Embed Size (px)
Citation preview

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 1 of 23
DEPARTMENT OF GENERAL MEDICINE CARDIOLOGY DIVISION
WYCOMBE HOSPITAL
Dr C P Clifford Dr S Firoozan
Tel: 01494 425004 Tel: 01494 425070 Fax: 01494 425908
Wycombe Hospital
Queen Alexandra Road High Wycombe
Buckinghamshire
HP11 2TT Tel: 01494 526161
Patient Information Sheet
Study Title: An OPen-label, Randomized, Controlled, Multicenter Study ExplorIng TwO TreatmeNt
StratEgiEs of Rivaroxaban and a Dose-Adjusted Oral Vitamin K Antagonist
Treatment Strategy in Subjects With Atrial Fibrillation Who Undergo
Percutaneous Coronary Intervention - PIONEER AF-PCI Study Number: RIVAROXAFL3003
Principal Investigator: Doctor Christopher Piers Clifford Telephone Number: 01494 425004 Doctor Clifford is doing this study for Janssen Scientific Affairs. The study has been approved by an Independent Ethics Committee/Institutional Review Board.
Janssen-Cilag International (NV) (Sponsor) Project Management and Site Monitoring provided by: Quintiles Ltd, 500 Brook Drive, Green
Park, Reading, Berks, RG2 6UU Local Oversight provided by: Global Clinical Operations, Janssen Research & Development
(Division of Janssen-Cilag Ltd), 50-100 Holmers Farm Way, High Wycombe, Bucks, HP12 4DP
Please read this information carefully You are invited to be in a research study because you have atrial fibrillation (AF). Taking part in a
research study is voluntary. Before you decide, you should know why the research is being done and
what it involves. Please read this information carefully and take your time to decide. Ask the study
doctor (or his or her staff) any questions you may have. You may take an unsigned copy of this form
home with you to read again. Take your time to think and talk about it with your family and friends
before making your decision. Being in a research study is not part of your routine medical care. A description of this clinical trial will be available at http://www.ClinicalTrials.gov as required by U.S.
Law. This website will not include information that can identify you. At most, the website will include a
summary of the study results. You can search this website at any time.

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 2 of 23
This information sheet is split into two parts. Part 1 (sections 1-14) tells you the purpose of the study and what will happen
to you if you take part. Part 2 (sections 15-24) gives you more information on the conduct of the
study. Ask your study doctor if there is anything that is not clear or if you would like
more information. Take time to decide whether or not you want to take part.
Patient Name: CRF ID:
You will be given a copy of this information sheet to keep

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 3 of 23
Patient Information Sheet Part 1
1. What is the purpose of the study? The investigational drug used in this study is called Rivaroxaban. Although approved for use
within the EU for the treatment of atrial fibrillation, the use of rivaroxaban within this study is
considered experimental as it is being investigated within a different patient group and at an
alternative dose. Rivaroxaban is being tested to see if it may be useful in treating patients who have an
irregular heart beat (called atrial fibrillation), and who have also had a nonsurgical procedure
used to restore blood flow to blocked arteries, called a percutaneous coronary intervention
(PCI). In particular this study is looking at patients who required the placement of a stent (a wire
mesh tube or „scaffold‟ used to keep the artery open) in at least one of the hearts arteries
during the PCI procedure. Patients that have atrial fribrillation, and/or have undergone a PCI with stent placement, are
put on medication to prevent the formation of blood clots (blockages) in the hearts arteries.
This study will compare the effects (both good and bad) of rivaroxaban to those of a vitamin
K antagonist. Rivaroxaban and Vitamin K Antagonists both act as blood thinners to reduce
the formation of blood clots inside the hearts arteries. The purpose of the study is to determine which treatment combination, rivaroxaban or
Vitamin K Antagonists, taken along with other antiplatelet therapies, is the most
effective and safe option to use in patients with atrial fibrillation who have also
received stents. Rivaroxaban was approved by the European Medicines Agency in December 2011 and in the United States by the Food and Drug Administration (FDA) in November 2011 to reduce the risk of stroke and systemic embolism (blood clots outside of the brain or spinal cord) in
patients with nonvalvular atrial fibrillation. Rivaroxaban is sold as XARELTO®
. Rivaroxaban
is also approved to reduce the risk of blood clots after total hip or knee replacement surgery, for treatment of Pulmonary Emboli (blood clots in the lungs) and Deep Vein Thrombosis (blood clots in the veins of legs), and is being studied in several other medical conditions. Vitamin K Antagonists are an older class of blood thinner drug used to reduce the risk of blood
clots in patients with atrial fibrillation and in patients undergoing a PCI. Vitamin K Antagonist
drugs include drugs such as warfarin, fluindione, phenprocoumon and acenocoumarol. If you are
assigned to take a Vitamin K Antagonist, you can ask the study doctor the name of the drug you
will receive; in the UK you are most likely to receive warfarin 2. Why have I been invited? You are being invited to take part in this study because you have been diagnosed with atrial
fibrillation and you now also have the need to have a stent placed in your heart artery. You
have also met the other requirements for becoming a participant in this study. Should you
wish, your study doctor can describe these other requirements to you. For your medical condition, your doctor would usually give you some type of drug that will

