Embed Size (px)
Citation preview
AUTOSOMAL & SEX CHROMOSOMAL DISORDERS

Autosomal Disorders
Cytogenetics- study of chromosomes and their abnormalities
Abnormalities of autosomes:Alteration in number (Aneuploidy)Alteration in structure
DeletionTranslocationRing chromosomeShiftInversion

Autosomal Chromosomal Disorders
Approximately 1 of every 160 liveborn infants has a demontrable chromosomal abnormality
Associated with 50% – 60% of first trimester spontaneous abortions
5% of stillbirths

Chromosomal Syndromes
Diagnosis based solely on clinical characteristic possible but not reliable because:
1. No single anomaly is pathognomonic for a given syndrome
2. Patterns of anomalies for different chromosomal syndromes share common features
3. Phenotypic variation exists among people with apparently identical abnormal karyotypes
4. Malformations associated with chromosomal aberrations also can be associated with normal karyotypes (etiologic heterogenecity)

Chromosomal Syndromes
Characteristics
1.Multiple malformations (craniofacial, skeletal, cardiac and genitourinary)
2.Abnormal facies, low set or malform ears, and certain digital anomalies (eg. Clinodactyly, polydactyly, syndactyly and single palmar creases)
3.Mental retardation
4. Intrauterine and postnatal growth retardation

Autosomal Disorders
A. Alteration in number (Aneuploidy)1. Down’s syndrome (Trisomy 21)2. Pataus syndrome (Trisomy 13)3. Edwards syndrome (Trisomy 18)
B. Alteration in structure1. Cri-du-chat syndrome (Deletion of chr.5)2. Wolf’s syndrome (Deletion of chr.4)
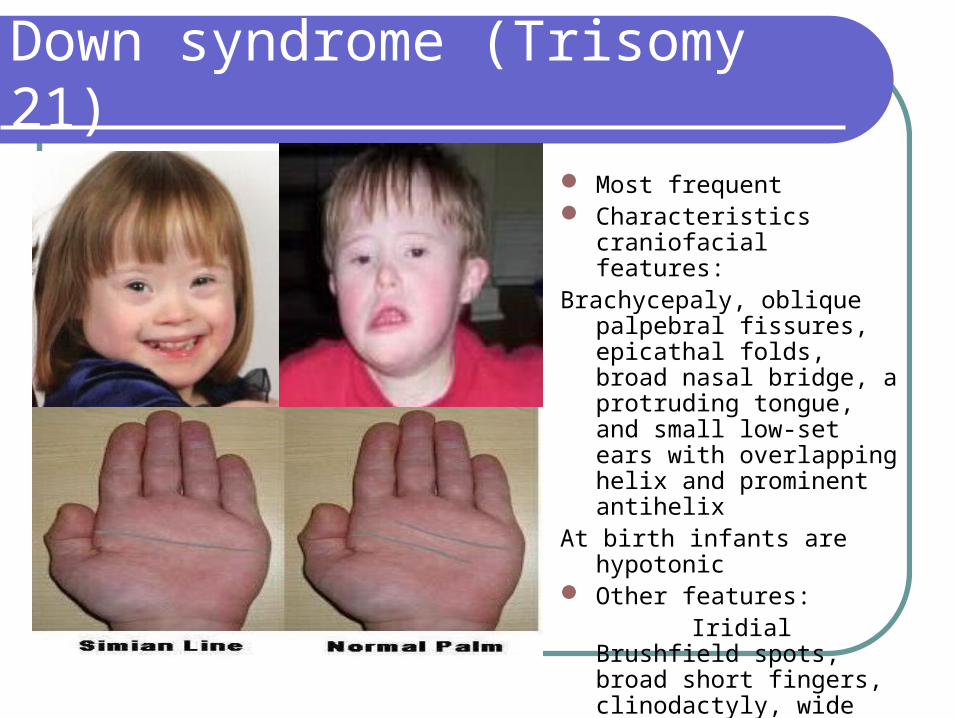
Down syndrome (Trisomy 21)
Most frequent Characteristics craniofacial
features:Brachycepaly, oblique palpebral
fissures, epicathal folds, broad nasal bridge, a protruding tongue, and small low-set ears with overlapping helix and prominent antihelix
At birth infants are hypotonic Other features: Iridial Brushfield spots, broad
short fingers, clinodactyly, wide space between first two toes
Simian Line-Present in only 30%

