Embed Size (px)
Citation preview

Proc. Nati. Acad. Sci. USAVol. 86, pp. 8063-8067, October 1989Medical Sciences
Autocrine mechanism for v-sis transformation requires cellsurface localization of internally activated growthfactor receptors
(cell transformation/platelet-derived growth factor receptors/anti-phosphotyrosine/suramin)
TIMOTHY P. FLEMING, TOSHIMITSU MATSUI, CHRISTOPHER J. MOLLOY, KEITH C. ROBBINS*,AND STUART A. AARONSONLaboratory of Cellular and Molecular Biology, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892
Communicated by Bernhard Witkop, July 14, 1989 (received for review June 9, 1989)
ABSTRACT v-sis represents a prototype for the class ofoncogenes that encode growth factors. Whether its platelet-derived growth factor (PDGF)-like product functionally acti-vates its receptors within the cell or at the cell surface haspotential implications in efforts to intervene with the v-sis-transformed phenotype. We demonstrate that intracellular aswell as cell surface forms of two PDGF receptor gene productsare tyrosine phosphorylated in v-sis transformants. In a chem-ically defred medium in which cell growth was dependent onv-sis expression, proliferation was partially inhibited by PDGFneutralizing antibody but completely blocked by suramin.Suramin treatment resulted in a marked reduction in tyrosinephosphorylated cell surface PDGF receptors but had no effecton the level of tyrosine phosphorylation of intracellular recep-tor species. All of these findings demonstrate that the v-sis-encoded mitogen can bind and activate its receptors inter-nally but that activated receptors must achieve a cell surfacelocation in order to functionally couple with intracellularmitogenic signaling pathways.
Genes encoding growth factors and their receptors have beenimplicated in the regulation of normal cell growth and devel-opment. There is also increasing evidence that genetic alter-ations affecting expression of such genes can contribute toaltered cell growth associated with malignancy. The v-sisoncogene of simian sarcoma virus encodes a growth factorhomologous to the B chain ofhuman platelet-derived growthfactor (PDGFB) (1, 2). Moreover, the normal homologues ofother oncogenes code for membrane-spanning growth factorreceptors (3-6). Genes that act early in intracellular path-ways of growth factor signal transduction have been impli-cated as oncogenes as well (7-11).
Intervention with the pathogenic expression of this impor-tant subset of genes might be most readily approached in thecase of growth factors, if functional activation of theirreceptor targets in transformed cells were confined to the cellsurface. Although not generally expressed by the same cells,growth factors and their receptors are often processedthrough the same secretory pathway (12, 13). Moreover, inthe case of the v-sis/PDGFB product, immature dimericforms present only within the cell have been shown to havemitogenic potential (14). Efforts to date to localize the criticalsite(s) for functional interaction ofthe v-sis-encoded mitogenwith PDGF receptors (PDGFRs) have led to contradictoryconclusions. Available evidence argues either that mitogeni-cally active v-sis gene products bind and activate PDGFRsduring processing within the cell (15-17) or that functional
interaction occurs only after these molecules achieve a cellsurface location (18, 19). Our present studies resolve thiscontroversy by establishing the specific sites of receptoractivation and functional coupling with mitogenic signaltransduction pathways. These findings have general impli-cations concerning localization within the cell of criticaltargets ofgrowth factor receptor action as well as approachestoward intervention with autocrine-associated malignancies.
MATERIALS AND METHODSCells and Reagents. NIH 3T3 mouse embryonic fibroblasts
(20) and the transformed derivatives sis/3T3 (21) and erbB-2(22) were established in this laboratory. PDGF BB ho-modimer (PDGF-BB) was purchased from Amgen Biologi-cals. Suramin was obtained from the Division of CancerTreatment, National Cancer Institute.DNA Synthesis Assay. DNA synthesis was measured as
described elsewhere (23) with a few modifications. Twenty-four-well Costar plates were precoated with human fibronec-tin (Collaborative Research) at 1 ug/cm2 prior to seeding thecells. Serum-free growth media consisted of a mixture ofMCDB 401 medium (Irvine Scientific), Leibovitz's L-15medium (GIBCO), and Ham's F12 medium (BioFluids, Rock-ville, MD) containing 10 nM selenium (GIBCO) and 10 tug oftransferrin per ml (Collaborative Research).Colony Formation in Soft Agar. Around 104 cells were
plated in 0.4% agarose suspension (SeaPlaque, FMC) inDulbecco's modified Eagle's medium as described elsewhere(24). Colonies were scored at 14 days.
