Embed Size (px)
Citation preview

RESEARCH Open Access
Bradymonabacteria, a novel bacterialpredator group with versatile survivalstrategies in saline environmentsDa-Shuai Mu1,2, Shuo Wang2, Qi-Yun Liang2, Zhao-Zhong Du2, Renmao Tian3, Yang Ouyang3, Xin-Peng Wang2,Aifen Zhou3, Ya Gong1,2, Guan-Jun Chen1,2, Joy Van Nostrand3, Yunfeng Yang4, Jizhong Zhou3,4 andZong-Jun Du1,2*
Abstract
Background: Bacterial predation is an important selective force in microbial community structure and dynamics.However, only a limited number of predatory bacteria have been reported, and their predatory strategies andevolutionary adaptations remain elusive. We recently isolated a novel group of bacterial predators, Bradymonabacteria,representative of the novel order Bradymonadales in δ-Proteobacteria. Compared with those of other bacterialpredators (e.g., Myxococcales and Bdellovibrionales), the predatory and living strategies of Bradymonadales are still largelyunknown.
Results: Based on individual coculture of Bradymonabacteria with 281 prey bacteria, Bradymonabacteriapreyed on diverse bacteria but had a high preference for Bacteroidetes. Genomic analysis of 13 recentlysequenced Bradymonabacteria indicated that these bacteria had conspicuous metabolic deficiencies, but theycould synthesize many polymers, such as polyphosphate and polyhydroxyalkanoates. Dual transcriptomeanalysis of cocultures of Bradymonabacteria and prey suggested a potential contact-dependent predationmechanism. Comparative genomic analysis with 24 other bacterial predators indicated that Bradymonabacteriahad different predatory and living strategies. Furthermore, we identified Bradymonadales from 1552 publiclyavailable 16S rRNA amplicon sequencing samples, indicating that Bradymonadales was widely distributed andhighly abundant in saline environments. Phylogenetic analysis showed that there may be six subgroups inthis order; each subgroup occupied a different habitat.
Conclusions: Bradymonabacteria have unique living strategies that are transitional between the “obligate” and the so-called facultative predators. Thus, we propose a framework to categorize the current bacterial predators into 3 groups:(i) obligate predators (completely prey-dependent), (ii) facultative predators (facultatively prey-dependent), and (iii)opportunistic predators (prey-independent). Our findings provide an ecological and evolutionary framework forBradymonadales and highlight their potential ecological roles in saline environments.
Keywords: Bacterial predator, Bradymonadales, Metabolic deficiencies, Comparative genomic analysis, Biogeographicanalysis
© The Author(s). 2020 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License,which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you giveappropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate ifchanges were made. The images or other third party material in this article are included in the article's Creative Commonslicence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commonslicence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtainpermission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to thedata made available in this article, unless otherwise stated in a credit line to the data.
* Correspondence: [email protected] Key Laboratory of Microbial Technology, Institute of MicrobialTechnology, Shandong University, No. 72, Jimo Binhai Road, Jimo, Qingdao266237, China2Marine College, Shandong University, Weihai 264209, ChinaFull list of author information is available at the end of the article
Mu et al. Microbiome (2020) 8:126 https://doi.org/10.1186/s40168-020-00902-0

BackgroundBacterial predators have been proposed as an indispens-able selective force in bacterial communities [1–4]. Pre-dation by bacteria can release nutrients [5] and affectbiogeochemical cycling. In contrast to phages, bacterialpredators do not need to be present in high concentra-tions to drive significant bacterial mortality in the envir-onment [6, 7]. In addition, bacterial predators havehigher prey-killing efficiency in low-nutrient mediumthan phages [8]. However, these studies have mostlybeen based on Bdellovibrio and like organisms (BALOs),and little is known of the ecological roles of other bac-terial predators.Predatory bacteria are classified into two categories, ob-
ligate or facultative predators, based on their prey-independent or prey-dependent living strategies [9]. Obli-gate predators include several genera collectively knownas BALOs [10]. These predatory bacteria can attack theirprey by penetrating the cell wall [11], dwelling in the peri-plasm, and then killing their host [12]. Therefore, theirlifestyle depends on the presence of their prey in the nat-ural environment, and BALOs lose viability within severalhours if the prey is not available [8, 13, 14]. Facultativepredators also include several genera [9], such asMyxococ-cus, Lysobacter, and Herpetosiphon [15]. These predatorskill their prey by secreting antimicrobial substances intothe surrounding environment [9, 16]. In general, the so-called facultative predators have been considered to bethose that can be maintained as pure bacterial culturesand be free living without their prey in natural environ-ments. However, due to the lack of additional types ofpredators, no assessment could be made with respect tohow dependent they were on their prey. Furthermore,whether there is a transitional type between obligate andso-called facultative predators is unclear.Bradymonabacteria are representative of the novel order
Bradymonadales, which are phylogenetically located inthe δ-Proteobacteria [17]. The first type species of Brady-monadales, Bradymonas sediminis FA350T, was isolatedin 2015 [17]. To date, 9 strains within the Bradymona-dales have been isolated and found to belong to 7 candi-date novel species; these Bradymonabacteria are bacterialpredators [18]. Interestingly, the phylum Proteobacteriacontains three orders of predatory bacteria. Among them,Myxococcales and Bradymonadales belong to δ-Proteo-bacteria, while Bdellovibrionales were classified as Oligo-flexia in 2017 [19]. Myxococcales and Bdellovibrionales areso-called facultative and obligate predators, respectively.Additionally, they have different distribution patterns inthe environment. Myxococcales are mainly found in soiland sediment niches [20, 21], while Bdellovibrionales areaquatic. However, how Bradymonadales adapt to preda-tory lifestyles and whether they have specific living strat-egies or ecological importance remain largely unknown.
Here, we analyzed the predation range of Bradymona-dales on diverse bacteria and their predatory morpho-logical and physiological characteristics. By usingcomparative genomic analysis of Bradymonadales andother predatory bacteria, we revealed the genetic andmetabolic potential of this group. To assess the diversityand frequency of occurrence of the various ribotypes ofknown predators (Bradymonadales, Myxococcales, andBdellovibrionales) on a global scale, we surveyed pub-lished 16S rRNA gene amplicon datasets from a numberof ecosystems representing a broad range of geographiclocations, climatic zones, and salinities. Our study pro-vides an ecological and evolutionary framework for Bra-dymonadales and highlights their potential ecologicalroles in predation.
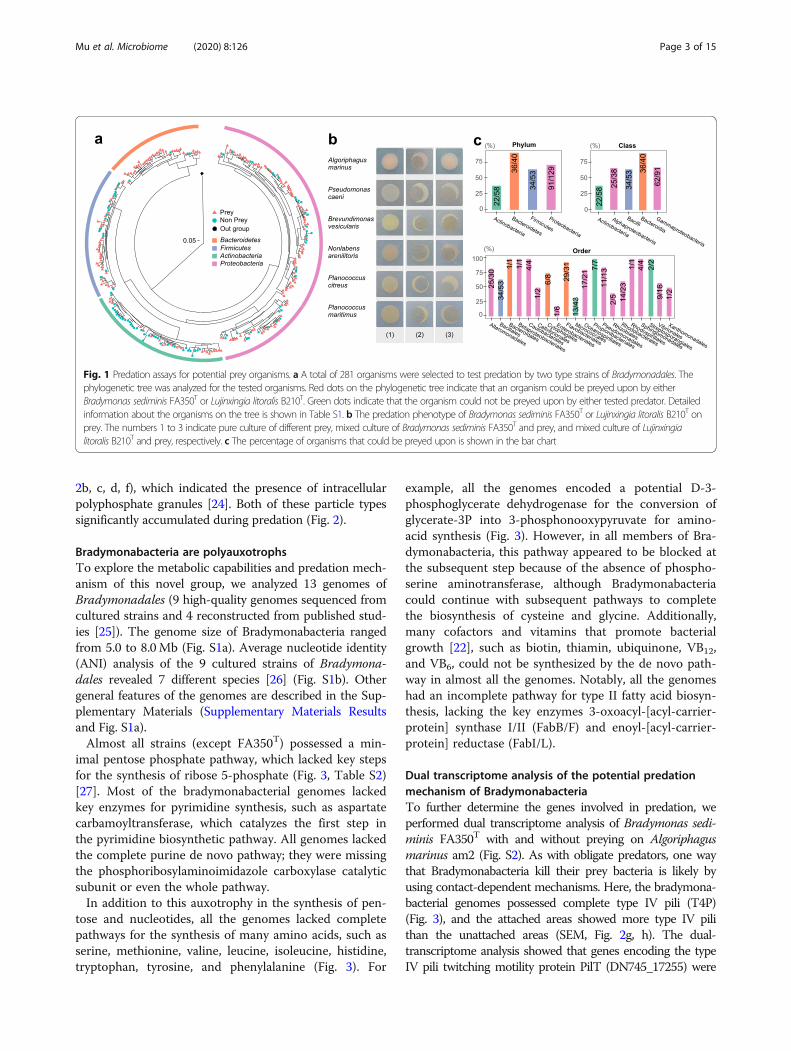
ResultsBradymonabacteria are efficient predators of diverse preybacteriaIn total, 9 strains of bacteria in the novel order Brady-monadales were isolated using the enrichment culturemethod [22]. Among these strains, eight strains wereisolated from costal sediment sampled in Weihai, China,while strain YN101 was isolated from a Gaodao saltern(36° 54′ N, 122° 14′ E) in Weihai, China. Strains FA350T
[17, 18] and B210T [23] are the two type strains for dif-ferent genera of Bradymonadales. Both these type strainswere used to investigate the predator-prey range of Bra-dymonabacteria. A total of 281 isolated bacteria werecocultured with Bradymonabacteria FA350T [17, 18] orB210T [23] as lawns in individual Petri dishes (Fig. 1a,Table S1). Zones of predation were measured (Fig. 1b),and the results showed that the Bradymonabacteriapreyed on diverse bacteria but showed a strong prefer-ence for Bacteroidetes (90% of tested bacteria could bepreyed on) and Proteobacteria (71% of tested bacteriacould be preyed on) (Fig. 1c). Predation on bacteria inthe orders Flavobacteriales, Caulobacterales, Propioni-bacteriales, and Pseudomonadales was broadly distrib-uted, with a mean predation percentage greater than90%, while predation of Micrococcales and Enterobacter-iales was less efficient.Transmission electron microscopy (TEM) and scanning
electron microscopy (SEM) analyses were performed tounderstand the mechanism of predation of strain FA350T
on the subcellular level. Lysis of the prey cells (Fig.2a) was detected near strain FA350T in both the TEM andSEM analyses (Fig. 2). Strain FA350T was found to havepili (Fig. 2b, g) and outer membrane vesicle (OMV)-likestructures (Fig. 2d, e, f, h). In addition, FA350T cells con-tained intracellular particles with low electron density(Fig. 2b, c, d, f), which were shown to contain polyhydrox-yalkanoates (PHAs) by Nile blue A staining. FA350T cellsalso contained several electron-dense (black) spots (Fig.
