Embed Size (px)
Citation preview

7/23/2019 Benzene Quantum Systems
http://slidepdf.com/reader/full/benzene-quantum-systems 1/10
Benzene:
1
2
3
4
5
6
We will solve Schodinger equation for this molecule by consider ing only p-orbitals of six carbons under the Huckel
approximation. Huckel approximation, though quite crude, provides very useful results.
Since a-bonding in planar molecules has different symmetry from the ^-bonding, corresponding molecular orbitals
separate in the Hamiltonian -do not have cross off-diagonal element and thus can be solved separately. Besides,
a-bonding is much stronger than ^-bonding, ^- and ^ D molecular orbitals lie within the a ? a D gap. Thus HOMO and
LUMO orbitals are either of ^,^D-type or nonbonding n-type. All that allows considering only p z -orbitals in
conjugated molecules for treating their spectroscopic features, while s and p x ,p y orbitals will be responsible
primarily for a-bonding, i.e. molecule’s shape.
Huckel approximation can be summarized with following statements:
1) < p i | p j >= N ij - all overlap integrals are zero
2) < p i | H | p i >= J - diagonal elements equal atomic energies of carbon p z electrons
3) < p i | H | p j >= KÝneighborsÞ - the interaction energy is nonzero only for neighboring carbons.
Both, J and K, are negative and can be paraterized for a typical conjugated carbon and carbon-carbon bond. Such
a parameterization can be a lso introduced for hetero atoms, S, O, N
If we write the secular equation for benzene
det
J ? E K 0 0 0 K
K J ? E K 0 0 0
0 K J ? E K 0 0
0 0 K J ? E K 0
0 0 0 K J ? E K
K 0 0 0 K J ? E
= 0, : ?4K6+ 9K4ÝJ ? E Þ 2 ? 6K2ÝJ ? E Þ 4
+ ÝJ ? E Þ 6= 0.
The solutions can be found E = J + K, E = J ? 2K, E = J ? K, E = J + 2K. For an arbitrary cyclic molecule with identical
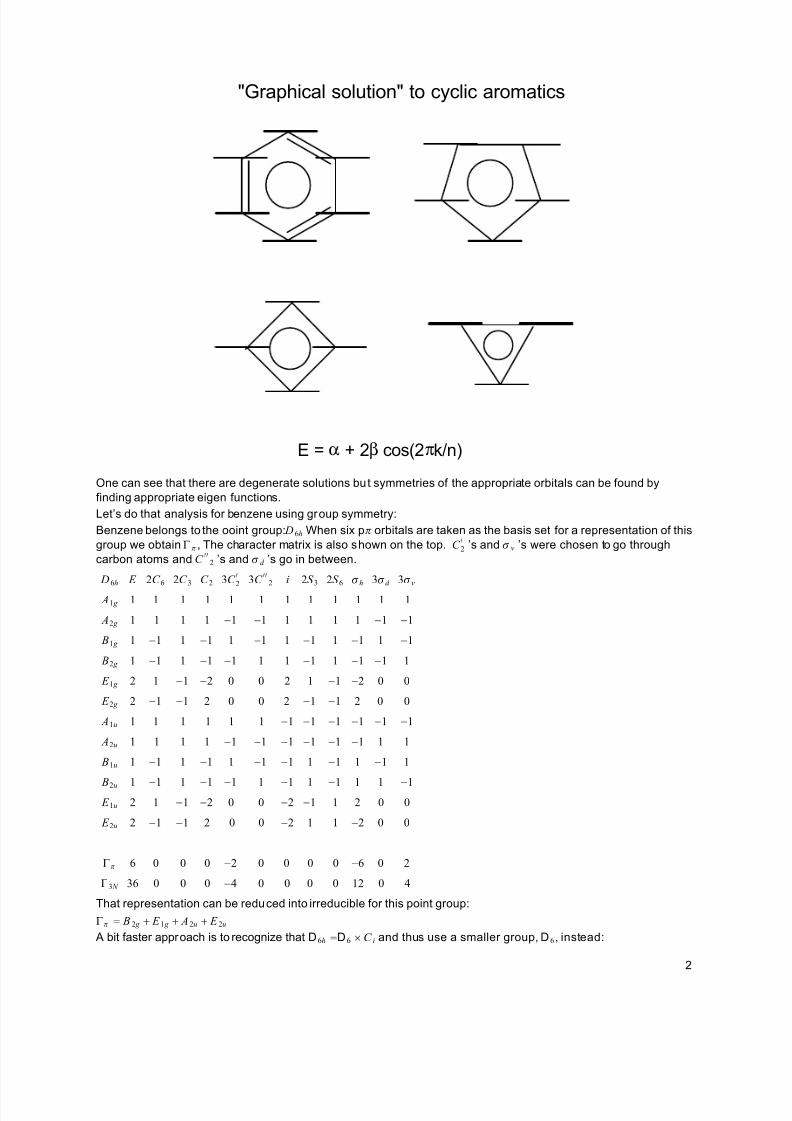
conjugated bonds, the solutions can be written as E = J + 2Kcos 2^n k and graphically represented as enegries
horizontally leveled at the corners of the appropriate polygon symmetrically placed on one of its corners:
1
7/23/2019 Benzene Quantum Systems
http://slidepdf.com/reader/full/benzene-quantum-systems 2/10
"Graphical solution" to cyclic aromatics
E = α + 2β cos(2πk/n)
One can see that there are degenerate solutions but symmetries of the appropriate orbitals can be found by
finding appropriate eigen functions.
Let’s do that analysis for benzene using group symmetry:
Benzene belongs to the ooint group: D 6h When six p^ orbitals are taken as the basis set for a representation of this
group we obtain @ ^ , The character matrix is also shown on the top. C 2v ’s and a v ’s were chosen to go through
carbon atoms and C vv
2 ’s and a d ’s go in between.
D6h E 2C 6 2C 3 C 2 3C 2v 3C vv
2 i 2S 3 2S 6 ah 3ad 3av
A1 g 1 1 1 1 1 1 1 1 1 1 1 1
A2 g 1 1 1 1 ?1 ?1 1 1 1 1 ?1 ?1
B1 g 1 ?1 1 ?1 1 ?1 1 ?1 1 ?1 1 ?1
B2 g 1 ?1 1 ?1 ?1 1 1 ?1 1 ?1 ?1 1
E 1 g 2 1 ?1 ?2 0 0 2 1 ?1 ?2 0 0
E 2 g 2 ?1 ?1 2 0 0 2 ?1 ?1 2 0 0
A1u 1 1 1 1 1 1 ?1 ?1 ?1 ?1 ?1 ?1
A2u 1 1 1 1 ?1 ?1 ?1 ?1 ?1 ?1 1 1
B1u 1 ?1 1 ?1 1 ?1 ?1 1 ?1 1 ?1 1
B2u 1 ?1 1 ?1 ?1 1 ?1 1 ?1 1 1 ?1 E 1u 2 1 ?1 ?2 0 0 ?2 ?1 1 2 0 0
E 2u 2 ?1 ?1 2 0 0 ?2 1 1 ?2 0 0
@^ 6 0 0 0 ?2 0 0 0 0 ?6 0 2
@3 N 36 0 0 0 ?4 0 0 0 0 12 0 4
That representation can be reduced into irreducible for this point group:
@^ = B 2 g + E 1 g + A2u + E 2u
A bit faster approach is to recognize that D6h =D6 × C i and thus use a smaller group, D6, instead:
2

7/23/2019 Benzene Quantum Systems
http://slidepdf.com/reader/full/benzene-quantum-systems 3/10
D6 E 2C 6 2C 3 C 2 3C 2v 3C vv
2
A1 1 1 1 1 1 1
A2 1 1 1 1 ?1 ?1
B1 1 ?1 1 ?1 1 ?1
B2 1 ?1 1 ?1 ?1 1
E 1 2 1 ?1 ?2 0 0
E 2 2 ?1 ?1 2 0 0
@^ 6 0 0 0 ?2 0
That representation can be reduced into irreducible for this point group:
@^ = B 2 + E 1 + A 2 + E 2 which can be easily ass igned g and u subscr ipt based on the required negative character
with respect tooperation a h in D 6h. Thus @ ^ = B2 g + E 1 g + A 2u + E 2u
We have not found the eigen functions nor energies yet, just their symmetries. Now the task is easier than before.
