodium ch
actical exom chalk or lthe hardness e salt is addly divided prhe manufact
ng out procSoaps are soy of the soapeased ionic s
ckish and obehavior o
odium ions t
g effect (SH
r solution cAddition of th, if bothsodie and ionizes complete
ng to Le Chathe ionizati
ion of the an of an acid
NaCH3CO2(sCH3CO2H (aq
This will decwill be less a
ion of the pH
to calculate unt of the comoles or in
ression of th
H)
sparingly soalt. For instahloride is add
xample ulimestone aqof the wated to preciprecipitate of ture of toothp
ess used inodium saltsp salts. The sstrength.
other waters of soap bthe solubility
)
contains an he conjugateium acetate ze to producely in solutiatelier's prinion of aceticacetic acid wor a base is
s) → Na+(aq)q) H+(aq)
crease the hyacidic than a
H of a Buffer
the pH of thonjugate basmolarities.
he degree of
oluble salt iance, the solded to a susp
used veryquifers is theter. In the w
pitate out spaf calcium carpaste.
n the manuf of fatty acsoaps precip
that containbecause of y of soap sal
acid and ite ion will reand acetic ace acetate ioion. Acetic nciple, the adc acid and swill decreaselimited by th
) + CH3CO2
) + CH3CO2
ydrogen ion solution con
r Solution
he buffer soe combined The Ka of th
f dissociatio
is reduced ilubility of sipension of si
y widelye addition of water treatmaringly solubrbonate that
facture of socids. Additiopitate due to
n appreciabcommon
lts is reduced
ts conjugatesult in a cha
acid are dissoons. Sodium
acid is a wddition of ashift its equie and the phe presence -(aq) -(aq)
concentrationtaining only
olution you nto make the
he acid also n
n of a weak
n a solutionilver chloridilver chlorid
y in arf sodium carbment processble calcium is generated
oaps benefitson of sodiua combinati
ble amount oion effect
d, making th
e base or aange of pH oolved in the
m acetate is weak acid so acetate ions filibrium to tH of the soof its conjug
on and thus y acetic acid
need to knowe solution. Tneeds to be k
k electrolyte
n that contade in water ise in water.[2]
reas drawbonate to thes, highly socarbonate. T
d is a valuab
s from the um chlorideion of comm
of Na+ inter. In the
he soap less e
a base and of the buffere same solut
a strong eleit only ion
from sodiumthe left. Thuolution will gate base or
the commond.
w the amounThese amouknown.
e containing
ains an ion is reduced if ]
wing drinkine raw water toluble sodiumThe very purble by-produ
common ioe reduces thmon ion effe
rfere with thpresence o
effective.
its conjugatr solution. Foion they botectrolyte so nizes slightlym acetate wius the percenincrease. Thacid.
n-ion solutio
nt of acid anunts should b
a
in f a
ng to m re ct
on he ct
he of
te or th it y. ill nt he
on
nd be

Example: A buffer solution was made by dissolving 10.0 grams of sodium acetate in 200.0 mL of 1.00 M acetic acid. Assuming the change in volume when the sodium acetate is not significant, estimate the pH of the acetic acid/sodium acetate buffer solution. The Ka for acetic acid is 1.7 x 10-5.
• First, write the equation for the ionization of acetic acid and the Ka expression. Rearrange the expression to solve for the hydronium ion concentration.
CH3COOH(aq) + H2O(l) --> H3O+(aq) + CH3COO-(aq)
[H3O+] = Ka[CH3COOH]
[CH3COO-]
• Second, determine the number of moles of acid and of the conjugate base.
(1.00 M CH3COOH)(200.0 mL)(1 L/1000 mL) = 0.200 mol CH3COOH
(10.0 g NaCH3COO)(1 mol/82.03 g) = 0.122 mol NaCH3COO
• Substitute these values, along with the Ka value, into the above equation and solve for the hydronium ion concentration. Convert the hydronium ion concentration into pH.
[H3O+] = (1.7 x 10-5)(0.200/0.122) = 2.79 x 10-5 pH = 4.56
Example: Calculate the ratio of ammonium chloride to ammonia that is required to make a buffer solution with a pH of 9.00. The Ka for ammonium ion is 5.6 x 10-10.
• First, write the equation for the ionization of the ammonium ion in water and the corresponding Ka expression. Rearrange the equation to solve for the hydronium ion concentration.
NH4+(aq) + H2O(l) --> H3O+(aq) + NH3(aq)
Ka = [H3O+][NH3]
[NH4+]
[H3O+] = Ka[NH4+]
[NH3]

• Second, convert the pH back into the hydronium ion concentration and then substitute it into the above equation along with the Ka. Solve for the ratio of ammonium ion to ammonia.
[H3O+] = 1 x 10-9 M
1 x 10-9 = 5.6 x 10-10(NH4+/NH3)
(NH4+/NH3) = 1.786/1
A ratio of 1.768 moles of ammonium ion for every 1 mole of ammonia or 1.768 M ammonium ion to 1 M ammonia.
Calculation of the pH of a Buffer Solution after Addition of a Small Amount of Acid
When a strong acid (H3O+) is added to a buffer solution the conjugate base present in the buffer consumes the hydronium ion converting it into water and the weak acid of the conjugate base.
A-(aq) + H3O+(aq) --> H2O(l) + HA(aq)
This results in a decrease in the amount of conjugate base present and an increase in the amount of the weak acid. The pH of the buffer solution decreases by a very small amount because of this ( a lot less than if the buffer system was not present). An "ICE" chart is useful in determining the pH of the system after a strong acid has been added.
Example: 50.0 mL of 0.100 M HCl was added to a buffer consisting of 0.025 moles of sodium acetate and 0.030 moles of acetic acid. What is the pH of the buffer after the addition of the acid? Ka of acetic acid is 1.7 x 10-5.
• First, write the equation for the ionization of acetic acid in water and the related Ka expression rearranged to solve for the hydronium ion concentration.
CH3COOH(aq) + H2O(l) --> H3O+(aq) + CH3COO-(aq)
[H3O+] = Ka[CH3COOH]
[CH3COO-]
• Second, make an "ICE" chart. Let "x" represent the hydronium ion concentration once equilibrium has been re-established. We will assume that all of the added acid is consumed.

CH3COOH(aq) H3O+(aq) CH3COO-(aq)
Initial Amount 0.030 moles (0.0500 L)(0.100 M) = 0.0050 moles 0.025 moles
Change in Amount + 0.005 moles -0.005 moles - 0.005 moles
Equilibrium Amount 0.035 moles x 0.020 moles
• Substitute into the Ka expression and solve for the hydronium ion concentration. Convert the answer into pH.
[H3O+] = (1.7 x 10-5)(0.035/0.020) = 2.975 x 10-5 pH = 4.53
Top
Calculation of the pH of a Buffer Solution after Addition of a Small Amount of Strong Base
When a strong base (OH-) is added to a buffer solution, the hydroxide ions are consumed by the weak acid forming water and the weaker conjugate base of the acid. The amount of the weak acid decreases while the amount of the conjugate base increases. This prevents the pH of the solution from significantly rising, which it would if the buffer system was not present.
OH-(aq) + HA(aq) --> H2O(l) + A-(aq)
The process for finding the pH of the mixture after a strong base has been added is similar to the addition of a strong acid shown in the previous section.
Example: Calculate the pH of a buffer solution that initially consists of 0.0400 moles of ammonia and 0.0250 moles of ammonium ion, after 20.0 mL of 0.75 M NaOH has been added to the buffer. Ka for ammonium ion is 5.6 x 10-10.
• First, write the equation for the ionization of the ammonium ion and the related Ka expression solved for the hydronium ion concentration.
NH4+(aq) + H2O(l) --> H3O+(aq) + NH3(aq)
[H3O+] = Ka[NH4+] [NH3]
• Second, make an "ICE" chart. Let "x" be the concentration of the hydronium ion at equilibrium. The change in the amount of the ammonium ion will be equal to the amount of strong base added (075 M x 0.0200 L = 0.0015 mol).
NH4+(aq) H3O+(aq) NH3(aq)

Initial Amount 0.0250 moles * not needed 0.0400 moles
Change in Amount - 0.0015 moles * not needed + 0.0015 moles
Equilibrium Amont 0.0235 moles X 0.0415 moles
• Third, substitute into the Ka expression and solve for the hydronium ion concentration. Convert the answer into pH.
[H3O+] = (5.6 x 10-10)(0.0235/0.0415) = 3.17 x 10-10 pH = 9.50
Calculation of the Buffer Capacity
The buffer capactity refers to the maximum amount of either strong acid or strong base that can be added before a significant change in the pH will occur. This is simply a matter of stoichiometry. The maximum amount of strong acid that can be added is equal to the amount of conjugate base present in the buffer. The maximum amount of base that can be added is equal to the amount of weak acid present in the buffer.
Example: What is the maximum amount of acid that can be added to a buffer made by the mixing of 0.35 moles of sodium hydrogen carbonate with 0.50 moles of sodium carbonate? How much base can be added before the pH will begin to show a significant change?
• First, write the equation for the ionization of the weak acid, in this case of hydrogen carbonate. Although this step is not truly necessary to solve the problem, it is helpful in identifying the weak acid and its conjugate base.
HCO3-(aq) + H2O(l) --> H3O+(aq) + CO3
2-(aq)
• Second, added strong acid will react with the conjugate base, CO32-. Therefore, the
maximum amount of acid that can be added will be equal to the amount of CO32-, 0.50
moles. • Third, added strong base will react with the weak acid, HCO3
-. Therefore, the maximum amount of base that can be added will be equal to the amount of HCO3
-, 0.35 moles.


Review questios -:
1. What is ionization? Explain with example.
2. What do you understand by degree of ionization?
3. Explain ionization equilibrium in aqueous solution.
4. What are the factors which affecting the ionization?
5. Explain- common ion effect, solubility effect, buffering effect.
6. Define Acid, base and their strength.
7. What is PH value? What is the PH value for acid and base.
8. Define the method of acid base titration.
9. What are the parameters for choice of indicator.
10. What do you mean by ionization constant.
11. Define hydrolysis.
12. Define Buffer solution.
13. Explain concept of electrolysis.
14. What is the Faraday’s law of electrolysis?
15. What are the industrial applications of electrolysis? Explain electroplating.
16. Write short notes on-:
• electrometallurgy, • electroplating & electro refining)

Chapter-4 INTRODUCTION
A water molecule contains one oxygen and two hydrogen atoms connected by covalent bonds. Water is a liquid at standard ambient temperature and pressure,but it often co-exists on Earth with its solid state, ice, and gaseous state , Water also exists in a liquid crystal state near hydrophili surfaces.
Water covers 71% of the Earth's surface, and is vital for all known forms of life. On Earth, 96.5% of the planet's water is found in seas and oceans, 1.7% in groundwater, 1.7% in glaciers and the ice caps of Antarctica and Greenland, a small fraction in other large water bodies, and 0.001% in the air as vapor, clouds (formed of solid and liquid water particles suspended in air), and precipitation. Only 2.5% of the Earth's water is freshwater, and 98.8% of that water is in ice and groundwater. Less than 0.3% of all freshwater is in rivers, lakes, and the atmosphere, and an even smaller amount of the Earth's freshwater (0.003%) is contained within biological bodies and manufactured products.
Water on Earth moves continually through water.the watercycle of evaporation and transpiration (evapotranspiration), condensation,precipitation, and runoff, usually reaching the sea. Evaporation and transpiration contribute to the precipitation over land.
Safe drinking water is essential to humans and other life forms even though it provides no calories or organic nutrients. Access to safe drinking water has improved over the last decades in almost every part of the world, but approximately one billion people still lack access to safe water and over 2.5 billion lack access to adequate sanitation.There is a clear correlation between access to safe water and GDP per capita. However, some observers have estimated that by 2025 more than half of the world population will be facing water-based vulnerability. A recent report (November 2009) suggests that by 2030, in some developing regions of the world, water demand will exceed supply by 50%. Water plays an important role in the world economy, as it functions as a solvent for a wide variety of chemical substances and facilitates industrial cooling and transportation. Approximately 70% of the freshwater used by humans goes to agriculture.
Chemical and physical properties (H)
Water is the chemical substance with chemical formula H 2O: one molecue of water has two hydrogen atomscovalently bonded to a single oxygen atom.
Water appears in nature in all three common states of matter (solid, liquid, and gas) and may take many different forms on Earth: water vapor and clouds in the sky, seawater in the oceans, icebergs in the polar oceans, glaciers and rivers in the mountains, and the liquid in aquifers in the ground.
The major chemical and physical properties of water are:

