Embed Size (px)
Citation preview

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
1 of 11
C A N C E R
Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3Dionysos Slaga*, Diego Ellerman*, T. Noelle Lombana*, Rajesh Vij, Ji Li, Maria Hristopoulos, Robyn Clark, Jennifer Johnston, Amy Shelton, Elaine Mai, Kapil Gadkar, Amy A. Lo, James T. Koerber, Klara Totpal, Rodney Prell, Genee Lee, Christoph Spiess, Teemu T. Junttila†
A primary barrier to the success of T cell–recruiting bispecific antibodies in the treatment of solid tumors is the lack of tumor-specific targets, resulting in on-target off-tumor adverse effects from T cell autoreactivity to target- expressing organs. To overcome this, we developed an anti-HER2/CD3 T cell–dependent bispecific (TDB) antibody that selectively targets HER2-overexpressing tumor cells with high potency, while sparing cells that express low amounts of HER2 found in normal human tissues. Selectivity is based on the avidity of two low-affinity anti-HER2 Fab arms to high target density on HER2-overexpressing cells. The increased selectivity to HER2-overexpressing cells is expected to mitigate the risk of adverse effects and increase the therapeutic index. Results included in this manuscript not only support the clinical development of anti-HER2/CD3 1Fab–immunoglobulin G TDB but also introduce a potentially widely applicable strategy for other T cell–directed therapies. The potential of this discov-ery has broad applications to further enable consideration of solid tumor targets that were previously limited by on-target, but off-tumor, autoimmunity.
INTRODUCTIONEarly clinical results using T cell–retargeting approaches for treat-ment of hematological malignancies have generated broad excite-ment. Clinical trials using T cells engineered to recognize CD19 on B cells have shown extraordinarily high (70 to 90%) response rates and durability of response in acute and chronic leukemia (1–4). Early clinical data with B cell maturation antigen (BCMA)–targeting chi-meric antigen receptor (CAR) T cell therapy in multiple myeloma cells further illustrate the transformative potential of T cell redirec-tion: 95% of the patients reached complete response (CR) or near CR status without a single event of relapse in a median follow-up of 6 months, with an encouraging safety profile (5). The first-generation CD3/CD19 bispecific T cell–redirecting antibody fragment blinatumomab has demonstrated impressive clinical potency, inducing partial and complete tumor regressions at a 0.015-mg dose (6, 7). Blinatumom-ab is indicated for the treatment of relapsed or refractory B cell pre-cursor acute lymphoblastic leukemia and pioneers the way for clinical development of full-length immunoglobulin G (IgG)–based CD3 bispe-cific antibodies with serum half-life improvement (8, 9). Although clinical proof of concept for T cell retargeting has been established in hematological malignancies and such modalities have the potential to revolutionize cancer therapy in these diseases, demonstration of clear clinical efficacy in solid tumors is still lacking. Several aspects contribute to the success of T cell retargeting in hematological malig-nancies, particularly the availability of lineage-specific targets such as CD19 and CD20 (10, 11) in B cell malignancies, as well as FcRH5 and BCMA in multiple myeloma (12, 13). These targets are homogeneous-ly expressed in cancer cells with high prevalence. Depletion of normal B and plasma cells, the only normal cells that express these targets, can be tolerated.
Redirecting T cell activity to eradicate solid tumors is substantial-ly more challenging. T cell trafficking and access to the tumor may be limited, and the number of T cells in the tumor may restrict the ther-
apeutic potential (14). Similarly, the solid tumor environment may pose structural barriers for distribution and penetration of therapeu-tic antibodies to the tumor (15, 16). A dominant immunosuppres-sive tumor microenvironment (17) may limit the effectiveness of T cell retargeting in solid tumors. Multiple immune cell types (such as regulatory T cells, myeloid-derived suppressor cells, macrophages, and fibroblasts) and tumor cells may contribute to the generation of T cell hostile environment, for example, by providing co-inhibitory signals such as programmed death ligand 1 (PD-L1) or by secreting soluble molecules [including indoleamine 2,3-dioxygenase (IDO) and trans-forming growth factor– (TGF)] that may attenuate T cell activity. Additional hurdles for solid tumor targeting are paucity of costimu-latory signals, heterogeneous target expression in tumors, and, often, suboptimal prevalence of target expression in any given tumor type.
However, the primary barrier to successful treatment of solid tumors with T cell–retargeting therapeutics is the lack of tumor-restricted antigens. This results in on-target off-tumor adverse effects caused by T cell reactivity to organs expressing the antigen. Clinical on-target toxicity has been reported widely for engineered T cell approaches targeting solid tumors. Carcinoembryonic antigen–targeting T cells induced severe inflammatory colitis (18). Melanoma antigen gp100- targeting T cells caused adverse effects in all melanocyte-containing tissues (skin, ears, and eyes), resulting in rash, hearing loss, and uve-itis (19). MAGE-A3–targeting T cell cross-reactivity to a peptide de-rived from the muscle protein titin, expressed in cardiomyocytes, and resulted in fatal toxicity against cardiac tissue (20). Epidermal growth factor receptor (EGFR)/CD3 bispecific T cell engager (BiTE) molecule induced severe toxicities in preclinical safety studies (21). Damage of EGFR-expressing tissues was detected with doses as low as 31 g/kg per day. Diarrhea detected in patients treated with epithelial cellular adhesion molecule (EpCAM)/CD3 BiTE may be accounted for by the high expression of the target in the gastrointestinal tract (22).
We have previously generated an anti-HER2/CD3 T cell–dependent bispecific (TDB) antibody that efficiently eradicates HER2-amplified tumors in preclinical models (23). Anti-HER2/CD3 TDB redirects T cells to kill HER2-expressing cells. A very low receptor occupancy is sufficient for anti-HER2/CD3–mediated T cell triggering. We have
Genentech Inc., 1 DNA Way, South San Francisco, CA 94080, USA.*These authors contributed equally to this work.†Corresponding author. Email: [email protected]
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
2 of 11
established a clear positive preclinical therapeutic index for anti- HER2/CD3 TDB based on higher potency against HER2-amplified/overexpressing cells. However, the anti-HER2/CD3 TDB can also elicit T cell responses against normal tissues that express low amounts of HER2 if administered at sufficiently high doses. HER2 is expressed in low amounts in a wide variety of vitally important normal tissues, and therefore, tolerability is a potential concern for HER2- targeting T cell therapies. HER2-targeting therapeutics with similar mechanisms of action (CD3 bispecifics and CAR T cells) tested in clinical trials have demonstrated dose-limiting toxicities at alarm-ingly low doses (24, 25).
The goal of this study was to develop an anti-HER2/CD3 TDB that would further increase the selectivity and target the HER2-amplified tumor cells with high potency, while sparing cells that express low amounts of HER2 similar to normal human tissues. Increased selec-tivity against HER2-amplified cells is ex-pected to mitigate the risk of on-target off- tumor adverse effects and increase the therapeutic index of anti-HER2/CD3 TDB.
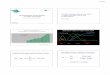
RESULTSHER2 affinity of the anti-HER2/CD3 TDB has a direct effect on killing activity, but not on selectivity against HER2-overexpressing cellsPreviously, we have shown that the anti- HER2/CD3 IgG TDB (4D5 IgG TDB) not only has robust activity in HER2- overexpressing breast cancer models in vitro and in vivo but also retains low activity against cell lines expressing low amounts of HER2 similar to normal HER2- positive human epithelial cells (23). The goal of this work was to increase the selectivity of 4D5 IgG TDB to HER2- overexpressing cells to minimize the risk of on-target off-tumor activity and there-by increase the therapeutic index of the HER2-targeting TDB. Initially, we tested whether lowering the affinity to HER2 provided selectivity of the TDB to HER2- overexpressing cells and generated mul-tiple variants of the anti-HER2 4D5 with monovalent affinities ranging from KD 0.3 to 353 nM (Fig. 1A). In the case of all variants, the main driver for the reduced affinity was an increase in dissociation rate constant (Kd), whereas the associa-tion rate constant (Ka) remained rela-tively unaltered.