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 4 of 23
help to prevent blood clots, such as a Vitamin K Antagonist (e.g. warfarin) or rivaroxaban (or dabigatran) . A Vitamin K Antagonist with a combination of two antiplatelet medicines is
considered the current „standard of care‟ for this condition, but exact practice varies between
hospitals and are also tailored to suit each individual patient‟s specific needs. In this study you will receive either a Vitamin K Antagonist (e.g. warfarin) or rivaroxaban,
along with other antiplatelet medicines (aspirin and/or clopidogrel, or similar). Your doctor
will be able to discuss with you what the current standard of care treatment option is at your
centre and how this might differ from the treatment you will receive on this study. 3. Do I have to take part? No. It is up to you to decide whether or not to take part. If you do, you will be given this
information sheet to keep and be asked to sign a consent form. You are still free to withdraw
at any time and without giving a reason. A descision to withdraw at any time, or a decision
not to take part, will not affect the current or potential future standard of care you will receive
from your doctor. It is anticipated that about 2100 subjects will take part in this worldwide study. Within the UK it expected that 123 subjects will participate. While you take part in this study, you may not take part in any other medical research
studies. 4. What will happen to me if I take part?
You will be in the study for about 12 months. During the study, you will visit your study doctor
approximately 9 times and you may have telephone call(s) from staff at your study site.
There are certain study tests, procedures and clinic visits you will be required to do, to
complete the study. Each study timepoint is listed below along with each of the things you
will need to do at each study timepoint or clinic visit. The study visits are also shown on the
Study Schedule of Procedures Table below. The Screening Phase Screening visit (may happen up to 10 days before, or soon after, stent placement) After reading this consent form and discussing it with your doctor(s) and if you agree to take part in the study, the study procedures listed below will be performed. Your study doctor will ask you questions, answer your questions and do tests to see if you are suitable for this study. You will have the following screening tests:
• After all your questions have been answered to your satisfaction, you will be asked to
sign this informed consent. You will be given a signed copy of this consent form to
keep for your records. • The study requirements, to make sure you qualify or are eligible for the study, will be
reviewed by your study doctor.

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 5 of 23
• Your age, gender, race etc will be recorded.
You will be asked questions about your medical history, including previous
electrocardiograms (ECG) showing you have atrial fibrillation, any other treatments you may have received to treat your atrial firbrillation, and all drugs that you are
taking now and took within 30 days before you started the study. You may also have another ECG, whereby sticky patches will be placed on your chest. These patches
are connected to a machine that shows the electrical activity of your heart. • You may undergo Ejection Fraction testing, to measure your heart‟s capacity to pump
blood, if this has not already been done within the last 6 months. This testing will only be performed if you have a history of heart failure or have just had a current episode of heart failure.
• You weight, height and waist circumference will be measured. • A needle will be used to draw about 2 teaspoons (10 mLs) of blood from a vein in
your arm for laboratory tests related to the study. • A urine pregnancy test will be done, only for women who can become pregnant.
Pregnant women may not take part in this research study. Pre-randomisation (after stent placement) These procedures will be performed after your doctor completes the stent procedure.
• About 2 teaspoonfuls (10 mLs) of blood will be drawn again from a vein in your arm
for laboratory tests. • Your blood clotting status will also be monitored after the stent procedure. You will
have a blood test, called an INR, to check on your blood clotting status. This test will be performed to ensure if it is safe for you to start the study medication.
• After the study doctor reviews the results of all information obtained and tests done and determines that you are eligible for this study, you will start to receive study medication. If you were taking other drugs before the study begins, the study doctor or study staff will tell you when to stop taking those drugs and when to start taking the assigned study drug. The study staff will also tell you how and when to take the study drug and when you will need to come to the study site for any blood tests and other assessments to see how you are doing.
The Treatment Phase What treatment will I receive? You will either receive rivaroxaban or a Vitamin K Antagonist. You will be put into a treatment group randomly or by chance (like flipping a coin). There are 3 treatment groups in this study: • the rivaroxaban 15 mg group (taken with one antiplatelet medicine) • the rivaroxaban 2.5 mg group (taken with two antiplatelet medicines) • the Vitamin K Antagonist group (taken with two antiplatelet medicines) Not everyone in the study will get rivaroxaban. The chance that you will be given rivaroxaban
is 2 out of every 3 patients, or approx. 66 percent. In this study patients are being treated for two conditions, atrial fibrillation and Percutaneous
Coronary Intervention with stent placement. This combination of conditions is difficult to treat

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 6 of 23
because patients have a dual risk of bleeding and stroke. This requires careful selection of
anticoagulant regimens by your study doctor to ensure both risks are minimized. Therefore,
in this study we are studying two different doses of rivaroxaban (15 mg/10 mg or 2.5 mg) in combination with one or two antiplatelet drugs to explore which regimen will be safest to treat
both conditions. These two regimens of rivaroxaban are being compared with the third arm
(VKA and two antiplatelet drugs), which is a current standard of treatment in patients with
both conditions. While the study is ongoing, you and your study doctor will openly know if you are receiving rivaroxaban or a Vitamin K Antagonist (e.g. warfarin). The antiplatelet medicines that will be given alongside rivaroxaban and the Vitamin K
Antagonist also work to prevent clot formation. Platelets are the key components of your
blood involved in clot formation; when a blood vessel is damaged platelets rush to that area
to form a clump. This clumping can narrow the blood vessels and lead to the formation of a
blockage (a clot). Antiplatelet therapies stop the platelets from clumping together. What happens to the medication that I’m currently taking for my atrial fibrillation? You may currently be on a Vitamin K Antagonist treatment and if you choose to participate in this study you may be either switched to rivaroxaban, or remain on your current treatment, depending on which treatment group you are allocated to. If your participation in the study requires you to be switched from a Vitamin K Antagonist to rivaroxaban, your study doctor will carefully watch you to be sure the treatment switch happens as safely as possible. In addition, if you are not already on antiplatelet medications these will be added alongside your anticoagulant medication. You will receive one or two antiplatelet medicines, depending on which treatment group you are in. If you are already receiving antiplatelet medication this may need to be switched to an alternative type, again depending on which treatment group you are in. Your study doctor will clearly discuss all of these treatment changes with you and guide you through the process. DAY 1 VISIT - the day you begin taking study drug (which may be the same day your stent procedure is completed)
• Blood clotting test (INR) may be performed to ensure if it is safe for you to start the
study medication. • You will take your first dose of study medication with other antiplatelet medications
(which also helps to prevent clots), such as clopidogrel, or prasugrel, or ticagrelor, and aspirin.
• The study staff will examine you and ask if you are having any specific study related
side effects. This visit will take about 1 hour to complete. Your study team will be able to advise you of specific timelines when scheduling your visits with you. Study Visits Day 10, Day 30, Month 3, Month 6 and Month 9
• The study staff will examine you for and ask specific questions about study related
side effects.
• The study staff may review certain medications you are taking.