Down syndrome (Trisomy 21)
Common internal anomalies (cardiac lesions and duodenal atresia)
Mental retardation-mean IQ 50 (range 25-70) if IQ is 70-80 mosaicism is suspected
Growth retardation and hypotonia persist and older patients opened become obese
Females are fertile male are not
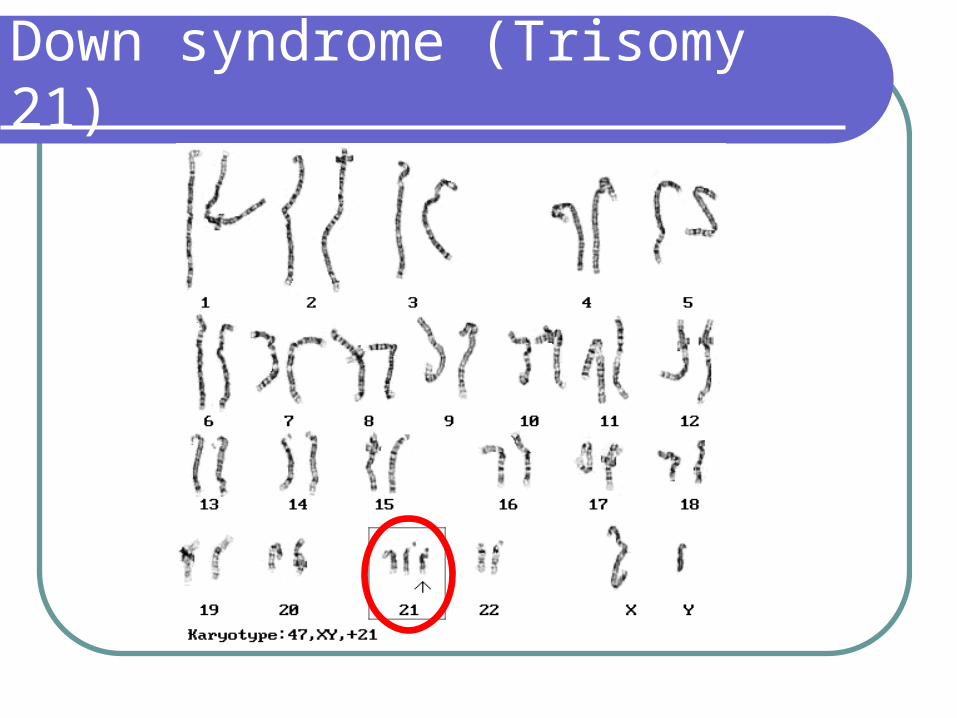
Down syndrome (Trisomy 21)
Down Syndrome
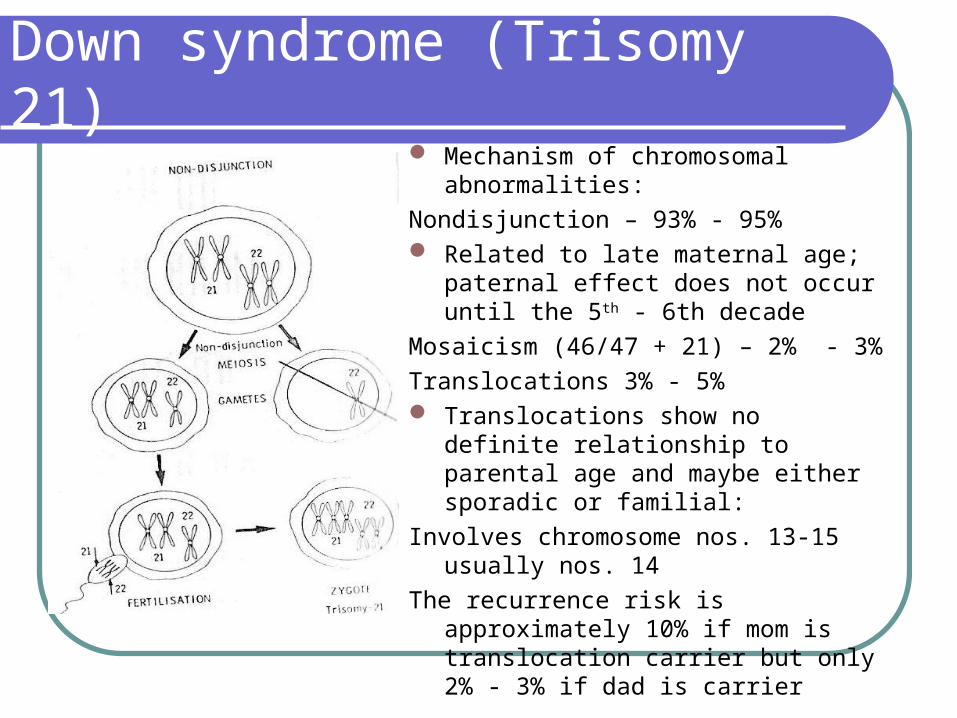
Mechanism of chromosomal abnormalities:
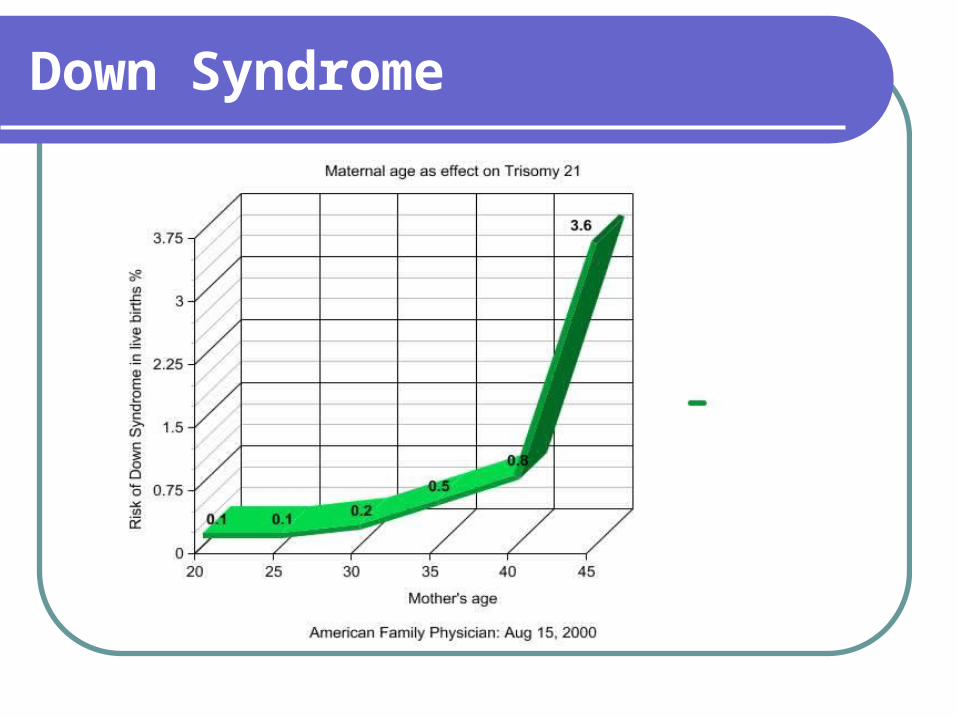
Nondisjunction – 93% - 95% Related to late maternal age; paternal
effect does not occur until the 5th - 6th decade
Mosaicism (46/47 + 21) – 2% - 3%
Translocations 3% - 5% Translocations show no definite
relationship to parental age and maybe either sporadic or familial:
Involves chromosome nos. 13-15 usually nos. 14
The recurrence risk is approximately 10% if mom is translocation carrier but only 2% - 3% if dad is carrier
Down syndrome (Trisomy 21)
Edwards syndrome (Trisomy 18)
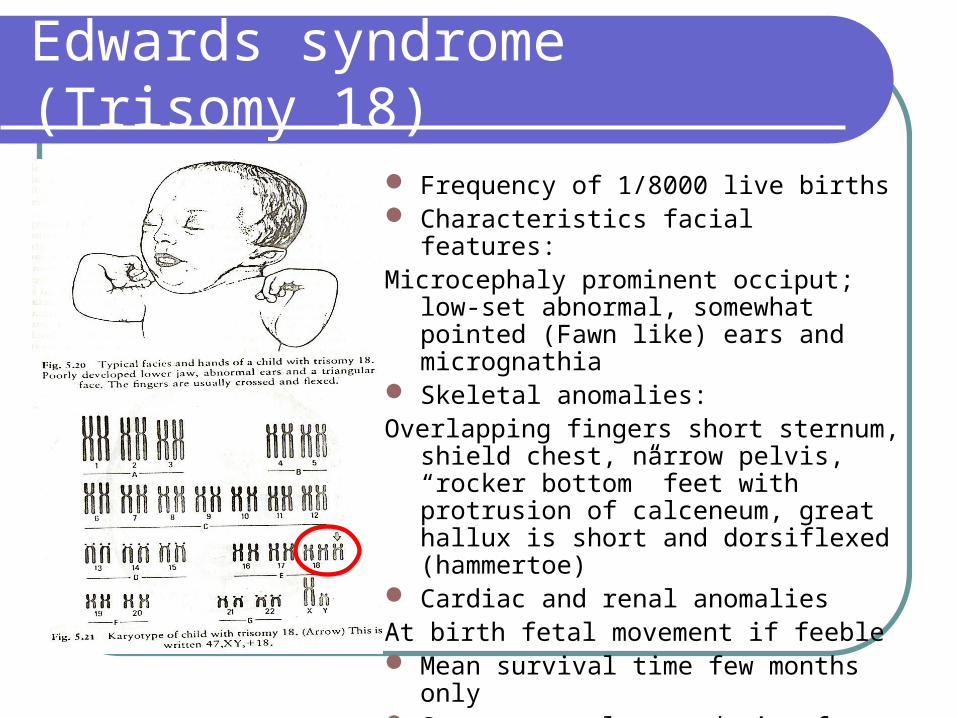
Frequency of 1/8000 live births Characteristics facial features:Microcephaly prominent occiput; low-set
abnormal, somewhat pointed (Fawn like) ears and micrognathia
Skeletal anomalies:Overlapping fingers short sternum, shield
chest, narrow pelvis, “rocker bottom” feet with protrusion of calceneum, great hallux is short and dorsiflexed (hammertoe)
Cardiac and renal anomalies At birth fetal movement if feeble Mean survival time few months only Severe mental retardation for those who
survives