To test primary selectivity, we selected SKBR3 and MCF7 as model cell lines. SKBR3 is a HER2-overexpressing breast cancer cell line, and MCF7 is a breast can-cer cell line expressing a low amount of HER2 (Fig. 1B), similar to noncancerous cells such as cultured cardiomyocytes (23). Cell membrane HER2 copy number in
SKBR3 and MCF7 has been previously reported to be 2 million and 15,000, respectively (26). Lowering the affinity of HER2 binding by previously described hu4D5 variants (27) gradually reduced the cyto-toxic activity of the anti-HER2/CD3 IgG TDB on both cell lines (Fig. 1C). Killing activity was practically abolished with TDBs having an anti-HER2 affinity of KD 49 nM or lower. A similar activity de-crease was seen in both SKBR3 and MCF7 cells, indicating that the anti-HER2 affinity plays a key role for the activity of the anti-HER2/CD3 TDB but does not provide a strategy to selectively target HER2-overexpressing cells.
Bivalent low-affinity binding to HER2 results in TDBs that are selective for HER2-overexpressing cellsWe hypothesized that the lack of binding and cell killing by the low-affinity TDBs could be rescued by bivalent HER2 engagement,
Fig. 1. HER2 affinity of the anti-HER2/CD3 IgG TDB has a direct effect on killing activity, but not selectivity for HER2-overexpressing cells. (A) Binding kinetics and affinities of anti-HER2 (hu4D5) Fab and variants. (B) HER2 West-ern blot of SKBR3 and MCF7 breast cancer lines. (C) Cytotoxic activity mediated by anti-HER2/CD3 IgG TDBs (monova-lent for HER2 and CD3) on SKBR3 (blue) and MCF7 (red) cells. Data are represented as means ± SD (n = 3).
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
3 of 11
or avidity. Because avidity-mediated binding depends on the receptor density, we speculated that it may provide a strategy to distinguish between cells that overexpress HER2 and cells with low expression of HER2. We designed TDB molecules with bivalent HER2 binding and monovalent CD3 binding by a genetic fusion of an anti-HER2 Fab to the N terminus of the hu4D5.knob heavy chain (1Fab-IgG; Fig. 2A) (28). The affinity of monovalent CD3 arm was 70.7 nM for hu-man CD3 (fig. S1A). The same CD3 arm was used in all tested 1Fab-IgG TDB and IgG TDB constructs. The molecules were tested for their cell binding (Fig. 2B and fig. S1B) and their ability to mediate apop-tosis and killing of SKBR3 and MCF7 cells (Fig. 2, C and D, and fig. S1C). 1Fab-IgG TDBs bivalent for HER2 were compared to the 4D5–wild-type (WT) IgG TDB with the trastuzumab-based HER2 arm (monovalent high-affinity HER2 binding) and identical CD3 arm. 1Fab-IgG TDBs that were made using anti-HER2 affinity variants with monovalent Fab affinity of KD 0.32 to 49 nM demonstrated similar binding to SKBR3 cells compared to the 4D5-WT IgG TDB (Fig. 2B). Thus, the bivalent 1Fab-IgG TDB format rescues binding of the 4D5-H91A variant to HER2 high-expressing cells. In contrast, HER2 Fab affinity lower than 49 nM (4D5- Y55E.H91A, KD = 172 nM; 4D5-Y100Aa, KD = 213 nM; 4D5-N30A.H91A, KD = 353 nM) was not sufficient for full re-covery of SKBR3 binding and killing in 1Fab-IgG format (fig. S1, B and C). Be-cause of the loss of activity, affinity vari-ants with very low HER2 affinity (KD > 50 nM) were not pursued further.
Next, we tested binding and killing of low HER2-expressing MCF7 cells by all 1Fab-IgG variants. Variants assembled from Fabs with monovalent affinities of KD 0.3 and 2.6 nM (4D5-Y102V and 4D5- Y55E.Y102V, respectively) resulted in similar binding to MCF7 cells compared to 4D5-WT IgG TDB (Fig. 2B). In contrast, 1Fab-IgG TDBs built from Fabs with KD values of 23 and 49 nM (4D5-D98A.F100A.Y102V and 4D5-H91A, respective-ly) demonstrated minimal binding to MCF7 cells (Fig. 2B). Considering that binding of 1Fab-IgG TDB variants, in-cluding Fabs with KD values of 23 and 49 nM, to SKBR3 was similar to 4D5-WT IgG TDB, and binding to MCF7 was sub-stantially lower compared to 4D5-WT IgG TDB, selectivity of these variants to HER2-overexpressing cells may be im-proved. Consistent with cellular bind-ing, 1Fab-IgG TDBs made from Fab variants with KD values of 23 and 49 nM were efficient in inducing T cell–mediated killing of SKBR3 cells (Fig. 2, C and D) but had no activity when MCF7 cells were targeted. However, 1Fab-IgG TDB variants with monovalent HER2 affin-ities ≤23 nM demonstrated substan-tial potency in killing of MCF7 cells, suggesting that these variants do not
sufficiently improve selectivity toward HER2-overexpressing cells (Fig. 2, C and D).
To understand the contribution of bivalent HER2 engagement, or avidity, to cell-based affinity in greater detail, we determined the apparent affinities of the 4D5-WT 1Fab-IgG TDB and 4D5-H91A 1Fab-IgG TDB molecules to SKBR3, MCF7, and cells transfected to express cynomolgus monkey (cyno) HER2 at a copy number com-parable to MCF7 (fig. S2). Despite the 164-fold difference between 4D5 and the affinity-attenuated 4D5-H91A variant as monovalent Fab (Fig. 1A), the cell-based binding affinities of the 1Fab-IgG mol-ecules to SKBR3 cells were comparable, as determined by either di-rect cell binding or competition binding assays (fig. S2A). Apparent affinity of 4D5-H91A 1Fab-IgG TDB for HER2 expressed on SKBR3 cells was in the range of 1.5 to 5 nM, which is ~10-fold higher com-pared to its monovalent Fab affinity measured by Biacore. This result suggests that the low monovalent Fab affinity can be compen-sated by avidity that is mediated by bivalent binding to HER2 in a cellular context. In contrast, 4D5-H91A 1Fab-IgG TDBs demon-strate only low apparent affinity to MCF7 or cyno HER2-expressing
Fig. 2. Avidity of low-affinity anti-HER2 binders results in anti-HER2/CD3 1Fab-IgG TDBs that are selective for HER2-overexpressing cells. (A) hu4D5 affinity variants [red, H91A (KD = 49 nM); blue, D98A.F100A.Y102V (KD = 23 nM); cyan, Y55E.Y102V (KD = 2.6 nM); magenta, Y102V (KD = 0.32 nM)] were converted to 1Fab-IgG TDB (bivalent HER2 binding and monovalent CD3 binding), and the activity was compared to anti-HER2/CD3 IgG TDB (monovalent for HER2 and CD3), with high-affinity (KD = 0.30 nM) anti-HER2 binding (black). (B) Binding to SKBR3 and MCF7 cells was determined by flow cytometry. MFU, mean fluorescence unit. (C) Induction of apoptosis (50 ng/ml) was deter-mined using IncuCyte assay for Caspase3/7 activity (n = 3). (D) Cytotoxicity was determined by CellTiter-Glo assay after 48 hours of treatment with 50 ng/ml (left and middle) or 50 g/ml (right) dose of TDBs (n = 3). (C and D) Data in all panels are represented as means ± SD. Welch’s t test was used for the statistical analysis.
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
4 of 11
cells (KD value of > 100 nM for both in direct cell binding), suggest-ing that the lower HER2 copy number on these cells is not sufficient for bivalent HER2 engagement.
Trastuzumab interferes with constitutive ligand-independent phos-phatidylinositol 3-kinase (PI3K) signaling in HER2-overexpressing cells, causing inhibition of PI3K pathway activity and downstream cell proliferation (29, 30). This direct effect on tumor cells may also be important in clinical activity of trastuzumab-based antibody- drug con-jugate ado-trastuzumab emtansine (31). Anti-HER2/CD3 IgG TDB does not inhibit the proliferation of HER2-overexpressing cells due to mon-ovalent binding (23). Treatment with anti- HER2/CD3 1Fab-IgG TDB (bivalent for HER2) resulted in transient reduction of phosphorylated AKT, without detectable ef-fect on HER3 phosphorylation (fig. S3) or substantial effect on proliferation/viability of SKBR3 cells, demonstrating that T cell–independent antisignaling effect of trastu-zumab is attenuated in 1Fab-IgG TDB with bivalent low affinity binding to HER2.
