Embed Size (px)
Citation preview

122
CHAPTER 3
RESULTS AND DISCUSSION
3.1 ULLMANN CONDENSATION OF HALOTHIOPHENE -
CARBOXYLIC ACIDS WITH SODIUM BISULPHITE
3.1.1 Observation Against Literature Information
While studying the Ullmann type nucleophilic substitution reactions
of chloro- and bromothiophene carboxylic acids with sodium bisulphite under
aqueous conditions, it was observed that 3-bromothiophene-2-carboxylic acid
3b underwent a facile substitution compared to 3-chlorothiophene-2-
carboxylic acid 3a (Scheme 3.1).
S
X
COOH
X= Cl (3a)
X= Br (3b)
NaOH / NaHSO3
CuCl / Reflux
pH 7.5- 7.7
S
SO3Na
COONa HCl / KClS
SO3K
COOH
(5a)
100 °C
(4a)
Scheme 3.1 Ullmann condensation with sodium bisulphite
The cuprous chloride mediated substitution of bromo acid 3b was
completed in 2 h at 100°C under standard conditions (entry 3, Table 3.1).
Interestingly no ligand was employed for this reaction. The conversion in the
case of chloro acid 3a was only 3.5% at 100°C (entry 1, Table 3.1). The
chloro acid 3a required heating of the content to 140- 143°C under autoclave
condition for 16 h for the nucleophilic substitution (entry 2, Table 3.1). The
123
nucleophilic substitution did not take place without cuprous chloride catalyst
(entry 4, Table 3.1).
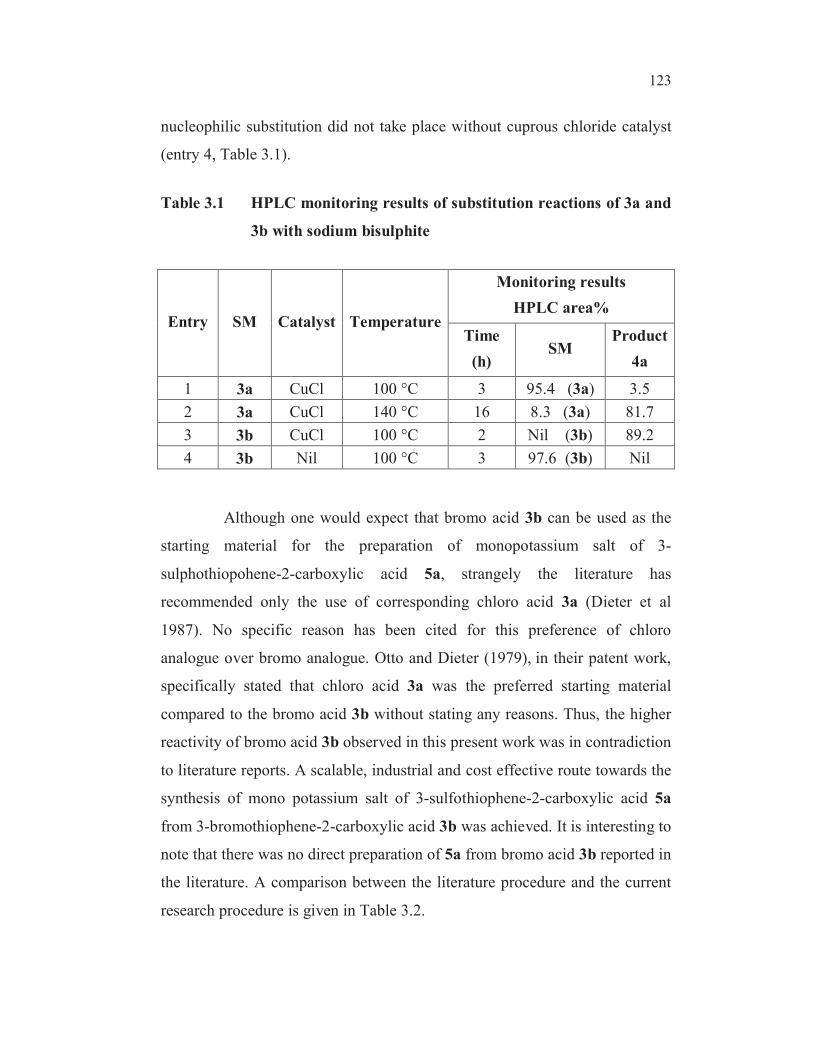
Table 3.1 HPLC monitoring results of substitution reactions of 3a and
3b with sodium bisulphite
Entry SM Catalyst Temperature
Monitoring results
HPLC area%
Time
(h)SM
Product
4a
1 3a CuCl 100 °C 3 95.4 (3a) 3.5
2 3a CuCl 140 °C 16 8.3 (3a) 81.7
3 3b CuCl 100 °C 2 Nil (3b) 89.2
4 3b Nil 100 °C 3 97.6 (3b) Nil
Although one would expect that bromo acid 3b can be used as the
starting material for the preparation of monopotassium salt of 3-
sulphothiopohene-2-carboxylic acid 5a, strangely the literature has
recommended only the use of corresponding chloro acid 3a (Dieter et al
1987). No specific reason has been cited for this preference of chloro
analogue over bromo analogue. Otto and Dieter (1979), in their patent work,
specifically stated that chloro acid 3a was the preferred starting material
