Embed Size (px)
Citation preview

APPUNTI DELLE LEZIONI DI
FISICA DEI LIQUIDI
Prof. Mauro Rovere
Corso di Laurea in FisicaUniversita di Roma Tre
c© Questa opera e pubblicata sotto una Licenza Creative Commonshttp://creativecommons.org/licenses/by-nc-nd/2.5/it/

Indice
1 Introduzione alla Fisica dei Liquidi 11.1 Stato liquido della materia . . . . . . . . . . . . . . . . . . . . 11.2 Sistemi e modelli microscopici . . . . . . . . . . . . . . . . . . 41.3 Teorie approssimate e metodi esatti . . . . . . . . . . . . . . . 51.4 Metodi sperimentali e funzioni di correlazione . . . . . . . . . 6
2 Richiami di Termodinamica 72.1 Funzioni estensive ed intensive . . . . . . . . . . . . . . . . . 72.2 Primo principio della termodinamica . . . . . . . . . . . . . . 82.3 Vincoli e trasformazioni . . . . . . . . . . . . . . . . . . . . . 82.4 Il secondo principio della termodinamica e l’entropia . . . . . 92.5 Definizione della temperatura . . . . . . . . . . . . . . . . . . 112.6 Condizioni di equilibrio . . . . . . . . . . . . . . . . . . . . . 122.7 Potenziale chimico ed equilibrio chimico . . . . . . . . . . . . 142.8 Equazioni di stato e condizioni di equilibrio . . . . . . . . . . 142.9 Funzioni intensive e quantita molari . . . . . . . . . . . . . . 152.10 Relazione di Gibbs-Duhem . . . . . . . . . . . . . . . . . . . . 162.11 Trasformate di Legendre e potenziali termodinamici . . . . . 172.12 Relazioni di Maxwell e alcune conseguenze . . . . . . . . . . . 192.13 Le funzioni risposta macroscopiche . . . . . . . . . . . . . . . 192.14 Condizioni di stabilita per un sistema . . . . . . . . . . . . . 202.15 Equilibrio delle fasi . . . . . . . . . . . . . . . . . . . . . . . . 222.16 Transizioni di fase e loro classificazione . . . . . . . . . . . . . 232.17 Equazione di Van der Waals . . . . . . . . . . . . . . . . . . . 252.18 Principio degli stati corrispondenti . . . . . . . . . . . . . . . 29
3 Richiami di Meccanica Statistica 313.1 Teoria degli ensembles . . . . . . . . . . . . . . . . . . . . . . 313.2 Ensemble microcanonico e legame con la termodinamica . . . 323.3 Vari tipi di ensemble . . . . . . . . . . . . . . . . . . . . . . . 33
3.3.1 Canonico . . . . . . . . . . . . . . . . . . . . . . . . . 343.3.2 Gran-canonico . . . . . . . . . . . . . . . . . . . . . . 343.3.3 Isobarico . . . . . . . . . . . . . . . . . . . . . . . . . 35
i

3.4 Sviluppi delle formule per i sistemi classici . . . . . . . . . . . 353.4.1 Ensemble Canonico . . . . . . . . . . . . . . . . . . . . 353.4.2 Ensemble gran-canonico . . . . . . . . . . . . . . . . . 363.4.3 Fluttuazioni del numero di particelle . . . . . . . . . . 38
ii

Indice
iii

Capitolo 1
Introduzione alla Fisica deiLiquidi
La Fisica dei Liquidi, nell’ accezione che intendiamo dargli in questo cor-so, e lo studio di come la composizione microscopica e l’interazione fra gliatomi che costituiscono i sistemi in fase liquida siano collegati alle proprietamacroscopiche.
La Fisica dei Liquidi si puo considerare come un importante campo diapplicazione della Meccanica Statistica alla Fisica della Materia.
1.1 Stato liquido della materia
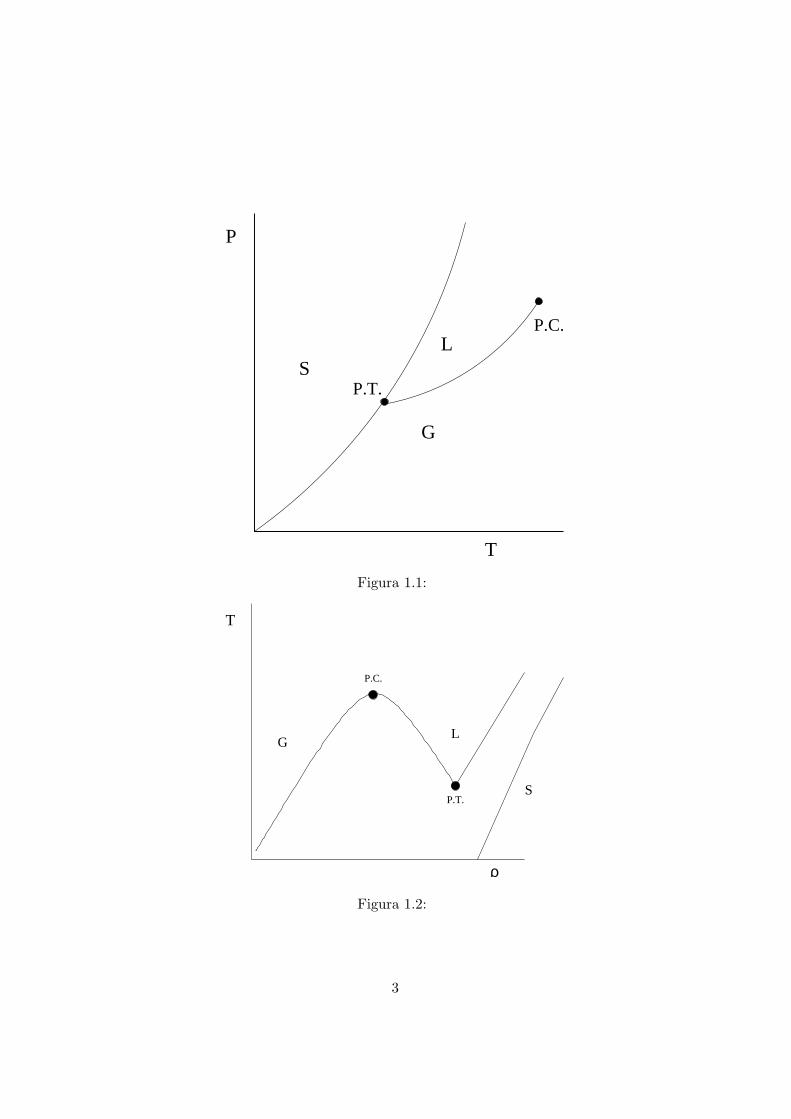
Lo stato liquido della materia presenta caratteristiche peculiari notevoli.Guardiamo al diagramma di fase di una sostanza semplice, assumendo perora la definizione tautologica che una sostanza e semplice se ha un diagram-ma di fase come nelle figure 1.1 e 1.2
Vediamo che sia nel piano (T, p), sia in quello (ρ, T ), la regione della faseliquida e molto ristretta. Essa e caratterizzata dalle curve di coesistenzaliquido-gas e liquido-solido. La curva liquido-gas termina nel punto critico(P.C.), sopra il quale abbiamo un generico stato fluido. La curva liquido-solido invece non presenta un punto critico e puo estendersi indefinitamente.In accordo alla regola delle fasi di Gibbs, per il nostro sistema monoatomicoesiste un punto, dove le tre fasi coesistono, detto punto triplo (P.T.).
La presenza di un punto critico fa in modo che si possa passare concontinuita dallo stato di gas a quello di liquido, seguendo un cammino nelpiano termodinamico, che parta dal gas, giri intorno al punto critico e finiscanel liquido. Partendo dal solido, invece, dovremo sempre attraversare lacurva di coesistenza per giungere al liquido, questo implica che non potremomai evitare una discontinuita nel passaggio di fase. E un’indicazione di comeci sia una maggiore affinita di comportamento fra il liquido e il gas, piuttostoche fra il solido e il liquido. Anche se a livello macroscopico ci possono essere
1

differenze notevoli fra gas e liquido, a livello microscopico le differenze nonsono sempre evidenti.
Si puo quindi parlare di uno stato fluido della materia, caratterizzato dauna struttura microscopica omogenea e isotropa. Esso coesiste con una fasedetta solida o meglio cristallina, caratterizzata da un ordine traslazionalenelle posizioni degli atomi. Lo stato fluido e unico sopra il punto criti-co, mentre al di sotto si presenta degenere in due stati, liquido e gassoso,coesistenti alla stessa temperatura e pressione, ma con densita diverse. Latransizione fra i due stati fluidi puo avvenire con continuita, mentre la tran-sizione fra fasi fluide e fase solida avviene sempre con un salto di densita.Le differenze piu evidenti fra il solido e il fluido, a livello microscopico, sonoriscontrabili se si guarda alle posizioni medie degli atomi e alla loro capa-cita di diffusione. Si potra notare la mancanza di ordine spaziale nel fluido,mentre la diffusione degli atomi nel solido e praticamente nulla confrontatacon quella degli atomi nei fluidi.
Lo studio della Fisica dei Liquidi e rilevante per diversi motivi. Dal unpunto di vista applicativo l’interesse e originato dall’importanza che i fluidihanno in processi tecnologici, che riguardano soprattutto l’industria chimi-ca. Fra i motivi di interesse ha acquistato sempre piu rilevanza il fatto chelo stato liquido, sebbene ristretto in una zona piccola dello spazio termo-dinamico, e contiguo a stati, che sono considerati anomali, quali i cristalliliquidi e i vetri. Questi ultimi sono materiali solidi amorfi, privi dell’ordinepresente nei cristalli, che si possono ottenere portando un sistema fluido inuno stato metastabile sottoraffreddato in modo da impedirgli di andare nellafase stabile cristallina. A livello microscopico la struttura spaziale e similea quella dei liquidi, ma diffusione degli atomi e viscosita sono simili a quelledei cristalli.
Altro motivo di interesse nello studio della Fisica dei liquidi risiede nelfatto che metodi sperimentali e teorici sviluppati per i liquidi vengono ap-plicati oggi per studiare macromolecole di interesse per la biologia e sistemicolloidali di grande interesse applicativo. Sono queste le nuove frontiere del-la ricerca nel campo della Fisica dei Liquidi che quindi ha iniziato ad avereun’importante sovrapposizione con quella che viene chiamata soft-matter.
Dal punto di vista fondamentale, i sistemi fluidi costituiscono un campodi prova notevole per modelli e metodi di Meccanica Statistica. Un fluidoalle densita tipiche dei liquidi si trova nello spazio termodinamico lontanosia dal gas ideale, sia dal dal cristallo armonico, vale a dire da modelli facilida studiare. Manca quindi un sistema imperturbato, al quale fare riferimen-to per sviluppare una teoria perturbativa. La mancanza poi di un ordinetraslazionale e la difficolta di prevedere la relazione di dispersione fra energiadei modi propri e vettore d’onda rende piu impegnativi gli esperimenti suquesti sistemi, rispetto agli analoghi effettuati sui solidi.
2
P.T.
G
LS
T
P.C.
P
Figura 1.1:
SP.T.
T
ρ
L
P.C.
G
Figura 1.2:
3

