Embed Size (px)
Citation preview

Proceedings of the 2015 ASEE Gulf-Southwest Annual Conference
Organized by The University of Texas at San Antonio
Copyright © 2015, American Society for Engineering Education
A Bio-network Based Pathway Extension Approach for Cancer Prognosis
Yaohang Yang, Shichen Zhao, Cunzhi Zhao, Chengwei Lei
Computer Science Department
McNeese State University
[email protected], [email protected],
[email protected], [email protected]
Abstract
There are 1,660,290 new cancer cases and 580,350 deaths of cancer occurred in the United States
in 2013. With a better and earlier prognosis of cancer, thousands of people’s lives can be saved
each year. Recently, a lot of studies have been done towards estimating cancer prognosis based on
pathway information, which improve the understanding of cancer at a systemic level. However,
the small size of genes in each pathway limits the performance of many classification algorithms
and further analysis of cancer.
In this paper, we introduce an approach to extend the cancer pathway based on biology network
topology analysis. With further research on the new extended pathway generated by our method,
we found that the newly added genes are highly correlated to the target cancer, which means the
accuracy of cancer prognosis will have a significant improvement. To test the performance, we
applied our method to the prostate cancer related pathway, and verified our output genes with
NCBI PubMed database. The results showed that our approach significantly improve the size of
pathway with very limited false positive genes involved.
Introduction
Nowadays, people’s understanding of cancer have enlarged by many researches, however, it is still
considered as one of incurable and deadly diseases in this decade (Jemal A, et al., 2011).
Particularly, the mortality of cancer in developing countries is much more than developed
countries and cancer patients in developed countries live longer than those in developing countries
(Jemal A, et al., 2011).
In recent years, the morbidity of prostate cancer (PCa) is rising in the world. In terms of clinical
practice, the traditional treatment effects for hormone refractory prostate cancer are often
unsatisfactory. With the development of microscopic techniques such as molecular biology, gene
level research gradually becomes the current hot spot. Cancer researchers are dedicated to find
right treatment, and they think cancer pathway expansion is one of the breaking points. By
detecting and localizing prostate tumors at their early stage, patients’ lives can be significantly
prolonged, which mainly attributes to prognosis of cancer pathway. Prostate cancer pathway is
based on biological network, which is widely used in biological research. Biological network is a
system that can be linked with any networks of biologic nodes and edges, which are the basic
components of a network, such as protein-protein interaction (PPI) networks (Proulx, S.R. et al.,
2005). However, previous researches were not cogent enough, because many genes and pathways
were excluded.

Proceedings of the 2015 ASEE Gulf-Southwest Annual Conference
Organized by The University of Texas at San Antonio
Copyright © 2015, American Society for Engineering Education
The higher accuracy of the cancer prognosis is, the more people’s lives will be saved from the
cancer. Using both cancer pathway extension method and biological networks can improve the
accuracy of prostate cancer prognosis. Meanwhile, new biomarker genes will be generated after
applying our method and algorithm. A biomarker is a substance that indicates a particular process,
and used in many biological field (Zimmer, Carl, 2015). The goal is to improve the accuracy of
cancer prognosis, eliminate unrelated genes, and minimize the error. In this article, we present how
our method reaches the goal and how the result verifies the expectation.
Methods
Given a biology pathway including K proteins, and a Protein-Protein correlation matrix S which
was generated from the biology network (Lei. C. and Ruan. J., 2013). Let S = k x n, where n is the
dimension of the core data set. Secondly, let Rn = ∑ 𝑆𝑖 ∗ 𝑛𝑘1 , which is an one by n matrix that
contains all the relations between pathways and target proteins. In order to find most correlative
proteins to the pathway, those proteins that already exist in the pathway have to be excluded.
Finally, we sorted all the genes based on their total correlation to the entire pathway, and pick the
most related genes as the candidates for the pathway extension. It is important to know that the
number of proteins in pathways differs from one to hundreds, so finding a suitable cutoff value
also has to be considered.
