Embed Size (px)
Citation preview

Catestatin (Chromogranin A352–372) and Novel Effects onMobilization of Fat from Adipose Tissue through Regulationof Adrenergic and Leptin Signaling*
Received for publication, December 20, 2011, and in revised form, April 24, 2012 Published, JBC Papers in Press, April 25, 2012, DOI 10.1074/jbc.M111.335877
Gautam K. Bandyopadhyay‡§1, Christine U. Vu§, Stefano Gentile§, Howon Lee§, Nilima Biswas§, Nai-Wen Chi‡§,Daniel T. O’Connor‡§, and Sushil K. Mahata‡§2
From the ‡Veterans Affairs San Diego Healthcare System, San Diego, California 92161 and the §Department of Medicine, Universityof California, San Diego, California 92093-0838
Background: Mice lacking the neurosecretory protein Chromogranin A are obese, presumably because of resistance tocatecholamines and leptin.Results: Catestatin (CST) reduces adiposity, an effect likely mediated by restoring leptin sensitivity and modulating adrenergicsignaling.Conclusion: CST promotes lipolysis by blocking �-AR signaling and stimulating fatty acid oxidation.Significance:We propose CST as a candidate antiobesity agent.
Chromogranin A knock-out (Chga-KO) mice displayincreased adiposity despite high levels of circulating cat-echolamines and leptin. Consistent with diet-induced obesemice, desensitization of leptin receptors caused by hyperlep-tinemia is believed to contribute to the obese phenotype of theseKO mice. In contrast, obesity in ob/ob mice is caused by leptindeficiency. To characterize the metabolic phenotype, Chga-KOmice were treated with the CHGA-derived peptide catestatin(CST) that is deficient in these mice. CST treatment reduced fatdepot size and increased lipolysis and fatty acid oxidation. Inliver, CST enhanced oxidation of fatty acids as well as theirassimilation into lipids, effects that are attributable to the up-regulation of genes promoting fatty acid oxidation (Cpt1�,Ppar�,Acox, andUcp2) and incorporation into lipids (Gpat andCD36). CST did not affect basal or isoproterenol-stimulatedcAMP production in adipocytes but inhibited phospholipase Cactivation by the �-adrenergic receptor (AR) agonist phenyleph-rine, suggesting inhibition of�-AR signaling by CST. Indeed, CSTmimicked the lipolytic effect of the�-ARblockerphentolamineonadipocytes. Moreover, CST reversed the hyperleptinemia ofChga-KO mice and improved leptin signaling as determined byphosphorylation of AMPK and Stat3. CST also improved periph-eral leptin sensitivity in diet-induced obese mice. In ob/ob mice,CST enhanced leptin-induced signaling in adipose tissue. In con-clusion, our results implicate CST in a novel pathway that pro-motes lipolysis and fatty acidoxidationbyblocking�-ARsignalingas well as by enhancing leptin receptor signaling.
Chromogranin A (CHGA in humans, Chga in mice), a48-kDa acidic secretory proprotein (1–3), gives rise to severalpeptides of biological importance, including the dysglycemichormone pancreastatin (CHGA250–301) (4, 5), the vasodilatorvasostatin (CHGA1–76) (6), and the antihypertensive peptidecatestatin (CST; CHGA352–372),3 which inhibits nicotine-in-duced catecholamine release (7–9). Initially identified as aphysiological brake in catecholamine secretion (7), CST hasbeen established as a pleiotropic hormone having effects onpromoting angiogenesis (10), lowering of blood pressure (8, 11,12), and cardiac contractility (13–15), as well as enhancingbaroreflex sensitivity (16, 17) and heart rate variability (18).In addition to the above cardiovascular functions, CSThas an
antimicrobial activity (19, 20) and also regulates mast cellmigration, cytokine production and release (21), smooth mus-cle cell proliferation (22), and monocyte migration (23). CSTcan act both extracellularly and intracellularly because the pep-tide can cross the cell membrane (24, 25).Fat cell functions are regulated by catecholamines through
four types of adrenergic receptors (AR): �1, �2, �3, and �2 (26,27). Activation of the three �-ARs is positively coupled toadenylyl cyclase by stimulatory GTP-sensitive proteins, result-ing in enhanced production of cyclic AMP. Cyclic AMP acti-vates protein kinase A, which in turn phosphorylates hormone-sensitive lipase, leading to hydrolysis of triglycerides (lipolysis).In contrast, �2-AR activation has the opposite effects on lipol-ysis because it is coupled to inhibitory GTP-sensitive proteins(28–31). Therefore, the net action of catecholamines dependson the balance between�- and�-ARs (27). Sustained activationof sympathetic nervous system or increased plasma cat-echolamines is often associated with desensitization of �-AR(32). In vivo studies have shown that the lipolytic action of cat-echolamines is blunted in obese subjects (33, 34). Cate-
* This work was supported, in whole or in part, by National Institutes of HealthGrant R01 DA011311 (to S. K. M.). This work was also supported by grantsfrom the Department of Veterans Affairs (to S. K. M., D. T. O., and N.-W. C.).
1 To whom correspondence may be addressed: Dept. of Medicine, Universityof California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093-0838. Tel.:858-552-8585,Ext.2637;Fax858-642-6425;E-mail:[email protected].
2 To whom correspondence may be addressed: Dept. of Medicine, Universityof California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093-0838. Tel.:858-552-8585, Ext. 2637; Fax 858-642-6425; E-mail: [email protected].
3 The abbreviations used are: CST, catestatin; DIO, diet-induced obese; AR,adrenergic receptor; NEFA, nonesterified fatty acid(s); PLC, phospholipaseC; AMPK, 5�-adenosine-monophosphate-activated protein kinase; Stat3,signal transducer and activator of transcription 3.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 287, NO. 27, pp. 23141–23151, June 29, 2012Published in the U.S.A.
JUNE 29, 2012 • VOLUME 287 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 23141
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

cholamine-induced regulation of lipolysis through �-ARdesensitization has also been demonstrated in vitro (32, 35).Repeated treatmentwith epinephrine results in the suppressionof basal and epinephrine-stimulated lipolysis in lean and obesesubjects (36). Even the in vivo lipolytic response to epinephrineis desensitized by prior exposure to epinephrine (37). In view ofthe above,we hypothesize that the increased fatmass in hypera-drenergic Chga-KO mice (38) reflects �-AR desensitization byincreased plasma catecholamines (8). Because catecholaminesare known to inhibit leptin secretion (39–41), �-AR desensiti-zation may prevent such inhibition and lead to increased leptinlevel along with the increased adipose mass as found inChga-KOmice and other obese models. Chronic hyperleptine-mia in turn may desensitize leptin receptor and perpetuate theobese phenotype. Therefore, we reasoned that CST could breakthis cycle and reduce obesity by restoring AR and leptin recep-tor sensitivity through normalization of catecholamine and lep-tin levels. Indeed, we found that chronic CST administration toobese Chga-KO mice resulted in a dramatic lean phenotype.CST treatment also reduced body weight and adipose mass inDIO mice without reducing food intake. Interestingly, CSTcould enhance leptin effects on adipose tissue metabolism andsignaling in both DIO and leptin-deficient ob/ob mice. Ourfindings suggest that the reduction in fat mass after chronicCST treatment is due to increased lipolysis and lipid mobiliza-tion through CST action on �2-AR and leptin receptor. In linewith this, CST promoted fatty acid oxidation and leptinsignaling.
EXPERIMENTAL PROCEDURES
Animals—Adult male (7 months old) WT (31.8 � 1.2 g) andChga-KO (39.2 � 1.5 g) mice in the mixed genetic background(129SvJ � C57BL/6) were studied. Both genotypes were gener-ated from the original founder carrying mixed genotype (50%129SvJ, 50% C57BL/6) and were maintained by sibling mating.The animals were kept in a 12-h dark/light cycle and fed stand-ard chow ad libitum. Male C57BL/6mice, 8 weeks old, were feda 60% high fat diet (D12492; Research Diets, Inc., New Bruns-wick, NJ) for 16 weeks before using for experiments. Male lep-tin-deficient (C57BL/6J-ob/ob) mice from the Jackson Labora-toryweremaintained on a standard chowdiet. The institutionalanimal care and utilization committee approved all procedures.Chga-KO,DIO, and ob/obmicewere treated dailywith saline orCST (5 �g/g of body weight intraperitoneally, for 12 days forChga-KO mice and 16 days for DIO and ob/obmice).Measurement of Glycerol, Adipokine, Lipid, and CST Levels
in Blood and in ConditionedMedia—Mice were fasted for 12 hprior to blood draw. Triglycerides and nonesterified fatty acids(NEFA) were assayed using kits fromWako Diagnostics (Rich-mond, VA). Glycerol was assayed using a kit fromSigma.Mediafrom the explant cultures and mouse serum were analyzed forglycerol and NEFA as a measure of lipolysis. ELISA kits wereused to determine plasma levels of leptin, adiponectin (Milli-pore, Billerica, MA), and CST (Bachem, Torrance, CA). ForCST assay, plasma samples and reference standards werepassed through mini C18 columns, and the flow-through frac-tions were assayed. The same kits were used for measurementsin culture media.
