Embed Size (px)
DESCRIPTION
article
Citation preview

BIFUNCTIONAL CHEMISTRY • “Organic Chemistry”, J. Clayden, N. Greeves, S. Warren and P. Wothers, Oxford
University Press. Mainly Chapters 21, 26, 27, 28, 10, 29. • Background / revision: Chapters 6, 12, 14 (reactions of aldehydes, ketones and
esters – lectures from year 1) • Similar chapters will be found in all “Organic Chemistry” texts, e.g. T. Solomons
& C. Fryhle, F. Carey etc. • Also recommended is “Chemisty Of The Carbonyl Group, A Programmed
Approach To Organic Reaction Mechanisms”, S. Warren (Wiley, 1974) - but only one copy in the library and now out of print!
• Bifunctional Compounds, RS Ward, OP (Oxford Primer No. 17): useful in parts
CONTENTS OVERVIEW Tautomers
Reactions of enols: C-Hal and C-N=O bond formation Reactions of enols and enolates with aldehydes and ketones Reactions of enolates with acylating agents Reactions of enolates with alkylating agents Reactions of enamines Conjugate addition reactions
1. Tautomers and evidence for tautomerism 2. Carbonyl group tautomers 3. Factors affecting tautomeric stability 4. Stable enols and other tautomers 5. Reactions implicating tautomers 6. Acid and base catalysis of enolisation 7. Implications of tautomersisation: racemisation and C=C isomerisation 8. Stable “enol equivalents” 9. Halogenation of enols and enolates 10. Nitrosation of enols 11. Base and acid catalysed aldol reactions 12. Dehydration of β-hydroxy carbonyl compounds 13. Crossed condensations: Knoevenagel condensation 14. The Henry (nitroaldol) reaction 15. The Claisen and Dieckmann condensations 16. Decarboxylation reactions 17. Problems with crossed condensations and alkylations 18. Lithium enolates 19. Malonate and β-keto esters (1,3-dicarbonyls) 20. Alkylation of nitriles and nitronates 21. Conjugate addition reactions 22. The Michael reaction – conjugate addition reactions with enolates 23. Conjugate additions reactions with enamines, nitronates and nitrile anions 24. The Robinson annulation 25. Acylation and alkylation of enamines

How can a "pure" compound be a mixture of two (or more) molecules?
acetyl acetone(pentan-2,4-dione) H3C
CCH2
C
O O
CH3
H3CC
CH2
C
O O
CH3 H3CC
CH
C
O O
CH3 H3CC
CH
C
O O
CH3
H H
16% "keto" form 84% "enol" form
2.24δ 2.24δ3.60δ 2.05δ5.50δ
14.5δ
These molecules exist in a dynamic equilibrium. Neither can be isolated on its own.
These molecules are referred to as tautomers
acidic OHH-C=
-CH-CH3-
H3CC
CC
O O
CH3
H
H

(NB for the life of me I cannot get this picture to paste in properly. You have the picture in the lecture handouts!)
1H-NMR spectroscopy using D2O as co-solvent
NMR using CDCl3 as solvent
OO
2.6
3.5
2.2
8.2
1.1
5.5
OHO
H
1.0
2.2
246 0
246 0
add D2Oshake sample tube, wait....re-reun NMR spectrum
D2O
For the C-H to be acidic enough to be lost easily it must be α to ("next door") a carbonyl group. If it is α to two different carbonyl groups it will be even more acidic.
acidic OH
OOODO
D D D
α
CH2 α to two C=O
α

Tautomers
Compounds whose structures differ markedly in arrangement of atoms, but which exist in equilibrium are called TAUTOMERS. This is not to be confused with resonance which refers to movement of electrons only.
The atoms that 'move' are always protons. A variety of factors will determine the stability of each tautomer and therefore the relative abundance of each. Consideration of acid-base reactions can be useful for simple examples.
Keto-enol tautomerism
HC
CCH3
O
HH
HC
CCH3
OH
H
Unsymmetrical ketones
H3CC
CCH2
O
HH
H3CC
CCH3
OH
H
H3CC
CCH2
OH
HH
H
Energy(kJ/mol)
enol
keto
C H C H O H OHKa1 Ka2
pKa1 pKa2
more substituted C=C less substituted C=C
Ea
∆H
Ea
keto form - weaker acid enol form - stronger acid
keto form still more stable than both enols
reflects difference in energy between tautomers
reflects rate of interconversion between tautomers
the keto tautomer for a simple ketone is ~85 kJmole-1 more stable than its enol (can be thought of as reflecting difference in sum of bond energies).
>~

