Embed Size (px)
Citation preview

REVIEW
Biology of A b Amyloid in Alzheimer’s Disease
Thomas Wisniewski,*,1 Jorge Ghiso,† and Blas Frangione†
*Department of Neurology and †Department of Pathology, New York University MedicalCenter, 550 First Avenue, New York, New York 10016
Received May 1, 1997; accepted for publication June 20, 1997
INTRODUCTION
Alzheimer’s disease (AD) is the most common causeof late-life dementia (Wisniewski and Frangione, 1996).AD can be divided into an early-onset form (onset ,60years) and the more common late-onset (.60 years)form. So far, three genes have been linked to early-onset AD including the b-amyloid precursor protein(bPP) on chromosome 21 (Levy et al., 1990; Goate et al.,1991), presenilin 1 (PS1) on chromosome 14 (Schellen-berg, 1992; Sherrington et al., 1995), and presenilin 2 onchromosome 1 (PS2) (Levy-Lahad et al., 1995; Rogaevet al., 1995; Li et al., 1995). The majority of early-onsetAD is thought to be related to mutations in PS1 and 2,while for late-onset AD an association with the inheri-tance of the apolipoprotein (apo) E allele E4 has beendescribed (Corder et al., 1993; Strittmatter et al., 1993).The presence of the apoE4 allele appears to be predomi-nantly a risk factor for patients with an onset of ADbetween the ages of 60 and 70 (Blacker et al., 1997).ApoE4 is also a risk factor for the related condition ofcongophilic angiopathy (Premkumar et al., 1996; Green-berg et al., 1996), as well as multi-infarction dementia(Shimano et al., 1989; Slooter et al., 1997). Neuropatho-logically, each of these subtypes of AD is characterizedby four major lesions: (a) intraneuronal, cytoplasmicdeposits of neurofibrillary tangles (NFT); (b) parenchy-mal amyloid deposits called neuritic plaques; (c) cere-brovascular amyloidosis; and (d) synaptic loss. Themajor constituent of the neuritic plaques and congo-philic angiopathy is amyloid b (Ab), although thesedeposits also contain other proteins (Wisniewski &Frangione, 1996). The amyloid deposits in AD share a
number of properties with all the other cerebral amyloi-doses such as the prion-related amyloidoses, as well asthe systemic amyloidoses. These characteristics are: (1)being relatively insoluble; (2) having a high b-sheetsecondary structure, which is associated with a ten-dency to aggregate or polymerize; (3) ultrastructurallythe deposits are mainly fibrillary; (4) the presence ofcertain amyloid-associated proteins such as amyloid Pcomponent, proteoglycans, and apolipoproteins; (5)the deposits show a characteristic apple-green birifrin-gence when viewed under polarized light, after Congored staining. All of the proteins with linkage to ADhave now been found as components of neuriticplaques (Masters et al., 1985; Wisniewski et al., 1995b,c).It remains to be determined whether all these proteinsare involved in the same or different pathologicalpathways and which of these proteins is the mostimportant for the most common, late-onset form of AD.
AMYLOID b, AMYLOID PRECURSORPROTEIN MUTATIONS, AND THEAMYLOID CASCADE HYPOTHESIS
Ab is a mainly 40- to 42-amino-acid peptide that isheterogeneous at both its amino and its carboxyltermini (Masters et al., 1985; Prelli et al., 1988; Joachimet al., 1988; Miller et al., 1993; Wisniewski et al., 1994b).The amino-terminal sequence of this peptide was firstdetermined by the seminal work of Dr. G. Glenner in1984 (Glenner & Wong, 1984). Neuritic plaque amyloidwas first sequenced in 1985 (Masters et al., 1985) andwas found to extend mainly to Ab residue 42/43.Vascular amyloid was initially reported as Ab1–28(Glenner & Wong, 1984), and later it was found to
1 To whom correspondence should be addressed. Fax: (212) 263-6751. E-mail: [email protected].
Neurobiology of Disease 4, 313–328 (1997)
Article No. NB970147
313
0969-9961/97 $25.00Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

extend mainly to Ab1–39/40 (Prelli et al., 1988; Joachimet al., 1988). This heterogeneity at the carboxyl termi-nus of Ab was attributed to differences in local tissueprocessing. More recent reports, using different tech-niques, have also found Ab1–42 in vascular amyloiddeposits (Roher et al., 1993). The Ab peptide is alsofound at low concentrations as a normal constituent ofbiological fluids, where it is known as soluble Ab(sAb) (Haass et al., 1992; Seubert et al., 1992; Shoji et al.,1992; Busciglio et al., 1993). sAb is predominantlyAb1–40, although shorter and longer sequences exist,including Ab1–28 and Ab1–42 (Vigo-Pelfrey et al.,1993). sAb is also found in the water-soluble fraction ofnormal and AD brains (Tabaton et al., 1994; Kuo et al.,1996). Brain sAb appears to be elevated in AD and theelevation precedes amyloid plaque formation inDown’s syndrome (DS) brains (Teller et al., 1996). BothAb and sAb are degradative fragments from a largeramyloid precursor protein (bPP) (Kang et al., 1987;Goldgaber et al., 1987; Robakis et al., 1987; Tanzi et al.,1987). The bPP gene is found on chromosome 21. Itcontains at least 19 exons, spanning 400 kb of DNA,with more than 10 isoforms of bPP mRNA that can begenerated by alternative splicing (Kitaguchi et al., 1988;Tanzi et al., 1988; Golde et al., 1990; Lemaire et al., 1989;Konig et al., 1992). The four major Ab-containingproducts are proteins of 695, 714, 751, and 770 aminoacids. The bPP has a predicted structure of a multido-main, transmembrane cell surface receptor (Kang et al.,1987; Goldgaber et al., 1987; Robakis et al., 1987; Tanzi etal., 1987). The Ab sequence arises from portions ofexons 16 and 17; therefore Ab cannot be generated byalternative splicing of bPP but requires proteolyticcleavage at both its N and its C termini. One of the firstdiscovered metabolic processing pathways of bPPinvolves a cleavage at Ab residue 16 by the so-calleda-secretase to release a large soluble protein containingonly the N-terminal sequence of Ab (Esch et al., 1990;van Nostrand et al., 1990; Sisodia et al., 1990; Wang etal., 1991). Two other secretases, known as b- andg-secretases, are thought to exist, which cleave bPP atthe N and C termini, respectively, of the Ab peptide.These processing pathways give rise to both normalsAb and the Ab in amyloid deposits. The longerAb1–42 has been found in vitro to be more fibrillogenicand has been suggested to be particularly importantfor the initiation of Ab deposits and cytotoxicity(Jarrett et al., 1993; Hilbich et al., 1991; Younkin, 1995;Iwatsubo et al., 1994). However, biochemical studieshave shown that a major component of preamyloidlesions, which are the Ab deposits appearing first inDS patients, is Ab17–42 (Gowing et al., 1994; Wisniewski
et al., 1996b; Lalowski et al., 1996). These preamyloidlesions also contain Ab1–42 and other Ab peptides, butbiochemical purifications suggest that these peptidesare present at lower concentrations. Amyloid depositsalso contain Ab1–40, while sAb in part extends to Abresidue 42. Since the amino acid sequence of sAb issimilar or identical to Ab, it is likely that AD amyloidcan be at least partially derived from sAb either fromthe brain or from a systemic source.
The origin of sAb remains a controversial topic. Itmay be from cells within the central nervous system,the peripheral circulation, or a mixture of these twosources. The last possibility seems very likely since ithas been shown that Ab peptides can cross the blood–brain barrier in monkey, mouse, and guinea pig modelsystems (Zlokovic et al., 1993; Maness et al., 1994;Ghilardi et al., 1996). The major difference between sAband Ab is thought to be conformational, with Abhaving a predominantly b-sheet content that is associ-ated with its relative insolubility and resistance todegradation. A number of different factors are likely tobe involved in the conformational change betweensAb and Ab. A small subset of patients with early-onset familial AD (FAD) have been shown to be linkedto a number of different mutations in the bPP gene(Hardy, 1996). It is probable that some of these muta-tions influence the conformational change of sAb toAb. The first bPP mutation to be described occursamong families with hereditary cerebral hemorrhagewith amyloidosis, Dutch type (HCHWA-D) (Levy etal., 1990). Pathologically this condition is characterizedby the deposition of amyloid mainly in cerebral ves-sels, as well as parenchymal Ab immunoreactive le-sions which are noncongophilic (Timmers et al., 1990).This heavy Ab deposition in cerebral vessels is associ-ated with strokes as a major clinical symptom.HCHWA-D is associated with a substitution of gluta-mine for glutamic acid at codon 693 (bPP770 number-ing), corresponding to residue 22 of Ab (Levy et al.,1990). A number of studies have shown in vitro that Abpeptides containing this mutation have a greater fibril-logenic potential (Wisniewski et al., 1991; Clements etal., 1993; Castano et al., 1995b). Other in vitro studies,utilizing transfected cells, have also given a possibleindication of how some of the other FAD mutations areassociated with AD (Citron et al., 1992; Cai et al., 1993;Suzuki et al., 1994).
Ab accumulation, according to many AD research-ers, is considered a central part of the pathogenesis ofAD, which directly or via a number of downstreamevents is ‘‘causative’’ in the disease. This notion is partof the amyloid cascade hypothesis (Hardy, 1996). In
314 Wisniewski, Ghiso, and Frangione
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

current versions of this hypothesis, it is suggested thatAb peptides which extend to residue 42 (Ab42) arefirst deposited in preamyloid lesions (or diffuseplaques). These are amorphous, roughly sphericalareas that are immunoreactive with anti-Ab antibod-ies, have irregular borders, and are associated with fewor no dystrophic neurites. Preamyloid deposits, unlikeamyloid, are not stained by Congo red or thioflavin S(Yamaguchi et al., 1988, 1991; Tagliavini et al., 1988;Wisniewski et al., 1989; Yamazaki et al., 1991). Ultrastruc-turally, these deposits are mainly nonfibrillar. Exten-sive numbers of preamyloid lesions can be found inaged individuals, with no reported clinical symptoms(Crystal et al., 1993; Davies et al., 1988; Delaere et al.,1990). In Down’s syndrome (DS) patients, in whomthere are three copies of the bPP gene, preamyloidlesions can appear as early as the age of 12 years(Wisniewski et al., 1994; Kida et al., 1995a; Lemere et al.,1996a). These lesions are thought to become com-pacted over a period of many years, at which pointthey acquire the characteristics of amyloid and areassociated with neuronal damage and NFT in the formof neuritic plaques (Hardy, 1996). Congo red-positive,neuritic plaques begin to appear in DS typically by theend of the third decade of life (Wisniewski et al., 1994;Kida et al., 1995a; Lemere et al., 1996a).
In addition, to the above DS data at least four otherpieces of evidence have been used to substantiate theamyloid cascade hypothesis and are responsible for itspopularity: (1) Clinically normal aged individuals candevelop extensive preamyloid deposits which mayherald the later development of AD pathology (Polvi-koski et al., 1995). (2) In vitro studies using Ab syntheticpeptides have shown that toxicity is dependent on thepresence of a fibrillar, predominantly b-sheet conforma-tion (Kosik & Coleman, 1992; Pike et al., 1993; Simmonset al., 1994; Ueda et al., 1994; Lorenzo & Yankner, 1994).(3) As already mentioned, some rare early-onset FADpedigrees are linked to bPP mutations. Studies usingtransfected cells with the ‘‘Swedish’’ double mutationat codons 670/671 of bPP have shown that this leads tohigher levels of total sAb peptides, compared withcells expressing wild-type constructs (Citron et al.,1992, 1993). On the other hand, cells expressing bPPmutations found at codon 717 do not appear to secretehigher total levels of sAb, but produce a higherproportion of the longer, more hydrophobic, Ab pep-tides that extend to residue 42 (Suzuki et al., 1994). (4)Transgenic mice expressing high levels of mutanthuman bPP show the development of diffuse andamyloid plaques (Games et al., 1995; Hsiao et al., 1996).However, other evidence suggests that additional fac-
tors may be as important as Ab in the pathogenesis ofAD and that the cascade hypothesis is an oversimplifi-cation. For example, studies of sAb measurement indifferent brain regions of the homozygous transgenicmice of Games et al. (1995), where FAD mutant bPP isoverexpressed, show elevations even in areas such asthe thalamus where amyloid deposition does not occur(or is rare) (Johnson-Wood et al., 1997). The sAb levelsin the thalamus of homozygous animals are essentiallythe same as the levels in the hippocampus of heterozy-gous transgenic mice, where amyloid does occur. Thisindicates that reaching a threshold concentration ofsAb is not enough for deposition and that otherregional brain-specific factors are also needed (Johnson-Wood et al., 1997). In addition, transgenic animals donot develop NFTs, a key feature of AD, suggesting thatthey are a good model of cerebral amyloidosis, ratherthan of AD. Two recent studies of neuronal andsynaptic density in bPP-overexpressing transgenic micemodels has provided further blows to the amyloidcascade hypothesis (Irizarry et al., 1997a; Irizarry et al.,1997b). These mice do not develop neuronal loss, lossof synaptophysin immunoreactivity, or loss of messen-ger RNA for neuronal synaptic, cytoskeletal, or meta-bolic proteins, despite extensive amyloid deposition.Furthermore, certain biochemical and immunohisto-chemical studies of preamyloid deposits have sug-gested that these lesions do not necessarily progress toneuritic plaque formation and that other factors inaddition to the presence of Ab42 may be important forneuritic plaque formation. These studies are discussedin the next section. The shortcomings of the amyloidcascade hypothesis have raised a number of otherpossibilities for the roles of Ab and bPP in AD. Abtoxicity may not be dependent on fibrillization oramyloid fibril formation. However, certain precursorsof fibrils, such as sAb peptides in a b-pleated conforma-tion or Ab peptides in oligomeric aggregates, maymediate neuronal toxicity. Alternatively Ab fibril forma-tion may be a protective or reactive process that is theend product of a number of distinct neuropathologicalevents. The Ab-only hypothesis for the etiology of ADmay need to be replaced by an Ab-and-something-elsehypothesis.
THE BIOCHEMISTRY OF PREAMYLOID
Three studies have investigated the biochemistry ofpreamyloid lesions (Gowing et al., 1994; Wisniewski etal., 1996b; Lalowski et al., 1996). These have examinedpreamyloid from different locations including ADcortex, where there was a mixture of preamyloid and
Biology of Ab Amyloid in Alzheimer’s Disease 315
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

