Embed Size (px)
Citation preview

1
Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen
Bioinformatik für Biochemiker
Oliver Kohlbacher WS 2009/2010
13. Ab-initio-Vorhersage
Gliederung
• Ab-Initio-Vorhersage – Konformationsraum – Systematische Suche – Stochastische Methoden – Metropolis-Monte-Carlo-Methode
• ROSETTA – Fragment Assembly – Bewertungsfunktion – Algorithmus – Ergebnisse
• ROBETTA
Ab-Initio-Vorhersage • Modellierung auf homologe Strukturen schlägt dann fehlt, wenn
ein Protein einer völlig neuen Faltungsklasse angehört
• Threading findet in diesem Fall keine passende Schablonenstruktur
• Abhilfe schaffen ab-initio-Verfahren, die die Struktur „aus ersten Prinzipien“, d.h. ohne Zuhilfenahme homologer Strukturen vorhersagen
• Dieses Problem ist wesentlich schwieriger als Threading, entsprechend sind die Erfolgsquoten schlechter
• Ab-initio-Methoden müssen Konformationsraum der Sequenz durchmustern und darin die native (d.h. in der Natur vorliegende) Struktur identifizieren (über eine geeignete Energiefunktion)

2
Suche im Konformationsraum
• Energiehyperflächen beschreiben Energie des Proteins als Funktion der Konformation
• Minima – entsprechen günstigen Konformationen
(Konformeren)
– sind meist lokale Minima!
• Globales Minimum ist Lösung des Vorhersageproblems
• Kann man die Energiehyperfläche systematisch durchmustern?
Systematische Suche
• Geht nur für kleine Anzahl Freiheitsgrade (kombinatorische Explosion)
• Beispiel – Protein mit 1000 Atomen – Koordinaten in Würfel von 20 Å Seitenlänge – Diskretisierung mit 0.2 Å Abstand ) 100 mögliche Werte für jede der 3000 Koordinaten