These results suggest that TDB selec-tivity toward HER2-overexpressing cells can be engineered by leveraging avidity and incorporating two low-affinity anti- HER2 binding sites. The narrow window for optimal affinity of the individual anti- HER2 binding sites ranges from KD ~20 to 50 nM because anti-HER2 variants with higher affinity did not improve se-lectivity, and variants with lower affinity compromised the potency of the TDB.
1Fab-IgG format improves the in vitro selectivity for HER2-overexpressing cells more than 1000-foldThe 4D5-H91A 1Fab-IgG TDB variant was selected to further study the in vitro activity (Fig. 3). A dose-response study demonstrated that 4D5-H91A 1Fab-IgG TDB activity on SKBR3 cells was almost identical compared to that of the high- affinity 4D5-WT IgG TDB [EC50 (median effective concentration), 6.9 and 6.3 pM, respectively; Fig 3A). In contrast to IgG TDB, no activity was seen against MCF7 cells at any doses. Selectivity studies were extended to include cell lines with a wide range of HER2 expression. The expres-sion of HER2 RNA was determined in a panel of 90 breast cancer cell lines (32), and cell lines were arbitrarily categorized into high [>400 normalized reads per kilo- base of transcript per million mapped reads (nRPKM)], medium (50 to 400 nRPKM), and low (<50 nRPKM) HER2-expressing cells (Fig. 3B). Cell lines with differential HER2 expression were chosen, and cyto-toxicity of the TDBs was studied at two
doses. The in vitro activity of monovalent high-affinity HER2 TDB has been previously reported to saturate at a concentration of ~10 ng/ml (23). At a high TDB dose (50 ng/ml), no clear correlation was seen between T cell activation and HER2 expression for the 4D5-WT IgG TDB (Fig. 3C, bottom). In notable contrast, T cell ac-tivation was induced only in the cell lines with the highest HER2 expression when 4D5-H91A 1Fab-IgG TDB was used. In agreement with these data, target cell killing was limited to high HER2 cell lines with the 4D5-H91A 1Fab-IgG TDB molecule, whereas 4D5-WT IgG TDB was broadly active and even induced killing of the low
Fig. 3. 1Fab-IgG format improves in vitro selectivity for HER2-overexpressing cells ≥1000-fold. In vitro activity of 4D5-H91A 1Fab-IgG TDB was compared to 4D5-WT IgG TDB. (A) Cytotoxicity dose-response in SKBR3 and MCF7 cells (n = 3). (B) RNA sequencing analysis of HER2 RNA expression in 90 breast cancer cell lines. Cell lines were arbi-trarily divided into high (red), medium (magenta), and low (blue) HER2-expressing cell lines. (C) Cytotoxicity and T cell activation mediated by high TDB doses in 14 cell lines with a range of HER2 expression (n = 2). Left: Treatment with 4D5-WT IgG TDB. Right: Treatment with 4D5-H91A 1Fab-IgG TDB. HER2 protein expression was confirmed by West-ern blot. Data are represented as means ± SD.
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
5 of 11
HER2-expressing cells (Fig. 3C). The TDB activity was lower at a concentration of 50 g/ml compared to a concentration of 50 ng/ml, suggesting that saturating concentration has been reached and that 4D5-H91A 1Fab-IgG TDB will likely not induce killing of low HER2- expressing cells even at higher concentration. The reduction in ac-tivity with supratherapeutic doses is likely due to a high-dose hook effect (33). Considering that the 50 ng/ml dose of 4D5-WT IgG TDB induced T cell activation and killing of low HER2 cells, where-as 50 g/ml of 4D5-H91A 1Fab-IgG TDB did not affect viability of low HER2-expressing cells, the selectivity to HER2-overexpressing cells was improved at least 1000-fold. We also tested the activity in human noncancer cell lines derived from multiple tissues such as lung, skin, and breast (fig. S4). Low HER2 expression was confirmed in all tested noncancerous cells by flow cytometry. 4D5-H91A 1Fab- IgG demonstrated no apoptotic activity against any of the seven tested cell lines at concentrations ≤5 g/ml. Next, we investigated the threshold of HER2 ex-pression that is required for the activity of 4D5-H91A 1Fab-IgG TDB (Fig. 4). Robust killing activity was detected on all cell lines expressing HER2 RNA ≥489 nRPKM (Fig. 4A). Minimal activity was detected on cell lines that expressed HER2 RNA ≤260 nRPKM. We further measured cell surface copy number of HER2 using a quantitative flow cytometry–based assay (Fig. 4A and fig. S5). None of the cells that express <200,000 HER2 per cell were sensitive to 1Fab-IgG TDB, whereas all cell lines with >200,000 HER2 were read-ily killed by the 1Fab-IgG TDB. An intra-cellular pool of HER2 protein detectable by Western blot, but not by flow cytom-etry, may explain the high Western blot signal detected in OE-33 cells (Fig. 4A). The activity was further benchmarked to HER2 diagnostic tests that are clinically validated and used for patient selection. Substantial killing activity by 4D5-H91A 1Fab-IgG TDB was only seen in immuno-histochemistry (IHC) 3+, fluorescence in situ hybridization (FISH) + cell lines (Fig. 4, B and C).
The results demonstrate that the in vitro potency of the 4D5-H91A 1Fab-IgG TDB is similar to that of monova-lent high-affinity HER2 TDB in killing of high HER2- expressing cell lines. However, 1Fab-IgG had no detectable ac-tivity on any tested low HER2-expressing cell lines, suggesting that it is selective for HER2- overexpressing cells.
4D5-H91A 1Fab-IgG TDB improves in vivo selectivity for HER2-overexpressing tumorsWe evaluated the translation of selec-tivity to in vivo tumor models. The ac-
tivity of the 4D5-H91A 1Fab-IgG TDB and 4D5-WT IgG TDB was compared in HER2 high and low tumor models. NOD SCID gamma (NSG) mice supplemented with human peripheral blood mononu-clear cells (PBMCs) were grafted with either HER2-amplified KPL4 tumors or HT55 tumors, which express about the same amount of HER2 as MCF7 (fig. S6A). HT55 tumors were used to model in vivo on-target activity on cells/tissues that express a similar quantity of HER2 to normal human tissues. As expected, 4D5-H91A 1Fab-IgG TDB has potent activity in killing the HER2-amplified KPL4 cells in vitro but did not induce HT55 killing (fig. S6B). The 4D5-H91A 1Fab-IgG TDB induced regressions after a single dose of anti-body (0.5 or 5 mg/kg; Fig. 5A), whereas the 4D5-WT IgG TDB in-duced regressions already at a dose of 0.05 mg/kg, indicating that the 4D5-H91A 1Fab-IgG TDB is ~10-fold less active in vivo. As
Fig. 4. A total of 2 × 105 HER2 per cell is the expression limit for 4D5-H91A 1Fab-IgG TDB activity. (A) In vitro cytotoxic activity of 4D5-H91A 1Fab-IgG TDB (middle) was compared to 4D5-WT IgG TDB (top). Cells were treated with a concentration of 1 g/ml for 48 hours (n = 3). HER2 protein was detected using Western blot analysis, and HER2 cell surface copy number was determined by flow cytometry using Quantum Alexa647 MESF Beads. Data points represent two individual experiments (n = 3). (B and C) Cell lines were categorized by HER2 diagnostic tests in clinical use [representative HER2 FISH (scale bars, 10 m) and IHC images (scale bars, 50 m) presented in (B)] and subjected to 4D5-H91A 1Fab-IgG TDB (50 ng/ml) for 48 hours. Cytotoxicity was determined by CellTiter-Glo (n = 3) (C). Data are represented as means ± SD. Welch’s t test was used for the statistical analysis.