Pataus syndrome Trisomy 13
1/20,000 live births 50% affected children died in the first
month Fewer than 5% survives past three
years of age Characteristic features:
holoprosencephaly, eye anomalies (microphthalmia, anopthalmia or coloboma), cleaf lip and palate, polydactyly and cardiac defects
Other features: cutaneous scalp defects, hemangiomata on face and forehead, low-set ears with abnormal helix, and rocker botton feet (convex soles and protruding heels)

80% associated with nondisjunction (47,+13)
Translocation occurs in 20% and involves chromosomes nos. 13-15
If either parent is carrier recurrence risk of an affected offspring increases but less than for Down syndrome
Pataus syndrome Trisomy 13

Cri-du-chat syndrome
Deletion of short arm of chromosome 5 (5p)
Peculiar cry (meowing of a cat) due to softening of larynx
During infancy facies become rounded (“moonlike”) because of microcephaly hypertelorism broad nasal bridge downward slanting of palpebral fissures, micrognathia and low-set ears

As patients ages facies become elongated and philtrum shorter
Hypertonicity Severe mental and
growth retardationCan reach adulthood
Deletion of chr.4 (Wolf’s Syndrome)
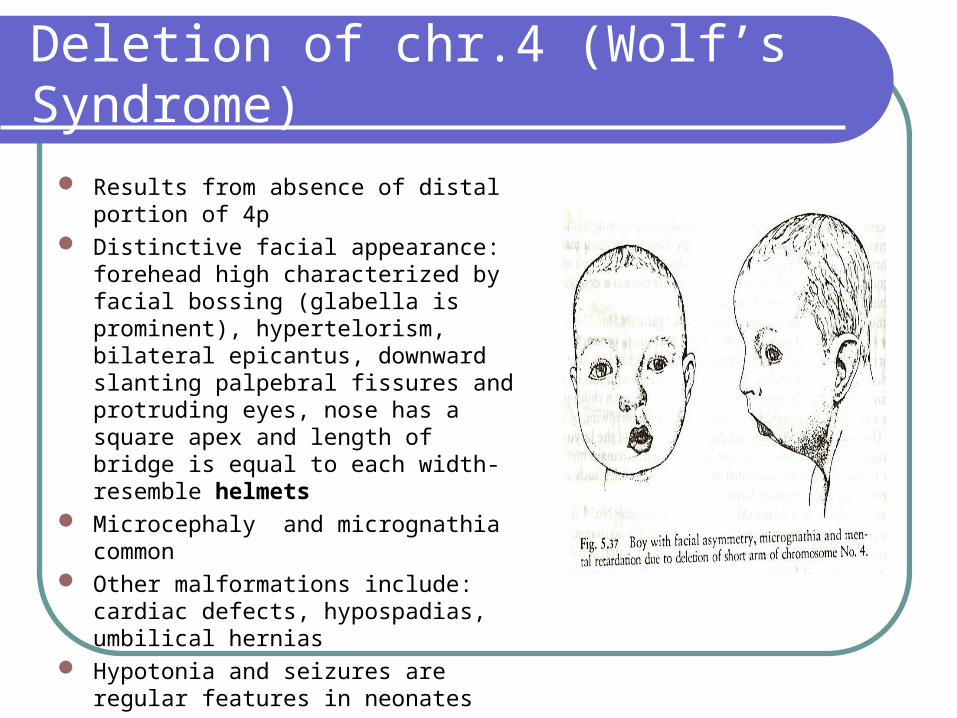
Results from absence of distal portion of 4p
Distinctive facial appearance: forehead high characterized by facial bossing (glabella is prominent), hypertelorism, bilateral epicantus, downward slanting palpebral fissures and protruding eyes, nose has a square apex and length of bridge is equal to each width-resemble helmets
Microcephaly and micrognathia common Other malformations include: cardiac
defects, hypospadias, umbilical hernias Hypotonia and seizures are regular
features in neonates Severe mental retardation

