Embed Size (px)
Citation preview

TRANSLOCATION OF mRNA CODONS, I. THE PREPARATIONAND CHARACTERISTICS OF A HOMOGENEOUS ENZYME
By PHILIP LEDER, LAWRENCE E. SKOGERSON, * AND MARION M. NAU
LABORATORY OF BIOCHEMISTRY, NATIONAL CANCER INSTITUTE,NATIONAL INSTITUTES OF HEALTH
Communicated by C. B. Anfinsen, November 27, 1968
Abstract. This report describes a convenient scheme for the further purifica-tion of an E. coli enzyme which is required for the translocation step in proteinbiosynthesis. The homogeneous enzyme translocase appears to be a relativelylarge monomeric protein, having a molecular weight of approximately 72,000,and acts in a catalytic fashion during protein synthesis. It is also one of themajor soluble macromolecular constituents of rapidly growing E. coli, comprisingmore than 2 per cent of the protein in ribosome-free extracts. Further, the rateof in vitro protein synthesis is linearly dependent upon the concentration of thepure enzyme until approximately one molecule of translocase is present perribosome in reaction mixtures.
One of the soluble enzymes involved in protein biosynthesis is required to makesucceeding mRNA codons available for translation.1 This enzyme, factor G ofNishizuka and Lipmann,2 also stabilizes the complex formed between nascentpeptidyl-tRNA and ribosome.1 Both observations are consistent with theparticipation of the enzyme in the translocation of mRNA and peptidyl-tRNAduring protein synthesis, as suggested earlier.2 In order to study the transloca-tion process more precisely, we have extended the purification scheme of Lucas-Lenard and Lipmann3 by two simple steps and purified this enzyme, convenientlyreferred to as translocase, to apparent homogeneity. A quite different purifica-tion scheme has been outlined recently by Parmeggiani.4 This report describesthe preparation and certain of the characteristics of the pure enzyme and demon-strates its catalytic role in protein biosynthesis as well as its relative abundanceamong the soluble proteins of rapidly growing E. coli.
Materials and Methods.-Poly U was obtained from Miles Chemical Company; 14CQPhe, from New England Nuclear; y-32P-GTP, from International Chemical and Nuclear;E. coli B tRNA, from General Biochemicals; acrylamide and N,N'-methylenebisacryl-amide from Eastman; agarose, from Bausch and Lomb; and microgranular DEAE-cellu-lose, from Whatman. Recrystallized guanidinium-HCl was the generous gift of Mr. LouisDobkin.
Purification of the enzyme: All steps were carried out at 4°. By the procedure of Lucas-Lenard and Lipmann,3 translocase was purified through the DEAE-Sephadex step fromthe S100 supernatant of early log phase of E. coli MRE-600.5 This fraction was dialyzedfor 18 hr against buffer A (0.05 M potassium cacodylate, pH 6.3; 0.05 M KC1, and 10-4dithiothreitol) and applied to a 1.5 X 30-cm column of DEAE-cellulose equilibrated withthe same buffer. The enzyme was eluted by a 400-ml linear gradient between 0.05 and0.4 M KCl in buffer A, and 4-ml fractions were collected every 5 min. The translocase-containing fractions were identified by complementation in polyphenylalanine synthesisor by the formation of a precipitin line when tested against a specific antitranslocase anti-serum.6 The most active fractions were pooled, precipitated with 70% (NH4)2S04, andthe precipitate was back-eluted with 50, 45, 40, and 35% (NH4)2S04 according to the
454

BIOCHEMISTRY: LEDER, SKOGERSON, AND NAU
method of Jakoby.7 These fractions were stable when stored at 40 for several months,and storage frequently led to the formation of small (0.1-mm) crystals and their aggre-gates as described by Jakoby.7 These could be brought into solution by reducing the(NH4)2SO4 concentration. The 45 and 40% eluates represented pure material and wereemployed for these studies. Protein was determined by the method of Murphy and Kies.8