Mu et al. Microbiome (2020) 8:126 Page 2 of 15
2b, c, d, f), which indicated the presence of intracellularpolyphosphate granules [24]. Both of these particle typessignificantly accumulated during predation (Fig. 2).
Bradymonabacteria are polyauxotrophsTo explore the metabolic capabilities and predation mech-anism of this novel group, we analyzed 13 genomes ofBradymonadales (9 high-quality genomes sequenced fromcultured strains and 4 reconstructed from published stud-ies [25]). The genome size of Bradymonabacteria rangedfrom 5.0 to 8.0Mb (Fig. S1a). Average nucleotide identity(ANI) analysis of the 9 cultured strains of Bradymona-dales revealed 7 different species [26] (Fig. S1b). Othergeneral features of the genomes are described in the Sup-plementary Materials (Supplementary Materials Resultsand Fig. S1a).Almost all strains (except FA350T) possessed a min-
imal pentose phosphate pathway, which lacked key stepsfor the synthesis of ribose 5-phosphate (Fig. 3, Table S2)[27]. Most of the bradymonabacterial genomes lackedkey enzymes for pyrimidine synthesis, such as aspartatecarbamoyltransferase, which catalyzes the first step inthe pyrimidine biosynthetic pathway. All genomes lackedthe complete purine de novo pathway; they were missingthe phosphoribosylaminoimidazole carboxylase catalyticsubunit or even the whole pathway.In addition to this auxotrophy in the synthesis of pen-
tose and nucleotides, all the genomes lacked completepathways for the synthesis of many amino acids, such asserine, methionine, valine, leucine, isoleucine, histidine,tryptophan, tyrosine, and phenylalanine (Fig. 3). For
example, all the genomes encoded a potential D-3-phosphoglycerate dehydrogenase for the conversion ofglycerate-3P into 3-phosphonooxypyruvate for amino-acid synthesis (Fig. 3). However, in all members of Bra-dymonabacteria, this pathway appeared to be blocked atthe subsequent step because of the absence of phospho-serine aminotransferase, although Bradymonabacteriacould continue with subsequent pathways to completethe biosynthesis of cysteine and glycine. Additionally,many cofactors and vitamins that promote bacterialgrowth [22], such as biotin, thiamin, ubiquinone, VB12,and VB6, could not be synthesized by the de novo path-way in almost all the genomes. Notably, all the genomeshad an incomplete pathway for type II fatty acid biosyn-thesis, lacking the key enzymes 3-oxoacyl-[acyl-carrier-protein] synthase I/II (FabB/F) and enoyl-[acyl-carrier-protein] reductase (FabI/L).
Dual transcriptome analysis of the potential predationmechanism of BradymonabacteriaTo further determine the genes involved in predation, weperformed dual transcriptome analysis of Bradymonas sedi-minis FA350T with and without preying on Algoriphagusmarinus am2 (Fig. S2). As with obligate predators, one waythat Bradymonabacteria kill their prey bacteria is likely byusing contact-dependent mechanisms. Here, the bradymona-bacterial genomes possessed complete type IV pili (T4P)(Fig. 3), and the attached areas showed more type IV pilithan the unattached areas (SEM, Fig. 2g, h). The dual-transcriptome analysis showed that genes encoding the typeIV pili twitching motility protein PilT (DN745_17255) were
Fig. 1 Predation assays for potential prey organisms. a A total of 281 organisms were selected to test predation by two type strains of Bradymonadales. Thephylogenetic tree was analyzed for the tested organisms. Red dots on the phylogenetic tree indicate that an organism could be preyed upon by eitherBradymonas sediminis FA350T or Lujinxingia litoralis B210T. Green dots indicate that the organism could not be preyed upon by either tested predator. Detailedinformation about the organisms on the tree is shown in Table S1. b The predation phenotype of Bradymonas sediminis FA350T or Lujinxingia litoralis B210T onprey. The numbers 1 to 3 indicate pure culture of different prey, mixed culture of Bradymonas sediminis FA350T and prey, and mixed culture of Lujinxingialitoralis B210T and prey, respectively. c The percentage of organisms that could be preyed upon is shown in the bar chart
Mu et al. Microbiome (2020) 8:126 Page 3 of 15
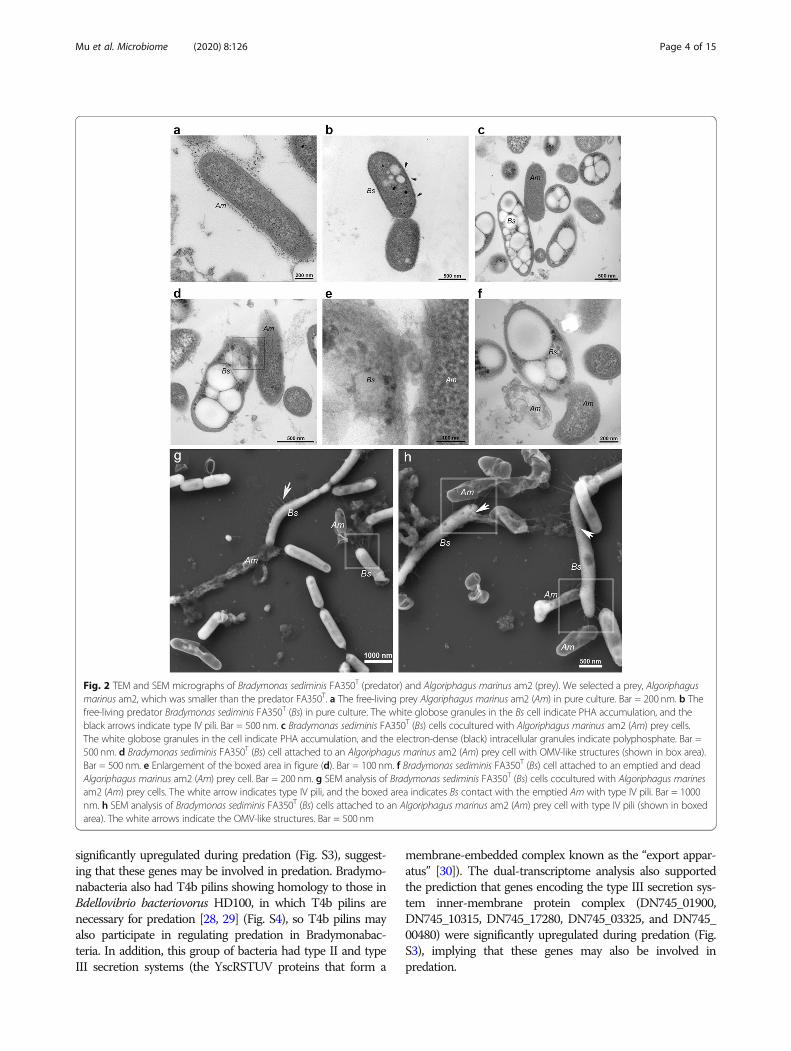
significantly upregulated during predation (Fig. S3), suggest-ing that these genes may be involved in predation. Bradymo-nabacteria also had T4b pilins showing homology to those inBdellovibrio bacteriovorus HD100, in which T4b pilins arenecessary for predation [28, 29] (Fig. S4), so T4b pilins mayalso participate in regulating predation in Bradymonabac-teria. In addition, this group of bacteria had type II and typeIII secretion systems (the YscRSTUV proteins that form a
membrane-embedded complex known as the “export appar-atus” [30]). The dual-transcriptome analysis also supportedthe prediction that genes encoding the type III secretion sys-tem inner-membrane protein complex (DN745_01900,DN745_10315, DN745_17280, DN745_03325, and DN745_00480) were significantly upregulated during predation (Fig.S3), implying that these genes may also be involved inpredation.
Fig. 2 TEM and SEM micrographs of Bradymonas sediminis FA350T (predator) and Algoriphagus marinus am2 (prey). We selected a prey, Algoriphagusmarinus am2, which was smaller than the predator FA350T. a The free-living prey Algoriphagus marinus am2 (Am) in pure culture. Bar = 200 nm. b Thefree-living predator Bradymonas sediminis FA350T (Bs) in pure culture. The white globose granules in the Bs cell indicate PHA accumulation, and theblack arrows indicate type IV pili. Bar = 500 nm. c Bradymonas sediminis FA350T (Bs) cells cocultured with Algoriphagus marinus am2 (Am) prey cells.The white globose granules in the cell indicate PHA accumulation, and the electron-dense (black) intracellular granules indicate polyphosphate. Bar =500 nm. d Bradymonas sediminis FA350T (Bs) cell attached to an Algoriphagus marinus am2 (Am) prey cell with OMV-like structures (shown in box area).Bar = 500 nm. e Enlargement of the boxed area in figure (d). Bar = 100 nm. f Bradymonas sediminis FA350T (Bs) cell attached to an emptied and deadAlgoriphagus marinus am2 (Am) prey cell. Bar = 200 nm. g SEM analysis of Bradymonas sediminis FA350T (Bs) cells cocultured with Algoriphagus marinesam2 (Am) prey cells. The white arrow indicates type IV pili, and the boxed area indicates Bs contact with the emptied Am with type IV pili. Bar = 1000nm. h SEM analysis of Bradymonas sediminis FA350T (Bs) cells attached to an Algoriphagus marinus am2 (Am) prey cell with type IV pili (shown in boxedarea). The white arrows indicate the OMV-like structures. Bar = 500 nm
Mu et al. Microbiome (2020) 8:126 Page 4 of 15

Another way that Bradymonabacteria kill their preybacteria is likely by secreting antimicrobial substancesinto the surrounding environment. As in most faculta-tive bacterial predators, a few potential antimicrobialclusters for secondary metabolite synthesis, such asLasso peptide [31], were identified in almost all genomesof Bradymonabacteria (Fig. 3). Genes involved in outermembrane vesicle (OMV) biosynthesis were alsodetected in most genomes, such as ompA (cell envelopebiogenesis protein), envC (Murein hydrolase activator),and tolR (envelope stability) [32]. Vesicle membrane-related genes (DN745_03865, DN745_02930, andDN745_07125) were significantly upregulated duringpredation (Table S4, Fig. S3). However, the fermentationsupernatant of Bradymonabacteria showed no antibac-terial activity.