We have two options:
a)Trained eye can draw the nodal planes and assign the molecular orbitals by analysis of their characters with
respect to appropriate symmetry operations as shown below.I prematuraly put energies next to the MO, but note
that encreaing number of the nodal planes corresponds to increasing energy of MO. Also, a2u with energy J + 2K is
the lowest because both, J and K, are negative. The approach is handy but might be confusing when trying to find
those c1 and c2 coefficients to finish the construction.
b2g α − 2β
e2u α − β
e1g α + β
a2u α + 2β
c1
c1
-c1
-c1
c1
c1
-c1
-c1
c1
c2
-c1
-c2
-c1
c1
+
+ +
+
+
+
+ +
+
-
-
-
--
-
-c2
- -
c1
c1
c1
c1
c1 c1
c1
-c1-c1
-c1
-c2
b) More straight forward (also a bi t more tedious) approach is by using the projection operator:
P W => R
e WÝ RÞ DO R
Again, we’l l resort to a smaller group, D6, instead of D6h. Then projection operators would look shorter :0). For
example: P A 2 = 1O E + 1OC 6 + 1OC 6?1 + 1OC 3
?1 + 1OC 3 + 1OC 2 ? 1OC 2v Ý1Þ ? 1OC 2
v Ý2Þ ? 1OC 2v Ý3Þ ? 1OC 2
vv Ý1Þ ? 1OC 2vv Ý2Þ ? 1OC 2
vv Ý3Þ.
Using which on p 1 results in
P A 2 p1 = p 1 + p 2 + p 6 + p 3 + p 5 + p 4 + p 1 + p 5 + p 3 + p 2 + p 4 + p 6 = 2Ý p1 + p 2 + p 3 + p 4 + p 5 + p 6Þ
3

7/23/2019 Benzene Quantum Systems
http://slidepdf.com/reader/full/benzene-quantum-systems 4/10
After normalizing we obtain:S 4Ýa2uÞ = 1
6
Ý p1 + p 2 + p 3 + p 4 + p 5 + p 6Þ. The same way it can be dobe with others.
Some attention is required for doubly degenerate species. The projection operator for E 2 :
P E 2 = 2O E ? 1OC 6 ? 1OC 6?1 ? 1OC 3
?1 ? 1OC 3 + 2OC 2 .when applied to say p 1 gives:
P E 2 p1 = 2 p1 ? p 2 ? p 6 ? p 5 ? p 3 + 2 p4 .That gives only one symmetry adapted orbital,
S 5 = 1
2 3
Ý2 p1 ? p 2 ? p 6 ? p 5 ? p 3 + 2 p4Þ
Another has to be found by generating a similar one after applying P E 2 say to p2:
P E 2 p2 = 2 p2 ? p 3 ? p 1 ? p 6 ? p 4 + 2 p5
and making it or thogonal to S 5:
S 6 = 2 p2 ? p 3 ? p 1 ? p 6 ? p 4 + 2 p5 ? S 5 < S 5 |2 p2 ? p 3 ? p 1 ? p 6 ? p 4 + 2 p5 >=
= 2 p2 ? p 3 ? p 1 ? p 6 ? p 4 + 2 p5 ? 1
2 3
2
Ý2 p1 ? p 2 ? p 6 ? p 5 ? p 3 + 2 p4Þ ?6 =
= 3
2 Ý p2 ? p 3 + p 5 ? p 6 Þ
Analogously: P E 1 = 2O E + 1OC 6 + 1OC 6?1 ? 1OC 3
?1 ? 1OC 3 ? 2OC 2 and
P E 1 p1 = 2 p1 + p 2 + p 6 ? p 5 ? p 3 ? 2 p4 ; =>S 2 = 1
2 3
Ý2 p1 + p 2 ? p 3 ? 2 p4 ? p 5 + p 6Þ
P E 1 p2 = 2 p2 + p 3 + p 1 ? p 6 ? p 4 ? 2 p5 and
S 3 = 2 p2 + p 3 + p 1 ? p 6 ? p 4 ? 2 p5 ? 1
2 3
2
Ý2 p1 + p 2 ? p 3 ? 2 p4 ? p 5 + p 6ÞÝ6Þ =
= 3
2 p2 + 3
2 p3 ?