Polyacrylamide gel electrophoresis: The general methods described by Davis9 wereused. Translocase preparations in 40% sucrose were layered under electrophoresis buffer(0.038 M glycine-Tris, pH 8.3) directly on top of gel. The gels were 7.51%7O acrylamide(0.5 X 5.0 cm) in 0.038 M Tris-HCl, pH 8.9. Light green dye was used for tracking.Samples were electrophoresed at a constant current of 5 ma/tube for 2-21/2 hr. Proteinbands were detected by being stained with amido schwartz. Electrophoresis in sodiumdodecylsulfate (SDS) polyacrylamide after reduction in 1.0% jB-mercaptoethanol-1.0%SDS was carried out according to the procedure of Shapiro et al.10 Results were com-pared to those obtained with ovalbumin, human serum albumin, and myeloma protein;the last was a gift from Dr. E. Kuff.Immunodiffusion assays: Standard immunodiffusion techniques were employed." The
diffusion agar contained 1% agarose, 0.1 M KPO4, pH 7.0, and 1 M NaCl. Preparationand specificity of the antitranslocase antiserum is as described.6 Quantitative immuno-diffusion was carried out in diffusion agar containing 0.5 ml antiserum per 12 ml agar, asdescribed by Mancini et al."2
Polyphenylalanine synthesis and GTPase activity: 14C-polyphenylalanine synthesis wasmeasured as hot 5% trichloroacetic acid (TCA)-insoluble material retained by a nitro-cellulose filter and counted in the liquid scintillation counter as described by Nirenberg.'3Release of 32p from y_-2P-GTP was measured as noncharcoal-adsorbable radioactivityin the filtrate of reactions stopped by the addition of 1 ml 5% TCA containing Norite A(1% w/v) and washed with an additional 1 ml of water.5Equilibrium centrifugation: The molecular weight of translocase factor was determined
by the high-speed equilibrium method of Yphantis'4 in a Spinco model E ultracentrifugeequipped with an automatic scanning absorption system. Measurements of the concen-tration distribution of protein in buffer and in guanidinium-HCl were made at 16,000,24,000, and 30,000 rpm in double-sector cells with sapphire windows. The cell containingbuffer was used as a counterbalance for the cell containing the guanidinium solution in anAn-H rotor. Data are presented as the logarithm of the optical density at 280 mu of theprotein solution versus the radius squared. The weight-average molecular weights werecalculated from the slope of the best line through the points, assuming a partial specificvolume of 0.73 cc/gm.
Protein synthetic elements: E. coli MRE-600 ribosomes, seven times washed in 1 MNH4Cl, and binding factor T (containing Tu and Ts) were prepared by the methods ofLucas-Lenard and Lipmann3 as modified by Erbe, Nau, and Leder.5 14C-Phe-tRNA(23.4 /uqmoles/A'm"; specific activity, 366) was prepared according to Leder and Burzstyn.5
Results.-Purification: As indicated in Table 1, there is only a 29-fold increase
TABLE 1. Purification of translocase.Specific activity Recovery
Fraction (units*/,ug protein) (% activity)S100 supernatant 19 10045-60% (NH4,SO4 30 48DEAE-Sephadex 100 39DEAE-cellulose 300 1345% (NH4)2SO4 eluate 550 9
Each 0.05-ml reaction mixture contained 0.05 M Tris-acetate, pH 7.2; 10 mM magnesium ace-tate; 0.05 M NH4Cl; 9 Ajumoles 14C-Phe-tRNA; 8.6 pg binding factor T; 0.13 A260 unit ribosomes;20 mjumoles poly U (as base); and a rate-limiting amount (0.1-0.002 psg) of each translocase prepara-tion. Incubation was at 230 for 10 min, and reactions were assayed as indicated in Materials andMethods.
* One unit corresponds to 1 psmole phenylalanine polymerized/standard reaction/10 min.
