Embed Size (px)
Citation preview

78
CHAPTER 3
MATERIALS AND METHODS
3.1 GENERAL
Established laboratory procedures as recommended by Purvis et al
(1966), Tuite (1969) were followed for the preparation of media, inoculation
and maintenance of cultures.
3.2 GLASSWARES
All the glasswares (Borosil or Corning) were immersed in cleaning
solution for a few hours. Then, the glasswares were washed thoroughly with
water, followed by detergent solution and finally rinsed with distilled water.
The cleaned glasswares were dried in hot air oven and stored.
Cleaning solution (Mahadevan and Sridhar, 1996)
Potassium dichromate - 60 g
Conc H2SO4 - 60 mL
Distilled water - 1000 mL
Potassium dichromate was dissolved in warm water, cooled and
sulphuric acid was added slowly. It was mixed thoroughly and used for
cleaning glasswares.

79
3.2.1 Sterilization
Dried glassware and media were sterilized in an autoclave for 20
min at 15 psi pressure.
3.3 CHEMICALS
Analytical grade chemicals supplied by Loba, Hi-Media, S.D.Fine
Chemicals, E-Merck and Sigma Chemicals (USA) were used.
3.4 SENNA SIAMEA (Lam) PLANT SEED COLLECTION
Senna siamea plant is easily available in medium size tree to 15-20
cm tall, with a straight trunk up to 30 cm in diameter, bole short, crown
usually dense and rounded at first, later become irregular and spreading with
dropping branches. Plant seeds collected in Anna university campus, Chennai,
were sterilized and used for further processing
3.4.1 Preparation of Plant Seed Aqueous Extract
Collected seeds of Senna siamea plant (Fig 3.1) was washed
several times with deionized water. Fresh 50grams seeds of Senna siamea
plants were collected and washed with double distilled water. These were
then cut into small pieces, transferred to 500 mL beaker and boiled with
300 mL of distilled water for 10 min up to 700C temperature using magnetic
stirrer. Obtained mixture was cooled and filtered through Whatman No. 1
filter paper. The boiled extract was refrigerated and used for experimental
procedures.

80
Figure 3.1 Synthesis of plant seed aqueous extract
3.4.2 Synthesis of Silver Nanoparticles
1 mM (1×10-3
) solution of silver nitrate (AgNO3) stock solution
was prepared using sterile deionized triple distilled water. A series of
different volumes (2-10 mL) of plant seed extract was added to 5 mL of
1×10-3
M aqueous silver nitrate (AgNO3) solution followed by addition of
distilled water to acquire a volume of 15 mL. After 3 hours of incubation
period at ambient conditions, color change from transparent light yellow to
brown color indicating the formation of silver nanoparticles. The occurrence
of brown color is a primary indication of the formation of silver nanoparticles
(AgNPs).
3.4.3 Synthesis of Gold Nanoparticles
1 mM (1×10-3
) solution of gold chloride (HAuCl4) stock solution
was prepared using sterile deionized triple distilled water. A series of
different volumes (2-10 mL) of plant seed extract was added to 5 mL of
1×10-3
M aqueous HAuCl4 solution followed by a addition of disttiled water

81
being added to acquire a volume of 15 mL. After 3 hours incubation period at
ambient conditions, color change from transparent light yellow to violet color
indicating the formation of gold nanoparticles. The occurrence of violet color
is a primary indication of the formation of gold nanoparticles (AuNPs).
3.5 IN VITRO ANTIBACTERIAL SENSITIVITY
DETERMINATION TEST
The in vitro activities of the antibiotics by the disc diffusion
method, (as recommended by the National Committee for Clinical Laboratory
Standards) and test compounds were determined by modified well diffusion
method as proposed by Magaldi (1997) were determined
3.5.1 Preparation of Test Extracts
The stock solution of different concentrations were prepared. The
seed aqueous extracts, silver and gold nanoparticles from the stock solution
with 50 μL, 100 μL and 200 μL concentrations were immediately dispensed
into each agar wells of culture inoculated in muller hinton agar (MHA) plates
using sterilized micropipette.
Group: 1 Test group: consisted of the organism plus different
concentrations of the plant seed aqueous extract, silver and gold nano
particles (this group determined whether the extract was effective as
antibacterial agents).
Group: 2 Positive controls: organism plus a known antibiotic (This
ensured that utilized organisms are susceptible to common chemotherapeutics
and not resistant strains).
Group: 3 Pure cultures: Only the organism in the absence of
antibiotics or plant seed aqueous extract, silver and gold nano particles. This