Evidence for the existence of tautomers
Effect of solvent
CH2
CCH3
O
CH3C
O
CC
CH3
O
CH3C
OH
H
Solvent
water
acetonitrile
(neat liquid)
hexane
% Enol
15%
58%
76%
92%
Observations
Multiple H-bonding opportunities between ketone tautomer and solvent
Solvent forms a non-polar cage around the molecule. Stabilization from intramolecular H-bonding only.
H3CC
CH
C
O O
CH3
H
H3CC
CH
C
O O
CH3
base
H3CC
CH
C
O O
CH3
H3CC
HC
C
O O
CH3
Cu2+Cu(II)
The tautomeric mixture may be affected by solvent. A hydrogen bond can be worth 20-30 kJmol-1
CH2
CCH3
O
CH2C
OH
ACAC anion
The ACAC anion is a good bidentate ligand for making
metal complexes
polar protic
non- polar
polar aprotic
H-bonding
conjugation
acidic H easily removed by a weak base
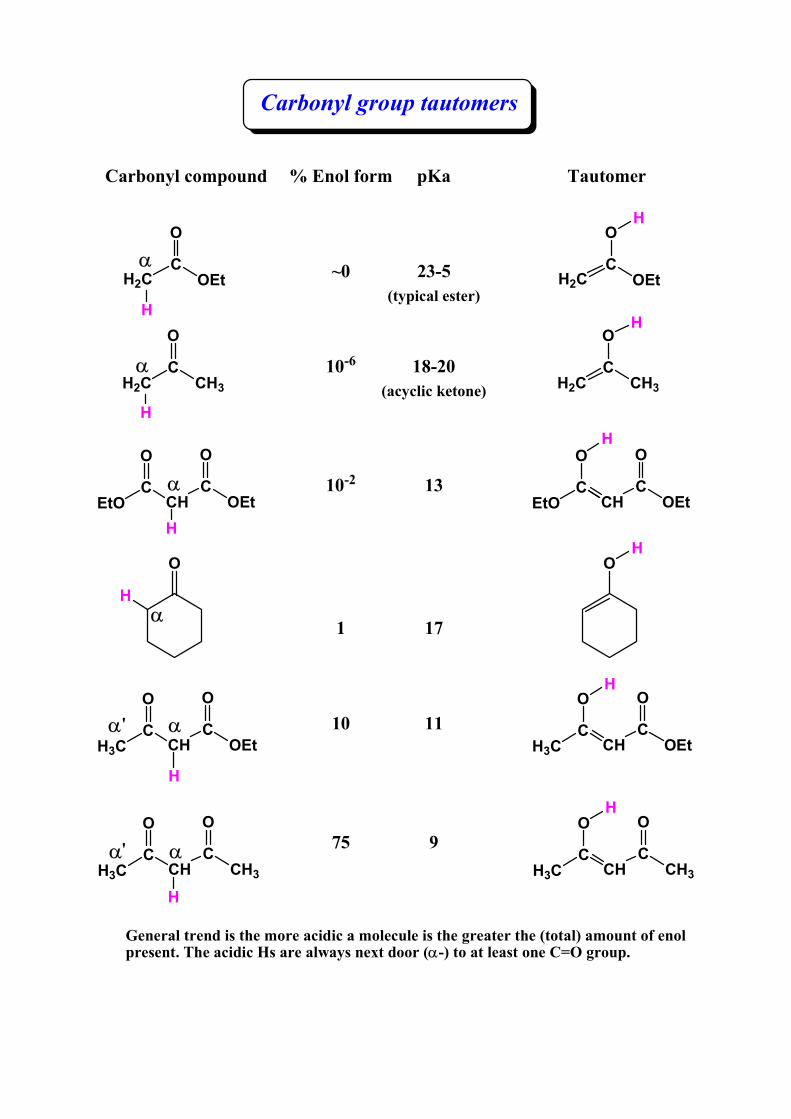
Carbonyl group tautomers
Carbonyl compound % Enol form
~0
10-6
10-2
1
10
75
Tautomer
H2CC
CH3
O
CHC
OEt
O
CEtO
O
O
CHC
OEt
O
CH3C
O
CHC
CH3
O
CH3C
O
H2CC
OEt
O
pKa
23-5(typical ester)
18-20(acyclic ketone)
13
17
11
9
H
H
H
H
H
H
General trend is the more acidic a molecule is the greater the (total) amount of enol present. The acidic Hs are always next door (α-) to at least one C=O group.
H2CC
CH3
O
CHC
OEt
O
CEtO
O
O
CHC
OEt
O
CH3C
O
CHC
CH3
O
CH3C
O
H2CC
OEt
OH
H
H
H
H
H
α
α
α
α
αα
αα'
'

Tautomeric Stability
Carbonyl compound Tautomer
CH2
COEt
O
CN
CH2
CCH3
O
CH3C
O
CC
OEt
O
CHN
H
CH2
COEt
O
CH3C
O
CC
OEt
O
CH3C
OH
H
CC
CH3
O
CH3C
OH
H
Notes
CC
CH3
O
CH3C
OH
H
CH2
CCH2
O
CH3C
OH
IRO
O
double bonds brought into conjugation
H-bonding and conjugation
1720cm-1 1740cm-1 unsaturated ester ν = 1640cm-1
H-bonding and conjugation
H-bonding but no conjugation = less stable enol

Stable enols
Vitamin C (ascorbic acid) - antioxidant protects body from stray reactive oxygen species
A77-1726 - the active metabolite of arthritis drug leflunomide (AravaTM)
Phenol
OH
O
OHHO
HN
O
O
N
CH3
Leflunomide (AravaTM)
O
OHHO
H
Some molecules exist entirely in their enol form.
Loss of aromatic stabilization in keto form - a big energy penalty.(the keto form is however implied as important intermediates in some reactions - see Yr 3)
reduced form
O
OO
O
OHHO
H
oxidised form
keto forms
p-CF3C6H4
HN
O
OH
N
C
CH3
A77-1726 - the active drug species
in vivo
[oxidation]
p-CF3C6H4
O
H
O
OHO
O
OHHO
H
O
OHO
O
OHHO
H
H
H
H
p-CF3C6H4
HN
O
O
N
C
CH3H
α
hydrogen is α- to three C-X multiple bonds - very acidic