) 106000 mögliche Energien (Das Universum enthält ca. 1080 Teilchen!)
Systematische Suche
• Unabhängige Betrachtung der
Koordinaten ist naiv
• Flexibilität wird überwiegend durch
Torsionen bestimmt
• Wesentlich geringere Anzahl
Freiheitsgrade (ca. 2-7 pro AS)
• Wenige Minima in den Torsionen
) grobe Rasterung (0/120/240°)
• Vermeidet Betrachtung physikal.
unsinniger Konformationen
Beispiel:
• 1000 Atome ~ 50 AS
) 200 Torsionen
• 3200 ¼ 1095 Konformationen
) immer noch viel!
3
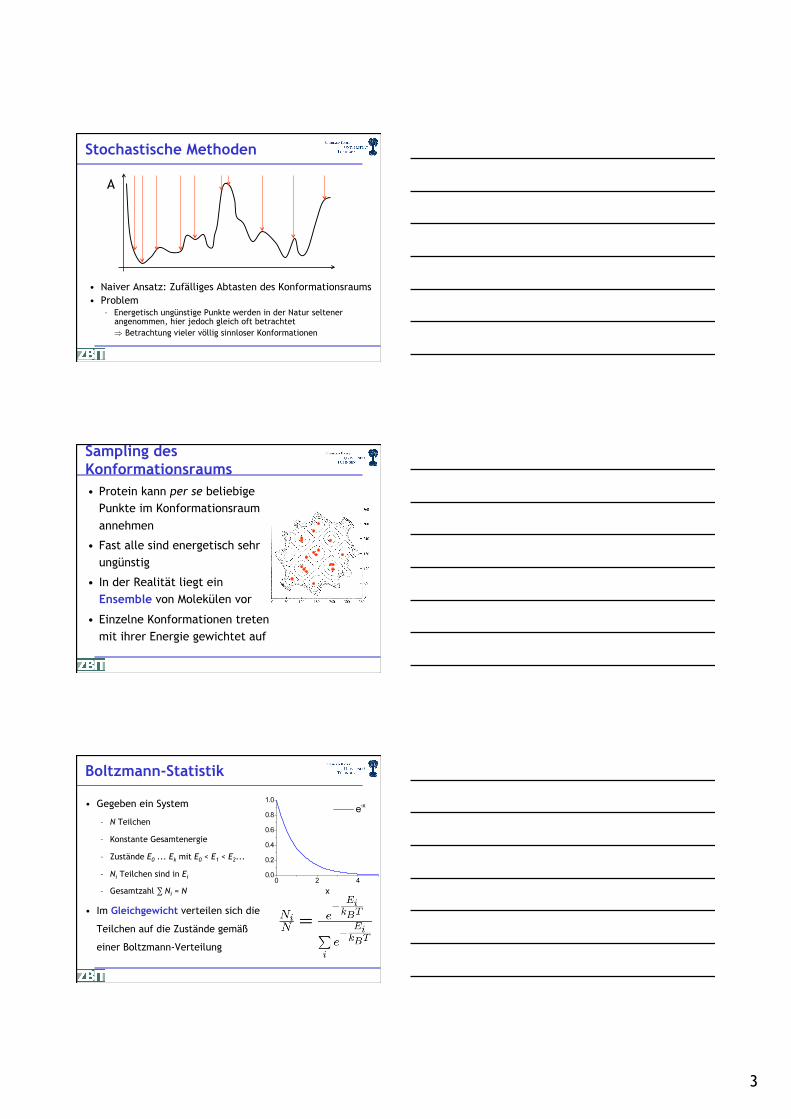
Stochastische Methoden
• Naiver Ansatz: Zufälliges Abtasten des Konformationsraums • Problem
– Energetisch ungünstige Punkte werden in der Natur seltener angenommen, hier jedoch gleich oft betrachtet
) Betrachtung vieler völlig sinnloser Konformationen
A
Sampling des Konformationsraums • Protein kann per se beliebige
Punkte im Konformationsraum annehmen
• Fast alle sind energetisch sehr ungünstig
• In der Realität liegt ein Ensemble von Molekülen vor
• Einzelne Konformationen treten mit ihrer Energie gewichtet auf
Boltzmann-Statistik
• Gegeben ein System
– N Teilchen
– Konstante Gesamtenergie
– Zustände E0 ... Ek mit E0 < E1 < E2...
– Ni Teilchen sind in Ei
– Gesamtzahl ∑ Ni = N
• Im Gleichgewicht verteilen sich die
Teilchen auf die Zustände gemäß
einer Boltzmann-Verteilung
4
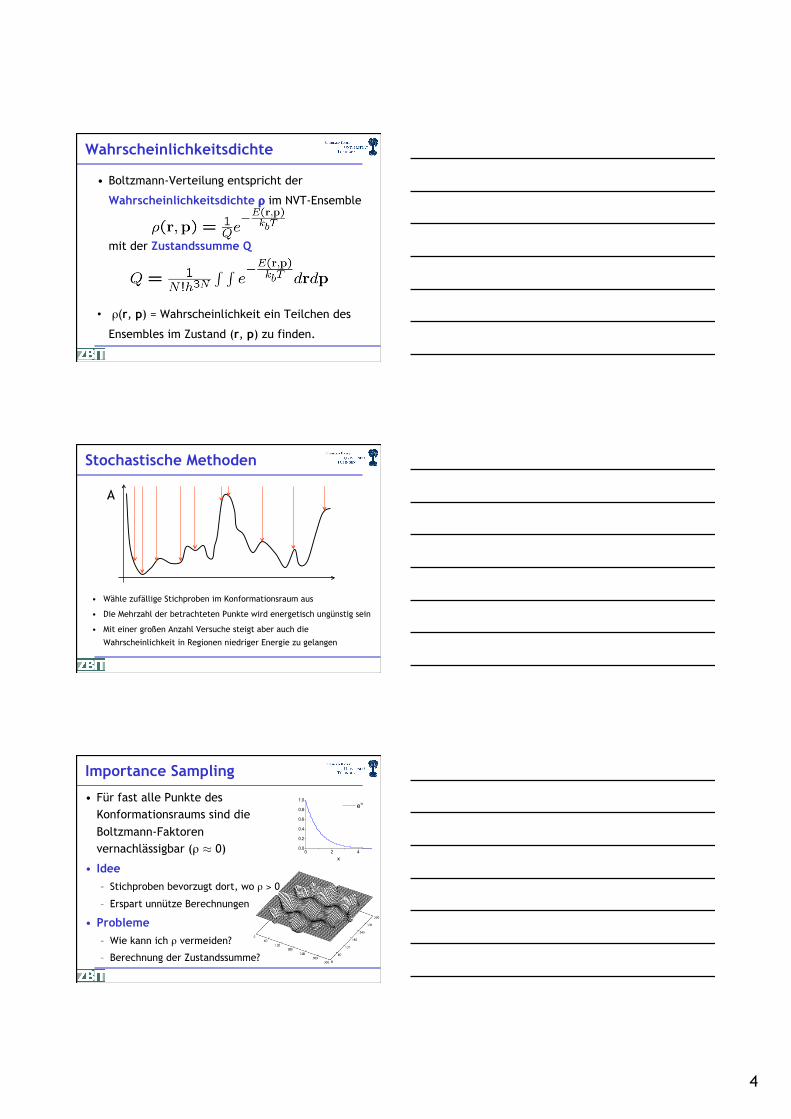
Wahrscheinlichkeitsdichte
• Boltzmann-Verteilung entspricht der
Wahrscheinlichkeitsdichte ρ im NVT-Ensemble
mit der Zustandssumme Q
• ρ(r, p) = Wahrscheinlichkeit ein Teilchen des