1.2 Sistemi e modelli microscopici
I sistemi che consideriamo si possono classificare in base al tipo di forze, cheagiscono fra gli atomi. Per forze microscopiche intendiamo forze efficaci, inquanto l’unico tipo di interazione fondamentale presente e quella coulom-biana fra nuclei ed elettroni che compongono il sistema. Dato che sarebbeimpossibile risolvere esattamente il problema quantistico, si procede a ridur-lo a quello di particelle costituenti, atomi o molecole, che interagiscono conun potenziale efficace. Tali costituenti vengono considerati in genere comeparticelle classiche e solo in pochi casi la loro natura quantistica appare inalcune proprieta. L’eccezione piu importante e l’elio, che rimane liquido an-che a temperature vicine allo zero assoluto, per gli altri fluidi le temperaturee densita, alle quali le fasi fluide sono stabili, rendono possibile lavorare inapprossimazione classica.
La lunghezza d’onda di de Broglie e definita a partire dall’impulso p diuna particella come
λ =h
p(1.1)
Il sistema puo essere considerato classico se la λ associata alle particelle e piupiccola delle lunghezze fisiche caratteristiche, che intervengono nel problema,in particolare la distanza media fra le particelle stesse. Tale distanza, cheindichiamo con a, e determinata dalla densita ρ in modo che
4π
3ρa3 = 1 (1.2)
D’altra parte l’energia media associata alle particelle sara dell’ordine dikBT , con kB costante di Boltzmann, quindi la lunghezza d’onda risultera in-versamente proporzionale a
√2mkBT , per essere piu precisi si usa introdurre
la lunghezza d’onda termica di de Broglie come
Λ =
√h2
2mπkBT(1.3)
L’approssimazione classica e giustificata se risulta
Λa¿ 1 (1.4)
Dato che Λ ∝ (mT )−1/2 e a ∝ ρ−1/3, la combinazione di massa, temperaturae densita determina la validita dell’approssimazione. La tabella riporta ivalori per alcuni liquidi monoatomici:
Λ Λ /aH2 3.3 0.97Ne 0.78 0.26Li 0.31 0.11Ar 0.30 0.083Na 0.19 0.054
4

Come si vede sistemi come argon e sodio liquido possono essere trat-tati come classici, per gli altri ci aspettiamo correzioni quantistiche e perl’idrogeno l’approssimazione classica e discutibile.
Una volta stabilito il modello di interazione fra gli atomi, si calcolanole proprieta del sistema con i metodi della Meccanica Statistica. I sistemisemplici sono quelli per i quali e possibile trovare un potenziale efficace acoppie che consenta di raggiungere un buon accordo con la fenomenologia.In genere questo e possibile per gli atomi a shell chiuse, come i gas rari. Glielettroni riempiono completamente i livelli atomici e la distribuzione di cari-ca risulta sferica. L’interazione e costituita da una parte attrattiva alla Vander Waals e da una repulsione a corte distanze, dovuta all’impossibilita disovrapporre le funzioni d’onda elettroniche. Per questi sistemi il potenzialedipende da pochi parametri ed e trasferibile dal solido al liquido.
Diverso e il caso dei sistemi a legame covalente, come il silicio, dove ladirezionalita del legame gioca un ruolo fondamentale. In questi casi non sipossono trovare buoni potenziali a due corpi. Bisogna peraltro ricordare cheil silicio, come anche il germanio, diventa metallico in fase liquida, quindi,nell’andare dal solido al liquido, cambia la natura delle forze interatomiche.
I sistemi, che sono metallici in fase solida, rimangono tali anche in faseliquida. Come nel solido, anche nel liquido gli elettroni di conduzione gioca-no un ruolo importante per determinare le proprieta del sistema. Per tenerconto di questo e spesso necessario introdurre opportuni potenziali efficaci,che hanno forme piu complesse rispetto alla semplice forma dell’interazionealla Van der Waals.
Una categoria a parte e costituita dai liquidi molecolari. In fase fluidagli atomi conservano il legame e quindi le unita costitutive, a livello mi-croscopico, sono le molecole. Esistono liquidi molecolari semplici, in generecostituiti da molecole omonucleari, come l’azoto, o quasi sferiche come ilCH4. Il liquido piu diffuso, l’acqua, e difficile da classificare, si puo dire chepresenta alcuni aspetti da liquido semplice ma offre anche una fenomenologiapeculiare che lo rende molto diverso dagli altri fluidi semplici.
Per i fluidi composti da macromolecole naturalmente il tipo di modellomicroscopico dal quale partire e molto piu complesso anche se come dettosopra per molti di questi sistemi si possono usare metodi simili a quellielaborati per la Fisica dei Liquidi.
1.3 Teorie approssimate e metodi esatti
Il calcolo delle quantita osservabili con metodi analitici richiede sempre op-portune approssimazioni, la cui verifica e spesso rimandata a posteriori. Leragioni principali delle difficolta, che si incontrano nello studio dei liquidio dei fluidi densi, derivano dal fatto che si tratta di sistemi dove, proprioper l’alta concentrazione degli atomi rispetto ai gas diluiti, ci sono frequen-
5

ti processi collisionali e forte correlazione fra le particelle. D’altra parte leinterazioni non sono cosı forti da stabilizzare il sistema in una fase con unordine configurazionale a lungo range, come accade per i cristalli.
Non esiste quindi un modello ideale, esattamente risolubile, al quale fa-re riferimento per sviluppare una teoria adeguata, come il gas ideale pertrattare i gas poco densi o il cristallo armonico per le proprieta dei solidi.
Metodi esatti di calcolo sono stati sviluppati dal dopoguerra ad oggi esono i metodi di simulazione al calcolatore. Hanno avuto grande sviluppo apartire dagli anni settanta. Il calcolo delle proprieta di un modello, attra-verso la simulazione numerica, equivale ad una sorta di esperimento su unsistema, del quale si conoscono esattamente le interazioni microscopiche frale particelle. Si ha cosı la possibilita di eseguire verifiche dettagliate delleteorie, ma anche di costruire una fenomenologia di modelli, da confrontarecon quella sperimentale.
1.4 Metodi sperimentali e funzioni di correlazione
Le funzioni di correlazione giocano un ruolo fondamentale nello studio dei si-stemi fluidi, esse ci rappresentano come la fluttuazione di una certa quantita,in un determinato punto dello spazio, ad un dato tempo, sia collegata allafluttuazione di un’altra quantita (o della stessa), in un altro punto dello spa-zio, ad un tempo diverso. A partire dalle funzioni di correlazione si possonoricavare tutte le proprieta dinamiche del sistema, mentre nel limite staticoesse contengono le informazioni sulle proprieta statiche e la termodinamicadel fluido.
La diffrazione dei raggi X e usata da lungo tempo per studiare la strut-tura dei liquidi. I raggi X hanno lunghezze d’onda dell’ordine delle distanzeinteratomiche e le loro energie sono molto piu alte di quelle proprie delsistema, consentendo una diffrazione elastica. Da alcuni anni pero ha acqui-stato sempre piu rilevanza la tecnica di spettroscopia neutronica. I neutroniinteragiscono con i nuclei del fluido e vedono le fluttuazioni della densitaatomica. Opportunamente accelerati e poi moderati diffondono nel siste-ma con uno scattering anelastico, dalla sezione d’urto e possibile ricavarela funzione di correlazione spazio-temporale delle densita. Questa funzionecontiene molta informazione sul comportamento dinamico e sulla strutturadel fluido.
6

Capitolo 2
Richiami di Termodinamica
I fenomeni macroscopici, che osserviamo nei materiali, derivano da flut-tuazioni del sistema a livello microscopico. I sistemi sono composti da unnumero enorme di particelle, circa 1023 in una mole, non possiamo quinditenere sotto controllo tutte le variabili in gioco, anche se in principio po-tremmo scrivere le equazioni del moto per ciascuna particella, cosı i metodiche possiamo usare sono statistici. La Meccanica Statistica e proprio lateoria matematica, che ci consente di trattare le fluttuazioni in termini divalori piu probabili o valori medi. Spesso pero i processi di misura sono cosılunghi, rispetto ai tempi atomici caratteristici ( ≈ 10−15s ), e si effettuanosu scale di lunghezza cosı grandi rispetto a quelle interatomiche (≈ 10−8cm)che il sistema ci appare in equilibrio statico, vale a dire le sue osservabilinon cambiano nel tempo. La Termodinamica si fonda su questo tipo di os-servazioni. Essa studia gli stati di equilibrio e i passaggi da uno stato diequilibrio ad un altro. Questi passaggi, o trasformazioni, hanno luogo perl’interazione del sistema con l’esterno, che avviene attraverso trasferimentidi energia. L’energia trasferita puo essere di tipo meccanico ed e allora as-sociata al cambiamento di una variabile macroscopica, come per esempio ilvolume, oppure essa viene trasferita alle variabili microscopiche, sotto formadi calore.
2.1 Funzioni estensive ed intensive
Nella termodinamica e importante distinguere le quantita estensive da quelleintensive. Le prime dipendono linearmente dal volume del sistema. Vediamole definizioni esatte.
Una funzione si dice omogenea di grado (od ordine) n se
f(λx) = λnf(x) (2.1)
Una funzione di piu variabili e omogenea di ordine n se
f(λx1, λx2, . . .) = λnf(x1, x2, . . .) (2.2)
7