HC
CR
N
HH
HC
CR
N
H
R1 H R1
R = H, aldimineR = alkyl, ketimine
Imine-enamine tautomerism
Nitro-acid tautomerism
More Tautomers plus relative stability for simple examples
Amide-iminol tautomerism
HC
CN
O
HH
R HC
CN
OH
HH
R HC
CNH
OH
H
R
H
HC
NO
HH
O
HC
NO
H
OH
this is very stable as the nitrogen lone pair is
delocalised (resonance) into the carbonyl group (without
H moving of course)
The NH is more acidic than the CH as nitrogen is more electronegative than carbon.
very similar to keto-enol tautomerisation
in the nitro form the O is resonance delocalized between two oxygens (rather than O and C) so is more stable that the aci tautomer.
GENERAL CASE
YZ
X
YZ
X
H
HX,Y,Z = C, N, O, S, P.....
COMPOUNDS MUST BE IN EQUILIBRIUM TO BE CONSIDERED AS TAUTOMERS
>stability
enamine
nitro
>stability
aci
>>stability
>
HC
CN
O
HH
R
H
amide iminol enol
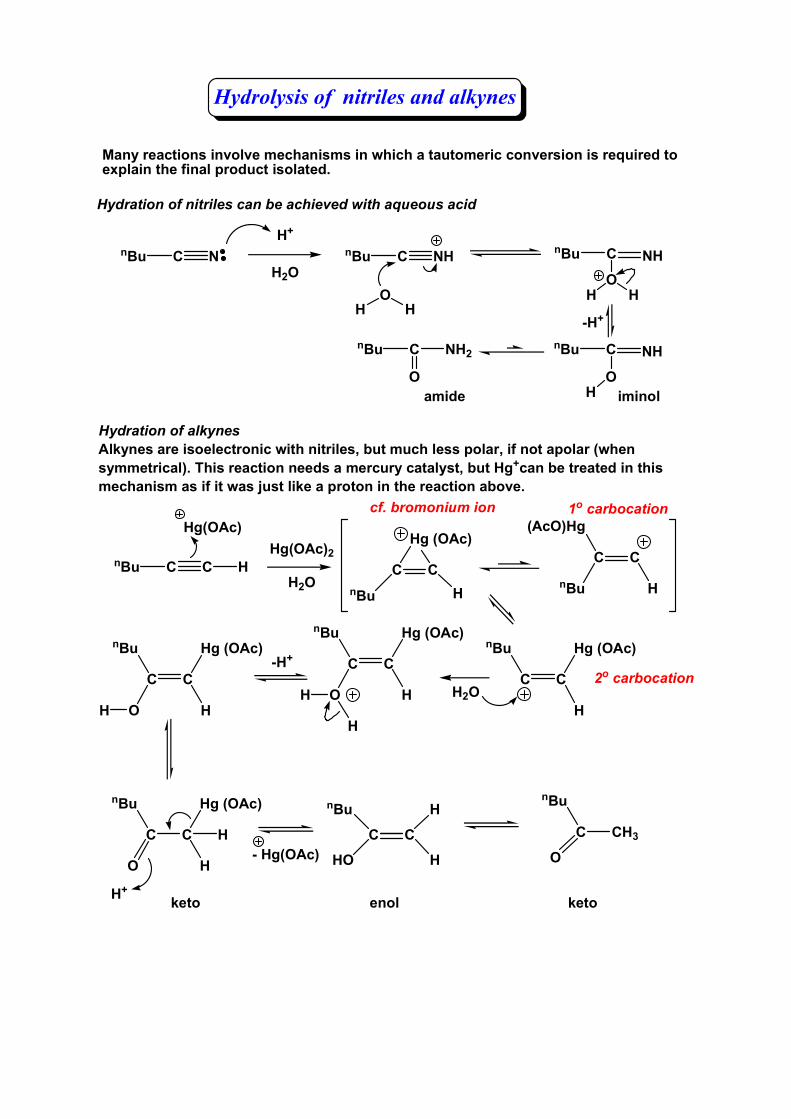
Hydrolysis of nitriles and alkynes
Hydration of alkynesAlkynes are isoelectronic with nitriles, but much less polar, if not apolar (when symmetrical). This reaction needs a mercury catalyst, but Hg+can be treated in this mechanism as if it was just like a proton in the reaction above.
C C HnBu C CHnBu
C C
Hg (OAc)Hg(OAc)2
H2O
H
Hg (OAc)nBu
C C
(AcO)Hg
HnBu
Hg(OAc)
Many reactions involve mechanisms in which a tautomeric conversion is required to explain the final product isolated.
2o carbocation
1o carbocation
H2O
C C
H
Hg (OAc)nBu
O
H
H
C NnBu
Hydration of nitriles can be achieved with aqueous acid
H+
H2OC NHnBu C NHnBu
OH H
C NHnBu
OH
C C
H
Hg (OAc)nBu
OH
-H+
cf. bromonium ion
HO
H-H+
iminol
C NH2nBu
Oamide
C C
H
Hg (OAc)nBu
O
H
H+
C C
H
nBu
HO
H
- Hg(OAc)C CH3
nBu
O
keto enol keto

Chemistry of Enols and Enolates
Acid catalysis
HC
CCH3
O
HH
The rate of interconversion between keto and enol tautomerscan be increased by acid or base catalysis (lowers Ea), howeverthe position of the equilibrium does not change. The enoltautomeric form (or more reactive enolate) is often implicated in organic reactions and used extensively in reaction mechanisms.
HC
CCH3
O
HH
HH
Base catalysis
HC
CCH3
O
HH
HC
CCH3
O
H
HO
H3CC
CC
O
H H
H
HH
OH
H
Energy (kJ/mol)
enol
keto
Ea
ENOLATE - more reactive than the enol due to its charge.
more lesssubstituted
Unsymmetrical ketones
Neither enol is formed 100% exclusively under all conditions. It is a question of which is favoured and to what extent.
HC
CCH3
O
H
H
HC
CCH3
O
H
HOH
HC
CCH3
O
H
H
H3CC
CCH3
O
HH
H
H3CC
CCH3
O
H
H
moresubstituted
H3CC
CCH2
O
HH
H3CC
CCH2
O
HH
H3CC
CCH2
O
H
H
Hless
substitutedgeneration of δ− on less substituted carbon preferred as no +I Me group.
fastest formed (kinetic) enolate

Implications of enolisation
Racemisationif the α-carbon is a sterogenic centre, then enolisation can result in complete racemisation
Isomerisation of double bondsAs well as the normal acid-catalysed migration of double bonds (middle example), isomerisation can be achieved via the enol (acidic conditions) or enolate (basic conditions).
Ph
H3C H
O
OMe
OH
H
R
AcHN H
O
OH
heating in AcOH
during recrystallization
NaOMe
Base catalysed
Acid catalysed
H
H
OH
Ph
CH3
O
OMe
OHH
H
OH
H
O
H
H
O
H
O
H
H
O
H
H
O
H
O
H
H
H
Ph
NHAc
OH
OH
H OMePh
H3C H
O
OMe
Ph
H3C H
O
OMe
+
Ph
NHAc
O
OH
H+ source
HHO
H OH2

Enol ethers and Enamines
Enol ethers - "protection" of aldehydes and ketones
O
H H+
EtOH
Enol ethers and enamines are stable, "protected" forms of aldehydes and ketones. They are reactive towards acid and water, which returns the starting carbonyl compound.
XC
CR1
R2
R3
R4
X = O, R1 = H, X = NH or NR
Enamines -can be made by reaction of aldehydes/ketones with a secondary amine. Enamines are useful compounds for a wide range of transformations. They can be used in place of enols and enolates as will be illusrated later on in the course
O
HN H+ catalyst
OH
H
OH
OEt
H
EtOH+ H+
-H+
OH2
OEt
OEt
OEt
OEt
H
OEt
OEt H
+ H+
-H+
OEt
-EtOH - remove by distillation
HOEt
ENOL ETHER
DIETHYL ACETAL
rememberH+/ROH/RCHO > acetalH+/H2O/RCH(OR)2 > aldehyde
OH HN HO HN
H2O NN-H2O
(removed by Dean Stark trap
to drive eqbm. to product)
IMINIUM ION
H
N
ENAMINE