• Water is a liquid at standard temperature and pressure. It is tasteless and odorless. The intrinsic colour of water and ice is a very slight blue hue, although both appear colorless in small quantities. Water vapour is essentially invisible as a gas.[12]
• Water is transparent in the visible electromagnetic spectrum. Thus aquatic plants can live in water becausesunlight can reach them. Infrared light is strongly absorbed by the hydrogen-oxygen or OH bonds.
• Since the water molecule is not linear and the oxygen atom has a higher electronegativity than hydrogen atoms, it carries a slight negative charge, whereas the hydrogen atoms are slightly positive. As a result, water is a polar molecule with an electrical dipole moment. Water also can form an unusually large number of intermolecular hydrogen bonds (four) for a molecule of its size. These factors lead to strong attractive forces between molecules of water, giving rise to water's high surface tension[13] and capillary forces. The capillary action refers to the tendency of water to move up a narrow tube against the force of gravity. This property is relied upon by all vascular plants, such as trees.[14]
• Water is a good polar solvent and is often referred to as the universal solvent. Substances that dissolve in water, e.g., salts, sugars, acids, alkalis, and some gases – especially oxygen, carbon dioxide (carbonation) are known as hydrophilic (water-loving) substances, while those that areimmiscible with water (e.g., fats and oils), are known as hydrophobic (water-fearing) substances.
• Most of the major components in cells (proteins, DNA and polysaccharides) are also dissolved in water.
• Pure water has a low electrical conductivity, but this increases with the dissolution of a small amount of ionic material such as sodium chloride.
• The boiling point of water (and all other liquids) is dependent on the barometric pressure. For example, on the top of Mt. Everest water boils at 68 °C(154 °F), compared to 100 °C (212 °F) at sea level. Conversely, water deep in the ocean near geothermal vents can reach temperatures of hundreds of degrees and remain liquid.
• At 4181.3 J/(kg·K), water has a high specific heat capacity, as well as a high heat of vaporization (40.65 kJ·mol−1), both of which are a result of the extensive hydrogen bonding between its molecules. These two unusual properties allow water to moderate Earth's climate by buffering large fluctuations in temperature.
• The maximum density of water occurs at 3.98 °C (39.16 °F).[15] It has the anomalous property of becoming less dense, not more, when it is cooled to its solid form, ice. During freezing, the 'open structure' of ice is gradually broken and molecules enter cavities in ice-like structure of low temperature water. There are two competing effects: 1) Increasing volume of normal liquid and 2) Decrease overall volume of the liquid. Between 0 and 3.98 °C, the second effect will cancel off the first effect so the net effect is

shrinvolumin ice
• The d917 k
• Watesinglusualvapo
• Wate
• Wate
• As acompfuel, waterenerg
On Earth
A graphi
nkage of volme in this soebergs.
density of likg/m3 .
er is miscible homogenelly forming r is complet
er forms an a
er can be spl
an oxide ofpounds burnit is an end
r into hydrogy that can bh
cal distribut
lume with iolid state, w
quid water
le with maneous liquid.
layers accoely miscible
azeotrope wi
it by electro
f hydrogen, or react witd-product ofogen and oxbe collected w
ion of the lo
increasing tewhich accoun
is 1,000 kg/
ny liquids, On the oth
ording to ine with air.
ith many oth
lysis into hy
water is foth oxygen orf the combuxygen byelecwhen the hy
ocations of w
emperature.[
nts for the fa
/m3 (62.43 lb
such as ethaher hand, wcreasing den
her solvents.
ydrogen and
formed whenr oxygen-co
ustion of hyctrolysis or
ydrogen and
water on Eart
[16] It expandact of ice flo
b/cu ft) at 4
anol, in allwater and nsity from t
oxygen.
n hydrogen ontaining comdrogen. Theany other moxygen reco
th.
ds to occupoating on liq
4 °C. Ice has
l proportionmostoils arethe top. As
or hydrogmpounds. We energy reqmeans is greombine.
y 9% greatequid water, a
s a density o
ns, forming e immiscibl
a gas, wate
en-containinWater is not quired to spleater than th
er as
of
a le, er
ng a
lit he

Water coThe Anta
HydrologEarth. Thmovemenis limnolhydrolog
The collethe hydro1,338,00
Liquid ocean, sewater. Was ground
Water is the presresponsibEarth, walesser butransportmany typ
Water cy
overs 71% oarctic ice she
gy is the stuhe study of tnt of grounogy and di
gy are in focu
ective massosphere. Ear0,000 km3 (3
water ea, lake, rive
Water is also dwater in aq
important inssure of thble for the mater is imporut still signit that occurspes of sedim
ycle(SH)
of the Eartheet, which co
udy of the mhe distributindwater is histribution us of ecohyd
of water forth's approxi321,000,000
is fouer, stream, capresent in th
quifers.
n many geolis groundwmelt that prortant in both ficant exten on the surf
mentary rocks
h's surface; ontains 61%
movement, dion of water hydrogeologof oceans
drology.
found on, unimate water 0 mi3).[5]
und in banal, pond, ohe atmosphe
logical procewater affectsoduces volcachemical an
nt, ice, are aface of the s, which mak
the oceans of all fresh
distribution, is hydrograp
gy, of glaciis oceanog
nder, and ovvolume (th
bodies oor puddle. Tere in solid,
esses. Grouns patterns anoes atsubdnd physical walso responsearth. Depo
ke up the geo
contain 96.5water on
and qualityphy. The stuiers is glacioraphy. Eco
ver the surfe total wate
of waterhe majority liquid, and v
ndwater is pof faulting.
duction zoneweathering psible for a lsition of traologic record
5% of the E
y of water thudy of the disology, of i
ological pro
face of a plaer supply of
, such of water on
vapor states.
present in moWater in
es. On the sprocesses. Warge amoun
ansported sedd of Earth hi
Earth's wate
hroughout thstribution aninland wateocesses wit
anet is callethe world)
as an Earth is se. It also exis
ost rocks, anthe mantle
surface of thWater and, tont of sedimendiment formistory.
er.
he nd rs th
ed is
an ea sts
nd is
he a nt
ms

Water cy
The wateexchangewater, gr
Water mfollowing
• evapoplant
• preci• runof
Most waland at thtranspiratland, hafrom fog water vapthe loweincrease.
Water ruto simultransportopportuncreating establishmis covere
ycle
er cycle (kne of waterroundwater,
moves perpetg transfer pr
oration fromts and animaipitation, froff from the l
ater vapor ovhe same ratetion contribu
as several fand dew.[33
por meets a est, just befo[34] Condens
unoff often cate river or
t model. Somnity for trav
river valleyment of poped with wa
nown scientir within thand plants.
tually throuocesses:
m oceans andals into air. m water vapand usually
ver the oceae as runoff inute another 7forms: most] Dew is smcool surface
fore sunrise sed water in
collects overr stream flome of water vel andcommys and deltaspulation centater. It is w
ifically as the hydrosphe
ugh each of
d other wate
por condensireaching the
ans returns tnto the sea, 72 Tt per yet commonlyall drops of e. Dew usuaand when the air may
r watershedsow and calcis diverted t
merce. Thros which proters. A flood
when a rive
the hydrologere, betwee
f these regio
er bodies int
ing from the e sea.
to the oceanabout 47 Ttar. Precipitay rain, snowwater that a
ally form in the temperaalso refract
flowing intoculate waterto irrigationough erosionovide rich d occurs wheer overflows
gic cycle) ren theatmos
ons in the w
to the air an
air and falli
ns, but windper year. Ov
ation, at a rat, and hail,
are condensethe morning
ature of the sunlight to p
o rivers. A mr quality pafor agricultu
n, runoff ssoil and
en an area os its banks
refers to thsphere, soil w
water cycle
nd transpirati
ing to earth o
ds carry watever land, evte of 119 Tt with some
ed when a hig when the t earth's sur
produce rainb
mathematicaarameters isure. Rivers ashapes the
level grouf land, usuaor flood f
he continuouwater, surfac
consisting o
ion from lan
or ocean.
er vapor oveaporation anper year ovecontributio
igh density otemperature face starts tbows.
al model uses hydrologicand seas offe
environmenund for thlly low-lyin
from the se
us ce
of
nd
er nd er on of is to
ed al
fer nt he g, a.

A drought is an extended period of months or years when a region notes a deficiency in its water supply. This occurs when a region receives consistently below average precipitation.
Fresh water storage(SH)
The Bay of Fundy at high tide (left) and low tide (right)
Some runoff water is trapped for periods of time, for example in lakes. At high altitude, during winter, and in the far north and south, snow collects in ice caps, snow pack and glaciers. Water also infiltrates the ground and goes into aquifers. This groundwater later flows back to the surface in springs, or more spectacularly in hot springs and geysers. Groundwater is also extracted artificially in wells. This water storage is important, since clean, fresh water is essential to human and other land-based life. In many parts of the world, it is in short supply.
Sea water(sH)
Sea water contains about 3.5% salt on average, plus smaller amounts of other substances. The physical properties of sea water differ from fresh water in some important respects. It freezes at a lower temperature (about −1.9 °C) and its density increases with decreasing temperature to the freezing point, instead of reaching maximum density at a temperature above freezing. The salinity of water in major seas varies from about 0.7% in the Baltic Sea to 4.0% in theRed Sea.
Tide(SH)
Tides are the cyclic rising and falling of local sea levels caused by the tidal forces of the Moon and the Sun acting on the oceans. Tides cause changes in the depth of the marine

and estuachangingand Sunlocal baththe intert
Effects o
An oasis
Overview(CO2
From a bproliferatallowing forms of solutes dMetabolifrom molarger m
arine water bg tide producn relative hymetry. Thtidal zone, is
on life(SH)
is an isolate
w of photosy
biological sttion of life torganic com
f life dependdissolve andism is the suolecules (thromolecules (e
bodies and ced at a giveto the Ear
he strip of seas an importan
ed water sour
ynthesis and
andpoint, wthat set it mpounds to d on water. Wd as an essum total of ough energye.g. starche
produce osen location isrth coupledashore that int ecologica
rcewith vege
drespiration.
water has mapart from react in wa
Water is vitasential part anabolism a
y requiring es, triglycer
scillating cus the result od with theis submergedl product of
etation in a d
Water (at r
many distinctother subs
ays that ultial both as aof many m
and catabolienzymatic crides and p
urrents knowof the changi
effects of d at high tideocean tides.
desert
right), toget
t properties stances. It cmately allowsolvent in w
metabolic proism. In anabchemical reaproteins for
wn as tidal ing positions
Earth rotae and expose.
ther with ca
that are crcarries out w replication
which many ocesses withbolism, wateactions) in or storage o
streams. Ths of the Mooation and thed at low tid
arbon dioxid
ritical for ththis role b
n. All knowof the body
hin the bodyer is removeorder to growof fuels an
he on he
de,
de
he by wn y's y. ed w
nd

informatimoleculeother pur
Water is energy tofrom air oxidize thprocess (
Water is (H+, thathydroxidlog of while bas
Aquatic l
Some of
Some ma
ion). In cates (e.g. glucrposes). With
fundamentao split off waor water) to
he hydrogen(cellular resp
also centralt is, a proto
de ion (OH−)the hydrog
ses have valu
life forms(SH
the biodiver
arine diatom
tabolism, waose, fatty achout water, t
al to photosater's hydrogo form glucn and carbonpiration).
l to acid-bason) donor, to form watgen ion coues greater t
H)
rsity of acora
ms – a keyphy
ater is usedcids and amthese particu
synthesis angen from oxycose and relen to capture t
se neutralitycan be neuter. Water isoncentrationthan 7.
al reef
ytoplankton
d to break mino acids toular metaboli
nd respirationygen. Hydroease oxygenthe sun's ene
y and enzymutralized by s considered n) of 7. Ac
group
bonds in oro be used foic processes
n. Photosynogen is combn. All living ergy and refo
me function. a base, a
to be neutracids have p
rder to genor fuels for e
could not ex
nthetic cells bined with C
cells use suform water an
An acid, a proton acce
al, with a pHpH values
nerate smalleenergy use oxist.
use the sunCO2 (absorbeuch fuels annd CO2 in th
hydrogen ioeptor such a
H (the negativless than
er or
n's ed nd he
on as ve 7

Earth surall fish lias dolphilives in wthe basisocean foo
Aquatic have gillsboth. MaperiodicaInvertebrincludinginvertebrrespiratio
Effects o
Water fou
Civilizatiso-calledthe anLarge meAires, Shaccessibilike Singthe Middmajor fac
Health an
rface watersve exclusivins and whalwater and pos for some od chain.
vertebrates s instead of
arine mammally to breathrates exhibitg breathing rate life evoon in water.
on human ci
untain
ion has histod cradle of cncient soetropolises lihanghai, Tokility via wat
gapore, havedle East, whctor in huma
nd pollution(
s are filled wvely in wateles. Some kiortions on la
underwater
must obtainf lungs, althmals, such he air. Somt a wide rantubes (see in
olved in an
vilization(S
orically flourcivilization, ociety of ike Rotterdakyo, Chicagoter and the e flourished here water isan developm
(SH)
with life. Ter, and therinds of animand. Plants sr ecosystem
n oxygen tohough some
as dolphime amphibiannge of modifnsectand mo
n aquatic ha
SH)
rished arounwas situated
the Egypam, London,o, and Hongresultant exfor the sam
s more scarcment.
he earliest lre are man
mals, such assuch as kelp
ms. Plankton
survive, ane species oins, whalesns are able tfications to ollusc siphonabitat most
nd rivers andd between tptians depenMontreal, P
g Kong owexpansion of me reason. ce, access to
life forms any types of s amphibian
p and algae gis generally
nd they do sof fish, sucs, otters, anto absorb oxsurvive in pns) and gillshave little
d major watethe major riv
nded entirParis, New their succetrade. IslandIn places s
o clean drin
appeared in marine ma
s, spend porgrow in the wy the found
so in variouch as thelu
nd seals needxygen throupoorly oxyges (Carcinus).or no spec
erways; MesversTigris anrely upon
York ess in part ds with safesuch as Nortnking water
water; nearlammals, sucrtions of thewater and ardation of th
us ways. Fisungfish, havd to surfacugh their skinenated water. However acialisation fo
opotamia, thnd Euphraten the NilCity, Buenoto their eas
e water portth Africa anwas and is
ly ch eir re he
sh ve ce n. rs as or
he es; le. os sy ts, nd
a