Treatment of Fat Pad Explants with CST and Leptin—Adi-pose tissue explants were prepared as described (42). Epididy-mal fat pads from 12-h fasted WT, Chga-KO, DIO, and ob/obmice with or without CST treatment were collected in Krebs-Ringer phosphate buffer containing 10 mM Hepes and 0.5%BSA. The tissues were minced to 1–2mm size and treated with100 nM CST, 1 �M leptin, or saline for 30 min (for signalinganalysis) or 3 h (for lipolysis and �-oxidation assays). Explantswere also treated acutely with CST, leptin, a combination, orsaline. At the end, incubation media were saved for analysis ofglycerol and fatty acids. The explants were washed prior tohomogenization for immunoblotting and analysis of fatty acidoxidation.Preparation of Primary Adipocytes—Primary adipocytes
were isolated from epididymal fat pads essentially as described(43). Adipose tissues from WT and Chga-KO mice wereminced in Krebs-Ringer bicarbonate-Hepes buffer, pH 7.4,containing 10 mM bicarbonate, 30 mM Hepes, 200 nM adeno-sine, 2.5 mM glucose, and 1% fatty acid-free BSA, and digestedfor 30–40 min with Type I collagenase (10 mg/g tissue; Invit-rogen) with gentle swirling in a 37 °C incubator. The digestionmixture was then filtered through a nylon strainer and centri-fuged at 400� g for 1min. The oily layer (released from brokencells) above floating fat cells was skimmed off, and fat cells wererecovered from the top and washed three times with warmKrebs-Ringer bicarbonate-Hepes.Immunoblotting of Signaling Molecules—Adipose tissue
explants after treatments ex vivo and tissues from mice treatedin vivo were homogenized in a buffer containing phosphataseand protease inhibitors (20 mM Tris/HCl, pH 7.5, 250 mM
sucrose, 2 mM EDTA, 2 mM EGTA, 2 mMNa3VO4, 10 mMNaF,2 mM Na4P2O7, 1 mM PMSF, 20 �g/ml leupeptin, 10 �g/mlaprotinin, and 1 �M microcystin-LR) as described (38, 44).Homogenates were subjected to SDS-PAGE and immuno-blotted. Primary antibodies forAMPKand Stat3were fromCellSignaling Technology (Beverly, MA). The chemiluminescencekit was from Pierce.Incorporation and Oxidation of Fatty Acid in Vivo—The
mice were injected with saline or CST (5 �g/g of body weightintraperitoneally, twice daily) for 12 days. One hr after the finalinjection, [U-14C]palmitate (5�Ci, 100�l, 0.2mM)was injectedintraperitoneally, and the mice were sacrificed 3 h later. Liverand adipose tissues (�100 mg) were homogenized in 0.8 ml of3.5 N perchloric acid and extracted by vortexing in 3 ml of amixture of methanol and chloroform (2:1, v/v). To the finalhomogenate, 1.2 ml of 3.5 N perchloric acid was added. Themixture was vortexed and centrifuged. The lower (chloroform)layer contained all the lipids derived from the incorporation of[14C]palmitate, whereas the upper acidic layer contained par-tially oxidized acid-soluble metabolites of [14C]palmitate. Thelower layer was further fractionated by thin layer chromatogra-phy on silica gel plates using a hexane:diethyl ether:acetic acid(79:20:1, v/v/v) mixture as the developing solvent. Lipogenesisfrom palmitate was determined based on the radioactivity ofthe free palmitic acid band compared with other lipids (phos-pholipids, triglycerides, diacylglycerol, etc.) on the TLC plate.Complete oxidation of [14C]palmitate was measured in cul-tured cells but not in mice.
Catestatin and Lipid Mobilization
23142 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 27 • JUNE 29, 2012
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Fatty Acid Oxidation in Explants and Cultured Cells—Oxi-dation of radiolabeled palmitate in the homogenates of adiposetissue explants as described previously (45). For oxidation stud-ies of cultured cells, HepG2 (hepatocytes) and 3T3-L1 preadi-pocytes were obtained fromATCC and cultured following sup-plier’s protocol. Confluent 3T3-L1 cultures were differentiatedinto adipocytes by treating with a mixture of dexamethasone(100 nM), isobutylmethylxanthine (1 �M) and insulin (100 nM)for 10 days. The media were then switched to serum-freeDMEMwith 1% BSA. Hepatocytes were assayed for lipogenesisand fat oxidation in response to CST treatment. Serum-freecultures were treated with CST (100 nM) for 24 h followed by a2-h incubation with 0.2 mM [U-14C]palmitate (0.5 �Ci/ml).Fatty acid oxidation in cells was determined by modifying apublished method (46). Specifically, the culture media in24-well plates were acidified with 10% HClO4 after incubationwith the labeled fatty acid and immediately coveredwith a thickfilter paper sheet (cut to size) soaked in 2 N NaOH and placedunderneath the plastic lid. The whole plate was sealed withparafilm. After further incubation for 2 h, the filter paper sheetwas marked as circles around the rim of the wells, and then thecircles were excised. The filter discs were counted to determinethe amount of 14CO2 absorbed in the papers. The cells in theculture wells were lysed in 1 N NaOH, and protein content wasassayed using Folin’s reagent (Bio-Rad).Real Time PCR—RNA was extracted using a kit (RNeasy
Plus; Qiagen) according to the manufacturer’s specifications.After DNase digestion, 100 ng of RNA was transcribed intocDNA in a 20-�l reaction using a high capacity cDNAkit (Invit-rogen), analyzed, and amplified. PCR was performed in a 25-�lreaction containing 5 �l of cDNA (one-fifth diluted), 2� SYBRGreen PCR Master Mix, and the primers were described inTable 1 (400 nM each). Differences in cycle threshold values(�Ct) between target and the housekeeping geneGAPDH wereused to calculate the levels of expression.Statistics—The data are expressed as themeans� S.E. Curve
fittingwas accomplished using the programKaleidagraph (Syn-ergy Software, Reading, PA). Statistical analyses were done byStudent’s t test or one-way analysis of variance followedbyBon-ferroni’s post hoc test. Statistical significancewas defined as p�0.05.
RESULTS
