Embed Size (px)
Citation preview

20
CHAPTER 2
CRYSTAL GROWTH METHODS
2.1 INTRODUCTION Growth of better and better quality crystals has been a fascinating research problem benefiting scientific, industrial and technological fields. This is accomplished by adopting a particular crystal growth technique suitable and appropriate to the basic characteristics of the material to be investigated. The art of crystallization stretches far back in the past and antedates than the written history of mankind1). Crystal growth, in principle, as we know today, involves control of a phase change. It may be categorized into three crystal growth processes: Solid growth – S→S process involving solid- solid phase transitions. Melt growth – L→S process involving liquid-solid phase transitions. Vapour growth – V→S process involving gas - solid phase transitions. Accordingly, there are three general categories of crystal growth methods, viz., (1) growth from melt, (2) growth from solution, and (3) growth from vapour. Melt growth can be further subdivided2-6) into (i) growth with crucible7-10) and (ii) growth without crucible11-15). Growth with crucible can be Verneuil Flame fusion method16,17), Float Zone method18,19), Chemical dissolution and zone movement20,21). There are also many growth methods known by their inventors viz. Czochralski8), Bridgemann-Stockbarger12,13), Kapitza14) etc. and by the methodology involved in the system, e.g. floatzone22), vertical gradient freeze23), zone-refining24), liquid phase epitaxy25), directional solidification26), growth under micro- and hyper-gravity27,28). For a monocomponent system, S→S processes are rarely used except for certain metals where strain annealing, sintering, devitrification, polymorphic phase change occurs. The L→S processes are two types: conservative and nonconservative. Directional solidification (Stockbarger-Bridgemann), Cooled seed (Kyropoulos) and Pulling (Czochralski) are conservative techniques. Zoning (horizontal, vertical, float zone, growth on a pedestal) and Verneuil (flame fusion, plasma and arc discharge) are nonconservative techniques. The V→S process is based on sublimation-condensation. For a polycomponent system, S→S processes involve precipitation from solid solution (exsolution). The L→S processes are two types: (1) Growth from solution (evaporation, slow cooling, temperature differential, and solution transport – thermal gradient zone melting) by use of aqueous solutions, organic solutions or molten salt solutions and (2) Growth by reaction (chemical

Ch.2.Crystal growth methods
21
reaction or electro-chemical reaction). In the V→S category crystal grows by reaction-van Arkel, Epitaxial and Gas phase growth of inorganic substances. The choice of a method depends on characteristics of the materials required. Most of the basics of crystal growth techniques and their modifications are described at length by Gilman29), Pamplin30), Brice31), Peiser32), Mullin33) and Vere34). Melting, sublimation, solution and vapor mechanisms are commonly used state changes with temperature variation, and sometimes pressure, as the control element. The important techniques of crystal growth are summarized as follows. 2.2 GROWTH FROM MELT Melt growth is the most widely applied method, especially for the growth of not too high melting point substances32). 2.2.1 CZOCHRALSKI CRYSTAL PULLING TECHNIQUE A versatile Czochralski method8) developed in 1918 for investigating the rate of growth of metal crystals is basically the modification of the technique known as Kyropoulos method10,35) which is even today used for growing solid-state materials like silicon. The process involved in this method is termed as ‘crystal pulling’, since it involves relative motion between a seed and the melt so that crystal is literally pulled out from the melt. The crystal pulling is applicable only to materials that melt congruently. The melt is first raised to a temperature a few degrees above melting point. Then the seed crystal, rotating slowly, is brought slowly into contact with the melt surface, and then lowering is stopped. After getting the desired length, the seeded crystal is slowly and carefully pulled out from the melt The crystal can be observed as it grows and adjustment in both temperature and the growth rate can be made as needed. With suitable precautions, the material withdrawn from the melt solidifies as a large cylindrical crystal. The practical aspects of the method have been discussed at length by Draper36). Fig. 2.1 illustrates schematically the basic principle of the technique. 2.2.2 BRIDGEMANN - STOCKBARGER TECHNIQUE The technique, originated by Bridgemann12) in 1925 and later modified by Stockbarger13), is shown schematically in Fig. 2.2(a). The material to be crystallized is placed in a cylindrical, conical shaped crucible, which can be lowered through a two-zone vertical furnace where the temperatures of upper and lower zones are respectively above and below the melting point of the eventual material. The temperature profile of the growth chamber is shown in Fig. 2.2(b). In some cases the