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 7 of 23
• You will be given study medication. • You need to return any unused study drug and all packaging so that the study staff
can see how many pills you actually took.
• At the day 30 visit, approximately 1 –2 teaspoonfuls (5 - 10 mL) of blood will be
drawn from a vein in your arm for laboratory tests related to the study. • If you are randomized to take a Vitamin K Antagonist, you will have INR tests (blood
clotting test) done. This blood test will occur at different times during the study as the study doctor feels that they are needed. The study doctor may adjust the dose of Vitamin K Antagonist that you receive based on these INR blood results.
These visits will take 30 minutes to 1 hour to complete. Your study team will be able to advise you of specific timelines when scheduling your visits with you. Month 12 or End of Study Treatment/Early Withdrawal (if you withdraw early from the study)
• The study staff will examine you for and ask specific questions about study related
side effects.
• You need to return any unused study drug and all packaging so that the study staff
can see how many pills you actually took. • The staff may review certain medications you are taking. • You will stop taking the study drug and you and the study doctor will discuss what
drugs you will take after the study. • A urine pregnancy test will be done only for women who can become pregnant.
Pregnant women may not take part in this research study.
• If you were randomized to take a Vitamin K Antagonist, you will have an INR test
(blood clotting test) done. • If you withdraw early, study staff will contact you at Month 12 (starting from your
study day 1) to monitor your health status and to find out how you are doing. If they are unable to contact you, they may ask your GP about your health status and to find out if you had any heart related problems after you stopped taking the study medicine.
This visit will take about 1 hour to complete. Your study team will be able to advise you of specific timelines when scheduling your visits with you. In addition to the visits and tests noted above, the study doctor will ask you to have other
tests as part of the “usual care” you would receive if you were not in the study and were
having a stent placement. At every visit your study doctor (and staff) will ask questions and review your medications with you. He or she will also ask about any side-effects you may have. Side-effects are any unexpected, unwanted or sometimes unpleasant reactions that may result from taking a drug or having a procedure. The table below shows what is done at each visit:
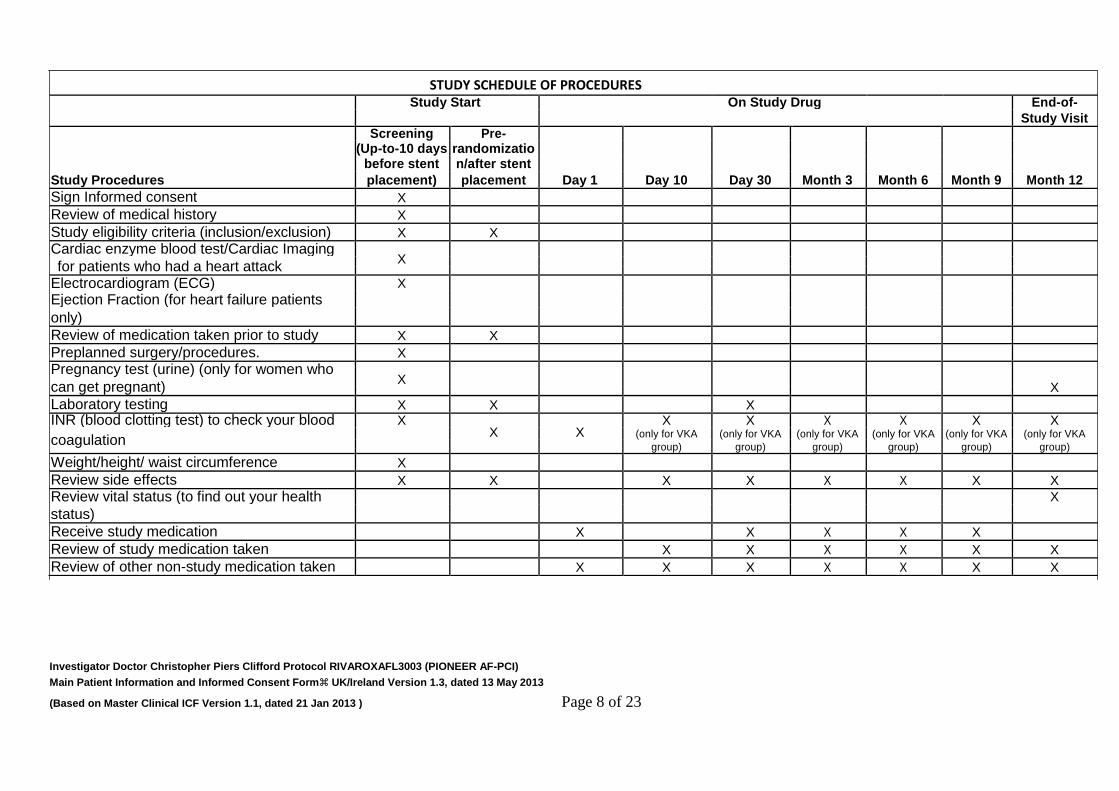
Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 8 of 23
Study Start On Study Drug End-of-
Study Visit
Screening Pre-
(Up-to-10 days randomizatio
Study Procedures before stent n/after stent
placement) placement Day 1 Day 10 Day 30 Month 3 Month 6 Month 9 Month 12
Sign Informed consent X
Review of medical history X
Study eligibility criteria (inclusion/exclusion) X X
Cardiac enzyme blood test/Cardiac Imaging X
for patients who had a heart attack
Electrocardiogram (ECG) X
Ejection Fraction (for heart failure patients
only)
Review of medication taken prior to study X X
Preplanned surgery/procedures. X
Pregnancy test (urine) (only for women who X
X
can get pregnant)
Laboratory testing X X X
INR (blood clotting test) to check your blood X X X
X X X X X X
coagulation (only for VKA (only for VKA (only for VKA (only for VKA (only for VKA (only for VKA
group) group) group) group) group) group)
Weight/height/ waist circumference X
Review side effects X X X X X X X X
Review vital status (to find out your health X
status)
Receive study medication X X X X X
Review of study medication taken X X X X X X
Review of other non-study medication taken X X X X X X X
STUDY SCHEDULE OF PROCEDURES