Bradymonabacteria are novel predators different fromthe so-called obligate or facultative predatorsComparative genomic analysis with other bacterial predatorswas performed to explore whether Bradymonabacteria have
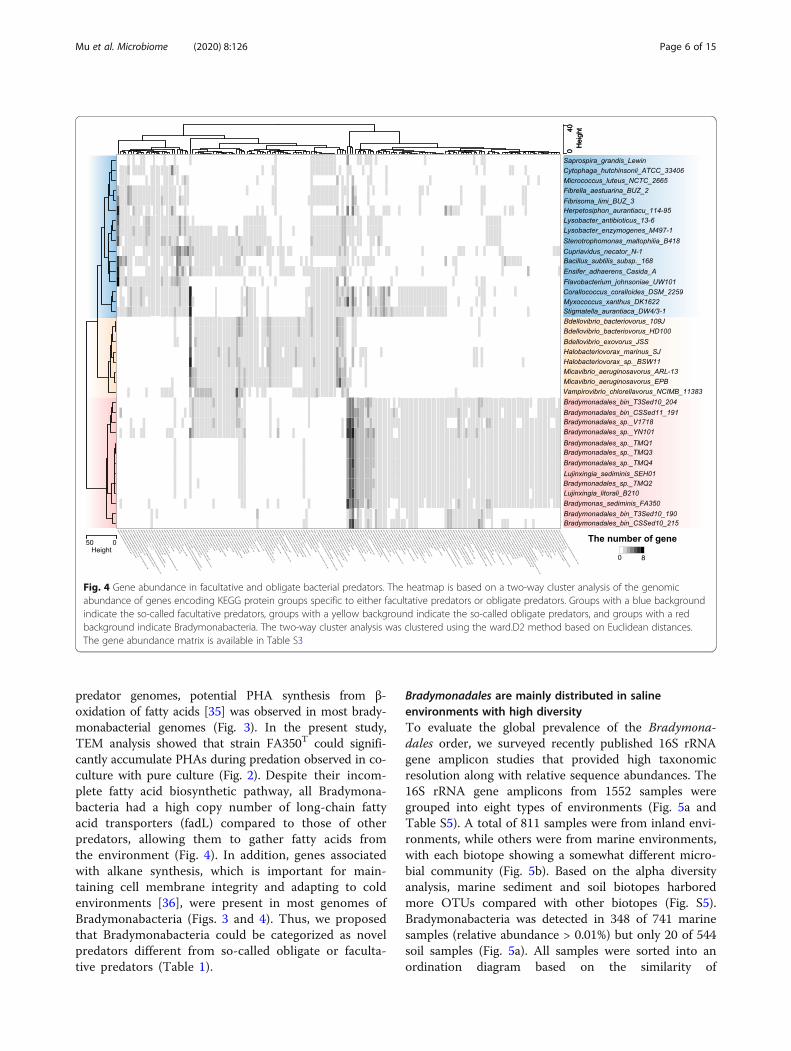
a unique living strategy. Two-way cluster analysis showedthat bradymonabacterial genomes contained features differ-ent from those of either obligate or facultative predators,which were phylogenetically located in a different branch(Fig. 4). The specific multiple metabolic deficiencies of Brady-monabacteria had some similarities to those of most obligatepredators. For example, both Bradymonabacteria and obli-gate predators possessed a minimal pentose phosphate path-way, lacked key enzymes for pyrimidine synthesis, and lackedcomplete pathways for the synthesis of many amino acids,cofactors, and vitamins (Fig. 4). However, Bradymonabacteriawith multiple auxotrophies could grow on common media(such as marine agar medium), though at a low growth rate[33], unlike obligate predators.Unlike most obligate predators, the polyphosphate accu-
mulation pathway, containing a pair of genes (polyphos-phate kinase and exopolyphosphatase) associated withboth polyphosphate formation and degradation [34], waspresent in most Bradymonabacteria (Fig. 4). Polyphos-phate accumulation was also detected in FA350T cellsduring predation (Fig. 2). In contrast to most of the other
Fig. 3 Metabolic capabilities of Bradymonabacteria. Metabolic predictions were mainly generated by referring to the KEGG and SEED databaseinterface. Each subgroup of Bradymonabacteria is depicted as a colored circle (see figure legend). Functional genes (abbreviation according toKEGG) encoding the relevant proteins/enzymes are labeled for each metabolic step where colored circles (that is, Bradymonabacteria strains) aredepicted to show the potential functions of each subgroup if any. The gray arrows indicate the corresponding genes detected for the pathwaysin almost all the genomes, while the red arrows indicate the corresponding genes missing from the pathways. The red “no entry” signs indicatethe many key genes in pathways that are missing. All putative transporters and F0F1 ATPases are shown as well as secretion systems, type IV pili,and predicted components of flagella. The process of starvation and stringent-responsive system remodeling is mediated by the production ofthe alarmones guanosine pentaphosphate, pppGpp, and guanosine tetraphosphate, ppGpp. The key metabolic predictions are supported by thegene information in Table S2
Mu et al. Microbiome (2020) 8:126 Page 5 of 15
predator genomes, potential PHA synthesis from β-oxidation of fatty acids [35] was observed in most brady-monabacterial genomes (Fig. 3). In the present study,TEM analysis showed that strain FA350T could signifi-cantly accumulate PHAs during predation observed in co-culture with pure culture (Fig. 2). Despite their incom-plete fatty acid biosynthetic pathway, all Bradymona-bacteria had a high copy number of long-chain fattyacid transporters (fadL) compared to those of otherpredators, allowing them to gather fatty acids fromthe environment (Fig. 4). In addition, genes associatedwith alkane synthesis, which is important for main-taining cell membrane integrity and adapting to coldenvironments [36], were present in most genomes ofBradymonabacteria (Figs. 3 and 4). Thus, we proposedthat Bradymonabacteria could be categorized as novelpredators different from so-called obligate or faculta-tive predators (Table 1).
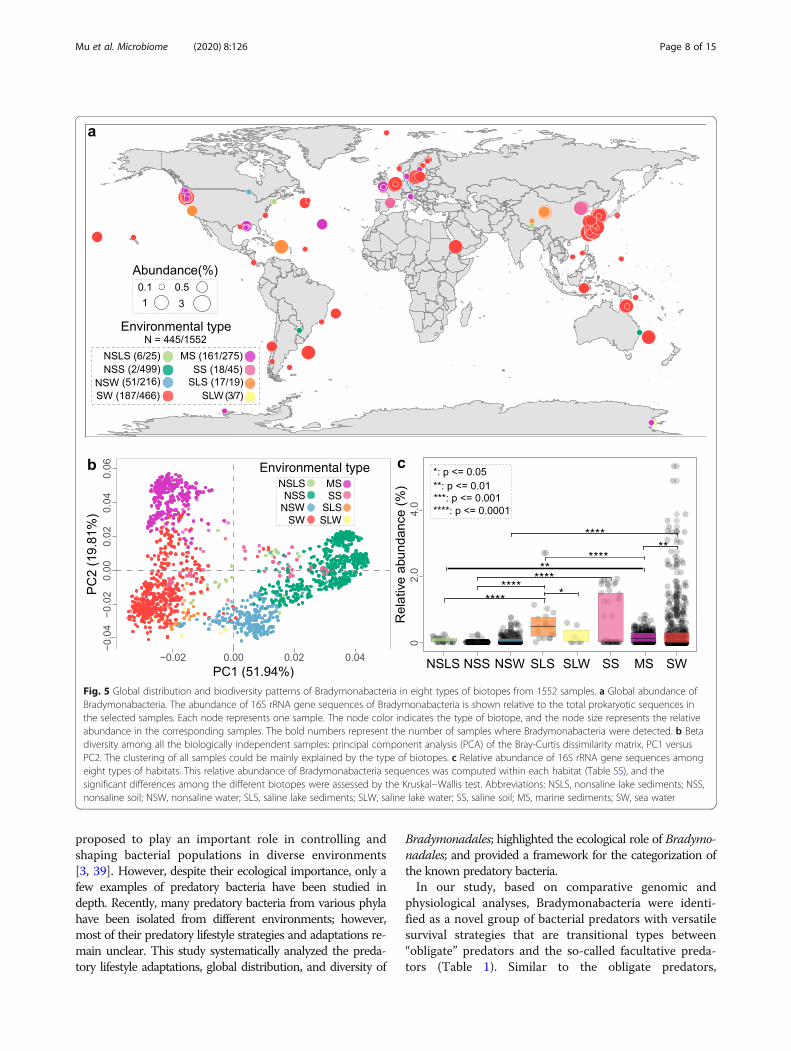
Bradymonadales are mainly distributed in salineenvironments with high diversityTo evaluate the global prevalence of the Bradymona-dales order, we surveyed recently published 16S rRNAgene amplicon studies that provided high taxonomicresolution along with relative sequence abundances. The16S rRNA gene amplicons from 1552 samples weregrouped into eight types of environments (Fig. 5a andTable S5). A total of 811 samples were from inland envi-ronments, while others were from marine environments,with each biotope showing a somewhat different micro-bial community (Fig. 5b). Based on the alpha diversityanalysis, marine sediment and soil biotopes harboredmore OTUs compared with other biotopes (Fig. S5).Bradymonabacteria was detected in 348 of 741 marinesamples (relative abundance > 0.01%) but only 20 of 544soil samples (Fig. 5a). All samples were sorted into anordination diagram based on the similarity of
Fig. 4 Gene abundance in facultative and obligate bacterial predators. The heatmap is based on a two-way cluster analysis of the genomicabundance of genes encoding KEGG protein groups specific to either facultative predators or obligate predators. Groups with a blue backgroundindicate the so-called facultative predators, groups with a yellow background indicate the so-called obligate predators, and groups with a redbackground indicate Bradymonabacteria. The two-way cluster analysis was clustered using the ward.D2 method based on Euclidean distances.The gene abundance matrix is available in Table S3
Mu et al. Microbiome (2020) 8:126 Page 6 of 15

communities (Fig. 5b). Saline biotopes were clearly sepa-rated from nonsaline biotopes (Fig. S6), suggesting thatsalinity was a significant factor in shaping microbialcommunities. For each biotope, the relative abundanceof Bradymonadales in the saline environments (i.e., sea-water and saline lake sediment) was significantly higherthan that in the nonsaline environments (i.e., nonsalinesoil and nonsaline water) (P ≤ 0.0001, Fig. 5c). The dis-tribution analysis was consistent with the genomic fea-ture analysis (Fig. 2), in which several genes encodingsodium symporters and Na+/H+ antiporters were foundin the genomes, suggesting a beneficial effect of salinityon Bradymonabacteria.In addition, we compared the relative abundance of
Bradymonadales with those of two orders of well-knownpredatory bacteria, Bdellovibrionales and Myxococcales[12, 37, 38]. We found that Myxococcales and Bdellovi-brionales were also globally distributed (Fig. S7); how-ever, Myxococcales were more commonly distributed insoil and sediment environments, while Bdellovibrionaleswere more likely to be found in freshwater and seawater(Fig. S7). The total relative abundances of Bradymona-dales, Bdellovibrionales, and Myxococcales ranged from0.7 to 6.4% of the total prokaryotic microbes in all 1552samples (Fig. S8a). The mean relative abundance of Bra-dymonadales (0.5%) was similar to that of Bdellovibrio-nales (0.6%) when both were detected in environmentalsamples (Fig. S8b). In contrast, Bradymonadales was oneof the most abundant known predatory bacteria in salinelake sediment and saline lake water (Fig. S8c).To further determine how salinity affected the relative