neuritic plaques (Gowing et al., 1994) and two sourcesof relatively pure preamyloid included the aged caninemodel of AD (Wisniewski et al., 1996b) and DS cerebel-lar tissue (Lalowski et al., 1996). Each of these studiesagreed that a major component of preamyloid isAb17–42, as well as other Ab peptides which extend toresidue 42 including Ab1–42, 8–42, 4–42, and 2–42. Theaged canine model has been used to study AD pathol-ogy, since the sequence of canine Ab is the same as thatin human and the preamyloid deposits found in theaged canine brain are very similar to human diffuseplaques. Extensive preamyloid deposition can occur inthe aged dog in the absence of neuritic plaques(Cummings et al., 1993; Wisniewski et al., 1990), and itis unusual for these lesions to mature. Similarly,extensive cerebellar preamyloid deposits that, through-out the life span of the patient, fail to convert intofibrillar deposits or do so very rarely can occur in DSpatients (Wisniewski et al., 1994; Kida et al., 1995b). Thebiochemical finding that Ab peptides extending toresidue 42 are the major constituent of preamyloid inlocations where plaque maturation does not occur orrarely occurs clearly suggests that the presence ofAb42 is not by itself sufficient for neuritic plaqueformation. Similar conclusions can be drawn from animmunohistochemical study, using antibodies specificto the carboxyl terminus of Ab42, of hereditary cere-bral hemorrhage with amyloidosis, Dutch type(HCHWA-D) patient tissue (Castano et al., 1996). Thesepatients develop extensive parenchymal preamyloiddeposits which throughout their lifetime do notprogress to neuritic plaques, while at the same timethey develop extensive cerebral vessel amyloid depos-its. Despite several in vitro studies which have shownthat synthetic peptides homologous to Ab whichcontain the HCHWA-D mutation have acceleratedfibrillogensis (Wisniewski et al., 1991; Clements et al.,1993; Castano et al., 1995b), the parenchymal deposi-tion of these Ab42 peptides in brain parenchyma doesnot lead to neuritic plaque formation. Hence each ofthese studies suggests that brain-specific factors arealso important in determining neuritic plaque forma-tion.
There are several possible roles for Ab17–42 inplaque development. It may act to prevent maturationof Ab deposits; alternatively, it may act as a nidus forthe further deposition of other Ab peptide species andform part of the amyloid fibril. The biochemical stud-ies of preamyloid have raised the possibility thatpreamyloid or certain types of preamyloid lesionshave a separate origin from neuritic plaques. Each ofbiochemical studies of preamyloid found Ab17–42 to
be a major component (Gowing et al., 1994; Wisniewskiet al., 1996b; Lalowski et al., 1996), while this peptidewas found to be a very minor component of neuriticplaques (Lalowski et al., 1996). These discrepancies inthe Ab content of these lesions suggests a distinctorigin for neuritic plaques, a possibility which hasbeen previously raised based on morphological stud-ies (Wisniewski et al., 1996). However, these biochemi-cal studies of preamyloid do not rule out that a subsetof preamyloid can progress to neuritic plaques or thatthe apparent lack of Ab17–42 in neuritic plaques isrelated to the methods used. In order to distinguishbetween each of the above possibilities further bio-chemical studies are needed of all the components ofthe insoluble and more soluble fractions of preamyloidand neuritic plaque amyloid.
ALZHEIMER’S DISEASE ANDAPOLIPOPROTEIN E
ApoE has been identified as one of the multiple,amyloid-associated proteins (Namba et al., 1991;Wisniewski & Frangione, 1992). ApoE is a 34-kDaproduct of a 4-exon gene located on the long arm ofchromosome 19, which is central to cholesterol metabo-lism (Mahley, 1988). In addition to its function incholesterol transport, apoE is important in local cir-cuits of lipid turnover that are involved in membranerepair. This is especially relevant to the nervous sys-tem, in which apoE has been shown to participate incholesterol redistribution during membrane remodel-ing after injury both in peripheral nerves and in brain.In humans, the apoE gene is polymorphic, leading tothree major apoE isoforms, namely E2, E3, and E4.ApoE3, the most common isoform, has a cysteine atposition 112 and an arginine at position 158; apoE2 isthe least frequent isoform, with cysteine at both posi-tions, whereas apoE4 presents arginine at both sites(Mahley, 1988).
A strong association between the inheritance of theapoE allele E4 and both sporadic AD and late-onsetFAD has been described (Corder et al., 1993; Strittmat-ter et al., 1993). These studies have shown that: (1)carriers of the E4 allele have an increased risk ofdeveloping AD in an allele-dose-dependent manner;(2) the apoE4 genotype modulates the age of onset ofthe disease (Corder et al., 1993; Strittmatter et al., 1993);and (3) the inheritance of the E4 allele correlates withincreased deposition of Ab in blood vessels and plaquesand a greater density of senile plaques in the cerebral
316 Wisniewski, Ghiso, and Frangione
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.
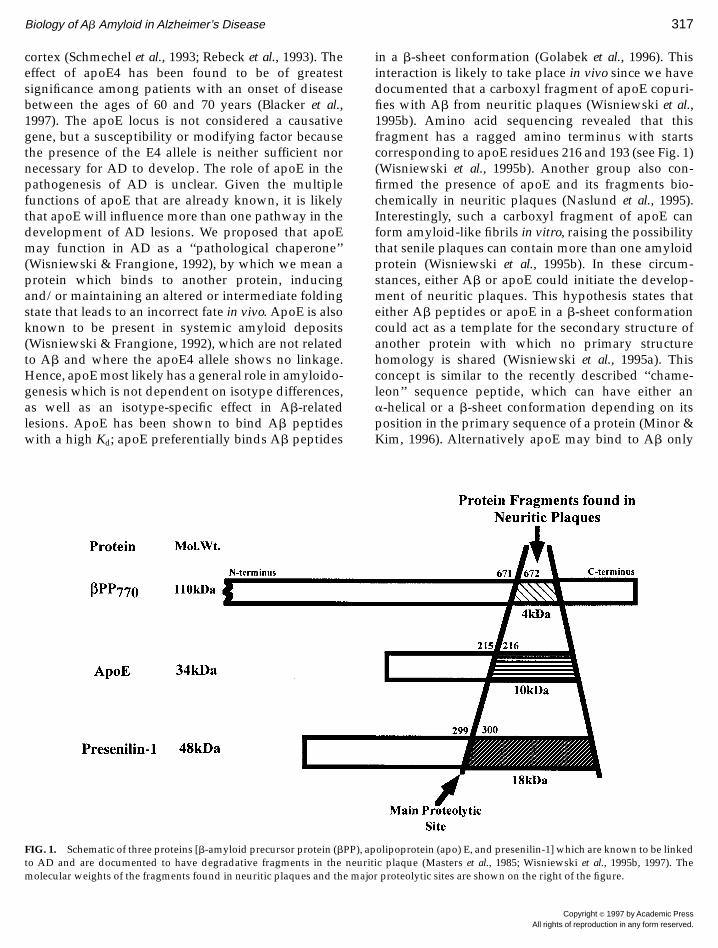
cortex (Schmechel et al., 1993; Rebeck et al., 1993). Theeffect of apoE4 has been found to be of greatestsignificance among patients with an onset of diseasebetween the ages of 60 and 70 years (Blacker et al.,1997). The apoE locus is not considered a causativegene, but a susceptibility or modifying factor becausethe presence of the E4 allele is neither sufficient nornecessary for AD to develop. The role of apoE in thepathogenesis of AD is unclear. Given the multiplefunctions of apoE that are already known, it is likelythat apoE will influence more than one pathway in thedevelopment of AD lesions. We proposed that apoEmay function in AD as a ‘‘pathological chaperone’’(Wisniewski & Frangione, 1992), by which we mean aprotein which binds to another protein, inducingand/or maintaining an altered or intermediate foldingstate that leads to an incorrect fate in vivo. ApoE is alsoknown to be present in systemic amyloid deposits(Wisniewski & Frangione, 1992), which are not relatedto Ab and where the apoE4 allele shows no linkage.Hence, apoE most likely has a general role in amyloido-genesis which is not dependent on isotype differences,as well as an isotype-specific effect in Ab-relatedlesions. ApoE has been shown to bind Ab peptideswith a high Kd; apoE preferentially binds Ab peptides
in a b-sheet conformation (Golabek et al., 1996). Thisinteraction is likely to take place in vivo since we havedocumented that a carboxyl fragment of apoE copuri-fies with Ab from neuritic plaques (Wisniewski et al.,1995b). Amino acid sequencing revealed that thisfragment has a ragged amino terminus with startscorresponding to apoE residues 216 and 193 (see Fig. 1)(Wisniewski et al., 1995b). Another group also con-firmed the presence of apoE and its fragments bio-chemically in neuritic plaques (Naslund et al., 1995).Interestingly, such a carboxyl fragment of apoE canform amyloid-like fibrils in vitro, raising the possibilitythat senile plaques can contain more than one amyloidprotein (Wisniewski et al., 1995b). In these circum-stances, either Ab or apoE could initiate the develop-ment of neuritic plaques. This hypothesis states thateither Ab peptides or apoE in a b-sheet conformationcould act as a template for the secondary structure ofanother protein with which no primary structurehomology is shared (Wisniewski et al., 1995a). Thisconcept is similar to the recently described ‘‘chame-leon’’ sequence peptide, which can have either ana-helical or a b-sheet conformation depending on itsposition in the primary sequence of a protein (Minor &Kim, 1996). Alternatively apoE may bind to Ab only
FIG. 1. Schematic of three proteins [b-amyloid precursor protein (bPP), apolipoprotein (apo) E, and presenilin-1] which are known to be linkedto AD and are documented to have degradative fragments in the neuritic plaque (Masters et al., 1985; Wisniewski et al., 1995b, 1997). Themolecular weights of the fragments found in neuritic plaques and the major proteolytic sites are shown on the right of the figure.
Biology of Ab Amyloid in Alzheimer’s Disease 317
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