Sex Chromosome Disorders
Turner’s Syndrome - XOKlinefelter’s Syndrome - XXY Triple X female - XXXDouble Y Male - XYY
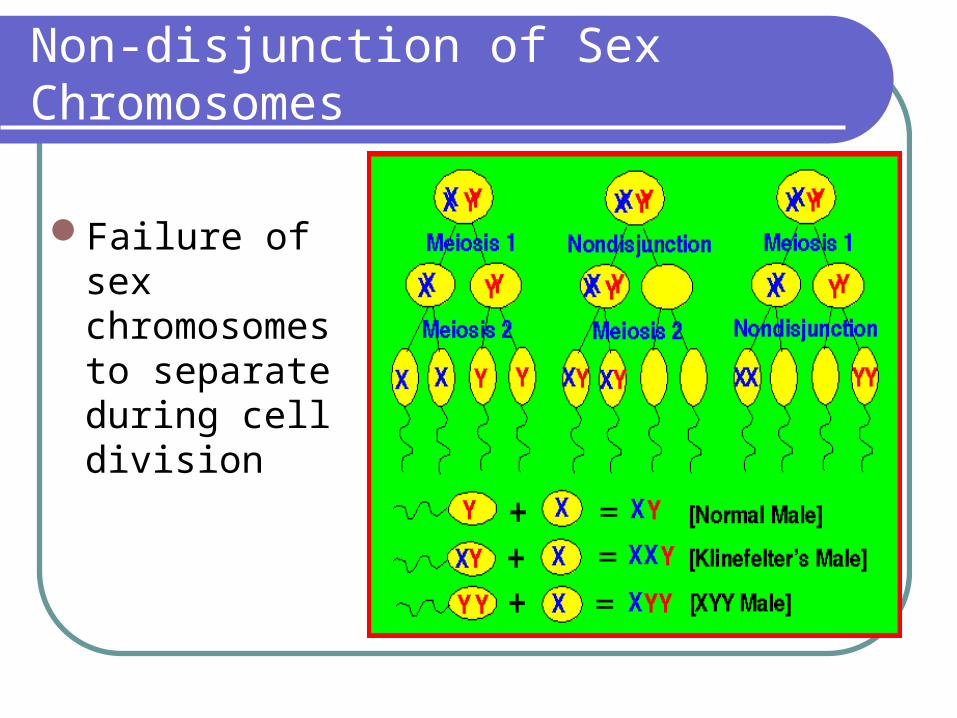
Non-disjunction of Sex Chromosomes
Failure of sex chromosomes to separate during cell division

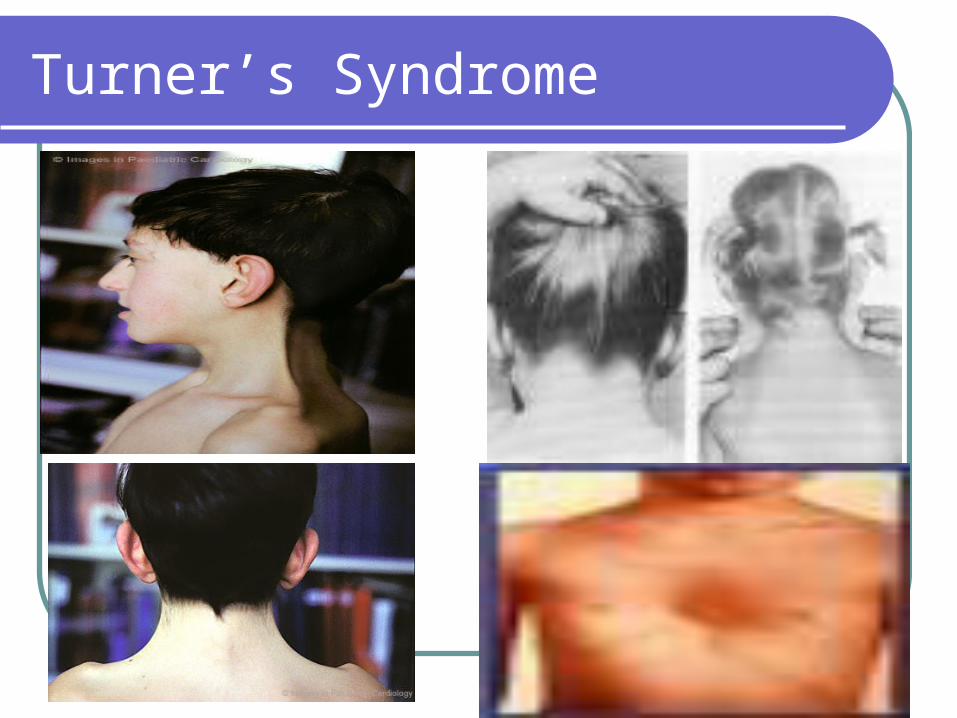
Turner’s Syndrome
Monosomy X Somatic anomalies
Short stature – less than 4’10”
Webbing of neck
Shield chest
CVS (coarctation of aorta/VSD)
Renal (horseshoe kidneys)
Bilateral lymphedema of feet due to hypoplasia of superficial vessels
Skeletal – Cubitus valgus (54%)
At birth: decreased birth weight, birth length close to 50th percentile
Normal intelligence
Turner’s Syndrome
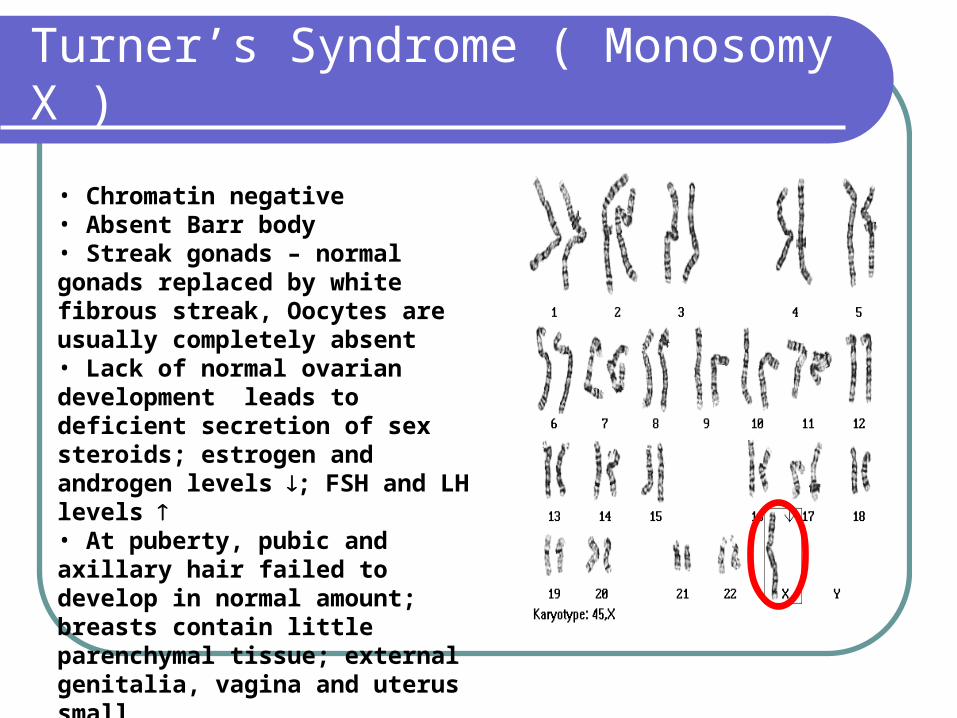
Turner’s Syndrome ( Monosomy X )
• Chromatin negative• Absent Barr body• Streak gonads – normal gonads replaced by white fibrous streak, Oocytes are usually completely absent• Lack of normal ovarian development leads to deficient secretion of sex steroids; estrogen and androgen levels ; FSH and LH levels • At puberty, pubic and axillary hair failed to develop in normal amount; breasts contain little parenchymal tissue; external genitalia, vagina and uterus small