Ensembles im Zustand (r, p) zu finden.
Stochastische Methoden
• Wähle zufällige Stichproben im Konformationsraum aus
• Die Mehrzahl der betrachteten Punkte wird energetisch ungünstig sein
• Mit einer großen Anzahl Versuche steigt aber auch die
Wahrscheinlichkeit in Regionen niedriger Energie zu gelangen
A
Importance Sampling
• Für fast alle Punkte des Konformationsraums sind die Boltzmann-Faktoren vernachlässigbar (ρ ¼ 0)
• Idee – Stichproben bevorzugt dort, wo ρ > 0
– Erspart unnütze Berechnungen
• Probleme – Wie kann ich ρ vermeiden?
– Berechnung der Zustandssumme?

5
Monte-Carlo-Methode
• Die Monte-Carlo-Methode hat ihren
Namen aus der Verwendung von
Zufallszahlen
• 1949: Metropolis und Ulam
verwenden den Begriff zum ersten
Mal
• 1953 Metropolis-Algorithmus
• 1970 und 1995 von Hastings und
Green zur Metropolis-Hastings-Green-Methode generalisiert
Metropolis, Ulam. The Monte Carlo method, J. Am. Statist. Assoc. (1949), 44 (247), 335-341
Systematische Suche vs. MC
Frenkel, Smit: Understanding Molecular Simulation, p. 24
Metropolis-Monte-Carlo
• Zustandssumme (und damit ρk) ist aufwändig zu berechnen
• Leicht dagegen: ρi/ρj
• Metropolis-Monte-Carlo-Algorithmus erzeugt nun diese einzelnen Zustände mit ihren relativen Wahrscheinlichkeiten, d.h. energetisch günstigere Zustände werden häufiger durchlaufen als ungünstige
6
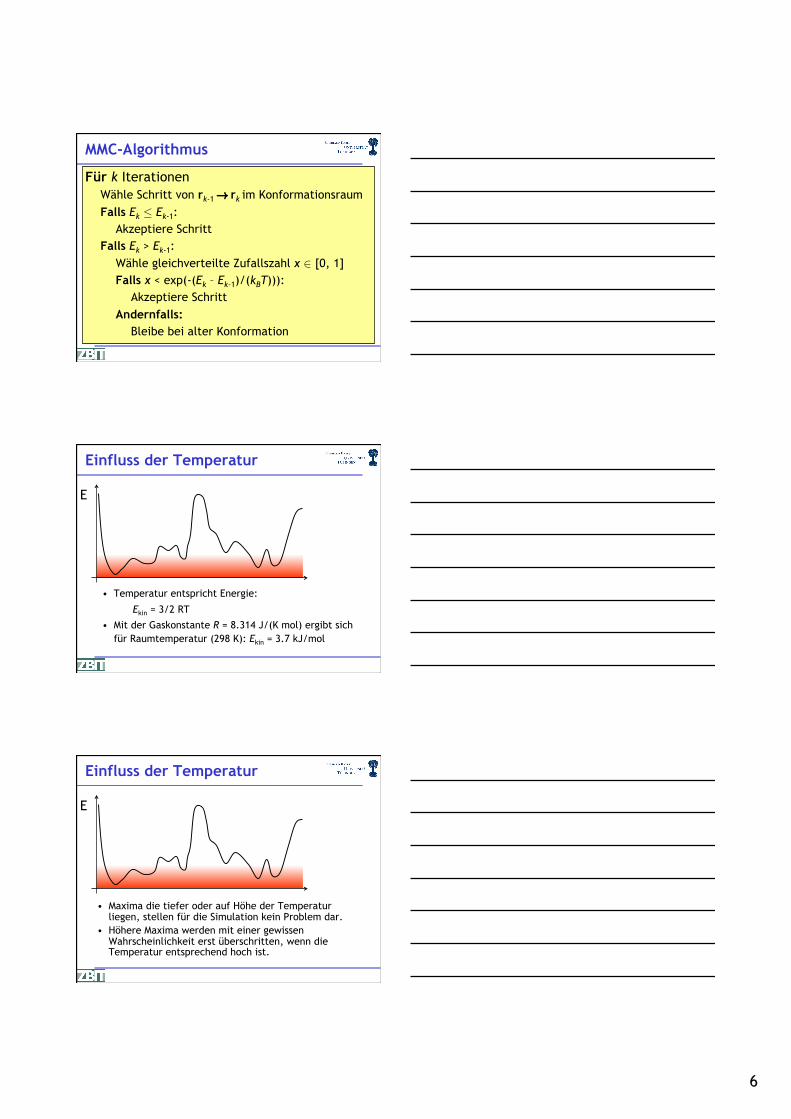
MMC-Algorithmus
Für k Iterationen Wähle Schritt von rk-1 ! rk im Konformationsraum Falls Ek · Ek-1:
Akzeptiere Schritt Falls Ek > Ek-1:
Wähle gleichverteilte Zufallszahl x 2 [0, 1] Falls x < exp(-(Ek – Ek-1)/(kBT))):