Effects of CST on Adiposity, Plasma Lipid, and Leptin Levelsin Overweight Chga-KOMice—Plasma CST concentration was3.8 ng/ml in WT mice fed with a normal chow diet (Fig. 1A).Administration of CST (5 �g/g of body weight intraperitone-ally/day for 12 days) to these mice raised CST concentration to7.0 ng/ml (Fig. 1A), a level maintained for at least 4 h (data notshown). High fat diet (60% fat, for 16 weeks) decreased CSTlevels to 2.8 ng/ml, which increased to 5.8 ng/ml upon CSTadministration. CST administration to CST-deficientChga-KOmice achieved a lowerCST level (2.3 ng/ml) thanWTmice. Chronic CST administration (5 �g/g of body weightintraperitoneally/day for 12 days) to Chga-KO mice reducedepididymal fat pad size to WT level (�25% reduction withrespect to body weight of Chga-KO mice) without affectingbody weight (Fig. 1, B and C) or liver weight (saline: 1.20 �0.08 g versus CST: 1.27 � 0.07 g). Interestingly, CST decreasedplasma triglyceride levels in Chga-KO mice (Fig. 1D). Thisdecrease in overall lipid content may be caused in part byincreased lipolysis as shown by increased glycerol and NEFAlevels in plasma (Fig. 1, E and F). We found that Chga-KOmicehave higher leptin levels thanWT (Fig. 1G), consistent with theestablished consequence of pancreastatin deficiency in theChga-KO mice (38). Interestingly, CST treatment of Chga-KOmice lowered plasma leptin to a level belowWT (Fig. 1G), sug-gesting that leptin at subphysiological levels is sufficient tomaintain the Chga-KO mice in a lean state. CST also inhibitedleptin production in cultured 3T3-L1 adipocytes (Fig. 1H), sug-gesting a direct effect on leptin secretion independent of othercirculating factors. Although leptin is known to facilitate fatoxidation anddecrease food intake (47, 48), sustained hyperlep-tinemia may desensitize its receptor and lead to obesity as seenin DIO models (49, 50). We therefore reasoned that CSTrestored leptin action inChga-KOmice by reversing the desen-sitization effect of chronic leptin excess. CST did not increasefood intake ofChga-KOmice (data not shown), suggesting thatCST, despite lowering leptin levels, preserved leptin signalingin the brain. Leptin-deficient ob/ob mice with sensitive leptinreceptors responded to short term CST treatment by reducingfood intake, whereas DIO mice, with peripheral leptin resist-ance and with a barrier against circulating leptin for hypotha-lamic action, failed to respond (as discussed later in Fig. 6,A andB).Effects of CST on Lipogenesis, Fatty AcidOxidation, andGene
Expression in Chga-KO Mice—In Chga-KO mice treated withCST, we found tissue-specific effects on [14C]palmitate incor-poration into lipids. The incorporation was decreased by CSTin adipose tissue but enhanced in liver (Fig. 2, A and B). Incontrast, CST stimulated palmitate oxidation into acid-solublemetabolites in both adipose tissue and liver (Fig. 2, C and D).The effect ofCSTon [14C]palmitate oxidation in cultured hepa-tocytes (HepG2) and adipocytes (3T3-L1) was measured basedon 14CO2 formation (Fig. 2E). Given that adipose tissue in CST-treated mice showed increased palmitate oxidation butdecreased incorporation into lipids, we conclude that CSTinhibits the expansion of adipose tissue and also promotes fattyacid uptake in liver for oxidation. Liver mRNA analyses
TABLE 1Primers for quantitative RT-PCR
Acox1 Forward GTC GAC CTT GTT CGC CAReverse GGT TCC TCA GCA CGG CTT
CD36 Forward TCC AGC CAA TGC CTT TGCReverse TGG AGA ATT ACT TTT TCA GTG CAG AA
Cpt1� Forward CAG GAT TTT GCT GTC AAC CTCReverse GAG CAT CTC CAT GGC GTA G
Gapdh Forward TAT GTC GTG GAG TCT ACT GGT GTReverse GTC ATC ATA CTT GGC AGG TTT CT
Gpat4 Forward TGT CTG GTT TGA GCG TTC TGReverse TTC TGG GAA GAT GAG GAT GG
Ppar� Forward GGG CTC TCC CAC ATC CTTReverse CCC ATT TCG GTA GCA GGT AGT C
Ppar�1 Forward GAG TGT GAC GAC AAG ATT TGReverse GGT GGG CCA GAA TGG CAT CT
Srebp-1c Forward GGA GCC ATG GAT TGC ACA TTReverse GCT TCC AGA GAG GAG GCC AG
Ucp2 Forward CAG CCA GCG CCC AGT ACCReverse CAA TGC GGA CGG AGG CAA AGC
Catestatin and Lipid Mobilization
JUNE 29, 2012 • VOLUME 287 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 23143
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from
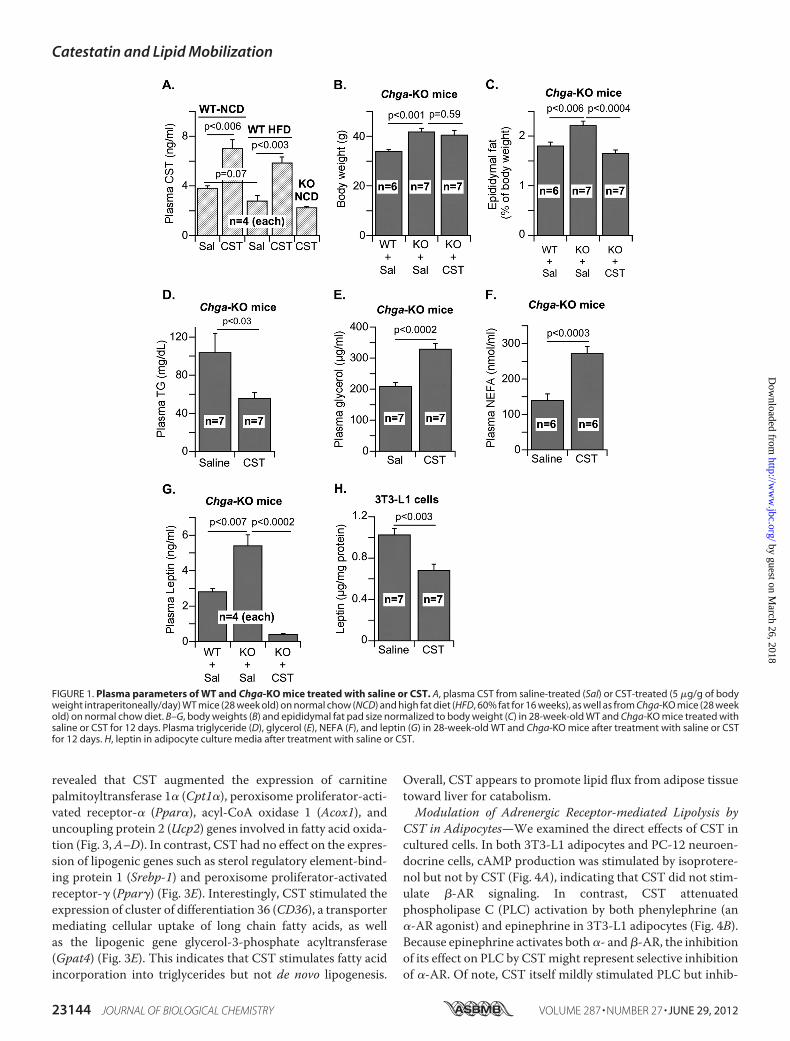
revealed that CST augmented the expression of carnitinepalmitoyltransferase 1� (Cpt1�), peroxisome proliferator-acti-vated receptor-� (Ppar�), acyl-CoA oxidase 1 (Acox1), anduncoupling protein 2 (Ucp2) genes involved in fatty acid oxida-tion (Fig. 3,A–D). In contrast, CST had no effect on the expres-sion of lipogenic genes such as sterol regulatory element-bind-ing protein 1 (Srebp-1) and peroxisome proliferator-activatedreceptor-� (Ppar�) (Fig. 3E). Interestingly, CST stimulated theexpression of cluster of differentiation 36 (CD36), a transportermediating cellular uptake of long chain fatty acids, as wellas the lipogenic gene glycerol-3-phosphate acyltransferase(Gpat4) (Fig. 3E). This indicates that CST stimulates fatty acidincorporation into triglycerides but not de novo lipogenesis.
Overall, CST appears to promote lipid flux from adipose tissuetoward liver for catabolism.Modulation of Adrenergic Receptor-mediated Lipolysis by
CST in Adipocytes—We examined the direct effects of CST incultured cells. In both 3T3-L1 adipocytes and PC-12 neuroen-docrine cells, cAMP production was stimulated by isoprotere-nol but not by CST (Fig. 4A), indicating that CST did not stim-ulate �-AR signaling. In contrast, CST attenuatedphospholipase C (PLC) activation by both phenylephrine (an�-AR agonist) and epinephrine in 3T3-L1 adipocytes (Fig. 4B).Because epinephrine activates both�- and�-AR, the inhibitionof its effect on PLC by CSTmight represent selective inhibitionof �-AR. Of note, CST itself mildly stimulated PLC but inhib-
FIGURE 1. Plasma parameters of WT and Chga-KO mice treated with saline or CST. A, plasma CST from saline-treated (Sal) or CST-treated (5 �g/g of bodyweight intraperitoneally/day) WT mice (28 week old) on normal chow (NCD) and high fat diet (HFD, 60% fat for 16 weeks), as well as from Chga-KO mice (28 weekold) on normal chow diet. B–G, body weights (B) and epididymal fat pad size normalized to body weight (C) in 28-week-old WT and Chga-KO mice treated withsaline or CST for 12 days. Plasma triglyceride (D), glycerol (E), NEFA (F), and leptin (G) in 28-week-old WT and Chga-KO mice after treatment with saline or CSTfor 12 days. H, leptin in adipocyte culture media after treatment with saline or CST.
Catestatin and Lipid Mobilization
23144 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 27 • JUNE 29, 2012
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from
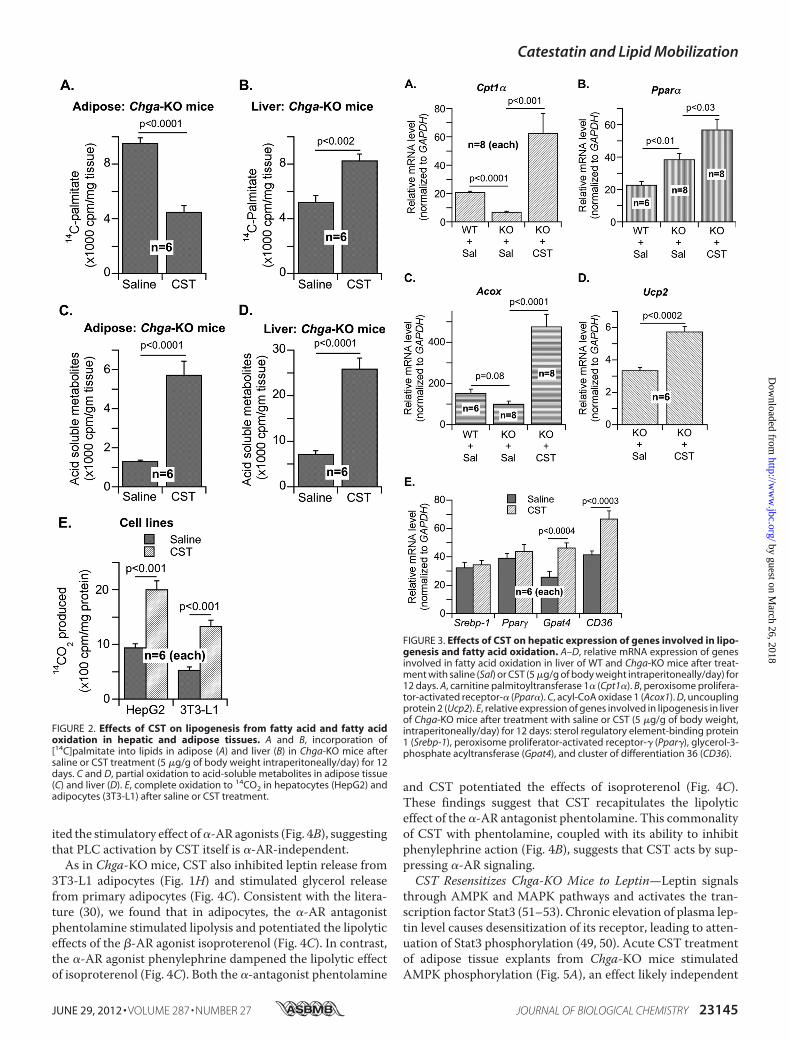
ited the stimulatory effect of�-AR agonists (Fig. 4B), suggestingthat PLC activation by CST itself is �-AR-independent.