Klinefelter’s Syndrome
1/1000 live born males Testicular feminization syndrome Androgen deficiency (lack of normal secondary sexual development) increasedgonadotropin levels (FSH LH) gynecomastia Hyalinization of seminiferous tubules Infertility External genitalia well differentiated although in neonates, may show
micro-penis Scrotum well developed, vas deferens normal, prostate small Slightly taller than normal, legs are relatively long compared with their
trunk and arms More likely to be retarded (IQ 50-85) or socially maladjusted, often
passive poorly motivated because of poor self-image
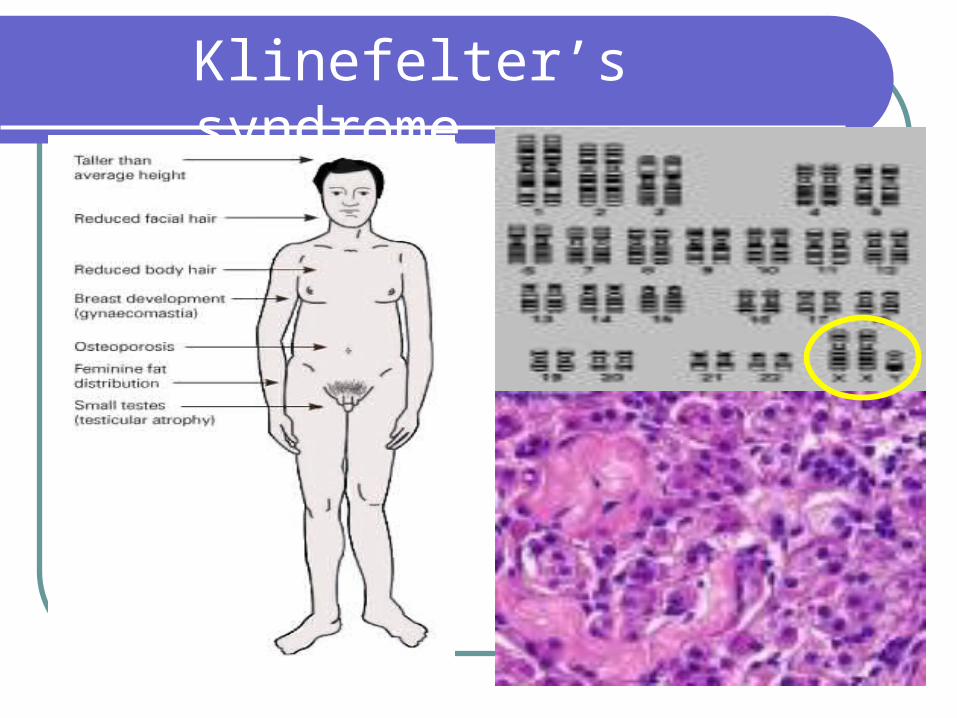
Klinefelter’s syndrome
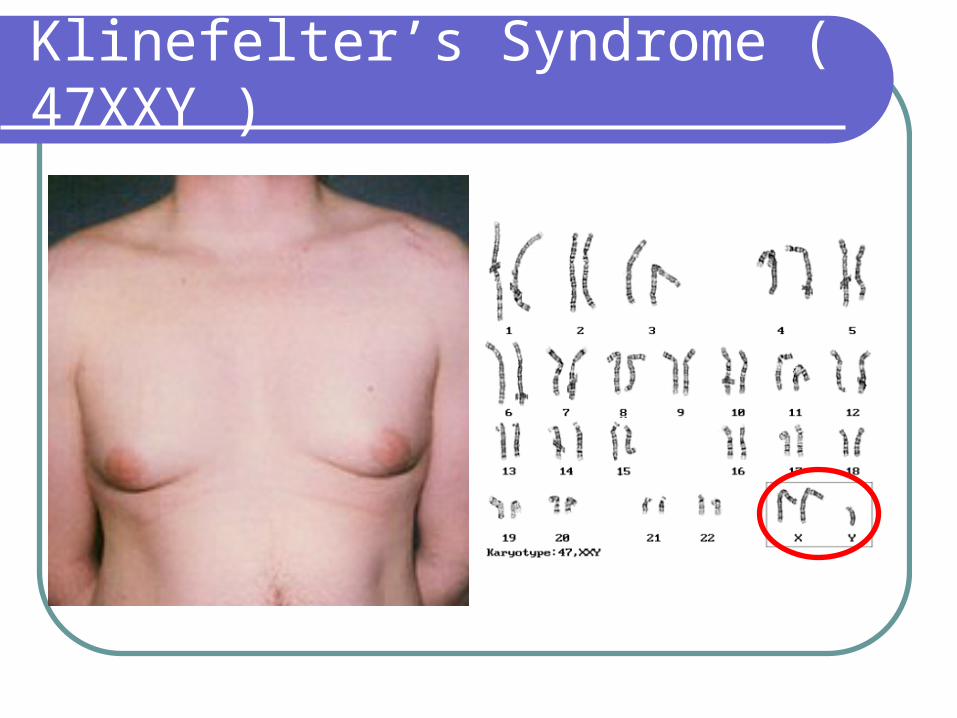
Klinefelter’s Syndrome ( 47XXY )