To test the performance of our approach, we first obtained the extended pathway genes based on
the biological network information. After that, we did a literature research of each gene in the
NCBI PubMed database. By searching the candidate gene name and cancer name together in
Title/Abstract, we could use the published paper numbers as the general evaluation of the
performance.
Experiment
In our experiment, we used the KEGG PATHWAY Database (KEGG Database) to test the
performance. The program pseudo code is attached in appendix. The gene relationship is
represented by a 9205 by 9205 matrix, which was generated from the Human PPI Network. We
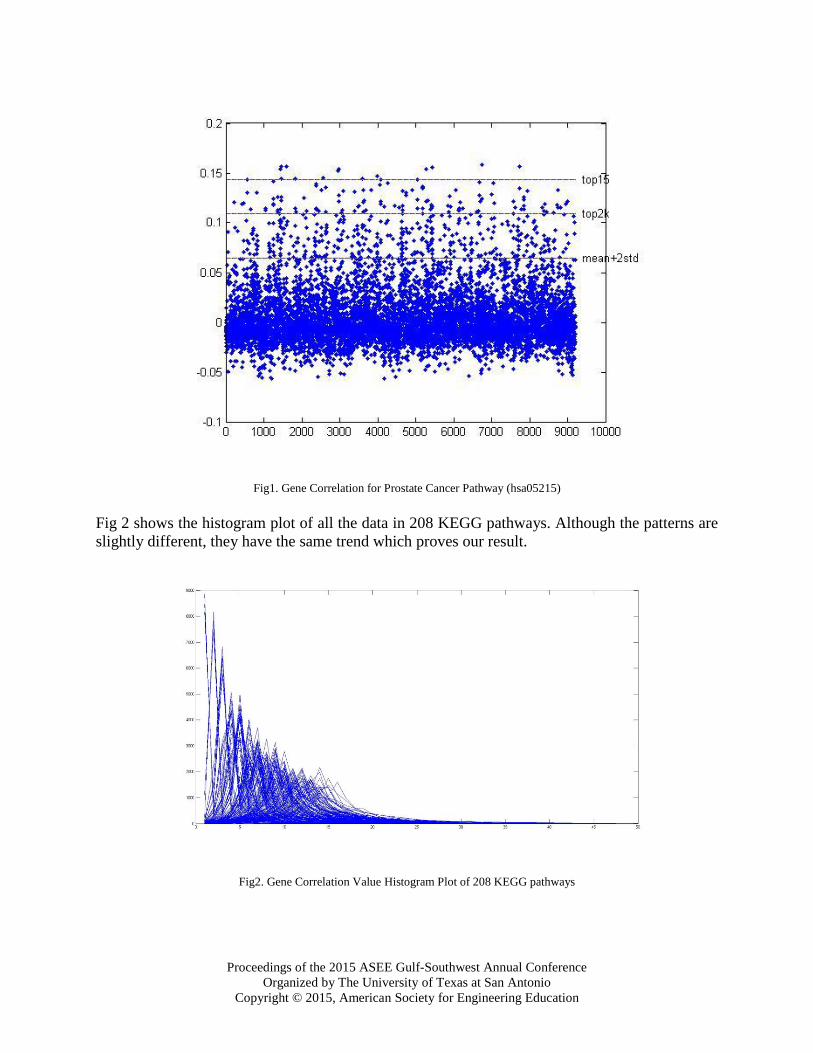
applied three cutoff strategies based on visualizing, doubling the length of the pathway, and
calculating statistically. First strategy is picking the top15 genes with the highest values. The
second cutoff value was set up as top2k where k is the number of proteins in the pathway, because
different pathways have different number of proteins. With the new critical value, the range of the
pathway is doubled. Lastly, we used the statistical method as our third cutoff value. Let c stand for
the critical value, then c =∑ 𝑆𝑖∗𝑛𝑘1
𝑘+ 2 ∗ 𝑆𝑇𝐷(
∑ 𝑆𝑖∗𝑛𝑘1
𝑘). By using this method, the result is twice the
value of standard deviation away from the mean. All three implementations are shown in Fig 1.