Akzeptiere Schritt Andernfalls:
Bleibe bei alter Konformation
Einfluss der Temperatur
• Temperatur entspricht Energie:
Ekin = 3/2 RT
• Mit der Gaskonstante R = 8.314 J/(K mol) ergibt sich für Raumtemperatur (298 K): Ekin = 3.7 kJ/mol
E
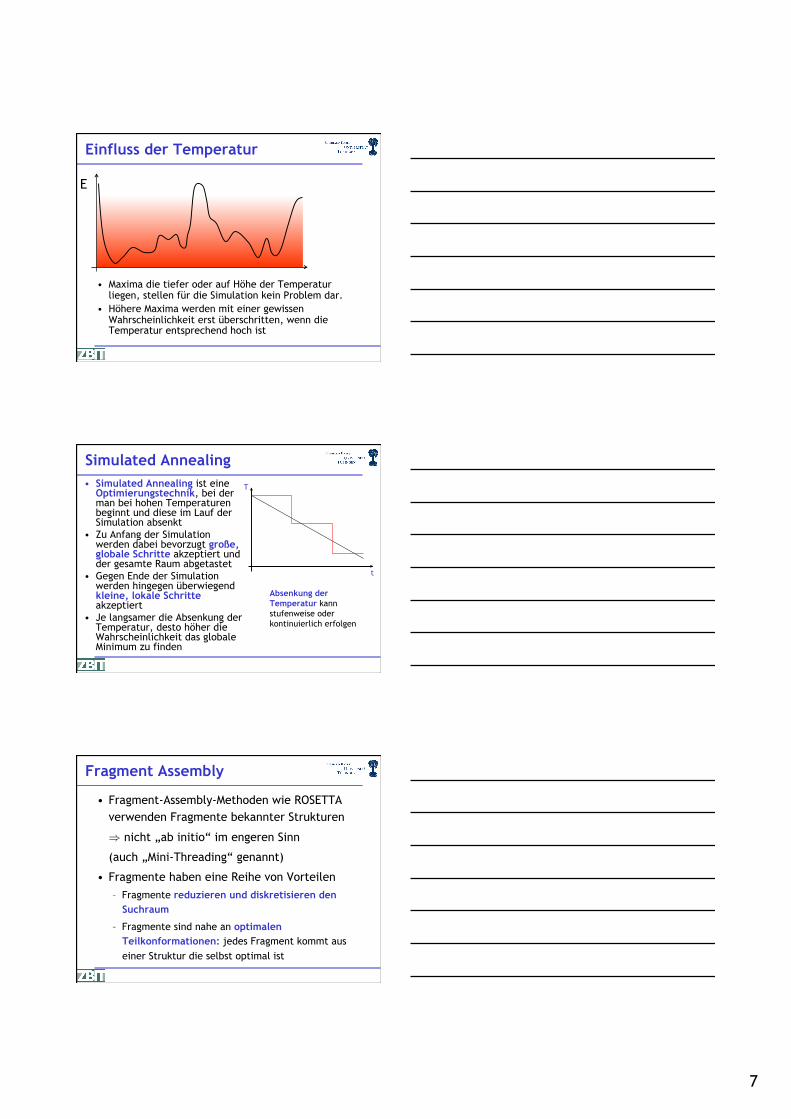
Einfluss der Temperatur
• Maxima die tiefer oder auf Höhe der Temperatur liegen, stellen für die Simulation kein Problem dar.
• Höhere Maxima werden mit einer gewissen Wahrscheinlichkeit erst überschritten, wenn die Temperatur entsprechend hoch ist.
E
7
Einfluss der Temperatur
• Maxima die tiefer oder auf Höhe der Temperatur liegen, stellen für die Simulation kein Problem dar.
• Höhere Maxima werden mit einer gewissen Wahrscheinlichkeit erst überschritten, wenn die Temperatur entsprechend hoch ist
E
Simulated Annealing • Simulated Annealing ist eine
Optimierungstechnik, bei der man bei hohen Temperaturen beginnt und diese im Lauf der Simulation absenkt
• Zu Anfang der Simulation werden dabei bevorzugt große, globale Schritte akzeptiert und der gesamte Raum abgetastet
• Gegen Ende der Simulation werden hingegen überwiegend kleine, lokale Schritte akzeptiert
• Je langsamer die Absenkung der Temperatur, desto höher die Wahrscheinlichkeit das globale Minimum zu finden
Absenkung der Temperatur kann stufenweise oder kontinuierlich erfolgen
T
t
Fragment Assembly
• Fragment-Assembly-Methoden wie ROSETTA verwenden Fragmente bekannter Strukturen
) nicht „ab initio“ im engeren Sinn
(auch „Mini-Threading“ genannt)
• Fragmente haben eine Reihe von Vorteilen – Fragmente reduzieren und diskretisieren den