Una funzione estensiva e una funzione omogenea del primo ordine.Una funzione intensiva e una funzione omogenea di ordine zero delle
variabili estensive, la funzione intensiva p sara caratterizzata dalla proprieta
p(x1, x2, . . .) = p(λx1, λx2, . . .) (2.3)
2.2 Primo principio della termodinamica
Nella termodinamica un ruolo essenziale e giocato dall’energia. All’equi-librio, l’energia interna, definita come la somma delle energie di tutte leparticelle, che compongono il sistema, rimane costante ed e misurabile ma-croscopicamente.
Per quello che riguarda gli stati di equilibrio, essi si possono definire inbase al postulato che gli stati di equilibrio macroscopico di un sistema sonoquelli caratterizzati completamente dalle variabili energia interna E, volumeV e numero di particelle (o moli) delle diverse componenti N1, N2,. . .
A priori non e garantito che le variabili siano sufficienti per descriverelo stato del sistema, se esso si trovasse in un campo esterno, dovremmoaggiungere altre variabili, per esempio il dipolo totale, se si trattasse di uncampo elettrico. Solo l’osservazione sperimentale ci garantira che abbiamoincluso tutte le variabili necessarie.
Il primo principio della termodinamica si puo formulare in termini dipostulati sull’energia interna E.
PE-1 L’energia interna E e una funzione estensiva del volume e del numerodi particelle delle diverse componenti.
PE-2 Un cambiamento infinitesimo dell’energia interna durante un trasfor-mazione e dato da
dE = δQ + δW (2.4)
vale a dire il cambio infinitesimo di energia e determinato dal calore infini-tesimo δQ trasferito al sistema, e dal lavoro δW infinitesimo compiuto sulsistema. Da notare che dE, differenziale esatto, non deve essere consideratola somma di δQ e di δW , che in generale possono non essere differenzialiesatti.
2.3 Vincoli e trasformazioni
Un sistema termodinamico puo subire una trasformazione se riceve o cedeverso l’esterno lavoro e/o calore e/o particelle. Se pensiamo che il con-tatto con l’esterno avvenga attraverso delle pareti, che lo racchiudono essecostituiscono dei vincoli.
Un vincolo puo essere:
8

a) adiabatico se impedisce lo scambio di calore
b) rigido se non permette la variazione di volume
c) impermeabile se non consente lo scambio di particelle
Un sistema puo essere anche suddiviso in sottosistemi da pareti interneanche vincoli interni. La parete in figura divide il sistema in due parti convolumi V1 e V2. Se la parete puo muoversi, potremo avere una variazionedei volumi interni, anche se il volume totale V = V1 + V2 rimane costante.
V V1 2
Uno stato e di equilibrio compatibilmente con i vincoli applicati. Unatrasformazione da uno stato A ad uno stato B puo avvenire rimuovendo unoo piu vincoli applicati al sistema quando e nello stato A. Lo stato B saraquello di equilibrio, compatibilmente con i vincoli rimasti.
In termodinamica si definiscono delle trasformazioni quasi-statiche, otte-nute attraverso un processo ideale, nel quale la trasformazione avviene concambiamenti cosı lenti, che il sistema va da uno stato A ad uno stato B at-traverso una successione di stati di equilibrio. Il concetto di trasformazionequasi statica e strettamente legato alla possibilita che essa sia anche rever-sibile. Se infatti si va da uno stato A ad uno stato B, attraverso successivistati di equilibrio, e possibile pensare di percorrere il processo inverso nellostesso modo. Ad essere rigorosi che una trasformazione sia quasi statica euna condizione solo necessaria perche sia reversibile, ma per semplificare leconsidereremo equivalenti.
2.4 Il secondo principio della termodinamica e l’en-tropia
La (2.4) ci dice che durante una trasformazione l’energia si puo convertirein diverse forme, ma non predice in che modo si evolvera il sistema. Se essosi trova in uno stato A in presenza di vincoli, se rimuoviamo uno o piu ditali vincoli, possiamo domandarci in quale nuovo stato B di equilibrio andraa finire. Per poter trattare questo problema occorre introdurre la funzioneentropia.
PS-1 Esiste una funzione entropia S, che ha le seguenti proprieta:
• e una funzione estensiva di E, V,N1, N2, . . .
9

• e una funzione continua, differenziabile e monotona crescentedell’energia interna E
PS-2 Se uno stato B di equilibrio e raggiungibile da uno stato A di equi-librio, in modo adiabatico, rimuovendo vincoli interni, allora si avraSB ≥ SA, dove il segno di eguaglianza vale se B e accessibile da A inmodo reversibile.
Per questo ultimo postulato, nei processi naturali, che avvengono insistemi isolati, avremo che ∆S = SB − SA sara sempre positiva, o piu ingenerale
(∆S)adiabatica ≥ 0 (2.5)
dove il segno di uguale vale se la trasformazione e reversibile.Dai postulati sull’entropia (PS-1,2) si puo derivare che esiste un principio
di minimo per l’energia interna. Per vederlo, consideriamo un sistema isola-to, composto da due sottosistemi Xa e Xb, separati da una parete isolante,con energie E0
a e E0b rispettivamente. L’energia totale sara
E0 = E0a + E0
b
e l’entropia sara la somma delle entropie dei due sottosistemi
S(E0, Va + Vb, N1a + N1b, . . . , ξ
)=
S(E0
a, Va, N1a, . . . , ξ)
+ S(E0
b , Va, N1b, . . . , ξ)
Rimuoviamo ora il vincolo adiabatico interno, si avranno nuove energiedi equilibrio
Ea = E0a −∆E Eb = E0
b + ∆E
dove ∆E e l’energia scambiata. Per quanto stabilito sopra avremo chel’entropia finale sara maggiore di quella iniziale ad energia totale costante
S(E0
a + E0b , . . . , ξ
)< S (Ea + Eb, . . . , ξ = 0)
Ma l’entropia in base al PS-1 e una funzione monotona crescente dell’e-nergia interna, quindi esistera un’energia
E < Ea + Eb
tale cheS
(E0
a + E0b , . . . , ξ
)= S (E, . . . , ξ = 0)
Ad entropia costante, rimuovendo i vincoli, avremo quindi
E(S, . . . , ξ = 0) < E0(S, . . . , ξ) (2.6)
10

Da (PS-1), tenendo per semplicita le Ni costanti, abbiamo
dS =(
∂S
∂E
)
VdE +
(∂S
∂V
)
EdV (2.7)
Grazie a (PS-1) inoltre possiamo invertire la relazione fra S ed E
E = E (S, V, N1, N2, . . .)
e quindi
dE =(
∂E
∂S
)
VdS +
(∂E
∂V
)
SdV (2.8)
con (∂S
∂E
)−1
V=
(∂E
∂S
)
V(2.9)
L’entropia e importante per stabilire le condizioni di equilibrio di unsistema, ma anche perche attraverso essa possiamo collegare la termodi-namica ai concetti statistici. D’altra parte, una volta che sia avvenuta latrasformazione, che essa sia stata provocata dal nostro intervento o sia avve-nuta spontaneamente, come avviene nei processi naturali, non fa differenza.Sappiamo che in natura esistono dei processi che avvengono solo in una di-rezione e sono quindi irreversibili. Le trasformazioni spontanee provengonodalle fluttuazioni microscopiche, che hanno luogo nel sistema, quindi la com-prensione della direzione in cui avvengono, della loro irreversibilita, e legataalla natura statistica di tali fluttuazioni. L’evoluzione spontanea verso unostato termodinamico si puo interpretare come l’evoluzione verso uno stato,che ha piu probabilita di essere realizzato. Boltzmann elaboro questi con-cetti e pose le basi della meccanica statistica legando la funzione entropiaalla probabilita dello stato macroscopico.
2.5 Definizione della temperatura
Lungo una trasformazione reversibile
dE = (δQ)rev − pdV
Dalla (2.8) abbiamo quindi
(δQ)rev =(
∂E
∂S
)
VdS
Possiamo definire la temperatura T come
T =(
∂E
∂S
)
V(2.10)
11

dato che S e monotona crescente di E, la temperatura (2.10) e semprepositiva. Essa e il fattore integrante del calore infinitesimo scambiato lungouna trasformazione reversibile
(δQ)rev = TdS
Dalla (2.8) abbiamo anche che
p = −(
∂E
∂V
)
S(2.11)
Dalla (2.7) inoltreTdS = dE + pdV
da cui ricaviamop = T
(∂S
∂V
)
E(2.12)
2.6 Condizioni di equilibrio
Le condizioni di equilibrio possono essere stabilite a partire dal principio dimassimo dell’entropia (o di minimo dell’energia). Per semplificare la nota-zione assumiamo che il sistema abbia una sola componente. Consideriamo lostato di equilibrio in assenza di vincoli interni e perturbiamo il sistema conuna piccola variazione virtuale di alcuni vincoli interni, il cambio di entropiasara dato da
(∆S)E,V,N = S (E, V, N, ξi)− S (E, V, N, ξi = 0) ≤ 0 (2.13)
dato che l’entropia deve essere massima all’equilibrio. Sviluppando in seriedi Taylor abbiamo
(∆S)E,V,N ≈ dS (ξi) + d2S (ξi) ≤ 0 (2.14)
condS =
∑
i
(∂S
∂ξi
)dξi (2.15)
d2S =∑
ij
(∂2S
∂ξi∂ξj
)dξidξj (2.16)
La condizione di massimo per l’entropia nello stato di equilibrio comportache
dS = (ξi → 0) = 0 (2.17)
d2S = (ξi → 0) ≤ 0 (2.18)
12