Ch.2.Crystal growth methods
22
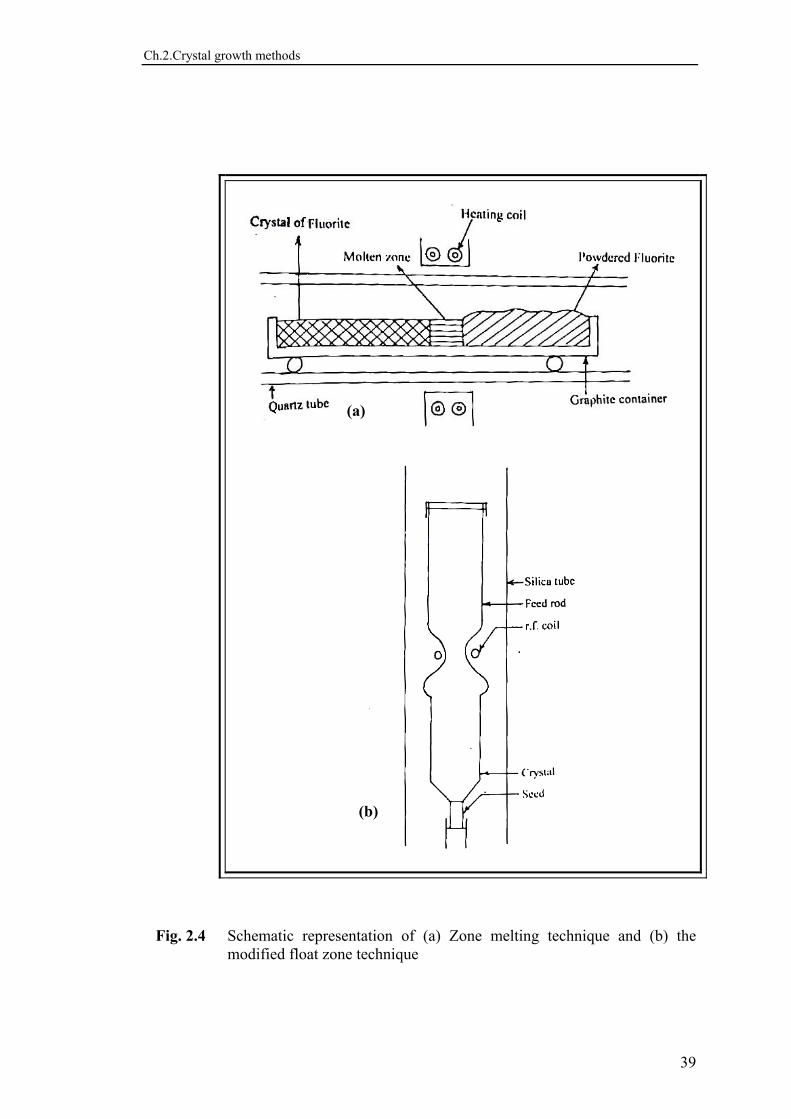
crucible is raised through a furnace. The basic requirement for this procedure is that the freezing isotherm should move systematically through the molten charge, and this can be satisfied by moving the crucible or the furnace, or by changing the furnace temperature. The tip of cone allows restricted nucleation and therefore, under favorable conditions, the material is almost entirely transformed into a large single crystal whose diameter is equal to the internal diameter of the conical crucible. The method is useful in preparation of crystals of metals and semiconductors, alkali and alkaline earth halides, and complex ternary fluorides of alkali and transition metals. This method is, however, not appropriate to materials, which expand on solidification, e.g. aluminium tungstate. 2.2.3 VERNEUIL FLAME FUSION TECHNIQUE This technique, developed by Verneuil in 190216,37), is mainly used to grow crystals with high melting point, like ZrO2 (2700oC), SrO (2400oC) etc. An oxy-hydrogen or oxy-acetylene flame is established and is used for heating purpose. The feed powder of the material to be crystallized is shaken mechanically or electrically from the hopper through a sieve, using a small vibrator with a low amplitude capacity. The flame is made to impinge on a pedestal where a small pile of partly fused alumina quickly builds up. As the pile rises, it reaches into the hotter part of the flame so that the tip becomes completely molten. The molten region increases in size and starts to solidify at the lower end. As more and more powder arrives, the solidifying region broadens into a crystal growing in length. Such a crystal is called boule. The method has been schematically illustrated in Fig. 2. 3. The largest use of this method has been for the growth of gem - quality ruby and emeralds with high melting point and for which no suitable crucible is found. Keck and Gulay18) introduced floating zone variant to produce ultra pure silicon. 2.2.4 ZONE-MELTING TECHNIQUE This technique, discovered by Pfann38) in 1852 was originally used for the purification of semiconductor materials. But since the product is usually crystalline, the technique is also used for growing single crystals. Zone refining technique is the most important zone melting method, where numbers of molten zones are passed along the charge in one direction either horizontally or vertically. This technique is illustrated in Fig. 2.4(a). By moving either the boat or the coil, the molten zone is moved along the boat, thus melting the material in the front portion and solidifying at the back to form the crystalline material. If the conditions are suitable, then the resultant material will be single crystalline. Fig. 2.4(b) shows a modification of the float zone technique, devised by Keck and Gulay18). In this method the material to be

Ch.2.Crystal growth methods
23
purified or grown is arranged in a vertical compacted rod. The molten zone floats below the two solid parts of the rod held in place by surface tension. Each zone carries a fraction of impurities to the end of the charge, thereby purifying the remainder. This technique is used for growing crystals as well, in addition to purifying several metals and compounds. 2.3 GROWTH FROM VAPOUR PHASE Crystals with good perfection can be obtained from the vapour phase. This method is used for production of thin films and those materials with van der Waal bonds. The growth may be due to any of the following processes. 2.3.1 SUBLIMATION This method is carried out in either static or a floating gas system. In a static system, the material is sealed in a tube and placed in a furnace with thermal gradient. The sublimation takes place in hotter portion of the furnace and crystal growth by condensation in the cooler portion. In a flow system, an inert gas is passed through the tube over the material in the hot zone, carrying the gaseous species into the cooler zone where it deposits. The method may be applicable to a material that has reasonably high vapour pressure at higher temperature up to 1000 oC. 2.3.2 CHEMICAL VAPOUR TRANSPORT This process occurs by chemical reactions in which a solid phase reacts with a gas like iodine, bromine, NH4Cl etc. to form vapour phase products. These vapour phase products then undergo reverse reactions, resulting in the reformation of the original solid phase. A large number dichalcogenide crystals have been grown with this procedure39-41). 2.3.3 VAPOUR DECOMPOSITION This method utilizes the irreversible decomposition of a gaseous compound by either chemical or thermal means, such as hydrolysis, to achieve crystal growth. For example, gaseous metal halide can be reduced to a metal in a hydrogen atmosphere or on a hot wire (1500 oC). Crystals grown by this technique are usually of small size but can be of high perfection. 2.4 GROWTH FROM SOLUTION
This is the simplest and one of the oldest methods42) of growing crystals in which the material to be crystallized is dissolved in a solvent to the desired degree of