Acid-catalysed Halogenation of Enols
RCH2C
CH3
OR
CC
CH3
O
H
H
H
AcOH
Ph
O 0.75 eq. AlCl3
Br2
Me3SiO
Halogenation with Lewis acid catalysis. Under these conditions the enol is more reactive than the benzene ring towards electrophilic subsitution.
Bromination of silyl enol ethers can be acheived with just bromine
Tautomerisation with a Bronsted acid catalysis produces the more substituted enol. Further iodination is hampered by steric hindrance of the corresponding iodo-enol.
Br BrMe3SiO
Br
O
Br
Me3SiBr
(NB Markovnikov-like: addition to give more stable cation)
O
Br
The Hell-Volhard Zelinsky reaction is a useful way to derivatise carboxylic acids via an enol.
ROH
O
RBr
O
Br2, PPBr3 H3PO3
RBr
O
Br Br
H
RBr
O
Br
RBr
OR
OH
O
Br
RBr
O
Br
Br2, P (cat.)-H3PO3
overallprocess
R OH
Obromide exchange with
unreacted acid
I I
I2
RCHC
CH3
O
I
Ph
OAlCl3
Br Br
Ph
O
Br
made in situ or added
PBr3
-Me3SiBr

Base-catalysed halogenation of Enolates
OH
CH3CH2C
CH3
O
CH3CH2C
CH2
O
The Iodoform Reaction (a visual test for methyl ketones)
RC
CH3
O aq. NaOHexcess I2
The base catalysed halogenation of ketones occurs through the more reactive enolate (i.e. not the enol). In unsymmetrical ketones the less hindered, more acidic proton is removed preferentially.
The product α-bromo ketone possesses an α-CH more acidic than in the starting material due to the presence of the electron withdrawing bromine. This means the product is more easily converted to the enolate. Despite steric hindrance, polyhalogenation can occur (it is impossible to isolate the mono bromo product in good yield) and this is the basis for the iodoform test.
enolate
CH3CH2C
CH2
OHenol
enolate more reactive than enol due to charge
Br BrCH3CH2
CCH2
O
Br
reactionalso possible with iodine
= faster
RC
C
O
Imech.
as above HH
>
-I halogen increases acidity of remaining α-Hs
repeat x 2R
CC
O
I
II
HO
RC
CI3
O
OHR
CO
O
CHI3
RC
OH
O
CI3
soluble carboxylate salt plus preciptate of yellow iodoform

Nitrosation of enols
HC
CR
N
HH
HC
CR
N
H
OHONitroso-oxime tautomerism
H
H
H3CC
CH2
CO2Et
O
NaNO2, HCl NO+
H3CC
CH
CO2Et
OHClH
Nitrosation of enols can be achived using conditions that generate NO (cf diazonium salt formation)
NaNO2
O O
NO
H O
N
OH
HCl
NaNO2
Synthesis of 1,2-diketones by oxime hydrolysis
HCl
H2O, heat
Synthesis of α-amino acids by oxime reduction
EtOC
CH2
C
OHCl
NaNO2
O
OEt EtOC
CC
O O
OEtN
O
EtOC
CC
O O
OEt
NHO
EtOC
CHC
O O
OEt
NH2
Hg-Al -or- sodium dithionite
O
O
H
<
nitroso oxime
{c.f. aldimine/ketimine: can have aldoximes (R=H) and ketoximes (R=alkyl/aryl)}
N O
H3CC
CCO2Et
O
H N
O
H3CC
CC
O
NOH
note the extended conjugation
O
OEt
HON
O
H2ON
OH+
-H2O
(oxime hydrolysis gives a ketone)
(oxime reduction)

The aldol reaction
EtH
O
EtH
O
H H H
OH
Base-catalyzed aldol reaction: It is important to remember that the aldehyde hydrogen is not acidic!
Another example of the base-catalyzed aldol reaction (tBu- = Me3C-)
tBuC
CH3
O
tBuC
CH2
OOH
The aldol reaction is a base- or acid-catalyzed reaction between two aldehydes or ketones. In simple cases the two reacting partners are the same molecule - i.e. a dimerization.
CC
CH3
OH3C
CH3
H3C
MeCH2
CCH3
OOH
Base-catalyzed aldol reaction: what happens with more than one type of acidic hydrogen?
no α-H here
MeCH
CCH3
OMe
CH2
CCH2
Ominor major+
O O
EtH
O
EtH
O
H
O
Et
HEt
H
O
HHO
Et
H
H OH
αβ
β−hydroxyaldehyde
tBuH3C
O
tBuH3C
OtBu
H3CHO
H OH
αβ
O
tBu
O
tBu
β−hydroxyaldehyde
O
O
OH
O
OHO
O O

H3CC
CH3
OH
CC
CH3
O
H
+HH3C
CCH3
OH
The acid catalyzed aldol reaction uses the less reactive enol (as opposed to enolate), but the aldehyde group can be "activated" (made more reactive) by protonation.
H
Acid-catalyzed reaction with a dialdehyde: an intramolecular reaction results in cyclisation.
O
O
The Aldol Reaction: acid catalysis
-H
OHC-(CH2)4-CHO
An unsymmetrical aldehyde will produce two enols and therefore two possible products.
OH
H O
O
O
R
R
OH
R
O
H+O
OH
R
O
R
OH
+
It does not matter which aldehyde enolises as the molecule is symmetrical
less reactive than enolate
protonated ketone more reactive electrophile
H3CC
CH3
OH
HC
CCH3
O
H
H3C CH3OH
O
OH
H O
OH
α
β
α
β
α
βα
β
1
234
56
7
1
234
56
7
1
234
567
123
45
67
123
45
67

The Aldol Reaction: synthesis of α,β-unsaturated carbonyl compounds
Acid-catalyzed dehydration
Base-catalysed elimination: This follows the E1cb mechanism as OH is a poor leaving group. "cb" stands for conjugate base and the reaction rate is proportional to [OH]x[ketone]. In some texts this process is incorrectly shown as a concerted process with proton abstraction and hydroxide leaving in a concerted fashion.
OH
Dehydration of an aldol product may follow the condensation reaction to produce α,β-unsaturated carbonyl compounds. This is normally inevitable and rapid with acid catalysis but will depend on the substrate and conditions in base catalysed reactions. Loss of water is (essentially) irreversible.
HCH3
OOH
O
OH
H O
OH
O
OH2
HO
H
O
HCH3
O
H
O
HCH3
O
H
O
HCH3
OH
H
HOH
OH
H
O
HCH3
OH
H
O
CH3
E2 (or E1) eliminationnot likely with poor
leaving group
fasterslower
-H2O
-H2O