abundance of Bradymonadales, we used the Gaodaomultipond salterns as a model and applied 16S rRNAgene amplicon, fluorescence in situ hybridization(FISH) (Tables S6 and S7), and real-time PCR analyses(Figs. S8d and S9). The results showed that Bradymona-dales appeared in all the tested multipond saltern data-sets, accounting for an average of 0.74% of all bacterialsequences and more than 1.0% relative abundancewithin the range of 80 and 265 g/L salinity (Fig. S8d),
significantly higher than those of Bdellovibrionales andMyxococcales. Bradymonadales may exhibit differentcorrelations with prey at different abundances, a possi-bility that will require further study. In addition, fluores-cence in situ hybridization (FISH) (Tables S6 and S7)and real-time PCR experiments were performed, and theresults showed a relative cell abundance of Bradymona-dales of up to 0.6% and a gene copy numbers ratio ashigh as 1.96% in sediments with salinity 80 g/L (Fig. S9).These findings support those of the global analysis (Fig.S8c) and suggest that Bradymonadales may be a dominantbacterial predator in some specific saline environments.To explore the diversity and distinct evolution of bra-
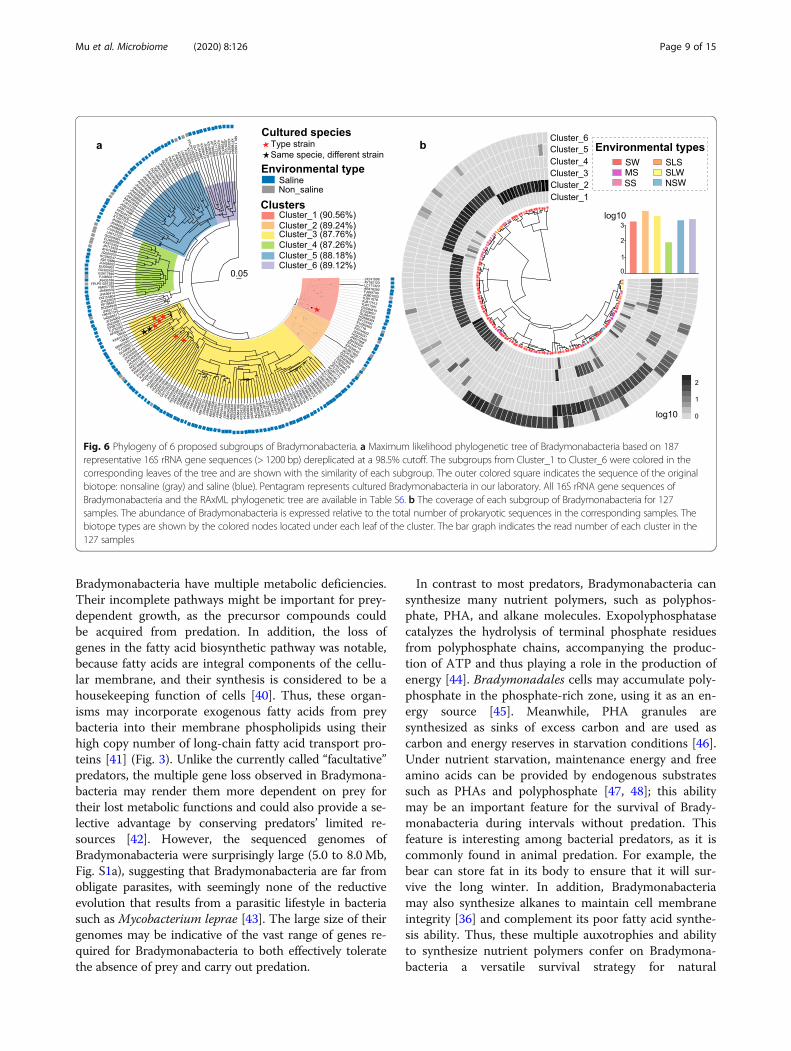
dymonabacterial subgroups in different biotopes, we per-formed a phylogenetic analysis of nearly full-length 16SrRNA gene sequences of diverse origin by maximumlikelihood inference (Table S6). A total of 187 OTUswere detected and found to form six sequence clusters(Fig. 6a). Almost 87.2% of the representative sequencesoriginated from saline biotopes (such as seawater, mar-ine sediments, salterns, corals, and saline lakes). Sincebradymonabacterial subgroups may be selectively dis-tributed in local biotopes, we investigated the relativeabundance of each subgroup throughout the 127 repre-sentative samples in which the relative abundance ofBradymonadales was above 1% of total 16S rRNA genereads (Fig. 6b). Five of the 6 bradymonabacterial sub-groups showed significantly higher abundance in salineenvironments. Cluster-2 and cluster-6 were mainly ob-served in seawater biotopes, whereas cluster-3 wasmainly observed in marine sediment and saline lakesediment (Fig. 6b), consistent with the environments ofthe cultured strains. Cluster-1 and cluster-4 were bothdetected in marine sediment and seawater biotopes.Cluster-5 lineages tended to occur in both freshwaterand seawater biotopes (Fig. 6b).
DiscussionIn all ecosystems, predation is an important interactionamong living organisms. Bacterial predators are
Table 1 The features of 3 different types of bacterial predators
Current predatortype
Redefinedpredatortype
Preydependent/independent
Metabolicpathwaydeficiencies
Pure-culturecultivable
Storing nutrients aspolymers
Predationstrategy
Predationspecificity
Obligate Obligate Completelyprey-dependent
High Extremelydifficult
None Contact-dependent
Gram-negative
Bradymonabacteria Facultative Facultativelyprey-dependent
High Difficult Polyhydroxyalkanoates,polyphosphate, andalkanes
Contact-dependent
Gram-negativeand Gram-positive
Facultative Opportunistic Prey-independent
Low Normal Polyphosphatea Mostlycontact-independent
Gram-negativeand Gram-positive
aA polyphosphate accumulation pathway was found in genomes but not determined by experiments
Mu et al. Microbiome (2020) 8:126 Page 7 of 15
proposed to play an important role in controlling andshaping bacterial populations in diverse environments[3, 39]. However, despite their ecological importance, only afew examples of predatory bacteria have been studied indepth. Recently, many predatory bacteria from various phylahave been isolated from different environments; however,most of their predatory lifestyle strategies and adaptations re-main unclear. This study systematically analyzed the preda-tory lifestyle adaptations, global distribution, and diversity of
Bradymonadales; highlighted the ecological role of Bradymo-nadales; and provided a framework for the categorization ofthe known predatory bacteria.In our study, based on comparative genomic and
physiological analyses, Bradymonabacteria were identi-fied as a novel group of bacterial predators with versatilesurvival strategies that are transitional types between“obligate” predators and the so-called facultative preda-tors (Table 1). Similar to the obligate predators,
Fig. 5 Global distribution and biodiversity patterns of Bradymonabacteria in eight types of biotopes from 1552 samples. a Global abundance ofBradymonabacteria. The abundance of 16S rRNA gene sequences of Bradymonabacteria is shown relative to the total prokaryotic sequences inthe selected samples. Each node represents one sample. The node color indicates the type of biotope, and the node size represents the relativeabundance in the corresponding samples. The bold numbers represent the number of samples where Bradymonabacteria were detected. b Betadiversity among all the biologically independent samples: principal component analysis (PCA) of the Bray-Curtis dissimilarity matrix, PC1 versusPC2. The clustering of all samples could be mainly explained by the type of biotopes. c Relative abundance of 16S rRNA gene sequences amongeight types of habitats. This relative abundance of Bradymonabacteria sequences was computed within each habitat (Table S5), and thesignificant differences among the different biotopes were assessed by the Kruskal−Wallis test. Abbreviations: NSLS, nonsaline lake sediments; NSS,nonsaline soil; NSW, nonsaline water; SLS, saline lake sediments; SLW, saline lake water; SS, saline soil; MS, marine sediments; SW, sea water
Mu et al. Microbiome (2020) 8:126 Page 8 of 15
Bradymonabacteria have multiple metabolic deficiencies.Their incomplete pathways might be important for prey-dependent growth, as the precursor compounds couldbe acquired from predation. In addition, the loss ofgenes in the fatty acid biosynthetic pathway was notable,because fatty acids are integral components of the cellu-lar membrane, and their synthesis is considered to be ahousekeeping function of cells [40]. Thus, these organ-isms may incorporate exogenous fatty acids from preybacteria into their membrane phospholipids using theirhigh copy number of long-chain fatty acid transport pro-teins [41] (Fig. 3). Unlike the currently called “facultative”predators, the multiple gene loss observed in Bradymona-bacteria may render them more dependent on prey fortheir lost metabolic functions and could also provide a se-lective advantage by conserving predators’ limited re-sources [42]. However, the sequenced genomes ofBradymonabacteria were surprisingly large (5.0 to 8.0Mb,Fig. S1a), suggesting that Bradymonabacteria are far fromobligate parasites, with seemingly none of the reductiveevolution that results from a parasitic lifestyle in bacteriasuch as Mycobacterium leprae [43]. The large size of theirgenomes may be indicative of the vast range of genes re-quired for Bradymonabacteria to both effectively toleratethe absence of prey and carry out predation.