Ch.2.Crystal growth methods
24
supersaturation. The solution is then slowly cooled or evaporated. If a suitable solvent is found, crystals can be grown at temperatures much below the melting point of the eventual crystal. The low temperatures involved here indeed relieve demand on expensive furnaces and power supplies. Crystal growth from aqueous solutions has been extensively and phenomenologically studied by measuring the concentration and temperature gradient around crystals growing in two-dimensional cell at the growth interface. The growth rate of the crystals is mostly found to be proportional to the normal component of the gradients43). 2.4.1 GROWTH FROM WATER SOLUTION This method is extensively used for obtaining single crystals of organic and inorganic materials. Two basic methods (cooling and evaporation) are used to grow large crystals from water solution. In both the cases, a saturated solution is prepared and the seed crystal is inserted. In one of the methods, temperature is lowered slowly so as to reduce the solubility and produce crystallization, while in the other method, the temperature is held constant and the solvent is made to evaporate isothermally to induce crystallization. Crystals like alkali halides44), sodium borate45), barium strontium nitrate46), Rochelle salt47), potassium and ammonium dihydrogen phosphate48-50), Ammonium Oxalate51,52), Potassium Hydrogen tartrate53), potash alum54), oxalic acid have been grown from water solution. 2.4.2 HYDROTHERMAL METHOD This method of crystal growth, schematically illustrated in Fig. 2.5, using aqueous solution at high temperature and pressure, was first used by Spezia55) to grow quartz hydrothermally, and quartz is still the prime material grown commercially hydrothermally on a large scale. To obtain even a low solubility of quartz in water, the temperature of water well above boiling point is necessary. To prevent the water from the boiling away, necessary pressure is applied. As this solubility is not sufficient for satisfactory growth, a mineralizer is added to the system. The method is carried out using a sealed high pressure vessel known as autoclave or bomb. Special, strong, corrosion-resistant and chemically inert material is used for the construction of an autoclave to withstand high pressure and temperature. It is kept at two different temperature regions. In the upper cooler part, seed material is supported while in the lower hotter part, feed material is used. The rate of growth depends on the temperature difference between top and bottom of the autoclave, pressure and the amount of mineralizer present. When hot solution from the bottom rises into the cooler part of the autoclave on account of convection, excess material gets deposited on the seed, which then grows in size.

Ch.2.Crystal growth methods
25
2.5 GROWTH FROM FLUX It is a high temperature solution growth method56) by which a wide range of materials in the form of single crystals57) are grown. In this method slow cooling or evaporation of the solvent from homogeneous mass has been mainly employed. In both these cases crystallization occurs by spontaneous nucleation. Control of the growth is usually restricted to external temperature region. Since the process is normally carried out in the sealed or partly sealed platinum crucibles, we cannot measure the growth kinetics. The components of the desired material are dissolved in a suitably chosen solvent, the so called ‘flux’, to form the high temperature solution which throws out the insoluble, desired phase by enforcing either slow cooling, or isothermal solvent evaporation, or gradient transport. The method is applicable for the growth of such materials which undergo profound structural transitions at temperature near their melting points which are usually much above the growth temperature. Elwell56), Wanklyn58), Brice59) and Scheel60) have given excellent reviews about practical aspects of the technique. 2.6 GEL GROWTH For certain materials with high or/and incongruent melting points, and those which are insoluble in water, and which decompose before melting at atmospheric pressure, and for which a suitable solvent is not available, gel method offers an attractive advantage. Since cadmium oxalate is one such material that has the above-mentioned limitations, gel method is applicable for it. 2.6.1 HISTORICAL The origin of the gel technique may be traced back to 1896 when the German colloidal chemist and photographer R. E. Liesegang61-63) observed periodic precipitation of slightly soluble salts in gelatin. He applied a thin layer of gelatin on a glass plate impregnated with potassium chromate, and then added above it a drop of silver nitrate. Consequently, silver chromate got precipitated out in the form of a number of well–defined concentric rings. This observation inspired workers like Ostwald64) Hatschek65), Holmes66-68), Rayleigh69), Bradford70), Fells and Firth71), Morse and Donnay72) etc. who experimented on reactions in various colloidal media. Fisher and Simons73,74) first claimed that gels could form excellent media for crystal growth. Interest in gel growth, due to its immense potential and usefulness was revived by Stong75). Soon thereafter, Vand et al76) published a communication describing results of their preliminary crystal growth study. A comprehensive work on

Ch.2.Crystal growth methods
26
the gel method by Henisch and co-workers77-81) gave momentum to crystal growers the whole world over. A decade later, the excellent review by Arora82) appeared which critically imparted useful information by classifying systematically different variants and modifications of the method. 2.6.2 GELS: NATURE, PREPARATION AND PROPERTIES Gel is a highly viscous two-component system of semi-solid nature, rich in liquid and having fine pores in it, It is also referred to as hydrogel or waterglass or silica gel or agar agar gel. The effective pore diameter of silica hydrogel is of the order of 50 – 160 Å. The gel is made of polymeric chains, either entangled in the case of physical gels or cross linked in the case of chemical gels. For physical gels, the gelling process is achieved through a variation of physical parameters while for chemical gels the gelling process results from a polymerization reaction. The various types of physical and chemical gels are: Sodium metasilicate (also known as silica hydrogel, waterglass or silicate glass) Agar gel (a carbohydrate polymer derived from seaweeds) Gelatin gel (resembling protein structure) Clay gel Soap fluid Polyacrylamide gel Dense solutions of metal hydroxides Polyvinyl alcohol Certain oleates, stearates, aluminates, etc. The gels for crystal growth have been reported in the litrature83-88) to be prepared by use of a number of treatments like warming, cooling, chemical reaction, addition of external reagent for gelation. Silica hydrogel has been the most commonly used due to its far better stability than all organic gels89,90), though in certain specific cases gelatin gel91,92), agar agar gel93-101) and polyacrylamide gel102,103) have been preferred. In some other cases104), both inorganic and organic gels have been found equally competitive for crystal growth. Silica hydrogels are prepared from a solution of Na2SiO3 ; Na+ cations are exchanged over an acidic resine (amberlite) and the resulting solution of H2SiO3
is left for gelling. To obtain silica hydrogel medium of desired pH, requisite amount of a suitable acid is normally added to the constantly agitated aqueous solution of Na2SiO3·9H2O. The acidity of the resulting solution determines the course and rate of polymerization105). The gelling process and the