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
6 of 11
expected from our in vitro results, reduced activity was observed in treatment of HT55 tumors (Fig. 5B). Monovalent high-affinity HER2 TDB induced HT55 tumor regressions at a single dose of 0.1 mg/kg. The 4D5-H91A 1Fab- IgG TDB did not have any antitumor activity in the treatment of HT55 tumors at doses up to 50 mg/kg (Fig. 5B). No higher doses were tested. In summary, these results demonstrate that both molecules are highly potent in the treatment of HER2- overexpressing tumors and support a widened therapeutic index for the 4D5-H91A 1Fab-IgG TDB, which selectively targets HER2- overexpressing cells. TDB in vivo selectivity to HER2-overexpressing tumors was improved greater than 100-fold.
Monovalent HER2 affinity KD values of 25 to 50 nM maximize selectivity of 1Fab-IgG TDBTwo 4D5 variants that demonstrated improved selectivity in the 1Fab-IgG format, D98A.F100A.Y102V and H91A (monovalent KD values, 23 and 49 nM, respectively), were selected for extended char-acterization to identify optimal affinity to achieve maximal selectiv-ity to HER2-overexpressing cells. In vitro dose response in SKBR3 killing demonstrated near-identical activity between the variants (fig. S7, A and B). Both molecules demonstrated robust antitumor activity at dose of 0.5 mg/kg in treatment of HER2-amplified KPL4 tumor xenografts (Fig. 5 and fig. S8). In vitro activity assays were extended by treating multiple cell lines expressing high, medium, or low amounts of HER2 (fig. S9) with a high concentration of the 1Fab-IgG variants (50 ng/ml). Responses of the cell lines were gen-erally similar between both molecules [P = not significant (NS); fig. S7B]. The potency of the D98A.F100A.Y102V variant appeared to
be slightly higher compared to the H91A variant, but the difference was not significant.
The length of the linker sequence between the anti-HER2 Fabs in the heavy chain may affect the protease resistance and stability as well as binding geometry, flexibility, and spatial reach of bivalent HER2 binding. We tested the effect of two alternative linker sequences in the anti-HER2 Fabs. All molecules in previous figures included the extended linker sequence (DKTHTGGGGSGG), consisting of the nat-ural DKTHT sequence derived from the upper IgG1 hinge of the distal Fab (34), followed by a GGGGSGG extension. The extended linker sequence was compared to a 1Fab-IgG molecule without the GGGGSGG extension (fig. S10A). The impact of the linkers was only tested in the H91A variant 1Fab-IgG context. No difference was seen in binding to SKBR3 or MCF7 cells (fig. S10A). In vitro dose response in SKBR3 killing as well as a broader cell line activity screen demonstrated near- identical activity between the variants (P = NS; fig. S10). In sum-mary, our data demonstrate that 1Fab-IgG TDBs that include anti- HER2 Fabs with monovalent affinities ranging from KD 23 to 49 nM display very similar activity profiles. Varying the linker sequence be-tween anti-HER2 Fabs had no effect on the activity or selectivity profile.
High dose of 4D5-H91A 1Fab-IgG TDB does not result in HER2-independent T cell activationFc receptor (FcR) binding has been attenuated in the TDBs by N297G substitution in the Fc region, which reduces relative binding affinity of human IgG1 to all human FcR by more than 500- to 1000-fold (35). To confirm that TDBs do not induce target-independent T cell activation at high concentration, we used a Jurkat reporter
Fig. 5. 4D5-H91A 1Fab-IgG TDB has improved in vivo selectivity for HER2-overexpressing tumors. Individual tumor volume response of HER2-amplified KPL4 breast cancer xenografts (A) or low HER2-expressing HT55 xenografts (B) to 4D5-H91A 1Fab-IgG and 4D5-WT IgG TDB in NSG mice supplemented with human PBMCs. Mice with established tumors received a single intravenous dose at day 0. Trellis plots of individual and fitted tumor volumes are presented. Each panel in the trellis depicts one dose group (n = 8; panel headers indicate group numbers). Bold, solid black lines indicate the fitted tumor volume for each dose group. Dashed blue lines indicate the fitted tumor volume for the control group (vehicle: histidine buffer). Gray lines indicate individual animals present through the course of the study. Red lines indicate mice that were removed from the study. Dashed horizontal red lines indicate tumor volume of 500 mm3.
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
7 of 11
cell line (Fig. 6A). A robust signal was detected when reporter cells were stimulated with TDB in the presence of HER2-expressing cells. Neither 4D5-WT IgG TDB nor 4D5-H91A 1Fab-IgG TDB activated T cells in the absence of HER2. Next, we spiked high concentration of TDB into human PBMCs (Fig. 6B). Bivalent anti- CD3 (OKT3) induced T cell activation at a concentration of ≥10 ng/ml, whereas no T cell activation was detected at a concentration of 100 g/ml. Last, we repeated the experiment using purified FcRI-positive mono-cytes (fig. S11) in the absence of human IgG and without evidence of target-independent T cell activation mediated by Fc interactions. Together, these results indicate that neither TDB format induces HER2-independent T cell activation at high concentrations.
4D5-H91A 1Fab-IgG TDB has limited pharmacological activity in cynos and is well tolerated up to 20 mg/kgCyno tissues express low amounts of HER2, similar to normal hu-man tissues. 4D5-H91A 1Fab-IgG TDB binds to cyno and human CD3 with comparable affinity (fig. S1A) and to cyno HER2-expressing cells with similar apparent affinity compared to MCF7 (fig. S2A). We demonstrated that as expected, 4D5-H91A 1Fab-IgG TDB does not induce apoptosis of cells that express low-moderate quantity of cyno HER2 (fig. S2, B and C). To test the tolerability of 4D5-H91A 1Fab-IgG TDB, we designed a single-dose tolerability study to analyze the pharmacodynamic (PD) activity and pharmacokinetics (PK) in cyno. Female monkeys were dosed with slow intravenous infusion
Fig. 6. 4D5-H91A 1Fab-IgG TDB has limited pharmacological activity in cyno and is well tolerated up to 20 mg/kg. TDB-induced HER2-independent T cell activation at high concentration was tested using (A) a Jurkat reporter cell line and (B) high concentration of TDB spiked into healthy donor PBMCs. RLU, relative light unit. (C and D) Female monkeys were dosed with slow intravenous infusion of 4D5-H91A 1Fab-IgG TDB at doses of 3 mg/kg (n = 3) and 20 mg/kg (n = 5). (C) Blood markers of inflam-mation (CRP), T cell activation (lymphocyte margination), and liver damage (ALT and AST) were measured 1 and 7 days after dosing. (D) Blood samples were collected at the indicated time points, and human IgG was detected by enzyme-linked immunosorbent assay (ELISA) to determine the PK parameters presented in the table. CL, clearance. (E) Cyno serum was harvested from one animal 7 and 14 days after dosing and subjected to healthy donor CD8+ and SKBR3 cells for 24 hours using the indicated serum dilutions (n = 3). Parallel experiments were carried out using dilutions of fresh 4D5-H91A 1Fab-IgG TDB (Control). Data are represented as means ± SE (A, B, and E) or means ± SEM (C and D). Welch’s t test was used for the statistical analysis.
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
8 of 11
at doses of 3 mg/kg (n = 3) and 20 mg/kg (n = 5). Tolerability/PK/PD readouts included clinical observations, clinical chemistry, hema-tology, cytokines, and PK. Histopathology was not included in the analysis. No or minimal test article-related changes were detected in clinical observations (body weight, heart rate, temperature, and re-spiratory rate). Typical TDB-associated pharmacological and clini-cal pathology responses (6, 9, 12, 21) were notably absent (Fig. 6C). C-reactive protein (CRP; an acute-phase reactant and a marker for inflammation) remained at the background. No lymphocyte margin-ation, increase of liver enzymes [alanine aminotransferase (ALT) and aspartate aminotransferase (AST)], or elevation of peripheral blood cytokines was detected despite the high TDB dose. Together, 4D5-H91A 1Fab-IgG TDB was well tolerated at high doses and demonstrated limited pharmacological activity in cyno.