Analysis of Tyrosine Phosphorylated a and (3 PDGFRs.Western analysis of cell lysates was performed as described(25). Briefly, cells were lysed with Staph-A buffer (10 mMsodium phosphate, pH 7.5/100 mM NaCl, 1% Triton X-100/0.1% SDS, 0.5% deoxycholate/0.1% aprotinin/1 mM phe-nylmethylsulfonyl fluoride/i mM sodium orthovanadate)and clarified by centrifugation at 10,000 x g for 30 min.Lysates were immunoprecipitated with peptide antisera spe-cific for either a or P PDGFRs or anti-phosphotyrosine serumas described (25). Immunoprecipitates were subjected toSDS/polyacrylamide gel electrophoresis, blotted onto nitro-cellulose filters, and treated with affinity-purified anti-phosphotyrosine serum (25). Immune complexes were iden-tified by using 125I-labeled protein A and visualized byautoradiography.
Abbreviations: PDGF, platelet-derived growth factor; PDGFR,PDGF receptor; PDGF-BB, PDGF homodimer; PDGFB, B chain ofPDGF.*Present address: Laboratory of Cellular Development and Oncol-ogy, National Institute of Dental Research, Bethesda, MD 20892.
8063
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.
8064 Medical Sciences: Fleming et al.
RESULTSExogenous PDGF Transiently Mimics the v-sis-Transformed
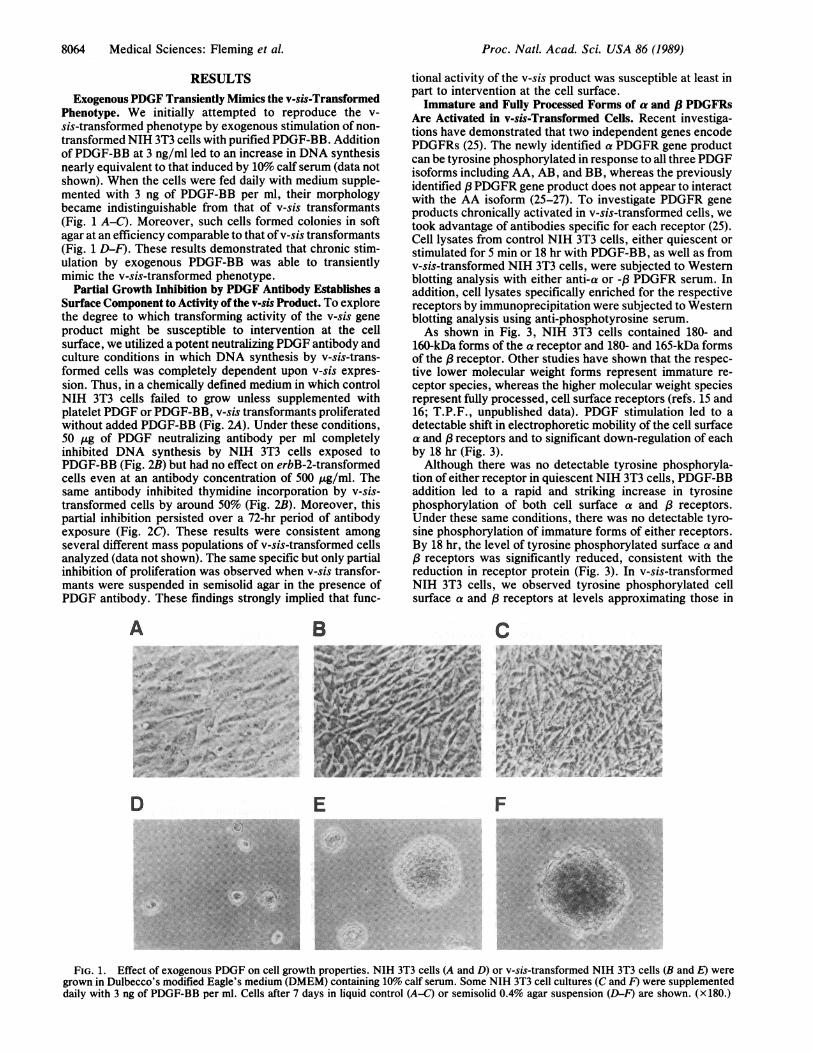
Phenotype. We initially attempted to reproduce the v-sis-transformed phenotype by exogenous stimulation of non-transformed NIH 3T3 cells with purified PDGF-BB. Additionof PDGF-BB at 3 ng/ml led to an increase in DNA synthesisnearly equivalent to that induced by 10% calf serum (data notshown). When the cells were fed daily with medium supple-mented with 3 ng of PDGF-BB per ml, their morphologybecame indistinguishable from that of v-sis transformants(Fig. 1 A-C). Moreover, such cells formed colonies in softagar at an efficiency comparable to that of v-sis transformants(Fig. 1 D-F). These results demonstrated that chronic stim-ulation by exogenous PDGF-BB was able to transientlymimic the v-sis-transformed phenotype.