The aldol reaction: crossed condensations
HC
H
O
RC
H
O
RC
R
O> >
Order of Reactivity: due to steric and inductive effects, different carbonyl groups will show differing levels of reactivity towards enolates, i.e. they will have different electrophilicity.
CH
O
H3CC
Ph
O
H2CC
Ph
O+
O2N
For successful crossed condensations only one carbonyl reactant must be "enolizable" and the other reactant must be a more reactive electrophile than the one which enolizes.
O
H
CH2OK2CO3
Reactions with formaldehyde (methanal) are often tricky as it is so reactive. Double aldol reactions can result.
ArC
H
O
increasing steric hindrance at electrophilic carbon
decreasing δ+ due to inductive effects of R
δ+ δ+ δ+
H2CC
Ph
O
Ar C H
O
H2CC
Ph
O
Ar C H
OH H OH
HCC
Ph
O
CHAr
-H2O
E1cb
Ar
O
H
HO
H
O
H
HO H O
H
O
H
OH OH
faster addition than HO elimination

More aldol reactions and crossed condensations
aq. KOH
aq. KOH
2 mol. eq. PhCHO
O
H
O
O
Intramolecular aldol reaction: ring size may control which product forms. Here a five membered ring forms faster than a three membered ring. If the three membered aldol product does form it is unlikely to eliminate to form the strained cyclopropene, but can be converted back to the enolate through a retro aldol reaction.
"Double" aldol reactions
The Knoevenagel condensation is a reaction of malonic acid ester with an aldehyde. The Doebner modification uses malonic acid and results in a decarboxylated product (see later and in organic labs).
EtO
O
OEt
O
EtO
O
OEt
ONH
HR
O
EtO
O
OEt
O
RON
H HH H
O
H
O
O
H
O
+
major minor
O
O
-H2O
E1cb
O
OO
ring strain means retro aldol is more likely than elimination to cyclopropene
no α-Hs
O
Ph
O
PhPh
(c.f. dibenzylidene acetone experiment in lab)
acidic α-H
EtO
O
OEt
O
RHO
EtO
O
OEt
O
RHO
H
NH
-H2O
E1cbEtO
O
OEt
O
R

The nitro aldol reaction: The Henry reaction
Nitro-aci tautomerism
RCH2
N
RC
NO OO HO
H
Generation of a nitronate (cf. enolate) anion
The hydrogen atoms α- to a nitro group are very acidicas C-H groups go, and much more so than simple ketones or aldehydes. Organo nitro compounds can thereforeundergo a reaction analogous to the aldol reaction. Also,the nitro group is not electrophilic under these reactionconditions and so no self condensation takes place.
The pKa of nitroethane (R
= Me) is ~9
H2C
NOOO
OHH
CHO
H3CN
O
O NaOH, MeOH
The nitro tautomer isthermodynamically more stable than the aci form, but the Eafor interconversion is relativelylarge (compared to keto-enol).The acid isomer thefore takestime to convert back to theequilibrium position (it is"kinetically semi-stable").
Energy (kJ/mol)
acinitro
Ea
O
CH2
NOO
O
NO
O
OH
NO
O
OHH
NOO
NO
O
nitrostyrene
nitroalkene
-H2O

The Claisen Condensation
COEtO
The Claisen condensation is similar to the aldol reaction except that it is a reaction between esters and not aldehydes or ketones. The Claisen reaction has can be considered to be the acylation of an enolate
RC
COEt
O
H HOEt
RC
COEt
O
H
RC
COEt
O
H
EtOH
PhC
COEt
O
H H
EtO
PhC
COEt
O
H
EtO-H
General reaction
+
Specific example
R
Due to the acidity of the product a whole molar equivalent of NaOEt is needed. When equilbrium is reached the ethanol is removed by distillation to shift the equilibrium towards the side of the salt, whereupon aqueous acid is added to liberate the product.
pKa = 25
pKa = 16
RC
COEt
O
HCOEtO
R
RC
COEt
O
HC
O
R OEt
RC
COEt
O
C
O
R
NB
RCH
COEt
O
CO
R
aq. HCl work upEtOH
+
COEtO
Ph
PhC
COEt
O
HCOEtO
Ph
PhC
COEt
O
HCO
Ph OEt
PhC
COEt
O
CO
Ph
PhCH
COEt
O
CO
Ph
aq. HCl work up
EtOH+
stable enolate
pKa = 11

