Embed Size (px)
Citation preview

Hydrolethalus Syndrome
Hydrolethalus syndrome was coined by Salonen et al.
in 1981 in patients with severe CNS malformations,
hydrocephalus, micrognathia, cleft lip/palate, lung
hypoplasia club foot, and polydactyly, distinguishable
from the Meckel syndrome by the absence of polycys-
tic kidneys (Salonen et al. 1981). The majority of cases
were reported from Finland.
Genetics/Basic Defects
1. Inheritance: autosomal recessive
2. Hydrolethalus syndrome locus: assigned to 11q23-
q25 in Finnish families
3. HYLS1 gene mutation
a. Responsible for hydrolethalus syndrome in the
Finnish population, HYLS1
b. c.632A > G (D211G) mutation: the common
mutation carried in the Finnish population
Clinical Features
1. A lethal condition: stillborn or died within a few
hours of birth in majority of cases
2. Hydramnios
3. Severe midline CNS malformations
a. A midline cerebral cleft: more common
b. Absence of the corpus callosum
c. Absence of septum pellucidum
d. Hydrocephalus secondary to aqueductal
stenosis
e. Dorsal midline defect of the foramen magnum,
forming a keyhole-shaped opening
(occipitoschisis)
f. Cerebellar heterotopias
g. Brain stem malformations
h. Cerebral gyral abnormalities
i. Absent olfactory lobes
j. Hypothalamic hamartomas
k. Dandy-Walker malformation
4. Craniofacial features
a. Microphthalmia
b. Nasal anomalies
c. Small mandible
d. Cleft lip/palate
e. Small tongue
f. Low-set, malformed ears
5. Pulmonary anomalies
a. Abnormal larynx, trachea, and bronchi
b. Pulmonary hypoplasia/agenesis
c. Defective lung lobulation
6. Congenital heart defects
a. Ventricular septal defect
b. Open foramen ovale
c. Common atrioventricular canal
7. Renal anomalies
8. Duplex/bicornis uterus
9. A typical “keyhole” occipital bone defect
10. Polydactyly
a. Hands: always postaxial
b. Feet: almost always preaxial
11. Differential diagnosis
a. Acrocallosal syndrome
i. Macrocephaly
ii. Craniofacial anomalies
iii. Hallux duplication
iv. Postaxial polydactyly
v. Absence of corpus callosum
vi. Mental retardation
H. Chen, Atlas of Genetic Diagnosis and Counseling, DOI 10.1007/978-1-4614-1037-9_122,# Springer Science+Business Media, LLC 2012
1081

b. Smith-Lemli-Opitz syndrome (severe form)
i. Hydrocephalus
ii. Cerebellar hypoplasia
iii. Cardiac anomalies
iv. Genital anomalies
v. Polydactyly
c. Orofacialdigital syndrome, type VI
i. Cerebellar anomalies
ii. Hypertelorism
iii. Micrognathia
iv. Midline cleft lip
v. Cleft palate
vi. A small tongue with lingual nodules and
multiple frenula
vii. Laryngeal anomalies
viii. Cardiac anomalies
ix. Preaxial polysyndactyly of hands and feet
x. Postaxial polysyndactyly of the feet
d. Pallister-Hall syndrome
i. Hypertelorism
ii. Micrognathia
iii. Midline cleft lip
iv. Cleft palate
v. A small tongue with lingual nodules and
multiple frenula
vi. Laryngeal anomalies
vii. Lung segmental anomalies
viii. Cardiac anomalies
ix. Postaxial polysyndactyly of the feet
e. Walker-Warburg syndrome
i. Overlapping features
a) Encephalocele
b) Agenesis of midline brain structures
c) Hydrocephalus
ii. Features usually not seen
a) Eye abnormalities
b) Polydactyly
Diagnostic Investigations
1. Radiologic studies
a. Skull
i. Microcephaly
ii. Occipital bone defect continuous with fora-
men magnum
iii. A keyhole shaped defect at the base of skull
b. Digital anomalies
i. Postaxial polydactyly of the hands
ii. Preaxial polydactyly or hallux duplication of
the feet
2. Ultrasound or MRI of the brain for delineation of
brain anomalies
3. Echocardiogram for delineation of congenital heart
defects
4. Molecular genetic study: target mutation analysis of
HYLS1 gene (c.632A > G)
Genetic Counseling
1. Recurrence risk
a. Patient’s sib
i. Recurrence risk: 25%
ii. Unaffected sibs of a proband: two thirds
chance of being heterozygotes
b. Patient’s offspring: a lethal entity not surviving
to reproduction
2. Prenatal diagnosis
a. Possible by prenatal ultrasonography in at-risk
families
b. Possible for pregnancies at risk when the dis-
ease-causing mutation in the family is known
3. Management
a. Supporting care
b. No treatment available for the underlying lethal
disorder
References
Adetoro, O. O., Komolafe, F., & Anjorin, A. (1984).