VOL. 62., 1969 455

456 BIOCHEMISTRY: LEDER, SKOGERSON, AND NAU
in the specific activity of the enzyme as purified from the S100 supernatant.Polyacrylamide gel electrophoresis at each step of the translocase purification(Fig. 1, 1-4) indicates that several impurities persist in material obtained fromDEAE-Sephadex (Fig. 1, 2). These are eliminated by the DEAE-cellulose and(NH4)2SO4 elution steps, the last of which yields a preparation homogeneous bythis criterion (Fig. 1, 4), as well as by reduced SDS-gel electrophoresis (Fig. 1, 5).In each gel pattern, the band corresponding to translocase may be identified conHe~~~~~glo
2 a- a3-4b 1
FIG. 1.-Polyacrylamide gel electrophoresis of translocase preparations. (1)S100 supernatant (split gel); (2) DEAE-Sephadex fraction; (3) DEAE-cellulosefraction; (4) 45% (NH4)2SO4 eluate (split gel). These analyses were carried outby discontinuous electrophoresis (buffer, pH 8.9; gel, pH 8.3) with approxi-mately 50 jig of protein as described under Materials and Methods. The lowerband in 2, 3, and 4 is a marker dye. The arrows in 1 and 4 indicate translocaseprotein synthetic and antigenic activity. Gel 5 represents f3-mercaptoethanol-SDS reduced translocase electrophoresed at pH 7.2 (buffer and gel) in 0.1% mer-captoethanol 0.1% SDS as described in Materials and Methods.
veniently by using specific antitranslocase antiserum. As shown in Figure 2,split gels corresponding to Figure 1, 1 and 4 (S100 and 45% (NH4)2SO4 eluate,respectively) were tested against antitranslocase by immunodiffusion. Thesingle precipitin arcs identify the band corresponding to translocase in each geland provide further evidence of the purity of the enzyme. The presence oftranslocase in the reactive band was confirmed by the ability of band eluates tocomplement for polyphenylalanine synthesis (not shown). Thus, the trans-locase-containing bands are as indicated by the arrows in Figure 1, 1 and 4. Ascan be seen in Figure 1, 1, translocase migration corresponds to one of the majorprotein fractions in the crude soluble fraction.The prominent nature of the translocase band and modest increase in specific
activity of the enzyme during purification (cf. Parmeggiani4) prompted us toquantitate the enzyme among the soluble proteins of E. coli. This was doneusing specific antitranslocase antiserum and a quantitative radial immunodiffu-sion technique'2 16, 17 which is based upon the observation that the diameter of aprecipitin ring formed by an antigen solution in antibody-containing agar is alinear function of the logarithm of the antigen concentration. With this method
PRoc. N. A. S.

BIOCHEMISTRY: LEDER, SKOGERSON, AND NAU 4
FIG. 2.-Identification of translocase in disc gelelectrophoretic patterns by immunodiffusion. 1 and4 are split gels corresponding to S100 supernatant(Fig. 1,1) and 45% (NH4)2SO4 eluate (Fig. 1, 4), re-spectively. The split gel is immersed in immunodif-fusion agar (see Materials and Methods), and theantibody well contains antitranslocase antiserum.The plate was incubated at room temperature for3 days.
and the 40 per cent (NH4)2S04 eluate as pure translocase (antigen standard), therelative concentration of translocase in a crude extract may be determined. Anexample of the method is shown in Figure 3. The results of several determina-tions (Table 2) show that translocase comprises somewhat more than 2 per centof the soluble protein of log-phase E. coli.
Ribosomal-dependent GTPase: A ribosome-dependent GTPase activity, asreported by Nishizuka and Lipmann,2 is apparent in translocase purified throughthe DEAE-Sephadex step (Table 3). A potent GTPase activity (independent ofribosomes, however) is also present in the S100 supernatant and continues tocontaminate the DEAE-Sephadex fraction. This nonspecific GTPase activityis absent from the homogeneous 45 per cent (NH4)2S04 eluate, but the ribosome-dependent GTPase activity is retained, entirely uncoupled from protein synthe-SiS. 2
Molecular weight and lack of subunit structure: The molecular weight of thepurified enzyme was determined by equilibrium centrifugation in the presence
FIG. 3.-Translocase con-centration as determined byantibody-agar ring diffusion.Duplicate wells containedthe following protein solu-tions: (1) 40% (NH4)2SO4eluate (upper, 0.37 mg/ml;lower, 0.20 mg/ml); (4)E. coli S100 supernatant(upper, 16.3 mg/ml; lower,0.2 mg/ml). Preparation ofthe agar plates, conditions 4of incubation, and measure-ment are described underMaterials and Methods.