Per l’energia potremo fare un analogo ragionamento, essa dovra essereminima per lo stato di equilibrio
(∆E)S,V,N = E (S, V,N, ξi)− E (S, V,N, ξi = 0) ≥ 0 (2.19)
le condizioni di minimo ora sono
dE = (ξi → 0) = 0 (2.20)
d2E = (ξi → 0) ≥ 0 (2.21)
Per ora consideriamo le condizioni al primo ordine (2.17,2.20), sullecondizioni al secondo ordine torneremo in seguito.Equilibrio termico
Consideriamo un sistema isolato, composto di due sottosistemi X1 e X2,separati da una parete rigida, adiabatica e impermeabile. L’energia totale
E = E1 + E2
sara costante. Pensiamo di rilasciare il vincolo di adiabaticita della pareteinterna, ci sara uno scambio di energia fra i due sottosistemi. Dato chel’energia totale deve restare costante
dE1 = −dE2 (2.22)
All’equilibrio dovra essere soddisfatta la condizione (2.17) e quindi
dS =(
∂S1
∂E1
)
V1,N1
dE1 +(
∂S2
∂E2
)
V2,N2
dE2 = 0 (2.23)
Dalla (2.22) e ricordando la (2.10), la (2.23) diventa(
1T1− 1
T2
)dE1 = 0 (2.24)
che si deve verificare per ogni valore di dE1, quindi la condizione di equilibriosara data da
T1 = T2 (2.25)
Il sistema risulta in equilibrio, se i due sottosistemi, da cui e composto,hanno la stessa temperatura.Equilibrio meccanico
Se nel sistema, che abbiamo considerato ora, rilasciamo non solo il vinco-lo di adiabaticita ma permettiamo alla parete di non essere rigida, il volumedei due sottosistemi variera in modo da lasciare costante il volume totaleV = V1 + V2. In aggiunta alla (2.22) avremo anche
dV1 = −dV2 (2.26)
13

La condizione di equilibrio (2.17) diventa ora(
1T1− 1
T2
)dE1 +
(p1
T1− p2
T2
)dV1 = 0 (2.27)
Il sistema sara in equilibrio se oltre alla (2.25) abbiamo anche soddisfatta lacondizione di equilibrio meccanico
p1 = p2 (2.28)
2.7 Potenziale chimico ed equilibrio chimico
Abbiamo finora considerato costante il numero di particelle delle diversecomponenti. Se vogliamo variare la quantita di particelle dobbiamo compiereun lavoro; questo lavoro viene chiamato chimico. Se variamo il numero diparticelle in modo quasi statico, esso sara dato da
δWc =n∑
i=1
µidNi (2.29)
dove e stato definito il potenziale chimico della specie i-esima µi. Dal primoprincipio (2.4) avremo che
dE = TdS − pdV +n∑
i=1
µidNi (2.30)
Il potenziale chimico e quindi dato anche dalla derivata
µi =(
∂E
∂Ni
)
S,V,Nk 6=i
(2.31)
Si puo ora ricavare un’altra condizione di equilibrio. Consideriamo ilsolito sistema isolato, separato in due sottosistemi X1 e X2, ma questavolta rilasciamo anche il vincolo di impermeabilita della parete interna. Siverifica facilmente che la condizione di equilibrio, perche non ci sia flusso(macroscopico) di particelle da una regione all’altra, e data dall’eguaglianzadei potenziali chimici
µ(1)i = µ
(2)i (2.32)
e quello che si chiama equilibrio chimico o materiale.
2.8 Equazioni di stato e condizioni di equilibrio
Ricordiamo che le condizioni di equilibrio (2.25),(2.28),(2.32), sono statericavate dividendo il sistema isolato in due sottosistemi. Dato che i due sot-tosistemi sono generici, e chiaro che un sistema e in equilibrio se le variabiliintensive T, p, µ soddisfano alle condizioni:
14

Equil. termico T uguale in tutti i puntiEquil. meccanico p uguale in tutti i puntiEquil. materiale µ uguale in tutti i punti
Le quantita intensive sono derivate dell’energia rispetto alle variabiliestensive, tali relazioni costituiscono le equazioni di stato del sistema:
T =(
∂E
∂S
)
V,Ni
(2.33)
p = −(
∂E
∂V
)
S,Ni
(2.34)
µi =(
∂E
∂N
)
S,V,Nj 6=i
(2.35)
2.9 Funzioni intensive e quantita molari
Riprendiamo le proprieta delle funzioni intensive descritte al paragrafo (2.1).Per esempio consideriamo la pressione
p (S, V,N1, . . . , Nn) = p (λS, λV, λN1, . . . , λNn) (2.36)
Se N = N1 + . . . + Nn, definiamo le concentrazioni o (frazioni molari) come
xi =Ni
N(2.37)
esse non sono indipendenti in quanto
n∑
i=1
xi = 1 (2.38)
Se nella (2.36) poniamo λ = 1/N , abbiamo
p = p
(S
N,V
N, x1, . . . , xn−1
)(2.39)
Le funzioni intensive dipendono da un parametro in meno.Si possono definire per le variabili estensive delle corrispondenti quantita
per particella o per mole del tipo
s =S
N
se N e il numero di particelle (o moli),s sara l’entropia per particella (o permole). Per una sola componente le equazioni di stato si possono riscriverecome
T = T (s, v) p = p(s, v) µ = µ(s, v) (2.40)
15

2.10 Relazione di Gibbs-Duhem
Il differenziale di una funzione a molte variabili e
df =n∑
i=1
(∂f
∂xi
)
xj 6=i
dxi (2.41)
E facile dimostrare il teorema di Eulero per le funzioni omogenee delprimo ordine
f (x1, . . . , xn) =n∑
i=1
(∂f
∂xi
)
xj 6=i
xi (2.42)
Applichiamo la (2.42) all’energia interna E
E = E (S, V, N1, . . . , Nn)
che possiamo scrivere come
E =(
∂E
∂S
)
V,Ni
S +(
∂E
∂V
)
S,Ni
V +∑
i
(∂E
∂Ni
)
S,V,Nj
Ni (2.43)
quindiE = TS − pV +
∑
i
µiNi (2.44)
Il differenziale totale della (2.44) sara
dE = TdS + SdT − pdV − V dp +∑
i
(µidNi + Nidµi)
ma il differenziale dell’energia dalla (2.8) e dalla (2.31) risulta essere
dE = TdS − pdV +∑
i
µidNi (2.45)
quindi deve essere soddisfatta la relazione, detta di Gibbs-Duhem
SdT − V dp +∑
i
Nidµi = 0 (2.46)
Questa relazione e importante perche collega fra loro le tre variabili intensive,che non sono quindi indipendenti. Nel caso ad una componente dalla (2.46)ricaviamo
dµ = −sdT + vdp (2.47)
16

2.11 Trasformate di Legendre e potenziali termo-dinamici
Consideriamo una funzione di n variabili f = f(x1, . . . , xn), che abbia undifferenziale esatto
df =n∑
i=1
uidxi (2.48)
dove leui =
(∂f
∂xi
)
xj
(2.49)
sono dette varaiabili coniugate alle xi.Vogliamo sostituire ad alcune delle variabili indipendenti xi le corrispon-
denti ui, per semplicita le ordiniamo, spostando alla fine quelle che vogliamosostituire
x1, . . . , xm, xm+1, . . . , xn → x1, . . . , xm, um+1, . . . , un
Definiamo la trasformata di Legendre della funzione f come
g = f −n∑
i=m+1
uixi (2.50)
la quale e una funzione g = g(x1, . . . , xm, um+1, . . . , un). Il suo differenzialesara dato da
dg =m∑
i=1
uidxi +n∑
i=m+1
(−xi)dui (2.51)
Vediamo l’applicazione delle (2.50)-(2.51) alle funzioni termodinamiche.Partiamo dall’energia interna E = E(S, V,N1, . . . , Nn), richiamando (2.45)
dE = TdS − pdV + µ1dN1 + . . . + µndNn (2.52)
Le variabili indipendenti sono quindi S, V, N1, . . . , Nn. L’entropia non esempre comoda da usare come variabile indipendente, supponiamo di volerlasostituire con la sua coniugata, la temperatura
T =(
∂E
∂S
)
V,Ni
(2.53)
dobbiamo considerare la trasformata di Legendre della E, che in accordoalla (2.50) e data dalla funzione
A(T, V, N1, . . . , Nn) = E − TS (2.54)
La nuova funzione viene chiamata energia libera di Helmholtz e il suo diffe-renziale e dato da
dA = −SdT − pdV +n∑
i=1
µidNi (2.55)
17

Con l’uso delle trasformate di Legendre si possono introdurre diversipotenziali termodinamici, in base alle variabili indipendenti che vogliamousare. Se nell’energia interna vogliamo sostituire il volume con la pressione,ricordando che
p = −(
∂E
∂V
)
S,Ni
dobbiamo sostituire a V → −p e otteniamo l’entalpia
H(S, p, N1, . . . , Nn) = E + pV (2.56)
Un potenziale termodinamico molto importante e l’energia libera diGibbs, ottenuta dalla E sostituendo (S, V ) → (T,−p)
G(T, p, N1, . . . , Nn) = E − TS + pV (2.57)
con
dG = −SdT + V dp +n∑
i=1
µidNi (2.58)
La funzione G e molto usata, perche le variabili T, p sono, in genere, le piusemplici da fissare sperimentalmente.
Negli sviluppi teorici si usa spesso il potenziale gran canonico, ottenutoda E, sostituendo (S,Ni) → (T, µi)
Φ(T, V, µ1, . . . , µn) = E − TS −∑
i
µiNi (2.59)
condΦ = −SdT − pdV −
∑
i
Nidµi (2.60)
In base al teorema di Eulero tutti i potenziali termodinamici sono fun-zioni lineari delle variabili estensive. Vediamo una conseguenza per l’energialibera di Gibbs, che si puo scrivere come
G =n∑
i=1
(∂G
∂Ni
)
Nj
Ni
D’altra parte dalla (2.44) abbiamo
G =∑
i
µiNi (2.61)
da cuiµi =
(∂G
∂Ni
)
Nj
(2.62)
Notiamo che sostituendo la (2.61) nella (2.59), otteniamo
Φ = −pV (2.63)
Per una sola componente abbiamo naturalmente
G = µN (2.64)
e il potenziale chimico risulta essere l’energia libera di Gibbs per particella.
18