after it is deposited and acts as a ‘‘glue’’ to fosterfurther plaque maturation, making it resistant to degra-dation, similar to the suggested role of glycosaminogly-cans and the serum amyloid P component in amyloiddeposits. Such a role is consistent with immunohisto-chemical studies on apoE distribution among differenttypes of amyloid deposits (Sheng et al., 1996; Gallo etal., 1994). Since the carboxyl terminus of apoE is thesame in each of the apoE isotypes and apoE carboxylfragments have also been copurified from systemicamyloidoses where the presence of apoE4 is not a riskfactor (Castano et al., 1995a), this role for apoE is likelyto be independent of isotype differences. A number ofhypotheses have been put forward to explain theisotype-specific role of apoE in AD. These include: (1)ApoE4 may be less of a neurotrophic factor than apoE3(Nathan et al., 1994, 1995; Puttfarcken et al., 1997). (2)ApoE4, but not apoE3 or E2, has been shown topotentiate Ab activation of complement in vitro (Mc-Geer et al., 1997). (3) Differences of ApoE isoformfunction in brain lipid transport may be associatedwith a reduced ability to undergo compensatory sprout-ing and synaptic remodeling among ApoE4 homozy-gous and heterozygous patients (Poirier, 1994). (4)Under certain in vitro conditions apoE4 has beenshown to promote Ab peptide fibrillogenesis morethan apoE3 or 2 (Wisniewski et al., 1994a; Ma et al.,1994; Sanan et al., 1994); however, others have shownthe opposite (Evans et al., 1995) using different condi-tions. If either of these in vitro effects is significant invivo, then apoE can be viewed as a chaperone proteinto Ab peptides, acting to determine its conformationalstate. However, the observation that the amyloid bur-den is increased among apoE4 AD patients (Schmechelet al., 1993; Rebeck et al., 1993) is more consistent withthis apoE isotype promoting fibrillogenesis in vivo.ApoE has been shown to be up-regulated in responseto many forms of nervous system injury. Under theseconditions, apoE, as well as other amyloid-associatedproteins like glycosaminoglycan, may bind to brain Abpeptides, which are in a b-sheet conformation, actingto maintain this abnormal conformation and leading toa resistance to proteolysis. It has been shown thatamong AD patients with the apoE4 isotype, the CSFAb42 levels are significantly decreased compared tothose of controls and AD patients with other apoEisotypes (Seubert et al., 1997). This reduction of Ab42CSF levels is thought to reflect deposition of thispeptide on amyloid plaques. If this is true, it wouldagain suggest that the presence of the apoE4 allele isdirectly or indirectly promoting Ab fibrillogenesis invivo. (4) Differential binding of different apoE isotypes
to Ab peptides may influence sAb clearance from thebrain. The binding of delipidated apoE3 and E4 to Abhas the same Kd (Golabek et al., 1996), while lipidatedapoE3 binds to Ab peptides better than lipidatedapoE4 (LaDu et al., 1994, 1995; Yang et al., 1997). Thelatter finding has been used to suggest that apoE3,with its better binding to Ab, may clear sAb morequickly from the brain, via interactions with apoE-specific receptors (LaDu et al., 1994). (5) ApoE4 linkageto AD may be related to its effects on cerebral vascularpathology. ApoE4 has been linked to congophilic angiopa-thy (independent of other AD pathology) (Premkumaret al., 1996; Greenberg et al., 1996), as well as multi-infarction dementia (Shimano et al., 1989; Slooter et al.,1997) and strokes (Pedro-Botet et al., 1992). Further workis needed to clarify why apoE4 is linked to late-onset AD.
ApoE does not appear to be the only apolipoproteininvolved in AD. It has also been shown that senileplaques contain apoJ and apoA1 (Choi-Miura et al.,1992; Wisniewski et al., 1995a). The latter apolipopro-tein is also known to be amyloidogenic (Wisniewski etal., 1995a). Plasma sAb has been shown to be partiallycomplexed to HDL particles and apoJ, which facilitatesthe passage of Ab peptides across the blood–brainbarrier (Koudinov et al., 1994; Matsubara et al., 1995;Zlokovic et al., 1994) (see below). Hence, severaldifferent apolipoproteins could be involved in neuriticplaque formation, in a situation analogous to thecomplex role of apolipoproteins in the genesis ofatherosclerotic plaques. In addition to the apolipopro-teins, other amyloid-associated proteins are likely toplay a role in plaque development including serumamyloid P component, glycosaminoglycans, heparin-binding growth-associated molecule, and a1-anti-chymotrypsin (Coria et al., 1988; Snow et al., 1987;Wisniewski et al., 1996a; Abraham et al., 1988). Chronicinflammation may also be important in the progres-sion of disease. In particular, activation of the classicalcomplement pathway may be a factor in this inflamma-tory response since AD lesions have been foundimmunohistochemically to contain proteins of thispathway (Eikelenboom & Stam, 1982; McGeer & Rog-ers, 1992). Neuritic plaque formation is likely to be amultiple-step process occurring over many years, in-volving the participation of several different proteins.
CLEARANCE OF A b PEPTIDES
The levels of sAb have been shown conclusively todiffer from controls in the plasma and CSF only amongsome subtypes of early-onset FAD patients, while insporadic AD patients no consistent differences have
318 Wisniewski, Ghiso, and Frangione
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

been documented in these biological fluids (Scheuneret al., 1996; Kosaka et al., 1997). However, the localbrain levels of sAb have been shown by ELISA inlate-onset AD to be significantly raised compared tonormal, age-matched controls (Tabaton et al., 1994;Tamaoka et al., 1994; Harigaya et al., 1995; Kuo et al.,1996). In a study among DS patients of varying ages itwas found that this elevation of brain sAb precedesamyloid plaque formation (Teller et al., 1996). Thiselevation of brain sAb can be a consequence of in-creased production and/or decreased clearance. Experi-ments done in a rat model have shown that syntheticAb1–40 is normally cleared very quickly from thebrain (Ghersi-Egea et al., 1996). It was found thatfollowing a brief infusion of 125I-sAb peptide into onelateral ventricle of a normal rat, by 3.5 min 30% wascleared from ventricular CSF into blood. Another 30%was removed over the next 6.5 min (Ghersi-Egea et al.,1996). These results suggest that clearance of sAbpeptide across the blood–brain barrier may be oneimportant mechanism for the regulation of brain sAblevels. The passage of Ab peptides has also beenstudied in the opposite direction: from the systemiccirculation into the brain in guinea pigs (Zlokovic et al.,1993; Zlokovic, 1996), squirrel monkeys (Ghilardi et al.,1996), mice (Maness et al., 1994), and rats (Saito et al.,1995). In these studies it was found that human sAbpeptides cross the BBB well in those species, such asguinea pigs and monkeys, that have the same Abamino acid sequence as that of humans. The impor-tance of species specificity is highlighted by a recentstudy in which it was shown that in rat, human Abpeptides cross the BBB poorly, while rat Ab peptidescross well (Curran et al., 1996). In serum and CSF aportion of sAb has been found to be complexed to apoJ(Ghiso et al., 1993; Matsubara et al., 1995); in addition,other binding proteins have been suggested such astransthyretin (TTR) and albumin (Schwarzman et al.,1994; Biere et al., 1996). High-affinity interactions in thelow nanomolar range have been demonstrated for theformation of Ab–apoJ complexes (Matsubara et al.,1995); however, the Kd values obtained for the bindingbetween TTR–Ab and albumin–Ab are in the micromo-lar range (unpublished observations). High-affinitytransport has been documented for apoJ–Ab com-plexes via gp330/megalin receptors, with a KM of 0.2nM (Zlokovic et al., 1996). In addition to complexing toapoJ, TTR, and albumin, sAb and Ab have been shownto bind to apoE in the brain (Tabaton et al., 1996;Wisniewski et al., 1995b; Naslund et al., 1995). Signifi-cantly apoE alone and apoE–Ab complexes have beenshown to cross the BBB poorly (Martel et al., 1996).
Hence which carrier protein brain sAb is bound to andthe integrity of the BBB may have significant implica-tions in determining its clearance from the brain.ApoE–Ab complexes can be ligands for the LRP or theVLDL receptors (Rebeck et al., 1993; Christie et al.,1996). LRP receptors are present on neurons, choroidplexus, and reactive astrocytes, while the VLDR recep-tor is present on microglia and some neurons (Christieet al., 1996). Since Ab binds to lipidated apoE3 betterthan lipidated apoE4 (LaDu et al., 1994, 1995; Yang etal., 1997), it is possible that there is impaired brain clear-ance of sAb–apoE4 complexes. There are also other recep-tor candidates which may be important for both themediation of someAb peptide effects and its clearance.Abhas been shown to interact with a cell surface receptor foradvanced glycation end products in neurons, microglia,and vascular endothelium (Yan et al., 1996). This interac-tion may mediate cell adhesion to Ab and the induction ofoxidative stress in microglia. Similarly another cell surfacereceptor, class A scavenger receptor (SR), mediatesadhesion of microglia to Ab fibrils and is associatedwith secretion of reactive oxygen species (El Khoury etal., 1996). The importance of these receptor-Ab andreceptor–Ab complex interactions in the pathogenesisof AD is the subject of extensive investigation cur-rently, since these can be sites of therapeutic interven-tion. These possible Ab–receptor interactions under-score the possible importance of free radical damage inAD. Studies from several laboratories have suggestedthat oxygen free radicals are related to the cytotoxicproperties of Ab (Behl et al., 1992; Butterfield et al.,1994; Goodman & Mattson, 1994). Whether or not Abis directly involved in mediating these effects, markersof oxidative injury have been shown to be topographi-cally associated with the neuropathological lesions ofAD, suggesting their importance for disease progres-sion (Pappolla et al., 1992; Smith et al., 1994; Hensley etal., 1995). These observations have led to the notionthat antioxidants are possible therapeutic agents. Re-cent clinical trials have suggested that there is poten-tial in this therapeutic approach (Sano et al., 1997).
PRESENILINS ANDALZHEIMER’S DISEASE
The PS1 gene on chromosome 14 was identifiedusing a positional cloning strategy (Sherrington et al.,1995; Schellenberg, 1992), while the PS2 gene onchromosome 1 was found based on its homology toPS1 (Levy-Lahad et al., 1995; Rogaev et al., 1995; Li etal., 1995). These two genes have 67% amino acididentity. The majority of early-onset AD is thought to
Biology of Ab Amyloid in Alzheimer’s Disease 319
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

be related to mutations in PS1 and 2. To date over 36mutations have been reported in PS1 and 2 in PS2among familial early-onset AD pedigrees (Lendon etal., 1997; Kwok et al., 1997). Recently we have found anadditional PS1 mutation in a Polish FAD kindred ofP117L (unpublished observations). Since the neuro-pathological features of PS-linked familial AD (FAD)are similar to those of the more common late-onset AD,it is presumed that understanding the role of PS inFAD will have implications for elucidating the pathol-ogy of all forms of AD. The PS1 and PS2 genes encodepredicted proteins of 467 and 448 amino acids, respec-tively, with seven to nine putative transmembranedomains (Lendon et al., 1997). Recent studies of PS1topology have indicated either six or eight transmem-brane domains, with both the amino and the carboxyltermini located in the cytoplasm (Doan et al., 1996; Li &Greenwald, 1996; Lehmann et al., 1997). Both PS1mRNA and PS2 mRNA have been found in manydifferent tissues and cell lines, with high levels in thebrain and neurons (Sherrington et al., 1995; Cribbs etal., 1996; Kovacs et al., 1996). In the normal brainimmunohistochemical studies have shown the pres-ence of this protein predominantly in neuronal cells,where it is preferentially concentrated in the cytoplasmand dendrites (Uchihara et al., 1996; Giannakopoulos etal., 1997; Elder et al., 1996; Weber et al., 1997). Theseimmunocytochemical studies have also suggested apredominant localization to the endoplasmic reticu-lum and Golgi (Kovacs et al., 1996; Lah et al., 1997). Inaddition, a recent study has localized PS to the nuclearmembrane, interphase kinetochores, and centrosomes,suggesting a role in chromosome segregation (Li et al.,1997). Investigations of PS1 processing in cell lines andin brain tissue have shown that a portion of PS1undergoes endoproteolytic cleavage to at least twomajor fragments: a 18-kDa carboxyl fragment and an,28-kDa amino-terminal fragment (Thinakaran et al.,1996). The normal biological role of these proteinsremains unknown; however, clues may be found in theknown functions of two Caenorhabditis elegans proteinswith which PS has homology. These proteins are sel-12(50% identity to PS) (Levitan & Greenwald, 1996) andspe-4 (25% identity to PS) (L’Hernault & Arduengo,1992). Over 80% of the known FAD PS mutations occurin residues that are conserved in sel-12. This protein isknown to be a facilitator of lin-12, a member of theNotch family of receptors involved in intercellularsignaling associated with determining cell fate in thenematode (Levitan & Greenwald, 1996). Sel-12 may bea coreceptor for lin-12 or play a role in receptortrafficking and recycling. Such a role in cellular traffick-
ing or protein processing is consistent with the pro-posed function for spe-4. Loss-of-function mutationsin spe-4 disrupt delivery of proteins to spermatidsduring spermatogenesis in the nematode (L’Hernault& Arduengo, 1992). Whatever the normal role of PS, itis clear from experiments with PS1 knockout mice thatPS is important for central nervous system develop-ment (Wong et al., 1997). These knockout mice dieduring late gestation due to massive hemorrhages thatare limited to the brain and spinal cord. This hemor-rhage is present beneath the primordial dura and lepto-meninges, as well as within the ventricles and in the brainparenchyma (Wong et al., 1997). Hence loss of PS functiondoes not appear to be compatible with life. The promi-nence of cerebral vessel pathology in these knockout miceis of interest, in that PS is known to be present both innormal cerebral vessels and in congophilic angiopathy(Wisniewski et al., 1997; Levey et al., 1997).
How PS is involved in the pathogenesis of AD isunknown; however, some evidence has suggested apossible interaction with Ab and/or bPP. PS mutationsmay influence the production of the more highlyamyloidogenic Ab1–42 form of Ab. Higher levels ofAb42 have been found in the plasma and culturedfibroblast media of PS1 and 2 mutation carriers(Scheuner et al., 1996). This increase has also beennoted in brains of transgenic mice and transfected cells(Duff et al., 1996; Citron et al., 1997; Borchelt et al., 1996).Furthermore immunohistochemical studies using anti-bodies for the carboxyl terminus of Ab42 have shownan increase of Ab42 deposits in the brains of ADpatients with PS1 mutations (Lemere et al., 1996b). Ithas also been recently shown that in cells which aretransfected with both PS-2 and bPP, stable complexescan form between these two gene products (Weide-mann et al., 1997), again suggesting that PS mayinteract with bPP metabolism. There are also reportsthat recombinant forms of PS2 can regulate apoptosisin lymphocytes and PC12 cells (Vito et al., 1996;Wolozin et al., 1996). However, it remains unclear howspecific the above observations are, since they arebased largely on data from transfected cells and trans-genic animals. Another piece of evidence suggestingan interaction between PS and Ab is the localization ofPS epitopes within neuritic plaques. In 1995 we re-ported that an antibody raised to the carboxyl termi-nus of PS1 immunoreacted with some neuritic plaques(Wisniewski et al., 1995c). This immunoreactivity wasfound in neuritic plaques of both PS1-linked FADpatients and in late-onset, sporadic AD, suggesting ageneral role for presenilin in AD. Since then additionalreports using other antibodies have immunohisto-
320 Wisniewski, Ghiso, and Frangione
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