As in Chga-KO mice, CST also inhibited leptin release from3T3-L1 adipocytes (Fig. 1H) and stimulated glycerol releasefrom primary adipocytes (Fig. 4C). Consistent with the litera-ture (30), we found that in adipocytes, the �-AR antagonistphentolamine stimulated lipolysis and potentiated the lipolyticeffects of the �-AR agonist isoproterenol (Fig. 4C). In contrast,the �-AR agonist phenylephrine dampened the lipolytic effectof isoproterenol (Fig. 4C). Both the �-antagonist phentolamine
and CST potentiated the effects of isoproterenol (Fig. 4C).These findings suggest that CST recapitulates the lipolyticeffect of the �-AR antagonist phentolamine. This commonalityof CST with phentolamine, coupled with its ability to inhibitphenylephrine action (Fig. 4B), suggests that CST acts by sup-pressing �-AR signaling.CST Resensitizes Chga-KO Mice to Leptin—Leptin signals
through AMPK and MAPK pathways and activates the tran-scription factor Stat3 (51–53). Chronic elevation of plasma lep-tin level causes desensitization of its receptor, leading to atten-uation of Stat3 phosphorylation (49, 50). Acute CST treatmentof adipose tissue explants from Chga-KO mice stimulatedAMPK phosphorylation (Fig. 5A), an effect likely independent
FIGURE 2. Effects of CST on lipogenesis from fatty acid and fatty acidoxidation in hepatic and adipose tissues. A and B, incorporation of[14C]palmitate into lipids in adipose (A) and liver (B) in Chga-KO mice aftersaline or CST treatment (5 �g/g of body weight intraperitoneally/day) for 12days. C and D, partial oxidation to acid-soluble metabolites in adipose tissue(C) and liver (D). E, complete oxidation to 14CO2 in hepatocytes (HepG2) andadipocytes (3T3-L1) after saline or CST treatment.
FIGURE 3. Effects of CST on hepatic expression of genes involved in lipo-genesis and fatty acid oxidation. A–D, relative mRNA expression of genesinvolved in fatty acid oxidation in liver of WT and Chga-KO mice after treat-ment with saline (Sal) or CST (5 �g/g of body weight intraperitoneally/day) for12 days. A, carnitine palmitoyltransferase 1� (Cpt1�). B, peroxisome prolifera-tor-activated receptor-� (Ppar�). C, acyl-CoA oxidase 1 (Acox1). D, uncouplingprotein 2 (Ucp2). E, relative expression of genes involved in lipogenesis in liverof Chga-KO mice after treatment with saline or CST (5 �g/g of body weight,intraperitoneally/day) for 12 days: sterol regulatory element-binding protein1 (Srebp-1), peroxisome proliferator-activated receptor-� (Ppar�), glycerol-3-phosphate acyltransferase (Gpat4), and cluster of differentiation 36 (CD36).
Catestatin and Lipid Mobilization
JUNE 29, 2012 • VOLUME 287 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 23145
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from
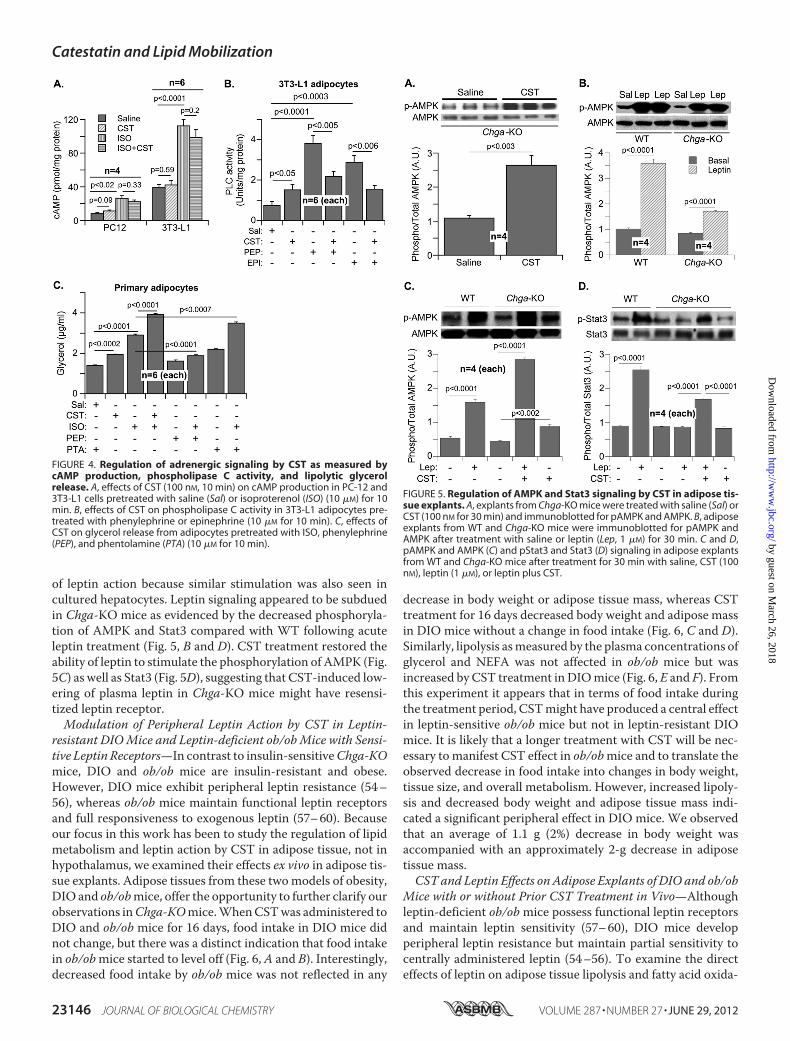
of leptin action because similar stimulation was also seen incultured hepatocytes. Leptin signaling appeared to be subduedin Chga-KO mice as evidenced by the decreased phosphoryla-tion of AMPK and Stat3 compared with WT following acuteleptin treatment (Fig. 5, B and D). CST treatment restored theability of leptin to stimulate the phosphorylation of AMPK (Fig.5C) as well as Stat3 (Fig. 5D), suggesting that CST-induced low-ering of plasma leptin in Chga-KO mice might have resensi-tized leptin receptor.Modulation of Peripheral Leptin Action by CST in Leptin-
resistant DIOMice and Leptin-deficient ob/obMice with Sensi-tive Leptin Receptors—In contrast to insulin-sensitiveChga-KOmice, DIO and ob/ob mice are insulin-resistant and obese.However, DIO mice exhibit peripheral leptin resistance (54–56), whereas ob/ob mice maintain functional leptin receptorsand full responsiveness to exogenous leptin (57–60). Becauseour focus in this work has been to study the regulation of lipidmetabolism and leptin action by CST in adipose tissue, not inhypothalamus, we examined their effects ex vivo in adipose tis-sue explants. Adipose tissues from these twomodels of obesity,DIO and ob/obmice, offer the opportunity to further clarify ourobservations inChga-KOmice.WhenCSTwas administered toDIO and ob/ob mice for 16 days, food intake in DIO mice didnot change, but there was a distinct indication that food intakein ob/obmice started to level off (Fig. 6, A and B). Interestingly,decreased food intake by ob/ob mice was not reflected in any
decrease in body weight or adipose tissue mass, whereas CSTtreatment for 16 days decreased body weight and adipose massin DIO mice without a change in food intake (Fig. 6, C and D).Similarly, lipolysis asmeasured by the plasma concentrations ofglycerol and NEFA was not affected in ob/ob mice but wasincreased by CST treatment in DIOmice (Fig. 6, E and F). Fromthis experiment it appears that in terms of food intake duringthe treatment period, CSTmight have produced a central effectin leptin-sensitive ob/ob mice but not in leptin-resistant DIOmice. It is likely that a longer treatment with CST will be nec-essary to manifest CST effect in ob/obmice and to translate theobserved decrease in food intake into changes in body weight,tissue size, and overall metabolism. However, increased lipoly-sis and decreased body weight and adipose tissue mass indi-cated a significant peripheral effect in DIO mice. We observedthat an average of 1.1 g (2%) decrease in body weight wasaccompanied with an approximately 2-g decrease in adiposetissue mass.CST and Leptin Effects on Adipose Explants of DIO and ob/ob
Mice with or without Prior CST Treatment in Vivo—Althoughleptin-deficient ob/obmice possess functional leptin receptorsand maintain leptin sensitivity (57–60), DIO mice developperipheral leptin resistance but maintain partial sensitivity tocentrally administered leptin (54–56). To examine the directeffects of leptin on adipose tissue lipolysis and fatty acid oxida-
FIGURE 4. Regulation of adrenergic signaling by CST as measured bycAMP production, phospholipase C activity, and lipolytic glycerolrelease. A, effects of CST (100 nM, 10 min) on cAMP production in PC-12 and3T3-L1 cells pretreated with saline (Sal) or isoproterenol (ISO) (10 �M) for 10min. B, effects of CST on phospholipase C activity in 3T3-L1 adipocytes pre-treated with phenylephrine or epinephrine (10 �M for 10 min). C, effects ofCST on glycerol release from adipocytes pretreated with ISO, phenylephrine(PEP), and phentolamine (PTA) (10 �M for 10 min).