3
2 p5 ?
3
2 p6 = 3
2 Ý p2 + p 3 ? p 5 ? p 6 Þ
B2 g S 1 = 1
6
Ý p1 ? p 2 + p 3 ? p 4 + p 5 ? p 6Þ
E 1 g S 2 = 1
2 3
Ý2 p1 + p 2 ? p 3 ? 2 p4 ? p 5 + p 6Þ
S 3 = 1
2 Ý p2 + p 3 ? p 5 ? p 6 Þ
A2u S 4 = 1
6
Ý p1 + p 2 + p 3 + p 4 + p 5 + p 6Þ
E 2u S 5 = 1
2 3
Ý2 p1 ? p 2 ? p 3 + 2 p4 ? p 5 ? p 6Þ
S 6 = 1
2 Ý p2 ? p 3 + p 5 ? p 6 Þ
So we found the eigen functions which graphically are represented below with shaded areas corresponding to a
negative sign and the size of each circle resembl ing the value of coefficient in the eigen function.
4

7/23/2019 Benzene Quantum Systems
http://slidepdf.com/reader/full/benzene-quantum-systems 5/10
a2u
e1g
e2u
b2g
The energies of these MOcan be found by evaluating appropriate < S i | H |S i >:
E(b2 g ) = < S 1 | H |S 1 >=< 1
6
Ý p1 ? p 2 + p 3 ? p 4 + p 5 ? p 6Þ| H | 1
6
Ý p1 ? p 2 + p 3 ? p 4 + p 5 ? p 6Þ >=
= 1
6
< p1 | H |Ý p1 ? p 2 + p 3-p 4 +p 5 ? p 6Þ > ? < p2 | H |Ý p1 ? p 2 + p 3 -p 4 +p 5-p 6
Þ > +
+ < p3 | H |Ý p 1 ? p 2 + p 3 ? p 4 + p 5-p 6
Þ > ? < p4 | H |Ý p 1-p 2 + p 3 ? p 4 + p 5 ? p 6
Þ > +
< p5 | H |Ý p 1-p 2 +p 3 ? p 4 + p 5 ? p 6Þ > ? < p6 | H |Ý p1 -p 2 +p 3-p 4
+ p 5 ? p 6Þ >
=
matrix element for highlighted orbitals are zero
= 1
6
< p1 | H |Ý p1 ? p 2 ? p 6Þ > ? < p2 | H |Ý p1 ? p 2 + p 3Þ > + < p3 | H |Ý? p2 + p 3 ? p 4Þ > ?
? < p4 | H |Ý p3 ? p 4 + p 5Þ > + < p5 | H |Ý? p4 + p 5 ? p 6Þ > ? < p6 | H |Ý p1 + p 5 ? p 6Þ >
=
= 1
66 J ? 2K = J ? 2K
Energies of other orbitals are calculated the same way:
E Ýb2 g Þ S 1 = 1
6
Ý p1 ? p 2 + p 3 ? p 4 + p 5 ? p 6Þ
E Ýe1 g Þ S 2 = 1
2 3
Ý2 p1 + p 2 ? p 3 ? 2 p4 ? p 5 + p 6Þ
S 3 = 1
2 Ý p2 + p 3 ? p 5 ? p 6 Þ
E Ýa2uÞ = S 4 =
1
6 Ý p1 + p 2 + p 3 + p 4 + p 5 + p 6Þ
E Ýe2uÞ S 5 = 1
2 3
Ý2 p1 ? p 2 ? p 3 + 2 p4 ? p 5 ? p 6Þ
S 6 = 1
2 Ý p2 ? p 3 + p 5 ? p 6 Þ
5

7/23/2019 Benzene Quantum Systems
http://slidepdf.com/reader/full/benzene-quantum-systems 6/10
a1u
2β : a1u
= 0.408(p1+ p
2+p
3+p
4+p
5+p
6)
α
e1g
β : e1g
(1) = 0.289(2p1+ p
2−p
3−2p
4−p
5+p
6); e
1g
(2) = 0.5( p2+p
3−p
5−p
6)
b2g
-2β : b2g
= 0.408(p1− p
2+p
3−p
4+p
5−p
6)
e2u
-β : e2u
(1) = 0.289(2p1− p
2−p
3+2p
4−p
5−p
6); e
2u
(2) = 0.5( p2−p
3+p
5−p
6)
We see that MOenergies are symmetric with respect tovalue of J. All planar conjugated hydrocarbons with suchproperty are called alternant. Alternant hydrocarbons can be recognized by their ability to have all carbons labeled
in two colors alternatingly (thus the name), i.e. when each neighbor has a different color.