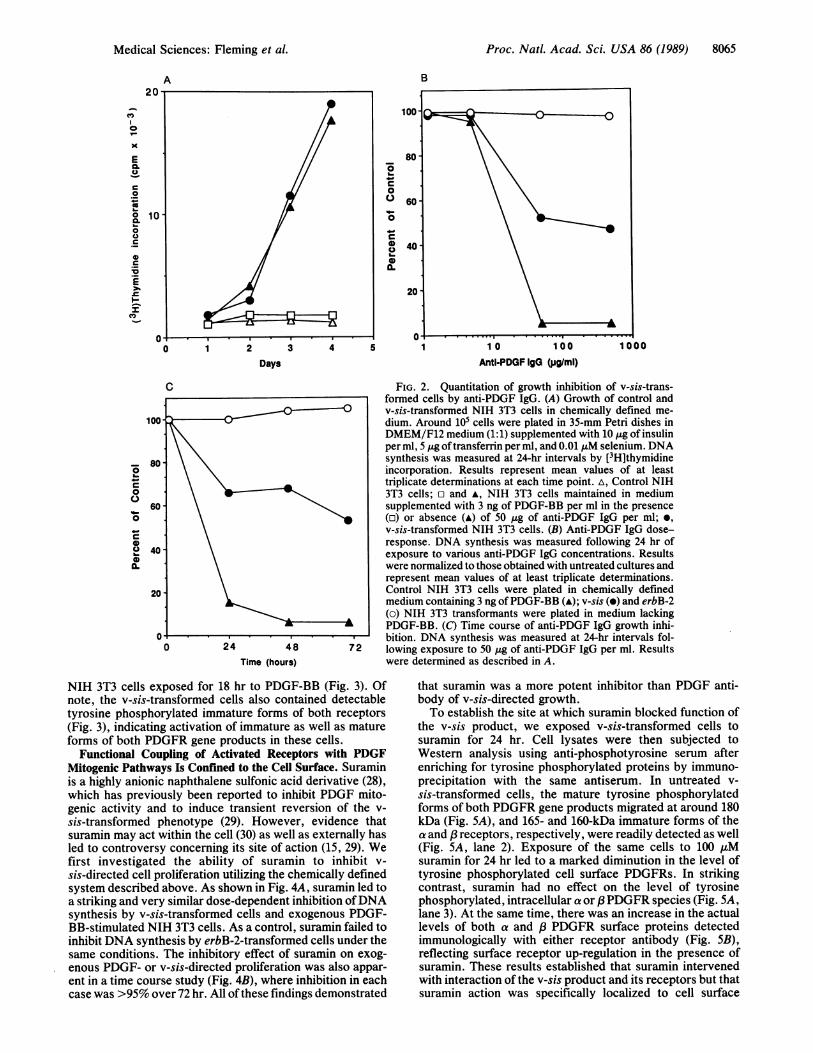
Partial Growth Inhibition by PDGF Antibody Establishes aSurface Component to Activity of the v-sis Product. To explorethe degree to which transforming activity of the v-sis geneproduct might be susceptible to intervention at the cellsurface, we utilized a potent neutralizing PDGF antibody andculture conditions in which DNA synthesis by v-sis-trans-formed cells was completely dependent upon v-sis expres-sion. Thus, in a chemically defined medium in which controlNIH 3T3 cells failed to grow unless supplemented withplatelet PDGF or PDGF-BB, v-sis transformants proliferatedwithout added PDGF-BB (Fig. 2A). Under these conditions,50 gg of PDGF neutralizing antibody per ml completelyinhibited DNA synthesis by NIH 3T3 cells exposed toPDGF-BB (Fig. 2B) but had no effect on erbB-2-transformedcells even at an antibody concentration of 500 gg/ml. Thesame antibody inhibited thymidine incorporation by v-sis-transformed cells by around 50% (Fig. 2B). Moreover, thispartial inhibition persisted over a 72-hr period of antibodyexposure (Fig. 2C). These results were consistent amongseveral different mass populations of v-sis-transformed cellsanalyzed (data not shown). The same specific but only partialinhibition of proliferation was observed when v-sis transfor-mants were suspended in semisolid agar in the presence ofPDGF antibody. These findings strongly implied that func-
A
_ 4. .4-
D-g|. :@*........ e, ,:
E
tional activity of the v-sis product was susceptible at least inpart to intervention at the cell surface.Immature and Fully Processed Forms of a and .8 PDGFRs
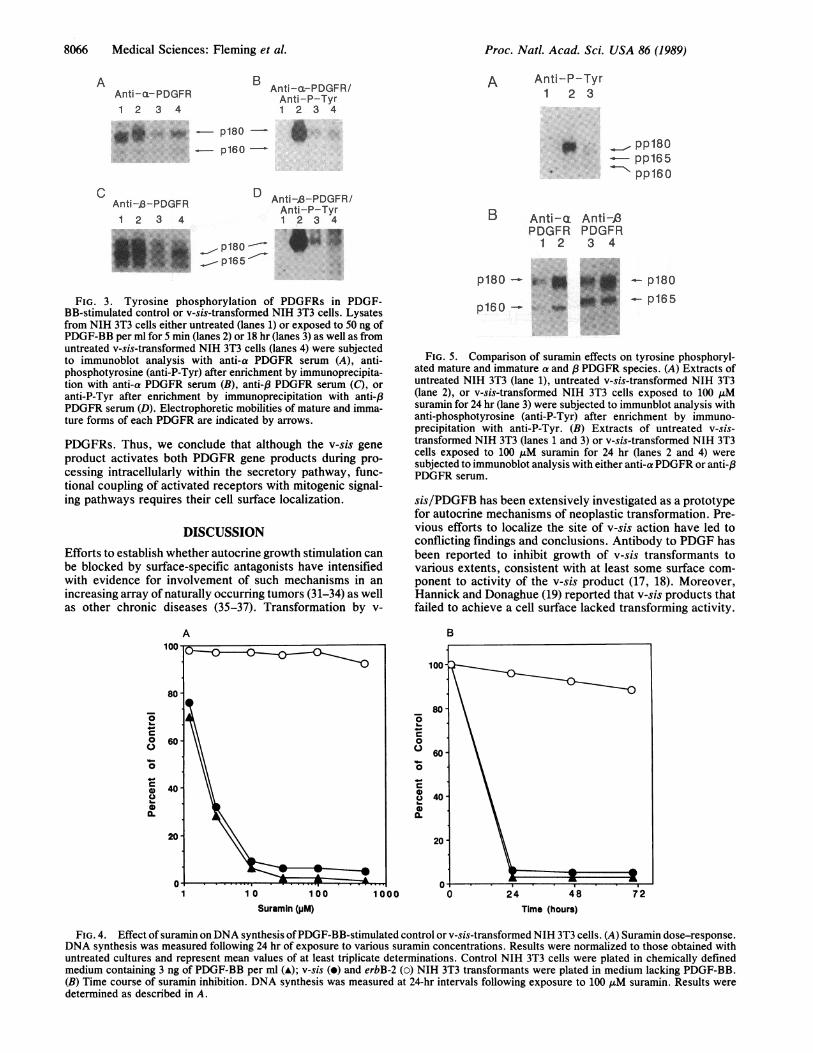
Are Activated in v-sis-Transformed Cells. Recent investiga-tions have demonstrated that two independent genes encodePDGFRs (25). The newly identified a PDGFR gene productcan be tyrosine phosphorylated in response to all three PDGFisoforms including AA, AB, and BB, whereas the previouslyidentified 3PDGFR gene product does not appear to interactwith the AA isoform (25-27). To investigate PDGFR geneproducts chronically activated