82
is to ensure that the organism was growing properly under the defined
laboratory conditions. This was necessary to distinguish poor growth from
inhibition of growth.
Group: 4 Negative controls: Organism plus AgNO3 and HAuCl4
solution (this was necessary to prove that the extraction solvent had no
inhibitory action of its own).
All quantitative data determinations were carried out in duplicate
with error < ±0.1.
3.5.2 Microbial Type Culture Collection (MTCC) Cultures
Five MTCC cultures of clinically important bacterial pathogens
which were obtained from Microbial culture collection, Chandigarh, India
were obtained as follows: Two-gram positive (Staphylcoccus aureus and
Bacillus subtilis) and four gram negative (Pseudomonas aeruginosa,
Klebsiella pneumoniae and Escherichia coli) were used. All strains were
recently purchased, collected between 2011 and 2012. The cultures were
stored frozen in skimmed milk 50% glycerol at -70ºC.
3.5.3 Inoculum Preparation
Bacterial inoculums were prepared using gram positive and gram-
negative bacterial pathogens from a 24 hours old culture on brain heart
infusion agar. With a sterile loop, the tops of four to five colonies were
transferred to a tube containing 5 mL of Mueller Hinton broth or Brain Heart
infusion broth. The tube was incubated at 35ºC for 24 hours. The turbidity of
the culture suspension was adjusted with broth or a sterile saline solution
(0.85 – 0.9%). The density of this culture was adjusted with 0.5 McFarland
standard and final inoculum size was approximately of 5x 105 CFU/mL.

83
3.5.4 Turbidity standards for inoculum preparation
To standardize the inoculum density for a susceptibility test, a BaSO4
turbidity standard, equivalent to a 0.5 McFarland standard or its optical
equivalent (e.g., latex particle suspension), should be used. A BaSO4 0.5
McFarland standard may be prepared as follows:
1. A 0.5-mL aliquot of 0.048 mol/L BaCl2 (1.175% w/v BaCl2
.2H2O) was added to 99.5 mL of 0.18 mol/L H2SO4 (1% v/v)
with constant stirring to maintain a suspension.
2. The correct density of the turbidity standard was verified by
using a spectrophotometer with a 1-cm light path and matched
cuvette to determine the absorbance. The absorbance at 625 nm
was 0.008 to 0.10 for the 0.5 McFarland standard.
3. The Barium sulfate suspension was transferred in 4 to 6 ml
aliquots into screw-cap tubes of the same size as those used in
growing or diluting the bacterial inoculum.
4. These tubes were tightly sealed and stored in the dark at room
temperature.
5. The barium sulfate turbidity standard was vigorously agitated
on a mechanical vortex mixer before each use and inspected for
a uniformly turbid appearance. If large particles appeared, the
standard was replaced. Latex particle suspensions were mixed
by inverting gently, not on a vortex mixer
6. The barium sulfate standards were be replaced or their densities
verified monthly.