The ground state configuration is Ýa2uÞ2Ýe1 g Þ
4=
æ X
1 A1 g . Note that this ground state energy is lower than that of
cyclohexatriene (benzene with three localized double bonds). The effect of sharing ^-electrons, the so -called
delocalization or resonance energy, equals 2K.
The lowest excited state configuration is Ýa2uÞ2Ýe1 g Þ
3Ýe2uÞ1 (with energy 2K above the ground state) from which
following states can be constructed:
Ýe1 g Þ × Ýe2uÞ = B1u + B2u + E 1u .each of which can be either a singlet or a triplet, i.e.:
b2g
e2u
e1g
a1u
b2g
e2u
e1g
a1u
b2g
e2u
e1g
a1u
X:1 A1g
1B2u
, 1B1u
, 1E1u
3B2u
, 3B1u
, 3E1u
At this level of comlexity we cannot choose which excited state is the lowest energy, 1 B1u , 1 B2u , or 1 E 1u (all we know
is that they each have corresponding triplet of lower energy 3 B1u < 1 B1u , 3 B2u < 1 B2u , and 3 E 1u < 1 E 1u). Wehave toanalyse other higher energy one-electron excited state configurations and find if there are any of the sam e
symmetry as the three under consideration. Configurational interaction between configurations of the same
symmetry lowers the appropriate lower energy state while increasing the corresponding high energy state energy.
The next one electron excited state configurations (with energy 3K) are:Ýa2uÞ2Ýe1 g Þ
3Ýb2 g Þ1 and Ýa2uÞ
1Ýe1 g Þ4Ýe2uÞ
1 .
Neither of them has a proper symmetry. The former makes: e1 g × b 2 g = e 2 g , while the latter: a2u × e 2u = e 2 g
6

7/23/2019 Benzene Quantum Systems
http://slidepdf.com/reader/full/benzene-quantum-systems 7/10
E2g
E2g
B2u
3β 3β 4β
We shall include even higher one electron excited state configuration with energy 4K. There is only one,
Ýa2uÞ1Ýe1 g Þ
4Ýb2 g Þ1 , which produces a 2u × b 2 g = b 2u. Thus, out of the three states, 1 B1u, 1 B2u , and 1 E 1u, only 1 B2u has
two configurations. Consequently, it will be the lowest energy state.
7

7/23/2019 Benzene Quantum Systems
http://slidepdf.com/reader/full/benzene-quantum-systems 8/10
Since in the D 6h group the dipole moment components behave like E1u (x,y) and A2u (z), the only dipole allowed
transition is C 1 E 1u ?
æ X
1 A1 g which indeed shows a high extinction coefficient, while the lower energy transitions, 1 B1u
and 1 B2u, are electronically forbidden. They are observed anyway but due to so call vibronic coupling.
Before that let’s consider electric dipole allowed transitions first. The matrix element for the transition moment is:
M evvve"v" =< fe v v v
D |W|fe vv v vv >=< fe v
D |W|fe vv >< fv v
D |fv vv >= M eve" < fv 1
v
D |fv 1vv >< f
v 2v
D |fv 2vv >.. .
The selection rules in this case are defined by the selection rules for electronic part M eve" and the Franck-Condon
factor, < fv v
D |fv vv >2 , for vibrational part. The latter would be nonzero if that integrand has the totally symmetric
species, which means that for totally symmetric vibrations all vibrations are allowed:
Av = 0, ±1, ±2, ±3,...
For all other vibrations the totally symmetric integrand appears only for even difference in vibrational number, i.e.
when:
Av = 0, ±2, ±4,...