in v-sis-transformed cells, wetook advantage of antibodies specific for each receptor (25).Cell lysates from control NIH 3T3 cells, either quiescent orstimulated for 5 min or 18 hr with PDGF-BB, as well as fromv-sis-transformed NIH 3T3 cells, were subjected to Westernblotting analysis with either anti-a or -(3 PDGFR serum. Inaddition, cell lysates specifically enriched for the respectivereceptors by immunoprecipitation were subjected to Westernblotting analysis using anti-phosphotyrosine serum.As shown in Fig. 3, NIH 3T3 cells contained 180- and
160-kDa forms of the a receptor and 180- and 165-kDa formsof the /3 receptor. Other studies have shown that the respec-tive lower molecular weight forms represent immature re-ceptor species, whereas the higher molecular weight speciesrepresent fully processed, cell surface receptors (refs. 15 and16; T.P.F., unpublished data). PDGF stimulation led to adetectable shift in electrophoretic mobility of the cell surfacea and 3receptors and to significant down-regulation of eachby 18 hr (Fig. 3).Although there was no detectable tyrosine phosphoryla-
tion of either receptor in quiescent NIH 3T3 cells, PDGF-BBaddition led to a rapid and striking increase in tyrosinephosphorylation of both cell surface a and /3 receptors.Under these same conditions, there was no detectable tyro-sine phosphorylation of immature forms of either receptors.By 18 hr, the level of tyrosine phosphorylated surface a andP receptors was significantly reduced, consistent with thereduction in receptor protein (Fig. 3). In v-sis-transformedNIH 3T3 cells, we observed tyrosine phosphorylated cellsurface a and 83 receptors at levels approximating those in
C
F
FIG. 1. Effect of exogenous PDGF on cell growth properties. NIH 3T3 cells (A and D) or v-sis-transformed NIH 3T3 cells (B and E) were
grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% calf serum. Some NIH 3T3 cell cultures (C and F) were supplemented
daily with 3 ng of PDGF-BB per ml. Cells after 7 days in liquid control (A-C) or semisolid 0.4% agar suspension (D-F) are shown. (x 180.)