FIGURE 5. Regulation of AMPK and Stat3 signaling by CST in adipose tis-sue explants. A, explants from Chga-KO mice were treated with saline (Sal) orCST (100 nM for 30 min) and immunoblotted for pAMPK and AMPK. B, adiposeexplants from WT and Chga-KO mice were immunoblotted for pAMPK andAMPK after treatment with saline or leptin (Lep, 1 �M) for 30 min. C and D,pAMPK and AMPK (C) and pStat3 and Stat3 (D) signaling in adipose explantsfrom WT and Chga-KO mice after treatment for 30 min with saline, CST (100nM), leptin (1 �M), or leptin plus CST.
Catestatin and Lipid Mobilization
23146 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 27 • JUNE 29, 2012
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from
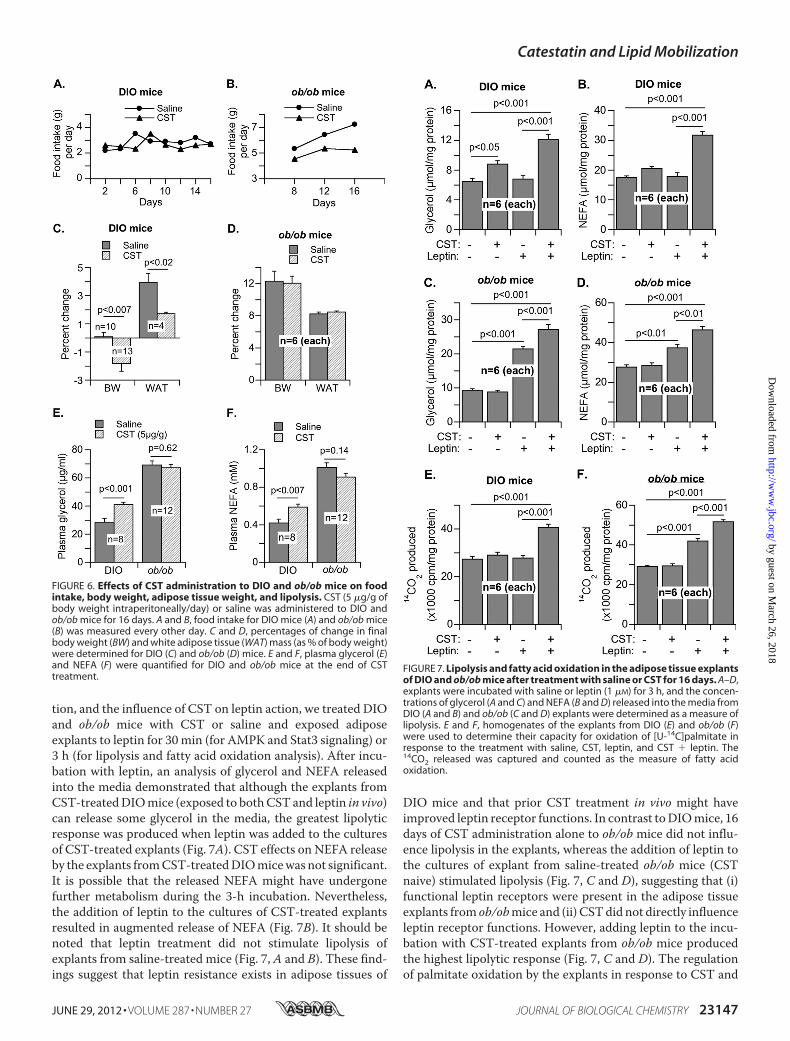
tion, and the influence of CST on leptin action, we treated DIOand ob/ob mice with CST or saline and exposed adiposeexplants to leptin for 30 min (for AMPK and Stat3 signaling) or3 h (for lipolysis and fatty acid oxidation analysis). After incu-bation with leptin, an analysis of glycerol and NEFA releasedinto the media demonstrated that although the explants fromCST-treatedDIOmice (exposed to bothCST and leptin in vivo)can release some glycerol in the media, the greatest lipolyticresponse was produced when leptin was added to the culturesof CST-treated explants (Fig. 7A). CST effects on NEFA releaseby the explants fromCST-treatedDIOmicewas not significant.It is possible that the released NEFA might have undergonefurther metabolism during the 3-h incubation. Nevertheless,the addition of leptin to the cultures of CST-treated explantsresulted in augmented release of NEFA (Fig. 7B). It should benoted that leptin treatment did not stimulate lipolysis ofexplants from saline-treated mice (Fig. 7, A and B). These find-ings suggest that leptin resistance exists in adipose tissues of
DIO mice and that prior CST treatment in vivo might haveimproved leptin receptor functions. In contrast toDIOmice, 16days of CST administration alone to ob/ob mice did not influ-ence lipolysis in the explants, whereas the addition of leptin tothe cultures of explant from saline-treated ob/ob mice (CSTnaive) stimulated lipolysis (Fig. 7, C and D), suggesting that (i)functional leptin receptors were present in the adipose tissueexplants from ob/obmice and (ii) CSTdid not directly influenceleptin receptor functions. However, adding leptin to the incu-bation with CST-treated explants from ob/ob mice producedthe highest lipolytic response (Fig. 7, C and D). The regulationof palmitate oxidation by the explants in response to CST and
FIGURE 6. Effects of CST administration to DIO and ob/ob mice on foodintake, body weight, adipose tissue weight, and lipolysis. CST (5 �g/g ofbody weight intraperitoneally/day) or saline was administered to DIO andob/ob mice for 16 days. A and B, food intake for DIO mice (A) and ob/ob mice(B) was measured every other day. C and D, percentages of change in finalbody weight (BW) and white adipose tissue (WAT) mass (as % of body weight)were determined for DIO (C) and ob/ob (D) mice. E and F, plasma glycerol (E)and NEFA (F) were quantified for DIO and ob/ob mice at the end of CSTtreatment.
FIGURE 7. Lipolysis and fatty acid oxidation in the adipose tissue explantsof DIO and ob/ob mice after treatment with saline or CST for 16 days. A–D,explants were incubated with saline or leptin (1 �M) for 3 h, and the concen-trations of glycerol (A and C) and NEFA (B and D) released into the media fromDIO (A and B) and ob/ob (C and D) explants were determined as a measure oflipolysis. E and F, homogenates of the explants from DIO (E) and ob/ob (F)were used to determine their capacity for oxidation of [U-14C]palmitate inresponse to the treatment with saline, CST, leptin, and CST � leptin. The14CO2 released was captured and counted as the measure of fatty acidoxidation.
Catestatin and Lipid Mobilization
JUNE 29, 2012 • VOLUME 287 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 23147
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from
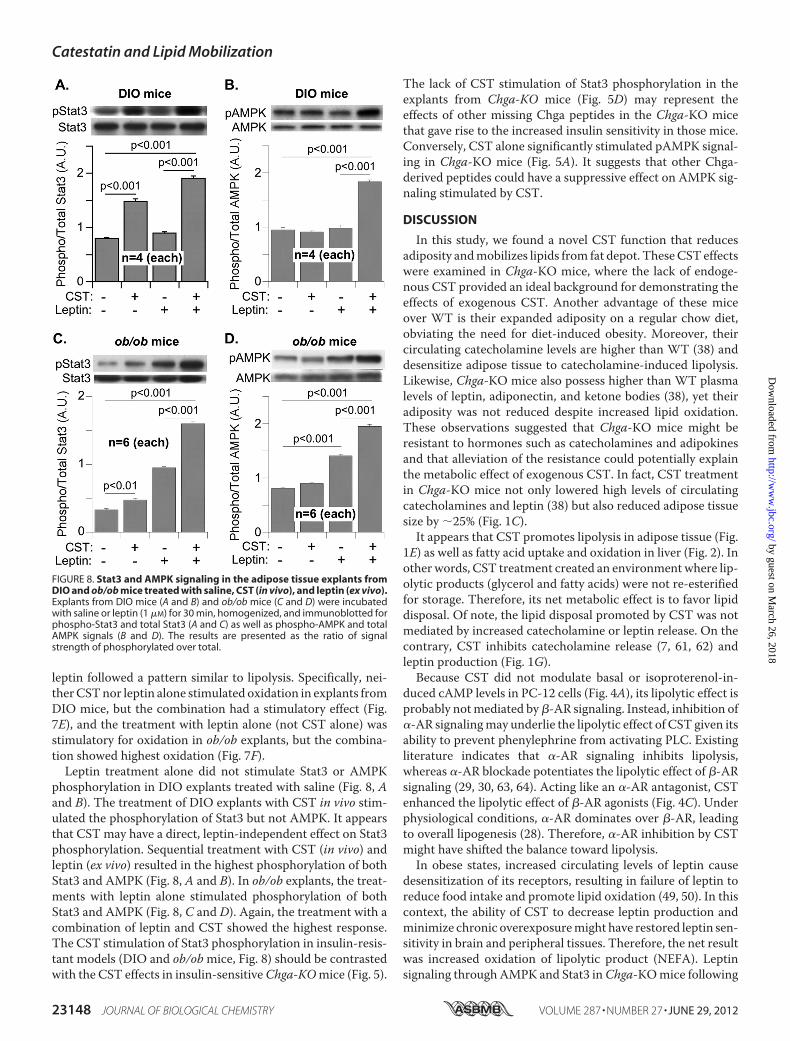
leptin followed a pattern similar to lipolysis. Specifically, nei-therCSTnor leptin alone stimulated oxidation in explants fromDIO mice, but the combination had a stimulatory effect (Fig.7E), and the treatment with leptin alone (not CST alone) wasstimulatory for oxidation in ob/ob explants, but the combina-tion showed highest oxidation (Fig. 7F).Leptin treatment alone did not stimulate Stat3 or AMPK
phosphorylation in DIO explants treated with saline (Fig. 8, Aand B). The treatment of DIO explants with CST in vivo stim-ulated the phosphorylation of Stat3 but not AMPK. It appearsthat CST may have a direct, leptin-independent effect on Stat3phosphorylation. Sequential treatment with CST (in vivo) andleptin (ex vivo) resulted in the highest phosphorylation of bothStat3 and AMPK (Fig. 8, A and B). In ob/ob explants, the treat-ments with leptin alone stimulated phosphorylation of bothStat3 and AMPK (Fig. 8, C and D). Again, the treatment with acombination of leptin and CST showed the highest response.The CST stimulation of Stat3 phosphorylation in insulin-resis-tant models (DIO and ob/obmice, Fig. 8) should be contrastedwith the CST effects in insulin-sensitiveChga-KOmice (Fig. 5).