OMe
NaOEt
MeO
EtO
The intramolecular Claisen condensation: The Dieckmann cyclisation
intramolecular cyclisation of a diester is useful for making 5,6 or 7-membered rings
EtOH
The Claisen condensation is between two different molecules of an ester
O OEt
OEt
O
OEt
H
O
OEt
O
EtO
O
OEt
O
O
MeO
OH
OMe
O
MeO
O OMe
O
O
OMeO OMe
O
OMeO
H
HH
HO
OMeO
aq. HCl work up
O
OMeO
H
123
45 123
4 5
α
no α−CH
O OEt
OEt
O
12
345
6
O O
OEt
H
O O
OEt
O O
OEt
H
aq. HCl work up
NaOEt 16
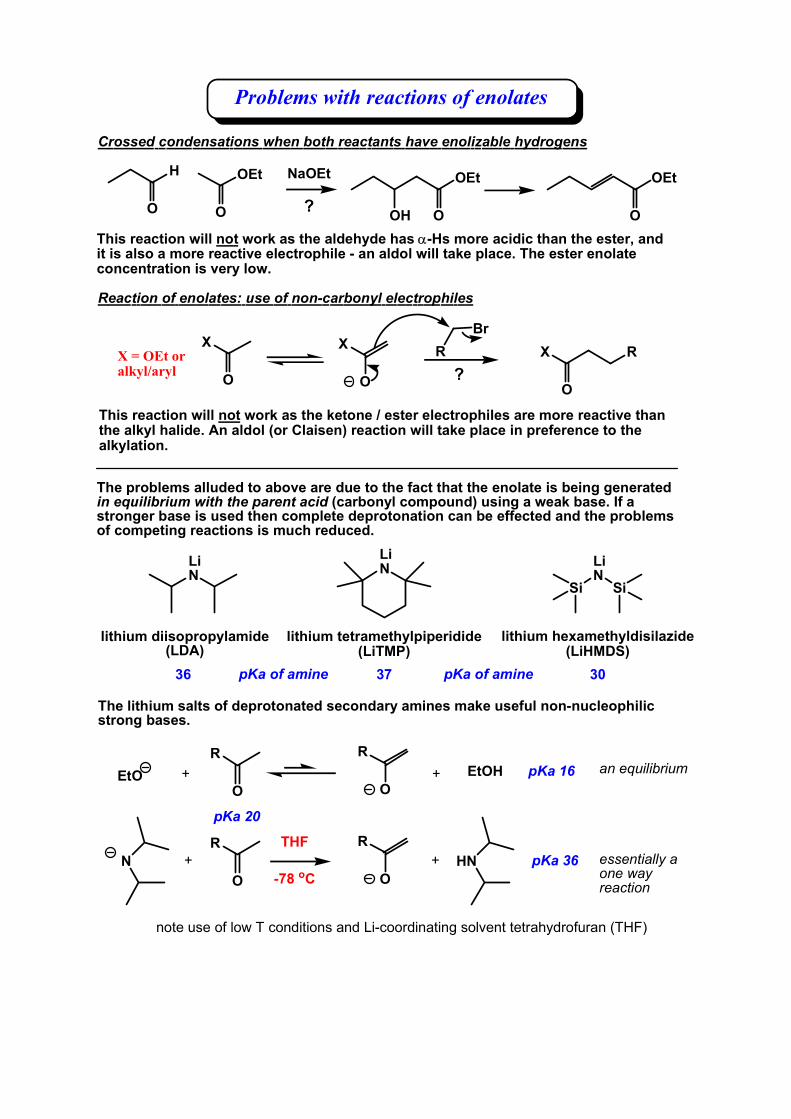
Crossed condensations when both reactants have enolizable hydrogens
This reaction will not work as the aldehyde has α-Hs more acidic than the ester, and it is also a more reactive electrophile - an aldol will take place. The ester enolate concentration is very low.
H
O
OEt
O OH O
OEtNaOEt
?
Reaction of enolates: use of non-carbonyl electrophiles
X
O
X
O
RX
O
This reaction will not work as the ketone / ester electrophiles are more reactive than the alkyl halide. An aldol (or Claisen) reaction will take place in preference to the alkylation.
Problems with reactions of enolates
X = OEt or alkyl/aryl
O
OEt
R
O
R
OEtOHEtO
R
O
R
ON HN
+ +
++
pKa 16
pKa 36
pKa 20
LiNLi
NSi
LiN
Si
lithium diisopropylamide(LDA)
lithium tetramethylpiperidide(LiTMP)
lithium hexamethyldisilazide(LiHMDS)
pKa of amine36 37 30
The problems alluded to above are due to the fact that the enolate is being generated in equilibrium with the parent acid (carbonyl compound) using a weak base. If a stronger base is used then complete deprotonation can be effected and the problems of competing reactions is much reduced.
The lithium salts of deprotonated secondary amines make useful non-nucleophilic strong bases.
THF
-78 oC
note use of low T conditions and Li-coordinating solvent tetrahydrofuran (THF)
pKa of amine
BrR
?
an equilibrium
essentially a one way reaction

Lithium enolates
R
ON THF
-78 oC
Deprotonation is initited by coordination of the lithium ion to the carbonyl oxygen.
Li
H R
OLi ONTHF
-78 oC
Li
H
OLi
Steric factors mean that unsymmetrical ketones may produce one enolate faster (kinetic enolate) even if it is not the most stable enolate (thermodynamic enolate).
However generation of Li enolates from aldehydes suffer from some problems:
Me Me
kinetic enolate
O OLi
OEt OEt
OtBu
O
OtBu
OLi
MeI
-78 to 0oC
n-BuI
-78 to 0oC
O O
Ph
O
Ph
O
Li
Li
Reaction of Li enolates with aldehydes
Reaction of Li enolates with alkyl halides
LDA-78 oC
LDA, THF
-78 oC
LDA, THF
-78 oC
unsymmetrical ketone
H
O LDA
-78 oC H
OLi
R R
H
O
Rfast
-78 oC
fast too!H
O
R
OLiR
N
OLi LDA
-78 oCR
aldol productaddition product
O
HPh
O
HEt
O
Ph
OLi
Ph
O
Et
OLi
O
Ph
OH
Ph
O
Et
OHEtCHO
PhCHO
-78 to 0oC
H+
H+
O
OEt
OtBu
O
two geometric isomers two diastereoisomers

β-Dicarbonyl compounds: properties and decarboxylation
MeO
O O
OMeH3C
O O
OEt
Ethyl acetoacetateethyl 3-oxobutanoate
Dimethyl malonate (diester of malonic acid)
made in the Claisen condensation of ethyl acetate
made in the crossed condensation of methyl acetate and dimethyl carbonate
MeO
O O
OMeH3C
O O
OEt
MeO
O O
OMeH3C
O O
OEt
H3C CH2
O
H3C CH2
O
MeO CH2
O
MeO CH2
O
H H H H
HH
H H
R
O O
OEtH H
β-Keto acids undergo facile decarboxylation. 1,3-Dicarboxylic acids will also lose CO2 if pushed a bit harder.
R
O O
O NaH H
R
O O
OH H
H
NaOH HCl (aq)
The presence of a β-carbonyl group increases the acidity of an α−hydrogen
In conclusion, the β-carboxylic ester: 1) can be used to control and direct deprotonation; 2) can be used to produce an enolate in "quantitative yield" (though not isolated);3) can be removed by the process of ester hydrolysis followed by decarboxylation.
T oCT = RT-100 oC for R = alkyl or arylT = >150 o C for R = OH
O
OMeMeO
pKa ~ 20 pKa ~ 11 pKa ~ 25 pKa ~ 13
EtOH
EtO EtO
EtOH EtOHEtOHpKa ~ 16 pKa ~ 16
R
OH
C
O
OH
HR
O
H
HH
ester hydrolysis
enol