B
Ag A'ts
Proc. Natl. Acad. Sci. USA 86 (1989)
Proc. Natl. Acad. Sci. USA 86 (1989) 8065
A B
C
0.x
CL
06
0
._
F0o
E
I-C,) 0
100
_ 80
0c
00
60
0
0o0 4
0 1 2 3 4Days
C
48Time (hours)
NIH 3T3 cells exposed for 18 hr to PDGF-BB (Fig. 3). Ofnote, the v-sis-transformed cells also contained detectabletyrosine phosphorylated immature forms of both receptors(Fig. 3), indicating activation of immature as well as matureforms of both PDGFR gene products in these cells.
Functional Coupling of Activated Receptors with PDGFMitogenic Pathways Is Confined to the Cell Surface. Suraminis a highly anionic naphthalene sulfonic acid derivative (28),which has previously been reported to inhibit PDGF mito-genic activity and to induce transient reversion of the v-sis-transformed phenotype (29). However, evidence thatsuramin may act within the cell (30) as well as externally hasled to controversy concerning its site of action (15, 29). Wefirst investigated the ability of suramin to inhibit v-sis-directed cell proliferation utilizing the chemically definedsystem described above. As shown in Fig. 4A, suramin led toa striking and very similar dose-dependent inhibition ofDNAsynthesis by v-sis-transformed cells and exogenous PDGF-BB-stimulated NIH 3T3 cells. As a control, suramin failed toinhibit DNA synthesis by erbB-2-transformed cells under thesame conditions. The inhibitory effect of suramin on exog-enous PDGF- or v-sis-directed proliferation was also appar-ent in a time course study (Fig. 4B), where inhibition in eachcase was >95% over 72 hr. All of these findings demonstrated
'aC
00
0
0
0.
1 10 100 1000
Antl-PDGF IgG (pglml)
FIG. 2. Quantitation of growth inhibition of v-sis-trans-formed cells by anti-PDGF IgG. (A) Growth of control andv-sis-transformed NIH 3T3 cells in chemically defined me-dium. Around 105 cells were plated in 35-mm Petri dishes inDMEM/F12 medium (1:1) supplemented with 10 gg of insulinper ml, 5 Mg of transferrin per ml, and 0.01 AuM selenium. DNAsynthesis was measured at 24-hr intervals by [3H]thymidineincorporation. Results represent mean values of at leasttriplicate determinations at each time point. A, Control NIH3T3 cells; E and A, NIH 3T3 cells maintained in mediumsupplemented with 3 ng of PDGF-BB per ml in the presence(o) or absence (A) of 50 tug of anti-PDGF IgG per ml; *,v-sis-transformed NIH 3T3 cells. (B) Anti-PDGF IgG dose-response. DNA synthesis was measured following 24 hr ofexposure to various anti-PDGF IgG concentrations. Resultswere normalized to those obtained with untreated cultures andrepresent mean values of at least triplicate determinations.Control NIH 3T3 cells were plated in chemically definedmedium containing 3 ng ofPDGF-BB (A); v-sis (e) and erbB-2(o) NIH 3T3 transformants were plated in medium lackingPDGF-BB. (C) Time course of anti-PDGF IgG growth inhi-bition. DNA synthesis was measured at 24-hr intervals fol-lowing exposure to 50 ug of anti-PDGF IgG per ml. Resultswere determined as described in A.
that suramin was a more potent inhibitor than PDGF anti-body of v-sis-directed growth.To establish the site at which suramin blocked function of
the v-sis product, we exposed v-sis-transformed cells tosuramin for 24 hr. Cell lysates were then subjected toWestern analysis using anti-phosphotyrosine serum afterenriching for tyrosine phosphorylated proteins by immuno-precipitation with the same antiserum. In untreated v-sis-transformed cells, the mature tyrosine phosphorylatedforms of both PDGFR gene products migrated at around 180kDa (Fig. SA), and 165- and 160-kDa immature forms of thea and ,3 receptors, respectively, were readily detected as well(Fig. SA, lane 2). Exposure of the same cells to 100 juMsuramin for 24 hr led to a marked diminution in the level oftyrosine phosphorylated cell surface PDGFRs. In strikingcontrast, suramin had no effect on the level of tyrosinephosphorylated, intracellular a or PPDGFR species (Fig. 5A,lane 3). At the same time, there was an increase in the actuallevels of both a and P PDGFR surface proteins detectedimmunologically with either receptor antibody (Fig. 5B),reflecting surface receptor up-regulation in the presence ofsuramin. These results established that suramin intervenedwith interaction of the v-sis product and its receptors but thatsuramin action was specifically localized to cell surface
Medical Sciences: Fleming et al.