An envir
Water fitpotable m
Water thbathing icalled safis used tomonitoreand 1–2 pmay be such as c
In the Uas greywand blackrequire furesidentia(sinks, shmay have
This natusocial andrink unhpeople wEvian suhalf a biladequatedeaths Organizafrom diar
Water, hprecipitatTherefore
onmental sc
t for human may be made
at is not fit is called by fe water, or o make wate
ed by governppm of chlomaintained
chlorine or oz
USA, non-powater, whichkwater, whurther treatmal wastewhowers and ke different m
ural resourcnd economichealthy wate
worldwide wummit.[35] Evllion people sanitation. a year a
ation estimatrrhea each y
however, is tion in quae, it is the re
ience progra
consumptioe potable by
for drinkingvarious nam"safe for baer safe for bnment regulaorine not yet
in satisfactozone or by th
otable formh is treatabhich generament in ordewater genkitchen runo
meanings in o
e is becomic concern. Cer. Most cou
who do not hven if this diwithout accePoor water
are causedtes that safear.[36]
not a finiantities manyelatively sm
am - a studen
n is called dfiltration or
g but is not hmes other thathing". Chlobathing or drations (typicreacted withory microbihe use of ult
ms of wastewble and th
ally container to be ma
nerated byoff, but nottoother countri
ing scarcer Currently, abuntries accephave access ifficult goal ess to safe dr
quality andd by polfe water c
ite resourcey degrees oall quantity
nt from Iowa
drinking watdistillation,
harmful for han potable orine is a skirinking. Its ucally 1 part ph impurities iological contraviolet ligh
water generathus easily ns sewage anade reusabley a hooilets, whichies and cultu
in certain pbout a billiopted the goato safe watis met, it wrinking wate
d bad sanitalluted drincould prev
, but ratherof magnitudof water in
a State Univ
ter or potableor by a rang
humans whor drinking in and mucouse is highlyper million (for bathing ndition usin
ht.
ted by humable to be
nd other fe. Greywate
ousehold's h generate bures.
laces, and in people aroal of halvinger and sanita
will still leaveer and over aation are denking watevent 1.4
r re-circulatde higher threserve in th
versity sampl
e water. Wage of other m
en used for water, and
ous membrany technical a(ppm) for drwater). Wat
ng chemical
mans may be made poforms of wer composes
sanitation blackwater.)
its availabiliound the wog by 2015 thation duringe more thana billion witheadly; someer. The Wmillion c
ted as potahan human he earth (abo
ling water
ater that is nomethods.
swimming ois sometime
ne irritant thand is usuallrinking wateter for bathin
disinfectan
be referred totable againwaste whics 50–80% o
equipmen) These term
ity is a majoorld routinelhe number o
g the 2003 Gn an estimatehout access t
e five millioWorld Healt
child death
able water iconsumptionout 1% of ou
ot
or es at ly er, ng nts
to n, ch of nt
ms
or ly of
G8 ed to on th hs
in n. ur

drinking water supply, which is replenished in aquifers around every 1 to 10 years), that is a non-renewable resource, and it is, rather, the distribution of potable and irrigation water which is scarce, rather than the actual amount of it that exists on the earth. Water-poor countries use importation of goods as the primary method of importing water (to leave enough for local human consumption), since the manufacturing process uses around 10 to 100 times products' masses in water.
HARD AND SOFT WATER (H) • Water that does not form an immediate lather with soap is called hard water. Hardness
of water is due to the presence of soluble calcium, magnesium or iron compounds. The most common compounds are calcium bicarbonate Ca(HCO3)2, magnesium bicarbonate Mg(HCO3)2, calcium sulphate CaSO4 and magnesium sulphate MgSO4. The addition of soap forms an insoluble scum. The scum consists of insoluble salts of these metals. Removal of these salts from the solution makes the water soft. Water that forms an immediate lather with soap is called soft water. Such water does not have dissolved salts of calcium, magnesium and iron.
Types of Hardness: (MH)
Depending upon the behaviour of water towards soap, hardness is divided into two types.
1. Temporary hardness: (H)
Hardness of water due to the presence of soluble bicarbonates of calcium and Magnesium is called temporary hardness. When water containing dissolved carbon dioxide passes over solid carbonates (chalk or limestone deposits etc.), these compounds get dissolved in water. Rainwater and distilled water are always soft because they do not have dissolved (soluble) salts.
Temporary hardness is removed in the following ways:
1. carbonate. 2. Chemical methods.
By adding slaked lime [Ca(OH)2] to hard water, insoluble carbonates are formed. The insoluble calcium carbonate is the ‘fur’ (or scale) formed in kettles, boilers, pipes, etc.
2. Permanent hardness: (H)
This is due to the presence of chlorides and sulphates of calcium and magnesium. Such a hardness can be removed by the addition of washing soda. This removes both the temporary and the permanent hardness of can be removed by following processes.
Removal of hardness of water by soda lime process (H)

Basic principle:
• Lime [Ca(OH)2] and soda [Na2CO3] are the reagents used to precipitate the dissolved salts of
Ca+2 and Mg+2 as CaCO3 and Mg(OH)2.
• The precipitated CaCO3 and Mg(OH)2 are filtered off.
• Lime reacts with temporary hardness, CO2, acids, bicarbonates and alums.
• Lime cannot remove the calcium permanent hardness which should be removed by soda.
• The precipitation reactions with lime and soda are very slow.
• Only calculated amounts of lime and soda are to be added. Excess amount of lime & soda
causes boiler troubles like caustic embrittlement.
• Calculation of lime & soda required for the process:
♣ Amount of lime required for softening = 74/100 [temp. hardness of Ca+2 + 2 x temp.
hardness of Mg+2 + permanent hardness of Mg+2 + CO2 + ½ HCl + H2SO4 + ½ NaHCO3
+ ½ KHCO3 + FeSO4 + {3 x Al2(SO4)3} – ½ NaAlO2]
♣ Amount of soda required for softening = 106/100 [permanent hardness of Ca+2 +
permanent hardness of Mg+2 + ½ HCl + H2SO4 + FeSO4 + {3 x Al2(SO4)3}- ½ NaHCO3 –
½ KHCO3]
* All in terms of chemical equivalents of CaCO3.
• The above mentioned formulae are based on the chemical reactions involved in the
process as mentioned below.
Treatment with lime:
1. Removal of temporary hardness of Ca+2
Ca(HCO3)2 + Ca(OH)2 2CaCO3 + 2H2O
2. Removal of Temporary hardness of Mg+2
Mg(HCO3)2 + 2Ca(OH)2 Mg(OH)2 + 2CaCO3 + 2H2O
3. Removal of Permanent hardness of Mg+2
MgCl2 + Ca(OH)2 Mg(OH)2 + CaCl2

MgSO4 + Ca(OH)2 Mg(OH)2 + CaSO4
Mg(NO3)2 + Ca(OH)2 Mg(OH)2 + Ca(NO3)2
4. Removal of CO2
CO2 + Ca(OH)2 CaCO3 + H2O 5. Removal of acids
2HCl + Ca(OH)2 CaCl2 + 2H2O
H2SO4 + Ca(OH)2 CaSO4 + 2H2O
6. Removal of bicarbonates of Na+
and K+
2NaHCO3 + Ca(OH)2 CaCO3 + Na2CO3 + 2H2O
2KHCO3 + Ca(OH)2 2CaCO3 + K2CO3 + 2H2O
7. Removal of alums
FeSO4 + Ca(OH)2 CaSO4 + Fe(OH)2
Al2(SO4)3 + 3Ca(OH)2 3CaSO4 + 2Al(OH)3
NaAlO2 + 2H2O Al(OH)3 + NaOH
2NaOH + CaCl2 Ca(OH)2 + 2NaCl
Treatment with soda:
Removal of Permanent hardness of Ca
+2
CaCl2 + Na2CO3 CaCO3 + 2NaCl
CaSO4 + Na2CO3 CaCO3 + Na2SO4
Ca(NO3)2 + Na2CO3 CaCO3 + 2NaNO3
Precipitation softening processes are used to reduce raw water hardness, alkalinity, silica, and other constituents. This helps prepare water for direct use as cooling tower makeup or as a first-stage treatment followed by ion exchange for boiler makeup or process use. The water is treated with lime or a combination of lime and soda ash (carbonate ion). These chemicals react with the hardness and natural alkalinity in the water to form insoluble compounds. The compounds precipitate and are removed from the water by sedimentation and, usually, filtration. Waters with moderate to high hardness and alkalinity concentrations (150-500 ppm as CaCO3) are often treated in this fashion.

Chemistry of Precipitation Softening (SH)
In almost every raw water supply, hardness is present as calcium and magnesium bicarbonate, often referred to as carbonate hardness or temporary hardness. These compounds result from the action of acidic, carbon dioxide laden rain water on naturally occurring minerals in the earth, such as limestone. For example:
Precipitation softening processes are used to reduce raw water hardness, alkalinity, silica, and other constituents. This helps prepare water for direct use as cooling tower makeup or as a first-stage treatment followed by ion exchange for boiler makeup or process use. The water is treated with lime or a combination of lime and soda ash (carbonate ion). These chemicals react with the hardness and natural alkalinity in the water to form insoluble compounds. The compounds precipitate and are removed from the water by sedimentation and, usually, filtration. Waters with moderate to high hardness and alkalinity concentrations (150-500 ppm as CaCO3) are often treated in this fashion.
Chemistry of Precipitation Softening (SH)
In almost every raw water supply, hardness is present as calcium and magnesium bicarbonate, often referred to as carbonate hardness or temporary hardness. These compounds result from the action of acidic, carbon dioxide laden rain water on naturally occurring minerals in the earth, such as limestone. For example:
CO2 + H2O = H2CO3
carbon dioxide
Water
carbonic acid
H2CO3 + CaCO3 ¯ = Ca(HCO3)2
carbonic acid
calcium carbonate
calcium bicarbonate
Hardness may also be present as a sulfate or chloride salt, referred to as noncarbonate or permanent hardness. These salts are caused by mineral acids present in rain water or the solution of naturally occurring acidic minerals.

The significance of "carbonate" or "temporary" hardness as contrasted to "noncarbonate" or "permanent" hardness is that the former may be reduced in concentration simply by heating. In effect, heating reverses the solution reaction:
Ca(HCO3)2 + Heat = CaCO3 ¯ + H2O + CO2
calcium bicarbonate
calcium carbonate
water
carbon dioxide
Reduction of noncarbonate hardness, by contrast, requires chemical addition. A combination of lime and soda ash, along with coagulant and flocculant chemicals, is added to raw water to promote a precipitation reaction. This allows softening to take place.
Soda lime process (H)-: There are two types of soda lime process.
(1) Cold lime process (2) Hot lime process
Cold Lime Softening (SH)

Precipitation softening accomplished at ambient temperatures is referred to as cold lime softening. When hydrated lime, Ca(OH)2, is added to the water being treated, the following reactions occur:
CO2 + Ca(OH)2 = CaCO3 ¯ + H2O
carbon dioxide calcium hydroxide calcium carbonate waterIf the proper chemical control is maintained on lime feed, the calcium hardness may be reduced to 35-50 ppm. Magnesium reduction is a function of the amount of hydroxyl (OH-) alkalinity excess maintained. Figures 7-1 and 7-2 show these relationships.
Noncarbonate or permanent calcium hardness, if present, is not affected by treatment with lime alone. If noncarbonate magnesium hardness is present in an amount greater than 70 ppm and an excess hydroxyl alkalinity of about 5 ppm is maintained, the magnesium will be reduced to about 70 ppm, but the calcium will increase in proportion to the magnesium reduction.
For example, in cold lime treatment of a water containing 110 ppm of calcium, 95 ppm of magnesium, and at least 110 ppm of alkalinity (all expressed as calcium carbonate), calcium could theoretically be reduced to 35 ppm and the magnesium to about 70 ppm. However, an additional 25 ppm of calcium would be expected in the treated water due to the following reactions:
MgSO4 + Ca(OH)2 = Mg(OH)2 ¯ + CaSO4
magnesium sulfate
calcium hydroxide
magnesium hydroxide
calcium sulfate
MgCl2 + Ca(OH)2 = Mg(OH)2 ¯ + CaCl2
magnesium chloride
Calcium
magnesium hydroxide
calcium chloride
To improve magnesium reduction, which also improves silica reduction in cold process softening, sodium aluminate may be used. The sodium aluminate provides hydroxyl ion (OH-) needed for improved magnesium reduction, without increasing calcium hardness in the treated water. In addition, the hydrolysis of sodium aluminate results in the formation of aluminum hydroxide, which aids in floc formation, sludge blanket conditioning, and silica reduction. The reactions are as follows:
Na2Al2O4 + 4H2O = 2Al(OH)3 ¯ + 2NaOH

sodium aluminate
water
aluminum hydroxide
sodium hydroxide
Mg [ SO4 ] + 2NaOH = Mg(OH)2¯ + [
Na2SO4 ]
Cl2 2NaCl
magnesium
sulfate/ chloride
sodium hydroxide
magnesium hydroxide
sodium sulfate/ chloride
Soda ash (Na2CO3) may be used to improve hardness reduction. It reacts with noncarbonate calcium hardness according to the following:
CaSO4 + Na2CO3 = CaCO3 ¯ + Na2SO4
calcium sulfate
sodium carbonate
calcium carbonate
sodium sulfate
CaCl2 + Na2CO3 = CaCO3 ¯ + 2NaCl
calcium chloride
sodium carbonate
calcium carbonate
sodium chloride
However, noncarbonate magnesium hardness reduction in cold process softening requires added lime. The reactions are as follows:
MgSO4 + Ca(OH)2 + Na2CO3 = Mg(OH)2 ¯ + CaCO3 ¯ + Na2SO4
magnesium sulfate
calcium hydroxide
sodium carbonate
magnesium hydroxide
calcium carbonate
sodium sulfate