TDB exposure was confirmed for both doses tested, and PK char-acteristics were as expected (Fig. 6D). Exposures were close to dose- proportional and maintained until the last evaluated time point (14 days). To confirm that the TDB is not inactivated in cyno, we recovered aliquots of serum from samples harvested 7 and 14 days after dosing with TDB (20 mg/kg) and used serum dilutions to mediate killing of SKBR3 cells in vitro using healthy donor human T cells (Fig. 6E). Mea-sured TDB serum concentrations in undiluted samples were 193 and 65 g/ml for day 7 and day 14 samples, respectively. One hundred–fold dilution of serum 14 days after dosing induced similar SKBR3 killing compared to maximal activity achieved in a parallel killing assay using fresh TDB at a high concentration of 1 g/ml (Fig. 6E), which is typically sufficient to saturate activity (23). Together, these data suggest that the low HER2 expression in normal cyno tissues is not sufficient to trigger T cell activation or off-tumor T cell activity in normal tissues that express low amounts of HER2.
DISCUSSIONSeveral anti-CD3 bispecific antibodies or antibody fragment–based molecules are in clinical development, and clinical proof of concept has been established by blinatumomab in hematological malignan-cies (6). In contrast, clinical efficacy and safety in the treatment of solid tumors remain unclear. Multiple aspects elevate the bar to suc-cess in solid tumor indications, most notable of which is the lack of “clean” tumor-restricted antigens. We have previously generated a highly efficacious anti-HER2/CD3 TDB that demonstrated ro-bust potency in HER2-overexpressing tumor models (23). The ther-apeutic index of the molecule is based on differential sensitivity to the TDB, correlating with the HER2 expression on tumor cells. De-spite the established positive preclinical therapeutic index at high concentrations, the high-affinity anti-HER2/CD3 TDB can trigger T cells to attack cells that express lower amounts of HER2, indicat-ing a risk for on-target off-tumor adverse effects. Therapeutics with similar mechanisms of action (CD3 bispecific antibodies and CAR T cells), none of which have been approved for clinical use, have il-lustrated the apparent risk of redirecting T cell activity to HER2- positive tumors when tested in clinical trials (24, 25). In addition to on-target toxicities on HER2-expressing tissues, some of the toxici-ties detected in the trials may be related to cytokine release. For exam-ple, ertumaxomab includes a functional Fc, which may result in HER2-independent immune cell activation. In the current study, our goal was to mitigate this risk by generating a TDB that would be able to redirect T cells selectively to HER2-overexpressing cancer cells while sparing the normal tissues.
A simple adjustment of HER2 affinity shifted the activity of HER2 TDB but was not sufficient to improve selectivity for overexpressing cells. In several preclinical reports, optimization of tumor antigen binding (“affinity tuning”) of CAR T cells has been demonstrated to result in selectivity to high tumor antigen density. Lowering the CAR binding affinity for EGFR, HER2, and CD38 demonstrated reduced risk of on-target off-tumor adverse effects and may there-fore be an appropriate mitigation strategy for targets that are over-expressed by tumor cells (36–38). This improved therapeutic index by affinity tuning was likely mediated by the multivalent display of the CAR on T cells. However, it is not clear whether the interaction of two CARs is sufficient for the avidity-based interaction between the T cell and the tumor cell or the interaction of multiple CAR molecules is required. In the case of HER2 TDB, HER2 affinity ad-justment combined with gain of avidity resulted in increased pre-clinical therapeutic index.
In the case of trastuzumab, a substantial 75- to 160-fold affinity reduction was needed to generate the 1Fab-IgG molecule with se-lectivity to HER2-overexpressing cells. In the cellular context, avid-ity of 1Fab-IgG enhanced binding to overexpressing cells by 10-fold (5 nM), whereas very low binding was detected to low HER2-expressing cells. Overall, the window of opportunity was fairly restricted, with optimal affinity ranging between KD 25 and 50 nM. Molecules with lower affinity lost substantial amounts of activity, and molecules with higher affinity failed to gain selectivity advantage. The optimal affinity range is likely dependent on multiple variables, including target copy number and biology. Thus, translation of our findings to another solid tumor antigen will require exploring best affinities de novo. In addition, the geometry of the molecule and locations of different binding modules are also likely to play a role in generating a molecule with optimal binding and activity profile.
Similar to our results, valence of HER2 binding has been shown not to be sufficient to affect selectivity without appropriately tuning the monovalent affinities (39). A related concept exploiting “cross-arm binding efficiency” is to increase tumor selectivity by targeting two dif-ferent tumor antigens (40). In this model, binding of the bispecific antibody to the second receptor is enhanced after it binds to the first receptor, causing an increase in apparent affinity to tumor cells. The therapeutic index in this strategy is based on lack of simultaneous ex-pression of the two tumor targets in normal cells. The two-target ap-proach has been tested preclinically using several target pairs (41–43).
A very high HER2 expression (HER2 IHC3+, ISH+) was required for the avidity effect and TDB-mediated killing. As a result, patient selection and the optimal target patient population for anti-HER2/CD3 1Fab-IgG will likely be similar to other HER2-targeted thera-pies including trastuzumab and ado-trastuzumab emtansine. The very high expression threshold for HER2 implies that molecules with less stringent selectivity profile (23) may be able to capture a larger patient population if tolerated at therapeutic doses. The requirement for high target expression also raises the question of how broadly ap-plicable the avidity-based selectivity strategy can be for other tumor antigens beyond HER2. Furthermore, HER2 amplification is a distinct driver event in tumors, which is not a common feature for tumor antigens. The mechanism of action of anti-HER2/CD3 1Fab-IgG TDB is independent of HER2 signaling (in contrast to trastuzumab), and HER2 pathway activation status should not influence the activity of the molecule.
Despite our findings, we acknowledge limitations in our study that should be considered when interpreting the data. The avidity
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
9 of 11
hypothesis was tested only using a single tumor target with a very large dynamic range in expression. Translation to other tumor antigens may therefore require substantial optimization of the parameters de-pending on the target biology. In addition, lack of species cross- reactivity of the antibodies generates limitations to in vivo model systems used in our study. Optimally, preclinical therapeutic index would be determined using a model with both safety and efficacy readout in the same individual, but this is not currently available. In addition, human PBMC–supplemented NSG mice may not optimal-ly recapitulate the immune contexture of human tumors.
In summary, to mitigate potential on-target off-tumor adverse effects of HER2 TDB, we developed anti-HER2/CD3 1Fab-IgG TDB that retains potent antitumor activity and has very low binding to low HER2-expressing cells (fig. S12). Anti-HER2/CD3 1Fab-IgG TDB in-duces CRs in treatment of HER2-amplified tumors at a dose of 0.5 mg/kg and is well tolerated in cynos at a dose of 20 mg/kg. In notable con-trast, HER2-targeting therapeutics with similar mechanisms of action (CD3 bispecifics and CAR T cells) tested in clinical trials have demon-strated toxicities at alarmingly low doses. Trastuzumab-based CAR T cells trial ended quickly in grade 5 (lethal) lung toxicity (25). Clin-ical trials of ertumaxomab, a HER2/CD3 TDB, defined 300 g flat dose as the maximum tolerated dose when using dose escalation (24). If administered to a 70-kg person, the tolerated dose is 0.004 mg/kg [4600-fold lower compared to the dose that was well tolerated in our study (20 mg/kg)]. In conclusion, these results support clinical development of the anti-HER2/CD3 1Fab-IgG TDB.
MATERIALS AND METHODSStudy designThe main objective of this study was to design and optimize an anti- HER2/CD3 TDB that selectively targets HER2-overexpressing tumor cells with high potency, while sparing cells that express low amounts of HER2 found in normal human tissues. Improved selectivity was demonstrated by treating cell lines in vitro and tumor xenografts in rodents. Tolerability of the TDB was demonstrated in primate studies. In vitro studies were based on one to three biological repeats, as spec-ified in the figure legends. For in vivo efficacy studies, 8 to 10 mice were randomized to each treatment group. For ethical reasons, group sizes in primate studies were limited to n = 3 to 5.
StatisticsWelch’s t test was used to test for differences between groups. Results were determined to be significant at P < 0.05. Statistical analyses were performed using GraphPad Prism version 7.