VOL. 62, 1969 457

458 BIOCHEMISTRY: LEDER, SKOGERSON, AND NAU
TABLE 2. Relative concentration of translocase among the soluble proteins of E. coli asdetermined by antibody-agar ring diffusion.
Precipitin ringdiameter
Protein (average of Translocaseconcentration duplicates) concentration Translocase
Preparation (mg/ml) (mm) (mg/ml) (%)S100 supernatant
1 16.3 7.5 0.34 2.12 16.3 8.3 0.44 2.73 8.2 5.9 0.18 2.24 8.2 5.7 0.18 2.2
Translocase (standard)1' 0.37 7.8 0.37 1002' 0.20 5.7 0.20 1003' 0.13 5.0 0.13 1004' 0.08 3.6 0.08 100
Antibody agar-ring diffusion plates were prepared as indicated in Materials and Method&. Proteinsolutions indicated in the table were added to 1.5-mm (diameter) wells in the agar, incubated 18-24hr at room temperature, and photographed with a Polaroid camera. Precipitin ring diameter mea-surements were made on the enlarged photograph, and the standard values were plotted on a semilogscale. The unknown values were determined from the measured precipitin ring diameters and thestandard function.
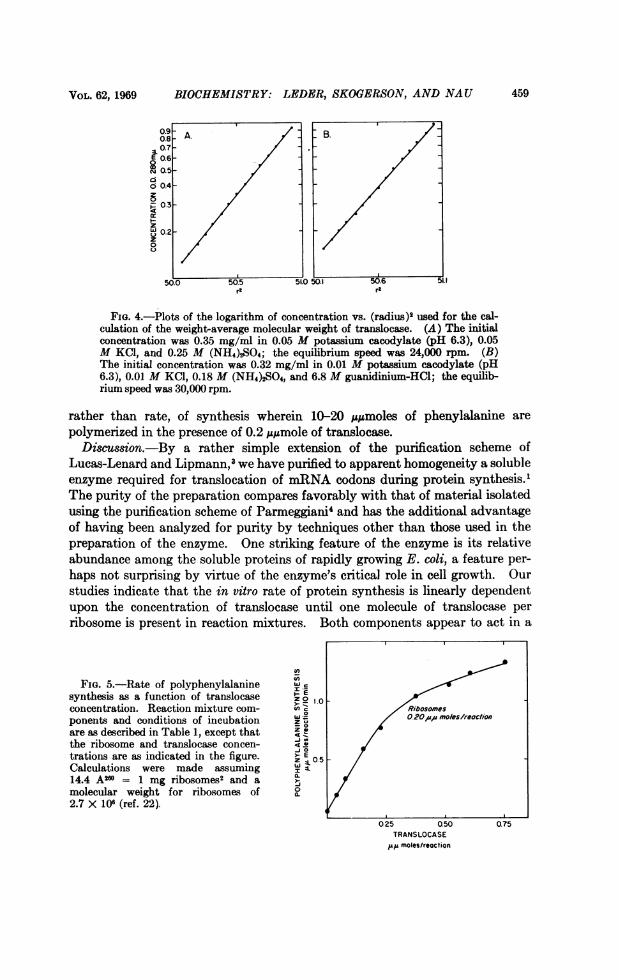
and absence of 6.8 M guanidine. Assuming a partial specific volume of 0.73cc/gm, the weight-average molecular weight of the enzyme is 72,000 i 5 percent. This determination is consistent with the electrophoretic migration of thereduced enzyme in SDS polyacrylamide gel as compared to standard proteins(Fig. 1, 5). The linear nature of the radius squared versus concentration plotfrom equilibrium centrifugation (Fig. 4) and single band observed in SDS-gelelectrophoresis further suggest the homogeneity of the enzyme.