2.12 Relazioni di Maxwell e alcune conseguenze
Se una funzione ha un differenziale esatto
df =(
∂f
∂x
)
ydx +
(∂f
∂y
)
x
dy
avremo che (∂
∂y
(∂f
∂x
)
y
)
x
=(
∂
∂x
(∂f
∂y
)
x
)
y
(2.65)
Relazioni di questo tipo, riferite a derivate seconde di potenziali termodi-namici, sono dette relazioni di Maxwell. Per esempio, se consideriamo ildifferenziale della A (2.55), abbiamo
(∂S
∂V
)
T,N=
(∂p
∂T
)
V,N(2.66)
Dal differenziale della G (2.58)(
∂S
∂p
)
T,N
= −(
∂V
∂T
)
p,N(2.67)
2.13 Le funzioni risposta macroscopiche
Ci sono delle funzioni termodinamiche che ci mostrano come il sistema ri-sponde, quando modifichiamo un parametro come la temperatura, la pres-sione etc. Una tipica funzione di questo tipo e il coefficiente di espansionetermica
αp =1V
(∂V
∂T
)
p(2.68)
Abbiamo poi la compressibilita isoterma
KT = − 1V
(∂V
∂p
)
T
(2.69)
e quella adiabatica
KS = − 1V
(∂V
∂p
)
S
(2.70)
Queste funzioni sono collegate alle capacita termiche. La capacita ter-mica a volume costante e data da
CV = T
(∂S
∂T
)
V(2.71)
mentre quella a pressione costante e data da
Cp = T
(∂S
∂T
)
p(2.72)
19

Dalle relazioni di Maxwell e possibile ricavare la relazione
Cp − CV = TVα2
p
KT(2.73)
e anche
KT −KS =TV α2
p
Cp(2.74)
2.14 Condizioni di stabilita per un sistema
Le condizioni equivalenti di massimo dell’entropia e di minimo dell’energiaci assicurano che il sistema isolato e in equilibrio. Se pero il sistema ein contatto con l’esterno, quali saranno le condizioni di equilibrio ? Essedipenderanno ovviamente dal tipo di scambio con l’esterno che il sistemapuo compiere. Supponiamo che un sistema σ possa scambiare calore con unbagno termico σ0, definito come un sistema che scambia calore mantenendouna temperatura costante. Il sistema totale σtot = σ + σ0 e isolato, quindil’entropia totale
Stot = S + S0
e costante e abbiamodS = −dS0 (2.75)
Il cambio di energia totale e
dEtot = dE + T0dS0 = dE − T0dS (2.76)
Se σ e σ0 sono in equilibrio T = T0 e la condizione di minimo per l’energiadi σtot diventa
d(E − TS)T=T0 = 0 (2.77)
Quindi per il nostro sistema σ la condizione di equilibrio diventa una condi-zione di minimo per l’energia libera di Helmholtz, a fissata temperatura; perdirlo meglio: gli stati di equilibrio di un sistema in contatto con un bagnotermico sono quelli che, avendo una temperatura uguale a quella del bagnotermico, minimizzano l’energia libera di Helmholtz.
Se sviluppiamo in serie di Taylor il cambio di energia totale, avremo
(∆E)Stot,Vtot,Ntot= (∆E + T0∆S0)Stot,Vtot,Ntot
≈ dE + T0dS0 + d2E ≥ 0(2.78)
e la condizione al secondo ordine e quindi
d2E ≥ 0 (2.79)
Se il sistema σ puo variare il proprio volume in contatto con un ba-gno termico, che garantisce una temperatura e una pressione costante, lacondizione di equilibrio diventa
(dG)T=T0,p=p0= d (E − TS + pV )T=T0,p=p0
= 0 (2.80)
20

quindi gli stati di equilibrio di un sistema in contatto con un bagno termico,che mantiene temperatura e pressione costanti, sono quelli che, avendo unatemperatura e una pressione uguali a quelle del bagno termico, minimizzanol’energia libera di Gibbs.
In modo analogo si puo procedere per gli altri potenziali termodinamici eritrovare le condizioni di equilibrio, per esempio, se il sistema puo scambiareanche particelle con l’esterno
(dΦ)T=T0,µ=µ0= 0 (2.81)
La condizione al secondo ordine (2.79) ci assicura la stabilita del siste-ma. La (2.79) si traduce nelle condizioni sulle derivate seconde dell’energiainterna (
∂2E
∂S2
)
V,N
=(
∂T
∂S
)
V,N≥ 0 (2.82)
(∂2E
∂V 2
)
S,N
= −(
∂p
∂V
)
S,N≥ 0 (2.83)
∂2E
∂S2
∂2E
∂V 2−
(∂2E
∂S∂V
)2
≥ 0 (2.84)
Dalle (2.82)-(2.84) si ottengono alcune importanti diseguaglianze
• compressibilita isotermaKT ≥ 0 (2.85)
• compressibilita adiabaticaKS ≥ 0 (2.86)
• capacita termica a volume costante
CV ≥ 0 (2.87)
Dalla (2.73) ricaviamo anche
Cp > CV (2.88)
Le condizioni sulle derivate seconde dell’energia interna ci dicono cheessa e una funzione convessa di S e V . L’entropia e invece una funzioneconcava.
Anche gli altri potenziali termodinamici hanno proprieta di convessitao concavita definita, rispetto ai parametri, dei quali sono funzioni. Perl’energia libera di Helmholtz abbiamo
(∂2A
∂V 2
)
T,N
= −(
∂p
∂V
)
T,N≥ 0 (2.89)
21

che e positiva per via della (2.85), mentre a causa della (2.87) abbiamo(
∂2A
∂T 2
)
V,N
= −(
∂S
∂T
)
V,N= −CV
T≤ 0 (2.90)
quindi la funzione A e concava in T e convessa in V .L’energia libera di Gibbs risulta essere una funzione concava di T e P .
La violazione di questo tipo di andamenti e il segnale di una instabilita nelsistema ed e tipica di regioni, dove si verificano transizioni di fase.
Prima di concludere sulla stabilita dei sistemi, notiamo che il segno po-sitivo delle funzioni risposta, come la compressibilita, esprimono il fatto chele fluttuazioni spontanee del sistema, in risposta alla variazione di un pa-rametro esterno (come la pressione), vanno nella direzione di ripristinare lecondizioni di equilibrio, annullando l’effetto della perturbazione. Questo echiamato in termodinamica principio di Le Chatelier, ma e un effetto pre-sente in tutta la Fisica, basta pensare alla legge di Lenz. I sistemi, per dirloin breve, tendono ad opporsi ai cambiamenti!
2.15 Equilibrio delle fasi
Se in un sistema sono presenti fasi diverse coesistenti, le condizioni di equi-librio sono le stesse ottenute in 2.8. Per semplificare pensiamo di avere duefasi in equilibrio, indicate con a e b, avremo
T (a) = T (b) p(a) = p(b) µ(a)i = µ
(b)i (2.91)
In genere, quando si studia un sistema, si fissano dall’esterno T o p,le condizioni di eguaglianza dei potenziali chimici determinano le curve dicoesistenza delle fasi. Per esempio, se il sistema ha una componente, T e pdevono esssere gli stessi nelle due fasi, abbiamo
µ(a) (T, p) = µ(b) (T, p) (2.92)
che e un’equazione in due incognite, da essa quindi si puo ricavare unafunzione
pcoex = p (T )coex (2.93)
che definisce una curva nel piano (p, T ), la curva di coesistenza fra le duefasi.
Se abbiamo un sistema con una componente e tre fasi, che devonocoesistere, oltre alla (2.92), dovra essere soddisfatta la
µ(b) (T, p) = µ(c) (T, p)
quindi con due equazioni e due incognite otterremo una sola soluzione, cherappresenta il punto triplo, come quello che abbiamo nelle figure 1.1,1.2. Lageneralizzazione a piu componenti e fasi porta alla regola delle fasi di Gibbs.
22

T
µ
a
b
µ
µ
T 0
a
b
Figura 2.1:
T
µ
a
b
T 0
b
a
Figura 2.2:
2.16 Transizioni di fase e loro classificazione
Per semplicita consideriamo un sistema ad una componente con N particelle.Lo stato stabile sara quello di minima energia libera di Gibbs, per dati T ep. Cambiando una delle variabili libere o entrambe, possiamo cambiare lafase di equilibrio del sistema. Consideriamo il potenziale chimico, fissiamop e guardiamo all’andamento in funzione di T (figura 2.1)Vediamo che
• T < T0 −→ la fase stabile e la (a)
• T > T0 −→ la fase stabile e la (b)
Al punto T = T0 abbiamo una transizione di fase. La curva di equilibrio equindi quella continua nella figura 2.2.
23

p
T
fase a
fase b
Figura 2.3:
Al variare della pressione possiamo costruire una curva di coesistenza frale fasi a e b nel piano (T, p)
Si vede anche dalla figura 2.2 che mentre µ e continuo in T0, abbiamouna discontinuita nella derivata. Questo accade anche nell’andamento afissata T in funzione di p. Alla transizione di fase, quindi, abbiamo dellediscontinuita nelle derivate
s = −(
∂µ
∂T
)
pv =
(∂µ
∂p
)
T
(2.94)
Alla transizione ci sara una discontinuita nell’entropia
∆s = s(b) − s(a) (2.95)
e un cambio di volume∆v = v(b) − v(a) (2.96)
Le transizioni di questo tipo, con discontinuita nelle derivate prime delpotenziale termodinamico, sono dette del primo ordine.
Vi sono poi transizioni di fase dove le derivate prime sono continue e sihanno non analiticita nelle derivate seconde. Si parla allora di transizionidi fase del secondo ordine. Le transizioni di fase sono sempre caratterizza-te da un comportamento non analitico del potenziale termodinamico, checaratterizza il sistema.
In una transizione del primo ordine abbiamo visto che il volume e discon-tinuo, lungo un’isoterma, al punto di transizione dovranno essere soddisfattele condizioni
p(a)(T, v(a)
)= p(b)
(T, v(b)
)(2.97)
24