Ch.2.Crystal growth methods
27
mechanical properties of a gel depend on the density and on the conditions prevailing during gelling. Denser gels exhibit concoidal fracture surfaces similar to glass. 2.7 AGAR AGAR GELS Agar gel, more correctly known as agar-agar has been used in the East for several hundred years and certainly since the seventeen century. Agar is traditionally claimed to have been discovered by Tarazaemon Minoya in 1658 in Japan. The word "agar" comes from the Malay word agar-agar (meaning jelly). It is also known as kanten, China grass, or Japanese isinglass meaning "cold weather," referring to the fact that it is harvested in the winter months. The various species of alga or seaweed from which agar is derived are sometimes called Ceylon moss. Gracilaria lichenoides specifically is referred to as agar-agar or Ceylon agar. Agar or agar agar is a gelatinous substance derived from seaweed. Historically, it is chiefly used as an ingredient in deserts throughout Japan, but in the past century it has found extensive use as a solid substrate to contain culture medium for microbiological work. Commercially it is derived primarily from Gelidium amansii. Chemically, agar is a polymer made up of subunits of the sugar galactose i.e. a strong gelling agent , an unbranched polysaccharide (or agarose) obtained from the cell walls (membranes) of red algae, primarily from the genuses Gelidium and Gracilaria, or seaweed (sphaerococcus euchema). Nutrient agar is used throughout the world to provide a solid surface containing medium for the growth of bacteria and fungi.
2.7.1 STRUCTURE
Chemically, Agar is a repeat unit of 3-6, anhydro L-Galactose. It also contains sulphate esters in low levels and some methoxy groups. The original structure of agar was believed to be a simple sulphated poly galactose. However, Araki106) showed the evidence proving the heterogeneity of the agar by separating the agar into two different polysaccharides using the acetylation method. One is called agarose and other aparopectin. The agarose is a virtually neutral polymer, while the agaropectin is a charged (acidic) polymer. Later, he pointed out that agar is a mixture of a neutral, dominating polysaccharide and that the component having the greatest gelling tendency in agarose (shown in Fig.2.6a and b). Agarose, which is a major component of agar-agar, is ideally composed of (1-4) linked 3,6-anhydro-α-L-galactose and alternating with (1-3) linked -β-D- galactose107), as a linear polymer structure shown in Fig.2.6(b). The latter molecule forms a helix and interaction of the helix causes gel formation108,109). A agaropectin has the same repeating unit as agarose but weakly

Ch.2.Crystal growth methods
28
substituted by charged groups as sulfate, methyl, and pyruvic acid acetal groups110), which inhibit gel formation. The degree of methyl groups on the agarose depends on the red seaweed variety. Agarose is difficult to extract from agar and commercial agarose may therefore contain various substituents. However agar is actually a very complex polysaccharide. The structure of agar and agarose preparation varies considerably depending on the source and the method of extraction111). In summary agar can be considered to consist mainly of alternating β–(1-3)-D and α-(1-4)-L linked galactose residues. Most of the α-(1-4) residues are modified by the presence of a 3,6 anhydro bridge. The other modifications that can be found are mainly substituents of sulphate, pyruvate, uronate or methoxyl groups112). It may be noticed that the hydrogen bonding plays a major role in the gelation mechanism of agarose gels, and that it is the case in agar-agar gels since agarose plays a major role in the gel formation of agar-agar gels. An X-ray diffraction study113) of agarose gel as a function of concentration revealed that molecules are in the form of double helices inter connected with side by side association. 2.7.2 PROPERTIES Agar has the ability to form gels upon cooling of a hot solution to 30 – 40 °C and depending on the seaweed source, the setting temperate can be as low as 30 oC and is typically between 30 - 40 oC for a 1.5% solution. Agar melts at 90 oC and gelified from 30 - 40 oC (becomes gelatinous). At temperatures above the melting point of the gel, thermal agitation overcomes the tendency to form helices and the polymer exists in solution as a random coil. On cooling, a three-dimensional network builds up in which double helices form the junction points of the polymer chains. Further cooling leads to aggregation of these junction points. As the polymerization continues, during the process of gelation, water accumulating on top of the gel surface, referred to a phenomenon known as ‘syneresis’, oozes away gradually to result into a set gel. According to Rees108) agar forms antisymmetric double helices on cooling that hydrogen bond to form clumps of helices. These clumps can then form larger grouping that from a large porous gel structure, behaving like a sponge. An agar agar gel of a particular shape can be dried, and upon rehydration it will swell to its original size and shape. In order to obtain Agar gel medium of desired density, requisite amount of agar is taken and heated above 90 oC, then allowed to be gelified. The density of the resulting solution determines the course and rate of polymerization. The gelling process itself takes an amount of time which can vary widely from minutes to many days, depending on the nature of the raw material, its temperature and history. The

Ch.2.Crystal growth methods
29
mechanical properties of a fully developed gel can vary widely, depending on the density and on the precise conditions prevailing during gelling. A relation between weight loss and shrinkage114) during gel drying suggests the effect of microsyneresis to be more conspicuous at lower temperature, whence the gel becomes so turbid that the expulsion of liquid from the pores does not increase the fraction of solid phase in the gel. The viscosity of an agar solution at constant temperature and concentration is a direct function of the average molecular weight. The viscosity rarely exceeds 10–15 cp. at 1% concentration at 60–90 °C. Usually the viscosity is lower as the gel strength is greater for the agar solution. The average molecular weight of agar ranges from 8,000 to greater than 100,000. Helberstadt et al115) showed that the gel consists of sheet-like structure of varying degree of surface roughness and porosity, forming interconnected cells. The cell walls in dense gels have pores less than 0.1 to 0. 05µ while 0.1 to 4.0µ for low-density gels. Some factors affecting crystal growth in gel are: nature, strength and purity of agar agar gel, environmental temperature, ageing time, impurities and many other variables. 2.8 METHODS OF CRYSTALLIZATION FROM GELS With enormous flow of information regarding crystal growth in gels, the gel technique can be categorized into the following four basic methods, each one of them having specific merits: a) Reaction method b) Complex-decomplexion method c) Chemical reduction method d) Solubility reduction method a) REACTION METHOD Reaction method is the basis of all the methods of gel growth and therefore it is known as crystal growth by chemical reaction. Two soluble reactants are allowed to diffuse through a gel column where they react and form an insoluble or relatively less soluble crystalline product116). The chemical reaction taking place can be illustrated as: Ax + By → Ay + Bx (1) where A and B are cations and x and y are anions. The requirements to grow single crystals by this method are:

Ch.2.Crystal growth methods
30
1. The reactants employed must have a solvent (usually water) and the product crystal must be relatively less soluble in that solvent.
2. The gel must remain stable in presence of reactants and must not react with either the reactant or with the product.
Agar Agar gel has been used as the growth medium in the present study. The gel medium is prepared by dissolving agar agar power in distilled water above 90 oC and allowed to be set. The growth process involves controlled diffusion of divalent chloride or nitrate solution into agar agar gel embedded with oxalate ions. The general chemical reaction, which can be used for preparation of oxalate crystals, using chloride solution in general, can be expressed as
C2 H2O4 + XCl2 → XC2O4 + 2HCl (2)
Here X is a divalent cation such as Cd+2, Ba+2, Zn+2, Pb+2, Sr+2, Ni+2, etc. For the monovalent cation, chemical reaction is expressed as
C2 H2O4 + 2YCl → Y2 C2O4 + 2HCl (3)
where Y is a monovalent cation such as Rb+, Cs+, K+, Na+, Li+, NH4
+, etc. Likewise, for the hydrogen oxalate family, the chemical reaction is expressed as
C2 H2O4 + YCl2 → Y C2O4 +2 HCl (4)
If we use the nitrate salts, instead, the chemical reaction can be expressed as
C2 H2O4 + X(NO3)2 → XH C2O4 + HNO3 (5)
C2 H2O4 + 2Y NO3 → Y2 C2O4 + 2HNO3 (6)
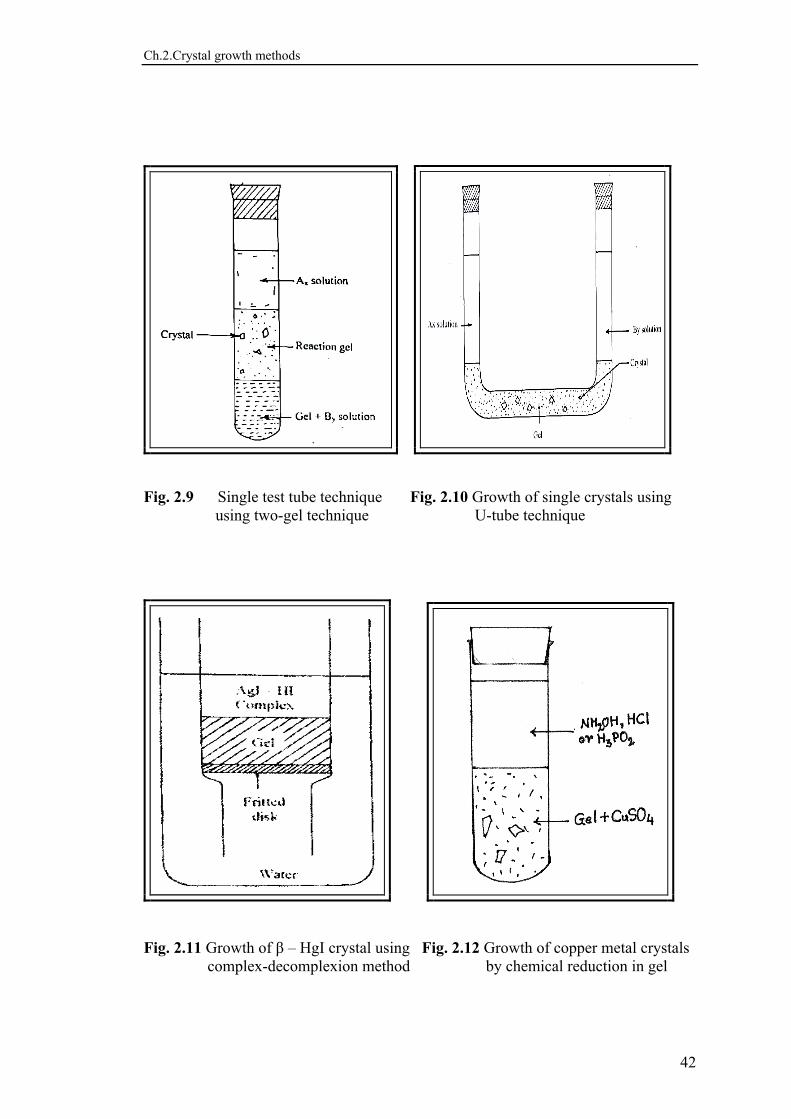
In the simplest way, one reactant is to be incorporated in gel mixture of proper pH before occurrence of gelation in a test tube or a beaker, and pour the other reactant on the set gel (Figs.2.7, 2.8, 2.9). The U-tube (Fig. 2.10) may be employed for neutral gelation and then the two reacting solutions are poured in the two vertical limbs. For greater degree of lateral diffusion of ions to growth sites, the modified apparatus117,118) were devised in this laboratory. The arrangement has successfully been employed to grow mixed single crystals of SrxCa1-xC4H4O6·4H2O, and this happens to be the first report118) in the literature for the growth of a series of mixed configuration in gel media. b) COMPLEX-DECOMPLEXION METHOD

Ch.2.Crystal growth methods
31
First reported by O’Connor et al119)., the method consists of first forming a chemical complex of the material of the eventual crystal to be grown with an aqueous solution of some suitable substance called complexing agent, in which the former is homogeneously miscible, and then providing externally a condition conducive to decomplex or dissociate the complex formed. A standard procedure120) adopted for decomplexion is to steadily increase the dilution while the complexed solution is diffusing through the gel. This method has been successively employed to grow good ionic conductors viz. β -AgI121), α-HgS and β-HgS122). The schematic diagram given in Fig. 2.11 illustrates fineness of the procedure. c) CHEMICAL REDUCTION METHOD This method is suitable for growing only metal crystals. Hatschek and Simons123) grew gold crystals by adding 8% oxalic acid solution over a set gel which contained gold chloride solution. Kratochvil et al124) grew gold crystals of triangular and hexagonal habits. Lead125), Copper126) and Cupric acid 127) crystals have been grown by this method, as schematically illustrated in Fig. 2.12 for copper. d) SOLUBILITY REDUCTION METHOD This method is particularly suitable for growing single crystals of water-soluble substances. Glocker and Soest128) first reported the growth of ammonium dihydrogen phosphate (ADP) single crystals by this method. The substance to be grown is dissolved in water and incorporated with the gel forming solution. After setting the gel, a solution which reduces the solubility of the substance is added over the set gel to induce crystallization. Crystals of KCl and NaCl crystals have been grown by adding concentrated HCl over the gel containing a saturated solution of potassium chloride and sodium chloride129). The crystallization occurs due to reduction of the solubility of potassium chloride and sodium chloride in the liquid phase by concentrated hydrochloric acid. Potassium dihydrogen phosphate crystals were grown130) by use of ethyl alcohol. 2.9 NUCLEATION AND GROWTH MECHANISM The graded neutral gel technique (GNGT) was evolved to control the nucleation in the gel growth of crystals131). To suppress nucleation, whether it is homogeneous or heterogeneous, and to stabilize the concentration gradients in the neighbourhood of growing crystals are the two known principal functions of gel. Mostly nucleation is observed, involving the concept of critical nucleus. As a result of statistical coincidence, a number of atoms or molecules can come together and form a rudimentary crystal.