8066 Medical Sciences: Fleming et al.
B Anti-cL-PDGFR/Anti-ac--PDGFR Anti-P-Tyr1 2 3 4 1 2 3 4
"*j.~~~~1p80-:: :::: ::p160-Anti -1-PDGFR
1 2 3 4
D Anti-J3-PDGFR/Anti-P-Tyr1 2 3 4
p180p165
A Anti-P-Tyr1 2 3
B Anti-aPDGFR
1 2
Anti-£3PDGFR3 4
FIG. 3. Tyrosine phosphorylation of PDGFRs in PDGF-BB-stimulated control or v-sis-transformed NIH 3T3 cells. Lysatesfrom NIH 3T3 cells either untreated (lanes 1) or exposed to 50 ng ofPDGF-BB per ml for 5 min (lanes 2) or 18 hr (lanes 3) as well as fromuntreated v-sis-transformed NIH 3T3 cells (lanes 4) were subjectedto immunoblot analysis with anti-a PDGFR serum (A), anti-phosphotyrosine (anti-P-Tyr) after enrichment by immunoprecipita-tion with anti-a PDGFR serum (B), anti-,8 PDGFR serum (C), oranti-P-Tyr after enrichment by immunoprecipitation with anti-,8PDGFR serum (D). Electrophoretic mobilities of mature and imma-ture forms of each PDGFR are indicated by arrows.
PDGFRs. Thus, we conclude that although the v-sis geneproduct activates both PDGFR gene products during pro-cessing intracellularly within the secretory pathway, func-tional coupling of activated receptors with mitogenic signal-ing pathways requires their cell surface localization.
DISCUSSIONEfforts to establish whether autocrine growth stimulation canbe blocked by surface-specific antagonists have intensifiedwith evidence for involvement of such mechanisms in anincreasing array of naturally occurring tumors (31-34) as wellas other chronic diseases (35-37). Transformation by v-
A100 -
80-
oC)0 60
0 4040
IL
p180 -
p160
p180
- p165
FIG. 5. Comparison of suramin effects on tyrosine phosphoryl-ated mature and immature a and 8 PDGFR species. (A) Extracts ofuntreated NIH 3T3 (lane 1), untreated v-sis-transformed NIH 3T3(lane 2), or v-sis-transformed NIH 3T3 cells exposed to 100 ,uMsuramin for 24 hr (lane 3) were subjected to immunblot analysis withanti-phosphotyrosine (anti-P-Tyr) after enrichment by immuno-precipitation with anti-P-Tyr. (B) Extracts of untreated v-sis-transformed NIH 3T3 (lanes 1 and 3) or v-sis-transformed NIH 3T3cells exposed to 100 ,uM suramin for 24 hr (lanes 2 and 4) weresubjected to immunoblot analysis with either anti-a PDGFR or anti-,8PDGFR serum.
sis/PDGFB has been extensively investigated as a prototypefor autocrine mechanisms of neoplastic transformation. Pre-vious efforts to localize the site of v-sis action have led toconflicting findings and conclusions. Antibody to PDGF hasbeen reported to inhibit growth of v-sis transformants tovarious extents, consistent with at least some surface com-ponent to activity of the v-sis product (17, 18). Moreover,Hannick and Donaghue (19) reported that v-sis products thatfailed to achieve a cell surface lacked transforming activity.
B
a-
0
0
0
a)
1 1 0 100 1000
Suramin (pM)48
Time (hours)
FIG. 4. Effect of suramin onDNA synthesis of PDGF-BB-stimulated control or v-sis-transformed NIH 3T3 cells. (A) Suramin dose-response.DNA synthesis was measured following 24 hr of exposure to various suramin concentrations. Results were normalized to those obtained withuntreated cultures and represent mean values of at least triplicate determinations. Control NIH 3T3 cells were plated in chemically definedmedium containing 3 ng of PDGF-BB per ml (A); v-sis (e) and erbB-2 (o) NIH 3T3 transformants were plated in medium lacking PDGF-BB.(B) Time course of suramin inhibition. DNA synthesis was measured at 24-hr intervals following exposure to 100 AM suramin. Results weredetermined as described in A.
A
C
.0:. ,0-1.- pp180- pp165- pp160
Proc. Natl. Acad. Sci. USA 86 (1989)

Proc. Natl. Acad. Sci. USA 86 (1989) 8067
In contrast, others have reported evidence that internal formsof the PDGFR are tyrosine phosphorylated and exhibitgreatly increased turnover in the absence of detectable ma-ture PDGFR species (15, 16). This has led to a so-called"internal autocrine" model, implying that receptor activa-tion and functional coupling with mitogenic pathways occurentirely within the cell (15, 16).