Proceedings of the 2015 ASEE Gulf-Southwest Annual Conference
Organized by The University of Texas at San Antonio
Copyright © 2015, American Society for Engineering Education
Fig1. Gene Correlation for Prostate Cancer Pathway (hsa05215)
Fig 2 shows the histogram plot of all the data in 208 KEGG pathways. Although the patterns are
slightly different, they have the same trend which proves our result.
Fig2. Gene Correlation Value Histogram Plot of 208 KEGG pathways

Proceedings of the 2015 ASEE Gulf-Southwest Annual Conference
Organized by The University of Texas at San Antonio
Copyright © 2015, American Society for Engineering Education
Based on NCBI PubMed database, the number of articles based on each gene was found. We
picked the top 50 genes in the extended prostate cancer pathway and plotted the number of related
articles found in the database.
Fig3. Number of Related Publications for the Top 50 Extended Genes
Rank 1 2 3 4 5 6 7 8
Name 'CEBPD' 'SPIB' 'NR3C1' 'FOXM1' 'FOS' 'IRF1' 'CEBPB' 'HMGA2'
Publications 6 1 4 >20 >20 4 4 11
Rank 9 10 11 12 13 14 15 Average
Name 'ATF2' 'MYBL2' 'JUN' 'KLF5' 'RUNX2' 'JDP2' 'E2F5'
Publications 11 7 >20 16 >20 1 5 10
Table1. Top 15 Genes Names in Extended Pathway and the Number of Related Publications
0
2
4
6
8
10
12
14
16
18
20
1 2 3 4 5 6 7 8 9 1011121314151617181920212223242526272829303132333435363738394041424344454647484950
Nu
mb
er
of
Art
icle
s
Genes

Proceedings of the 2015 ASEE Gulf-Southwest Annual Conference
Organized by The University of Texas at San Antonio
Copyright © 2015, American Society for Engineering Education
Discussion
In addition, with a further look on the top 5 ranked genes relevant to prostate cancer pathway, it is
interesting that 4 out of 5 of them have vital role in prostate cancer formation and development.
CEBPD is often known as involving in regulation of apoptosis and cell proliferation, which acts
as tumor suppressor (Gery, S., et al., 2005). Chuang CH, et al. also affirmed that CEBPD showed
an additive effect in triggering the apoptotic pathway and enhancing apoptosis in PrCa cells
(Chuang CH, et al., 2014).
SPIB is member of a subfamily of transcription factors. SPIB was found to affect binding by
SNP344, which existed in a large cohort of healthy individuals and among patients suffering from
ovarian, breast, endometrial and prostate cancer, Knappskog S, et al. detected no differences with
respect to SNP344 distribution between healthy individuals and cancer patients. (Knappskog S, et
al., 2012).
NR3C1 (nuclear receptor subfamily 3, group C, member 1) is also known as the glucocorticoid
receptor (GR, or GCR), is the receptor to which cortisol and other glucocorticoids bind. Isikbay M
et al verified GR activation can contribute to resistance to prostate cancer androgen receptor-
directed therapy (Isikbay, M., et al., 2014).
FOXM1 is proved to have the potential as a target for cancer therapies and diagnosis (Laoukili, J.,
et al., 2005). The study of Cheng XH et al revealed FOXM1 oncogene and demonstrated that this
crosstalk is required for tumor cell proliferation during progression of prostate cancer in vivo
(Cheng, XH., et al., 2014).
The information states a strong relevance between the extended genes and prostate cancer. This
can help researchers to further understand prostate cancer, and help biology scientist to target the
tumors precisely.
Conclusions
Our method was successfully applied and a solid result was obtained. Results showed that our
method can predict high quality candidate genes by computational method. It was firmly proved
by highly related publications found in NCBI PubMed database. The extended pathway could
enlarge the candidate genes for cancer detection, and increases the performance of the cancer
treatment. As shown in the example, the extended genes provided a solid correlation to prostate
cancer, which is also proved by other wet lab experiments. The high accuracy of the method will
significantly improve the prognosis of the cancer, and lower the risk of mistakes, which will save
thousands of people’s lives. With extended pathways, it also can help the biologist to understand
the cancer in a systematic way, and discover the biological principal behind the disease.