chemically confirmed the presence of PS in associationwith the amyloid of AD neuritic plaques (Giannakopou-los et al., 1997; Bouras et al., 1996; Uchihara et al., 1996;Levey et al., 1997). However, some investigators havealso reported just neuronal staining (Weber et al., 1997)or immunoreactivity in NFT (Murphy et al., 1996)without neuritic plaque staining. These divergent re-sults are likely to be related to differences in theepitopes used to produce the antibodies for each ofthese studies and to be related to other factors such asvariations in antibody titer, distinct antibody affinities,and the types of tissue fixation and pretreatment.Recently the observation that antibodies raised tocarboxyl epitopes of PS immunoreact with neuriticplaques has been confirmed biochemically (Wisniewskiet al., 1997). It was found that the 18-kDa carboxylfragment of PS was also present in partially purifiedneuritic plaque amyloid fractions and that this frag-ment appeared to bind to Ab. The identity of this PSfragment was confirmed by amino acid sequencingand found to start at residue 300 (see Fig. 1) (Wisniewskiet al., 1997), which is very similar to a recent studywhere the main cleavage site of PS1 was found to occurbetween residues 298 and 299 in cells transfected withhuman PS1 (Podlisny et al., 1997). These findings againpoint to a possible close association between PS andAb/bPP. Another, not mutually exclusive, possibilityis that the processing of PS may be important in AD.Some PS1 mutations and one of the PS2 mutationshave been shown to alter PS processing with an in-crease in the production of the PS carboxyl fragment(Tanzi et al., 1996; Merken et al., 1996; Thinakaran et al.,1996). However, this has not been a consistent findingwith all the PS1 FAD-linked mutations. For example,the Cys411Tyr PS1 mutation shows no quantitativealteration of the amino and carboxyl fragments in braintissue (Podlisny et al., 1997). A number of other possibleinvolvements of PS in AD have also been suggested. Forexample it has been reported that cells transfected withthe L286V PS1 FAD-linked mutation are sensitized todeath induced by Ab peptides (Guo et al., 1997). PSmutations may have a role in chromosomal missegre-gation during mitosis (Li et al., 1997). Given theprominence of cerebral vessel abnormalities in PS1knockout mice (Wong et al., 1997) and the presence ofPS immunoreactivity in congophilic angiopathy(Wisniewski et al., 1997), PS may also have a role indetermining AD cerebral vessel pathology.
The role of PS in the pathogenesis of AD still remainsunclear. Further experiments utilizing patient samplesas well as cell lines and transgenic animals willprovide insights into the possible role of PS in AD.
SUMMARY
The genetic associations with the pathological fea-tures of AD are diverse: A rapidly growing number ofmutations in presenilin 1 and 2 on chromosomes 14and 1, respectively, are found in many early-onset FADpatients (Lendon et al., 1997). In addition, bPP muta-tions are found in a small percentage of early-onsetFAD kindreds. The apoE4 allele on chromosome 19 isassociated with the presence of the most common formof AD, sporadic AD (Wisniewski & Frangione, 1992;Namba et al., 1991). However, it is clear that otherproteins are also involved in the pathogenesis of AD,since some early-onset FAD kindreds do not havelinkage to PS1, PS2, apoE, or bPP, while at least 50% oflate-onset AD is unrelated to apoE. Other proteinswhich have been implicated in the formation of senileplaques, but so far are not known to have any geneticlinkage to AD, include proteoglycans (Snow et al.,1987), apoA1 (Wisniewski et al., 1995a), a1-antichymo-trypsin (Abraham et al., 1988), HB-GAM (Wisniewskiet al., 1996a), complement components (McGeer &Rogers, 1992), acetylcholinesterase (Friede, 1965), andNAC (Ueda et al., 1993). Which of these proteins will bethe most important for the etiology of the mostcommon form of AD, late-onset sporadic AD, remainsan open question. Three of the genes which are nowknown to be linked to AD, including PS1, bPP, andapoE, have been established immunohistochemicallyand biochemically to be components of senile plaques(see Fig. 1). This raises at least two possibilities: eithereach of these proteins is part of one pathway withAb-related amyloid formation as a final causativepathogenic event or amyloid deposition in AD is areactive process related to dysfunction of a number ofdifferent CNS proteins. Whether or not amyloid forma-tion is directly causative in the pathogenesis of AD,current data suggest that new therapeutic approacheswhich may inhibit the aggregation and/or the confor-mational change of sAb to Ab fibrils (Soto et al., 1996)have the greatest likelihood to make a significantimpact on controlling amyloid accumulation in AD.
REFERENCES
Abraham, C. R., Selkoe, D. J., & Potter, H. (1988) Immunochemicalidentification of the serine protease inhibitor alpha 1-antichymo-trypsin in the brain amyloid deposits of Alzheimer’s disease. Cell52, 487–501.
Behl, C., Davis, J. B., Cole, G. M., & Schubert, D. (1992) Vitamin Eprotects nerve cells from amyloid protein toxicity. Biochem. Bio-phys. Res. Commun. 186, 944–950.
Biology of Ab Amyloid in Alzheimer’s Disease 321
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

Biere, A. L., Ostaszewski, B., Stimson, E. R., Hyman, B. T., Maggio,J. E., & Selkoe, D. J. (1996) Amyloid b-peptide is transported onlipoproteins and albumin in human plasma. J. Biol. Chem. 271,32916–32922.
Blacker, D., Haines, J. L., Rodes, L., Terwedow, H., Go, R. C. P.,Harrell, L. E., Perry, R. T., Bassett, S. S., Chase, G., Meyers, D.,Albert, M. S., & Tanzi, R. (1997) ApoE-4 and age at onset ofAlzheimer’s disease: The NIMH genetics initiative. Neurology 48,139–147.
Borchelt, D. R., Thinakaran, G., Eckman, C. B., Lee, M. K., Daven-port, F., Ratovitsky, T., Prada, C. M., Kim, G., Seekins, S., Yager, D.,Slunt, H. H., Wang, R., Seeger, M., Levey, A. I., Gandy, S. E.,Copeland, N. G., Jenkins, N. A., Price, D. L., & Younkin, S. G.(1996) Familial Alzheimer’s disease-linked presenilin 1 variantselevate Ab1-42/1-40 ratio in vitro and in vivo. Neuron 17, 1005–1013.
Bouras, C., Giannakopoulos, P., Schioi, J., Tezapsidis, N., & Robakis,N. K. (1996) Presenilin-1 polymorphism and Alzheimer’s disease.Lancet 347, 1185–1186.
Busciglio, J., Gabuzda, D. H., Matsudaira, P., & Yankner, B. A. (1993)Generation of b-amyloid in the secretory pathway in neuronal andnon-neuronal cells. Proc. Natl. Acad. Sci. USA 90, 2092–2096.
Butterfield, D. A., Hensley, K., Harris, M., Mattson, M. P., & Carney,J. M. (1994) Beta-amyloid peptide free radical fragments initiatesynaptosomal lipoperoxidation in a sequence-specific fashion:Implications to Alzheimer’s disease. Biochem. Biophys. Res. Com-mun. 200, 710–715.
Cai, X. D., Golde, T. E., & Younkin, S. G. (1993) Release of excessamyloid b protein from a mutant amyloid b protein precursor.Science 259, 514–516.
Castano, E. M., Prelli, F., Pras, M., & Frangione, B. (1995a) Apolipo-protein E carboxyl-terminal fragments are complexed to amyloidsA and L. Implications for amyloidogenesis and Alzheimer’sdisease. J. Biol. Chem. 270, 17610–17615.
Castano, E. M., Prelli, F., Wisniewski, T., Golabek, A. A., Kumar,R. A., Soto, C., & Frangione, B. (1995b) Fibrillogenesis in Alzhei-mer’s disease of amyloid beta peptides and apolipoprotein E.Biochem. J. 306, 599–604.
Castano, E. M., Prelli, F., Soto, C., Beavis, R., Matsubara, E., Shoji, M.,& Frangione, B. (1996) The length of amyloid-b in hereditarycerebral hemorrhage with amyloidosis, Dutch type—Implicationsfor the role of amyloid-b 1-42 in Alzheimer’s disease. J. Biol. Chem.271, 32185–32191.
Choi-Miura, N. H., Ihara, Y., Fukuchi, K., Takeda, M., Nakano, Y.,Tobe, T., & Tomita, M. (1992) SP-40, 40 is a constituent ofAlzheimer’s amyloid. Acta Neuropathol. 83, 260–264.
Christie, R. H., Chung, H., Rebeck, G. W., Strickland, D., & Hyman,B. T. (1996) Expression of the very low-density lipoprotein recep-tor (VLDL-r), an apolipoprotein-E receptor, in the central nervoussystem and in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 55,491–498.
Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y.,Seubert, P., Vigo-Pelfrey, C., Lieberburg, I., & Selkoe, D. J. (1992)Mutation of the beta-amyloid precursor protein in familial Alzhei-mer’s disease increases beta-protein production. Nature 360, 672–674.
Citron, M., Vigo-Pelfrey, C., Teplow, D. B., Miller, C., Schenk, D.,Johnston, J., & Winblad, B. (1993) Excessive production of amyloidb-amyloid precursor protein gene. Proc. Natl. Acad. Sci. USA 91,11993–11997.
Citron, M., Westaway, D., Xia, W. M., Carlson, G., Diehl, T.,Levesque, G., Johnson-Wood, K., Lee, M., Seubert, P., Davis, A.,
Kholodenko, D., Motter, R., Sherrington, R., Perry, B., Yao, H.,Strome, R., Lieberburg, I., Rommens, J., Kim, S., Schenk, D., Fraser,P., Hyslop, P. S., & Selkoe, D. J. (1997) Mutant presenilins ofAlzheimer’s disease increase production of 42-residue amyloidb-protein in both transfected cells and transgenic mice. NatureMed. 3, 67–72.
Clements, A., Walsh, D. M., Williams, C. H., & Allsop, D. (1993)Effects of the mutation Glu22 to Gln and Ala 21 to Gly on theaggregation of a synthetic fragment of the Alzheimer’s amyloidb/A4 peptide. Neurosci. Lett. 161, 17–20.
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E.,Gaskell, P. C., Small, G. W., Roses, A. D., Haines, J. L., &Pericak-Vance, M. A. (1993) Gene dose of apolipoprotein E type 4allele and the risk of Alzheimer’s disease in late onset families.Science 261, 921–923.
Coria, F., Castano, E. M., Prelli, F., Larrondo-Lillo, M., vanDuinen, S.,Shelanski, M. L., & Frangione, B. (1988) Isolation and characteriza-tion of amyloid P component from Alzheimer’s disease and othertypes of cerebral amyloidosis. Lab. Invest. 58, 454–457.
Cribbs, D. H., Chen, L. S., Bende, S. M., & LaFerla, F. M. (1996)Widespread neuronal expression of the presenilin-1 early-onsetAlzheimer’s disease gene in the murine brain. Am. J. Pathol. 148,1797–1806.
Crystal, H. A., Dickson, D. W., Sliwinski, M. J., Lipton, R. B., Grober,E., Marks-Nelson, H., & Antis, P. (1993) Pathological markersassociated with normal aging and dementia in the elderly. Ann.Neurol. 34, 566–573.
Cummings, B. J., Su, J. H., Cotman, C. W., White, R., & Russell, M. J.(1993) Beta-amyloid accumulation in aged canine brain: A modelof early plaque formation in Alzheimer’s disease. Neurobiol. Aging14, 547–560.
Curran, G. L., Haggard, J. J., Selkoe, D. J., & Poduslo, J. F. (1996)Permeability and residual plasma volume of human dutch variantand rat amyloid b-protein at the BBB. Soc. Neurosci. Abstr. 22, 1168.[Abstract]
Davies, P., Duyckaerts, C., Beyreuther, K., Masters, C. L., Peitte, F., &Hauw, J. J. (1988) A4 amyloid protein deposition and the diagnosisof Alzheime’s disease: Prevalence in aged brains determined byimmunohistochemistry compared with conventional neuropatho-logical techniques. Neurology 38, 1688–1693.
Delaere, P., Duyckaerts, C., Beyreuther, K., Masters, C. L., Peitte, F., &Hauw, J. J. (1990) Large amounts of neocortical bA4 depositswithout neuritic plaques or tangles in a psychometrically assessednondemented case. Neurosci. Lett. 116, 87–93.
Doan, A., Thinakaran, G., Borchelt, D. R., Slunt, H. H., Ratovitsky, T.,Podlisny, M. B., Selkoe, D. J., Seeger, M., Gandy, S. E., Price, D. L.,& Sisodia, S. S. (1996) Protein topology of presenilin 1. Neuron 17,1023–1030.
Duff, K., Eckman, C., Zehr, C., Yu, X., Prada, C. M., Perez-Tur, J.,Hutton, M., Buee, L., Harigaya, Y., Yager, D., Morgan, D., Gordon,M. N., Holcomb, L., Refolo, L., Zenk, B., Hardy, J., & Younkin, S.(1996) Increased amyloid-b42(43) in brains of mice expressingmutant presenilin 1. Nature 383, 710–713.
Eikelenboom, P., & Stam, F. C. (1982) Immunoglobulins and comple-ment factors in senile plaques. Acta Neuropathol. 57, 239–242.
El Khoury, J., Hickman, S. E., Thomas, C. A., Cao, L., Silverstein,S. C., & Loike, J. D. (1996) Scavenger receptor-mediated adhesionof microglia to b-amyloid fibrils. Nature 382, 716–719.
Elder, G. A., Tezapsidis, N., Carter, J., Shioi, J., Bouras, C., Li, H. C.,Johnson, J. M., Efthimiopoulos, S., Friedrich, V. L., & Robakis,N. K. (1996) Identification and neuron specific expression of the
322 Wisniewski, Ghiso, and Frangione
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