In contrast to most predators, Bradymonabacteria cansynthesize many nutrient polymers, such as polyphos-phate, PHA, and alkane molecules. Exopolyphosphatasecatalyzes the hydrolysis of terminal phosphate residuesfrom polyphosphate chains, accompanying the produc-tion of ATP and thus playing a role in the production ofenergy [44]. Bradymonadales cells may accumulate poly-phosphate in the phosphate-rich zone, using it as an en-ergy source [45]. Meanwhile, PHA granules aresynthesized as sinks of excess carbon and are used ascarbon and energy reserves in starvation conditions [46].Under nutrient starvation, maintenance energy and freeamino acids can be provided by endogenous substratessuch as PHAs and polyphosphate [47, 48]; this abilitymay be an important feature for the survival of Brady-monabacteria during intervals without predation. Thisfeature is interesting among bacterial predators, as it iscommonly found in animal predation. For example, thebear can store fat in its body to ensure that it will sur-vive the long winter. In addition, Bradymonabacteriamay also synthesize alkanes to maintain cell membraneintegrity [36] and complement its poor fatty acid synthe-sis ability. Thus, these multiple auxotrophies and abilityto synthesize nutrient polymers confer on Bradymona-bacteria a versatile survival strategy for natural
Fig. 6 Phylogeny of 6 proposed subgroups of Bradymonabacteria. a Maximum likelihood phylogenetic tree of Bradymonabacteria based on 187representative 16S rRNA gene sequences (> 1200 bp) dereplicated at a 98.5% cutoff. The subgroups from Cluster_1 to Cluster_6 were colored in thecorresponding leaves of the tree and are shown with the similarity of each subgroup. The outer colored square indicates the sequence of the originalbiotope: nonsaline (gray) and saline (blue). Pentagram represents cultured Bradymonabacteria in our laboratory. All 16S rRNA gene sequences ofBradymonabacteria and the RAxML phylogenetic tree are available in Table S6. b The coverage of each subgroup of Bradymonabacteria for 127samples. The abundance of Bradymonabacteria is expressed relative to the total number of prokaryotic sequences in the corresponding samples. Thebiotope types are shown by the colored nodes located under each leaf of the cluster. The bar graph indicates the read number of each cluster in the127 samples
Mu et al. Microbiome (2020) 8:126 Page 9 of 15

environments, which contrasts with that of the currentlyknown “obligate” or “facultative” predators.As bacterial predators, Bradymonabacteria have devel-
oped a wide range of mechanisms to attack their prey.Although genes involved in OMV-like biosynthesis weredetected in most genomes, the fermentation supernatantof Bradymonabacteria showed no antibacterial activity,suggesting that bradymonabacterial predation depends onthe cell contact, which is different from that of the cur-rently known facultative predators. Contact-dependentpredation mechanisms allow predators to attach to theprey and then carry out predation. This prey-dependentprocess has a relatively low energy cost and could preventsecretory virulence factors from being diluted by the sur-rounding environment [49]. Bradymonabacteria also hasT4P, which could pull adherent bacteria into close associ-ation with other bacteria [50]. T4P could also transportbound substrates such as DNA [51] into the periplasmand export exoproteins across the outer membrane [52].Contact-dependent type III secretion systems have alsobeen found in Bradymonabacteria and are reported to becapable of moving virulence factors across bacterial outermembranes and directly across the host cell membraneinto the cytoplasm of a host cell [53]. However, no reportshave indicated that the type III secretion system is in-volved in direct combat between bacteria. Whether typeIII secretory complexes could penetrate the bacterial cellwall is unknown. Further gene knockout experiments andsystematic TEM analysis should be performed to identifywhether and how the type III secretion system works dur-ing predation.Our biogeographic analysis suggested that Bradymona-
bacteria are mainly distributed in saline environments,and some other studies have also detected Bradymona-dales in hypersaline soda lake sediments [25], suggestingthat saline environments could be enriched in these bac-teria. Our genome analysis also showed that Bradymona-dales had many genes encoding sodium symporters andNa+/H+ antiporters to maintain osmotic pressure in salineenvironments. These findings supported the global ana-lysis (Figs. 5 and S8c), suggesting that Bradymonadalesmight be a dominant bacterial predator in some specificsaline environments compared with Bdellovibrionales andMyxococcales. The analysis of the complex intragroupphylogeny of the 6 subgroups of Bradymonabacteria re-vealed that distinct evolutionary bradymonabacterial sub-groups had arisen in different biotopes, suggesting theoccurrence of adaptive evolution specific to each habitat.Patterns related to salinity status also suggest that mostBradymonadales are halophiles [17].Bradymonabacteria had a very high predation effi-
ciency on bacteria within the phylum Bacteroidetes.Members of the phylum Bacteroidetes are one the mostabundant groups of bacteria in the ocean [54]. Thus, a
high predation efficiency on Bacteroidetes may indicatethat Bradymonabacteria has important roles in regulat-ing Bacteroidetes communities in oceans. Furthermore,Bradymonabacteria had a high predation efficiency onFlavobacteria and Proteobacteria, some of which arecommensal bacteria in fish [55], suggesting that Brady-monabacteria may be involved in fish microbiomedysbiosis. In addition, Bradymonadales were detected incoral samples, as Bradymonabacteria have a wide rangeof prey, including the coral pathogen Vibrio harveyi,suggesting that Bradymonabacteria may protect coralhosts by consuming potential pathogens [39]. The exactecological roles of this group in different environmentsshould be determined in further studies.
ConclusionThe unique metabolic pathways of Bradymonabacteria,which include conspicuous metabolic deficiencies similar tothose of obligate predators but with a more effective starva-tion stress response mechanism, provide these bacteria withtransitional survival models between “obligate” and so-calledfacultative predators. We suggest that Bradymonabacteria, asfacultative prey-dependent predators, can be renamed as fac-ultative predators. In addition, the currently used “facultative”predators term can be replaced by opportunistic predators,which are prey-independent predators. Thus, we propose aframework to categorize the current bacterial predators into3 groups: (i) obligate predators (completely prey-dependent),such as most of the BALOs; (ii) facultative predators (faculta-tively prey-dependent), such as the Bradymonabacteria cul-tured in the present study; and (iii) opportunistic predators(prey-independent), such as Myxobacteria and Lysobacter sp.(Table 1). This categorization replaces the currently known“facultative” predators with opportunistic predators and willbe helpful for further study of the different ecological import-ance of each type of bacterial predator. The evolution of bac-terial predation in these three groups of predators shouldalso be studied in the future to better understand the signifi-cance of predation to biological evolution.Our study highlights the ecological role of Bradymo-
nadales in saline environments. Given their substantialsequence and cell frequencies in the saline environmentand their storage of nutrients as polymers in cells duringpredation, Bradymonadales may have an alternative wayof regulating global nutrient cycling. To better under-stand the impact of bradymonabacterial predation onregulating biogeochemical cycling, predation mutantsand microcosms need to be developed in further studies.
MethodsPredation experimentsTo explore the predation of Bradymonabacteria, we usedBradymonas sediminis FA350T and Lujinxingia litoralisB210T as representative strains. All candidate prey strains
Mu et al. Microbiome (2020) 8:126 Page 10 of 15

were obtained from our laboratory. Cells were centrifuged,washed, and concentrated in seawater to a final OD600 of3.0 for predator strains and 6.0 for candidate prey strains.Drops of 5.0 μl of the predator strain suspensions were de-posited on the surfaces of agar plates and allowed to dry.Next, 20.0 μl drops of each different candidate prey strainsuspension were placed near the predator spot. The plateswere incubated at 33 °C, and images were taken after 48 hwith a digital camera. To detect PHA accumulation, thegranules were stained with the Nile red component of Nileblue A.
Genome sequencing and comparative genome analysesTo explore the potential metabolic capacity of bacterialpredators, we sequenced 3 complete genomes and 6draft genomes of all currently known Bradymonabacteriaisolate strains. The genomic DNA of all strains wasextracted with a DNA extraction kit (TaKaRa Bio)according to the manufacturer’s instructions. For strainsFA350T, V1718, and YN101, complete genome sequen-cing was performed by Nanjing CocoBio Co., China,using the Illumina HiSeq platform accompanied withthe SMRT platform to build Illumina PE and Pacbio li-braries. The single-molecule sequencing data assemblywas accomplished using SOAPdenovo v2.04 and CeleraAssembler v8.0, and the results were rectified throughBLAST searches in the BlastR database. The de novo as-sembly of the scaffolds was performed using Celera As-sembler 8.0, which were then overlapped and trimmedusing GapCloser v1.12 (SOAPdenovo-related software)[18]. Draft genome sequencing of the other 6 strains wasperformed by Shanghai Personal Biotechnology Co., Ltd.(Shanghai, China) using Solexa paired-end sequencingtechnology [2]. A library with an average fragmentlength of 400 bp was constructed, and the final genomeswere assembled using SOAPdenovo version 2.04 [56].We also retrieved 37 predator genomes from NCBI (in-cluding 4 metagenome-assembled genomes). tRNA andgene prediction were performed using tRNAscan andprodigal, respectively. The genome-based metabolic po-tential of the bacterial predators was predicted by Blas-tKOALA (https://www.kegg.jp/blastkoala/). The averagenucleic acid identities among the 9 cultured Bradymona-bacteria strains were calculated using pyani (https://github.com/widdowquinn/pyani), and the percentage ofconserved proteins (POCP) in each strain was calculatedas described previously by Qin et al. [57].