Triple X syndrome
1/1000 live born females Maybe associated with subnormal intelligence
– IQ is 16 points below those of their sibs, IQ is 45-70 (5%)
1/3 show some mental or behavioral problemsSuboptimal fertility Delayed menarche or premature ovarian
failure
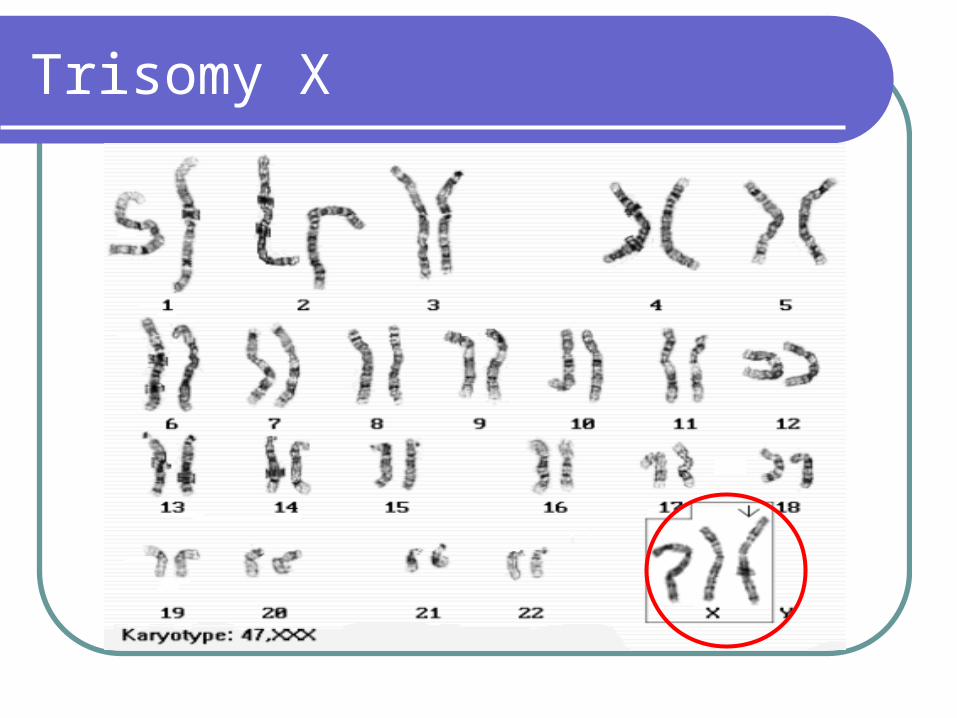
Trisomy X