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 9 of 23
5. Expenses and payments You will not be paid for taking part in this study. Your visits to the study doctor and the study-related medical tests that he/she will be
arranging for you will not cost you any money. The sponsor will not pay for visits to the
doctor, or other treatments or tests that are not part of this study. The sponsor will not pay for the stent procedure or tests related to that procedure, that would be done as part of „routine‟ care. The sponsor will pay for reasonable expenses incurred for study visits. These expenses include: • Your local travel/parking expenses to and from study visits. • Meals that you eat during study visits. You will need to keep receipts for these expenses. All receipts should be submitted to the
study nurse. 6. What will I have to do? While you are in the study you must: • Give correct and accurate information about your demographics (e.g. age, gender,
ethnicity), medical history and current medical condition • Tell the study doctor about any health problems you have during the study • Tell the study doctor about any new medicine or drug you want to take or that you must take
during the study (including vitamins and herbs or herbal supplements). • Do not take any other drugs or remedies unless the study doctor has approved them before
hand. This includes prescription and over-the-counter drugs (including vitamins and herbs or
herbal supplements). • Always take the study drug as instructed • Do not give your study drug to anyone else • You should not stop study medication without notifying your study doctor, because this can
increase your risk of having a stroke (a rapid loss of brain function due to a disturbance in
the blood supply to the brain) or a blood clot elsewhere. • Bring all study drug and all empty packages to your study doctor at each visit • Keep study drug out of the reach of children • It is very important that you come to all study appointments • Do not participate in other medical research studies while you are enrolled in this study • If relevant, do not get pregnant or cause your partner to become pregnant 7. How do I take the study medication? If you decide to take part in this study, you agree to take the study drug in the way you have been asked to take it by the study staff or doctor.

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 10 of 23
Randomized (Assigned by Chance) to take rivaroxaban 15 mg: The study doctor will take a blood test called an INR to see the status of your blood‟s ability to
clot. Once the INR is within a certain range, the study doctor will tell you when to start taking
rivaroxaban. Your study doctor may also prescribe you antiplatelet medication (clopidogrel or
prasugrel or ticagrelor) prior to starting your study drug. You will be asked to stop taking
aspirin. You will start to take one 15 mg rivaroxaban tablet by mouth, every day, with food (preferably
with your evening meal), at approximately the same time each day. Depending on your kidney function, you may be given a lower dose of rivaroxaban to take (10mg); this will be determined by your study doctor based on the results of blood tests that review your liver function. The study doctor will instruct you on the time of day you should take your study medication. You will take your first dose of rivaroxaban the day you receive the study drug or as determined by your INR value. You will take the rivaroxaban along with antiplatelet therapy (clopidogrel or
prasugrel or ticagrelor) for 12 months. Your study doctor may decide to stop antiplatelet therapy during this 12 month period and continue only with rivaroxaban 15 mg (10 mg rivaroxaban if you have lower kidney function). If you miss a dose of rivaroxaban you should take the dose as soon as possible on the same day, and then continue the next day with your usual dose of one 15mg (or 10mg) tablet a day. Randomized (Assigned by Chance) to take rivaroxaban 2.5 mg: The study doctor will take a blood test called an INR to see the status of your blood‟s ability to
clot. Once the INR is within a certain range, the study doctor will tell you when to start taking
rivaroxaban. You will start to take rivaroxaban 2.5 mg tablets, one tablet by mouth twice a day. You should
take one tablet in the morning and one tablet in the evening (about 12 hours apart), around the
same time each day. The study doctor will instruct you on the time of day you should take your
study medication. You will take your first dose of rivaroxaban the day you receive the study
drug or as determined by your INR value. This 2.5 mg rivaroxaban tablet may be taken with or
without food. If you miss a dose of rivaroxaban you should take the dose as soon as possible on the same
day. Do not double the next dose to make up for a missed dose. The next day after the missed
dose, you should continue with the usual dose of one 2.5 mg tablet twice a day as before. In addition to the twice a day rivaroxaban, you will take a low dose aspirin tablet and another antiplatelet tablet (clopidogrel or prasugrel or ticagrelor) every day. You will take rivaroxaban
twice a day plus the aspirin and antiplatelet tablet for 1, 6 or 12 months. The length of time you take these medications will be determined by your study doctor at the beginning of the study
based on what is best for your medical care. If at any time during this 1, 6 or 12 month
treatment period the study doctor determines that you must discontinue aspirin or other antiplatelet treatment, you will be prescribed with a higher dose of rivaroxaban (15 mg, or 10
mg if you have a lower kidney function). If you are pre -specified for 12 month treatment, then you will receive all 3 of these medications for the complete 12 months. However, if you are pre-specified for 1 or 6 months of rivaroxaban 2.5mg treatment, you will stop taking the antiplatelet tablet (clopidogrel or prasugrel or ticagrelor) at that pre-specified timepoint. You will continue to take one low-dose aspirin tablet

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 11 of 23
for the rest of the 12-months of the study. You will also start to take a different dose of rivaroxaban (either 15 mg or 10 mg tablet) only once a day by mouth. The study doctor will determine which dose of rivaroxaban you should take (15 mg or 10 mg) by reviewing the results of blood tests that measure your kidney function. You should take the first tablet of the new rivaroxaban dose about 12 hours after you took the last tablet of 2.5 mg rivaroxaban. It is important that you take the new rivaroxaban tablet once a day with food (ie, a meal) at the same time. Randomized (Assigned by Chance) to take Vitamin K Antagonist: If you were taking a Vitamin K Antagonist before the study and are randomized to take a
Vitamin K Antagonist during the study, you will keep taking this medication as directed by the
study doctor (see below). In the UK you are most likely to receive warfarin. You will have
periodic blood tests, called an INR, to check the status of your blood‟s ability to clot. The study
doctor may need to adjust the dose of the Vitamin K Antagonist that you are taking, based on
your INR value. The recommended INR range is 2 to 3. You will take the Vitamin K Antagonist as directed by the study doctor along with a low dose
aspirin tablet and another antiplatelet tablet (clopidogrel or prasugrel or ticagrelor) every day
for 1, 6 or 12 months. The length of time you take these medications will be determined by your
study doctor at the beginning of the study based on what is best for your medical care. If at any
time during this 1, 6 or 12 month treatment period the study doctor determines that you must
discontinue aspirin or other antiplatelet treatment, you will continue with your Vitamin K
Antagonist as directed by your study doctor. If you are pre -specified for 12 month treatment, then you will receive all 3 of these medications
for the complete 12 months. However, if you are pre-specified for 1 or 6 months Vitamin K
Antagonist treatment, you will stop taking the antiplatelet tablet (clopidogrel or prasugrel or
ticagrelor) at that pre-specified timepoint and will continue to take one low -dose aspirin tablet
for the rest of the 12-months of the study along with the Vitamin K Antagonist. Your study doctor will give you a prescription for a Vitamin K Antagonist to be filled at a pharmacy as part of usual care. You should take the study drug as directed by the study staff and your doctor. If you have any questions about taking the study drug, you should call the study site. 8. What are the alternatives for diagnosis or treatment? If you choose to not take part in this study, your doctor may treat you with one of several
standard therapies following stent placement which may include aspirin, or aspirin plus an additional anti-platelet medicine. Along with this, your doctor might also decide to treat you with
an approved blood thinner such as a Vitamin K Antagonist. The risks and benefits of these
other treatments have been explained to you. Your doctor will answer any questions you have about these other treatments. If after consideration of these potential benefits and risks you do
not wish to participate in this study, you will receive usual standard of care treatment. 9. What are the side effects of any treatment received when taking part?