SUPPLEMENTARY MATERIALSwww.sciencetranslationalmedicine.org/cgi/content/full/10/463/eaat5775/DC1Materials and MethodsFig. S1. Binding and cytotoxicity of anti-HER2/CD3 1Fab-IgG TDBs with monovalent HER2 affinity lower than 50 nM.Fig. S2. 4D5-H91A 1Fab-IgG TDB binding affinity for HER2 in cellular context.Fig. S3. T cell–independent antisignaling effect of 1Fab-IgG TDB with bivalent low affinity binding to HER2.Fig. S4. Activity of 4D5-H91A 1Fab-IgG TDB in HER2-expressing noncancer cells.Fig. S5. HER2 copy number analysis.Fig. S6. In vitro activity of 4D5-H91A 1Fab-IgG TDB targeting the in vivo efficacy models.Fig. S7. Cytotoxicity of 4D5-H91A 1Fab-IgG TDB and 4D5-D98A.F100A.Y102V 1Fab-IgG TDB.Fig. S8. In vivo efficacy of 4D5-D98A.F100A.Y102V 1Fab-IgG TDB.Fig. S9. HER2 expression in cancer cell lines.Fig. S10. Effect of linker length on anti-HER2/CD3 1Fab-IgG TDB activity.
Fig. S11. T cell activation in the presence of FcRI+ monocytes.Fig. S12. A schematic representation of the strategy that resulted in TDBs that selectively kill HER2-overexpressing cells.References (44–53)
REFERENCES AND NOTES 1. R. J. Brentjens, M. L. Davila, I. Riviere, J. Park, X. Wang, L. G. Cowell, S. Bartido, J. Stefanski,
C. Taylor, M. Olszewska, O. Borquez-Ojeda, J. Qu, T. Wasielewska, Q. He, Y. Bernal, I. V. Rijo, C. Hedvat, R. Kobos, K. Curran, P. Steinherz, J. Jurcic, T. Rosenblat, P. Maslak, M. Frattini, M. Sadelain, CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 5, 177ra38 (2013).
2. M. Kalos, B. L. Levine, D. L. Porter, S. Katz, S. A. Grupp, A. Bagg, C. H. June, T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 3, 95ra73 (2011).
3. S. L. Maude, N. Frey, P. A. Shaw, R. Aplenc, D. M. Barrett, N. J. Bunin, A. Chew, V. E. Gonzalez, Z. Zheng, S. F. Lacey, Y. D. Mahnke, J. J. Melenhorst, S. R. Rheingold, A. Shen, D. T. Teachey, B. L. Levine, C. H. June, D. L. Porter, S. A. Grupp, Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014).
4. C. J. Turtle, L.-A. Hanafi, C. Berger, M. Hudecek, B. Pender, E. Robinson, R. Hawkins, C. Chaney, S. Cherian, X. Chen, L. Soma, B. Wood, D. Li, S. Heimfeld, S. R. Riddell, D. G. Maloney, Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor–modified T cells. Sci. Transl. Med. 8, 355ra116 (2016).
5. F. Fan, W. Zhao, J. Liu, A. He, Y. Chen, X. Cao, Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J. Clin. Oncol. 35, LBA3001 (2017).
6. R. Bargou, E. Leo, G. Zugmaier, M. Klinger, M. Goebeler, S. Knop, R. Noppeney, A. Viardot, G. Hess, M. Schuler, H. Einsele, C. Brandl, A. Wolf, P. Kirchinger, P. Klappers, M. Schmidt, G. Riethmüller, C. Reinhardt, P. A. Baeuerle, P. Kufer, Tumor regression in cancer patients by very low doses of a T cell–engaging antibody. Science 321, 974–977 (2008).
7. H. Kantarjian, A. Stein, N. Gökbuget, A. K. Fielding, A. C. Schuh, J.-M. Ribera, A. Wei, H. Dombret, R. Foà, R. Bassan, Ö. Arslan, M. A. Sanz, J. Bergeron, F. Demirkan, E. Lech-Maranda, A. Rambaldi, X. Thomas, H.-A. Horst, M. Brüggemann, W. Klapper, B. L. Wood, A. Fleishman, D. Nagorsen, C. Holland, Z. Zimmerman, M. S. Topp, Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 376, 836–847 (2017).
8. E. J. Smith, K. Olson, L. J. Haber, B. Varghese, P. Duramad, A. D. Tustian, A. Oyejide, J. R. Kirshner, L. Canova, J. Menon, J. Principio, D. MacDonald, J. Kantrowitz, N. Papadopoulos, N. Stahl, G. D. Yancopoulos, G. Thurston, S. Davis, A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci. Rep. 5, 17943 (2015).
9. L. L. Sun, D. Ellerman, M. Mathieu, M. Hristopoulos, X. Chen, Y. Li, X. Yan, R. Clark, A. Reyes, E. Stefanich, E. Mai, J. Young, C. Johnson, M. Huseni, X. Wang, Y. Chen, P. Wang, H. Wang, N. Dybdal, Y.-W. Chu, N. Chiorazzi, J. M. Scheer, T. Junttila, K. Totpal, M. S. Dennis, A. J. Ebens, Anti-CD20/CD3 T cell–dependent bispecific antibody for the treatment of B cell malignancies. Sci. Transl. Med. 7, 287ra70 (2015).
10. B.-Z. Katz, Y. Herishanu, Therapeutic targeting of CD19 in hematological malignancies: Past, present, future and beyond. Leuk. Lymphoma 55, 999–1006 (2014).
11. G. Salles, M. Barrett, R. Foà, J. Maurer, S. O’Brien, N. Valente, M. Wenger, D. G. Maloney, Rituximab in B-cell hematologic malignancies: A review of 20 years of clinical experience. Adv. Ther. 34, 2232–2273 (2017).
12. J. Li, N. J. Stagg, J. Johnston, M. J. Harris, S. A. Menzies, D. DiCara, V. Clark, M. Hristopoulos, R. Cook, D. Slaga, R. Nakamura, L. McCarty, S. Sukumaran, E. Luis, Z. Ye, T. D. Wu, T. Sumiyoshi, D. Danilenko, G. Y. Lee, K. Totpal, D. Ellerman, I. Hötzel, J. R. James, T. T. Junttila, Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell 31, 383–395 (2017).
13. A. Seckinger, J. A. Delgado, S. Moser, L. Moreno, B. Neuber, A. Grab, S. Lipp, J. Merino, F. Prosper, M. Emde, C. Delon, M. Latzko, R. Gianotti, R. Lüoend, R. Murr, R. J. Hosse, L. J. Harnisch, M. Bacac, T. Fauti, C. Klein, A. Zabaleta, J. Hillengass, E. A. Cavalcanti-Adam, A. D. Ho, M. Hundemer, J. F. San Miguel, K. Strein, P. Umaña, D. Hose, B. Paiva, M. D. Vu, Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell 31, 396–410 (2017).
14. R. S. Herbst, J.-C. Soria, M. Kowanetz, G. D. Fine, O. Hamid, M. S. Gordon, J. A. Sosman, D. F. McDermott, J. D. Powderly, S. N. Gettinger, H. E. K. Kohrt, L. Horn, D. P. Lawrence, S. Rost, M. Leabman, Y. Xiao, A. Mokatrin, H. Koeppen, P. S. Hegde, I. Mellman, D. S. Chen, F. S. Hodi, Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014).
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
10 of 11
15. J. H. E. Baker, K. E. Lindquist, L. A. Huxham, A. H. Kyle, J. T. Sy, A. I. Minchinton, Direct visualization of heterogeneous extravascular distribution of trastuzumab in human epidermal growth factor receptor type 2 overexpressing xenografts. Clin. Cancer Res. 14, 2171–2179 (2008).
16. A. I. Minchinton, I. F. Tannock, Drug penetration in solid tumours. Nat. Rev. Cancer 6, 583–592 (2006).
17. T. F. Gajewski, S.-R. Woo, Y. Zha, R. Spaapen, Y. Zheng, L. Corrales, S. Spranger, Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr. Opin. Immunol. 25, 268–276 (2013).
18. M. R. Parkhurst, J. C. Yang, R. C. Langan, M. E. Dudley, D.-A. N. Nathan, S. A. Feldman, J. L. Davis, R. A. Morgan, M. J. Merino, R. M. Sherry, M. S. Hughes, U. S. Kammula, G. Q. Phan, R. M. Lim, S. A. Wank, N. P. Restifo, P. F. Robbins, C. M. Laurencot, S. A. Rosenberg, T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 19, 620–626 (2011).