In the present studies, we demonstrated that exogenousPDGF can induce growth alterations that mimic the v-sis-transformed state. These findings establish that PDGFRactivation at the cell surface is sufficient to induce transfor-mation despite receptor down-regulation that accompanieschronic exogenous growth factor stimulation. By developinga chemically defined culture system in which v-sis action wasrequired for cell proliferation, we further demonstrated par-tial growth inhibition by PDGF neutralizing antibody. Thesefindings establish a surface-accessible component to theaction of the v-sis-encoded mitogen but exclude a classicalautocrine mechanism, since the same antibody was able tocompletely block the proliferation of NIH 3T3 cells in re-sponse to exogenous PDGF.Immature, intracellular forms of two independent PDGFR
gene products as well as their respective mature, cell surfaceforms were shown to be chronically activated in v-sis-transformed cells. These findings establish that ligand bind-ing and receptor activation occur within the secretory path-way. Finally, we showed that suramin completely inhibitedv-sis-directed proliferation associated with its specific inhi-bition of the tyrosine phosphorylated state of surfacePDGFRs. The lack of any effect on tyrosine phosphorylationof internal, immature receptors implies not only that suraminspecifically acts at that level of the cell surface PDGFRs butthat activated surface receptors must be required to effec-tively transduce the mitogenic signal.Our findings do not exclude the possibility that some
growth factor-receptor interactions are first initiated at thecell surface. PDGF antibody and suramin were able toeffectively block exogenous PDGF stimulation. Yet, weobserved only partial inhibition of v-sis-directed growth byPDGF neutralizing antibody. If antibody only inhibits denovo ligand binding to receptors, the partial resistance ofv-sis-directed proliferation likely reflects antibody-inacces-sible ligand already bound to PDGFRs prior to their reachingthe cell surface. The total inhibition of v-sis-directed growthinduced by suramin suggests that this agent is also able tostrip ligands already bound to receptors and to down-regulateenzymatic activity of internally activated receptors as theyreach the cell surface.Our evidence that the functional coupling of activated
PDGFRs with intracellular mitogenic signaling pathways inNIH 3T3 cells requires their cell surface localization stronglysuggests that critical targets of receptor action may bespecifically located at or near the inner surface of the cellmembrane. Alternatively, events associated with internaliza-tion of activated receptors may be critical for effectivereceptor signaling. The v-fms product represents an activatedform of the colony-stimulating factor 1 (CSF-1) receptor (4,38), which is structurally related to the PDGFRs (25, 39).Certain glycosylation inhibitors have been reported to inducereversion of v-fms-transformed cells associated with a lack ofmaturation of the v-fms product (40). Thus, activated CSF-1receptors may also require a cell surface location in order toefficiently couple with mitogenic signaling pathways.Whether these findings can be generalized to other growthfactor receptors remains to be determined. If so, then surfaceacting molecules that can interfere with ligand-receptorinteractions may have applicability in efforts to interveneclinically in autocrine-associated malignancies as well as
other chronic diseases whose pathogenesis involves auto-crine growth stimulation.
1. Doolittle, R. F., Hunkapiller, M. W., Hood, L. E., Devare, S. G.,Robbins, K. C., Aaronson, S. A. & Antoniades, H. N. (1983) Science221, 275-277.
2. Waterfield, M. D., Scrace, G. T., Whittle, N., Stroobant, P., Johnsson,A., Wasteson, A., Westermark, B., Heldin, C.-H., Huang, J. S. & Deuel,T. F. (1983) Nature (London) 304, 35-39.
3. Downward, J., Yarden, Y., Mayes, E., Scrace, G., Totty, N., Stockwell,P., Ullrich, A., Schlessinger, J. & Waterfield, M. D. (1984) Nature(London) 307, 521-527.
4. Sherr, C. J., Rettenmier, C. W., Sacca, R., Roussel, M. F., Look, A. T.& Stanley, E. R. (1985) Cell 41, 665-676.
5. Yarden, Y., Kuang, W., Yang-Feng, T., Coussens, L., Munemitsu, S.,Dull, T. J., Chen, E., Schlessinger, J., Francke, U. & Ullrich, A. (1987)EMBO J. 6, 3341-3351.
6. Qui, F., Ray, P., Brown, K., Barker, P. E., Jhanwar, S., Ruddle, F. H.& Besmer, P. (1988) EMBO J. 7, 1003-1011.
7. Ralston, R. & Bishop, J. M. (1985) Proc. Natl. Acad. Sci. USA 82,7845-7849.
8. Greenberg, M. E. & Ziff, E. B. (1984) Nature (London) 311, 433-438.9. Berridge, M. B. & Irvine, F. F. (1984) Nature (London) 312, 315-318.
10. Persons, D. A., Wilkison, W. D., Bell, R. M. & Finn, D. J. (1988) Cell52, 447-458.
11. Housey, G. M., Johnson, M. K., Hsiao, W. L., O'Brian, C. A., Mur-phy, J. P., Kirschmeier, P. & Weinstein, I. B. (1988) Cell 52, 343-354.