The lack of CST stimulation of Stat3 phosphorylation in theexplants from Chga-KO mice (Fig. 5D) may represent theeffects of other missing Chga peptides in the Chga-KO micethat gave rise to the increased insulin sensitivity in those mice.Conversely, CST alone significantly stimulated pAMPK signal-ing in Chga-KO mice (Fig. 5A). It suggests that other Chga-derived peptides could have a suppressive effect on AMPK sig-naling stimulated by CST.
DISCUSSION
In this study, we found a novel CST function that reducesadiposity andmobilizes lipids from fat depot. TheseCST effectswere examined in Chga-KO mice, where the lack of endoge-nous CST provided an ideal background for demonstrating theeffects of exogenous CST. Another advantage of these miceover WT is their expanded adiposity on a regular chow diet,obviating the need for diet-induced obesity. Moreover, theircirculating catecholamine levels are higher than WT (38) anddesensitize adipose tissue to catecholamine-induced lipolysis.Likewise, Chga-KO mice also possess higher than WT plasmalevels of leptin, adiponectin, and ketone bodies (38), yet theiradiposity was not reduced despite increased lipid oxidation.These observations suggested that Chga-KO mice might beresistant to hormones such as catecholamines and adipokinesand that alleviation of the resistance could potentially explainthe metabolic effect of exogenous CST. In fact, CST treatmentin Chga-KO mice not only lowered high levels of circulatingcatecholamines and leptin (38) but also reduced adipose tissuesize by �25% (Fig. 1C).It appears that CST promotes lipolysis in adipose tissue (Fig.
1E) as well as fatty acid uptake and oxidation in liver (Fig. 2). Inother words, CST treatment created an environmentwhere lip-olytic products (glycerol and fatty acids) were not re-esterifiedfor storage. Therefore, its net metabolic effect is to favor lipiddisposal. Of note, the lipid disposal promoted by CST was notmediated by increased catecholamine or leptin release. On thecontrary, CST inhibits catecholamine release (7, 61, 62) andleptin production (Fig. 1G).Because CST did not modulate basal or isoproterenol-in-
duced cAMP levels in PC-12 cells (Fig. 4A), its lipolytic effect isprobably notmediated by�-AR signaling. Instead, inhibition of�-AR signalingmay underlie the lipolytic effect of CST given itsability to prevent phenylephrine from activating PLC. Existingliterature indicates that �-AR signaling inhibits lipolysis,whereas �-AR blockade potentiates the lipolytic effect of �-ARsignaling (29, 30, 63, 64). Acting like an �-AR antagonist, CSTenhanced the lipolytic effect of �-AR agonists (Fig. 4C). Underphysiological conditions, �-AR dominates over �-AR, leadingto overall lipogenesis (28). Therefore, �-AR inhibition by CSTmight have shifted the balance toward lipolysis.In obese states, increased circulating levels of leptin cause
desensitization of its receptors, resulting in failure of leptin toreduce food intake and promote lipid oxidation (49, 50). In thiscontext, the ability of CST to decrease leptin production andminimize chronic overexposuremight have restored leptin sen-sitivity in brain and peripheral tissues. Therefore, the net resultwas increased oxidation of lipolytic product (NEFA). Leptinsignaling through AMPK and Stat3 inChga-KOmice following
FIGURE 8. Stat3 and AMPK signaling in the adipose tissue explants fromDIO and ob/ob mice treated with saline, CST (in vivo), and leptin (ex vivo).Explants from DIO mice (A and B) and ob/ob mice (C and D) were incubatedwith saline or leptin (1 �M) for 30 min, homogenized, and immunoblotted forphospho-Stat3 and total Stat3 (A and C) as well as phospho-AMPK and totalAMPK signals (B and D). The results are presented as the ratio of signalstrength of phosphorylated over total.
Catestatin and Lipid Mobilization
23148 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 27 • JUNE 29, 2012
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

acute leptin treatment was subdued compared with WT. CSTtreatment restored leptin action in Chga-KO mice, suggestingresensitization of the leptin receptor.To further clarify the interactions between CST and leptin
pathways and to establish CST as an antiobesity factor, weexamined the effects of CST in leptin-resistant DIO mice andleptin-deficient ob/ob mice. DIO mice are known to exhibitperipheral leptin resistance (54–56).We observed leptin resist-ance in adipose tissue explants from DIO mice where acuteleptin treatment did not stimulate lipolysis, �-oxidation, orphosphorylation of Stat3 and AMPK. However, prior CSTtreatment of DIOmice for 16 days led to the sensitization of allacute leptin effects in the adipose tissue explants, suggestingsensitization of leptin receptor-like functions in adipose tissue.CST administration toDIOmice did not reduce food intake butactually caused amodest reduction in bodyweight proportionalto the loss of adiposemass. As a result, the products of lipolysis,glycerol and NEFA, were increased in serum.Unlike DIO mice, leptin-deficient ob/ob mice maintain lep-
tin sensitivity (57–60). As a result, acute treatment of adiposeexplants with leptin stimulated lipolysis, fatty acid oxidation,and phosphorylation of Stat3 and AMPK. The leptin effectswere enhanced in the explants from CST treated ob/ob mice,whereas CST treatment alone did not show significant periph-eral effects. It should be noted that the treatment of ob/obmicewith CST for 16 days started to reduce food intake (by 25%)resembling a leptin-like effect. This is in contrast to DIO mice,where CST treatment did not reduce food intake. One of thereasons for the failure of leptin to reduce food intake by activat-ing hypothalamic leptin receptors in DIOmice is limited deliv-ery of circulating leptin across the blood-brain barrier. Whenadministered through the intracerebroventricular route, leptincould activate hypothalamic receptor signaling in DIO mice(54–56). It is therefore highly possible that like leptin, intrac-erebroventricular administration of CST might reduce foodintake in DIOmice and enhance hypothalamic leptin response.Administration of CST to DIO mice by the intraperitonealroute, on the other hand, could generate only peripheralresponse and improve peripheral leptin sensitivity. It is alsopossible that CST acted centrally in ob/obmice to reduce foodintake, but longer treatment is required (6–8 weeks instead of16 days) to initiate a reduction of body weight and adipose tis-sue mass. We will address these possibilities in a future work.Restoration of leptin-mediated AMPK signaling and fatty acidoxidation inDIOmice byCSTwith concommitant reduction inbodyweight and fatmass suggests a crucial physiological role ofCST in fine-tuning lipid metabolism to prevent obesity. Wehave also seen that CST stimulates lipolysis by antagonizing�-AR functions. Is there an inverse relationship between�-AR and leptin receptor functions where CST could be akey regulator?Wewill explore these possibilities in future. Inconclusion, our data support an essential role of the endog-enous bioactive peptide CST in restoring homeostasisduring metabolic disorders by controlling catecholaminerelease and lipid disposal via modulation of adrenergic andleptin signaling.
REFERENCES1. Winkler, H., and Fischer-Colbrie, R. (1992) The chromogranins A and B.