Stoichiometry of the ribosome requirement for translocase and its catalytic actionduring protein synthesis: Having a pure enzyme of known molecular weight per-mits us to study the stoichiometry and catalytic nature of its involvement inprotein synthesis. In the experiment shown in Figure 5, the rate of polyphenyl-alanine synthesis is linearly dependent upon translocase concentration untilapproximately 0.2-0.25 ,lsmole of enzyme is present. This is so in reactions con-taining 0.2 ,uqumole of ribosomes and corresponds to a ribosome/translocase ratioof approximately 1. In experiments with a variety of ribosome concentrations,this ratio is consistently observed for that level of translocase which begins tosaturate with respect to the rate of the synthetic reaction. As also shown inFigure 5, both ribosomes and translocase are acting catalytically. This effectis more clearly demonstrated in experiments (not shown) which measure extent,
TABLE 3. Ribosome-dependent GTPase activity of translocase preparations.Ribosomes
Present AbsentAddition tY32P Released (e/moles)
S100 supernatant 0.08 ,ug 244 219DEAE-Sephadex 0.17 ,ug 106 1645% (NH4)2SO4 eluate 0.22 jug 261 0
Each 0.05-ml reaction mixture contained 0.05 M Tris-acetate, pH 7.2; 10 mM magnesium acetate;0.05M KCl; 0.6 A260 ribosomes; and 0.2 Mm .ye.32P-GTP and translocase as indicated in the table.Incubation was at 300 for 10 min. The assay is described under Material and Methods.
PROC. N. A. S.
VOL. 62, 1969 BIOCHEMISTRY: LEDER, SKOGERSON, AND NAU 459
0. A. B.
0.56-.0.206
2N0.
o 0.4 055. 01 ~ L
FIG. 4. Plots of the logarithm of concentration vs. (radiu)2 Bsed for the cal-culation of the weight-average molecular weight of tran~slocase. (A) The initialconcentration was 0.35 mg/ml in 0.05 M potassium cacodylate (pH 6.3), 0.05M KCI, and 0.25 M (NH4)2S04; the equilibrium speed was 24,000 rpm. (B)The initial concentration was 0.32 mg/ml in 0.01 M potassium cacodylate (pHI6.3), 0.01 M KCI, 0.18 M (NH4)2S~O, and 6.8 M guanidiniumHCI; the equilib-rium speed was 30,000 rpm.
rather than rate, of synthesis wherein 10-20 l;&Amoles of phenylalanine arepolymerized in the presence of 0.2 ,uIumole of tran~slocase.Discussion.-By a rather simple extension of the purification scheme of
Lucas-Lenard and Lipmann,3 we have purified to apparent homogeneity a solubleenzyme required for translocation of m.RNA codons during protein synthesis-'The purity of the preparation compares favorably with that of material isolatedusing the purification scheme of Parmeggiani4 and has the additional advantageof having been analyzed for purity by techniques other than those used in thepreparation of the enzyme. One striking feature of the enzyme is its relativeabundance among the soluble proteins of rapidly growing E. coli, a feature per-haps not surprising by virtue of the enzyme's critical role in cell growth. Ourstudies indicate that the in vitro rate of protein synthesis is linearly dependentupon the concentration of translocase until one molecule of translocase perribosome is present in reaction mixtures. Both components appear to act in a
j:u03
FIG. 5.-Rate of polyphenylalanine r
synthesis as a function of translocase Z 0 1.0 wconcentration. Reaction mixture corm U) c /Rbsomesponents and conditions of incubation Zut 2^Focsrotare as described in Table 1, except that Zz/the ribosome and translocase concen- < v
0~~~~~~~~~~
trations are as indicated in the figure. o E vs ( u
concentrtionwas0.35 mg/i in 0.0 ML poasumccdyae(p .),00
Calculations were made assuming I n 014.4 A2r0 = 1 mg ribosomeS2 and a30molecular weight for ribosomes ofp e2.7 X 1 0ff (ref. 22) /
TRANSLOCASE/L. moles/reaction

460 BIOCHEMISTRY: LEDER, SKOGERSON, AND NAU
catalytic fashion during protein synthesis. The role of ribosomal-dependentGTPase activity present in the pure enzyme but uncoupled from protein synthe-sis is difficult to evaluate. Clearly, GTP is involved in codon recognition,1' 18-20a step which precedes peptide bond formation and translocation during proteinsynthesis; but the point at which GTP hydrolysis occurs is still unclear.19' 21Possibly, intact GTP remains associated with the peptidyl-tRNA-mRNA-ribo-some complex until hydrolyzed during the translocation reaction. Studies usinghighly purified transfer components to test this possibility are in progress.