XYY syndrome
Approximately 1/1000 liveborn malesTall stature Increased tendency to psychiatric problemsAntisocial behavior and mental retardationUsually have grossly normal testis and
normal external genitalia; Spermatogenic arrest is seen in approximately half the cases
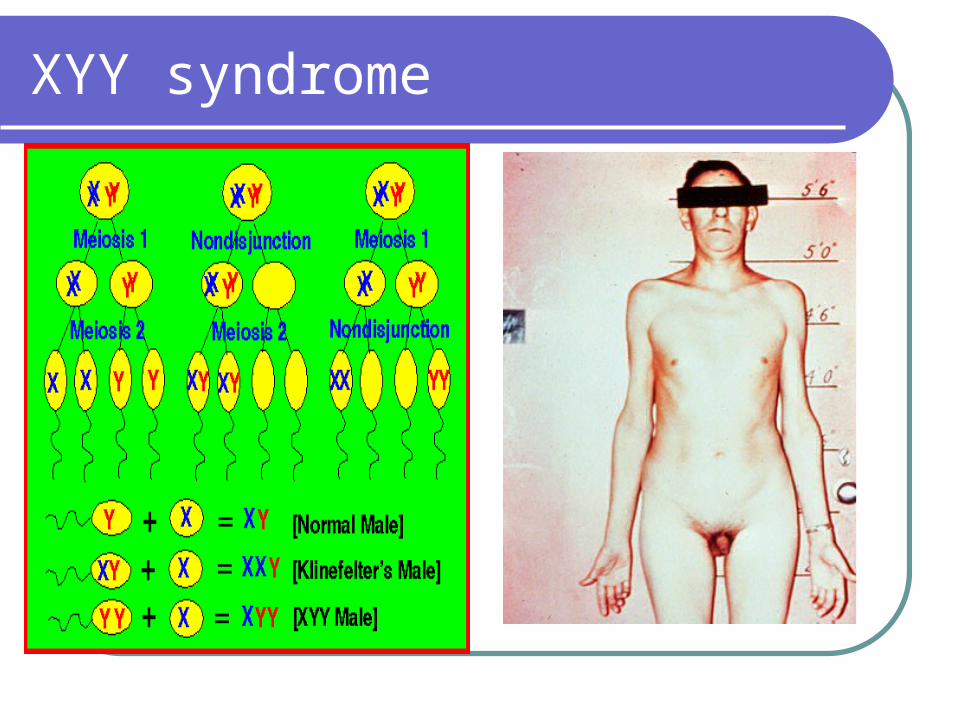
XYY syndrome
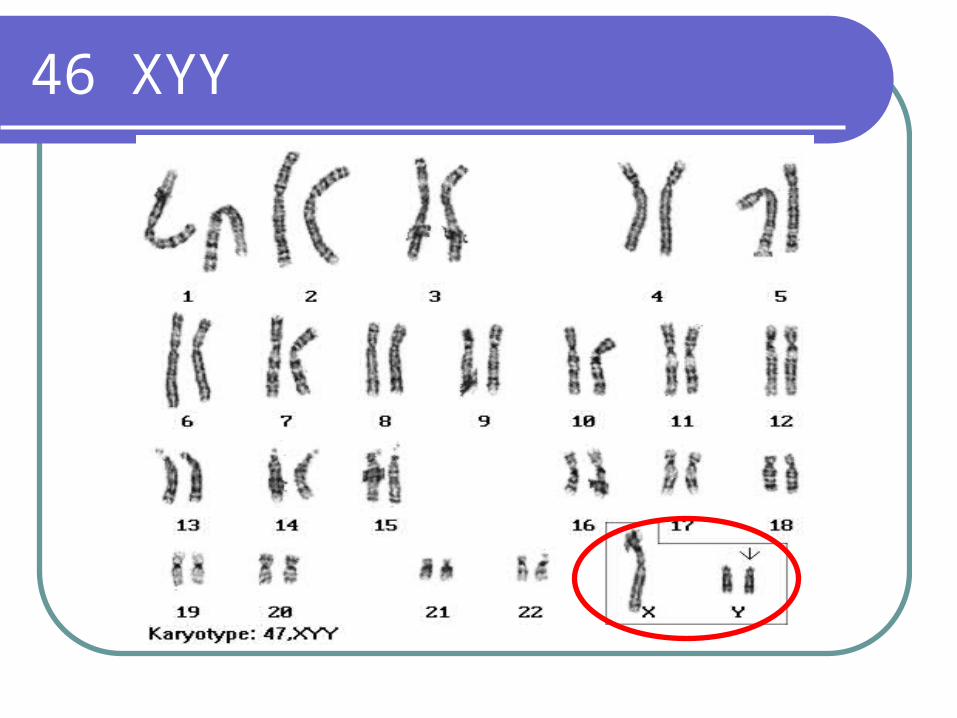
46 XYY








![Cognitive Deficits and Behavioral Disorders in Children: A ... · brain leading to cognitive, emotional or behavioral abnormalities [8]. Abnormalities of neurodevelopment, particularly](https://img.pdfslide.net/doc/110x75/5f26b465f6003d1f63082c87/cognitive-deficits-and-behavioral-disorders-in-children-a-brain-leading-to.jpg)