S182/presenilin I protein in human and rodent brains. J. Neurosci.Res. 45, 308–320.
Esch, F. S., Keim, P. S., Beattie, E. C., Blacker, R. W., Culwell, A. K.,Oltersdorf, T., McClure, D., & Ward, P. J. (1990) Cleavage ofamyloid b peptide during constitutive processing of its precursor.Science 248, 1122–1124.
Evans, K. C., Berger, E. P., Cho, C. G., Weisgraber, K. H., & Lansbury,P. T., Jr. (1995) Apolipoprotein E is a kinetic but not a thermody-namic inhibitor of amyloid formation: Implications for the patho-genesis and treatment of Alzheimer disease. Proc. Natl. Acad. Sci.USA 92, 763–767.
Friede, R. L. (1965) Enzyme histochemical studies of senile plaques.J. Neuropathol. Exp. Neurol. 24, 477–491.
Gallo, G., Wisniewski, T., Choi-Miura, N. H., Ghiso, J., & Frangione,B. (1994) Potential role of apolipoprotein-E in fibrillogenesis. Am.J. Pathol. 145, 526–530.
Games, D., Adams, D., Alessandrini, R., Barbour, R., Berthelette, P.,Blackwell, C., Carr, T., Clemens, J., Donaldson, T., Gillespie, F.,Guido, T., Hagoplan, S., Johnson-Wood, K., Kan, K., Lee, M.,Leibowitz, P., Lieberburg, I., Little, S., Masliah, E., McConlogue, L.,Montoya-Zavala, M., Mucke, L., Paganini, L., Penniman, E.,Power, M., Schenk, D., Seubert, P., Snyder, B., Soriano, F., Tan, H.,Vitale, J., Wadsworth, S., Wolozin, B., & Zhao, J. (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717Fb-amyloid precursor protein. Nature 373, 523–527.
Ghersi-Egea, J. F., Gorevic, P. D., Ghiso, J., Frangione, B., Patlak, C. S.,& Fenstermacher, J. D. (1996) Fate of cerebrospinal fluid-borneamyloid b-peptide: Rapid clearance into blood and appreciableaccumulation by cerebral arteries. J. Neurochem. 67, 880–883.
Ghilardi, J. R., Catton, M., Stimson, E. R., Rogers, S., Walker, L. C.,Maggio, J. E., & Mantyh, P. W. (1996) Intra-arterial infusion of125I-Ab1–40 labels amyloid deposits in the aged primate brain invivo. Neuroreport 7, 2607–2611.
Ghiso, J., Matsubara, E., Koudinov, A., Choi-Miura, N. H., Tomita,M., Wisniewski, T., & Frangione, B. (1993) The cerebrospinal-fluidsoluble form of Alzheimer’s amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem. J. 293, 27–30.
Giannakopoulos, P., Bouras, C., Kovari, E., Shioi, J., Tezapsidis, N.,Hof, P. R., & Robakis, N. K. (1997) Presenilin-1 immunoreactiveneurons are preserved in late-onset Alzheimer’s disease. Am. J.Pathol. 150, 429–436.
Glenner, G. G., & Wong, C. W. (1984) Alzheimer’s disease: initialreport of the purification and characterization of a novel cerebro-vascular amyloid protein. Biochem. Biophys. Res. Commun. 120,885–890.
Goate, A., Chartier-Harlin, M.-C., Mullan, M., Brown, J., Crawford,F., Fidani, L., Giuffra, L., Haynes, A., Irving, N., James, L., Mant,R., Newton, P., Rooke, K., Roques, P., Talbot, C., Pericak-Vance, M.,Roses, A., Williamson, R., Rossor, M., Owen, M., & Hardy, J. (1991)Segregation of a missense mutation in the amyloid precursorprotein gene with familial Alzheimer’s disease. Nature 349, 704–706.
Golabek, A. A., Soto, C., Vogel, T., & Wisniewski, T. (1996) Theinteraction between apolipoprotein E and Alzheimer’s amyloidb-peptide is dependent on b-peptide conformation. J. Biol. Chem.271, 10602–10606.
Golde, T. E., Estus, S., Usiak, M., Younkin, L. H., & Younkin, S. G.(1990) Expression of b-amyloid protein precursor mRNAs; recog-nition of a novel alternatively spliced form and quantitation inAlzheimer’s disease using PCR. Neuron 4, 253–267.
Goldgaber, D., Lerman, M. I., McBride, O. W., Saffiotti, U., &
Gajdusek, D. C. (1987) Characterization and chromosomal localiza-tion of a cDNA encoding brain amyloid of Alzheimer’s disease.Science 235, 877–880.
Goodman, Y., & Mattson, M. P. (1994) Secreted forms of beta-amyloid precursor protein protect hippocampal neurons againstamyloid beta-peptide-induced oxidative injury. Exp. Neurol. 128,1–12.
Gowing, E., Roher, A. E., Woods, A. S., Cotter, R. J., Chaney, M.,Little, S. P., & Ball, M. J. (1994) Chemical characterization of Ab
17–42 peptide, a component of diffuse amyloid deposits ofAlzheimer disease. J. Biol. Chem. 269, 10987–10990.
Greenberg, S. M., Briggs, M. E., Hyman, B. T., Kokoris, G. J., Takis, C.,Kanter, D. S., Kase, C. S., & Pessin, M. S. (1996) Apolipoprotein Ee4 is associated with the presence and earlier onset of hemorrhagein cerebral amyloid angiopathy. Stroke 27, 1333–1337.
Guo, Q., Furukawa, K., Sopher, B. L., Pham, D. G., Xie, J., Robinson,N., Martin, G. M., & Mattson, M. P. (1997) Alzheimer’s PS-1mutation perturbs calcium homeostasis and sensitizes PC12 cellsto death induced by amyloid b-peptide. Neuroreport 8, 379–383.
Haass, C., Schlossmacher, M. G., Hung, A. Y., Vigo-Pelfrey, C.,Mellon, A., Ostaszewski, B. L., Lieberburg, I., Koo, E. H., Schenk,D., Teplow, D. B., & Selkoe, D. J. (1992) Amyloid beta-peptide isproduced by cultured cells during normal metabolism. Nature 359,322–325.
Hardy, J. (1996) New insights into the genetics of Alzheimer’sdisease. Ann. Med. 28, 255–258.
Harigaya, Y., Shoji, M., Kawarabayashi, T., Kanai, M., Nakamura, T.,Iizuka, T., Igeta, Y., Saido, T. C., Sahara, N., Mori, H., & Hirai, S.(1995) Modified amyloid beta protein ending at 42 or 40 withdifferent solubility accumulates in the brain of Alzheimer’s dis-ease. Biochem. Biophys. Res. Commun. 211, 1015–1022.
Hensley, K., Hall, N., Subramaniam, R., Cole, P., Harris, M., Aksenov,M., Aksenova, M., Gabbita, S. P., Wu, J. F., Carney, J. M., Lovell, M.,Markesbery, W. R., & Butterfield, D. A. (1995) Brain regionalcorrespondence between Alzheimer’s disease histopathology andbiomarkers of protein oxidation. J. Neurochem. 65, 2146–2156.
Hilbich, C., Kisters-Woike, B., Reed, J., Masters, C. L., & Beyreuther,K. (1991) Aggregation and secondary structure of synthetic amy-loid beta A4 peptides of Alzheimer’s disease. J. Mol. Biol. 218,149–163.
Hsiao, K. K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y.,Younkin, S., Yang, F., & Cole, G. (1996) Correlative memorydeficits, Ab elevation and amyloid plaques in transgenic mice.Science 274, 99–102.
Irizarry, M. C., McNamara, M., Fedorchak, K., Hsiao, K. K., &Hyman, B. T. (1997a) Tg(HuAPP695.K670N-M671L)2576 micedevelop age-related Ab deposits and neuropil abnormalities, butno neuronal loss in CA1. J. Neuropathol. Exp. Neurol., 56, 965–973.
Irizarry, M. C., Soriano, F., McNamara, M., Page, K. J., Schenk, D.,Games, D., & Hyman, B. T. (1997b) Ab deposition is associatedwith neuropil change, but not overt neuronal loss in humanamyloid precursor protein V717 F(PD APP) transgenic mice. J.Neurosci. 17, 7053–7059.
Iwatsubo, T., Odaka, A., Suzuki, N., Mizusawa, H., Nukina, N., &Ihara, Y. (1994) Visualization of Ab 42(43) and Ab 40 in senileplaques with end-specific Ab monoclonals: Evidence that aninitially deposited species is Ab 42(43). Neuron 13, 45–53.
Jarrett, J. T., Berger, E. P., & Lansbury, P. T., Jr. (1993) The carboxyterminus of the beta amyloid protein is critical for the seeding ofamyloid formation: Implications for the pathogenesis of Alzhei-mer’s disease. Biochemistry 32, 4693–4697.
Joachim, C. L., Duffy, L. K., Morris, J. H., & Selkoe, D. J. (1988)
Biology of Ab Amyloid in Alzheimer’s Disease 323
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