compared to the bromo acid 3b without stating any reasons. Thus, the higher
reactivity of bromo acid 3b observed in this present work was in contradiction
to literature reports. A scalable, industrial and cost effective route towards the
synthesis of mono potassium salt of 3-sulfothiophene-2-carboxylic acid 5a
from 3-bromothiophene-2-carboxylic acid 3b was achieved. It is interesting to
note that there was no direct preparation of 5a from bromo acid 3b reported in
the literature. A comparison between the literature procedure and the current
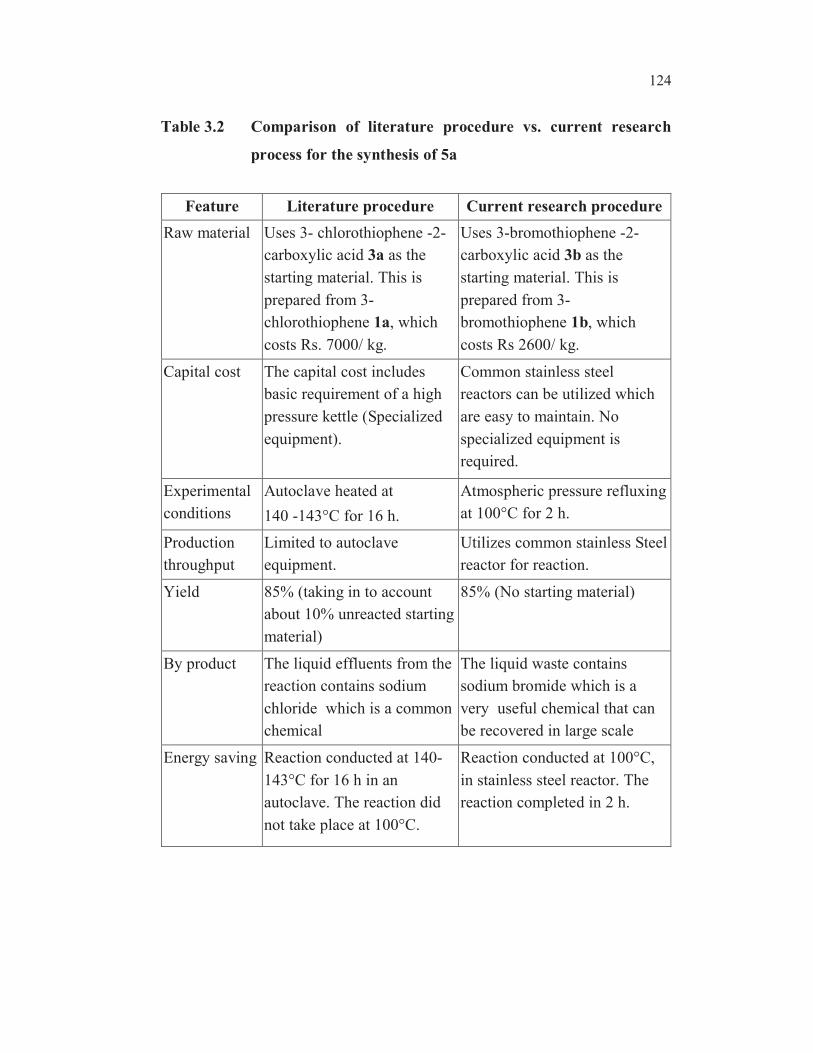
research procedure is given in Table 3.2.
124
Table 3.2 Comparison of literature procedure vs. current research
process for the synthesis of 5a
Feature Literature procedure Current research procedure
Raw material Uses 3- chlorothiophene -2-
carboxylic acid 3a as the
starting material. This is
prepared from 3-
chlorothiophene 1a, which
costs Rs. 7000/ kg.
Uses 3-bromothiophene -2-
carboxylic acid 3b as the
starting material. This is
prepared from 3-
bromothiophene 1b, which
costs Rs 2600/ kg.
Capital cost The capital cost includes
basic requirement of a high
pressure kettle (Specialized
equipment).
Common stainless steel
reactors can be utilized which
are easy to maintain. No
specialized equipment is
required.
Experimental
conditions
Autoclave heated at
140 -143°C for 16 h.
Atmospheric pressure refluxing
at 100°C for 2 h.
Production
throughput
Limited to autoclave
equipment.
Utilizes common stainless Steel
reactor for reaction.
Yield 85% (taking in to account
about 10% unreacted starting
material)
85% (No starting material)
By product The liquid effluents from the
reaction contains sodium
chloride which is a common
chemical
The liquid waste contains
sodium bromide which is a
very useful chemical that can
be recovered in large scale
Energy saving Reaction conducted at 140-
143°C for 16 h in an
autoclave. The reaction did
not take place at 100°C.
Reaction conducted at 100°C,
in stainless steel reactor. The
reaction completed in 2 h.