84
3.5.5 Test Medium
The well diffusion method was performed using Mueller Hinton
Agar (MHA) medium: Casein acid hydrolysate: 17.5 g; Beef Heart infusion: 2
g; Starch soluble 1.5 g; (PH
7.3 ± 0.2), Agar 17 g; H2O 1000 mL was used.
3.5.6 Well diffusion method
The well diffusion test (Bennet et al 1966, Janssen et al 1987,
Magaldi et al 2004) was performed using MHA. The medium was prepared
and autoclaved at 15 lbs pressure (121ºC) for 15 min followed by cooling in a
50-55ºC water bath after removal from the autoclave. The cooled medium
was poured into sterile petri plates to a uniform depth of 4 mm; this is
equivalent to approximately 25 mL in a 90 mm plate. Once the medium had
solidified, then the culture was inoculated on the medium. Within 15 min of
adjusting the density of the inoculum, a sterile cotton swab was dipped into
the standardized bacterial suspension or inoculated with 1 mL of the organism
suspension. The sterile swab was used to streak on the surface of the MHA
medium to ensure an even distribution of the inoculum. The plates were left
to undisturbed for 3 to 5 min to absorb the excess moisture. Sterilized 10 mm
cork borer was used to made agar wells and 50 μL, 100 μL and 200 μL of the
stocks solutions were placed into each wells. The plates were incubated at 35-
37ºC for 24 hours. However NCCLS disc diffusion and MIC standard
breakpoints were used for the interpretative results. Streptomycin antibiotic
used as standard for these studies. Streptomycin is an antibiotic that inhibits
both Gram-positive and Gram-negative bacteria and is therefore a useful
broad-spectrum antibiotic (Jan-Thorsten Schantz 2004).
The percentage of inhibition was calculated by the formula,
I (Diameter of the inhibition zone)
% of inhibition = × 100
90 (Diameter of the petri-plate in mm)