Ch.2.Crystal growth methods
32
This crystal is likely to dissolve again unless it reaches a certain critical size. Beyond this size, the energy relations do favour continued growth. Heterogeneous nucleation can occur in presence of foreign particles. Systematic experiments with filtering media to avoid heterogeneous nucleation could not succeed fully because the nuclei were too small and most of the filters appeared to add some particles, while they subtracted others. In gel growth systems, the crystals become increasingly scarce and more perfect with increasing distance from gel interface, since slow diffusion of ions at greater depths below the gel interface leads to perfection. As far as the mechanism by which crystals grow in gels is concerned, there have been evidences of two dimensional piling and spreading of growth layers predominantly taking place from one or more initiation centers132,133). The fact that the large crystals at times can be obtained in gels proves that the supersaturation near the surface of these crystals must be much higher in gel than in solution. The suppression of ionic velocities in gels prevents the development of heterogeneous nuclei. What, in fact, goes in the medium is the diffusion of dissolved matter as a consequence of the casual character of thermal motion of molecules. The molecules pass through an energy barrier ∆G, which is the free energy of activation for the molecular transport process. A molecule or an ion changes its place with a frequency given by,
( )kT∆Gexph
kTτ 1 −⋅=− (7)
But, according to Einstein134) the root mean square displacement λ in time τ is
λ = (2Dτ)1/2, (8) where D is the diffusion constant. Putting λ = d, the ionic diameter, the frequency of nucleation is
( )kT∆Gexph
kTd
2Dτf 21 −⋅=== − (9)
The energy ∆G is, of course, equivalent to Ec, the energy available for the creation of crystal nucleation and is given by
223
c Lπ
ρσ
316E ⋅⋅= (10)
where σ is the surface energy per unit area, ρ is the droplet density and L is the heat of desolvation. According to Fick’s law,
J = -D grad C (11)

Ch.2.Crystal growth methods
33
dC/dt = D∆2C, (12) where J is the quantity of matter transporting per unit area in the direction of the concentration gradient perpendicular to ∆C. Assuming that the precipitates are not too close to one another, as is true in gel growth, the concentration in space around the crystal of radius R is a function of the distance from the centre, and the boundary conditions are:
C(x, o) = Cα
C(R, t) = S (13)
C(∞,t) = C The solutions of equations (11) and (12) give the radius R as a function of speed V of the advancing growth fronts in gel:
R = [DV(C∞-S) t]1/2 (14) Since each particle is to be treated independently of the others, the factor V(C∞-S) is small and hence the equation (14) reduces to
R = (Dt)1/2 (15) The equation (15) is typical of one dimensional diffusion process and has been experimentally verified, for example, first by Liaw and Faust135). So, it is basically the directional diffusion which is found responsible for crystal growth in gels.
REFERENCES
1) Birringuccio, pirotechnica (1540) 2) W. Shockley and G. L. Pearson, Phys. Rev. 74 (1948) 74 3) L. T. Canham, Appl. Phys. Lett. 57 (1990) 1046 4) J. A. Sekhar, J. Cryst. Growth 109 (1991) 113 5) T. Ashley et al., SPIE Inst. Soc. Opt. Engg. (USA), 1361 (1991) 238 6) S. Kuppurao and J. J. Derby, J. Cryst. Growth 172 (1997) 350 7) R. Nacker and Neues Jahrab, Mineral Geol. 2 (1915) 133 8) J. Czochralski, Physik Z. Chem. 92 (1918) 219 9) G. Tamman, Metallography, Trans. Dean and Svenson (Chemical catalog Co.
New York, 1925) 10) S. Kyropoulos, Anaorg Z. Chem., 154 (1926) 308 11) J. Obreimov and L. W. Shubnikov, Physik. Z. 25 (1924) 31 12) P. W. Bridgemann, Proc. Am. Acad. Arts. and Sci. 60 (1925) 305 13) D. C. Stockbarger, Rev. Sci. Instr. 10 (1939) 205 14) P. Kapitza, Rev. Soc. (London) A119 (1928) 368