µ(a)(T, p(a)
)= µ(b)
(T, p(b)
)(2.98)
Da esse si ricavano i valori di v(a) e v(b).Lungo la curva di coesistenza si puo anche scrivere un’equazione diffe-
renziale, dalla (2.92) (dµ(a)
)coex
=(dµ(b)
)coex
(2.99)
Ricordando chedµ = −sdT + vdp
abbiamo [−s(a)dT + v(a)dp = −s(b)dT + v(b)dp
]coex
da cui si ricava l’equazione di Clausius-Clapeyron(
dp
dT
)
coex=
qλ
T∆v(2.100)
dove e stato introdotto il calore latente della transizione
qλ = T∆s (2.101)
2.17 Equazione di Van der Waals
Van der Waals introdusse nel 1873 un’equazione di stato con l’idea di tenerconto, in modo approssimato, dell’interazione fra le molecole di un gas. Lasua equazione riesce a descrivere la transizione liquido-vapore ed e alla basedi molte equazioni di stato empiriche, tuttora usate in impieghi pratici. Leidee, dalle quali e derivata l’equazione, sono d’altra parte ancora valide edanzi costituiscono le linee guida di molti sviluppi teorici successivi.
Si vuole scrivere un’equazione di stato, che ricordi quella del gas ideale,che richiamiamo
pV = NkBT
Si introducono due effetti: il primo e detto di volume escluso, due particelledel sistema avranno una repulsione a corta distanza, che impedisce la lorosovrapposizione (fig. 2.4)
Si introduce il parametro empirico b, che rappresenta la porzione di vo-lume escluso di ciascuna molecola. Il volume totale occupabile sara quindiV − nb e l’equazione di stato diventa
p (V −Nb) = NkBT
Si vede che per N e T finiti, quando p diventa molto grande, il volume non vaa zero come accade per il gas ideale, ma V → Nb, si ha quindi un massimoimpacchettamento possibile delle molecole.
25

Oltre all’effetto repulsivo, si avra anche una attrazione fra le particelle,che produrra una diminuizione della pressione. Nel gas ideale la pressioneera dovuta agli urti con le pareti, ora questi urti saranno diminuiti per effettodelle forze attrattive, che tenderanno a tenere una particella piu lontana dallepareti. La diminuizione sara proporzionale al numero di coppie di particellepresenti e quindi avremo un termine del tipo
aN2
V 2
dove a e un altro parametro empirico.
Figura 2.4:
L’equazione diventa quindi(
p + aN2
V 2
)(V −Nb) = NkBT (2.102)
Riscriviamola in termini di v = V/N , ci sono varie forme equivalenti;come polinomio di terzo grado in v diventa
v3 −(
b +kBT
p
)v2 +
a
pv − ab
p= 0 (2.103)
Nel limite di grandi T e p la (2.103) diventa
v3 − kBT
pv2 = 0 (2.104)
che non e altro che l’equazione del gas ideale.Dalla (2.102) si possono ricavare le isoterme p = p(v) nella forma
p =kBT
(v − b)− a
v2(2.105)
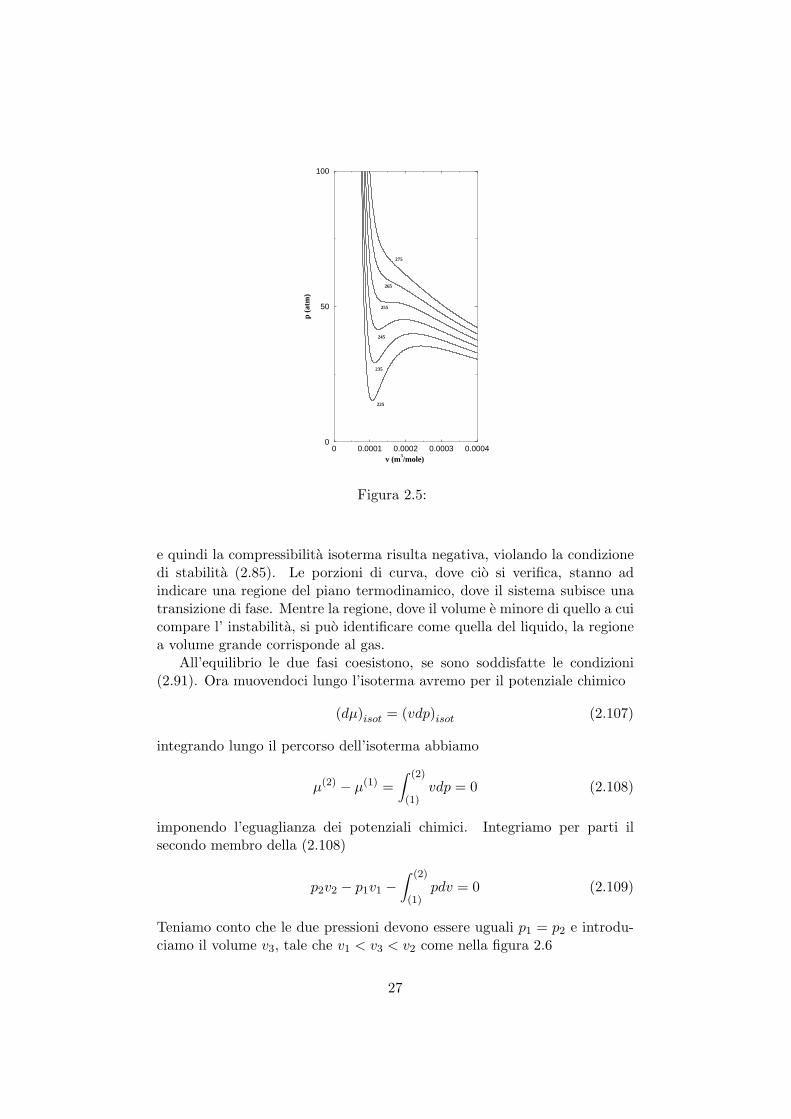
Le curve, che si ottengono, sono come quelle in figura 2.5 per diverse tem-perature.
Le temperature decrescono a partire dall’alto e al disotto di una certatemperatura cominciano ad apparire curve dove
(∂p
∂v
)
T> 0 (2.106)
26
0 0.0001 0.0002 0.0003 0.0004v (m
3/mole)
0
50
100
p (a
tm)
225
235
245
255
265
275
Figura 2.5:
e quindi la compressibilita isoterma risulta negativa, violando la condizionedi stabilita (2.85). Le porzioni di curva, dove cio si verifica, stanno adindicare una regione del piano termodinamico, dove il sistema subisce unatransizione di fase. Mentre la regione, dove il volume e minore di quello a cuicompare l’ instabilita, si puo identificare come quella del liquido, la regionea volume grande corrisponde al gas.
All’equilibrio le due fasi coesistono, se sono soddisfatte le condizioni(2.91). Ora muovendoci lungo l’isoterma avremo per il potenziale chimico
(dµ)isot = (vdp)isot (2.107)
integrando lungo il percorso dell’isoterma abbiamo
µ(2) − µ(1) =∫ (2)
(1)vdp = 0 (2.108)
imponendo l’eguaglianza dei potenziali chimici. Integriamo per parti ilsecondo membro della (2.108)
p2v2 − p1v1 −∫ (2)
(1)pdv = 0 (2.109)
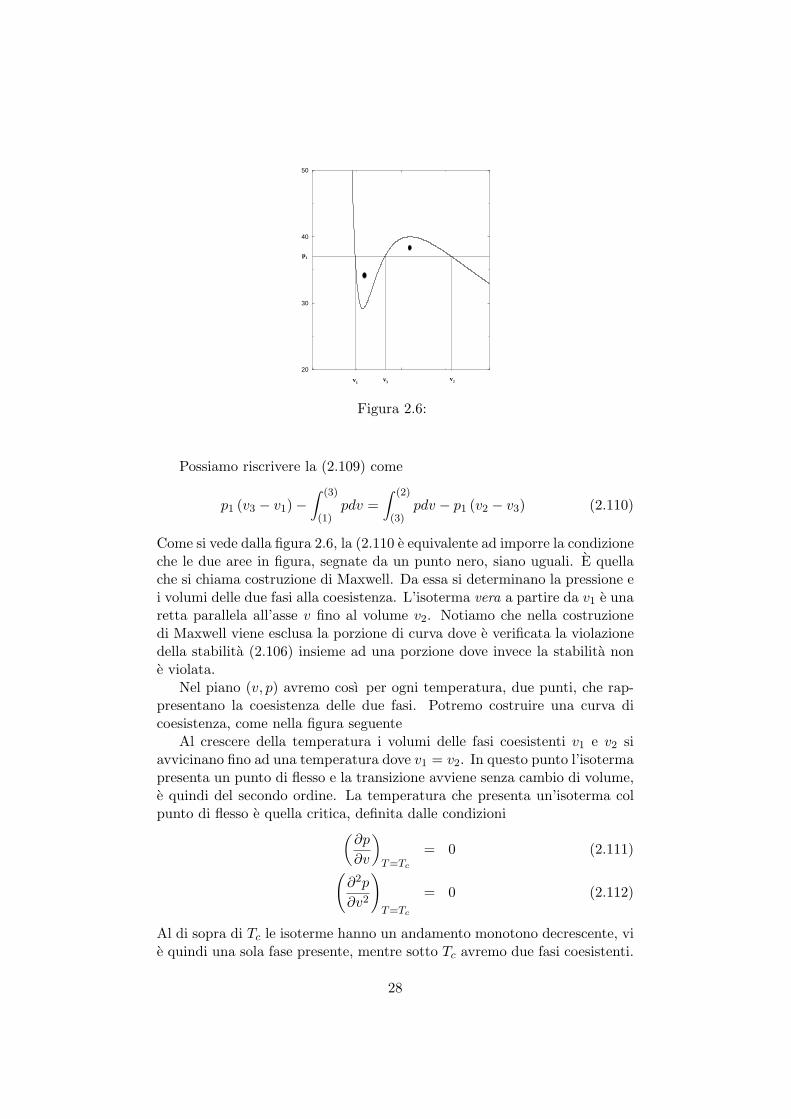
Teniamo conto che le due pressioni devono essere uguali p1 = p2 e introdu-ciamo il volume v3, tale che v1 < v3 < v2 come nella figura 2.6
27
20
30
40
50
p1
v1v3
v2
Figura 2.6:
Possiamo riscrivere la (2.109) come
p1 (v3 − v1)−∫ (3)
(1)pdv =
∫ (2)
(3)pdv − p1 (v2 − v3) (2.110)
Come si vede dalla figura 2.6, la (2.110 e equivalente ad imporre la condizioneche le due aree in figura, segnate da un punto nero, siano uguali. E quellache si chiama costruzione di Maxwell. Da essa si determinano la pressione ei volumi delle due fasi alla coesistenza. L’isoterma vera a partire da v1 e unaretta parallela all’asse v fino al volume v2. Notiamo che nella costruzionedi Maxwell viene esclusa la porzione di curva dove e verificata la violazionedella stabilita (2.106) insieme ad una porzione dove invece la stabilita none violata.
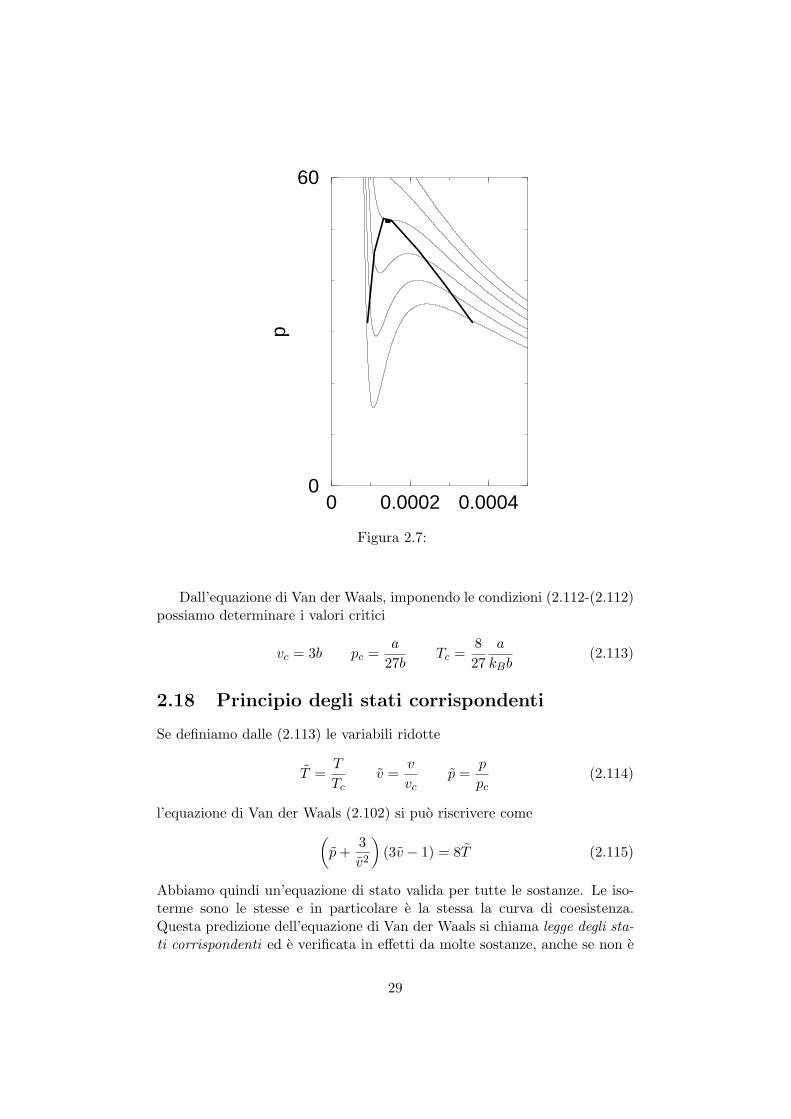
Nel piano (v, p) avremo cosı per ogni temperatura, due punti, che rap-presentano la coesistenza delle due fasi. Potremo costruire una curva dicoesistenza, come nella figura seguente
Al crescere della temperatura i volumi delle fasi coesistenti v1 e v2 siavvicinano fino ad una temperatura dove v1 = v2. In questo punto l’isotermapresenta un punto di flesso e la transizione avviene senza cambio di volume,e quindi del secondo ordine. La temperatura che presenta un’isoterma colpunto di flesso e quella critica, definita dalle condizioni
(∂p
∂v
)
T=Tc
= 0 (2.111)(
∂2p
∂v2
)
T=Tc
= 0 (2.112)
Al di sopra di Tc le isoterme hanno un andamento monotono decrescente, vie quindi una sola fase presente, mentre sotto Tc avremo due fasi coesistenti.
28
0 0.0002 0.00040
60
p
Figura 2.7:
Dall’equazione di Van der Waals, imponendo le condizioni (2.112-(2.112)possiamo determinare i valori critici
vc = 3b pc =a
27bTc =
827
a
kBb(2.113)
2.18 Principio degli stati corrispondenti
Se definiamo dalle (2.113) le variabili ridotte
T =T
Tcv =
v
vcp =
p
pc(2.114)
l’equazione di Van der Waals (2.102) si puo riscrivere come(
p +3v2
)(3v − 1) = 8T (2.115)
Abbiamo quindi un’equazione di stato valida per tutte le sostanze. Le iso-terme sono le stesse e in particolare e la stessa la curva di coesistenza.Questa predizione dell’equazione di Van der Waals si chiama legge degli sta-ti corrispondenti ed e verificata in effetti da molte sostanze, anche se non e
29