Suchraum
– Fragmente sind nahe an optimalen Teilkonformationen: jedes Fragment kommt aus einer Struktur die selbst optimal ist

8
ROSETTA
Kernideen • Betrachtung des
Konformationsraums für Teilsequenzen
• Teilsequenzen nehmen nur geringe Anzahl energetisch günstiger Konformationen an
• Diese Konformationen werden durch einen Satz Fragmente äquivalent repräsentiert
• Konformationen der Fragmente überwiegend durch lokale WW bestimmt
Simons et al., J. Mol. Biol. (1997), 268, 209 Simons et al., Proteins (1999), 34, 82
ROSETTA
• Modell – Torsionswinkelraum, reduziert auf
Fragmente – Seitenketten auf Cβ reduziert
• Potentialfunktion – Wahrscheinlichkeitsbasiert (Bayes-Ansatz)
• Algorithmus – Simulated Annealing: MMC mit linear sinkender Temperatur
– Feste Anzahl Schritte (10000)
Fragmentbibliothek
• Abgeleitet aus nicht-redundantem Teilsatz der PDB
• Aus den Strukturen werden alle 9-mere und 3-mere gesammelt
• Zu jeder Teilsequenz der Zielsequenz werden daraus die 25 nächsten Fragmente ausgewählt
• ROSETTA verwendet Fragmente der Längen 3 und 9

9
Bewertungsfunktion • Rosetta verwendet eine Bewertungsfunktion basierend auf
bedingten Wahrscheinlichkeiten, abgeleitet aus Strukturdatenbanken
• Prinzipiell nur ein elegante Art ein paarweises Potential für eine Sequenz x in einer Struktur y zu schreiben: – P(x|y) : Wahrscheinlichkeit, dass Sequenz x Struktur y annimmt – P(xi, xj|rij): Wahrscheinlichkeit für Reste xi, xj für einen Abstand rij – P(xi, xj): A-priori-Wahrscheinlichkeiten der Reste xi, xj
Unabhängig von Struktur Sippl-Potential!