Proceedings of the 2015 ASEE Gulf-Southwest Annual Conference
Organized by The University of Texas at San Antonio
Copyright © 2015, American Society for Engineering Education
References
1. C. Lei. and Ruan. J., 2013, A novel link prediction algorithm for reconstructing protein-
protein interaction networks by topological similarity, Bioinformatics, 29(3): 355-364.
2. Chuang CH, Wang WJ, Li CF, Ko CY, Chou YH, Chuu CP, Cheng TL, Wang JM
(2014). The combination of the prodrugs perforin-CEBPD and perforin-granzyme B
efficiently enhances the activation of caspase signaling and kills prostate cancer. Cell
Death Dis. doi: 10.1038/cddis.2014.106.
3. Cheng, XH, Black, M, Ustiyan, V, Le, T, Fulford, L, Sridharan, A, Medvedovic, M,
Kalinichenko, VV, Whitsett, JA, Kalin, TV. (2014). SPDEF inhibits prostate
carcinogenesis by disrupting a positive feedback loop in regulation of the Foxm1
oncogene. PLoS Genet. doi: 10.1371/journal.pgen.1004656.
4. Gery, S., Tanosaki, S., Hofmann, W., Koppel, A., & Koeffler, H. P. (2005). C/EBP delta
expression in a BCR-ABL-positive cell line induces growth arrest and myeloid
differentiation. Oncogene. doi:10.1038/sj.onc.1208393.
5. Isikbay, M., Otto, K., Kregel, S., Kach, J., Cai, Y., Vander Griend, DJ., Conzen, SD.,
Szmulewitz, RZ.. (2014). Glucocorticoid receptor activity contributes to resistance to
androgen-targeted therapy in prostate cancer. Horm Cancer. doi: 10.1007/s12672-014-
0173-2.
6. Jemal A, Bray, F, Center, MM, Ferlay, J, Ward, E, Forman, D (2011). "Global cancer
statistics". CA: a cancer journal for clinicians 61 (2): 69–90. doi:10.3322/caac.20107.
7. Knappskog S, Gansmo LB, Romundstad P, Bjørnslett M, Trovik J, Sommerfelt-Pettersen
J, Løkkevik E; Norwegian Breast Cancer Group trial NBCG VI, Tollenaar RA, Seynaeve
C, Devilee P, Salvesen HB, Dørum A, Hveem K, Vatten L, Lønning PE. (2012). MDM2
promoter SNP344T>A (rs1196333) status does not affect cancer risk. PLoS One. doi:
10.1371/journal.pone.0036263.
8. Laoukili, J., Kooistra, M. R., Brás, A., Kauw, J., Kerkhoven, R. M., Morrison, A.,
Medema, R. H. (2005). FoxM1 is required for execution of the mitotic programme and
chromosome stability. Nature Cell Biology. doi:10.1038/ncb1217.
9. Proulx, S.R. et al. (2005). "Network thinking in ecology and evolution". Trends in
Ecology and Evolution 20 (6): 345–353. doi:10.1016/j.tree.2005.04.004.
10. Zimmer, Carl (January 22, 2015). "Even Elusive Animals Leave DNA, and Clues,
Behind". New York Times. Retrieved January 23, 2015.

Proceedings of the 2015 ASEE Gulf-Southwest Annual Conference
Organized by The University of Texas at San Antonio
Copyright © 2015, American Society for Engineering Education
Appendix
Pseudo code
Input: pathways (M by N), fullcorr (M1 by N1)_
for i = 1:M
for j = 1:M1
ind_found = all indexes that found in names
end
for k = 1:length(ind_found)
corr = fullcorr(ind_found(k))
end
corr = mean (corr)
for k1 <= 10
C = maxvalue; IDNmax = index of max value
make C = min (corr)
if INDmax and index_found do not deplicate
result = [ result INDmax]
k1 = k1 +1
end
end
end





