MgCl2 + Ca(OH)2 + Na2CO3 = Mg(OH)2¯ + CaCO3 ¯ + 2NaCl
magnesium chloride
calcium hydroxide
sodium carbonate
magnesium hydroxide
calcium carbonate
sodium chloride
In these reactions, dissolved solids are not reduced because a solution reaction product (sodium sulfate or sodium chloride) is formed.
Warm Lime Softening (SH)
The warm lime softening process operates in the temperature range of 120-140°F (49-60°C). The solubilities of calcium, magnesium, and silica are reduced by increased temperature. Therefore, they are more effectively removed by warm lime softening than by cold lime softening. This process is used for the following purposes:
• To recover waste heat as an energy conservation measure. The water to be treated is heated by a waste stream, such as boiler blowdown or low-pressure exhaust steam, to recover the heat content.
• To prepare feed to a demineralization system. The lower levels of calcium, magnesium, and especially silica reduce the ionic loading on the demineralizer when warm lime-softened water is used rather than cold lime-softened water. This may reduce both the capital and operating costs of the demineralizer. However, most strong base anion resins have a temperature limitation of 140°F (60°C); therefore, additional increases in temperature are not acceptable for increasing the effectiveness of contaminant reduction.
• To lower the blowdown discharge from cooling systems. Cooling tower blowdown may be treated with lime and soda ash or caustic to reduce calcium and magnesium levels so that much of the blowdown may be returned to the cooling system. Silica levels in the recirculating cooling water are also controlled in this manner.
In any warm lime or warm lime-soda ash process, temperature control is critical because temperature variations of as little as 4°F/hr (2°C/hr) can cause gross carryover of the softener pricipitates.
Hot Process Softening (SH)
Hot process softening is usually carried out under pressure at temperatures of 227-240°F (108-116°C). At the operating temperature, hot process softening reactions go essentially to completion. This treatment method involves the same reactions described above, except that raw water CO2 is vented and does not participate in the lime reaction. The use of lime and

soda ash permits hardness reduction down to 0.5 gr/gal, or about 8 ppm, as calcium carbonate.
Magnesium is reduced to 2-5 ppm because of the lower solubility of magnesium hydroxide at the elevated temperatures.
Silica Reduction (SH)
Hot process softening can also provide very good silica reduction. The silica reduction is accomplished through adsorption of the silica on the magnesium hydroxide precipitate. If there is insufficient magnesium present in the raw water to reduce silica to the desired level, magnesium compounds (such as magnesium oxide, magnesium sulfate, magnesium carbonate, or dolomitic lime) may be used. . Magnesium oxide is the preferred chemical because it does not increase the dissolved solids concentration of the water.
CONVENTIONAL LIME-SODA ASH TREATMENT
When water has minimal magnesium hardness, only calcium needs to be removed. Only enough
lime and soda ash are added to water to raise pH to between 10.3 and 10.6, and calcium hardness
will be removed from the water (but minimal magnesium hardness will be removed).
EXCESS LIME TREATMENT
When magnesium hardness is more than about 40 mg/l as CaCO3, magnesium hydroxide scale
deposits in household hot-water heaters operated at normal temperatures of 140 to 150° F. To
reduce magnesium hardness, more lime must be added to the water. Extra lime will raise pH
above 10.6 to help magnesium hydroxide precipitate out of the water.
SPLIT TREATMENT
When water contains high amounts of magnesium hardness, split treatment may be used.
Approximately 80 percent of the water is treated with excess lime to remove magnesium at a pH
above 11, after which it is blended with 20 percent of the source water. Split treatment can
reduce the amount of carbon dioxide required to re-carbonate the water as well as offer a savings
in lime feed.

Since the fraction of the water that is treated contains an excess lime dose, magnesium is almost
completely removed from this portion. When this water is mixed with the water that does not
undergo softening, the carbon dioxide and bicarbonate in that water re-carbonates the final blend.
Split treatment reduces the amount of chemical needed to remove hardness from water by 20 to
25 percent (a significant savings).
DESIGN CONSIDERATIONS
In lime soda-ash softening plants, the softening process may be carried out by a sequence of
rapid mix, flocculation, and sedimentation or in a solids contactor. In the solids contactor the
rapid mix, flocculation, and sedimentation occur in a single unit. The process begins with the
mixing of the chemicals into the water, followed by violent agitation, termed rapid mixing. This
allows chemicals to react with, and precipitate calcium or magnesium hardness in the water. Lime Softening 3
Flocculation allows flocs to contact other flocs and grow large enough to settle in the
sedimentation stage. Water is mixed gently with a small amount of energy. Most flocculators are
compartmentalized, allowing for a tapered mix, so less energy must applied as the flocs grow in
size.
Detention time in the flocculator is important to allow particles to come in contact with each
other. The minimum time recommended is 30 minutes for conventional water softening.
Sludge returned to the head of the flocculator reduces the amount of chemical needed and
provides seed flocs for the precipitation. The estimated return sludge is 10 to 25 percent of the
source water.
Sedimentation follows flocculation. Settling rates for these tanks are a function of particle size

and density. Detention times in the settling basins range from 1.5 hours to 3.0 hours, and they
can be rectangular, square, or circular (some designs incorporate inclined tube settlers). Lime Softening 4
Sedimentation can also occur in the solids-contact unit, in which the water is mixed with
chemicals and flocculated in the center of the basin, then forced down and trapped for removal in
a sludge blanket in the bottom of the tank.
Sludge Removal
Residue created from lime-soda ash
softening is normally very high in
calcium carbonate or a mixture of
calcium carbonate, and magnesium
hydroxide. Calcium carbonate sludges
are normally dense, stable inert, and
dewater readily. Solids content in the
sludge range from 5 to 30 total solids
with a pH greater than 10.5.
Lime-soda ash sludges may be treated
with lagooning, vacuum filtration,
centrifugation, pressure filtration,
recalcination, or land application. The
most common method is storage of
sludge in lagoons and application to
farmland or landfills disposal.
Calculations
There are two methods for calculating lime and soda ash dosages (conventional dosage method
and conversion factor method). The conventional method, although much longer, is helpful in

understanding the chemical and mathematical relationships involved in softening. The
conversion factor method is simpler, quicker, and more practical for daily operations.
In both calculation methods, lime and soda ash dosages depends on carbonate and non-carbonate
hardness in the water. Lime is used to remove carbonate harness, and both lime and soda ash are
used to remove non-carbonate hardness. If total hardness is less than or equal to total alkalinity,
there is no non-carbonate hardness (only carbonate hardness). If total hardness is greater than
total alkalinity, non-carbonate hardness equals the difference between total hardness and total
alkalinity (and carbonate hardness equals total alkalinity).
If total hardness is equal to or less than total alkalinity, then:
Lime Dosage = the carbon dioxide concentration [CO2] + the total hardness concentration
[Total Hardness] + the magnesium concentration [Mg] + [Excess]
Optimum chemical dosages can be evaluated with a jar test. Lime Softening 5
Relationship between pH and alkalinity (HCO3
-
, CO3
2- and OH-
)
Alkalinity (mg/l as CaCO3) is the capacity of water to neutralize acids. This is determined by the
content of carbonate, bicarbonate and hydroxide. Alkalinity is a measure of how much acid can
be added to a liquid without causing any significant change in pH.
When pH is less than 8.3, all alkalinity is in the bicarbonate form and is commonly referred to as
natural alkalinity. When pH is above 8.3, alkalinity may consist of bicarbonate, carbonate, and

hydroxide. As pH increases the alkalinity progressively shifts to carbonate and hydroxide forms.
Total alkalinity is the sum of bicarbonate, carbonate, and hydroxide alkalinity. Various
chemicals effect water differently:
Lowers Alkalinity: Increases Alkalinity:
Aluminum sulfate Calcium hypochlorite
Carbon dioxide Caustic soda
Chlorine gas Hydrate lime
Ferric Chloride Soda ash
Ferric Sulfate Sodium Aluminate
Sulfuric acid
The following table gives molecular weights for common chemicals:
Quicklime (CaO)…………………… 56
Hydrate Lime (CaOH)………………74
Magnesium (Mg)……………………24.3
Carbon Dioxide (C02)……………….44
Magnesium Hydroxide (Mg(OH)2)... 58.3
Soda Ash (NaCO3)…………………. 106
Alkalinity (as CaCO3)……………… 100
Hardness (as CaCO3)………………..100 Lime Softening 6
Quicklime dosage can be calculated with the following formula:
Quicklime (CaO) mg/l = (A + B + C + D) + % EXCESS
purity of lime as a decimal
A = Carbon Dioxide in source water
= mg/l CO2 x (CaO/CO2)
= mg/l CO2 x 56/44
= mg/l CO2 x 1.27

B = Bicarbonate alkalinity removed
= mg/l as CaCO3 x (CaO/CaCO3)
= mg/l x 56/100
= mg/l alkalinity x .56
C = Hydroxide alkalinity in softener effluent
= mg/l hydroxide alkalinity x (CaO/CaCO3)
= mg/l hydroxide alkalinity x 56/100
= mg/l hydroxide alkalinity x .56
D = Magnesium removed in softening
= Mg/l as Mg2+ x (CaO/Mg(OH)2
= Mg/l as Mg2+ x 56/24.3
= Mg/l as Mg2+ x 2.30
If hydrated lime (CaOH) is used in place of quicklime, the molecular weight of quicklime of 56
should be replaced with the weight of hydrated lime (74).
When treating water that contains non-carbonate hardness, soda ash is required. The amount of
soda ash can be estimated by using the following formula:
Soda Ash (NaCo3) mg/l = mg/l Non Carbonate Hardness as CaCO3 x Na2
CO3 /CaCO3
= mg/l Non-Carbonate Hardness as CaCO3 x 106/100
= mg/l Non-Carbonate Hardness as CaCO3 x 1.06
After softening, pH of the water is generally above 10. If left at this pH, water will plate filter
sand and cause problems in the distribution system. Carbon dioxide (through re-carbonation), is
added to lower the pH. The amount of carbon dioxide (CO2) required can be estimated:
Total CO2 (mg/l) = Ca(OH)2 (mg/l) x CO2
+ Mg(OH)residual x CO2

Ca(OH)2 Mg(OH)2 Lime Softening 7
Total CO2 = Ca(OH)2 mg/l x 44/74 + Mg(OH)2 residual x 44/58.3
Total CO2 = Ca(OH)2 mg/l x .59 + Mg(OH)2 residual x .75
Conversion Method
Equivalent weight conversions required in the conventional method have been combined into
single factors shown in the table below. These factors, multiplied by the concentration of the
corresponding material, will give the lime or soda ash dosage needed to remove material in units
of milligrams per liter or pounds per million gallons. The total dosage is the sum of all material
removed from the water, such as the carbon dioxide, bicarbonate alkalinity, and the magnesium,
plus the amount of excess that is required to reduce the hardness in the water. The total soda-ash
dosage is found in the same manner by finding the sum of the amounts needed to remove the
non-carbonate material from the water. An additional calculation is needed to adjust for the
purity of the lime or soda-ash used.
Converting to:
Lime (CaO) Soda Ash
mg/l lb/MG mg/l lb/MG
Carbon Dioxide (mg/l as CaCO3) 1.27 10.63 --- ---
Bicarbonate Alkalinity (mg/l as CaCO3) 0.56 4.67 --- ---
Magnesium as Mg (mg/l as CaCO3) 2.31 19.24 --- ---
Excess as CaCO3 (mg/l as CaCO3) 0.56 4.67 --- ---
Non-carbonate Hardness (mg/l as CaCO3) --- --- 1.06 8.83
Excess Soda ash (mg/l as CaCO3) --- --- 1.06 8.83
Example:
The following test results were provided by the laboratory:

CO2 concentration 25 mg/l as CO2
HCO3 (bicarbonate) concentration 205 mg/l as CaCO3
Mg (magnesium) concentration 9 mg/l as Mg
non-carbonate hardness concentration 95 mg/l as CaCO3
Assuming no excess lime is added, find correct dosages for lime (containing 90% pure CaO) and
soda ash (containing 99% pure Na2
CO3) required to remove all hardness.
CO2 25 mg/l x 1.27 = 31.75 mg/l as CaO
HCO3 205 mg/l x 0.56 = 114.80 mg/l as CaO
Mg 9 mg/l x 2.31 = 20.79 mg/l as CaO
TOTAL = 167.34 mg/l as CaO Lime Softening 8
Then adjust lime dosage for purity:
Actual Lime Dose = 167.34 mg/l = 185.93 mg/l
.90
Next, find soda ash dosage:
Soda Ash Dosage = 95 mg/l x 1.0 = 100.7 mg/l as Na2
CO3
Actual Dosage = 100.7 mg/l = 101.7 mg/l as Na2
CO3
.99
Recarbonation
After adding lime and/or soda ash, treated water will generally have a pH greater than 10. It is
necessary to lower the pH to stabilize the water and prevent deposition of carbonate scale on
filter sand and distribution piping. Recarbonation is the most common process used to reduce
pH. This procedure adds carbon dioxide to water after softening. Generally, enough carbon
dioxide is added to reduce the pH of the water to less than 8.7. The amount of carbon dioxide

added is determined using a saturation index. The Langelier Index (LI) is the most common
stabilization index used, but some plants instead use the Rizner Index, (reciprocal of the
Langelier Index). The Langelier Index is expressed as pH of stabilization (pHs) minus actual pH
measured (pHs - pH). When the Langelier Index is positive, pipes tend to become coated with
scale. When it is negative, the water tends to be corrosive.
When low magnesium water is softened, no excess lime needs to be added. After softening,
water becomes supersaturated with calcium carbonate and has a pH between 10.0 and 10.6.
When carbon dioxide is added, the excess calcium carbonate is converted back to permanent
hardness or calcium bicarbonate by the following formula:
Ca2+ (calcium ion) + CO3
2- (carbonate ion) + CO2 (carbon dioxide) + H2
O (water)
= 2HCO3
-
(bicarbonate ions)
When high magnesium water is softened, excess lime needs to be added to raise the pH above
11, and magnesium hydroxide precipitates out. After treatment, enough carbon dioxide must be
added to neutralize the excess hydroxide ions, as well as convert carbonate ions to bicarbonate
ions. The first stage of this reaction reduces the pH to between 10.0 and 10.5. In this range,
calcium carbonate is formed and magnesium hydroxide that did not precipitate, or did not settle
out, is converted to magnesium carbonate.
Ca2+ (calcium ion) + 2 OH-
(hydroxyl ions) + CO2 (carbon dioxide) <----> CaCO3
(calcium carbonate) + H2

O (water)
Mg2+ magnesium ion) + 2 0H-
(hydroxyl ions) + CO2 (carbon dioxide) <----> MgCO3
(magnesium carbonate) + H20 (water) Lime Softening 9
Additional carbon dioxide needs to be added to lower the pH to between 8.4 and 8.6. The
previously formed calcium carbonate re-dissolves and carbonate ions are converted to
bicarbonate ions as shown below:
CaCO3 (calcium carbonate) + H20 (water) + CO2 (carbon dioxide) <----> Ca2+ (calcium
ion) + 2HCO3
- (bicarbonate ions)
Mg2+ (magnesium ion) + CO3
2+ (carbonate ion) + CO2 (carbon dioxide) + H20 (water)
<----> Mg2+ (magnesium ion) + 2 HCO3
-
(bicarbonate ions)
For treatment of low magnesium water (where excess-lime addition is not required) single-stage
recarbonation is used. The water is mixed with lime or soda ash in the rapid-mix basin, resulting
in a pH of 10.2 to 10.5. If non-carbonate hardness removal is required, soda ash will also be
added at this step. After rapid mixing, the resulting slurry is mixed gently for a period of 30 to 50
minutes to allow the solids to flocculate. After flocculation, the water is allowed to flow into a
sedimentation basin where the solids will be removed by sedimentation. Following
sedimentation the clear water flows to the recarbonation basin where carbon dioxide is added to
reduce the pH to between 8.3 and 8.6. Any particles remaining in suspension after recarbonation

are removed by filtration.
Two-Stage Softening
Two-stage softening is sometimes used for treatment of high magnesium water (where excess
lime is required). Excess lime is added in the first stage to raise pH to 11.0 or higher for
magnesium removal. Following first stage treatment, carbon dioxide is added to reduce the pH to
between 10.0 and 10.5, the best value for removal of calcium carbonate. If non-carbonate
hardness removal is needed, soda ash will be added at this point. After second stage treatment,
the water flows to a secondary recarbonation tank, where pH is reduced to between 8.3 and 8.6.
Single-Stage Softening
Single-stage recarbonation is the one most commonly practiced (Because of the high capital cost
for building this type of two-stage treatment train). There are some benefits to using the twostage method, including reduced operating cost since less carbon dioxide is needed. Better
finished water quality is usually obtained through the two-stage process. Lime Softening 10
Conclusions:
1. Half mole of lime is required for the removal of one mole of HCl and bicarbonates of
Na+
and K+ each.
2. One mole of lime is required for the removal of one mole of temporary hardness of
Ca+2, permanent hardness of Mg+2, CO2, FeSO4, H2SO4 each.
3. Two moles of lime is required for the removal of one mole of temporary hardness of
Mg+2
4. Three moles of lime is required for the removal of one mole of Al2(SO4)3.
5. NaOH produced by NaAlO2 reacts with hardness causing salts and produces lime.
Hence its hardness must be subtracted from the lime requirement.
6. During the treatment of lime to remove the permanent hardness of Mg+2, acids and

alums the permanent hardness of Ca+2 is generated in water. Hence they require both
lime and soda treatment.
Water Softeners (SH)
Before any discussion of water softeners, we must first define what hard water is and
why softening water (the removal of hardness minerals) is desirable.
Hard Water
Hard water is caused by “hardness ions” found in city water. Typically these hardness
ions are Calcium, Ca++
, and Magnesium, Mg++, but can also include Manganese Mn++
and Iron, Fe++ / Fe+++
.
Water is considered hard when it exceeds 3 GPG [(G)rains (P)er (G)allon] of hardness
ions. Since 1 GPG is equivalent to 17.1 ppm [(P)arts (P)er (M)illion], if city water
contains more than 50 ppm hardness, then you probably need a water softener for
just about any use.
Hard Water Problems
Hardness in water causes scale build up
in pipes and equipment. Hardness will
reduce the efficiency of any boiler or
heat exchanger. Cooling towers,
washers, fixtures, sinks and other
surfaces in contact with hard water will

require more frequent maintenance, and
will have a much shorter useful life.
Reverse osmosis membranes are
especially susceptible to hardness
fouling.
Hardness ions also interfere with many chemical processes such as chemical
compounding and aqueous cleaners.
Hard Water and Plumbing
The tendency of hard water to form precipitates when heated or partially evaporated,
as in a boiler or water heater can be a serious problem. The nature of the precipitate
depends upon the species of the anions present in the hard water. One of the most
common anions in natural water is the bicarbonate (HCO3
-
) ion, formed by the
reaction of atmospheric carbon dioxide with water. When a solution containing Ca++
and HCO3
-
ions is heated, a precipitate of calcium carbonate forms as a result of the
following net ionic reaction: 2 HCO3
-
+ heat CO3
=
+ CO2 (gas) + H2
O
The carbonate then reacts with calcium
Ca++ + CO3
=

CaCO3 (solid)
Ca++ + 2HCO3- CaCO3 (solid) + CO2 (gas) + H2
O
Boilers and Hot Water Systems
Hard water causes pipes to scale up. Scale collects on heating elements, shortening
their life and making them operate less efficiently. Hard water also contributes to
inefficient and costly operation of water-using equipment. Heated hard water forms a
scale of calcium and magnesium salts. Pipes can become clogged with scale that
reduces water flow and ultimately requires pipe replacement.
Thickness of Percentage Loss
Scale in inches of Thermal Efficiency
1/16 .....…............... 15%
1/8 ................…..... 25%
1/4 ....................... 39%
3/8 .........................55%
1/2 ........................ 70%
Reverse Osmosis membranes
Reverse osmosis works by passing a feed water across the membrane, allowing a
substantial fraction of the water to pass through the membrane, leaving the
concentrated contaminants behind. As pure water is removed, the contaminants build
up in concentration. As soon as that concentration exceeds the solubility of a
particular species, precipitation can occur resulting in scaling.
Ca++ (aq.con.) + CO3
=
CaCO3 (solid)
Because of the low solubility of hardness ions, saturation of those ions is exceeded at
even a modest recovery rate, causing scale formation. Scaling reduces the efficiency

of the membrane by reducing the effective surface area by both crystallization and the
tendency to co-precipitate other ions and insoluble particles, and by being a source of
contaminants.
Once hardness scale has formed on a membrane, it is difficult to remove during a
cleaning, again because of the low solubility of hardness ions. Even after cleaning,
sub-microscopic scale deposits on the membrane surface can provide nucleation sites
for the formation of new scale with the attendant co-precipitates. Laundering Problems
Hardness minerals combine with some soils to form insoluble salts, making them
difficult to remove from cloth. Continuous laundering of cloth in hard water can
damage fibers and shorten the life of fabrics by up to 40 percent. Hard water used to
make up cleaning solutions reduces the efficiency of the alkaline cleaners so that more
cleaner is required.
The use of extra cleaners results in a larger amount of chemical being released to the
environment. Removing hardness and other salts from process water reduces
chemical and maintenance costs. It will also improve the quality control and minimize
the environmental impact of processes.
Reactions - How Soap Scum is Formed
Hard water has many undesirable tendencies, one of which is to form a precipitate
with the most common type of soap which contains sodium stearate, NaC18H35O2, the
sodium salt of an organic acid. The calcium and magnesium salts of this acid are
insoluble in water. And example of this reaction is:
Ca++ + 2C18H35O2
-
Ca(C18H35O2)
2 (solid)
This reaction continues until nearly all the Ca++ or Mg++ ions are removed from the
water by forming the soap scum precipitate; only then does soap become effective as

a cleansing agent.
Water Softeners
Types of Water Softeners
Water softeners are not all equal. There are essentially two kinds of water softeners:
1) Those that work and, 2) those that don’t work.
Softeners that work include the standard ion exchange water softeners known
and used world wide, hot or cold lime softening, and eventually new divalent
rejector RO membranes, and some ED (Electro-Dialysis) softening processes.
“Softeners” that don’t work, (at least reliably) are anything magnetic or electromagnetic.
Standard Ion Exchange Water Softeners
The type of Water softener that will work on all city water chemistry, the type that
works the world over, is the type that operates on the ion exchange principle. When
you really need soft water, the only answer today is an ion exchange water softener.
(Someday RO and/or ED may be legitimate economic alternatives.)
The Ion Exchange Softening Process
In the ion exchange water softening process, water is passed through a bed of
sulfonated styrene-divynalbenzene spherical resin beads (about 1 mm in diameter).
The exchange sites created on the beads by the sulfonation process are then
saturated with sodium ions by passing a brine solution through the resin. The ion exchange “softening” process takes place when the hardness ions, Ca++ and Mg++
attach themselves to the resin beads causing the sodium ions on the resin beads to be
released into the water.
Using an Ion Exchange Column (R = Ion Exchange Resin Site)
Ca++ (aq) + Na2R(solid) CaR(solid) + 2Na+ (aq)
When the resin becomes saturated with calcium and magnesium, it is regenerated by
passing an NaCl brine solution through the resin. The high concentration of sodium
ions in the brine causes the reaction to be reversed with sodium replacing the calcium

and magnesium ions which are then discharged into the waste water.
Ion Exchange process. (H)
Ion exchange is an exchange of ions between two electrolytes or between an electrolytesolution and a complex. In most cases the term is used to denote the processes of purification, separation, and decontamination of aqueous and other ion-containing solutions with solidpolymeric or mineralic 'ion exchangers'.
Typical ion exchangers are ion exchange resins (functionalized porous or gel polymer),zeolites, montmorillonite, clay, and soil humus. Ion exchangers are either cation exchangersthat exchange positively charged ions or anion exchangers that exchange negatively charged ions . There are also amphoteric exchangers that are able to exchange both cations and anions simultaneously. However, the simultaneous exchange of cations and anions can be more efficiently performed in mixed beds that contain a mixture of anion and cation exchange resins, or passing the treated solution through several different ion exchange materials.
Ion exchangers can be unselective or have binding preferences for certain ions or classes of ions, depending on their chemical structure. This can be dependent on the size of the ions, their charge, or their structure. Typical examples of ions that can bind to ion exchangers are:
• H+ (proton) and OH− (hydroxide) • Single-charged monatomic ions like Na+, K+, and Cl− • Double-charged monatomic ions like Ca2+ and Mg2+ • Polyatomic inorganic ions like SO4
2− and PO43−
• Organic bases, usually molecules containing the amino functional group -NR2H+ • Organic acid, often molecules containing -COO− (carboxylic acid) functional groups • Biomolecules that can be ionized: amino acids, peptides, proteins, etc.
Along with absorption and adsorption, ion exchange is a form of sorption.