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 12 of 23
All medicines can cause unwanted effects, called side effects. General Risks of Rivaroxaban In previous studies of healthy subjects, the risk of minor side effects with rivaroxaban was
similar to placebo (inactive drug) . During early development for the approved uses, 16,236
patients were exposed to rivaroxaban and the safety of rivaroxaban has been evaluated. Liver enzyme abnormalities (indicating possible liver irritation) were the same or lower in patients treated with rivaroxaban compared to patients receiving a comparator drug or placebo (inactive drug). In a study of heart attack patients the frequency of increase of liver enzymes (ALT) in patients receiving 5 to 20 mg of rivaroxaban was 2.3%; these increases were comparable to an inactive medication (2.9%) thus showing a similar safety profile. In large studies of patients who had orthopedic (knee or hip) surgery, increased liver enzymes were seen in 2.4% of patients receiving rivaroxaban for up to 5 weeks and 3.7% of patients receiving a comparator treatment. In some studies there have been rare cases of patients who developed severe liver impairment that sometimes contributed to their death. These include patients who were receiving either rivaroxaban, medications other than rivaroxaban (active control medication), or placebo. In those cases where patients received rivaroxaban, other medical conditions were considered the likely causes of the liver impairment. If your liver enzyme levels increase, we will do more tests to learn if the abnormal levels are related to rivaroxaban or to another cause. Your study doctors will follow you closely until the tests are back to normal or return to the values before you started the study. During studies of rivaroxaban involving patients undergoing hip or knee replacement surgery, a
temporary rise in an enzyme in the pancreas called lipase (which breaks down fats that you
eat) occurred in less than 1% of the subjects receiving rivaroxaban and these levels returned to
normal even while rivaroxaban was continued. As rivaroxaban is still being tested for other uses, you must understand there may be side
effects that are not yet known and you may have other side effects that have not been reported
before. So, you must notify your study doctor or one of the members of the study staff of any
new symptoms that you may have. Bleeding Risks of Rivaroxaban Rivaroxaban is a blood thinner, and may make it more difficult for you to form blood clots that
prevent you from bleeding too much. Therefore, there is a risk of bleeding from anywhere in
your body either without injury or due to minor injury. This may include bleeding that is seen (such as bruising (black and blue marks)) or unseen (such as a low blood cell count a condition
of too little iron in the blood). Bleeding complications may show up as weakness, paleness (whitish skin color), dizziness, headache, chest pain or angina, shortness of breath, low blood
pressure, or unexplained swelling. In the study of heart attack patients where rivaroxaban was administered in the range of doses
from 5 to 20 mg daily for up to 6 months the most common types of bleeding events were: gum
bleeds (5.1%), nose bleedings (8.6%), and bruising (hematoma) (3.0%). Gastrointestinal
(1.0%) and rectal (0.8%) bleeding, melena (blood in stool) ( 0.4%), blood in urine (1.7%) and
bleeding in the surface of the outer eye membrane (conjunctiva) (0.6%) were less common. Cases of intracranial bleeding (brain bleeding) (especially in patients with high blood pressure,
the elderly, and those taking other blood thinning agents) and critical organ bleeding have been

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 13 of 23
reported. Some of these cases resulted in death. Cases of bleeding in the adrenal gland and
eye have occurred. Fatal bleeding, while rare, may occur while taking rivaroxaban. The risks of taking rivaroxaban are balanced by the benefit of preventing strokes in patients with atrial fibrillation. There is no specific antidote for reversing the effects of rivaroxaban. Should a significant
bleeding event occur there are a number of routine measures that can be used by your study
doctor to try and control the bleeding; temporary interruption of rivaroxaban may be sufficient
as the drug is rapidly cleared from your body once treatment is stopped. Risks of Taking Rivaroxaban with Other Drugs You may be taking aspirin and another similar drug (such as clopidogrel, prasugrel or ticagrelor) to block platelets from clumping together. Platelets are the part of the blood important for clotting. Aspirin and clopidogrel can also thin the blood and make it more difficult to form blood clots, just as rivaroxaban can. In the study of heart attack patients where rivaroxaban (from 5 to 20 mg daily) and aspirin, with or without such drug as clopidogrel, were
also administered to decrease the occurrence of a heart attack or stroke, the bleeding rate was higher in those who received all three medications (aspirin, clopidogrel and rivaroxaban). However, the majority (over 80%) of these bleeding events were mild bleedings such as gingival (gum) or nose bleeds, or bruising. If you are also taking a combination of these medications, this may increase your risk of bleeding. Within this study the doses of the medications within the rivaroxaban treatment arms have
been selected to provide the best possible protection from stroke, whilst keeping the bleeding
risk as low as possible. Caution should be used when such medications as naproxen, a drug often used to decrease
pain, temperature, or to treat arthritis (pain in joints accompanied by swelling and stiffness) are
administered with rivaroxaban, since these medications can also affect ability of blood to form
clots. Another medication in this group is ibuprofen; use of ibuprofen should be avoided. You
should also not take drugs which can affect the ability of blood clots to form. Since the liver breaks down rivaroxaban, certain medications that are also broken down the
same way can sometimes cause rivaroxaban blood levels to rise or fall depending on the other drug taken. Certain medications, such as ketoconazole (a drug used to treat fungal infection)
and many anti -HIV medications can cause rivaroxaban levels to rise. Other medications, such
as rifampicin (an antibiotic drug), phenytoin, Phenobarbital (both used to treat seizures), or St. John‟s wort (a vitamin), can cause rivaroxaban levels to fall. Please tell your study doctor about
any additional medications including over the counter medicines or herbal supplements you may be taking to make sure that they do not interfere with rivaroxaban levels. There may be risks with the use of rivaroxaban that are not yet known.
In summary, you may have a higher risk of bleeding if you take rivaroxaban and take other medications that increase your risk of bleeding, including:
• aspirin or aspirin containing products
• Non-steroidal anti-inflamatory drugs (NSAIDs; e.g. ibuprofen and naproxen)
• warfarin sodium