19. L. A. Johnson, R. A. Morgan, M. E. Dudley, L. Cassard, J. C. Yang, M. S. Hughes, U. S. Kammula, R. E. Royal, R. M. Sherry, J. R. Wunderlich, C.-C. R. Lee, N. P. Restifo, S. L. Schwarz, A. P. Cogdill, R. J. Bishop, H. Kim, C. C. Brewer, S. F. Rudy, C. VanWaes, J. L. Davis, A. Mathur, R. T. Ripley, D. A. Nathan, C. M. Laurencot, S. A. Rosenberg, Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114, 535–546 (2009).
20. B. J. Cameron, A. B. Gerry, J. Dukes, J. V. Harper, V. Kannan, F. C. Bianchi, F. Grand, J. E. Brewer, M. Gupta, G. Plesa, G. Bossi, A. Vuidepot, A. S. Powlesland, A. Legg, K. J. Adams, A. D. Bennett, N. J. Pumphrey, D. D. Williams, G. Binder-Scholl, I. Kulikovskaya, B. L. Levine, J. L. Riley, A. Varela-Rohena, E. A. Stadtmauer, A. P. Rapoport, G. P. Linette, C. H. June, N. J. Hassan, M. Kalos, B. K. Jakobsen, Identification of a Titin-derived HLA-A1–presented peptide as a cross-reactive target for engineered MAGE A3–directed T cells. Sci. Transl. Med. 5, 197ra103 (2013).
21. R. Lutterbuese, T. Raum, R. Kischel, P. Hoffmann, S. Mangold, B. Rattel, M. Friedrich, O. Thomas, G. Lorenczewski, D. Rau, E. Schaller, I. Herrmann, A. Wolf, T. Urbig, P. A. Baeuerle, P. Kufer, T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc. Natl. Acad. Sci. U.S.A. 107, 12605–12610 (2010).
22. W. M. Fiedler, M. Wolf, M. Kebenko, M. Goebeler, B. Ritter, A. Quaas, E. Vieser, Y. Hijazi, I. Patzak, M. Friedrich, P. Kufer, S. Frankel, R. Seggewiss-Bernhardt, S. Kaubitzsch, A phase I study of EpCAM/CD3-bispecific antibody (MT110) in patients with advanced solid tumors. J. Clin. Oncol. 30, 2504–2504 (2012).
23. T. T. Junttila, J. Li, J. Johnston, M. Hristopoulos, R. Clark, D. Ellerman, B.-E. Wang, Y. Li, M. Mathieu, G. Li, J. Young, E. Luis, G. Lewis Phillips, E. Stefanich, C. Spiess, A. Polson, B. Irving, J. M. Scheer, M. R. Junttila, M. S. Dennis, R. Kelley, K. Totpal, A. Ebens, Antitumor efficacy of a bispecific antibody that targets HER2 and activates T cells. Cancer Res. 74, 5561–5571 (2014).
24. N. Haense, A. Atmaca, C. Pauligk, K. Steinmetz, F. Marmé, G. M. Haag, M. Rieger, O. G. Ottmann, P. Ruf, H. Lindhofer, S.-E. Al-Batran, A phase I trial of the trifunctional anti Her2 × anti CD3 antibody ertumaxomab in patients with advanced solid tumors. BMC Cancer 16, 420 (2016).
25. R. A. Morgan, J. C. Yang, M. Kitano, M. E. Dudley, C. M. Laurencot, S. A. Rosenberg, Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843–851 (2010).
26. Z. Aguilar, R. W. Akita, R. S. Finn, B. L. Ramos, M. D. Pegram, F. F. Kabbinavar, R. J. Pietras, P. Pisacane, M. X. Sliwkowski, D. J. Slamon, Biologic effects of heregulin/neu differentiation factor on normal and malignant human breast and ovarian epithelial cells. Oncogene 18, 6050–6062 (1999).
27. R. F. Kelley, M. P. O’Connell, Thermodynamic analysis of an antibody functional epitope. Biochemistry 32, 6828–6835 (1993).
28. K. Miller, G. Meng, J. Liu, A. Hurst, V. Hsei, W.-L. Wong, R. Ekert, D. Lawrence, S. Sherwood, L. DeForge, J. Gaudreault, G. Keller, M. Sliwkowski, A. Ashkenazi, L. Presta, Design, construction, and in vitro analyses of multivalent antibodies. J. Immunol. 170, 4854–4861 (2003).
29. T. T. Junttila, R. W. Akita, K. Parsons, C. Fields, G. D. Lewis Phillips, L. S. Friedman, D. Sampath, M. X. Sliwkowski, Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 15, 429–440 (2009).
30. G. D. Lewis, J. A. Lofgren, A. E. McMurtrey, A. Nuijens, B. M. Fendly, K. D. Bauer, M. X. Sliwkowski, Growth regulation of human breast and ovarian tumor cells by heregulin: Evidence for the requirement of ErbB2 as a critical component in mediating heregulin responsiveness. Cancer Res. 56, 1457–1465 (1996).
31. T. T. Junttila, G. Li, K. Parsons, G. L. Phillips, M. X. Sliwkowski, Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 128, 347–356 (2011).
32. C. Klijn, S. Durinck, E. W. Stawiski, P. M. Haverty, Z. Jiang, H. Liu, J. Degenhardt, O. Mayba, F. Gnad, J. Liu, G. Pau, J. Reeder, Y. Cao, K. Mukhyala, S. K. Selvaraj, M. Yu, G. J. Zynda, M. J. Brauer, T. D. Wu, R. C. Gentleman, G. Manning, R. L. Yauch, R. Bourgon, D. Stokoe,
Z. Modrusan, R. M. Neve, F. J. de Sauvage, J. Settleman, S. Seshagiri, Z. Zhang, A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 33, 306–312 (2015).
33. C. Selby, Interference in immunoassay. Ann. Clin. Biochem. 36 (Pt. 6), 704–721 (1999). 34. R. J. Brezski, G. Georgiou, Immunoglobulin isotype knowledge and application to Fc
engineering. Curr. Opin. Immunol. 40, 62–69 (2016). 35. M. Lo, H. S. Kim, R. K. Tong, T. W. Bainbridge, J.-M. Vernes, Y. Zhang, Y. L. Lin, S. Chung,
M. S. Dennis, Y. J. Y. Zuchero, R. J. Watts, J. A. Couch, Y. G. Meng, J. K. Atwal, R. J. Brezski, C. Spiess, J. A. Ernst, Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J. Biol. Chem. 292, 3900–3908 (2017).
36. H. G. Caruso, L. V. Hurton, A. Najjar, D. Rushworth, S. Ang, S. Olivares, T. Mi, K. Switzer, H. Singh, H. Huls, D. A. Lee, A. B. Heimberger, R. E. Champlin, L. J. N. Cooper, Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent anti-tumor activity. Cancer Res. 75, 3505–3518 (2015).
37. E. Drent, M. Themeli, R. Poels, R. de Jong-Korlaar, H. Yuan, J. de Bruijn, A. C. M. Martens, S. Zweegman, N. van de Donk, R. W. J. Groen, H. M. Lokhorst, T. Mutis, A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol. Ther. 25, 1946–1958 (2017).
38. X. Liu, S. Jiang, C. Fang, S. Yang, D. Olalere, E. C. Pequignot, A. P. Cogdill, N. Li, M. Ramones, B. Granda, L. Zhou, A. Loew, R. M. Young, C. H. June, Y. Zhao, Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 75, 3596–3607 (2015).
39. Y. Cao, J. Y. Axup, J. S. Y. Ma, R. E. Wang, S. Choi, V. Tardif, R. K. V. Lim, H. M. Pugh, B. R. Lawson, G. Welzel, S. A. Kazane, Y. Sun, F. Tian, S. Srinagesh, T. Javahishvili, P. G. Schultz, C. H. Kim, Multiformat T-cell-engaging bispecific antibodies targeting human breast cancers. Angew. Chem. Int. Ed. Engl. 54, 7022–7027 (2015).
40. P. J. Carter, G. A. Lazar, Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 17, 197–223 (2018).
41. Y. Mazor, A. Hansen, C. Yang, P. S. Chowdhury, J. Wang, G. Stephens, H. Wu, W. F. Dall’Acqua, Insights into the molecular basis of a bispecific antibody’s target selectivity. MAbs 7, 461–469 (2015).