Alkylation of dimethyl malonate
Malonate derivatives (1,3-diesters in general) can be easily mono-alkylated and dialkylated
MeO OMe
O O
MeO OMe
O O
Br R1
NaOMe
NaOMe R2CH2Br
i) NaOH (aq),ii) HCl(aq) + heating
i) NaOH (aq),ii) HCl(aq) + heating
"Meldrum's acid" is a cyclic malonate ester. Both malonate and Meldrum's acid can also be acylated using acid chlorides or anhydrides. Can be used to make β-keto esters.
O
O
O
O O
O
O
O
OEt R Cl
O
O
O
O
O
R
Cl O
O
O
O
O
R
O
O
O
O
O
R
OH
-Cl
H
HH
stableenol
keto
favourable cannot self condense
MeO OMe
O O
H
R1
H
repeatMeO OMe
O O
R1 R2
if R1 = R2 use 2 eq. NaOEt and RBr in a one pot reaction
HO R1
OCO2+ HO R1
O
CO2+
R2
alkylation of a malonate ester followed by decarboxylation produces the same net result as alkylation of a simple enolate.
αEtOH
O
O
O
O
R
O
H
EtO
O
O
O
R
O
Et
H
H+ transfer
O
OC
O
O
R
OH
Et O
OO
REt
-CO2

Alkylation of ethyl acetoacetate
β-keto esters can be easily alkylated in the same fashion as malonate esters.
H3C OEt
O O
H3C OEt
O O
BrR1
NaOEt
NaOH (aq)
HCl(aq) heating
H3C OEt
O O
H3C OEt
O O
H3C
O
R2
R1R2R1H H
BrR1
BrR2
i, NaOH (aq)
ii, HCl(aq) heating
O
OEt
O
H H
A second alkylation can be achieved regiospecifically between the two C=O groups. R1 and R2 can be added sequentially (R1=R2) or in one go using 2 moles of base and halide (for R1=R2).
BrOEt
OEt
OO
ii, NaOEt
Dieckmann products (cyclic β-keto esters) can be alkylated just as easily.
i, Dieckmann
O
O
O
NaOH (aq)
HCl(aq) heating
O
O
OH
H3C OEt
O O
H R1
H3C ONa
O O
H R1
not normally isolated
H3C O
O O
H R1
H
H3C
OH
H3C
O
CH2R1CH2R1
mechanism via sequential deprotonation/alkylation even if 2 eq. of base / RBr used
O
O
OEt
OHO
-CO2
-CO2
OEt
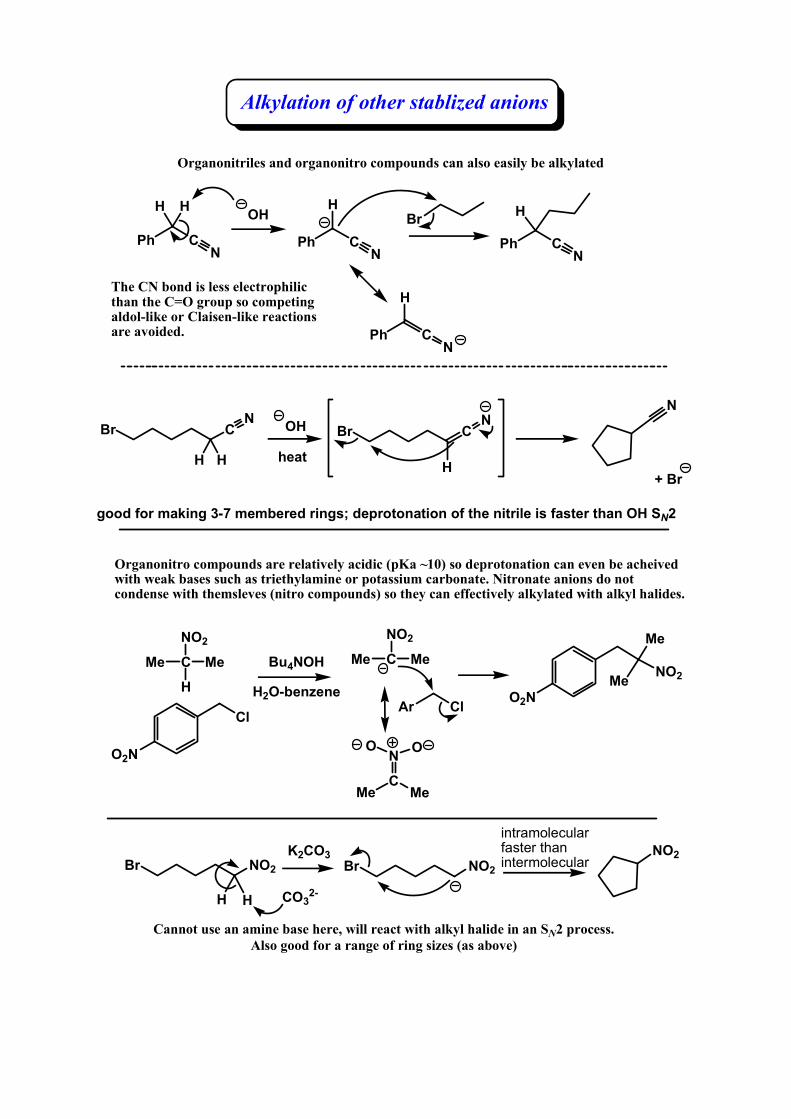
Alkylation of other stablized anions
Organonitriles and organonitro compounds can also easily be alkylated
Organonitro compounds are relatively acidic (pKa ~10) so deprotonation can even be acheived with weak bases such as triethylamine or potassium carbonate. Nitronate anions do not condense with themsleves (nitro compounds) so they can effectively alkylated with alkyl halides.
The CN bond is less electrophilic than the C=O group so competing aldol-like or Claisen-like reactions are avoided.
Ph CN
OHH H
Ph CN
HBr
Br CN
H H
OH
heat
Cl
O2N
Me C
NO2
Bu4NOH
H2O-benzene
Me
Br NO2K2CO3
Cannot use an amine base here, will react with alkyl halide in an SN2 process. Also good for a range of ring sizes (as above)
H
Ph CN
H
Ph CN
H
good for making 3-7 membered rings; deprotonation of the nitrile is faster than OH SN2
Br CN
H
N
+ Br
Me C
NO2
Me
Ar Cl O2N
Me
Me NO2
Br NO2
NO2
MeC
N
Me
O O
H H CO32-
intramolecularfaster than intermolecular