Ion exchange is a reversible process and the ion exchanger can be regenerated or loaded with desirable ions by washing with an excess of these ions.
Applications
Ion exchange is widely used in the food & beverage, hydrometallurgical, metals finishing, chemical & petrochemical, pharmaceutical, sugar & sweeteners, ground & potable water, nuclear, softening & industrial water, semiconductor, power, and a host of other industries.
Most typical example of application is preparation of high purity water for power engineering, electronic and nuclear industries; i.e.polymeric or mineralic insoluble ion exchangers are widely used for water softening, water purification, water decontamination, etc.
Ion exchange is a method widely used in household (laundry detergents and water filters) to produce soft water. This is accomplished by exchanging calcium Ca2+ and magnesium Mg2+ cations against Na+ or H+ cations . Another application for ion exchange in domestic water treatment is the removal of nitrate and natural organic matter.
Industrial and analytical ion exchange chromatography is another area to be mentioned. Ion exchange chromatography is achromatographical method that is widely used for chemical analysis and separation of ions. For example, in biochemistry it is widely used to separate charged molecules such as proteins. An important area of the application is extraction and purification of biologically produced substances such as proteins and DNA/RNA.
Ion-exchange processes are used to separate and purify metals, including separating uranium from plutonium and other actinides, including thorium, and lanthanum, neodymium, ytterbium, samarium, lutetium, from each other and the other lanthanides. There are two series of rare earth metals, the lanthanides and the actinides, both of whose families all have very similar chemical and physical properties. Using methods developed by Frank Spedding in the 1940s, ion-exchange used to be the only practical way to separate them in large quantities, until the advent of solvent extraction techniques that can be scaled up enormously.
A very important case is the PUREX process (plutonium-uranium extraction process), which is used to separate the plutonium and theuranium from the spent fuel products from a nuclear reacto, and to be able to dispose of the waste products. Then, the plutonium and uranium are available for making nuclear-energy materials, such as new reactor fuel and nuclear weapons.
The ion-exchange process is also used to separate other sets of very similar chemical elements, such as zirconium and hafnium, which is also very important for the nuclear industry. Zirconium is practically transparent to free neutrons, used in building reactors, but hafnium is a very strong absorber of neutrons, used in reactor control rods.
Ion exchangers are used in nuclear reprocessing and the treatment of radioactivewaste.
Ion exchange resins in the form of thin membranes are used in chloralkaliprocess, fuel cells and vanadium redox batteries. Ion exchange can also be used to remove hardness from

water by exchanging calcium and magnesium ions for sodium ions in an ion exchange column.
Liquid (aqueous) phase ion exchange desalination has been demonstrated . In this technique anions and cations in salt water are exchanged for carbonate anions and calcium cations respectively using electrokinetic shockwaves. Calcium and carbonate ions then react to form calcium carbonate, which then precipitates leaving behind fresh water. The desalination occurs at ambient temperature and pressure and requires no membranes or solid ion exchangers. Theoretical energy efficiency of this method is on par with electrodialysis and reverse osmosis.
Ion exchange softener
Permutit’s process. (H)

This is a modern method employed for the softening of hard water. hydrated sodium aluminium silicate (Na2Al2Si2O8.xH2O) is called permutit. These complex salts are also known as zeolites.
The permutit as loosely packed in a big tank over a layer of coarse sand. Hard water is introduced into the tank from the top. Water reaches the bottom of the tank and then slowly rises through the permutit layer in the tank. The cations present in hard water are exchanged for sodium ions. Therefore this method is also called ion exchange method.
Na2Z + Ca+2 ——→ CaZ + 2Na+
Sodium zeolite (From hard water) Cal. Zeolite
Na2Z + Mg+2 ——→ MgZ + 2Na+
Sodium zeolite (From hard water) Mag. Zeolite
where Z = Al2Si2O8.xH2O
Disadvatages of hard water in industry (MH)
Deposits are an insulating layer on heat transfer surfaces. This leads to more power being consumed or to the installation of heavier duty, more expensive heat exchangers to compensate. It is estimated that 40% more energy is needed to heat water in a system fouled with ¼ inch of calcium carbonate scale. As scale and other deposits generally form inside closed systems it is not always evident that deposition is occurring, but some clues can provide the evidence that is necessary.
The disadvantages of hard water include aesthetic issues, such unsightly stains, spots, and films on dishes, and dull, faded laundry. Hard water can also cause other, more serious problems with plumbing and appliances that could end up costing a home owner money. When people have hard water, it means that there is a naturally high concentration of minerals such as calcium carbonate, magnesium, and iron in the water itself. This condition does not necessarily pose any health risks. There are, however,

certain disadvantages of hard water that people find to be quite a nuisance. One of the biggest problems is the buildup the minerals leave behind. Depending upon the levels of concentration, the minerals contained in hard water often create visible residues. Many individuals notice brown or rust-colored stains in areas such as toilet bowls, bathroom sinks, and showers. These stains might be difficult to remove and, even after thorough cleaning, come back quickly. The stains generally continue to return unless the wateris treated so as to lessen the concentration of minerals. While some disadvantages of hard water are only nuisances, there are certain issues that could result in serious and expensive problems. For instance, one of the main concerns regardinghard water is the effect it has on the efficiency of appliances such as the water heater. Over time, the minerals build up in the water heater, forming a scaly layer, sometimes called lime scale. This layer, in turn, cuts back on the ability of the unit to heat the water as it should and thus decreases its overall efficiency. Over time, the heater uses more energy and works less effectively, costing the home owner money. Determination of hardness of water (MH)
Complexometric Titration
Permanent hardness is usually determined by titrating it with a standard solution of ethylenediamminetetraacetic acid, EDTA. The EDTA is a complexing, or chelating agent used to capture the metal ions. This causes the water to become softened, but the metal ions are not removed from the water. EDTA simply binds the metal ions to it very tightly.
EDTA
EDTA is a versatile chelating agent. A chelating agent is a substance whose molecules can form several bonds to a single metal ion. Chelating agents are multi-dentate ligands. A ligand is a substance that binds with a metal ion to form a complex ion. Multidentate ligands are many clawed, holding onto the metal ion to form a very stable complex. EDTA can form four or six bonds with a metal ion.

It is frequently used in soaps and detergents because it forms complexes with calcium and magnesium ions. These ions which are in hard water are bound to the EDTA and cannot interfere with the cleaning action of the soap or detergent.
EDTA is also used in foods. Certain enzymes are responsible for food spoilage. EDTA is used to remove metal ions from these enzymes. It is used to promote color retention in dried bananas, beans, chick peas, canned clams, pecan pie filling, frozen potatoes and canned shrimp. It is used to improve flavor retention in canned carbonated beverages, beer, salad dressings, mayonnaise, margarine, and sauces. It inhibits rancidity in salad dressings, mayonnaise, sauces and salad spreads. You are using EDTA with a molarity of .0080 for the titration.
Example (H)
You titrate 50.00 mL of water sample using 10.68 mL of EDTA.
What is the CONCENTRATION of Ca2+ ion?
What is the hardness?
Water for drinking purposes (H)
Drinking water or potable water is water safe enough to be consumed by humans or used with low risk of immediate or long term harm. In most developed countries, the water supplied to households, commerce and industry meets drinking water standards, even though only a very small proportion is actually consumed or used in food preparation. Typical uses (for other than potable purposes) include toilet flushing, washing and landscape irrigation.

Over large parts of the world, humans have inadequate access to potable water and use sources contaminated with disease vectos, pathogens or unacceptable levels of toxins or suspended solids. Drinking or using such water in food preparation leads to widespread acute and chronic illnesses and is a major cause of death and misery in many countries. Reduction of waterborne diseases is a major public health goal in developing countries.
Water has always been an important and life-sustaining drink to humans and is essential to the survival of all known organisms. Excluding fat, water composes approximately 70% of the human body by mass. It is a crucial component of metabolic processes and serves as a solvent for many bodily solutes.
Requirements (SH)
Although covering some 70% of the Earth's surface, most water is saline. Freshwater is available in almost all populated areas of the earth, although it may be expensive and the supply may not always be sustainable. Sources where water may be obtained include:
• ground sources such as groundwater, hyporheic zones and aquifers. • precipitation which includes rain, hail, snow, fog, etc. • surface water such as rivers, streams, glaciers • biological sources such as plants. • the sea through desalination • water supply network
Spring water is groundwater that rises to the ground surface. Springs are often used as sources for bottled water. Tap water, delivered by domestic water
systems in developed nations, refers to water piped to homes and delivered to a tap or spigot. For these water sources to be consumed safely they must receive adequate treatment and meet drinking water regulations.
The most efficient way to transport and deliver potable water is through pipes. Plumbing can require significant capital investment. Some systems suffer high operating costs. The cost to replace the deteriorating water and sanitation infrastructure of industrialized countries may be as high as $200 billion a year. Leakage of untreated and treated water from pipes reduces access to water. Leakage rates of 50% are not uncommon in urban systems.
Because of the high initial investments, many less wealthy nations cannot afford to develop or sustain appropriate infrastructure, and as a consequence people in these areas may spend a correspondingly higher fraction of their income on water. 2003 statistics from El Salvador, for example, indicate that the poorest 20% of households spend more than 10% of their total income on water. In the United Kingdom authorities define spending of more than 3% of one's income on water as a hardship.

SOLUTIONS & COLLOIDS (MH)
solution: (H)
homogenous mixture; uniform throughout
solvent: (H)
substance that does the dissolving; ** substance which is in greater quantity.
solute: (H)
substance that is dissolved; ** substance which is in smaller quantity.
Types of Solutions (SH)
saturated (SH) : when undissolved solute is in equilibrium with the dissolved solute.
unsaturated (SH): contains less than the saturated amount of solute for that temperature
supersaturated (SH) : contains more solute than a saturated solution can normally hold.
Solubility (SH) : the quantity of solute that will dissolve in a specified amount of solvent at a specific temperature.
Colloids (H)
A colloid is a substance microscopically dispersed throughout another substance.
The dispersed-phase particles have a diameter of between approximately 2 and 500 nanometers. Such particles are normally invisible in an optical microscope, though their presence can be confirmed with the use of an ultramicroscop or an electron microscope. Homogeneous mixtures with a dispersed phase in this size range may be called colloidal aerosols, colloidal emulsions, colloidal foams, colloidal dispersions, or hydrosols. The dispersed-phase particles or droplets are affected largely by the surface chemistry present in the colloid.
Some colloids are translucent because of the Tyndall effect, which is the scattering of light by particles in the colloid. Other colloids may be opaque or have a slight color.
Hydrocolloids (H)
A hydrocolloid is defined as a colloid system wherein the colloid particles are hydrophilic polymers dispersed in water. A hydrocolloid has colloid particles spread throughout water, and depending on the quantity of water available that can take place in different states, e.g., gel or sol (liquid). Hydrocolloids can be either irreversible (single-state) or reversible.

For example, agar, a reversible hydrocolloid of seaweed extract, can exist in a gel and solid state, and alternate between states with the addition or elimination of heat.
Many hydrocolloids are derived from natural sources. For example, agar-agar and carrageenan are extracted from seaweed, gelatin is produced by hydrolysis of proteins of bovine and fish origins, and pectin is extracted from citrus peel and apple pomace.
Gelatin desserts like jelly or Jell-O are made from gelatin powder, another effective hydrocolloid. Hydrocolloids are employed in food mainly to influence viscosity (e.g., a sauce).Hydrocolloid-based medical dressings are used for skin and wound treatment.
Other main hydrocolloids are xanthan gum, gum arabic, guar gum, locust bean gum, cellulose derivatives as carboxymethyl cellulose, alginate and starch.
Particle Size (H)
Colloidal particle is a small amount of matter having size typical for colloids and with a clear phase boundary . A group of such particles (aggregate, agglomerate) or being a macromolecule (e.g. solution of polymer molecules is a molecular colloid) or a molecular aggregate.
A colloidal sized particle is defined in diameter from 1-1000 nanometers. Soluble particles smaller than this will form a solution as opposed to a colloid.
Colloidal state (H)
Colloid science concerns systems in which one or more of the components has at least one dimension within the nanometre (10-9m) to micrometre (10-6m) range, i.e. it concerns, in the main, systems containing large molecules and/or small particles. The adjective 'microheterogeneous' provides an appropriate description of most colloidal systems. There is, however, no sharp distinction between colloidal and non-colloidal systems.
with particle size in the range of 10.7 – 10-4 cm are said to be in the
colloidal state. Such articles are too small to be visible to the naked eye. They
have particle size which is intermediate between that in a true solution and
particles in precipitate. A suspension of a colloidal particles in a liquid scatter
light so that the solution appears cloudy when seen at right angles to the beam
of light passing through it (i.e. scattering of light falling on large sized
particles).
In true solution, the solute particles are present as molecules or ions
giving a homogenous mixture consisting of a single phase.
In colloidal solution, the unit of particles of dissolved substance are either

very large molecules (starch, gelatin) or aggregate of large number of
molecules. The colloidal solution consists of dispersed phase (small
concentration) and dispersion medium (large concentration) like solvent.
Properties of colloids
1- The particles of the dispersed phase are relatively large, however they
pass through ordinary filter media.
2- The dispersed phase don't dissolve in the dispersion medium.
3- They scatter light (Tyndal effect).
4- Particles show random motion (Brownian motion), due to collision with
molecules of the dispersion medium.
5- Particles adsorb ions (its own ions in preference to others).
6- Particles may have an electrical charge which lead to repulsive forces
which stabilize the colloid dispersion and prevent its coagulation.
7- When the particles of the dispersion phase join together, they coagulate
and separate due to gravity.
8- Particles have large surface area.
9- Colloidal suspensions have negligible effects on colligative properties. 60
Terminology and Classification
1- Lyophilic colloids (solvent loving). They are so called because of
affinity of particles for the dispersion medium. Solutions of lyophiles are
prepared by simply dissolving the material in the solvent. Because f
attraction between the dispersed phase and dispersion medium, salvation
(hydration in case of water) of the particles occur. Most of these colloids
are organic n nature e.g. gelatin, acacia, insulin, albumin. The solution is
viscous because of strong affinity for water (called gels).
2- Lyophobic colloid The dispersed phase has little attraction to the solvent
(solvent hating). Their properties differ from the lyophilic (hydrophilic).