Hydrolethalus syndrome in consecutive African siblings.
Pediatric Radiology, 14, 422–424.Ammala, P., & Salonen, R. (1995). First-trimester diagnosis of
hydrolethalus syndrome. Ultrasound in Obstetrics &Gynecology, 5, 60–62.
Anyane-Yeboa, K., Collins, M., Kupsky, W., et al. (1987).
Hydrolethalus (Salonen-Herva-Norio) syndrome: Further
clinicopathological delineation. American Journal ofMedical Genetics, 26, 899–907.
Aughton, D. (1994). Sonographic detection of hydrolethalus
syndrome. Journal of Clinical Ultrasound, 22, 286–287.Aughton, D. J., & Cassidy, S. B. (1987). Hydrolethalus syn-
drome: Report of an apparent mild case, literature review,
and differential diagnosis. American Journal of MedicalGenetics, 27, 935–942.
Bachman, H., Clark, R. D., & Salahi, W. (1990).
Holoprosencephaly and polydactyly: A possible expression
of the hydrolethalus syndrome. Journal of Medical Genetics,27, 50–52.
1082 Hydrolethalus Syndrome

Chan, B. C., Shek, T. W., & Lee, C. P. (2004). First-trimester
diagnosis of hydrolethalus syndrome in a Chinese family.
Prenatal Diagnosis, 24, 587–590.Christensen, B., Blaas, H. G., Isaksen, C. V., et al. (2000). Sibs
with anencephaly, anophthalmia, clefts, omphalocele, and
polydactyly: Hydrolethalus or acrocallosal syndrome? Amer-ican Journal of Medical Genetics, 91, 231–234.
de Ravel, T. J., van der Griendt, M. C., Evan, P., et al. (1999).
Hydrolethalus syndrome in a non-Finnish family: Confirma-
tion of the entity and early prenatal diagnosis. PrenatalDiagnosis, 19, 279–281.
Dincsoy, M. Y., Salih, M. A., al-Jurayyan, N., et al. (1995).
Multiple congenital malformations in two sibs reminiscent
of hydrolethalus and pseudotrisomy 13 syndromes. Ameri-can Journal of Medical Genetics, 56, 317–321.
Hartikainen-Sorri, A. L., Kirkinen, P., & Herva, R. (1983).
Prenatal detection of hydrolethalus syndrome. PrenatalDiagnosis, 3, 219–224.
Herva, R., & Seppanen, U. (1984). Roentgenologic findings of
the hydrolethalus syndrome. Pediatric Radiology, 14, 41–43.Kivela, T., Salonen, R., & Paetau, A. (1996). Hydrolethalus:
A midline malformation syndrome with optic nerve
coloboma and hypoplasia. Acta Neuropathologica (Berlin),91, 511–518.
Krassikoff, N., Konick, L., & Gilbert, E. F. (1987). The
hydrolethalus syndrome. Birth Defects Original ArticleSeries, 23, 411–419.
Mee, L., Honkala, H., Kopra, O., et al. (2005). Hydrolethalus
syndrome is caused by a missense mutation in a novel gene
HYLS1. Human Molecular Genetics, 14, 1475–1488.Morava, E., Adamovich, K., & Czeizel, A. E. (1996). Dandy-
Walker malformation and polydactyly: A possible expres-
sion of hydrolethalus syndrome. Clinical Genetics, 49,211–215.
Muenke, M., Ruchelli, E. D., Rorke, L. B., et al. (1991). On
lumping and splitting: A fetus with clinical findings of the
oral-facial-digital syndrome type VI, the hydrolethalus syn-
drome, and the pallister-hall syndrome. American Journal ofMedical Genetics, 41, 548–556.
Norgard, M., Yankowitz, J., Rhead, W., et al. (1996). Prenatal
ultrasound findings in hydrolethalus: Continuing difficulties
in diagnosis. Prenatal Diagnosis, 16, 173–179.
Pryde, P. G., Qureshi, F., Hallak, M., et al. (1993). Two consec-
utive hydrolethalus syndrome-affected pregnancies in
a nonconsanguinous black couple: Discussion of problems
in prenatal differential diagnosis of midline malformation
syndromes. American Journal of Medical Genetics, 46,537–541.
Rakheja, D., Cimo, M. L., Ramus, R. M., et al. (2004).
Hydrolethalus syndrome, in contrast to Smith-Lemli-Opitz
syndrome, is not due to a defect in post-squalene cholesterol
biosynthesis: A case report. American Journal of MedicalGenetics, 129A, 212–213.