The first 25 years and future perspectives. Neuroscience 49, 497–5282. Taupenot, L., Harper, K. L., and O’Connor, D. T. (2003) The chromogra-
nin-secretogranin family. New Engl. J. Med. 348, 1134–11493. Montero-Hadjadje, M., Vaingankar, S., Elias, S., Tostivint, H., Mahata,
S. K., and Anouar, Y. (2008) Chromogranins A and B and secretogranin II.Evolutionary and functional aspects. Acta Physiol. (Oxf.) 192, 309–324
4. Tatemoto, K., Efendic, S., Mutt, V., Makk, G., Feistner, G. J., and Barchas,J. D. (1986) Pancreastatin, a novel pancreatic peptide that inhibits insulinsecretion. Nature 324, 476–478
5. Sánchez-Margalet, V., González-Yanes, C., Najib, S., and Santos-Alvarez,J. (2010) Metabolic effects and mechanism of action of the chromograninA-derived peptide pancreastatin. Regul. Pept. 161, 8–14
6. Aardal, S., Helle, K. B., Elsayed, S., Reed, R. K., and Serck-Hanssen, G.(1993) Vasostatins, comprising the N-terminal domain of chromograninA, suppress tension in isolated human blood vessel segments. J. Neuroen-docrinol. 5, 405–412
7. Mahata, S. K., O’Connor, D. T., Mahata, M., Yoo, S. H., Taupenot, L., Wu,H., Gill, B.M., and Parmer, R. J. (1997)Novel autocrine feedback control ofcatecholamine release. A discrete chromogranin a fragment is a noncom-petitive nicotinic cholinergic antagonist. J. Clin. Invest. 100, 1623–1633
8. Mahapatra, N. R., O’Connor, D. T., Vaingankar, S. M., Hikim, A. P., Ma-hata, M., Ray, S., Staite, E., Wu, H., Gu, Y., Dalton, N., Kennedy, B. P.,Ziegler, M. G., Ross, J., and Mahata, S. K. (2005) Hypertension from tar-geted ablation of chromogranin A can be rescued by the human ortholog.J. Clin. Invest. 115, 1942–1952
9. Mahata, S. K., Mahata, M., Fung, M. M., and O’Connor, D. T. (2010)Catestatin. A multifunctional peptide from chromogranin A. Regul. Pept.162, 33–43
10. Theurl, M., Schgoer, W., Albrecht, K., Jeschke, J., Egger, M., Beer, A. G.,Vasiljevic, D., Rong, S.,Wolf, A.M., Bahlmann, F.H., Patsch, J. R.,Wolf, D.,Schratzberger, P., Mahata, S. K., and Kirchmair, R. (2010) The neuropep-tide catestatin acts as a novel angiogenic cytokine via a basic fibroblastgrowth factor-dependent mechanism. Circ. Res. 107, 1326–1335
11. Fung, M. M., Salem, R. M., Mehtani, P., Thomas, B., Lu, C. F., Perez, B.,Rao, F., Stridsberg, M., Ziegler, M. G., Mahata, S. K., and O’Connor, D. T.(2010) Direct vasoactive effects of the chromogranin A (CHGA) peptidecatestatin in humans in vivo. Clin. Exp. Hypertens. 32, 278–287
12. Gaede, A. H., and Pilowsky, P. M. (2012) Catestatin, a chromogranin A-derived peptide, is sympathoinhibitory and attenuates sympathetic baro-sensitivity and the chemoreflex in rat CVLM. Am. J. Physiol. Regul. Integr.Comp. Physiol. 302, R365–R372
13. Angelone, T., Quintieri, A. M., Brar, B. K., Limchaiyawat, P. T., Tota, B.,Mahata, S. K., and Cerra, M. C. (2008) The antihypertensive chromogra-nin a peptide catestatin acts as a novel endocrine/paracrine modulator ofcardiac inotropism and lusitropism. Endocrinology 149, 4780–4793
14. Mazza, R., Gattuso, A., Mannarino, C., Brar, B. K., Barbieri, S. F., Tota, B.,and Mahata, S. K. (2008) Catestatin (chromogranin A344–364) is a novelcardiosuppressive agent. Inhibition of isoproterenol and endothelin sig-naling in the frog heart. Am. J. Physiol. Heart Circ. Physiol. 295,H113–H122
15. Imbrogno, S., Garofalo, F., Cerra, M. C., Mahata, S. K., and Tota, B. (2010)The catecholamine release-inhibitory peptide catestatin (chromograninA344–363) modulates myocardial function in fish. J. Exp. Biol. 213,3636–3643
16. Gayen, J. R., Gu, Y., O’Connor, D. T., and Mahata, S. K. (2009) Globaldisturbances in autonomic function yield cardiovascular instability andhypertension in the chromogranin a null mouse. Endocrinology 150,5027–5035
17. Gaede, A. H., and Pilowsky, P. M. (2010) Catestatin in rat RVLM is sym-pathoexcitatory, increases barosensitivity, and attenuates chemosensitiv-ity and the somatosympathetic reflex. Am. J. Physiol. Regul. Integr. Comp.Physiol. 299, R1538–R1545
18. Dev, N. B., Gayen, J. R., O’Connor, D. T., and Mahata, S. K. (2010) Chro-mogranin a and the autonomic system. Decomposition of heart rate var-iability and rescue by its catestatin fragment. Endocrinology 151,
Catestatin and Lipid Mobilization
JUNE 29, 2012 • VOLUME 287 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 23149
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

2760–276819. Briolat, J., Wu, S. D., Mahata, S. K., Gonthier, B., Bagnard, D., Chasserot-
Golaz, S., Helle, K. B., Aunis, D., and Metz-Boutigue, M. H. (2005) Newantimicrobial activity for the catecholamine release-inhibitory peptidefrom chromogranin A. Cell Mol. Life Sci. 62, 377–385
20. Radek, K. A., Lopez-Garcia, B., Hupe, M., Niesman, I. R., Elias, P. M.,Taupenot, L., Mahata, S. K., O’Connor, D. T., and Gallo, R. L. (2008) Theneuroendocrine peptide catestatin is a cutaneous antimicrobial and in-duced in the skin after injury. J. Invest. Dermatol. 128, 1525–1534
21. Aung, G., Niyonsaba, F., Ushio, H., Kajiwara, N., Saito, H., Ikeda, S.,Ogawa, H., and Okumura, K. (2011) Catestatin, a neuroendocrine antimi-crobial peptide, induces human mast cell migration, degranulation andproduction of cytokines and chemokines. Immunology 132, 527–539
22. Guo, X., Zhou, C., and Sun, N. (2011) The neuropeptide catestatin pro-motes vascular smooth muscle cell proliferation through the Ca2�-cal-cineurin-NFAT signaling pathway. Biochem. Biophys. Res. Commun. 407,807–812
23. Egger, M., Beer, A. G., Theurl, M., Schgoer, W., Hotter, B., Tatarczyk, T.,Vasiljevic, D., Frauscher, S.,Marksteiner, J., Patsch, J. R., Schratzberger, P.,Djanani, A. M., Mahata, S. K., and Kirchmair, R. (2008) Monocyte migra-tion. A novel effect and signaling pathways of catestatin.Eur. J. Pharmacol.598, 104–111
24. Sugawara, M., Resende, J. M., Moraes, C. M., Marquette, A., Chich, J. F.,Metz-Boutigue, M. H., and Bechinger, B. (2010) Membrane structure andinteractions of human catestatin by multidimensional solution and solid-state NMR spectroscopy. FASEB J. 24, 1737–1746
25. Helle, K. B. (2010) The chromogranin A-derived peptides vasostatin-I andcatestatin as regulatory peptides for cardiovascular functions.Cardiovasc.Res. 85, 9–16
26. Arner, P. (1999) Catecholamine-induced lipolysis in obesity. Int. J. Obes.Relat. Metab. Disord. 23, (Suppl. 1) 10–13
27. Arner, P. (2005) Human fat cell lipolysis. Biochemistry, regulation andclinical role. Best Pract. Res. Clin. Endocrinol. Metab. 19, 471–482
28. Lafontan,M., Barbe, P., Galitzky, J., Tavernier, G., Langin, D., Carpene, C.,Bousquet-Melou, A., and Berlan, M. (1997) Adrenergic regulation of adi-pocyte metabolism. Hum. Reprod. 12, (Suppl. 1) 6–20
29. Stich, V., de Glisezinski, I., Crampes, F., Suljkovicova, H., Galitzky, J., Riv-iere, D., Hejnova, J., Lafontan, M., and Berlan, M. (1999) Activation ofantilipolytic �2-adrenergic receptors by epinephrine during exercise inhuman adipose tissue. Am. J. Physiol. 277, R1076–1083
30. Stich, V., Pelikanova, T., Wohl, P., Sengenès, C., Zakaroff-Girard, A., La-fontan, M., and Berlan, M. (2003) Activation of �2-adrenergic receptorsblunts epinephrine-induced lipolysis in subcutaneous adipose tissue dur-ing a hyperinsulinemic euglycemic clamp in men. Am. J. Physiol. Endocri-nol. Metab. 285, E599–E607
31. Lafontan, M., and Langin, D. (2009) Lipolysis and lipid mobilization inhuman adipose tissue. Prog. Lipid Res. 48, 275–297
32. Mori, S., Nojiri, H., Yoshizuka, N., and Takema, Y. (2007) Rapid desensi-tization of lipolysis in the visceral and subcutaneous adipocytes of rats.Lipids 42, 307–314
33. Jensen, M. D. (1997) Lipolysis. Contribution from regional fat. Annu. Rev.Nutr. 17, 127–139
34. Bougnères, P., Stunff, C. L., Pecqueur, C., Pinglier, E., Adnot, P., and Ric-quier, D. (1997) In vivo resistance of lipolysis to epinephrine. A new fea-ture of childhood onset obesity. J. Clin. Invest. 99, 2568–2573
35. Lafontan, M., and Berlan, M. (1993) Fat cell adrenergic receptors and thecontrol of white and brown fat cell function. J. Lipid Res. 34, 1057–1091
36. Townsend, R. R., Klein, S., and Wolfe, R. R. (1994) Changes in lipolyticsensitivity following repeated epinephrine infusion in humans. Am. J.Physiol. 266, E155–E160
37. Stallknecht, B., Bülow, J., Frandsen, E., and Galbo, H. (1997) Desensitiza-tion of human adipose tissue to adrenaline stimulation studied by micro-dialysis. J. Physiol. 500, 271–282