85
3.6 ANTIOXIDANT ACTIVITY
3.6.1 2,2-Diphenyl-1-picrylhydrazyl (DPPH) free radical Scavenging
assay
Various concentrations of the stock solutions (concentrations of
100 to 500 µL) were mixed with 0.25 mM DPPH in ethanol, to produce a
final DPPH concentration of 0.1 mM. The mixture was vigorously shaken and
left to stand for 10 min in the dark and its absorbance was measured at
517 nm. L-Ascorbic acid was used as the control (McCune and Johns, 2002).
The radical-scavenging activities of samples, expressed as percentage
inhibition of DPPH, were calculated according to the formula:
Absorbance of Control – Absorbamce of sample
% of inhibition = × 100
Absorbance of control
3.7 IN VITRO ANTICANCER STUDY
3.7.1 Cell Line and Culture
A549 (lung carcinoma) cell line was obtained from National centre
for cell sciences Pune (NCCS). The cells were maintained in Minimal
Essential Media supplemented with 10% FBS, penicillin (100 U/ml), and
streptomycin (100 μg/ml) in a humidified atmosphere of 50 μg/ml CO2
at 37 °C.
3.7.2 Reagents
Minimal Essential Media (MEM) was purchased from Hi Media
Laboratories, Fetal bovine serum (FBS) was purchased from Cistron
laboratories and Trypsin, methylthiazolyl diphenyl- tetrazolium bromide
(MTT), and Dimethyl sulfoxide (DMSO) were purchased from Sisco research
86
laboratory chemicals Mumbai. All other chemicals and reagents were
procured from Sigma Aldrich Mumbai.
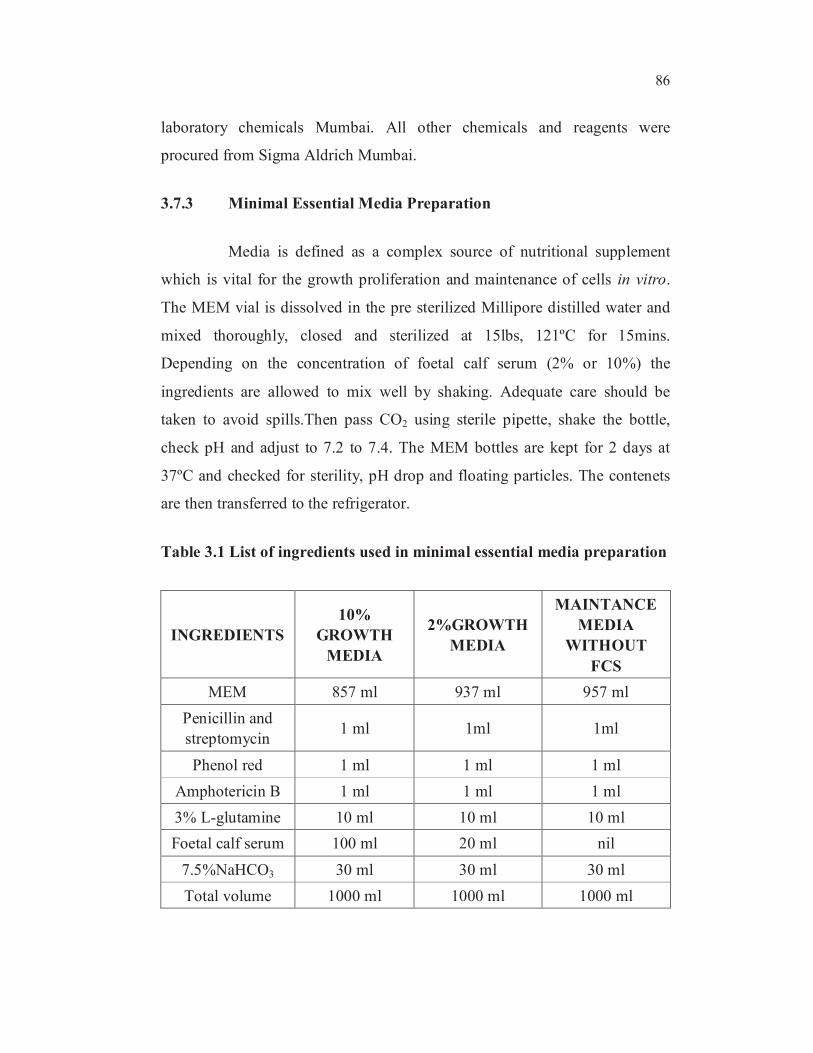
3.7.3 Minimal Essential Media Preparation
Media is defined as a complex source of nutritional supplement
which is vital for the growth proliferation and maintenance of cells in vitro.
The MEM vial is dissolved in the pre sterilized Millipore distilled water and
mixed thoroughly, closed and sterilized at 15lbs, 121ºC for 15mins.
Depending on the concentration of foetal calf serum (2% or 10%) the
ingredients are allowed to mix well by shaking. Adequate care should be
taken to avoid spills.Then pass CO2 using sterile pipette, shake the bottle,
check pH and adjust to 7.2 to 7.4. The MEM bottles are kept for 2 days at
37ºC and checked for sterility, pH drop and floating particles. The contenets
are then transferred to the refrigerator.
Table 3.1 List of ingredients used in minimal essential media preparation
INGREDIENTS
10%
GROWTH
MEDIA
2%GROWTH
MEDIA
MAINTANCE
MEDIA
WITHOUT
FCS
MEM 857 ml 937 ml 957 ml
Penicillin and
streptomycin 1 ml 1ml 1ml
Phenol red 1 ml 1 ml 1 ml
Amphotericin B 1 ml 1 ml 1 ml
3% L-glutamine 10 ml 10 ml 10 ml
Foetal calf serum 100 ml 20 ml nil
7.5%NaHCO3 30 ml 30 ml 30 ml
Total volume 1000 ml 1000 ml 1000 ml