12. Farquhar, M. G. (1985) Annu. Rev. Cell Biol. 1, 447-488.13. Robbins, K. C., Leal, F., Pierce, J. H. & Aaronson, S. A. (1985) EMBO
J. 7, 1783-1792.14. Leal, F., Williams, L. T., Robbins, K. C. & Aaronson, S. A. (1985)
Science 230, 327-330.15. Keating, M. & Williams, L. T. (1988) Science 239, 914-916.16. Huang, S. S. & Huang, J. S. (1988) J. Biol. Chem. 263, 12608-12618.17. Huang, S. I., Huang, S. S. & Deuel, T. F. (1984) Cell 39, 79-87.18. Johnsson, A., Betsholtz, C., Heldin, C. & Westermark, B. (1985) Nature
(London) 317, 438-440.19. Hannick, D. & Donoghue, D. J. (1988) J. Cell. Biol. 107, 287-298.20. Jainchill, J. L., Aaronson, S. A. & Todaro, G. J. (1969) J. Virol. 4,
549-553.21. Beckmann, M. P., Betsholtz, C., Heldin, C.-H., Westermark, B., Di-
Marco, E., DiFiore, P. P., Robbins, K. C. & Aaronson, S. A. (1988)Science 241, 1346-1349.
22. DiFiore, P. P., Pierce, J. H., Kraus, M. H., Segatto, O., King, C. R. &Aaronson, S. A. (1987) Science 237, 178-182.
23. Finzi, E., Fleming, T., Segatto, O., Pennington, C., Bringman, T. S.,Derynck, R. & Aaronson, S. A. (1987) Proc. Natl. Acad. Sci. USA 84,3733-3737.
24. DiFiore, P. P., Pierce, J. H., Fleming, T. P., Hazan, R., Ullrich, A.,King, C. R., Schlessinger, J. & Aaronson, S. A. (1987) Cell 51, 1063-1070.
25. Matsui, T., Heidarn, M., Miki, T., Popescu, N., La Rochelle, W., Kraus,M., Pierce, J. & Aaronson, S. A. (1989) Science 243, 800-804.
26. Heldin, C.-H., Backstrom, G., Ostman, A., Hammacher, A.,Ronnstrand, L., Rubin, K., Nister, M. & Westermark, B. (1988) EMBOJ. 7, 1387-1393.
27. Hart, C. E., Forstrom, J. W., Kelly, J. D., Seifert, R. A., Smith, R. A.,Ross, R., Murray, M. J. & Bowen-Pope, D. F. (1988) Science 240,1529-1531.
28. Ehrlich, P. & Shiga, K. (1904) Berl. Klin. Wochenschr. 41, 329, 392.29. Betsholtz, C., Johnsson, A., Heldin, C.-H. & Westermark, B. (1986)
Proc. Natl. Acad. Sci. USA 83, 6440-6444.30. Hanking, F. (1978) Adv. Pharmacol. Chemother. 15, 289-322.31. Cuttitta, F., Carney, D. E., Mulshine, J., Moody, T. W., Fedorko, J.,
Fischler, A. & Minna, J. D. (1985) Nature (London) 316, 823-826.32. Westermark, B. & Heldin, C.-H. (1986) Med. Oncol. Pharmacother. 3,
177-183.33. Igarashi, H., Rao, C. D., Siroff, M., Leal, F., Robbins, K. C. &
Aaronson, S. A. (1987) Oncogene 1, 79-85.34. Derynck, R., Goeddel, D. I., Ullrich, A., Guttermann, J. U., Williams,
R. D., Bringman, T. S. & Berger, W. H. (1987) Cancer Res. 47, 707-712.35. Ross, R. (1986) N. Engl. J. Med. 314, 488-500.36. Hamerman, D., Taylor, S., Kirschenbaum, I., Klagsbrun, M., Raines,
E. W., Ross, R. & Thomas, K. A. (1987) Proc. Soc. Exp. Biol. Med. 186,384-389.
37. Wahl, S. M., Malone, D. G. & Wilder, R. L. (1985) J. Exp. Med. 161,210-222.
38. Coussens, L., Van Beveren, C., Smith, D., Chen, E., Mitchell, R. L.,Isacke, C. M., Verma, I. M. & Ullrich, A. (1986) Nature (London) 320,277-280.
39. Yarden, Y., Escobedo, J. A., Kuang, W.-J., Yang-Feng, T. L., Daniel,T. O., Tremble, P. M., Chen, E. Y., Ando, M. E., Harkins, R. N.,Francke, U., Fried, V. A., Ullrich, A. & Williams, L. T. (1986) Nature(London) 323, 226-232.
40. Nichols, E. J., Manger, R., Hakomori, S., Herscovics, A. & Rohrschnei-der, L. R. (1985) Mol. Cell. Biol. 5, 3467-3475.
Medical Sciences: Fleming et al.





