Electron microscopy analysesWe selected Algoriphagus marinus am2, which is smallerthan the predator Bradymonas sediminis FA350T, as prey.Bradymonas sediminis FA350T and Algoriphagus marinusam2 were cultured separately to the exponential growth
phase, adjusted to the same OD value, mixed together,and cocultured on marine agar medium at 33 °C for 68 h.For TEM analysis, mixed culture samples were sup-
ported on carbon/formvar-coated copper grids. Thegrids were inverted over a drop of 1% uranyl acetate.Thin sections were prepared with the predator-prey co-cultures at 68 h incubation. The samples were mixedwith 0.5 ml of 2% glutaraldehyde in 0.1M sodium caco-dylate buffer, centrifuged and resuspended in 1 ml of thesame solution for 3 h. The cells were washed in cacody-late buffer, fixed with 1% osmium tetroxide, and encasedin agar. The agar-encased cells were then fixed in 2% ur-anyl acetate, dehydrated through an ethanol series, andembedded in Epon resin. Thin sections were cut andstained with uranyl acetate and lead citrate. Specimenswere examined with a JEM-1200EX electron microscopeoperated at 80 kV.For SEM analysis, mixed culture samples were washed
3 times with PBS and fixed for 1 h in 2.5% glutaralde-hyde in sodium cacodylate buffer (0.1 M, pH 7.2). Todehydrate the bacteria, the EM grids underwent a seriesof washes in increasing concentrations of ethanol (25,50, 75, and 96%) and placed in a vacuum overnight. Thesamples were coated with gold and observed using aNova NanoSEM 450.
Dual transcriptomic analysesTo determine the gene expression profiles of the typestrain FA350T, Algoriphagus marius am2T was used asprey due to its cell morphology being different from B.sediminis FA350T. Moreover, B. sediminis FA350T pre-dated A. marius am2T well, and the two species have adistant evolutionary relationship that was helpful for fur-ther gene mapping analysis. A pure culture of FA350T
and a coculture of FA350T with the prey A. marinusam2 were cultured on marine agar medium at 33 °C for0 h, 68 h, and 120 h, respectively. Mixed-culture cellssedimented by centrifugation and washed, and theresulting pellets stored at − 80 °C prior to RNA extrac-tion. Each time point was collected in triplicate (n = 3)for further transcriptomic analysis. RNA was extractedusing an miRNEasy mini kit (Qiagen 217004), rRNA wasremoved using a Ribo-Zero Magnetic kit (Bacteria) fromEpicentre (MRZB12424), and cDNA library constructionwas performed with a TruSeq Stranded mRNA librarypreparation kit from Illumina (RS-122-2101) [58]. Se-quencing was carried out on a HiSeq sequencer atNovogene Co., Ltd. (Beijing, China).
Transcriptome mapping and differential expressionanalysisFor transcriptomic analysis of mixed culture samples (dualtranscriptomic analysis), total RNA sequences were mappedto the complete genome of FA350T using the method
Mu et al. Microbiome (2020) 8:126 Page 11 of 15

reported by Westermann et al. [59]. Before downstream pro-cessing, trimmomatic was used to clean the reads to removeadaptor sequences and leading and trailing bases with qualitythresholds below 20, perform sliding window trimming (withparameters 4:15), and remove reads less than 36 bp inlength. After cleaning, the remaining paired reads weremapped to the respective genomes to calculate expressionvalues. The reference genomes used were B. sediminisFA350T (accession: CP030032.1) and A. marinus am2 (acces-sion: MSPQ00000000.1). The output fragments per kilobaseof transcript per million mapped reads (FPKM) were calcu-lated for further analysis. Significantly upregulated anddownregulated genes were defined using a false discoveryrate of less than 0.001, a P value of < 0.05, and a minimum 1log2(fold)-change of gene expression.
Phylogenetic analysis of bradymonabacterial type IV piliAn unrooted, maximum likelihood phylogeny shows re-lationships between the type IVa, type IVb, and type IVcpili and the archaellum (archaeal flagellum) and theT2SS and T4SS extension ATPases. The protein amino-acid sequences were aligned with mafft and used to esti-mate a maximum likelihood phylogeny with RAxMLunder the JTT substitution model with gamma-distributed rate variation. The protein amino-acid se-quences of Bradymonas were annotated by RAST (RapidAnnotation using Subsystem Technology) [60]. Theother protein amino acid sequences were obtained fromother research supplementary materials [61].
Biogeographic distribution database constructionAll the 16S rRNA gene sequences analyzed in this paperwere downloaded from the European Nucleotide Arch-ive (https://www.ebi.ac.uk/ena, ENA) during or beforeDecember 2018. As a result, we collected 1552 samplesfrom 102 projects or studies: 25 from nonsaline lake sed-iments (NSLS), 275 from marine sediments (MS), 499from nonsaline soil (NSS), 45 from saline soil (SS), 216from nonsaline water (NSW), 19 from saline lake sedi-ments (SLS), 466 from seawater (SW), and 7 from salinelake water (SLW) (Table S5).
Microbial community compositionThe raw 16S rRNA gene reads were filtered withUCHIME. Quality filtering, chimera detection, dereplica-tion, clustering into OTUs, and assigning taxonomic in-formation were performed using VSEARCH [62]. TheSILVA database Ref_SSU release 132 was used as a ref-erence taxonomic database (https://www.arb-silva.de/).Alpha diversity indices (Shannon, Simpson, Good’scoverage and Ace) detailing the microbial communitycomposition within each sample were calculated usingscikit-bio (http://scikit-bio.org/) in Python, and alpha di-versity indices (Chao1) were calculated using the
package fossil (https://www.rdocumentation.org/pack-ages/fossil) in R. For estimating community dissimilar-ities, the Bray–Curtis dissimilarity was calculatedbetween 1551 samples (total samples: 1552) by vegan inR based on the relative abundance of order taxonomylevel.
Phylogenetic analysesBoth RAxML [63] and FastTree [64] were employed toconstruct the Bradymonabacteria phylogenetic tree.Given both the topology of the phylogenetic tree and itsgood coverage of all Bradymonabacteria lineages, weestablished the phylogenetic tree using 187 representa-tive Bradymonabacteria 16S rRNA gene sequences,which were all longer than 1200 bp (at 98.5% cutoff).These sequences were aligned using mafft. The Brady-monabacteria subgroup designations were confirmedwhen one subgroup with > 10 representative sequenceswas monophyletic by two phylogenetic trees constructedby different programs using the maximum likelihood ap-proach [65]. The environmental type (i.e., saline andnonsaline) of each Bradymonabacteria sequence in thetree was collected from GenBank. A genome-based phyl-ogeny of bacterial predators and 9 cultured Bradymona-bacteria strains was constructed using core genes [66],and trees were constructed using RAxML [63]. Allphylogenetic trees were drawn using ggtree [67] in R.
Quantitative real-time PCRThe environmental DNA samples extracted in the previ-ous step were used for qPCR experiments in order todetect the abundance of bacteria and Bradymonadales ineach sample. The primer pair composed of 341F (5′-CCTACGGGAGGCAGCAG-3′) and 534R (5′-ATTACCGCGGCTGCTGGCA-3′) was used for quantificationof bacteria [22]. A Bradymonadales-specific primer setcomposed of qBRA1295F (5′-CTCAGTWCGGATY-GYAGTCTG-3′) and qBRA1420R (5′-GTCACYGACTTCTGGAGCAARYG-3′), which was designed in thepresent study and generated an amplicon of 148 bases,was used for quantification of Bradymonadales. Thespecificity and coverage test of the primers were de-scribed in the Supplementary Materials (SupplementaryMethods and Results, Fig. S10 and S11, Tables S8 andS9). Reactions for each sample were carried out in anABI StepOnePlus thermal cycler under the followingconditions: an initial denaturation step at 95 °C for 10min and then 40 cycles of 15 s at 95 °C and 30 s at 60 °C.The reaction was performed in a total volume of 20 μl,composed of 10 μl 2X Universal SYBR Green Fast qPCRMix (ABclonal), 0.4 μl of each primer (10 μM), 1 μl ofsample, and 8.2 μl of MiliQ water. The Plasmid DNAStandard was constructed by introducing the 16S rDNAgene amplified from Bradymonas sediminis FA350T into
Mu et al. Microbiome (2020) 8:126 Page 12 of 15

the pMD18-T Vector (TaKaRa) following the manufac-turer’s instructions. The plasmid was isolated and puri-fied using a MiniBEST Plasmid Purification Kit(TaKaRa). DNA copy number was determined by theconcentration and relative molecular weight of the Plas-mid DNA. For each QPCR assay, the plasmid aliquotwas serially diluted to produce concentrations rangingfrom 109 to 103 DNA copies/μl to generate calibrationcurves. Each sample was measured in triplicate, andnegative controls (no template NTC) were included.
Supplementary informationSupplementary information accompanies this paper at https://doi.org/10.1186/s40168-020-00902-0.
Additional file 1: Figure S1. The General features of bacterialpredators. Figure S2. General gene expression profiles of Bradymonassediminis FA350 during mix-culturing with prey Algoriphagus marinesam2. Figure S3. Gene expression profiles of Bradymonas sediminis FA350during mix-culturing with prey Algoriphagus marines am2. Figure S4.Phylogenetic analysis of secretion system machinery reveals a distinct tadtype IVb subtype of type IV pili. Figure S5. The alpha diversity in 1,552samples. Figure S6. Principal component analysis of different samples as-sociated with saline status. Figure S7. Global distribution of Myxococcalesand Bdellovibrionales in eight different biotopes from 1,552 samples. Fig-ure S8. The relative abundance of Bradymonadales, Myxococcales, andBdellovibrionale. Figure S9. Cell abundance and relative abundance ofgene copy number of Bradymonadales from solar saltern sediments. Fig-ure S10. Specificity test of the Quantitative real-time PCR primers. FigureS11. Quantitative real-time PCR amplification detection and the standardcurve.