They are usually inorganic n nature e.g. gold, silver, sulphur. In contrast
to lyophilic colloid, it is necessary to use special method to prepare
hydrophobic colloid.
a) Dispersion (reduce particle size to colloidal dimentions using high
density ultrasonic generators of colloidal mill etc.).
b) Condensation from materials in the subcolloidal dimentions which are
caused to aggregate into particles in the colloidal state or condensation
reactions e.g. hydrolysis, oxidation reduction etc. They are stabilized by
an acquiring a charge.
3) Association colloids (Amphophiles)
(Accumulate on surface between 2 fluids).
These are surfactants. Their molecules have two opposing solution
affinities (lypophilic part and hydrophilic part). When these are present in a
liquid at low concentration they exist separately in subcolloidal dimentions. If
the concentration is increased, aggregation occurs over a narrow range (50
monomer or more), thus forming what is called micelles. The concentration at
which micelle is formed is the critical micelle concentration. 61
Example of pharmaceutical colloids
Colloidal AgCl, AgI, Ag proteinate (effective germicide), colloidal
sulphur. Many natural and synthetic polymers are important n pharma-ceutical
practice.
Polymers: These are macromolecules formed by polymerization or
condensation of small non-colloidal molecules e.g. proteins, natural colloids,
plasma proteins which are responsible for binding certain drug molecules so
that the pharmacological action of the drug molecule is affected by them.
Starch and hydroxymethylallulose, cyclodeztrin are also examples.
Stability of colloidal system

The colloidal particles are not stable in the dispersion medium. The
particles of colloidal substance attain their stability in solution if they don't
adhere to each other and coagulate. The surface of the particle is covered by
different substances :
a) In the dispersed phase particles are lyophilic (hydrophile in case of
water). Stabilization is brought about by surrounding the particle with a
protective solvent sheath that prevents mutual adherence when the particle
collide as a result of Brownian motion.
b) If the dispersed phase is lyophobic e.g. (hydrophobe; heats water). The
particles are stable only in presence of an electrolyte that be adsorbed on the
surface in the form of either positive or negative charge, so as the particles
become similarly charged so they don't coagulate. The stability may
increase by collecting solvent molecules around them after adsorbing ions
(salvation) i.e. two stabilizing layers. The amount fo electrolyte must be of
certain value that is necessary for maximum adsorption (if less no stability). 62
If large amount of electrolyte is used this would cause coagulation of the
hydrophonic particles.
Examples of systems which are colloidal
Aerosols Foodstuffs
Agrochemicals Ink
Cement Paint
Cosmetics Paper
Dyestuffs Pharmaceuticals
Emulsions Plastics
Fabrics Rubber
Foams Soil

Examples of processes which rely heavily on the application of colloid/surface phenomena are:
Adhesion Ore flotation
Chromatography Precipitation
Detergency Road surfacing
Electrophoretic deposition Sewage disposal
Emulsion polymerization Soil conditioning
Food processing Sugar refining
Grinding Water clarification
Heterogeneous catalysis Water evaporation control
Ion exchange Water repellency
Lubrication Wetting
Oil-well drilling
Tyndall effect (MH)
The Tyndall effect, also known as Tyndall scattering, is light scattering by particles in a colloid or particles in a fine suspension. It is named after the 19th century physicist John Tyndall. It is similar to Rayleigh scattering, in that the intensity of the scattered light depends on the fourth power of the frequency, so blue light is scattered much more strongly than red light. An example in everyday life is the blue colour sometimes seen in the smoke emitted by motorcycles, particularly two stroke machines where the burnt engine oil provides the particles.
Under the Tyndall effect, the longer-wavelength light is more transmitted while the shorter-wavelength light is more reflected via scattering. An analogy to this wavelength dependency is that longwave electromagnetic waves such as radio waves are able to pass through the walls of buildings, while shortwave electromagnetic waves such as light waves are stopped and reflected by the walls. The Tyndall effect is seen when light-scattering particulate-matter

is dispersed in an otherwise light-transmitting medium, when the cross-section of an individual particulate is the range of roughly between 40 and 900 nanometers, i.e., somewhat below or near the wavelength of visible light (400–750 nanometers).
It is particularly applicable to colloidal mixtures and suspensions; for example, the Tyndall effect is commercially exploited to determine the size and density of particles in aerosols and other colloidal matter .
After a solute, such as salt, dissolves in water, the salt is gone, right? NO! It is said to be "in
solution". A solution is a mixture that is completely uniform throughout. In water, the salt
crystals dissolve by separating into ions, which are on the atomic level. These ions become
uniformly "mingled" with water molecules, producing a homogeneous mixture, one that is
uniform throughout.
Water mixtures are classified according to the size of particles dispersed in the water.
Suspensions are mixtures containing relatively large, easily-seen particles. The particles remain
suspended for a while after stirring, but then settle out or form layers within the liquid.
Suspensions are classified as heterogeneous mixtures because they are not uniform throughout.
Muddy water is a good example of a suspension: if the water sits, after time, the dirt will settle
out. In a suspension, the component particles are much larger than in a solution. Particles of a
size between those in a solution and those in a suspension are called colloidal.
A colloid is a mixture of water that contains colloidal particles. The properties of colloids differ
from those of solutions and suspensions. Many colloids are cloudy or milky in appearance but
look clear when they are very dilute. Unlike a suspension, the particles in a colloid are not large
enough to settle out. Homogenized milk is an example of a colloid. Colloidal mixtures exhibit
the Tyndall effect -- The scattering of visible light in all directions. You can see a beam of light
passed through a colloid just as you see a sunbeam in a dusty room. Suspensions also exhibit

the Tyndall effect, but solutions never do.
Examples:
The visible beam of headlights in fog is caused by the Tyndall effect. The water droplets scatter the light, making the headlight beams visible.
Brownian movement (MH)
Brownian motion or pedesis (from Greek: πήδησις P��e�s�s "leaping") is the presumably random moving of particles suspended in a fluid (a liquid or a gas) resulting from their bombardment by the fast-moving atoms or molecules in the gas or liquid. The term "Brownian motion" can also refer to the mathematical model used to describe such random movements, which is often called a particle theory.[1]
In 1827, the botanist Robert Brown, looking through a microscope at particles found in pollen grains in water, noted that the particles moved through the water but was not able to determine the mechanisms that caused this motion. Atoms and molecules had long been theorized as the constituents of matter, and many decades later, Albert Einstein published a paper in 1905that explained in precise detail how the motion that Brown had observed was a result of the pollen being moved by individual water molecules. This explanation of Brownian motion served as definitive confirmation that atoms and molecules actually exist, and was further verified experimentally by Jean Perrin in 1908. Perrin was awarded the Nobel Prize in Physics in 1926 "for his work on the discontinuous structure of matter" (Einstein had received the award five years earlier "for his services to theoretical physics" with specific citation of different research). The direction of the force of atomic bombardment is constantly changing, and at different times the particle is hit more on one side than another, leading to the seemingly random nature of the motion. This transport phenomenon is named after Robert Brown.
The mathematical model of Brownian motion has a few real-world applications. For instance,Stock market fluctuations are often cited, although Benoit Mandelbrot rejected its applicability to stock price movements in part because these are discontinuous.[2]
Brownian motion is among the simplest of the continuous-time stochastic (or probabilistic) processes, and it is a limit of both simpler and more complicated stochastic processes (seerandom walk and Donsker's theorem). This universality is closely related to the universality of the normal distribution. In both cases, it is often mathematical convenience rather than the accuracy of the models that motivates their use. This is because Brownian motion, whose time derivative is everywhere infinite, is an idealised approximation to actual random physical processes, which always have a finite time scale.
In 1827, the botanist Robert Brown, looking through a microscope at particles found in pollen grains in water, noted that the particles moved through the water but was not able to determine the mechanisms that caused this motion. Atoms and molecules had long been

theorized as the constituents of matter, and many decades later, Albert Einstein published a paper in 1905that explained in precise detail how the motion that Brown had observed was a result of the pollen being moved by individual water molecules. This explanation of Brownian motion served as definitive confirmation that atoms and molecules actually exist, and was further verified experimentally by Jean Perrin in 1908. Perrin was awarded the Nobel Prize in Physics in 1926 "for his work on the discontinuous structure of matter" (Einstein had received the award five years earlier "for his services to theoretical physics" with specific citation of different research). The direction of the force of atomic bombardment is constantly changing, and at different times the particle is hit more on one side than another, leading to the seemingly random nature of the motion. This transport phenomenon is named after Robert Brown.
The mathematical model of Brownian motion has a few real-world applications. For instance,Stock market fluctuations are often cited, although Benoit Mandelbrot rejected its applicability to stock price movements in part because these are discontinuous.[2]
Brownian motion is among the simplest of the continuous-time stochastic (or probabilistic) processes, and it is a limit of both simpler and more complicated stochastic processes (seerandom walk and Donsker's theorem). This universality is closely related to the universality of the normal distribution. In both cases, it is often mathematical convenience rather than the accuracy of the models that motivates their use. This is because Brownian motion, whose time derivative is everywhere infinite, is an idealised approximation to actual random physical processes, which always have a finite time scale.
Coagulation (MH)
Coagulation (thrombogenesis) is the process by which blood forms clots. It is an important part of hemostasis, the cessation of blood loss from a damaged vessel, wherein a damaged blood vessel wall is covered by a platelet and fibrin-containing clot to stop bleeding and begin repair of the damaged vessel. Disorders of coagulation can lead to an increased risk of bleeding (hemorrhage) or obstructive clotting (thrombosis).
Coagulation is highly conserved throughout biology; in all mammals, coagulation involves both a cellular (platelet) and a protein (coagulation factor) component. The system in humans has been the most extensively researched and is the best understood.[citation needed]
Coagulation begins almost instantly after an injury to the blood vessel has damaged the endothelium lining the vessel. Exposure of the blood to proteins such as tissue factor initiates changes to blood platelets and the plasma protein fibrinogen, a clotting factor. Platelets immediately form a plug at the site of injury; this is called primary hemostasis. Secondary hemostasis occurs simultaneously: Proteins in the blood plasma, called coagulation factors or clotting factors, respond in a complex cascade to form fibrin strands, which strengthen the platelet plug.