Ch.2.Crystal growth methods
34
15) F. Stober, Krist. Z. 61 (1925) 299 16) A. Verneuil, Compt. Rend., 135 (1902) 791 17) P. H. Keck et al., Rev. Sci. Instr. 25 (1959) 298 18) P. H. Keck and M. J. Gulay, Phys. Rev. 89 (1950) 1297 19) W. G. Pfann and K. M. Olsen, Phys. Rev. (1953) 322 20) F. H. Horn, J. Electrochem. Soc. 105 (1958) 393 21) W. C. Dash, J. Appl. Phys. 31 (1960) 736 22) H. C. Theurer, US Patent (1952) 3060123 23) J. Kang et al., J. Cryst. Growth 140 (1994) 435 24) N. L. Parr, Zone Refining and Allied Techniques, Newnes, London (1960) 25) J. C. Brice, Growth of Crystals from melt, vol.5, Solid State Physics, (P.
Wohlfarth Ed. North Holland, Amsterdam,1965) 26) D. T. J. Hurle, Handbook of Crystal Growth, Allied Pub. 12 (1994) ch. 15 27) E. Lendvay et al., J. Cryst. Growth 71 (1985) 538 28) L. L. Regel and O. V. Shumaev, J. Cryst. Growth 119 (1992) 70 29) J. J. Gilman, The Art and Science of Growing Crystals (Wiley, New York,
1963) 30) B. R. Pamplin, Crystal Growth (Pergamon Press, Oxford, 1980) 31) J. C. Brice, Crystal Growth Processes (Wiley, New York, 1986) 32) H. S. Peiser, Crystal Growth (Pergamon Press, Oxford, 1967) 33) J. W. Mullin, Crystallization 4th Ed., Nutterworth Heinmann pub. (2001) 34) A.W. Vere, Crystal Growth (Principles and Progress Plenum Press,1987) 35) Yoshio Kagebayashi, Yusuke Mori and Takatomo Sasaki, Bull. Mater. Sci. 22
(1999) 971 36) R. C. J. Draper, Crystal Growth, Ed. B. R. Pamplin, (Pergamon Press, Oxford, U.
K. 1975) 37) A. Verneuil, Ann. Chim. Phys. 3 (1904) 20 38) W. G. Pfann, Trans AIME, 194 (1952) 747 39) S. K. Srivastava, T. K. Mandal and B. K. Samantaray, Synthetic Metals 90
(1997) 135 40) H. Tributsch, J. Electrochem. Soc. 107 (1978) 1087 41) H. Tributsch, Beri Bunsenges, Phys. Chem. 81 (1977) 361 42) H. E. Buckely, Crystal Growth (Wiley: New York, 1951) 43) T. Ogawa, Prog. Cryst. Growth Charact. Mater (U. K. ) 25 (1992) 51 44) N. Kubota, H. Otaska, N. Doki, M. Yokota and A. Sato, J. Crystal Growth 220
(2000) 135 45) V. Surender and K. Krishan Rao, Bull. Mater. Sci. 18 (1995) 289 46) L. J. Wu, W. C. Chen, C. R. Li and A. Y. Xie, J. Mater. Research Bull. 35
(2000)145 47) K. Shimizu, T. Kikuta, R. Nozaki and Y. Shiozaki, Ferroelectrics
(Switzerland) 261 (2001) 245 48) Brijesh Amin, Ph.D. Thesis, S. P. University, Vallabh Vidyanagar (2006) 49) J. W. Mullin and G. Gaska, Canadian J. Chem. Eng. 47 (1969) 483 50) V. K. Dixit, B. V. Rodrigues and H. L. Bhat, Bull. Mater. Sci. 24 (2001) 455 51) E. Mielniczek-Brzoska and K. Sangwal, Cryst. Res. Technol 29 (1994) 1027 52) E. Mielniczek-Brzoska and K. Sangwal, Cryst. Res. Technol 30 (1995) 807 53) S. Veintemillas-Verdaguer and R. Rodriguez-Clemente, J. Crystal Growth 99
(1990) 211 54) Rolf Lacmann and Ulrike Tanneberger, J. Crystal Growth 147 (1995) 194 55) G. Spezia, Acad. Sci. Toriho Atti. 40 (1905) 254

Ch.2.Crystal growth methods
35
56) D. Elwell and H. J. Sheel, Crystal Growth from High-temperature solutions (Academic Press, New York,1975)
57) Bhupendra N. Chudasama, Ph.D. Thesis, S. P. University, Vallabh Vidyanagar (2008)
58) B. M. Wanklyn, Crystal Growth, Pergamon Press, Oxford 1 (1974) 717 59) J. C. Brice, The growth of crystals from liquids, (North-Holland, Amsterdam,
1974) 60) H. J. Scheel, Prog. Crystal Growth and Characterisation 5 (1982) 277 61) R. E. Liesegang, Nature Wochschr 11 (1896) 353 62) R. E. Liesegang, Z. Physic. Chem. 88 288 (1914) 1 63) R. E. Liesegang, Phot. Archv. 21 (1966) 221 64) W. Z. Ostwald, Z. Phys. Chem. 27 (1897) 365 65) E. Hatschek, Kolloid Z. 8 (1911) 13 66) H. N. Holmes, J. Phys. Chem. 21 (1917) 709 67) H. N. Holmes, J. Franklin. Inst. 184 (1917) 743 68) H. N. Holmes, Colloid Chem. (Ed. J Alexander) 1 (1926) 796 69) Lord Rayleigh, Phil. Mag. 38 (1919) 738 70) S. C. Bradford, Colloid Chem. Ed. J Alexander, 1(1926) 790 71) H. A. Fells and J. B. Firth, Proceedings of Royal Society. London, 112A
(1926) 468 72) H. V. Morse and J. D. H. Donnay, Bull. Society Frem. Mineralogie
54(1931)19 73) L. W. Fisher and F. L. Simons, Amer. Mineral. 11 (1926) 124 74) L. W. Fisher and F. L. Simons, Amer. Mineral. 11 (1926) 200 75) C. L. Stong, Sci. Amer. 206 (1962) 155 76) V. Vand, H. K. Henisch and J. W. Faust, Acta Cryst. 16 (1963) 137. 77) H. K. Henisch, J. Dennis and J. I. Hanoka, J. Phys. Chem. Solids 26 (1965)
493 78) H. K. Henisch, J. Dennis and J. I. Hanoka, J. Electrochem Soc. 112 (1965) 627 79) H. K. Henisch and C. Srinivas Gopalan, Solid State Commun. 4 (1966) 415 80) H. K. Henisch, Helv. Phys. Acta. (Switzerland) 41 (1968) 388 81) H. K. Henisch, Crystals Growth in Gels (The Pennsylvania State University
Press, University Park. Pa. USA, 1970) 82) S. K. Arora, Prog. Cryst. Growth Charact. 4 (1981) 345 83) D. J. Lloyd, Colloid Chem. (Ed. J. Alexander,1926) 84) E. S. Hedges, Colloids, (Edward Arnold and Co., London, 1931) 85) F. E. Bartell, Lab. Manual of Colloid and Surface Chem. (1979) 86) C. A. E. Alexander, Colloid Sci. (Clarendon Press, Oxford, 2, 1946) 87) C. J. Planck and L. C. Drake, J. Colloid Sci. 2 (1947) 399 88) C. J. Planck and L. C. Drake, J. Colloid Sci. 2 (1947) 413 89) C. Barta, Z. Zemelicka and V. Rane, J. Cryst. Growth 10 (1971) 158 90) M. Ohta and M. Tsutsumi, J. Cryst. Growth 47 (1979) 135 91) E. Blanks, R. Chianneli and F. Pintchovsky, J. Cryst. Growth 18 (1973) 185 92) A. R. Patel and A. V. Rao, Kristall und Technik 14 (1979) 51 93) E. Banks, R. Chianneli and F. Pinchovsky, J. Crystal Growth 18 (1973) 185 94) R. Leckbusch, J. Cryst. Growth 23 (1974) 74 95) T. Bandopadhyay and Ashok de, Ind. J. Earth Sci. 4 (1977) 95 96) N. Palaniandavar, F D Gnanam and P Ramasamy, Ind. J. Phys. 61A (1987)
455