Additional file 2: Table S1. Detailed information of organisms on thephylogenetic tree
Additional file 3: Table S2. Detailed genes information for figure 3.
Additional file 4: Table S3. Detailed genes information for figure 4.
Additional file 5: Table S4. Detailed key genes expression changedduring predation.
Additional file 6: Table S5. Detailed information of 16S rRNA genesamplicon samples used in this study.
Additional file 7: Table S6. Detailed information of the representative16S rRNA gene sequences.
Additional file 8: Table S7. Probes designed and optimized in thisstudy.
Additional file 9: Table S8. Specificity and coverage of primersqBRA1295F and qBRA1420R using the SILVA database SSU r138 Ref NR.
AcknowledgementsThis work was supported by the National Natural Science Foundation ofChina (31770002 and 41876166), National Science and TechnologyFundamental Resources Investigation Program of China (2019FY100700)
Consent for publicationsNot applicable
Authors’ contributionsDSM, GJC, JZ, and ZJD designed the study. SW carried out TEM, SEM, andtranscriptome analyses. ZZD carried out FISH and real-time PCR analysis.DSM, QYL, SW, XPW, and RT performed bioinformatic analyses. DSM and ZJDanalyzed data and wrote the paper. JYN, AZ, and YY improved the paperwriting. All authors read and approved the manuscript
Availability of data and materialsThe genomes of cultured bradymonabacteral isolates have been depositedin the NCBI database under GenBank accession numbers CP042467.1
(Bradymonadales strain V1718), CP042468.1 (Bradymonadales strain YN101),VOPX00000000.1 (Bradymonadales strain TMQ1), VOSL00000000.1(Bradymonadales strain TMQ2), QRGZ00000000.1 (Bradymonadales strainTMQ3), VOSM00000000.1 (Bradymonadales strain TMQ4), CP030032.1(Bradymonas sediminis FA350T), QHKO00000000.1 (Lujinxingia litorali B210T),and SADD00000000.1 (Lujinxingia sediminis SEH01T). The genomes ofuncultured Bradymonabacteria have been deposited in the NCBI databaseunder GenBank accession numbers PWKZ00000000.1 (Bradymonadales binCSSed10_215), PWTN00000000.1 (Bradymonadales bin CSSed11_191),PXAJ00000000.1 (Bradymonadales bin T3Sed10_204), and PWZZ00000000.1(Bradymonadales bin T3Sed10_190). The 16S rRNA gene data sets of Gaodaosalterns have been deposited in the Sequence Read Archive under accessionnumber SRP217756 for all the samples. The transcriptome sequences forpredation of FA350T have been deposited in the NCBI database underaccession numbers PRJNA559243 and PRJNA559253. All Bradymonabacteralisolates have been deposited at the Shandong Infrastructure of MarineMicrobial Resources hosted by the Laboratory of Marine Microbiology atShandong University (http://www.sdum.wh.sdu.edu.cn/search.html?itemId=14). All Bradymonabacteral isolates are available upon request. The bash, R,and python scripts for this study are available on the GitHub: https://github.com/2015qyliang/BradymonabacteriaAnalysis.
Ethics approval and consent to participateNot applicable
Competing interestsNo conflict of interest exists in the submission of this manuscript, and themanuscript has been approved by all authors for publication. The authorsdeclare that they have no competing interests.
Author details1State Key Laboratory of Microbial Technology, Institute of MicrobialTechnology, Shandong University, No. 72, Jimo Binhai Road, Jimo, Qingdao266237, China. 2Marine College, Shandong University, Weihai 264209, China.3Institute for Environmental Genomics, University of Oklahoma, Norman,Oklahoma 73019, USA. 4State Key Joint Laboratory of EnvironmentSimulation and Pollution Control, School of Environment, TsinghuaUniversity, Beijing 100084, China.
Received: 6 January 2020 Accepted: 27 July 2020
References1. Young KD. The selective value of bacterial shape. Microbiol Mol Biol R. 2006;
70(3):660–703.2. Chauhan A, Cherrier J, Williams HN. Impact of sideways and bottom-up
control factors on bacterial community succession over a tidal cycle. P NatlAcad Sci USA. 2009;106(11):4301–6.
3. Johnke J, Cohen Y, de Leeuw M, Kushmaro A, Jurkevitch E, Chatzinotas A.Multiple micro-predators controlling bacterial communities in theenvironment. Curr Opin Biotech. 2014;27:185–90.
4. Li HH, Chen C, Sun QP, Liu RL, Cai JP. Bdellovibrio and like organismsenhanced growth and survival of Penaeus monodon and altered bacterialcommunity structures in its rearing water. Appl Environ Microb. 2014;80(20):6346–54.
5. Martinez V, Jurkevitch E, Garcia JL, Prieto MA. Reward for Bdellovibriobacteriovorus for preying on a polyhydroxyalkanoate producer. EnvironMicrobiol. 2013;15(4):1204–15.
6. Richards GP, Fay JP, Dickens KA, Parent MA, Soroka DS, Boyd EF. Predatorybacteria as natural modulators of Vibrio parahaemolyticus and Vibriovulnificus in seawater and oysters. Appl Environ Microb. 2012;78(20):7455–66.
7. Williams HN, Lymperopoulou DS, Athar R, Chauhan A, Dickerson TL, Chen H,et al. Halobacteriovorax, an underestimated predator on bacteria: potentialimpact relative to viruses on bacterial mortality. ISME J. 2016;10(2):491–9.
8. Chen H, Laws EA, Martin JL, Berhane TK, Gulig PA, Williams HN. Relativecontributions of Halobacteriovorax and bacteriophage to bacterial cell deathunder various environmental conditions. Mbio. 2018;9(4):e01202–18.
9. Perez J, Moraleda-Munoz A, Marcos-Torres FJ, Munoz-Dorado J. Bacterialpredation: 75 years and counting! Environ Microbiol. 2016;18(3):766–79.
Mu et al. Microbiome (2020) 8:126 Page 13 of 15

10. Cabrera G, Perez R, Gomez JM, Abalos A, Cantero D. Toxic effects ofdissolved heavy metals on Desulfovibrio vulgaris and Desulfovibrio sp. strains.J Hazard Mater. 2006;135(1-3):40–6.
11. Im H, Dwidar M, Mitchell RJ. Bdellovibrio bacteriovorus HD100, a predator ofgram-negative bacteria, benefits energetically from Staphylococcus aureusbiofilms without predation. Isme J. 2018;12(8):2090–5.
12. Sockett RE. Predatory lifestyle of Bdellovibrio bacteriovorus. Annu RevMicrobiol. 2009;63:523–39.
13. Hespell RB, Thomashow MF, Rittenberg SC. Changes in cell compositionand viability of Bdellovibrio bacteriovorus during starvation. Arch Microbiol.1974;97(4):313–27.
14. Hobley L, Lerner TR, Williams LE, Lambert C, Till R, Milner DS, et al. Genomeanalysis of a simultaneously predatory and prey-independent, novelBdellovibrio bacteriovorus from the river Tiber, supports in silico predictionsof both ancient and recent lateral gene transfer from diverse bacteria. BMCGenomics. 2012;13:670.
15. Pasternak Z, Pietrokovski S, Rotem O, Gophna U, Lurie-Weinberger MN,Jurkevitch E. By their genes ye shall know them: genomic signatures ofpredatory bacteria. Isme J. 2013;7(4):756–69.
16. Tang BL, Yang J, Chen XL, Wang P, Zhao HL, Su HN, et al. A predator-preyinteraction between a marine Pseudoalteromonas sp and Gram-positivebacteria. Nat Commun. 2020;11:285.
17. Wang ZJ, Liu QQ, Zhao LH, Du ZJ, Chen GJ. Bradymonas sediminis gen. Nov.,sp. nov., isolated from coastal sediment, and description ofBradymonadaceae fam. Nov. and Bradymonadales Ord. Nov. Int J Syst EvolMicrobiol. 2015;65(5):1542–9.
18. Wang S, Mu DS, Zheng WS, Du ZJ. Complete genome sequence ofBradymonas sediminis FA350T, the first representative of the orderBradymonadales. Mar Genomics. 2019;46(4):62–5.
19. Hahn MW, Schmidt J, Koll U, Rohde M, Verbarg S, Pitt A, et al. Silvanigrellaaquatica gen. Nov., sp. nov., isolated from a freshwater lake, description ofSilvanigrellaceae fam. Nov. and Silvanigrellales Ord. Nov., reclassification ofthe order Bdellovibrionales in the class Oligoflexia, reclassification of thefamilies Bacteriovoracaceae and Halobacteriovoraceae in the new orderBacteriovoracales Ord. Nov., and reclassification of the familyPseudobacteriovoracaceae in the order Oligoflexales. Int J Syst Evol Microbiol.2017;67(8):2555–68.
20. Zhou XW, Li SG, Li W, Jiang DM, Han K, Wu ZH, et al. Myxobacterialcommunity is a predominant and highly diverse bacterial group in soilniches. Env Microbiol Rep. 2014;6(1):45–56.
21. Jiang DM, Kato C, Zhou XW, Wu ZH, Sato T, Li YZ. Phylogeographicseparation of marine and soil myxobacteria at high levels of classification.ISME J. 2010;4(12):1520–30.
22. Mu DS, Liang QY, Wang XM, Lu DC, Shi MJ, Chen GJ, et al.Metatranscriptomic and comparative genomic insights into resuscitationmechanisms during enrichment culturing. Microbiome. 2018;6(1):230.
23. Guo LY, Li CM, Wang S, Mu DS, Du ZJ. Lujinxingia litoralis gen. Nov., sp. nov.and Lujinxingia sediminis sp. nov., two new representatives in the orderBradymonadales. Int J Syst Evol Microbiol. 2019. https://doi.org/10.1099/ijsem.0.003556.