Protein chemical and immunocytochemical studies of meningovas-cular beta-amyloid protein in Alzheimer’s disease and normalaging. Brain Res. 474, 100–111.
Johnson-Wood, K., Lee, M., Motter, R., Hu, K., Gordon, G., Barbour,R., Khan, K., Gordon, M., Tan, H., Games, D., Lieberburg, I.,Schenk, D., Seubert, P., & McConlogue, L. (1997) Amyloid precur-sor protein processing and Ab42 deposition in a transgenic mousemodel of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 94,1550–1555.
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters,C. L., Grzeschik, K. H., Multhaup, G., Beyreuther, K., & Muller-Hill, B. (1987) The precursor of Alzheimer’s disease amyloid A4protein resembles a cell-surface receptor. Nature 325, 733–736.
Kida, E., Choi-Miura, N. H., & Wisniewski, K. E. (1995a) Depositionof apolipoproteins E and J in senile plaques is topographicallydetermined in both Alzheimer’s disease and Down’s syndromebrain. Brain Res. 685, 211–216.
Kida, E., Wisniewski, K. E., & Wisniewski, H. M. (1995b) Earlyamyloid-b deposits show different immunoreactivity to the amino-and carboxyl-terminal regions of b-peptide in Alzheimer’s diseaseand Down’s syndrome brain. Neurosci. Lett. 193, 105–108.
Kitaguchi, N., Takahashi, Y., Tokushima, Y., Shiojuiri, S., & Ito, H.(1988) Novel precursor of Alzheimer’s disease amyloid proteinshows protease inhibitory activity. Nature 331, 530–532.
Konig, G., Monning, U., Czech, C., Prior, R., Banati, R., Schreiter-Gasser, U., Bauer, J., Masters, C. L., & Beyreuther, K. (1992)Identification and differential expression of a novel alternativesplice isoform of the b A4 amyloid precursor protein (APP) mRNAin leukocytes and brain microglial cells. J. Biol. Chem. 267, 10804–10809.
Kosaka, T., Imagawa, M., Seki, K., Arai, H., Sasaki, H., Tsuji, S.,Asami-Odaka, A., Fukushima, T., Imai, K., & Iwatsubo, T. (1997)The bAPP717 Alzheimer mutation increase the presentage ofplasma amyloid-b protein ending at Ab42(43). Neurology 48,741–745.
Kosik, K. S., & Coleman, P. (1992) Is b-amyloid neurotoxic? Neurobiol.Aging 13, 535–627.
Koudinov, A., Matsubara, E., Frangione, B., & Ghiso, J. (1994) Thesoluble form of Alzheimer’s amyloid beta protein is complexed tohigh density lipoprotein 3 and very high density lipoprotein innormal human plasma. Biochem. Biophys. Res. Commun. 205,1164–1171.
Kovacs, D., Fausett, H. J., Page, K. J., Kim, T., Moir, R. D., Merriam,D. E., Hollister, R., Hallmark, O. G., Mancini, R., Felsenstein, K. M.,Human, B. T., Tanzi, R., & Wasco, W. (1996) Alzheimer associatedpresenilin 1 and 2: Neuronal expression in brain and localizationto intracellular membranes in mammalian cells. Nature Med. 2,224–229.
Kuo, Y. M., Emmerling, M. R., Vigo-Pelfrey, C., Kasunic, T. C.,Kirkpatrick, J. B., Murdoch, G. H., Ball, M. J., & Roher, A. E. (1996)Water-soluble Ab (N-40, N-42) oligomers in normal and Alzhei-mer disease brains. J. Biol. Chem. 271, 4077–4081.
Kwok, J. B. J., Taddel, K., Hallupp, M., Fisher, C., Brooks, W. S., Broe,G. A., Hardy, J., Fulham, M. J., Nicholson, G. A., Stell, R., St.George Hyslop, P., Fraser, P., Kakulas, B. A., Clarnette, R., Relkin,N. R., Gandy, S. E., Schofield, P., & Martins, R. N. (1997) Two novel(M233T and R278T) presenilin-1 mutations in early-onset Alzhei-mer’s disease pedigrees and preliminary evidence for associationof presenilin-1 mutations with a novel phenotype. Neuroreport, inpress.
L’Hernault, S. W., & Arduengo, P. M. (1992) Mutation of a putativesperm membrane protein in Caenorhabditis elegans prevents
sperm differentiation but not its associated meiotic divisions. J.Cell Biol. 119, 55–68.
LaDu, M. J., Falduto, M. T., Manelli, A. M., Reardon, C. A., Getz,G. S., & Frail, D. E. (1994) Isoform-specific binding of apolipopro-tein E to beta-amyloid. J. Biol. Chem. 269, 23403–23406.
LaDu, M. J., Pederson, T. M., Frail, D. E., Reardon, C. A., Getz, G. S.,& Falduto, M. T. (1995) Purification of apolipoprotein E attenuatesisoform-specific binding to beta-amyloid. J. Biol. Chem. 270, 9039–9042.
Lah, J. J., Heilman, C. J., Nash, N. R., Rees, H. D., Yi, H., Counts, S. E.,& Levey, A. I. (1997) Light and electron microscopic localization ofpresenilin-1 in primate brain. J. Neurosci. 17, 1971–1980.
Lalowski, M., Golabek, A. A., Lemere, C. A., Selkoe, D. J., Wisniewski,H. M., Beavis, R. C., Frangione, B., & Wisniewski, T. (1996) The‘‘non-amyloidogenic’’ p3 fragment (Ab17–42) is a major constitu-ent of Down syndrome cerebellar preamyloid. J. Biol. Chem. 271,33623–33631.
Lehmann, S., Chiesa, R., & Harris, D. A. (1997) Evidence for asix-transmembrane domain structure of presenilin 1. J. Biol. Chem.272, 12047–12051.
Lemaire, H. G., Salbaum, J. M., Multhaup, G., Kang, J., Bayney, R. M.,Unterbeck, A., Beyreuther, K., & Muller-Hill, B. (1989) The PreA4695 precursor protein of Alzheimer’s disease A4 amyloid isencoded by 16 exons. Nucleic Acids Res. 17, 517–522.
Lemere, C. A., Blusztajn, J. K., Yamaguchi, H., Wisniewski, T., Saido,T. C., & Selkoe, D. J. (1996a) Sequence of deposition of heteroge-neous amyloid b-peptides and APO E in Down syndrome:Implications for initial events in amyloid plaque formation.Neurobiol. Dis. 3, 16–32.
Lemere, C. A., Lopera, F., Kosik, K. S., Lendon, C. L., Ossa, J., Saido,T. C., Yamaguchi, H., Ruiz, A., Martinez, A. O., Madrigal, L.,Hincapie, L., Arango, J. C., Anthony, D. C., Koo, C. H., Goate, A., &Selkoe, D. J. (1996b) The E280A presenilin 1 Alzheimer mutationproduced Ab42 deposition and severe cerebellar pathology. Na-ture Med. 2, 1146–1150.
Lendon, C. L., Ashall, F., & Goate, A. M. (1997) Exploring theetiology of Alzheimer disease using molecular genetics. J. Am.Med. Assoc. 277, 825–831.
Levey, A. I., Heilman, C. J., Lah, J. J., Nash, N. R., Rees, H. D., Wakai,M., Mirra, S. S., Rye, D., Nochlin, D., Bird, T. D., & Mufson, E. J.(1997) Presenilin-1 protein expression in familial and sporadicAlzheimer’s disease. Ann. Neurol., 41, 742–753.
Levitan, D., & Greenwald, I. (1996) Facilitation of lin-12-mediatedsigalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’sdisease gene. Nature 377, 351–354.
Levy, E., Carman, M. D., Fernandez-Madrid, I., Lieberburg, I.,Power, M. D., vanDuinen, S. G., Bots, G. T. A. M., Luyendijk, W., &Frangione, B. (1990) Mutation of the Alzheimer’s disease amyloidgene in hereditary cerebral hemorrhage, Dutch type. Science 248,1124–1126.
Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J.,Pettingell, W. H., Chang-en, Y., Jondro, P. D., Schmidt, S. D., Wang,K., Crowley, A. C., Fu, Y. H., Guenette, S. Y., Galas, D., Nemens, E.,Wijsman, E. M., Bird, T. D., Schellenberg, G. D., & Tanzi, R. E.(1995) Candidate gene for the chromosome 1 familial Alzheimer’sdisease locus. Science 269, 973–977.
Li, J., Ma, J., & Potter, H. (1995) Identification and expression analysisof a potential familial Alzheimer disease gene on chromosome 1related to AD3. Proc. Natl. Acad. Sci. USA 92, 12180–12184.
Li, J., Xu, M., Zhou, H., Ma, J., & Potter, H. (1997) Alzheimerpresenilins in the nuclear membrane, interphase kinetochores,and centro-
324 Wisniewski, Ghiso, and Frangione
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