riprodotta esattamente dall’equazione (2.115). Se si rappresentano i puntidi coesistenza liquido-gas per diverse sostanze nel piano T/Tc vs. ρ/ρc, ipunti collassano tutti sulla stessa curva. Questa curva e diversa, soprattut-to vicino al punto critico, da quella di Van der Waals, pero la predizione diuniversalita di comportamento e verificata.
30

Capitolo 3
Richiami di MeccanicaStatistica
3.1 Teoria degli ensembles
Ad un macrostato con definiti N ,V ed energia interna E corrispondono unenorme numero di microstati a livello microscopico. La meccanica statisticasi occupa di calcolare le proprieta macroscopiche a partire dagli stati micro-scopici di un sistema, attraverso delle operazioni di media sulle fluttuazioni,che hanno luogo a livello microscopico.
Nelle misure sperimentali quello che osserviamo sono medie su tempimolto piu lunghi dei tempi atomici. Se A(t) e un generico operatore, definitosulle variabili dinamiche microscopiche, associato ad un osservabile, essoevolve nel tempo in accordo alle leggi di Newton. Una misura di A sara unamedia temporale del tipo
A = limt→∞
1t
∫ t
0dt′A
(t′
)(3.1)
Per calcolare queste medie in meccanica statistica si realizzano dellecopie mentali del sistema, equivalenti perche corrispondono allo stesso statomacroscopico. Si assume che tutti i microstati siano ugualmente probabili,e quello che si chiama: postulato della probabilita uguale a priori. Questoinsieme di copie mentali viene chiamato ensemble e la media temporale (3.1)viene rimpiazzata da una media sull’ensemble.
〈A〉 = A (3.2)
Un sistema classico in tre dimensioni e caratterizzato da 3N coordinate(q1, . . . , q3N ) e 3N impulsi (p1, . . . , p3N ). Nello spazio delle fasi a 6N di-mensioni un punto rappresenta uno stato microscopico. Il punto evolve inbase alle equazioni di Hamilton. Un ensemble e un insieme di questi punti,caratterizzati dal fatto che corrispondono ad uno stesso stato macroscopico.
31

Si evolveranno in un volume limitato e con un valore fissato dell’energia (seH e l’hamiltoniana)
H(q1, . . . , q3N , p1, . . . , p3N ) = E (3.3)
Per descrivere l’evoluzione nello spazio delle fasi, si introduce la funzio-ne densita ρ
(qN ,pN , t
), dove qN = (q1, . . . , q3N ) e pN = (p1, . . . , p3N ),
normalizzata in modo che∫
dqNdpNρ(qN ,pN , t
)= 1 (3.4)
essa descrive ad ogni istante la densita di punti nello spazio delle fasi equindi il modo in cui sono distribuiti i membri dell’ensemble. La densita diprobabilita soddisfa all’equazione di evoluzione temporale di Liouville
∂ρ
∂t= −iLρ
(qN ,pN , t
)(3.5)
dove L e l’operatore di Liouville, che agisce su una funzione f come leparentesi di Poisson
Lf = f, H =∑
i
(qi
∂f
∂qi+ pi
∂f
∂pi
)(3.6)
Le distribuzioni che ci interessano sono quelle di equilibrio per le quali
∂ρ
∂t= 0
La densita di probabilita dovra quindi essere un funzionale dell’hamiltonianaρ = ρ [H]. Con queste distribuzioni potremo calcolare le medie di equilibriosull’ensemble. Se α = (qN ,pN ) e un punto nello spazio delle fasi
〈A〉 =∑α
ρens [H(α)]A (α) (3.7)
Scegliendo diverse distribuzioni, avremo differenti tipi di ensemble.
3.2 Ensemble microcanonico e legame con la ter-modinamica
Con N,V, E fissati dobbiamo lavorare nell’ensemble microcanonico. Il pesostatistico wmic (H) dell’ensemble e definito da
wmic (N, V, E) = δ (E −H) (3.8)
La probabilita all’equilibrio sara
ρmic (H) =wmic
Zmic(3.9)
32

qui e stata introdotta la funzione di partizione che normalizza la ρmic
Zmic (N,V, E) =∑α
δ (E −H (α)) (3.10)
Il legame con la termodinamica e dato dall’entropia che risulta
S(N, V,E)kB
= lnZmic (3.11)
in accordo con l’idea di Boltzmann.
3.3 Vari tipi di ensemble
Ogni ensemble e caratterizzato da una densita di probabilita all’ equilibrioρens (H)
ρens (H) =wens (H)
Zens(3.12)
dove e wens (H) e il peso statistico mentre la funzione di partizione
Zens =∑α
wens (H) (3.13)
serve a soddisfare la condizione di normalizzazione (3.4). La media di unosservabile sull’ensemble e data dalla (3.7).
Il legame con la termodinamica si ottiene dalla funzione di partizione(3.13), per ogni tipo di ensemble abbiamo associato un potenziale termo-dinamico. Ogni densita (3.12) e determinata da fissate variabili esternex1, x2, . . ., dalle quali dipendera il relativo potenziale termodinamico (3.14)
Ψens (x1, x2, . . .) = − ln Zens (x1, x2, . . .) (3.14)
Per compiere una trasformazione da un ensemble all’altro si possonousare le trasformate di Legendre. Se, per esempio, vogliamo sostituire lavariabile x1 con la coniugata y1
y1 =(
∂Ψens
∂x1
)
x2,...(3.15)
il nuovo ensemble e definito da un peso statistico
w′ens = ex1y1wens (3.16)
con una funzione di partizione
Z ′ens =∫
dx1 ex1y1 Zens (3.17)
33