24. Kim M, Kang O, Zhang Y, Ren L, Chang X, Jiang F, et al. Sphingoaurantiacuspolygranulatus gen. Nov., sp. nov., isolated from high-Arctic tundra soil, andemended descriptions of the genera Sandarakinorhabdus, Polymorphobacterand Rhizorhabdus and the species Sandarakinorhabdus limnophila,Rhizorhabdus argentea and Sphingomonas wittichii. Int J Syst Evol Microbiol.2016;66(1):91–100.
25. Vavourakis CD, Andrei AS, Mehrshad M, Ghai R, Sorokin DY, Muyzer G. Ametagenomics roadmap to the uncultured genome diversity in hypersalinesoda lake sediments. Microbiome. 2018;6(1):168.
26. Jain C, Rodriguez RL, Phillippy AM, Konstantinidis KT, Aluru S. Highthroughput ANI analysis of 90 K prokaryotic genomes reveals clear speciesboundaries. Nat Commun. 2018;9(1):5114.
27. Gil R, Silva FJ, Pereto J, Moya A. Determination of the core of a minimalbacterial gene set. Microbiol Mol Biol Rev. 2004;68(3):518–37.
28. Avidan O, Petrenko M, Becker R, Beck S, Linscheid M, Pietrokovski S, et al.Identification and characterization of differentially-regulated type IVb pilingenes necessary for predation in obligate bacterial predators. Sci Rep. 2017;7(1):1013.
29. Duncan MC, Gillette RK, Maglasang MA, Corn EA, Tai AK, Lazinski DW, et al.High-throughput analysis of gene function in the bacterial predatorBdellovibrio bacteriovorus. MBio. 2019;10(3):e01040–19.
30. Dietsche T, Tesfazgi Mebrhatu M, Brunner MJ, Abrusci P, Yan J, Franz-Wachtel M, et al. Structural and functional characterization of the bacterialtype III secretion export apparatus. PLoS Pathog. 2016;12(12):e1006071.
31. Li J, Chen G, Wu H, Webster JM. Identification of two pigments and ahydroxystilbene antibiotic from Photorhabdus luminescens. Appl EnvironMicrobiol. 1995;61(12):4329–33.
32. Nevermann J, Silva A, Otero C, Oyarzun DP, Barrera B, Gil F, et al.Identification of genes involved in biogenesis of outer membrane vesicles(OMVs) in Salmonella enterica Serovar Typhi. Front Microbiol. 2019;10:104.
33. Liu C, Liu Y, Xu XX, Wu H, Xie HG, Chen L, et al. Potential effect of matrixstiffness on the enrichment of tumor initiating cells under three-dimensional culture conditions. Exp Cell Res. 2015;330(1):123–34.
34. Grote J, Schott T, Bruckner CG, Glockner FO, Jost G, Teeling H, et al.Genome and physiology of a model Epsilonproteobacterium responsible forsulfide detoxification in marine oxygen depletion zones. Proc Natl Acad SciU S A. 2012;109(2):506–10.
35. Fukui T, Shiomi N, Doi Y. Expression and characterization of (R)-specificenoyl coenzyme a hydratase involved in polyhydroxyalkanoate biosynthesisby Aeromonas caviae. J Bacteriol. 1998;180(3):667–73.
36. Sukovich DJ, Seffernick JL, Richman JE, Hunt KA, Gralnick JA, Wackett LP.Structure, function, and insights into the biosynthesis of a head-to-headhydrocarbon in Shewanella oneidensis strain MR-1. Appl Environ Microbiol.2010;76(12):3842–9.
37. Rendulic S, Jagtap P, Rosinus A, Eppinger M, Baar C, Lanz C, et al. Apredator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomicperspective. Science. 2004;303(5658):689–92.
38. Munoz-Dorado J, Marcos-Torres FJ, Garcia-Bravo E, Moraleda-Munoz A, PerezJ. Myxobacteria: moving, killing, feeding, and surviving together. FrontMicrobiol. 2016;7:781.
39. Welsh RM, Zaneveld JR, Rosales SM, Payet JP, Burkepile DE, Thurber RV.Bacterial predation in a marine host-associated microbiome. Isme J. 2016;10(6):1540–4.
40. Nayfach S, Shi ZJ, Seshadri R, Pollard KS, Kyrpides NC. New insights fromuncultivated genomes of the global human gut microbiome. Nature. 2019;568(7753):505–10.
41. Black PN, DiRusso CC. Transmembrane movement of exogenous long-chainfatty acids: proteins, enzymes, and vectorial esterification. Microbiol Mol BiolRev. 2003;67(3):454–72.
42. Morris JJ, Lenski RE, Zinser ER. The Black queen hypothesis: evolution ofdependencies through adaptive gene loss. Mbio. 2012;3(2):00036–12.
43. Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, et al.Massive gene decay in the leprosy bacillus. Nature. 2001;409(6823):1007–11.
44. Saunders AM, Mabbett AN, McEwan AG, Blackall LL. Proton motive forcegeneration from stored polymers for the uptake of acetate under anaerobicconditions. FEMS Microbiol Lett. 2007;274(2):245–51.
45. Kim KS, Rao NN, Fraley CD, Kornberg A. Inorganic polyphosphate is essentialfor long-term survival and virulence factors in Shigella and Salmonella spp. PNatl Acad Sci USA. 2002;99(11):7675–80.
46. Ratcliff WC, Kadam SV, Denison RF. Poly-3-hydroxybutyrate (PHB) supportssurvival and reproduction in starving rhizobia. FEMS Microbiol Ecol. 2008;65(3):391–9.
47. Moller L, Laas P, Rogge A, Goetz F, Bahlo R, Leipe T, et al. Sulfurimonassubgroup GD17 cells accumulate polyphosphate under fluctuating redoxconditions in the Baltic Sea: possible implications for their ecology. Isme J.2019;13(2):482–93x.
48. Kuroda A, Nomura K, Ohtomo R, Kato J, Ikeda T, Takiguchi N, et al. Role ofinorganic polyphosphate in promoting ribosomal protein degradation bythe Lon protease in E. coli. Science. 2001;293(5530):705–8.
49. Granato ET, Meiller-Legrand TA, Foster KR. The evolution and ecology ofbacterial warfare. Curr Biol. 2019;29(11):R521–R37.
50. Chamot-Rooke J, Mikaty G, Malosse C, Soyer M, Dumont A, Gault J, et al.Posttranslational modification of pili upon cell contact triggers N.meningitidis dissemination. Science. 2011;331(6018):778–82.
51. Ellison CK, Dalia TN, Ceballos AV, Wang JCY, Biais N, Brun YV, et al. Retraction ofDNA-bound type IV competence pili initiates DNA uptake during naturaltransformation in Vibrio cholerae. Nat Microbiol. 2018;3(7):773–80.
52. Craig L, Forest KT, Maier B. Type IV pili: dynamics, biophysics and functionalconsequences. Nat Rev Microbiol. 2019;17(7):429–40.
53. Ruhe ZC, Subramanian P, Song KH, Nguyen JY, Stevens TA, Low DA, et al.Programmed secretion arrest and receptor-triggered toxin export duringantibacterial contact-dependent growth inhibition. Cell. 2018;175(4):921–33.
Mu et al. Microbiome (2020) 8:126 Page 14 of 15

54. Fernandez-Gomez B, Richter M, Schuler M, Pinhassi J, Acinas SG, GonzalezJM, et al. Ecology of marine Bacteroidetes: a comparative genomicsapproach. Isme J. 2013;7(5):1026–37.
55. Legrand TPRA, Catalano SR, Wos-Oxley ML, Fran S, Matt L, Bansemer MS,et al. The inner workings of the outer surface: skin and gill microbiota asindicators of changing gut health in yellowtail kingfish. Front Microbiol.2018;8:2664.
56. Mu D, Zhao J, Wang Z, Chen G, Du Z. Draft genome sequence ofAlgoriphagus sp. strain NH1, a multidrug-resistant bacterium isolated fromcoastal sediments of the northern Yellow Sea in China. Genome Announc.2016;4(1):e01555–15.
57. Qin QL, Xie BB, Zhang XY, Chen XL, Zhou BC, Zhou JZ, et al. A proposedgenus boundary for the prokaryotes based on genomic insights. J Bacteriol.2014;196(12):2210–5.
58. Mu DS, Yu XX, Xu ZX, Du ZJ, Chen GJ, et al. Sci Rep. 2016;6:srep29953.59. Westermann AJ, Forstner KU, Amman F, Barquist L, Chao Y, Schulte LN, et al.
Dual RNA-seq unveils noncoding RNA functions in host-pathogeninteractions. Nature. 2016;529(7587):496–501.
60. Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEEDand the rapid annotation of microbial genomes using subsystemstechnology (RAST). Nucleic Acids Res. 2014;42(D1):D206–D14.
61. Ellison CK, Kan J, Dillard RS, Kysela DT, Ducret A, Berne C, et al. Obstructionof pilus retraction stimulates bacterial surface sensing. Science. 2017;358(6362):535–8.
62. Rognes T, Flouri T, Nichols B, Quince C, Mahe F. VSEARCH: a versatile opensource tool for metagenomics. Peerj. 2016;4:2584.
63. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
64. Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490.
65. Liu XB, Li M, Castelle CJ, Probst AI, Zhou ZC, Pan J, et al. Insights into theecology, evolution, and metabolism of the widespread Woesearchaeotallineages. Microbiome. 2018;6:102.
66. Na SI, Kim YO, Yoon SH, Ha SM, Baek I, Chun J. UBCG: up-to-date bacterialcore gene set and pipeline for phylogenomic tree reconstruction. JMicrobiol. 2018;56(4):280–5.
67. Yu GC, Smith DK, Zhu HC, Guan Y, Lam TTY. GGTREE: an R package forvisualization and annotation of phylogenetic trees with their covariates andother associated data. Methods Ecol Evol. 2017;8(1):28–36.
Publisher’s NoteSpringer Nature remains neutral with regard to jurisdictional claims inpublished maps and institutional affiliations.
Mu et al. Microbiome (2020) 8:126 Page 15 of 15





