87
3.7.4 Preparation of Ingredient
a) Penicillin and streptomycin: (concentration 100 IU of
penicillin and 100 µg of streptomycin)
Dissolve both antibiotics in sterile Millipore distilled water, so
as to give a final concentration 100 IU of penicillin and 100
µg of streptomycin/ml. Mix well and distribute in 1ml
aliquots. Store at -20ºC and check sterility.
b) Fungizone (amphotericin B) (20µg/ml)
Dissolve in sterile Millipore distilled water so as to give a
final concentration of 20 µg/ml and distribute in 1ml aliquots
in vials. Store at -20ºC. Check sterility before using.
c) L-glutamine (3%)
Weigh 3g of l-glutamine accurately and dissolve in 100 ml
sterile Millipore distilled water and mix well. Filter through
Millipore membrane filter 0.22µ and distribute in 5ml aliquots
in vials. Store at -20ºC. Check sterility.
d) 5% sodium-bi-carbonate
Weigh requisite quantity of sodium-bi-carbonate (to give
7.5% solution) accurately and dissolve in 100 ml of sterile
Millipore distilled water. Filter through What man filter paper
No.4, distribute into bottles at 121ºC, 15lbs, 15mins. Cool and
store at 4ºC.
e) Foetal calf serum
Bring FCS at room temperature. Inactivate at 56ºC in water
bath for½ hour and cool at room temperature. If floating

88
particles are seen filter through Seitz filter. Distribute in
100 ml, 50 ml, and 20 ml quantities in sterile bottles. Store at -
20ºC.
3.7.5 Trypsin, PBS, Versene, Glucose Solution (TPVG)
a) 2% trypsin: 100 ml
Weigh 2g of trypsin accurately; dissolve in 100 ml sterile
Millipore distilled water with magnetic stirrer for ½ hour.
Filter through membrane filter. Store at -20ºC
b) 0.2%EDTA (versene)
Weigh 200mg of EDTA accurately. Dissolve in 100 ml of
sterile Millipore distilled water and autoclave at
15 lbs/15mins.
c) 10% glucose-100 ml
Weigh 1g of glucose accurately. Dissolve in 100 ml of sterile
Millipore distilled water and filter through whatmann filter
paper and autoclave at 15lbs/15mins.
d) TPVG-100 ml
PBS - 840 ml
% trypsin - 50 ml
0.2% EDTA - 100 ml
10% glucose - 5 ml
Penicillin & streptomycin - 5ml

89
Mix all ingredients and adjust the pH to 7.4 with 0.1 N HCl or
0.1 N NaOH. Distribute in 100 ml aliquots. Store at -20ºC.
3.7.6 Subculturing and Maintenance of Cell Line
• Bring the medium and TPVG to room temperature.
• Observe the tissue culture bottles for growth, cell
degeneration, pH and turbidity.
• Select the bottles for splitting.
The following procedure is followed in sequence.
• Wipe the mouth of the bottle with cotton soaked in spirit.
• Remove the growth medium using 10 ml pipette.
• Then the cells in the bottle were gently rinsed with MEM
without FCS. The dead cells and FCS are washed out and then
the medium is discarded.
• 4-5 ml of TPVG was added over the cells. Allow TPVG to act
for 3-5 minutes and it was pipetted out.
• Incubate at 37oC for 3-5 minutes. The cells become individual
in nature as suspension.
• Add 5ml of 10% MEM with FCS by using serological pipette.
• Release carefully by using serological pipette and add 20 ml
of MEM and homogenize.

90
3.7.7 Seeding of Cells
After homogenization pour 4-5 ml in to 24 well plates. In each well
add 1ml of the suspension to the 24 well plates and keep in a dessicator under
5% CO2 atmosphere. After 2 days of incubation period observe the cells in
inverted microscope. If the cells became 80% confluent, then it is used for the
Cytotoxicity studies.
3.7.8 MTT assay for in vitro cytotoxicity study
MTT assay is a calorimetric assay for measuring cell viability, for
cellular proliferation and activation. It is also used to determine the
cytotoxicity of potential medical agents and other toxic materials.
a) Principle
MTT was first described by Mosmann in 1983. Yellow MTT (3-(4,
5-dimethyl thiazol-2-yl)-2, 5-diphenyltetrazolium bromide, a tetrazol) is
reduced to purple formazan in the mitochondria of the living cells. A
solubilization solution is added to dissolve the insoluble purple formazan
product into a colored solution. The absorbance can be quantified by
measuring at a certain wavelength (usually between 500 and 600 nm) by a
spectrophotometer.
The reduction takes place only when mitochondrial dehydrogenase
enzyme is active and therefore conversion is directly related to number of
viable cells. When the amount of purple formazan produced by cells treated
with an agent is compared with the amount of formazan produced by
untreated control cells, effectiveness of the agent causing death of cells can be
deduced