We are grateful to Dr. Michael G. Mage for patiently introducing us to a wide varietyof immunologic techniques, expertly carried out by Miss Barbara Loyd, and to Dr.Robert S. Adelstein for his advice and assistance in the equilibrium sedimentation studies.We are also grateful to Mrs. Elizabeth Stotler for her expert assistance in the preparationof this manuscript.
Abbreviations used: Phe, phenylalanine; poly U, polyuridylic acid; SDS, sodium dodecyl-sulfate; TCA, trichloroacetic acid.
* Supported by postdoctoral fellowship PF-461 of the American Cancer Society.'Erbe, R. W., and P. Leder, Biochem. Biophys. Res. Commun., 31, 798 (1968).2 Nishizuka, Y., and F. Lipmann, Arch. Biochem. Biophys., 116, 344 (1966).3 Lucas-Lenard, J., and F. Lipmann, these PROCEEDINGS, 55, 1562 (1966).4Parmeggiani, A., Biochem. Biophys. Res. Commun., 30, 613 (1968).6 Erbe, R. W., M. M. Nau, and P. Leder, J. Mol. Biol., in press.6Leder, P., L. E. Skogerson, and D. J. Roufa, these PROCEEDINGS, in press.7Jakoby, W. B., Anal. Biochem., in press.8 Murphy, J. B., and M. W. Kies, Biochem. Biophys. Ada, 45, 382 (1960).9 Davis, B. J., Ann. N. Y. Acad. Sci., 121, 404 (1964).10 Shapiro, A. L., E. Vifluela, and J. V. Maizel, Biochem. Biophys. Res. Commun., 28, 815
(1967).11 Ouchterlony, O., in Immunological Methods, ed. J. F. Ackroyd (Oxford: Scientific Publi-
cations, 1964), p. 55.12 Mancini, G., A. 0. Carbonara, and J. F. Heremans, Immunochemistry, 2, 235 (1965).1Nirenberg, M. W., in Methods in Enzymology, ed. S. P. Colowick and N. 0. Kaplan (New
York: Academic Press, 1963), vol. 6, p. 17.14Yphantis, D. A., Biochemistry, 3, 297 (1964).16 Leder, P., and H. Bursztyn, these PROCEEDINGS, 56, 1579 (1966).16Fahey, J. L., and E. M. McKelvey, J. Immunol., 94, 84 (1968).17 Hill, R. J., Immunochemistry, 5, 185 (1968).18 Ravel, J. M., these PROCEEDINGS, 57, 1811 (1967).19 Ertel, R., N. Brot, B. Redfield, J. E. Allende, and H. Weissbach, these PROCEEDINGS, 59,
861 (1968).20 Lucas-Lenard, J., and A.-L. Haenni, these PROCEEDINGS, 59, 554 (1968).21 Ravel, J. M., R. L. Shorey, S. Froehmer, and W. Shive, Arch. Biochem. Biophys., 125, 514
(1968).22 Tissibres, A., J. D. Watson, D. Schlessinger, and B. R. Hollingworth, J. Mol. Biol., 1, 221
(1959).
PRLOC. N. A. S.





![Presence HeatShockmRNAsin FieldGrown Soybeans'buffer (11 x SSC, 6% [v/v] formaldehyde, 20 mMMOPS[pH kD 92-68-12-a go_ 7.0]) andapplied to nitrocellulose previously equilibrated in](https://img.pdfslide.net/doc/110x75/61243c73979b8f05940596af/presence-heatshockmrnasin-fieldgrown-soybeans-buffer-11-x-ssc-6-vv-formaldehyde.jpg)