Incases like the lowest energy transition in benzene, which is electronically forbidden, the only way the matrix
element for the transition moment would be nonzero is to step back from the Born-Oppenheimer approximation
and consider mixing nuclear (vibrational) and electronic coordinates, so called vibronic coupling.
The electronic Hamiltonian parametrically depends on on vibrational coordinates:
H e = H e0
+ >i
/ H e/Q i
Q i +.. = H e0
+ H v
The excited state wavefunction f f 0 becomes accordinly mixed with other zero-order electronic states :
fe v = f f 0
+ >k c k fk
0
where the degree of mixing, ck
= <fk
0 | H v |f f 0
>
E f 0? E k 0
, depends on both, the vibronic coupling element < fk
0 | H v|f f
0> and the
energy separation between the states E f 0 ? E k
0 . The electronic transition moment becomes:
M eve" =< fe v |W|fe vv >=< f f 0 |W|f
e vv
0> +>
k c k < fk
0 |W|fe vv
0>
While the first term (0-0 transition) is zero, the other terms might be not. Then the intensity of transition would
become nonzero, or as it is often referred to as intensity is borrowed from a neigboring transition. That is why B1u
transition, beeing closer to the allowed E1u, is more intense than B2u. The energy factor E f 0 ? E k
0 is not the only
one responsible for nonzero c k , the other part , < fk 0 | H v |f f
0>, enforces additional selection rules:
< fk 0 | H v|f f
0>=
/ H e/Q i Q i =0
< fk 0 |Q i|f f
0>
or Av = ±1ݱ3,±5,...Þ for vibronicaly active mode, the mode for which the symmetry of @ fk 0
× @Q i matches that of the
ground stateæ X . The rules for other vibrational states would follow the rules of ordinary Franck-Condon factor
described above. Incase of benzene, vibronically allowed transition should be a 2u and e1u. For theæ A
1 B2u state it
translates to a reqire for either b 1 g b1 g × B2u = B2u or e2 g e2 g × B2u = E 1u . There are 12×3-6=30 vibrational
modes in benzene, which for the ground state would be realized in 20 fundamental frequencies:2a1 g + a 2 g + a 2u + 2b1u + 2b2 g + 2b2u + e 1 g + 3e1u + 4e2 g + 2e2u. From these modes only e2 g satisfy the selection rules
(X 15Ýe2 g Þ through X 18Ýe2 g Þ) for vibronic transition. By symmetry all four of them can contribute and those that have
the highest value of / H e/Q i
contribute the most. C-H vibrations do not affect the energies of ^-electrons very much,
it that should be a mode that shakes the molecular sceleton associated with ^-system. A likely candidate is C-C-C
in plane bending, X18Ýe2 g Þ, with energy ca. 500-600 cm ?1 (606 cm ?1 in the ground state and 522 cm ?1 in the
electronically first excited state). Thus the excited vibronic transitions that should be observed are:æ A
1 B2u18 0
1 - transition from vibrational ground state in theæ X
1 A1 g to
æ A
1 B2u at vibrationally excited state with v"=1 at
X 18Ýe2 g Þ.
Another possibility is to observe a hotæ A
1 B2u18 1
0 transition from the vibrationally excited with v’=1 at X 18Ýe2 g Þæ X
1 A1 g to
theæ A
1 B2u and v"=0 at X 18Ýe2 g Þ. The latter transition is very temperature dependent (hot). What about combinational
transitions? Again, the other vibrations have to be involved in r ing distortion vibrations (preferably C-C stretching
since it affects the Hamiltonian the most) and posses appropriate symmetry, which can be achived by combining
X 18Ýe2 g Þ with totally symmetric combinations of the ring distortion vibrations such as X 2Ýa1 g Þ (992 cm?1 in the ground
state and 923 cm ?1 in the electronically first excited state).
As a result , a series of vibronic transitions æ A
1 B2u20
m18 01 appears as a famous benzene spectrum near 260 nm.
8

7/23/2019 Benzene Quantum Systems
http://slidepdf.com/reader/full/benzene-quantum-systems 9/10
9

7/23/2019 Benzene Quantum Systems
http://slidepdf.com/reader/full/benzene-quantum-systems 10/10
10