somes suggests a role in chromosome segragation. Cell 90, 917–927.
Li, X., & Greenwald, I. (1996) Membrane topology of the C. elegansSEL-12 presenilin. Neuron 17, 1015–1021.
Lorenzo, A., & Yankner, B. A. (1994) Beta-amyloid neurotoxicityrequires fibril formation and is inhibited by congo red. Proc. Natl.Acad. Sci. USA 91, 12243–12247.
Ma, J., Yee, A., Brewer, H. B., Jr., Das, S., & Potter, H. (1994)Amyloid-associated proteins alpha 1-antichymotrypsin and apoli-poprotein E promote assembly of Alzheimer beta-protein intofilaments. Nature 372, 92–94.
Mahley, R. W. (1988) Apolipoprotein E: Cholesterol transport proteinwith expanding role in cell biology. Science 240, 622–630.
Maness, L. M., Banks, W. A., Podlisny, M. B., Selkoe, D. J., & Kastin,A. J. (1994) Passage of human amyloid beta-protein 1–40 across themurine blood–brain barrier. Life Sci. 55, 1643–1650.
Martel, C. L., Mackic, J. B., McComb, J. G., Ghiso, J., & Zlokovic, B. V.(1996) Blood–brain barrier uptake of the 40 and 42 amino acidsequences of circulating Alzheimer’s amyloid b in guinea pigs.Neurosci. Lett. 206, 157–160.
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDon-ald, B. L., & Beyreuther, K. (1985) Amyloid plaque core protein inAlzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA82, 4245–4249.
Matsubara, E., Frangione, B., & Ghiso, J. (1995) Characterization ofapolipoprotein J-Alzheimer’s A beta interaction. J. Biol. Chem. 270,7563–7567.
McGeer, P. L., Walker, D. G., Pitas, R. E., Mahley, R. W., & McGeer,E. G. (1997) Apolipoprotein E4 (ApoE4) but not apoE3 or apoE2potentiates b-amyloid protein activation of complement in vitro.Brain Res. 749, 135–138.
McGeer, P. L., & Rogers, J. (1992) Anti-inflammatory agents as atherapeutic approach to Alzheimer’s disease. Neurology 42, 447–449.
Merken, M., Takahashi, H., Honda, T., Sato, K., Murayama, M.,Nakazato, Y., Noguchi, K., Imahori, K., & Takashima, A. (1996)Characterization of human presenilin 1 using N-terminal specificmonoclonal antibodies: Evidence that Alzheimer mutations affectproteolytic processing. FEBS Lett. 389, 297–303.
Miller, D. L., Papayannopoulos, I. A., Styles, J., Bobin, S. A., Lin, Y. Y.,Biemann, K., & Iqbal, K. (1993) Peptide compositions of thecerebrovascular and senile plaque core amyloid deposits of Alzhei-mer’s disease. Arch. Biochem. Biophys. 301, 41–52.
Minor, D. L., & Kim, P. S. (1996) Context-dependent secondarystructure formation of a designed protein sequence. Nature 380,730–734.
Murphy, G. M., Jr., Forno, L. S., Ellis, W. G., Nochlin, D., Levy-Lahad,E., Poorkaj, P., Bird, T. D., Jiang, Z. L., & Cordell, B. (1996)Antibodies to presenilin proteins detect neurofibrillary tangles inAlzheimer’s disease. Am. J. Pathol. 149, 1839–1846.
Namba, Y., Tomonaga, M., Kawasaki, H., Otomo, E., & Ikeda, K.(1991) Apolipoprotein E immunoreactivity in cerebral amyloiddeposits and neurofibrillary tangles in Alzheimer’s disease andkuru plaque amyloid in Creutzfeldt–Jakob disease. Brain Res. 541,163–166.
Naslund, J., Thyberg, J., Tjernberg, L. O., Wernstedt, C., Karlstrom,A. R., Bogdanovic, N., Gandy, S. E., Lannfelt, L., Terenius, L., &Nordstedt, C. (1995) Characterization of stable complexes involv-ing apolipoprotein E and the amyloid beta peptide in Alzheimer’sdisease brain. Neuron 15, 219–228.
Nathan, B. P., Bellosta, S., Sanan, D. A., Weisgraber, K. H., Mahley,
R. W., & Pitas, R. E. (1994) Differential effects of apolipoproteins E3and E4 on neuronal growth in vitro. Science 264, 850–852.
Nathan, B. P., Chang, K. C., Bellosta, S., Brisch, E., Ge, N., Mahley,R. W., & Pitas, R. E. (1995) The inhibitory effect of apolipoproteinE4 on neurite outgrowth is associated with microtubule depolymer-ization. J. Biol. Chem. 270, 19791–19799.
Pappolla, M. A., Omar, R. A., Kim, K. S., & Robakis, N. K. (1992)Immunohistochemical evidence of oxidative stress in Alzheimer’sdisease. Am. J. Pathol. 140, 621–628.
Pedro-Botet, J., Senti, M., Nogues, X., Rubies-Prat, J., Roquer, J.,D’Olhaberriague, L., & Olive, J. (1992) Lipoprotein and apolipopro-tein profile in men with ischemic stroke. Role of lipoprotein(a),triglyceride-rich lipoproteins, and apolipoprotein E polymor-phism. Stroke 23, 1556–1562.
Pike, C. J., Burdick, D., Walencewicz, A. J., Glabe, C. G., & Cotman,C. W. (1993) Neurodegeneration induced by beta-amyloid pep-tides in vitro: The role of peptide assembly state. J. Neurosci. 13,1676–1687.
Podlisny, M. B., Citron, M., Amarante, P., Sherrington, R., Xia, W.,Zhang, J., Diehl, T., Levesque, G., Fraser, P., Haass, C., Koo, E. H.,Seubert, P., St. George Hyslop, P., Teplow, D. B., & Selkoe, D. J.(1997) Presenilin proteins undergo heterogeneous endoproteolysisbetween Thr291 and Ala299 and occur as stable N- and C-terminalfragments in normal and Alzheimer brain tissue. Neurobiol. Dis. 3,325–337.
Polvikoski, T., Sulkava, R., Haltia, M., Kainulainen, K., Vuorio, A.,Verkkoniemi, A., Niinisto, L., Halonen, P., & Kontula, K. (1995)Apolipoprotein E, dementia, and cortical deposition of b-amyloidprotein. N. Eng. J. Med. 333, 1242–1247.
Poirier, J. (1994) Apolipoprotein E in animal models of CNS injuryand in Alzheimer’s disease. Trends Neurosci. 17, 525–530.
Prelli, F., Castano, E. M., Glenner, G. G., & Frangione, B. (1988)Differences between vascular and plaque core amyloid in Alzhei-mer’s disease. J. Neurochem. 51, 648–651.
Premkumar, D. R. D., Cohen, D. L., Hedera, P., Friedland, R. P., &Kalaria, R. N. (1996) Apolipoprotein E-e4 alleles in cerebralamyloid angiopathy and cerebrovascular pathology associatedwith Alzheimer’s disease. Am. J. Pathol. 148, 2083–2095.
Puttfarcken, P. S., Manelli, A. M., Falduto, M. T., Getz, G. S., & LaDu,M. J. (1997) Effect of apolipoprotein E on neurite outgrowth andb-amyloid-induced toxicity in developing rat primary hippocam-pal cultures. J. Neurochem. 68, 760–769.
Rebeck, G. W., Reiter, J. S., Strickland, D. K., & Hyman, B. T. (1993)Apolipoprotein E in sporadic Alzheimer’s disease: Allelic varia-tion and receptor interactions. Neuron 11, 575–580.
Robakis, N. K., Ramakrishna, N., Wolfe, G., & Wisniewski, H. M.(1987) Molecular cloning and characterization of a cDNA encod-ing the neuritic plaque amyloid peptides. Proc. Natl. Acad. Sci.USA 84, 4190–4194.
Rogaev, E., Sherrington, R., Rogaeva, E. A., Levesques, G., Ikeda, M.,Llang, Y., Chi, H., Lin, C., Holman, K., Tsuda, T., Mar, L., Sorbi, S.,Nacmias, B., Placentini, S., Amaducci, L., Chumakov, I., Cohen, D.,Lannfelt, L., Fraser, P. E., Rommens, J. M., & St George-Hyslop, P.(1995) Familial Alzheimer’s disease in kindreds with missensemutations in a gene on chromosome 1 related to the Alzheimer’sdisease type 3 gene. Nature 376, 775–778.
Roher, A. E., Lowenson, J. D., Clarke, S., Woods, A. S., Cotter, R. J.,Gowing, E., & Ball, M. J. (1993) b-Amyloid-(1–42) is a majorcomponent of cerebrovascular amyloid deposits: Implications forthe pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 90,10836–10840.
Saito, Y., Buciak, J., Yang, J., & Pardridge, W. M. (1995) Vector-
Biology of Ab Amyloid in Alzheimer’s Disease 325
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

mediated delivery of 125I-labeled b-amyloid peptide Ab1–40 throughthe blood–brain barrier and binding to Alzheimer disease amyloidof the Ab1–40/vector complex. Proc. Natl. Acad. Sci. USA 92,10227–10231.
Sanan, D. A., Weisgraber, K. H., Russell, S. J., Mahley, R. W., Huang,D., Saunders, A., Schmechel, D., Wisniewski, T., Frangione, B.,Roses, A. D., & Strittmatter, W. J. (1994) Apolipoprotein E associ-ates with beta amyloid peptide of Alzheimer’s disease to formnovel monofibrils. Isoform apoE4 associates more efficiently thanapoE3. J. Clin. Invest. 94, 860–869.
Sano, M., Ernesto, C., Thomas, R. G., Klauber, M. R., Schafer, K.,Grundman, M., Woodbury, P., Growdon, J. H., Cotman, C.,Pfeiffer, E., Schneider, L. S., & Thal, L. J. (1997) A controlled trial ofselegiline, alpha-tocopherol, or both as treatment for Alzheimer’sdisease. N. Eng. J. Med. 336, 1216–1222.
Schellenberg, G. (1992) Genetic linkage for a novel familial Alzhei-mer’s disease locus on chromosome 14. Science 258, 868–871.
Scheuner, D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki,N., Bird, T. D., Hardy, J., Hutton, M., Kukull, W., Larson, E.,Levy-Lahad, E., Viitanen, M., Peskind, E., Poorkaj, P., Schellen-berg, G., Tanzi, R., Wasco, W., Lannfelt, L., Selkoe, D., & Younkin,S. (1996) Secreted amyloid b-protein similar to that in the senileplaques of Alzheimer’s disease is increased in vivo by the preseni-lin 1 and 2 and APP mutations linked to familial Alzheimer’sdisease. Nature Med. 2, 864–870.
Schmechel, D. E., Saunders, A. M., Strittmatter, W. J., Crain, B. J.,Hulette, C. M., Joo, S. H., Pericak-Vance, M. A., Goldgaber, D., &Roses, A. D. (1993) Increased amyloid beta-peptide deposition incerebral cortex as a consequence of apolipoprotein E genotype inlate-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 90, 9649–9653.
Schwarzman, A. L., Gregori, L., Vitek, M. P., Lyubski, S., Strittmatter,W. J., Enghilde, J. J., Bhasin, R., Silverman, J., Weisgraber, K. H.,Coyle, P. K., & Goldgaber, D. (1994) Transthyretin sequestersamyloid beta protein and prevents amyloid formation. Proc. Natl.Acad. Sci. USA 91, 8368–8372.
Seubert, P., Vigo-Pelfrey, C., Esch, F., Lee, M., Dovey, H., Davis, D.,Sinha, S., Schlossmacher, M., Whaley, J., Swindlehurst, C., McCor-mack, R., Wolfert, R., Selkoe, D. J., Lieberburg, I., & Schenk, D. B.(1992) Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature 359, 325–327.
Seubert, P., Motter, R., Schenk, D. B., Lieberburg, I., Kholodenko, D.,Galasko, D., Thomas, R., Chang, L., Miller, B., Clark, C., Knopman,D. S., Kaye, J., Green, R. C., Kertiles, L., Bashirzadeh, R., & Boss,M. A. (1997) ApoE genotype influences the CSF level of Ab42 inAlzheimer’s disease. Neurology 48, A379. [Abstract]
Sheng, J. G., Mrak, R. E., & Griffin, W. S. T. (1996) Apolipoprotein Edistribution among diffuse plaque types in Alzheimer’s disease:Implications for its role in plaque progression. Neuropathol. Appl.Neurobiol. 22, 334–341.
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesques,G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., Tsuda, T., Mar, L.,Foncin, J. F., Bruni, A. C., Montesi, M. P., Sorbi, S., Rainero, I.,Pinessi, L., Nee, L., Chumakov, I., Pollen, D., Brookes, A., Sanseau,P., Polinsky, R. J., Wasco, W., DaSilva, H. A. R., Haines, J. L.,Pericak-Vance, M. A., Tanzi, R. E., Roses, A. D., Fraser, P. E.,Rommens, J. M., & St George-Hyslop, P. (1995) Cloning of a genebearing missense mutations in early onset familial Alzheimer’sdisease. Nature 375, 754–760.
Shimano, H., Ishibashi, S., & Murase, T. (1989) Plasma apolipopro-
teins in patients with multi-infarct dementia. Atherosclerosis 79,257–260.
Shoji, M., Golde, T. E., Ghiso, J., Cheung, T. T., Estus, S., Shaffer,L. M., Cai, X. D., McKay, D. M., Tintner, R., Frangione, B., &Younkin, S. G. (1992) Production of the Alzheimer amyloid betaprotein by normal proteolytic processing. Science 258, 126–129.
Simmons, L. K., May, P. C., Tomaselli, K. J., Rydel, R. E., Fuson, K. S.,Brigham, E. F., Wright, S., Lieberburg, I., Becker, G. W., Brems,D. N., & Li, W. Y. (1994) Secondary structure of amyloid betapeptide correlates with neurotoxic activity in vitro. Mol. Pharmacol.45, 373–379.
Sisodia, S. S., Koo, E. H., Beyreuther, K., Unterbeck, A., & Price, D. L.(1990) Evidence that b-amyloid protein in Alzheimer’s disease isnot derived by normal processing. Science 248, 492–495.
Slooter, A. J. C., Ming-Xin, T., van Duijn, C. M., Stern, Y., Ott, A., Bell,K., Breteler, M. M. B., Van Broeckhoven, C., Taternichi, T. K.,Tycko, B., Hofman, A., & Mayeux, R. (1997) Apolipoprotein E e4and the risk of dementia with stroke. J. Am. Med. Assoc. 277,818–821.
Smith, M. A., Kutty, R. K., Richey, P. L., Yan, S. D., Stern, D., Chader,G. J., Wiggert, B., Petersen, R. B., & Perry, G. (1994) Hemeoxygenase-1 is associated with the neurofibrillary pathology ofAlzheimer’s disease. Am. J. Pathol. 145, 42–47.
Snow, A. D., Willner, J., & Kisilevsky, R. (1987) Sulphated gly-cosaminoglycans: a common constituent of all amyloid? Lab.Invest. 56, 120–124.
Soto, C., Kindy, M. S., Baumann, M., & Frangione, B. (1996)Inhibition of Alzheimer’s amyloidosis by peptides that preventb-sheet conformation. Biochem. Biophys. Res. Commun. 226, 672–680.
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance,M., Enghild, J., Salvesen, G. S., & Roses, A. D. (1993) Apolipopro-tein E: High-avidity binding to beta-amyloid and increasedfrequency of type 4 allele in late-onset familial Alzheimer disease.Proc. Natl. Acad. Sci. USA 90, 1977–1981.
Suzuki, N., Cheung, T. T., Cai, T. T., Cai, X. D., Odaka, A., Otvos, L.,Eckman, C., Golde, T. E., & Younkin, S. G. (1994) An increasepercentage of long amyloid b protein secreted by familial amyloidb protein precursor (b PP717) mutants. Science 264, 1336–1340.
Tabaton, M., Nunzi, M. G., Xue, R., Usiak, M., Autilio-Gambetti, L.,& Gambetti, P. (1994) Soluble amyloid beta-protein is a marker ofAlzheimer amyloid in brain but not in cerebrospinal fluid. Bio-chem. Biophys. Res. Commun. 200, 1598–1603.
Tabaton, M., Piccini, A., Dapino, D., Angelini, G., Gambetti, P., &Zaccheo, D. (1996) Type and amount of the apolipoproteinE/amyloid b protein complex in Alzheimer’s and control brain.Soc. Neurosci. Abstr. 22, 1696. [Abstract]
Tagliavini, F., Giaccone, G., Frangione, B., & Bugiani, O. (1988)Preamyloid deposits in the cerebral cortex of patients withAlzheimer’s disease and nondemented individuals. Neurosci. Lett.93, 191–196.
Tamaoka, A., Kondo, T., Odaka, A., Sahara, N., Sawamura, N.,Ozawa, K., Suzuki, N., Shoji, S., & Mori, H. (1994) Biochemicalevidence for the long-tail form (Ab1–42/43) of amyloid b protein as aseed molecule on cerebral deposits of Alzheimer’s disease. Bio-chem. Biophys. Res. Commun. 205, 834–842.
Tanzi, R. E., Gusella, J. F., Watkins, P. C., Bruns, G. A., St. GeorgeHyslop, P., VanKeuren, M. L., Patterson, D., Pagan, S., Kurnit,D. M., & Neve, R. L. (1987) Amyloid b-protein gene: cDNA,mRNA distribution and genetic linkage near the Alzheimer’slocus. Science 235, 880–884.
326 Wisniewski, Ghiso, and Frangione
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