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 14 of 23
• Any medicine that contain heparin
• Clopidogrel
• Other medicines to prevent or treat blood clots Tell your doctor if you take any of these medicines. Ask your doctor or pharmacist if you are not sure if your medicine is one listed above. General Risks of Vitamin K Antagonists
Other than the bleeding risks of Vitamin K Antagonists, other side effects are usually low, but they may include necrosis (cell death) of skin or other tissues, allergic reactions (rash, hives, itching, difficulty breathing, tightness in the chest, swelling of the mouth, face, lips, or tongue), jaundice (yellowing of skin, eyes, mucous membranes), increases in liver function blood tests, osteoporosis, fever, oedema (swelling), chest pain, abdominal pain and anaemia (low red blood cell count). Other infrequent side effects may include: cholesterol deposits in the feet that
may cause “purple toe syndrome” (a condition where small deposits of cholesterol (fat) break loose and flow into the blood vessels in the skin of the feet, which causes a blue purple color and may be painful), fatigue, weakness, nausea, vomiting, diarrhea, hair loss, and cold intolerance. Bleeding Risks of Vitamin K Antagonists A Vitamin K Antagonist is a blood thinner, which means it makes your blood less likely to form a clot. This may lead to excess bleeding, especially after an injury or surgery. Major bleeding or even fatal bleeding may occur, which is why your blood will be monitored frequently using a test called the Prothrombin Time which is reported as a number called the INR. The higher the INR, the higher is your risk of bleeding. Because of the bleeding risks you should not take Vitamin K Antagonists if you have had recent bleeding episodes, such as in your gastrointestinal system or inside your head. You should not take medications which can increase your risk of stomach bleeding, such as aspirin (additional aspirin should not be taken on top of the low dose aspirin already included in this study) and Nonsteroidal anti-inflammatory drugs [naproxen or ibuprofen]. Your study doctors will ask you questions about risk factors for severe bleeding while taking Vitamin K Antagonists. In this study the treatment combination within the Vitamin K Antagonist arm is considered the
„current standard of care‟ for treating this condition and the medications are selected to provide
the best possible protection from stroke, whilst keeping the bleeding risk as low as possible. As many medications may interact with Vitamin K Antagonists, you should check with the study
doctors before taking any other medication, whether it is prescribed or over-the-counter,
vitamins or herbal supplements. The more common bleeding risks of taking Vitamin K Antagonists include: nosebleeds, bruising in the skin, blood in the urine, heavy menstrual bleeding, and gastrointestinal bleeding
(vomiting blood and/or blood in the stool). The risks of taking Vitamin K Antagonists are
balanced by the benefit of preventing strokes in patients with atrial fibrillation. To minimize any risk of bleeding your study doctors will check your Prothrombin Time as frequently as needed
to keep the INR in the appropriate range. They will instruct you to adjust your dose of Vitamin K Antagonist according to the INR results. Risks of Stopping Study Drug

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 15 of 23
You should not stop study medication without notifying your study doctor. At whatever point
you stop the study and/or stop taking the study medication, the study doctor will ensure that
you are continued on blood thinner(s). Your study doctor may have you return for a few extra
visits to adjust your treatment after you stop study medication. If you stop the study medication and do not immediately see the study doctor or your regular
doctor to continue appropriate medical treatment, you may be at a much higher risk for
developing strokes or blood clots in your arteries from your atrial fibrilliation. In addition, you
may also be at higher risk for developing clots in your hearts arteries, including in the area where the stent was initially placed.
Side effects from Blood Tests Risks from having blood tests may include a small amount of pain where the needle enters the
vein. In addition, a temporary bruise or “black and blue mark” may develop. Very rarely, the
vein may become inflamed or infected. Fainting, and in rare cases infection, may occur.
Other During the study your condition may remain the same or get worse. Before participating you should consider if this will affect any insurance you have and seek advice if necessary.
10. What about Birth Control and Pregnancy During the Study? The risks of the study drug to the human embryo, foetus, or breastfed infant are unknown. If
you are a woman who can become pregnant, you will have a pregnancy test at your first study
visit. This pregnancy test must be negative for you to be in the study. If you are a woman:
• You must be postmenopausal (for at least 18 months), or surgically sterile (not able to
have children), or not heterosexually active. • Taking part in the study might harm your unborn child or breastfed baby. • You cannot take part in this study if you are pregnant or breastfeeding a child. • You must agree not to become pregnant while you are in this study. • If you are sexually active and able to get pregnant you must use birth control during the
study. Birth control methods that can be used while in this study include prescription oral
contraceptives, contraceptive injections, intrauterine device, double-barrier method,
contraceptive patch, and male partner sterilization. The type of birth control you use must
be discussed with the study doctor before you begin the study. The study doctor must
approve the method you use before you can enter the study. • If you get pregnant during the study, you must tell the study doctor immediately. You will
have to stop taking the study drug. The study doctor will advise you about your medical
care and will ask you to allow him/her to collect information about your pregnancy and the
health of your baby.