Algorithmus
Bestimme 25 nächste Nachbarn für jede Teilsequenz
Starte mit gestreckter Struktur
Für 10000 Iterationen:
Wähle zufällig eine Teilsequenz x´ aus x
Wähle zufällig x´´ aus den Fragmenten für x´
Ersetze die Torsionswinkel in x´ mit denen aus x´´
Wenn dadurch Atome überlappen, verwerfe Zug
Berechne Score
Akzeptiere Zug gemäß MMC-Kriterium
ROSETTA
ROSETTA-Trajektorie von 1UBI

10
ROSETTA – Ergebnisse CASP5
Verwendung von ROSETTA • ROSETTA erzeugt eine ganze Anzahl von Strukturen
• Diese Strukturen werden mit Hilfe der Scoring-Funktion bewertet: bester (negativster) Score = beste Struktur
• Je mehr Durchläufe (d.h. unabhängige Simulationen) durchgeführt werden, desto höher die Chance eine sehr gute Struktur zu finden
• Stochastische Methoden verwenden Zufallszahlen: jeder Lauf erzeugt andere Ergebnisse!
• Beispielausgabe (Details in der Übung):
filename score env pair vdw hs ss no_pdbfile_fail -71.86 -27.02 -18.88 1.99 -1.69 -22.96 aa2PTL0001.pdb -67.85 -24.50 -12.15 0.58 -4.43 -21.45 no_pdbfile_fail -64.30 -27.23 -16.53 1.07 -2.72 -17.13 aa2PTL0002.pdb -67.77 -18.90 -9.64 2.06 2.63 -33.28 no_pdbfile_fail -69.70 -31.18 -12.69 1.47 -1.76 -21.73 aa2PTL0003.pdb -91.94 -18.14 -13.14 2.01 -8.71 -40.83 no_pdbfile_fail -43.20 -11.10 -10.10 1.83 -7.51 -11.75 Beste Energie
Beste Struktur
ROSETTA – Ergebnis
ROSETTA-Ergebnis (rot) für 2PTL (gelb: PDB-Struktur)

11
ROBETTA
• ROBETTA ist ein vollautomatischer Online-Server zur Proteinstruktur-Vorhersage
• ROBETTA kombiniert – Domänenzerlegung
– Ab-initio-Vorhersage für kleine Domänen
– Threading für Domänen mit bekanntem Fold
• Diese Schritte werden bei ROBETTA vollautomatisch durchgeführt, heraus kommen recht passable Strukturen
• Nachteil: extrem lange Wartezeiten des Online-Servers machen diesen fast unbenutzbar
ROBETTA – Überblick
ROBETTA – Überblick

12
ROBETTA – Submission
ROBETTA – Status
Literatur + Links
Literatur • Simons, Kooperberg, Huang, Baker: Assembly of protein tertiary
structures from fragments with similar local sequences using simulate anealing and Bayesian scoring functions. J. Mol. Biol. (1997), 268, 209-25
• Kim, Chivian, Baker: Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. (2004), 32 Suppl 2, W526-31
Links • Bakerlab (Entwickler von ROSETTA) http://www.bakerlab.org • ROBETTA-Server
http://robetta.bakerlab.org





