42. Y. Mazor, K. F. Sachsenmeier, C. Yang, A. Hansen, J. Filderman, K. Mulgrew, H. Wu, W. F. Dall’Acqua, Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci. Rep. 7, 40098 (2017).
43. S. Zheng, S. Moores, S. Jarantow, J. Pardinas, M. Chiu, H. Zhou, W. Wang, Cross-arm binding efficiency of an EGFR x c-Met bispecific antibody. MAbs 8, 551–561 (2016).
44. P. Carter, L. Presta, C. M. Gorman, J. B. Ridgway, D. Henner, W. L. Wong, A. M. Rowland, C. Kotts, M. E. Carver, H. M. Shepard, Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. U.S.A. 89, 4285–4289 (1992).
45. M. Dillon, Y. Yin, J. Zhou, L. McCarty, D. Ellerman, D. Slaga, T. T. Junttila, G. Han, W. Sandoval, M. A. Ovacik, K. Lin, Z. Hu, A. Shen, J. E. Corn, C. Spiess, P. J. Carter, Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. MAbs 9, 213–230 (2017).
46. A. B. Bos, J. N. Duque, S. Bhakta, F. Farahi, L. A. Chirdon, J. R. Junutula, P. D. Harms, A. W. Wong, Development of a semi-automated high throughput transient transfection system. J. Biotechnol. 180, 10–16 (2014).
47. A. B. Bos, P. Luan, J. N. Duque, D. Reilly, P. D. Harms, A. W. Wong, Optimization and automation of an end-to-end high throughput microscale transient protein production process. Biotechnol. Bioeng. 112, 1832–1842 (2015).
48. C. Spiess, M. Merchant, A. Huang, Z. Zheng, N.-Y. Yang, J. Peng, D. Ellerman, W. Shatz, D. Reilly, D. G. Yansura, J. M. Scheer, Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat. Biotechnol. 31, 753–758 (2013).
49. B. Modrek, L. Ge, A. Pandita, E. Lin, S. Mohan, P. Yue, S. Guerrero, W. M. Lin, T. Pham, Z. Modrusan, S. Seshagiri, H. M. Stern, P. Waring, L. A. Garraway, J. Chant, D. Stokoe, G. Cavet, Oncogenic activating mutations are associated with local copy gain. Mol. Cancer Res. 7, 1244–1252 (2009).
50. C. O’Brien, G. Cavet, A. Pandita, X. Hu, L. Haydu, S. Mohan, K. Toy, C. S. Rivers, Z. Modrusan, L. C. Amler, M. R. Lackner, Functional genomics identifies ABCC3 as a mediator of taxane resistance in HER2-amplified breast cancer. Cancer Res. 68, 5380–5389 (2008).
51. T. H. Long, H. Lawce, C. Durum, S. R. Moore, S. B. Olson, K. Gatter, M. L. Troxell, The new equivocal: Changes to HER2 FISH results when applying the 2013 ASCO/CAP guidelines. Am. J. Clin. Pathol. 144, 253–262 (2015).
52. A. C. Wolff, M. E. H. Hammond, D. G. Hicks, M. Dowsett, L. M. McShane, K. H. Allison, D. C. Allred, J. M. S. Bartlett, M. Bilous, P. Fitzgibbons, W. Hanna, R. B. Jenkins, P. B. Mangu, S. Paik, E. A. Perez, M. F. Press, P. A. Spears, G. H. Vance, G. Viale, D. F. Hayes; American Society of Clinical Oncology; College of American Pathologists, Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 31, 3997–4013 (2013).
53. A. C. Wolff, M. E. H. Hammond, D. G. Hicks, M. Dowsett, L. M. McShane, K. H. Allison, D. C. Allred, J. M. S. Bartlett, M. Bilous, P. Fitzgibbons, W. Hanna, R. B. Jenkins, P. B. Mangu,
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Slaga et al., Sci. Transl. Med. 10, eaat5775 (2018) 17 October 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
11 of 11
S. Paik, E. A. Perez, M. F. Press, P. A. Spears, G. H. Vance, G. Viale, D. F. Hayes; American Society of Clinical Oncology; College of American Pathologists, Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Arch. Pathol. Lab. Med. 138, 241–256 (2014).
Acknowledgments: We would like to thank the Genentech Research Materials Group and Antibody Production Group for technical assistance in antibody expression and purification, B. Wilson for GRIP ELISA, L. Schutt for cyno data interpretation, and P. Carter for support for the project. Funding: All funding for the study was provided by Genentech Inc., a member of the Roche group. Author contributions: All authors contributed to the study design, data analysis, and interpretation. D.E. provided the original idea for the strategy. D.S., D.E., T.N.L., R.V., M.H., R.C., J.J., and E.M. performed the experiments. A.S., K.G., A.A.L., J.T.K., K.T., R.P., C.S., and T.T.J. provided the study oversight. D.S., C.S., and T.T.J. wrote the manuscript. G.L. and T.T.J. were responsible for the administration. Competing interests: All authors are employees of Genentech Inc., a member of the Roche Group, and hold stock and
options. A patent application, entitled “Bispecific antigen-binding molecules and methods of use,” relating to the subject matter of this manuscript, has been filed by Genentech Inc. Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. Investigators may request materials from Genentech by submitting a request form at www.gene.com/gene/reagents-program/request.do.
Submitted 13 March 2018Resubmitted 18 May 2018Accepted 13 September 2018Published 17 October 201810.1126/scitranslmed.aat5775
Citation: D. Slaga, D. Ellerman, T. N. Lombana, R. Vij, J. Li, M. Hristopoulos, R. Clark, J. Johnston, A. Shelton, E. Mai, K. Gadkar, A. A. Lo, J. T. Koerber, K. Totpal, R. Prell, G. Lee, C. Spiess, T. T. Junttila, Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci. Transl. Med. 10, eaat5775 (2018).
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from

by anti-HER2/CD3Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells
Lee, Christoph Spiess and Teemu T. JunttilaJohnston, Amy Shelton, Elaine Mai, Kapil Gadkar, Amy A. Lo, James T. Koerber, Klara Totpal, Rodney Prell, Genee Dionysos Slaga, Diego Ellerman, T. Noelle Lombana, Rajesh Vij, Ji Li, Maria Hristopoulos, Robyn Clark, Jennifer
DOI: 10.1126/scitranslmed.aat5775, eaat5775.10Sci Transl Med
high density of surface HER2 relative to healthy tissues.binds two HER2 molecules at a time, but with low affinity for each one, making it selective for tumors that have aredirect the T cells to recognize tumor cells. To improve treatment safety, the authors selected an antibody that
dependent bispecific antibody that binds to both HER2 and the CD3 protein on T cells, helping−developed a T cell . haveet alnormal tissues also express HER2, resulting in toxicity from HER2-targeted treatments. Slaga
HER2 is a receptor tyrosine kinase that is often overexpressed in breast cancer. Unfortunately, manyLess can be more for tumor targeting
ARTICLE TOOLS http://stm.sciencemag.org/content/10/463/eaat5775
MATERIALSSUPPLEMENTARY http://stm.sciencemag.org/content/suppl/2018/10/15/10.463.eaat5775.DC1
CONTENTRELATED
http://stm.sciencemag.org/content/scitransmed/11/508/eaax8861.fullhttp://stm.sciencemag.org/content/scitransmed/11/497/eaau7534.fullhttp://stm.sciencemag.org/content/scitransmed/11/476/eaav1620.fullhttp://stm.sciencemag.org/content/scitransmed/4/127/127rv2.fullhttp://stm.sciencemag.org/content/scitransmed/7/315/315ra188.fullhttp://stm.sciencemag.org/content/scitransmed/10/461/eaat1445.fullhttp://stm.sciencemag.org/content/scitransmed/9/401/eaam7049.full
REFERENCES
http://stm.sciencemag.org/content/10/463/eaat5775#BIBLThis article cites 53 articles, 19 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
registered trademark of AAAS. is aScience Translational MedicineScience, 1200 New York Avenue NW, Washington, DC 20005. The title
(ISSN 1946-6242) is published by the American Association for the Advancement ofScience Translational Medicine
of Science. No claim to original U.S. Government WorksCopyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement
by guest on January 24, 2020http://stm
.sciencemag.org/
Dow
nloaded from