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 16 of 23
11. What are the possible benefits of taking part? Rivaroxaban is believed to thin your blood to prevent blood clots from forming in the arteries of
your heart that cause heart attacks. When used with other standard medicines like aspirin and
clopidogrel it may reduce the chance that a heart attack will reoccur or reduce the risk of death
or stroke. There is no guarantee that you will personally benefit, however knowledge gained
from your participation may help others. 12. What happens when the research study stops? After the study is over, the sponsor will not continue to provide you with rivaroxaban study
drug. If you were assigned to receive rivaroxaban within the study you will be switched to the
current standard of care available at your hospital. Your doctor will work with you to ensure that
your risk of stroke, or blood clots elsewhere, is adequately managed as a part of your usual
care. If you were assigned to the Vitamin K Antagonist treatment group you may remain on your
current treatment as this is available as standard of care medication. Your study doctor will
advise you of any treatment changes that are required as part of your usual care. Your study doctor will discuss your future medical care options with you.
13. What if there is a problem? If you become ill or have side effects or injuries because of the study, talk to your doctor.
Your doctor will make sure that you get medical care and advice during and after the study
and will notify the sponsor of any potential compensation claims. The above statement does
not limit your legal rights. 14. Will my taking part in the study be kept confidential? Yes. We will follow ethical and legal practice and all information about you will be handled in confidence. The details are included in Part 2. This completes Part 1. If the information in Part 1 has interested you and you are
considering participation, please read the additional information in Part 2 before
making any decision.
Patient Information Sheet Part 2
15. What if relevant new information becomes available? There may be risks with the use of Rivaroxaban that are not yet known. Sometimes, during the
course of a study, your study doctor may learn new information about the treatment or study

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 17 of 23
drug that might change whether or not you want to continue in the study. If this happens, your
doctor will tell you about it in a timely manner. If you decide not to carry on, your study doctor will make arrangements for your continued medical care. If you decide to continue in the study you may be asked to sign an updated consent form. If the study is stopped for any reason, you will be told why and your continuing care will be arranged. 16. What will happen if I don’t want to carry on with the study? Your taking part in this study is completely voluntary. You do not have to be in this research
study. You can agree to be in the study now and change your mind later. You may stop your
participation at any time. Your decision will not affect your regular or usual care. It will not affect
your getting all the care, medicine, and equipment you should be getting. Your study doctor also has the right to take you out of the study at any time with or without your agreement. The sponsor has the right to direct your study doctor to take you out of the study at
any time with or without your agreement. These decisions will be made if: • It is in your best medical interest to stop your participation • You do not follow instructions • The study is cancelled Agreement to Follow-up After Stopping Study Drug/Early Withdrawal It is important that you remain in regular contact with your study doctor even if you no longer
want to be in the study so that he/she can monitor your health status. If you stop the study early, the study staff would like permission to contact you to ask how you
are doing. This contact will happen at around the time you would have completed the study
(about 12 months after your first study dose). If the study staff is unable to contact you, they
will ask your GPhow you are doing or for permission to review your medical records. As with all
information collected about you as part of the study, this information will remain confidential.
Please refer to the section 18 „Will my taking part in this study be kept confidential?‟ When you sign the consent form you will be asked if you give permission for the study staff
to contact you, to contact yourGP , or to review your medical records, at the end of the
study. You may refuse any of these contacts and still participate in the study. If you decide to leave the study at any time, the information collected about you up to that point
will have been processed and you must agree not to limit the use of your study data. However
this information will have been anonymised so nothing will identify you. You will continue to
receive the appropriate care for your condition. 17. What if there is a problem? In the event of serious injury shown to be caused by a drug being tested or administered as
part of the trial protocol or any test or procedure you received as part of the trial,
compensation may be available. Before participating you should consider if this will affect any
insurance you have and seek advice if necessary. Compensation for injuries that are serious and beyond those discussed with you or identified in writing before you agreed to take part in the study and are caused or probably were caused by

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 18 of 23
taking part in this study, will be considered in accordance with the „Clinical Trial Compensation Guidelines‟ issued by the Association of the British Pharmaceutical Industry (ABPI). A copy of these guidelines is available on request. Broadly speaking the ABPI guidelines recommend that the Sponsor should compensate you without you having to prove that it is their fault or go to court. This applies in cases where it is likely that such injury results from giving the study drug or any extra procedure carried out in accordance with the protocol for the study. The Sponsor will not compensate you where such injury results from any procedure carried out which is not in accordance with the protocol for the study or if the persons administering the Study have been negligent. Your right at law to claim compensation for injury where you can prove negligence is not affected. The Sponsor will not be bound by these guidelines to pay compensation if the injury resulted from a drug or procedure outside the trial protocol. If you become ill or suffer side effects or injuries because of the study, contact your doctor.
Your doctor will make sure that you get medical care and advice during and after the study and
will notify the sponsor of any potential compensation claims. You can contact the ABPI via their website ( www.abpi.org.uk), by telephone on 0870 890 4333, or by writing to: ABPI Head Office, 7th floor, Southside, 105 Victoria Street, London, SW1E 6QT. The above statements do not limit your legal rights.
18. Will my taking part in this study be kept confidential? The Sponsor will use the information collected about you for the purposes of the study and for
scientific research, such as study of your disease. The Sponsor may also use this information to
apply for permission to sell the drug in some countries. The information will be stored both on
paper and on computer, without identifying you by name. To protect your privacy, the information
will be labelled with a code number. If the results of the study are published, your identity will be
kept confidential. By signing this form, you are permitting this use of your information. Your study doctor will keep your personal medical records and a list that links your name to your code number for at least 15 years. Regulatory authorities, NHS R&D Office representatives, employees at the study site and
representatives of the Sponsor will be able to access this list in order to compare and check
the study information collected about you with information in your medical records. As far as
the law allows, your medical records will not be made public. By signing this form, you are
allowing direct access to your medical records by those who have legitimate reason to look at
them. The information collected may be sent to other members of the Sponsor‟s group of companies, to
contractors working for them and to regulatory authorities. None of this information will contain your
name. It may also be sent to some countries outside Europe that may not have the same levelof
data privacy protection as Europe. Data sent between Europe & the United States of America
share the same level of data privacy protection. The Sponsor will protect your privacy as far as
the law allows and will keep and supervise the information collected about you only for as long
as needed. You can arrange with your study doctor to see the information collected about you, and you