Cofactors
Various substances are required for the proper functioning of the coagulation cascade:
• Calcium and phospholipids (a platelet membrane constituent) are required for the tenase and prothrombinase complexes to function. Calcium mediates the binding of the complexes via the terminal gamma-carboxy residues on FXa and FIXa to the phospholipid surfaces expressed by platelets, as well as procoagulant microparticles or microvesicles shed from them. Calcium is also required at other points in the coagulation cascade.
• Vitamin K is an essential factor to a hepatic gamma-glutamyl carboxylase that adds a carboxyl group to glutamic acid residues on factors II, VII, IX and X, as well as Protein S, Protein C andProtein Z. In adding the gamma-carboxyl group to glutamate residues on the immature clotting factors Vitamin K is itself oxidized. Another enzyme, Vitamin K epoxide reductase, (VKORC) reduces vitamin K back to its active form. Vitamin K epoxide reductase is pharmacologically important as a target of anticoagulant drugs warfarin and related coumarins such asacenocoumarol, phenprocoumon, and dicumarol. These drugs create a deficiency of reduced vitamin K by blocking VKORC, thereby inhibiting maturation of clotting factors. Vitamin K deficiency from other causes (e.g., in malabsorption) or impaired vitamin K metabolism in disease (e.g., in hepatic failure) lead to the formation of PIVKAs (proteins formed in vitamin K absence) which are partially or totally non-gamma carboxylated, affecting the coagulation factors' ability to bind to phospholipid.
WHY THEY ARE USED (SH)
• All waters, especially surface waters, contain both dissolved and suspended particles. • Coagulation and flocculation processes are used to separate the suspended solids
portion from • the water. • The suspended particles vary considerably in source, composition charge, particle
size, shape, • and density. Correct application of coagulation and flocculation processes and
selection of the • coagulants depend upon understanding the interaction between these factors. The
small particles • are stabilized (kept in suspension) by the action of physical forces on the particles
themselves. • One of the forces playing a dominant role in stabilization results from the surface
charge present • on the particles. Most solids suspended in water possess a negative charge and, since
they have

• the same type of surface charge, repel each other when they come close together. Therefore, they
• will remain in suspension rather than clump together and settle out of the water. HOW THE PROCESSES WORK (SH)
• Coagulation and flocculation occur in successive steps intended to overcome the forces
• stabilizing the suspended particles, allowing particle collision and growth of floc. If step one is
• incomplete, the following step will be unsuccessful. COAGULATION (SH)
• The first step destabilizes the particle’s charges. Coagulants with charges opposite those of the
• suspended solids are added to the water to neutralize the negative charges on dispersed nonsettlable solids such as clay and color-producing organic substances.
• Once the charge is neutralized, the small suspended particles are capable of sticking together.
• The slightly larger particles, formed through this process and called microflocs, are not visible to
• the naked eye. The water surrounding the newly formed microflocs should be clear. If it is not,
• all the particles’ charges have not been neutralized, and coagulation has not been carried to
• completion. More coagulant may need to be added. • A high-energy, rapid-mix to properly disperse the coagulant and promote particle
collisions is • needed to achieve good coagulation. Over-mixing does not affect coagulation, but
insufficient • mixing will leave this step incomplete. Coagulants should be added where sufficient
mixing will • occur. Proper contact time in the rapid-mix chamber is typically 1 to 3 minutes.
FLOCCULATION (SH)
• Following the first step of coagulation, a second process called flocculation occurs. Flocculation,
• a gentle mixing stage, increases the particle size from submicroscopic microfloc to visible
• suspended particles. • The microflocs are brought into contact with each other through the process of slow
mixing. • Collisions of the microfloc particles cause them to bond to produce larger, visible
flocs called

• pinflocs. The floc size continues to build through additional collisions and interaction with
• inorganic polymers formed by the coagulant or with organic polymers added. Macroflocs are
• formed. High molecular weight polymers, called coagulant aids, may be added during this step to
• help bridge, bind, and strengthen the floc, add weight, and increase settling rate. Once the floc
• has reached it optimum size and strength, the water is ready for the sedimentation process.
• Design contact times for flocculation range from 15 or 20 minutes to an hour or more. • Coagulation 200 Operational Considerations • Flocculation requires careful attention to the mixing velocity and amount of mix
energy. To • prevent the floc from tearing apart or shearing, the mixing velocity and energy input
are usually • tapered off as the size of the floc increases. Once flocs are torn apart, it is difficult to
get them to • reform to their optimum size and strength. The amount of operator control available in • flocculation is highly dependent upon the type and design of the equipment.
SEDIMENTATION
• Design and Operating Considerations • Sedimentation basins are used in conventional plants. Direct-filtration plants skip the • sedimentation stage and go directly to filtration. Detention times for sedimentation are
in the • range of 1 to 4 hours. Inlets are designed to distribute water evenly and at uniform
velocities. • Overflow rates should not exceed 20,000 gallons per day per foot of weir length.
Velocity should • not exceed 0.5 feet per minute. • Sedimentation basins are used to settle out the floc before going to the filters. Some
type of • sludge collection device should be used to remove sludge from the bottom of the
basin. DESIGN CONSIDERATIONS: CONVENTIONAL PLANTS (SH)
• Conventional plant designs separate the coagulation, or rapid-mix, stage from the flocculation, or
• slow-mix, stage. Normally this is followed by a sedimentation stage, after which filtration takes
• place. Plants designed for direct filtration route the water directly from flocculation to filtration.

• These systems typically have a higher raw-water quality. Conventional designs can incorporate
• adjustable mixing speeds in both the rapid-mix and slow-mix equipment. Multiple feed points for
• coagulants, polymers, flocculants, and other chemicals can be provided. There is generally
• adequate space to separate the feed points for incompatible chemicals. • Conventional plant designs have conservative retention times and rise rates. This
usually results • in requirements for large process basins and a large amount of land for the plant site.
On-site • pilot plant evaluation of the proposed process, by a qualified engineer familiar with
the source of • the water, is advisable prior to selection and construction of the units. • Coagulation 202 Retention or detention time is the theoretical time in minutes that
water spends in a process. It is • calculated by dividing the liquid volume, in gallons, of a basin by the plant flow rate
in gallons • per minute. Actual detention time in a basin will be less than the theoretical detention
time • because of “dead areas” and short circuiting, which could be due to inadequate
baffling. • Retention time = basin volume (gallons) • gpm flow • The rise rate is calculated by dividing the flow in gallons per minute by the net upflow
area of • the basin in square feet. • Rise Rate = gpm flow • surface area
DESIGN CONSIDERATIONS: COMBINATION UNITS (SH)
• Some designs incorporate coagulation, flocculation, and sedimentation within a single unit.
• These designs can be separated into upflow solids contact units and sludge blanket units. Most
• solids contact designs use recirculation of previously formed floes to enhance floc formation and
• maximize usage of treatment chemicals. Sludge bed designs force the newly forming flocs to
• pass upward through a suspended bed of floc. In both styles of units, the cross-sectional surface
• of the basin increases from the bottom to top, causing the water flow to slow as it rises, and

• allowing the floc to settle out. The combination units generally use higher rise rates and shorter
• detention time than conventional treatment. Numerous manufacturers market proprietary units
• based on these design concepts. These units are more compact and require less land for plant site
• location. On-site pilot plant evaluation of the proposed process, by a qualified engineer familiar
• with the source water, is advisable prior to selection and construction of combined units.
• The choice of coagulant chemical depends upon the nature of the suspended solid to be removed,
• the raw water conditions, the facility design, and the cost of the amount of chemical necessary to
• produce the desired result. • Final selection of the coagulant (or coagulants) should be made following thorough
jar testing • and plant scale evaluation. Considerations must be given to required effluent quality,
effect upon • down stream treatment process performance, cost, method and cost of sludge handling
and • disposal, and net overall cost at the dose required for effective treatment. • Inorganic Coagulants • Inorganic coagulants such as aluminum and iron salts are the most commonly used.
When added • to the water, they furnish highly charged ions to neutralize the suspended particles.
The inorganic • hydroxides formed produce short polymer chains which enhance microfloc formation. • Inorganic coagulants usually offer the lowest price per pound, are widely available,
and, when • properly applied, are quite effective in removing most suspended solids. They are also
capable of • removing a portion of the organic precursors which may combine with chlorine to
form • disinfection by-products. They produce large volumes of floc which can entrap
bacteria as they • settle. However, they may alter the pH of the water since they consume alkalinity.
When applied • in a lime soda ash softening process, alum and iron salts generate demand for lime
and soda ash.

• They require corrosion-resistant storage and feed equipment. The large volumes of settled floc
• must be disposed of in an environmentally acceptable manner. • Coagulation 204 Inorganic Coagulant Reactions • Common coagulant chemicals used are alum, ferric sulfate, ferric chloride, ferrous
sulfate, and • sodium aluminate. The first four will lower the alkalinity and pH of the solution while
the • sodium aluminate will add alkalinity and raise the pH. The reactions of each follow: • ALUM • A12(SO4)3 + 3 Ca(HCO3)2 ------------> 2 Al(OH)3+ 3CaSO4 + 6 CO2
Aluminum + Calcium gives Aluminum + Calcium + Carbon • Sulfate Bicarbonate Hydroxide Sulfate Dioxide
(already in the water to treat)
FERRIC SULFATE • Fe2(SO4)3 + 3 Ca(HCO3)2 ------------> 2 Fe(OH)3+ 3CaSO4+ 6 CO2
Ferric + Calcium gives Ferric + Calcium + Carbon • Sulfate Bicarbonate Hydroxide Sulfate Dioxide
• FERRIC CHLORIDE
2 Fe Cl3 + 3 Ca(HCO3)2 ------------> 2 Fe(OH)3 + 3CaCl+ 6CO2 Ferric + Calcium gives Ferric + Calcium + Carbon
• Chloride Bicarbonate Hydroxide Chloride Dioxide
• FERROUS SULFATE • FeS04+ Ca(HCO3)2 ------------> Fe(OH)2+ CaS04 + 2CO2
Ferrous + Calcium gives Ferrous + Calcium + Carbon • Sulfate Bicarbonate Hydroxide Sulfate Dioxide
• SODIUM ALUMINATE • 2 Na2A12O4 + Ca(HCO3)2 ------------> 8 Al(OH)3 + 3 Na2CO3 + 6 H20
Sodium + Calcium gives Aluminum + Sodium + Water • Aluminate Carbonate Hydroxide Carbonate • • Na2Al2O4 + CO2 ------------> 2 Al(OH)3 + NaCO3 • Sodium + Carbon gives Aluminum + Sodium • Aluminate Dioxide Hydroxide Carbonate
• Na2Al2O4 + MgCo3 ------------> MgAl2O4 + Na2CO3

Sodium + Magnesium gives Magnesium + Sodium
• Aluminate Carbonate Aluminate Carbonate POLYMERS (SH)
• Polymers--long-chained, high-molecular-weight, organic chemicals--are becoming more widely
• used, especially as coagulant aids together with the regular inorganic coagulants. Anionic
• (negatively charged) polymers are often used with metal coagulants. Low-to-medium weight,
• positively charged (cationic) polymers may be used alone or in combination with the aluminum
• and iron type coagulants to attract the suspended solids and neutralize their surface charge. The
• manufacturer can produce a wide range of products that meet a variety of source-water
• conditions by controlling the amount and type of charge and relative molecular weight of the
• polymer. • Polymers are effective over a wider pH range than inorganic coagulants. They can be
applied at • lower doses, and they do not consume alkalinity. They produce smaller volumes of
more • concentrated, rapidly settling floc. The floc formed from use of a properly selected
polymer will • be more resistant to shear, resulting in less carryover and a cleaner effluent. • Polymers are generally several times more expensive in their price per pound than
inorganic • coagulants. Selection of the proper polymer for the application requires considerable
jar testing • under simulated plant conditions, followed by pilot or plant-scale trials. • All polymers must be approved for potable water use by regulatory agencies.
Regulators (SH)
Five mechanisms keep platelet activation and the coagulation cascade in check. Abnormalities can lead to an increased tendency toward thrombosis:
• Protein C is a major physiological anticoagulant. It is a vitamin K-dependent serine protease enzyme that is activated by thrombin into activated protein C (APC). Protein C is activated in a sequence that starts with Protein C and thrombin binding to a cell surface protein thrombomodulin. Thrombomodulin binds these proteins in such a way that it activates Protein C. The activated form, along with protein S and a phospholipid as

cofactors, degrades FVa and FVIIIa. Quantitative or qualitative deficiency of either may lead to thrombophilia (a tendency to develop thrombosis). Impaired action of Protein C (activated Protein C resistance), for example by having the "Leiden" variant of Factor V or high levels of FVIII also may lead to a thrombotic tendency.
• Antithrombin is a serine protease inhibitor (serpin) that degrades the serine proteases: thrombin, FIXa, FXa, FXIa, and FXIIa. It is constantly active, but its adhesion to these factors is increased by the presence of heparan sulfate (a glycosaminoglycan) or the administration of heparins (different heparinoids increase affinity to FXa, thrombin, or both). Quantitative or qualitative deficiency of antithrombin (inborn or acquired, e.g., in proteinuria) leads to thrombophilia.
• Tissue factor pathway inhibitor (TFPI) limits the action of tissue factor (TF). It also inhibits excessive TF-mediated activation of FVII and FX.
• Plasmin is generated by proteolytic cleavage of plasminogen, a plasma protein synthesized in the liver. This cleavage is catalyzed by tissue plasminogen activator (t-PA), which is synthesized and secreted by endothelium. Plasmin proteolytically cleaves fibrin into fibrin degradation products that inhibit excessive fibrin formation.
• Prostacyclin (PGI2) is released by endothelium and activates platelet Gs protein-linked receptors. This, in turn, activates adenylyl cyclase, which synthesizes cAMP. cAMP inhibits platelet activation by decreasing cytosolic levels of calcium and, by doing so, inhibits the release of granules that would lead to activation of additional platelets and the coagulation cascade.

Review Questions
1.What is the significance of water in our life? Explain with suitable examples.
2.Define Water cycle.
3.What do you mean by hardness of water? Give the causes of hardness.
4.What are the types of hardness?Explain.
5.Define the various processes for the treatment of water.
6. Explain soda lime process with suitable equations and sketch.
7.Explain ionexchange method for water treatment with suitable equations and sketch.
8.What is the difference between soda lime process and ion exchange method.
9. Explain cold lime process and hot lime process.
10.What are the industrial problems with hard water. Explain.
11. What is the concept of Tyndel effect? Explain.
12.Write short notes on-: solution, solvent, colloids, particle size, solute.
13.How can you estimate the degree of hardness of water?
14.What are the methods for expressing the degree of hardness.
15. Explain Brownian motion with neat sketch.
16. What do you mean by coagulation?
17. Define the different type of water softener.
18. Define the drawbacks of hard water.

Saroda Post, Dholka Taluka, District - Ahmedabad, Gujarat - 382260, India
www.raiuniversity.edu
“The lesson content has been compiled from various sources in public domain including but not limited to the internet for the convenience of the users. The university has no proprietary right on the same.”





