91
After the addition of the drug, cell death and cell viability were
estimated. The result is confirmed by additional metabolic intervention
experiment such as MTT assay.
b) Procedure
• After incubation, remove the medium from the wells for
MTT assay.
• In each well wash with MEM (w/o) FCS. And add 200µl of
MTT concentration of (5mg/ml).
• Incubate for 6-7hrs in 5% CO2 incubator.
• After incubation, 1ml of DMSO was added in each well and
mixed by pipette and leave for 45 sec and it shows the
purple color formation.
• The suspension is transferred in to the cuvette of
spectrophotometer and O.D values are read at 595 nm
In order to study the antitumor activity of a drug, it is important to
determine the cytotoxic concentration of the drug. Cytotoxicity tests define
the upper limit of the extract concentration, which is non-toxic to the cell line.
The concentration which is nontoxic to the cells is chosen for antitumor
assay.The Cytotoxicity of samples on A549 was determined by the MTT
assay (Mosmann et al 1983). Cells (1 × 105/well) were plated in 1ml of
medium/well in 24-well plates (Costar Corning, Rochester, NY). After 48
hours incubation, the cell reaches the confluence. Then, cells were incubated
in the presence of various concentrations of the samples in 0.1% DMSO for
48h at 37°C.

92
After removal of the sample solution and washing with phosphate-
buffered saline (pH 7.4), 200µl/well (5 mg/ml) of 0.5% 3-(4,5-dimethyl-2-
thiazolyl)-2,5-diphenyl--tetrazolium bromide cells(MTT) phosphate- buffered
saline solution was added. After 4h incubation, 0.04M HCl/ isopropanol was
added. Viable cells were determined by the absorbance at 570 nm.
Measurements were performed and the concentration required for a 50%
inhibition of viability (IC50) was determined graphically. The absorbance at
570 nm was measured with a UV-Spectrophotometer using wells without
sample containing cells as blanks. The effect of the samples on the
proliferation of A549 was expressed as the % cell viability, using the
following formula:
% cell viability = A549 of treated cells / A549 of control cells × 100%.
3.7.9 Drug Dilution
a) Stock drug concentration
1. 5 ml of extract was prepared with a concentration of
10 mg/ml.
2. 500 µl of MEM without FCS was taken in about 9 eppendroff
tubes.
3. 500 µl of the working concentration was added to the first
eppendroff tube, then same was transferred from first to last
tube by serial dilution to obtain the desired concentration of
the drug.

93
b) Sampling
• 48h monolayer culture of Vero cell line at a concentration of
one lakh /ml /well (10 cells / ml / well) was seeded in 24 well
titer plate.
• The plates were microscopically examined for confluent
monolayer, turbidity and toxicity.
• The growth medium (MEM) was removed using pipette. Care
was taken so that the tip of the pipette did not touch the cell
sheet.
• The cell monolayer was washed twice with MEM without
FCS.
• To the washed cell sheet, 1ml of the medium (without FCS)
containing defined concentration of the drug was added. .
• Then each dilution of the drug ranges from 1:1 to 1:256 and
they were added to the respective wells of the 24 well titer
plates.
• To the cell control wells add 1ml MEM (w/o) FCS control.
The plates were incubated at 37ºC in 5% CO2 environment and
observed for cytotoxicity using inverted microscope.





