Conjugate addition reactions
α,β-Unsaturated carbonyl compounds may react at the carbonyl carbon OR at the β-carbon depending on the choice of nucleophile and / or conditions.
O1. MeMgBr2. H2O
1. MeMgBr 1% Cu(I)Cl2. H2O
ONaCNHCN
5-10 oC
NaCNHCN
80 oC
1,2- or 'direct'
addition product
1,4- or 'conjugate'
addition product
In the case of CN the direct addition product is formed reversibly, so at high temps the more stable 1,4-addition product is formed. Direct addition gives the kinetic product (fastest formed), but conjugate addition gives the thermodynamic product (more stable).
Factors favouring conjugate addition
Thermodynamic control (forming the more stable product) in reversible reactions, higher T and longer reaction times
Less reactive or unreactive carbonyl compounds (esters, amides)
Unsubstituted β-carbon atom (less steric hindrance)
Soft nucleophiles which prefer to react with the soft electrophilic β-carbon.
Factors favouring direct addition
Kinetic control (faster formed product) in reversible reactions, lower T and shorter reaction times
More reactive carbonyl compounds (aldehydes, acid chlorides)
Substituted β-carbon atom (more steric hindrance)
Hard nucleophiles which prefer to react with the hard electrophilic C=Ο carbon.
SOFT NUCLEOPHILESHARD NUCLEOPHILES
O
CN
O
Me
OH
OH
NC
Me
δ+δ+
O O
OH, ROH, RMgBr(early row elements)
High charge density on nucleophilic atom. Property dominated by
electrostatics
I, RS, RSH, (later row elements)also stable enolates
Polarizable nucleophilic atom. Property dominated by orbital effects

Conjugate addition with enolates: the Michael Reaction
Stable enolates, such as those from malonates and β-keto esters, make excellent nucleophiles for conjugate addition reactions. They are soft nucleophiles and their stable enolate favours the retro aldol step in reaction shown above.
OMe
OMeONaMeO
O
MeO2C
MeOO
MeOO
As the final enolate is more basic than methoxide or hydroxide, this can be used to deprotonate the starting 1,3-dicarbonyl compound* and the reactions can be run with a catalytic amount of added base.
MeOH, ∆
Ph
Ph
OKOHcat.
H3CO
H3CO
H3CO
RT
H
O
R
O
OR
O
NR2
O
R1
O
O R2R1
O
R2
OOR1
O
R2
R1
O
R2
O
1,4- 1,2-
OHR1
O
R2
Cl
O
R
O
R
O
R
O
H3CO
DECREASING REACTIVITY OF CARBONYL GROUP
DECREASING STERIC HINDRANCE AT THE BETA CARBON
β β β
INCREASING PREFERENCE FOR CONJUGATE ADDITION
HRO
H OR
OR
Even though the 1,2 addition is the faster process (kinetic) it is reversible, so ultimately the 1,4- addition product forms.
*
MeO2C
MeO2C
OMe
O
CH3OC
CH3OC Ph
Ph
O
"H "
"H "
MeO2C
MeO2C
OMe
O
CH3OC
CH3OC Ph
Ph
O
"H " = MeOH or *

Conjugate addition of enolate-like nucleophiles
Nitroalkanes are excellent for conjugate addition reactions
Enamines
N
OHO
heat
OEt
ON
CH3
OHO ON
CH2
O O
EtOH, ∆
N OHO O
N
C
O O
EtOH, ∆H3C H3CCH3 CH3H
R=H, CH3
Organontrile compounds: the nitrile is not as reactive to direct attack in comparison to carbonyl compounds so unsaturated nitriles such as acrylonitrile are amongst the best known Michael acceptors.
O
O
C
O CN
Ph
CCH3
O
Ph
OH
H
N N
N
OH
OH
H
N
O
OH
H
OEt
OO2N
OEt
OO2N
O
O
NO2
O
O
NO2
EtO H
OEtH
CCH3
O
Ph
N
CNC
CH3
O
Ph
N
CN
"cyanoethylation"
H OH

Michael reactions can be followed by cyclization
Michael reactions can produce compounds that are eminently suitable for subsequent aldol or Claisen cyclisation reactions.
O
CO2MeH
O
CO2Me OMeO
O
CO2Me
OK2CO3MeOHheat
MeO
O
CO2MeO
MeO
O
CO2MeMeO
MeO
O
CO2Me
O
MeOMeOH
heat 2h
Michael then aldol - the "Robinson annulation"
Michael then Claisen - synthesis of dimedone
O
CO2Me
O
O
CO2Me
O
O
CO2Me
O
HO
CO2Me
O
CO2Me
O
E1cb - H2O
aldol
There are three possible enolates of this molecule. Can you work out why the other
two do not lead to a product?
MeO
O
CO2Me
O
MeO
O
CO2Me
O
CO2Me
O
O
CO2Me
O
O
CO2Me
O
O
O
O
H3O+
H OMe
HMeO
OMeH
H
MeO
H3O+
heat
-CO2-MeOH
dimedone
There are three possible enolates of this molecule. Can you work out why
the other two do not lead to a product?

Reactions of Enamines
Acylation reactions are Claisen-like, as the electrophile (anhydride or acid chloride) is in the same oxidation state as an ester. This reaction in effect enables ketones or aldehydes as nucleophiles (as enamines) to cross react with anhydrides or acid chlorides. C-acylation of enamines is straightforward as N-acylation is reversible.
Ph
NO
O
OO
O
O
Enamines also react at carbon with active alkyl halides (allylic, benzylic, α-haloketones) with heating. Less reactive halides on the other hand can also react on nitrogen forming an ammonium salt. This reaction is irreversible and causes problems with by-product formation.
NBrBr
heatMeCN
several h.
The enamine is described as an ambident nucleophile as it can react at either the N or C end.
Ph
N
H
O
Ph
N O
CH3CO2
Ph
NO
O2CCH3
N
H
N-H
N
Enamines can be hydrolysed in aqueous acid back to the corresponding carbonyl compound.
Ph
N O
Ph
O O
H+
H2O
Ph
N O
Ph
N O
Ph
N O
N
H+
H2O
O
HH+H2O
OH2
OH
H
+ H+
- H+
+ H+- H2O

Reactions of Enamines
Alkylation of enamines is particularly useful when applied aldehydes. Remember that enolates of aldehydes, even if generated with LDA, are very sensitive to aldol-like reactions.
HON
O
Br
heatMeCN
several h.
NHO
cat H
H3Oheat
morpholine
Enamines can also be useful nucleophiles for cyclisation reactions which involve a conjugate addition reaction followed by an aldol-like condensation.
O NR2
O
+HO NR2 O NR2
O NR2
O NR2 O NR2H H
RT, 4 h
8M HCl
overall
4o centre: cannot form enamine
H
H+
N
O
N
O
OH
H+
NH
OO+ H+ transfer
OH
H
H





