38. Gayen, J. R., Saberi, M., Schenk, S., Biswas, N., Vaingankar, S. M., Cheung,W. W., Najjar, S. M., O’Connor, D. T., Bandyopadhyay, G., and Mahata,S. K. (2009) A novel pathway of insulin sensitivity in chromogranin A nullmice. A crucial role for pancreastatin in glucose homeostasis. J. Biol.Chem. 284, 28498–28509
39. Fritsche, A.,Wahl, H.G.,Metzinger, E., Renn,W., Kellerer,M., Häring,H.,and Stumvoll, M. (1998) Evidence for inhibition of leptin secretion bycatecholamines in man. Exp. Clin. Endocrinol. Diabetes 106, 415–418
40. Scriba, D., Aprath-Husmann, I., Blum, W. F., and Hauner, H. (2000) Cat-echolamines suppress leptin release from in vitro differentiated subcuta-neous human adipocytes in primary culture via �1- and �2-adrenergicreceptors. Eur. J. Endocrinol. 143, 439–445
41. Couillard, C., Mauriège, P., Prud’homme, D., Nadeau, A., Tremblay, A.,Bouchard, C., and Després, J. P. (2002) Plasma leptin response to an epi-nephrine infusion in lean and obese women. Obes. Res. 10, 6–13
42. Thalmann, S., Juge-Aubry, C. E., andMeier, C. A. (2008) Explant culturesof white adipose tissue.Methods Mol. Biol. 456, 195–199
43. Karnieli, E., Zarnowski, M. J., Hissin, P. J., Simpson, I. A., Salans, L. B., andCushman, S. W. (1981) Insulin-stimulated translocation of glucose trans-port systems in the isolated rat adipose cell. Time course, reversal, insulinconcentration dependency, and relationship to glucose transport activity.J. Biol. Chem. 256, 4772–4777
44. Bandyopadhyay, G. K., Yu, J. G., Ofrecio, J., and Olefsky, J. M. (2005)Increased p85/55/50 expression and decreased phosphotidylinositol 3-ki-nase activity in insulin-resistant human skeletal muscle. Diabetes 54,2351–2359
45. Bandyopadhyay, G. K., Yu, J. G., Ofrecio, J., and Olefsky, J. M. (2006)Increased malonyl-CoA levels in muscle from obese and type 2 diabeticsubjects lead to decreased fatty acid oxidation and increased lipogenesis;thiazolidinedione treatment reverses these defects. Diabetes 55,2277–2285
46. Mao, X., Kikani, C. K., Riojas, R. A., Langlais, P., Wang, L., Ramos, F. J.,Fang, Q., Christ-Roberts, C. Y., Hong, J. Y., Kim, R. Y., Liu, F., and Dong,L. Q. (2006) APPL1 binds to adiponectin receptors and mediates adi-ponectin signalling and function. Nat. Cell Biol. 8, 516–523
47. Seeley, R. J., van Dijk, G., Campfield, L. A., Smith, F. J., Burn, P., Nelligan,J. A., Bell, S. M., Baskin, D. G., Woods, S. C., and Schwartz, M. W. (1996)Intraventricular leptin reduces food intake and body weight of lean ratsbut not obese Zucker rats. Horm. Metab. Res. 28, 664–668
48. Minokoshi, Y., Kim, Y. B., Peroni, O. D., Fryer, L. G., Müller, C., Carling,D., and Kahn, B. B. (2002) Leptin stimulates fatty-acid oxidation by acti-vating AMP-activated protein kinase. Nature 415, 339–343
49. Wang, M. Y., Orci, L., Ravazzola, M., and Unger, R. H. (2005) Fat storagein adipocytes requires inactivation of leptin’s paracrine activity. Implica-tions for treatment of human obesity. Proc. Natl. Acad. Sci. U.S.A. 102,18011–18016
50. Knight, Z. A., Hannan, K. S., Greenberg, M. L., and Friedman, J. M. (2010)Hyperleptinemia is required for the development of leptin resistance.PLoS One 5, e11376
51. Kim, Y. B., Uotani, S., Pierroz, D. D., Flier, J. S., and Kahn, B. B. (2000) Invivo administration of leptin activates signal transduction directly in insu-lin-sensitive tissues. Overlapping but distinct pathways from insulin. En-docrinology 141, 2328–2339
52. Morris, D. L., and Rui, L. (2009) Recent advances in understanding leptinsignaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 297,E1247–E1259
53. Vaisse, C., Halaas, J. L., Horvath, C. M., Darnell, J. E., Jr., Stoffel, M., andFriedman, J. M. (1996) Leptin activation of Stat3 in the hypothalamus ofwild-type and ob/ob mice but not db/db mice. Nat. Genet. 14, 95–97
54. El-Haschimi, K., Pierroz, D. D., Hileman, S.M., Bjørbaek, C., and Flier, J. S.(2000) Two defects contribute to hypothalamic leptin resistance in micewith diet-induced obesity. J. Clin. Invest. 105, 1827–1832
55. Lin, S., Thomas, T. C., Storlien, L. H., and Huang, X. F. (2000) Develop-ment of high fat diet-induced obesity and leptin resistance in C57Bl/6Jmice. Int. J. Obes. Relat. Metab. Disord. 24, 639–646
56. Van Heek, M., Compton, D. S., France, C. F., Tedesco, R. P., Fawzi, A. B.,Graziano, M. P., Sybertz, E. J., Strader, C. D., and Davis, H. R., Jr. (1997)Diet-induced obesemice develop peripheral, but not central, resistance toleptin. J. Clin. Invest. 99, 385–390
57. Pelleymounter, M. A., Cullen, M. J., Baker, M. B., Hecht, R., Winters, D.,Boone, T., and Collins, F. (1995) Effects of the obese gene product on bodyweight regulation in ob/ob mice. Science 269, 540–543
58. Koch, C., Augustine, R. A., Steger, J., Ganjam, G. K., Benzler, J., Pracht, C.,
Catestatin and Lipid Mobilization
23150 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 27 • JUNE 29, 2012
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Lowe, C., Schwartz, M. W., Shepherd, P. R., Anderson, G. M., Grattan,D. R., and Tups, A. (2010) Leptin rapidly improves glucose homeostasis inobese mice by increasing hypothalamic insulin sensitivity. J. Neurosci. 30,16180–16187
59. Frühbeck, G., Aguado, M., and Martínez, J. A. (1997) In vitro lipolyticeffect of leptin on mouse adipocytes. Evidence for a possible autocrine/paracrine role of leptin. Biochem. Biophys. Res. Commun. 240, 590–594
60. Frühbeck, G., Aguado, M., Gómez-Ambrosi, J., and Martínez, J. A. (1998)Lipolytic effect of in vivo leptin administration on adipocytes of lean andob/ob mice, but not db/db mice. Biochem. Biophys. Res. Commun. 250,99–102
61. Mahata, S. K., Mahata, M., Wakade, A. R., and O’Connor, D. T. (2000)Primary structure and function of the catecholamine release inhibitorypeptide catestatin (chromogranin A344–364). Identification of amino acid
residues crucial for activity.Mol. Endocrinol. 14, 1525–153562. Mahata, S. K., Mahata, M., Wen, G., Wong, W. B., Mahapatra, N. R.,
Hamilton, B. A., and O’Connor, D. T. (2004) The catecholamine release-inhibitory “catestatin” fragment of chromogranin A. Naturally occurringhuman variants with different potencies for multiple chromaffin cell nic-otinic cholinergic responses.Mol. Pharmacol. 66, 1180–1191
63. Gómez-Ambrosi, J., Frühbeck, G., Aguado, M., Milagro, F. I., Margareto,J., and Martínez, A. J. (2001) Divergent effects of an �2-adrenergic antag-onist on lipolysis and thermogenesis. Interactions with a �3-adrenergicagonist in rats. Int. J. Mol. Med. 8, 103–109
64. Polak, J., Moro, C., Bessière, D., Hejnova, J., Marquès, M. A., Bajzova, M.,Lafontan,M., Crampes, F., Berlan,M., and Stich, V. (2007)Acute exposureto long-chain fatty acids impairs �2-adrenergic receptor-mediated antili-polysis in human adipose tissue. J. Lipid Res. 48, 2236–2246
Catestatin and Lipid Mobilization
JUNE 29, 2012 • VOLUME 287 • NUMBER 27 JOURNAL OF BIOLOGICAL CHEMISTRY 23151
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Biswas, Nai-Wen Chi, Daniel T. O'Connor and Sushil K. MahataGautam K. Bandyopadhyay, Christine U. Vu, Stefano Gentile, Howon Lee, Nilimafrom Adipose Tissue through Regulation of Adrenergic and Leptin Signaling
) and Novel Effects on Mobilization of Fat372−352Catestatin (Chromogranin A
doi: 10.1074/jbc.M111.335877 originally published online April 25, 20122012, 287:23141-23151.J. Biol. Chem.
10.1074/jbc.M111.335877Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/287/27/23141.full.html#ref-list-1
This article cites 64 references, 14 of which can be accessed free at
by guest on March 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from











![AComparisonofSynaptophysin,Chromogranin,andL ......(CANCERRESEARCH50.6068-6074.September15.1990] AComparisonofSynaptophysin,Chromogranin,andL-DopaDecarboxylaseas MarkersforNeuroendocrineDifferentiation](https://img.pdfslide.net/doc/110x75/5fc9854286c02e7d25410f02/acomparisonofsynaptophysinchromograninandl-cancerresearch506068-6074september151990.jpg)







![Research Article Increased Chromogranin A Cell Density in ...downloads.hindawi.com/journals/grp/2015/823897.pdf · organ in the body [ ]. ese cells project specialized microvilli](https://img.pdfslide.net/doc/110x75/5fd64d49c22ac35b4b7b6b56/research-article-increased-chromogranin-a-cell-density-in-organ-in-the-body.jpg)