Tanzi, R. E., McClatchy, A. I., Lamperti, E. D., Villa-Kornaroff, L.,Gusella, J. F., & Neve, R. L. (1988) Protease inhibitor domainencoded by an amyloid precursor mRNA associated with Alzhei-mer’s disease. Nature 331, 528–530.
Tanzi, R. E., Kovacs, D., Kim, T., Moir, R. D., Guenette, S. Y., & Wasco,W. (1996) The gene defects responsible for familial Alzheimer’sdisease. Neurobiol. Dis. 3, 159–168.
Teller, J. K., Russo, C., DeBusk, L. M., Angelini, G., Zaccheo, D.,Dagna-Bricarelli, F., Scartezzini, P., Bertolini, S., Mann, D. M. A.,Tabaton, M., & Gambetti, P. (1996) Presence of soluble amyloidb-peptide precedes amyloid plaque formation in Down’s syn-drome. Nature Med. 2, 93–95.
Thinakaran, G., Borchelt, D. R., Lee, M., Slunt, H. H., Spitzer, L., Kim,G., Ratovitsky, T., Davenport, F., Nordstedt, C., Seeger, M., Hardy,J., Levey, A. I., Gandy, S. E., Jenkins, N. A., Copeland, N. G., Price,D. L., & Sisodia, S. S. (1996) Endoproteolysis of presenilin 1 andaccumulation of processed derivatives in vivo. Neuron 17, 181–190.
Timmers, W. F., Tagliavini, F., Haan, J., & Frangione, B. (1990)Parenchymal preamyloid and amyloid deposits in the brains ofpatients with hereditary cerebral hemorrhage with amyloidosisDutch type. Neurosci. Lett. 118, 223–226.
Uchihara, T., Hamid, H. K., Duyckaerts, C., Jean-Francois, F., Fraser,P. E., Levesque, L., St. George Hyslop, P., & Jean-Jacques, H. (1996)Widespread immunoreactivity of presenilin in neurons of normaland Alzheimer’s disease brains: Double-labeling immunohisto-chemical study. Acta Neuropathol. 92, 325–330.
Ueda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto,M., Otero, D. A., Kondo, J., Ihara, Y., & Saitoh, T. (1993) Molecularcloning of cDNA encoding an unrecognized component of amy-loid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 90, 11282–11286.
Ueda, K., Fukui, Y., & Kageyama, H. (1994) Amyloid beta protein-induces neuronal cell death: Neurotoxic properties of aggregatedamyloid beta protein. Brain Res. 639, 240–244.
van Nostrand, W. E., Schmaier, A. H., Farrow, J. S., & Cunningham,D. D. (1990) Protease nexin-II (amyloid b-protein precursor): Aplatelet alpha granule protein. Science 248, 745–748.
Vigo-Pelfrey, C., Lee, D., Keim, P., Lieberburg, I., & Schenk, D. B.(1993) Characterization of beta-amyloid peptide from humancerebrospinal fluid. J. Neurochem. 61, 1965–1968.
Vito, P., Lacana, E., & D’Adamio, L. (1996) Interfering with apopto-sis: Ca21-binding protein ALG-2 and Alzheimer’s disease geneALG-3. Science 271, 521–525.
Wang, R., Meschia, J. F., Cotter, R. J., & Sisodia, S. S. (1991) Secretionof the b/A4 amyloid precursor protein. J. Biol. Chem. 266, 16960–16964.
Weber, L. L., Leissring, M. A., Yang, A. J., Glabe, C. G., Cribbs, D. H.,& LaFerla, F. M. (1997) Presenilin-1 immunoreactivity is localizedintracellularly in Alzheimer’s disease brain, but not detected inamyloid plaques. Exp. Neurol. 143, 37–44.
Weidemann, A., Paliga, K., Durrwang, U., Czech, C., Evin, G.,Masters, C. L., & Beyreuther, K. (1997) Formation of stablecomplexes between two Alzheimer’s disease gene products: Pre-senilin-2 and b-amyloid precursor protein. Nature Med. 3, 328–332.
Wisniewski, H. M., Bancher, C., Barcikowska, M., Wen, G. Y., &Currie, J. (1989) Spectrum of morphological appearance of amy-loid deposits in AD. Acta Neuropathol. 78, 337–347.
Wisniewski, H. M., Wegiel, J., Morys, J., Baucher, C., Soltysiak, Z., &Kim, K. S. (1990) Alzheimer’s disease: Epidemiology, neuropathology,
neurochemistry and clinics (K. Maurer, D. Rieder, & H. Beckman,Eds.), pp. 151–168. Springer-Verlag, Berlin.
Wisniewski, H. M., Wegiel, J., & Popovitch, E. R. (1994) Age-associated development of diffuse and thioflavine-S-positiveplaques in Down syndrome. Dev. Brain Dysfunct. 7, 330–339.
Wisniewski, H. M., Wegiel, J., & Kotula, L. (1996) Some neuropatho-logical aspects of Alzheimer’s disease and its relevance to otherdisciplines. Neuropathol. Appl. Neurobiol. 22, 3–11.
Wisniewski, T., Ghiso, J., & Frangione, B. (1991) Peptides homo-logous to the amyloid protein of Alzheimer’s disease containinga glutamine for glutamic acid substitution have acceleratedamyloid fibril formation. Biochem. Biophys. Res. Commun. 179,1247–1254.
Wisniewski, T., Castano, E. M., Golabek, A. A., Vogel, T., & Frangi-one, B. (1994a) Acceleration of Alzheimer’s fibril formation byapolipoprotein E in vitro. Am. J. Pathol. 145, 1030–1035.
Wisniewski, T., Lalowski, M., Levy, E., Marques, M. R., & Frangione,B. (1994b) The amino acid sequence of neuritic plaque amyloidfrom a familial Alzheimer’s disease patient. Ann. Neurol. 35,245–246.
Wisniewski, T., Golabek, A. A., Kida, E., Wisniewski, K. E., &Frangione, B. (1995a) Conformational mimicry in Alzheimer’sdisease. Role of apolipoproteins in amyloidogenesis. Am. J. Pathol.147, 238–244.
Wisniewski, T., Lalowski, M., Golabek, A. A., Vogel, T., & Frangione,B. (1995b) Is Alzheimer’s disease an apolipoprotein E amyloido-sis? Lancet 345, 956–958.
Wisniewski, T., Palha, J. A., Ghiso, J., & Frangione, B. (1995c)S182 protein in Alzheimer’s disease neuritic plaques. Lancet 346,1366.
Wisniewski, T., Lalowski, M., Baumann, M., Rauvala, H., Raulo, E.,Nolo, R., & Frangione, B. (1996a) HB-GAM is a cytokine present inAlzheimer’s and Down’s syndrome lesions. Neuroreport 7, 667–671.
Wisniewski, T., Lalowski, M., Bobik, M., Russell, M., Strosznajder, J.,& Frangione, B. (1996b) Amyloid b 1–42 deposits do not lead toAlzheimer’s neuritic plaques in aged dogs. Biochem. J. 313,575–580.
Wisniewski, T., Dowjat, W., Permanne, B., Palha, J. A., Kumar, A.,Gallo, G., & Frangione, B. (1997) Presenilin is associated withAlzheimer’s disease amyloid. Am. J. Pathol. 151, 601–610.
Wisniewski, T., & Frangione, B. (1992) Apolipoprotein E: a pathologi-cal chaperone protein in patients with cerebral and systemicamyloid. Neurosci. Lett. 135, 235–238.
Wisniewski, T., & Frangione, B. (1996) Molecular biology of brainaging and neurodegenerative disorders. Acta Neurobiol. Exp. 56,267–279.
Wolozin, B., Iwasaki, K., Vito, P., Ganjei, J. K., Lacana, E., Sunder-land, T., Zhao, B., Kusiak, J. W., Wasco, W., & D’Adamio, L. (1996)Participation of presenilin 2 in apoptosis: Enhanced basal activityconferred by an Alzheimer mutation. Science 274, 1710–1713.
Wong, P. C., Zheng, H., Chen, H., Becher, M. W., Trumbauer, M. E.,Chen, H. Y., Price, D. L., Van der Ploeg, L. H., & Sisodia, S. S. (1997)Massive brain hemorrhage, abnormal skeletal development andlethality in presenilin 1 deficient embryos. Mol. Mech. Alzheimer’sDis. 14. [Abstract]
Yamaguchi, H., Hirai, S., Morimatsu, M., Shoji, M., & Harigaya, Y.(1988) Diffuse type of senile plaques in the brains of Alzheimer-type dementia. Acta Neuropathol. 77, 113–119.
Biology of Ab Amyloid in Alzheimer’s Disease 327
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

Yamaguchi, H., Nakazato, Y., Shoji, M., Takatama, M., & Hirai, S.(1991) Ultrastructure of diffuse plaques in senile dementia of theAlzheimer type: Comparison with primitive plaques. Acta Neuro-pathol. 82, 13–20.
Yamazaki, T., Yamaguchi, H., Okamoto, K., & Hirai, S. (1991)Ultrastructural localization of argyrophilic substances in diffuseplaques of Alzheimer-type dementia demonstrated by methena-mine silver staining. Acta Neuropathol. 81, 540–545.
Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H. J., Roher, A., Slattery, T.,Zhao, L., Nagashima, M., Morser, J., Migheli, A., Nawroth, P.,Stern, D., & Schmidt, A. M. (1996) RAGE and amyloid-b peptideneurotoxicity in Alzheimer’s disease. Nature 382, 685–691.
Yang, D. S., Smith, J. D., Zhou, Z. M., Gandy, S. E., & Martins, R. N.(1997) Characterization of the binding of amyloid-b peptide to cellculture-derived native apolipoprotein E2, E3, and E4 isoforms andto isoforms from human plasma. J. Neurochem. 68, 721–725.
Younkin, S. G. (1995) Evidence that Ab 42 is the real culprit inAlzheimer’s disease. Ann. Neurol. 37, 287–288.
Zlokovic, B. V., Ghiso, J., Mackic, J. B., McComb, J. G., Weiss, M. H., &Frangione, B. (1993) Blood–brain barrier transport of circulatingAlzheimer’s amyloid beta. Biochem. Biophys. Res. Commun. 197,1034–1040.
Zlokovic, B. V., Martel, C. L., Mackic, J. B., Matsubara, E., Wisniewski,T., McComb, J. G., Frangione, B., & Ghiso, J. (1994) Brain uptake ofcirculating apolipoproteins J and E complexed to Alzheimer’samyloid beta. Biochem. Biophys. Res. Commun. 205, 1431–1437.
Zlokovic, B. V. (1996) Cerebrovascular transport of Alzheimer’samyloid b and apolipoproteins J and E: Possible anti-amyloido-genic role of the blood–brain barrier. Life Sci. 59, 1483–1497.
Zlokovic, B. V., Martel, C. L., Matsubara, E., McComb, J. G., Zheng,G., McCluskey, R. T., Frangione, B., & Ghiso, J. (1996) Glyco-protein 330/megalin: Probable role in receptor-mediated trans-port of apolipoprotein J alone and in a complex with Alz-heimer disease amyloid b at the blood–brain and blood–cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. USA 93, 4229–4234.
328 Wisniewski, Ghiso, and Frangione
Copyright r 1997 by Academic PressAll rights of reproduction in any form reserved.

![Index [assets.cambridge.org]assets.cambridge.org/.../index/9781107011687_index.pdf · Index Aβ see amyloid-β ABILHAND questionnaire 591 abundance, in motor system 602–5 acarbose](https://img.pdfslide.net/doc/110x75/5f031f417e708231d407a572/index-index-a-see-amyloid-abilhand-questionnaire-591-abundance-in-motor.jpg)



