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 19 of 23
can ask for any mistakes to be corrected. If you decide to leave the study at any time, the
Sponsor may still use your information collected up to that point, as the law allows. 19. Involvement of the General Practitioner (GP) We would like your permission to contact the doctors or medical staff you see regularly to let
them know that you are taking part in this study. It is important for all of your doctors to know
that you may be taking an experimental drug. While you are in the study, the study doctor will
ask about your symptoms. If you have symptoms after the study ends or if you stop the study
early your other doctors may want to contact the study staff. 20. What will happen to any samples I give? Samples are any fluid (e.g., blood) collected from you in this study. The sponsor will use the samples collected from you for the purposes of the study. To protect your privacy, your samples will be labeled with a code number. The scientists doing the research will not know your identity. Your samples may be sent to other members of the Johnson & Johnson group of companies, to contractors working for them and to regulatory authorities. Your samples may also be shared with research partners for scientific research purposes.
Before sharing with research partners, your samples will be labeled with a code number that is
different from your study code number. Your samples will not contain any personal identifiers.
Your samples will not be sold, loaned or given to any other independent groups for their own
use. Research partners working with the sponsor are not allowed to share samples with
anyone who is not authorized by the sponsor. The sponsor will control what is done with your
samples. You will not be paid for any use of your samples, results, or inventions made from research on
them. You are providing your samples, for use by the sponsor. The sponsor (and research
partners, where applicable) own the use of the results, treatments, or inventions that can be
made from this research. You will be informed of any analyses on blood samples that were not planned when you signed this form. You have the right to refuse further analyses of these samples. 21. What will happen to the results of the research study? The sponsor aims to publish the results of the study as soon as possible after the study has finished. If the results of the research are published, your identity will remain confidential. If reference to you is made, this will be done using code numbers. By signing this form, you are permitting this use of your information. Should you wish to discuss the results of the study, you should contact your Study Doctor.

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 20 of 23
22. Who is organising and funding the research? The Sponsor of this study will pay Buckinghamshire Healthcare NHS Trust, Cardiology Department for including you in this study. Nil conflicts of interest. 23. Who has reviewed the study?
All clinical research is looked at by an independent group of people, called a Research Ethics
Committee to protect your safety, rights, wellbeing and dignity. This study has been reviewed
and given favourable opinion by London Surrey Borders Research Ethics Committee (REC:
13/LO/0528).
24. Further information and contact details If you have any questions about the study, please contact: Nicola Bowers, 07956645035, Cardiac Research Sister If you feel that this study has caused you harm, please contact: Nicola Bowers, 07956645035, Cardiac Research Sister If you have any questions about your rights as a research subject, please contact: Nicola Bowers, 07956645035, Cardiac Research Sister

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 21 of 23
Consent Form
Study Title: An OPen-label, Randomized, Controlled, Multicenter Study ExplorIng TwO TreatmeNt
StratEgiEs of Rivaroxaban and a Dose-Adjusted Oral Vitamin K Antagonist
Treatment Strategy in Subjects With Atrial Fibrillation Who Undergo
Percutaneous Coronary Intervention - PIONEER AF-PCI
Study Number: RIVAROXAFL3003
Principal Investigator:
Doctor Christopher Piers Clifford
Patient
CRF ID:
name:
Please read the following statements and put your initials in the box to
Please initial
show that you have read and understood them and that you agree with
them. each box
1
I confirm that I have read and understand the information sheet dated
13/May/2013 for the above study. I have had the opportunity to consider
the information, ask questions and have had these answered satisfactorily.
2
I understand that my involvement is voluntary and that I am free to withdraw
at any time, without giving any reason and without my medical care or legal
rights being affected.
3
I understand that relevant sections of any of my medical notes and data
collected during the study may be looked at by responsible individuals from
the Sponsor or authorised by the Sponsor, from regulatory authorities or
from the NHS Trust, where it is relevant to my taking part in this research. I
give permission for these individuals to have access to my records.
4 I agree to my GP being informed of my participation the study.

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 22 of 23
The study site has my permission to contact me to collect information
or to review publically available records (if available and allowed by
local law) on how I am doing at what would have been the end of the
study.
The study site has my permission to contact my GP (General
Practitioner) who will review my medical records and tell the study
staff how I am doing at what would have been the end of the study.
6 In the event that I withdraw from the study early, I understand that the information collected about me up to that point will continue to be used by the sponsor. I agree to my data being used if I withdraw from the study early.
Please read the following statements and put your initials in the box to Please initial show that you have read and understood them and that you agree with
them. each box
5 I understand that if I stop the study medication and leave the study early, or
the study staff are unable to contact me, the study site (study doctor and/or
staff) would like permission to make the following contacts. PLEASE TICK
THE BOXES BELOW:

Investigator Doctor Christopher Piers Clifford Protocol RIVAROXAFL3003 (PIONEER AF-PCI) Main Patient Information and Informed Consent Form UK/Ireland Version 1.3, dated 13 May 2013 (Based on Master Clinical ICF Version 1.1, dated 21 Jan 2013 ) Page 23 of 23
To be filled in by the patient I agree to take part in the above study
Your name Date (Day/Month/Year) Signature e.g. 14/July/2006) To be filled in by the person obtaining consent (investigator) I confirm that I have explained the nature, purposes and possible effects of the research
study to the person whose name is printed above. They agreed to take part by signing and
dating above.
Name of Investigator
Date (Day/Month/Year) Signature
(or person obtaining consent if different e.g. 14/July/2006)
from Investigator)
Impartial Witness At least one impartial witness is mandatory when the patient, is unable to read or write. An
impartial witness must be present during the entire informed consent discussion. I confirm that the information in the consent form was accurately explained to, and apparently understood by, the patient, and that consent was freely given by the patient.
Name of Impartial Witness Date (Day/Month/Year) Signature e.g. 14/July/2006)
Instructions to Study Staff If the study doctor signing this form is not the Principal Investigator, they must be authorised to take
consent on the Site Signature Log. Filing instructions:
• 1 (copy) for patient • 1 (copy) for medical notes • 1 (original) to be filed in the Trial Centre File





