Ch.2.Crystal growth methods
36
97) B. P. Agrawal, K. M. Chauhan and Mohan M. Bhadbhade, Ind. J. Pure & Appl. Phys. 37 (1999) 395
98) B. Wani, F. Ahmed and P. N. Kotru, J. Crys. Growth (2003) 99) E. V. Petrova, N. V. Gvozdev and L. N. Rashkovich, J. Optoelect. & Adv.
Mat. 6 (2004) 261 100) P. V. Dalal and K. B. Saraf, Bull. Mater. Sci. 5 (2006) 421 101) Ballav Moni Borah, Hema Lakshmi and Gopal Das, Mater. Sci. and Eng. C 28
(2008) 1173 102) Z. Blank and W. Brenner, J. Cryst. Growth 11 (1971) 225 103) B. Brezina and M. Haurankova, J. Cryst. Growth 34 (1976) 248 104) G. N. Y. Van Rosmalen and W. G. J. Marchee, J. Cryst. Growth 35 (1976)
169 105) Ti Au-Pang, Scientia Scinica 12 (1963) 1311 106) C. Araki, J. Chem. Soc., Japan 58 (1937) 1214 107) C. Araki and K. Arai, ibid 30 (1957) 287 108) D. A. Rees, Adv. Carbohydr. Chem. Biochem. 24 (1969) 267 109) S. Arnott, A. Fulmer, W. E. Scoott, I. C. M. Dea, R. Moorhouse and D. A.
Rees, J. Mol. Biol. 90 (2) (1974) 269 110) C. Araki, Proc. Int. Seaweed. Symp. 5 (1966) 3 111) M. Lahaye and C. Rochas, Hydrobiologia 126 (1991) 137 112) J. Minghou, M. Lahaye and W. Yaphe, Chin. J. Oceanol. Limnol. 6(2) (1988)
87 113) T. K. Attwood and D. B. Seller, Biopolymers 29 (2004) 1325 114) S. Chakrabarti, J. Sahu, A. Biswas and H. N. Acharya, J. Mater. Sci. Lett. 11 (1992)
763 115) E. S. Helberstadt, H. K. Henisch, J. Nukel and F. W. White, J. Colloid and Interface 29
(1969) 469 116) A. F. Armington, J. J. Connor and M. A. Dipietro, Physical Science Research
Report, AFCR Lab., USA No. 325 (1967) 117) A. R. Patel and H. L. Bhatt, J. Cryst. Growth 12 (1972) 288 118) A. R. Patel and S. K. Arora, J. Cryst. Growth 37 (1977) 343 119) J. J. O'Connor, M. A. Dipietro, A. F. Armington and B. Rubin, Nature 212
(1966) 68 120) J. J. O'Connor, and A. F. Armington, J. Mater. Res. Bull. 3 (1968) 923 121) E. S. Helberstadt, Nature 216 (1967) 574 122) H. K. Suri, H. K. Henisch and J. W. Faust, J. Crystal Growth 7 (1970) 277 123) E. Hatschek and F. L. Simons, Kolloid Z. 10 (1912) 265 124) P. Kratochvil, B. Spursil and M. Heyrovsky, J. Cryst. Growth 3/4 (1968) 360 125) H. M. Liaw and Jr. J. W. Faust, J. Cryst. Growth 13/14 (1972) 471 126) J. M. McCaulay, R. Roy and M. Freund, ACCG (Gaithersburg, USA, 1969) 127) Z. Blank and W. Brenner, ACCG (Gaithersburg, USA, 1969) 128) D. A. Glocker and I. F. Soest, J. Chem. Phys. 51 (1969) 3142 129) C. C. Desai and A. N. Hanchinal, Kristall und Technik 20 (1985) 899 130) M. S. Joshi and A. V. Antony, J. Mater. Sci. 13 (1978) 939 131) G. Sivanesan, Cryst. Res. Technol 27 (1992) 1033 132) K. Sangwal and A. R. Patel, J. Crystal Growth 23 (1974) 282 133) K. Sangwal, Prog. in Crystal Growth and Characterisation 36 (1998) 163 134) A. Einstein, Ann. Physik. 19 (1906) 371 135) H. M. Liaw and Jr. J. W. Faust, J. Crystal Growth 8 (1971) 8

Ch.2.Crystal growth methods
37
Fig. 2.1 Schematic representation of Czochralski crystal pulling technique
Fig. 2.2 Schematic representation of Bridgemann-Stockbarger technique

Ch.2.Crystal growth methods
38
Fig. 2.3 Schematic representation of Verneuil flame fusion technique
Ch.2.Crystal growth methods
39
Fig. 2.4 Schematic representation of (a) Zone melting technique and (b) the
modified float zone technique
(a)
(b)

Ch.2.Crystal growth methods
40
Fig. 2.5 Schematic representation of Hydrothermal technique

Ch.2.Crystal growth methods
41
(a)
(b) Fig. 2.6 (a) Agar forming macroreticuated gels (b) Basic Structure of agar indicating
D-Galactose and 3,6 anhydro-L-Galactose repeat unit
Fig. 2.7 Single test tube containing Fig. 2.8 Single test tube using solid gel with
dissolved salt(By) salt (By) dissolved in water taken in a bag
Ch.2.Crystal growth methods
42
Fig. 2.9 Single test tube technique Fig. 2.10 Growth of single crystals using using two-gel technique U-tube technique
Fig. 2.11 Growth of β – HgI crystal using Fig. 2.12 Growth of copper metal crystals complex-decomplexion method by chemical reduction in gel





