Salonen, R., & Herva, R. (1990). Hydrolethalus syndrome.
Journal of Medical Genetics, 27, 756–759.Salonen, R., Herva, R., & Norio, R. (1981). The hydrolethalus
syndrome: Delineation of a “new”, lethal malformation
syndrome based on 28 patients. Clinical Genetics, 19,321–330.
Sharma, A. K., Phadke, S., Chandra, K., et al. (1992). Overlap
between Majewski and hydrolethalus syndromes: A report of
two cases. American Journal of Medical Genetics,43, 949–953.
Shotelersuk, V., Punyavoravud, V., Phudhichareonrat, S., et al.
(2001). An Asian girl with a ‘milder’ form of the
hydrolethalus syndrome. Clinical Dysmorphology,10, 51–55.
Siffring, P. A., Forrest, T. S., & Frick, M. P. (1991). Sonographic
detection of hydrolethalus syndrome. Journal of ClinicalUltrasound, 19, 43–47.
Toriello, H. V., & Bauserman, S. C. (1985). Bilateral pulmonary
agenesis: Association with the hydrolethalus syndrome and
review of the literature from a developmental field
perspective. American Journal of Medical Genetics,21, 93–103.
Verloes, A., Ayme, S., Gambarelli, D., et al. (1991).
Holoprosencephaly-polydactyly (‘pseudotrisomy 13’) syn-
drome: A syndrome with features of hydrolethalus and
Smith-Lemli-Opitz syndromes. A collaborative multicentre
study. Journal of Medical Genetics, 28, 297–303.Visapaa, I., Salonen, R., Varilo, T., et al. (1999). Assignment of
the locus for hydrolethalus syndrome to a highly restricted
region on 11q23-25. American Journal of Human Genetics,65, 1086–1095.
Hydrolethalus Syndrome 1083
c
ad
e
b
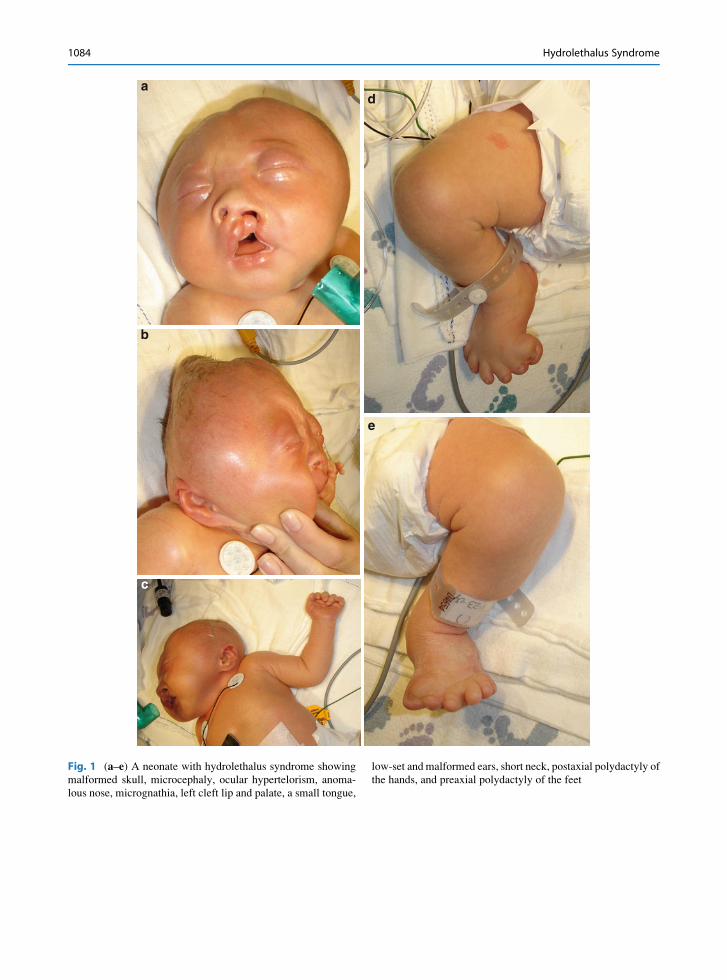
Fig. 1 (a–e) A neonate with hydrolethalus syndrome showing
malformed skull, microcephaly, ocular hypertelorism, anoma-
lous nose, micrognathia, left cleft lip and palate, a small tongue,
low-set and malformed ears, short neck, postaxial polydactyly of
the hands, and preaxial polydactyly of the feet
1084 Hydrolethalus Syndrome




