3.3.1 Canonico
Se vogliamo sostituire l’energia E dobbiamo considerare che il sistema sia incontatto con un bagno termico, che garantisce una temperatura costante. Inbase alla (3.14) e alla (3.15), abbiamo che la quantita coniugata all’energiae (
∂ (−S/kB)∂E
)
V= − 1
kB
(∂S
∂E
)
V= − 1
kBT= −β (3.18)
Otteniamo l’ensemble canonico con la funzione di partizione
Zcan =∫
dEe−βE∫
dΓδ(E −H) =∫
dΓe−βH (3.19)
Il potenziale termodinamico nel nuovo ensemble caratterizzato dalle va-riabili fissate N , V e T o β sara l’energia libera di Helmholtz
− S
kB+ βE = −βTS + βE = βA (3.20)
dalla (3.14) abbiamo quindi
βA(N, V, T ) = − ln Zcan(N, V, T ) (3.21)
3.3.2 Gran-canonico
A partire dal canonico, vogliamo rilasciare il vincolo del numero costantedi particelle. Il sistema e in contatto con un bagno termico che consente ditenere fissa la temperatura e il potenziale chimico e si puo avere scambio diparticelle. Quindi dobbiamo passare da (N, V, T ) a (µ, V, T ). Abbiamo
(∂βA
∂N
)
T,V= βµ (3.22)
e quindi la funzione di partizione diventa
ZGC =∞∑
N=0
eβµNZcan(N,V, T ) (3.23)
Il potenziale termodinamico associato
βΩ(µ, V, T ) = − ln ZGC(µ, V, T ) (3.24)
e dato daβΩ = βA− βµN = −βPV (3.25)
34

3.3.3 Isobarico
Se vogliamo lavorare a temperatura e pressione costante, dobbiamo lasciarefluttuare il volume, quindi si passa da (N, V, T ) a (N, p, T ); si vede facilmenteche la funzione di partizione sara
Zisob =∫
dV e−βpV Zcan(N, V, T ) (3.26)
e il potenziale termodinamico e l’energia libera di Gibbs
βG = βA + βPV (3.27)
3.4 Sviluppi delle formule per i sistemi classici
Nel seguito ci interessiamo di sistemi di particelle classiche e quindi ci con-centriamo sull’applicazione del metodo degli ensembles a un sistema classicocaratterizzato da una hamiltoniana
H =N∑
i=1
p2i
2m+ U (r1, . . . , rN) (3.28)
3.4.1 Ensemble Canonico
Possiamo scrivere la funzione di partizione canonica per un sistema classicoQN (V, T ) come
QN (V, T ) =1
N ! h3N
∫ ∫drNdpN exp
[−βH
(pN , rN
)](3.29)
Possiamo eseguire esattamente l’integrale sugli impulsi e abbiamo
QN (V, T ) =1
N ! Λ3NZN (V, T ) (3.30)
dove Λ e la lunghezza d’onda termica di De Broglie
Λ =
√h2
2πmkBT(3.31)
e abbiamo definito l’integrale configurazionale ZN (V, T )
ZN (V, T ) =∫
drNe−βU(r1,...,rN) (3.32)
Per il gas ideale la (3.32) diventa ZN (V, T )=V N e quindi dalla (3.30)
QidN (V, T ) =
V N
N ! Λ3N(3.33)
35

La funzione di partizione (3.30) si puo riscrivere come
QN (V, T ) = QidN (V, T )Qexc
N (V, T ) (3.34)
dove la funzione di partizione di eccesso e
QexcN (V, T ) =
ZN (V, T )V N
(3.35)
L’energia libera si puo dividere quindi in due parti: ideale e di eccesso
A = Aid + Aexc (3.36)
La parte ideale dell’energia libera e data da
βAid = − ln
[V N
N ! Λ3N
]≈ N [ln ρ + 3 ln Λ− 1] (3.37)
dove ρ e la densita ρ=N/V ; la parte di eccesso e invece
βAexc = − lnZN (V, T )
V N(3.38)
essa contiene il contributo che viene dall’interazione fra le particelle.La funzione densita di probabilita del canonico
ρcanN
(rN ,pN ) =1
N ! h3N
exp[−βH
(pN , rN
)]
QN (V, T )(3.39)
si puo a sua volta fattorizzare in una parte relativa agli impulsi
ρimp
N(pN ) =
Λ3N
h3Nexp
[−β
∑
i
p2i
2m
](3.40)
e una funzione di distribuzione configurazionale data da
ρN
(rN ) =exp [−βU (r1, . . . , rN )]
ZN (V, T )(3.41)
3.4.2 Ensemble gran-canonico
La funzione di partizione del gran-canonico si puo scrivere come
ZGC (µ, V, T ) =∞∑
N=0
eβµNQN (V, T ) (3.42)
con Q0 = 1La densita di probabilita e data da
36

ρGC(rN ,pN ) =1
N ! h3N
exp [βµN ] exp[−βH
(pN , rN
)]
ZGC(µ, V, T )(3.43)
Per un sistema classico con hamiltoniana (3.28)
ZGC (µ, V, T ) =∞∑
N=0
zN
N !ZN (V, T ) (3.44)
dove abbiamo definito l’attivita
z =eβµ
Λ3(3.45)
Possiamo quindi definire una densita di probabilita configurazionale grancanonica
ρGC(rN ) =1
ZGC
zN
N !exp
[−βU(rN )
](3.46)
Il valor medio di un operatore A(rN ) che dipende solo dalle coordinatesara dato da
〈A〉 =1
ZGC
∞∑
N=0
zN
N !
∫drNA(rN ) exp
[−βU(rN )
](3.47)
Poiche il numero di particelle fluttua e importante calcolare il valoremedio < N >. Dato che N non dipende dalle (rN ) dalla 3.47 abbiamo
〈N〉 =1
ZGC
∞∑
N=0
zN
N !NZN (V, T ) (3.48)
da cui
〈N〉 =z
ZGC
∞∑
N=0
zN−1
N !NZN (V, T )
e quindi
〈N〉 = z∂
∂zln ZGC (3.49)
Ricordando che−βpV = − ln ZGC (3.50)
si vede che combinando la (3.50) con la (3.49) per eliminare la z fra le due sipuo ottenere l’equazione di stato che collega pressione, volume, temperaturae numero di particelle. Per un gas ideale dalla (3.44) abbiamo
ZidGC (µ, V, T ) =
∞∑
N=0
zN
N !V N = exp (zV ) (3.51)
37

quindi−βpV = − ln Zid
GC = −zV (3.52)
d’altra parte dalla (3.49)
〈N〉 = z∂
∂z(zV ) = zV (3.53)
eliminando la z fra la (3.52) e la (3.53) si ottiene l’equazione dei gas perfetti.
3.4.3 Fluttuazioni del numero di particelle
Nell’ensemble gran-canonico si puo ricavare una relazione molto importan-te che collega la fluttuazione microscopica del numero di particelle con lacompressibilita isoterma.
Partiamo dal calcolo dello scarto quadratico medio
〈∆N2〉 = 〈N2〉 − 〈N〉2 (3.54)
Dalla formula (3.48) si vede che
〈∆N2〉 =1
ZGC
∞∑
N=0
zN
N !N2ZN (V, T )−
(1
ZGC
∞∑
N=0
zN
N !NZN (V, T )
)2
(3.55)
Da questa si ricava con pochi passaggi la relazione
〈∆N2〉 = z
(∂〈N〉∂z
)
T,V(3.56)
Ricordando la definizione di z (3.45) dato che
∂z
∂(βµ)= z
la (3.56) diventa
〈∆N2〉 = z
(∂〈N〉∂z
)
T,V
=∂z
∂(βµ)
(∂〈N〉∂z
)
T,V
= kBT
(∂〈N〉∂µ
)
T,V
(3.57)
La derivata termodinamica che appare al secondo membro della (3.57) sipuo collegare alla compressibilita isoterma
(∂〈N〉∂µ
)
T,V
= V
(∂ρ
∂µ
)
T
= V
(∂ρ
∂p
)
T
(∂p
∂µ
)
T
= V ρ
(∂ρ
∂p
)
T
(3.58)
38

dove abbiamo usato(
∂p
∂µ
)
T
= ρ
La compressibilita isoterma (2.88) si puo scrivere anche come
KT = − 1V
(∂V
∂p
)
T
=1ρ
(∂ρ
∂p
)
T
(3.59)
introdotta la (3.59) nella (3.56), la (3.55) ci porta alla relazione
〈∆N2〉 = kBT〈N〉2V
KT (3.60)
La (3.58) e molto importante perche ci fa vedere chiaramente il collegamentofra le fluttuazioni a livello microscopico descritte dalla meccanica statisti-ca e la termodinamica che appare in una quantita come la compressibilitaisoterma misurabile macroscopicamente. Come abbiamo visto la KT deveessere sempre positiva e ci fornisce l’indicazione della stabilita del sistema,che quindi viene collegata alle fluttuazioni microscopiche del numero di par-ticelle. Questa quantita e equivalente nel gran-canonico alle fluttuazionidella densita.
〈∆N2〉〈N〉2 =
kBT
VKT
statistica termodinamica (3.61)
Quando la compressibilita diverge, come accade al punto critico, dalla(3.58) vediamo che questo comporta che anche le fluttuazioni microscopichediventano enormi. La transizione di fase e associata a forti fenomeni difluttuazione della densita nel sistema fluido.
39





