125
The monopotassium salt of 3-sulphothiophene-2-carboxylic acid 5a
obtained from this research work was completely characterized by spectral
analysis (Section 2.5).
This procedure was scaled up successfully towards consistent
quality and yield of the monopotassium salt 5a (Table 3.3).
Table 3.3 Results of industrial scale preparation of 5a
EntryInput of 3b
(Kg)
Output 5a
(Kg)
HPLC Purity
(%)
1 175 165.6 99.4
2 177 165.8 99.6
3 175 160.1 99.7
3.1.2 Design of Experiments
In order to understand the higher reactivity of the bromo acid 3b
over that of chloro acid 3a and to study the mechanism of this nucleophilic
substitution, experiments were designed and conducted as shown in Table 3.4.
3-bromothiophene-2-carboxylic acid 3b was considered as the model
compound for the research studies under standard condition as given below.
Standard condition – The starting material (SM) was dissolved in one
equivalent of 10% aq. sodium hydroxide. Aqueous sodium bisulphite solution
(1.15 equiv. 37% aq.w/w), unless otherwise mentioned, was added and the pH
was adjusted to 7.5-7.7 using 30% aq. NaOH. Cuprous chloride (0.1 mol) was
added to the reaction mixture, unless otherwise mentioned, and the mixture
was heated to 100°C, unless otherwise mentioned.

126
Table 3.4 Design of experiments
S.No Experimental Design
1 Effect of different catalysts (AgNO3, ZnCl2, NiCl2, Lanthanum triflate,
Ytterbium triflate, Pd (OAc) 2, Pd (OAc) 2 with BINAP as ligand and
without catalyst).
2 Effect of different copper catalysts (CuCl, CuBr, CuI, CuO, Cu(OTf)2,
CuCl2, Cu(0), CuCl with TBAB as phase transfer catalyst)
3 Influence of temperature
4 Influence of pH
5 Optimal catalyst loading
6 Effect of co-solvents and additives (ethanol and 1,4-dioxan as co-
solvents and NaCl and 2,2’-bipyridyl as additives)
7 Different loading of nucleophile (sodium bisulphite)
8 Different nucleophiles (sodium sulphite , calcium bisulphite, cesium
bisulphite and no nucleophile)
9 Isomeric bromothiophenecarboxylic acids
10 Influence of different EWG substituent instead of carboxylic acid
( -COCH3 , -COOMe)
11 Effect of radical scavengers ( TEMPO, oxygen, p-dinitrobenzene,
THF, BHT)
12 Microwave conditions (microwave conditions, 80°C and 100W)
Samples were drawn at specified intervals from the reaction mixture
and analyzed by HPLC (mobile phase methanol: water: TFA:
TEA=1500:500:1:0.5 v/v; wavelength 254 nm and flow rate 1 mL per
minute.). The product was isolated wherever required, by acidification with
con. HCl followed by treatment with KCl as monopotassium salt of the
corresponding sulphothiophene carboxylic acid.

127
The required starting material viz., bromo acid 3b is known in the
literature (Tietze and Lohmann 2002, Kou-Yi and Ludwig 1975, Corral et al
1985, Masami Takahashi 1993 and Gol’dfarb and Vol’kenshteuin 1959). The
methods described in the literature for the preparation of bromo acid 3b either
employ chemicals that are hazardous and difficult to handle in large scale like
n-butyl lithium, grignard reagent or reagents like potassium permanganate,
potassium chlorate for oxidation of bromo ketone 2b. All these methods either
involve isolation issues or possess environment, safety and health (EHS)
concern. A simple two step route was envisaged for the synthesis of the
bromo acid 3b as outlined in Scheme 3.2.
S
Br(1b)
EDC/ AlCl3
S
Br(2b)
COCH3NaOCl
(oxidation)
S
Br(3b)
COOHCH3COCl
Scheme 3.2 Synthetic scheme for the preparation of 3-
bromothiophene-2-carboxylic acid 3b
Friedel-Crafts acetylation of commercially available
3-bromothiophene 1b, following a similar literature procedure (Pelly 2005)
afforded 3-bromo-2-acetylthiophene 2b, in 90% yield (Section 2.3). The
crude 2b as such was converted to the corresponding carboxylic acid 3b in
80% yield (Section 2.4) by haloform reaction using aqueous sodium
hypochlorite solution (10% w/w) following a similar literature reference
(Arnold, 1952). This simple and safe route which is amenable for large scale
manufacture of bromo acid 3b has not been exploited so for. This safe and
scalable process was successfully standardized to produce the bromo acid 3b
with consistent quality and yield in multi kilo levels (Table 3.5).

128
Table 3.5 Multi kilo level batches of 3b
Entry Input of 1b
(Kg)
Output of 3b
(Kg)
HPLC purity of 3b
(%)
1 100 97 99.5
2 100 98 99.5
3 100 97.8 99.5
3.1.3 Effect of Different Catalysts
It was observed that bromo acid 3b did not undergo substitution
with sodium bisulphite under standard conditions in the absence of cuprous
chloride, as monitored by HPLC. A number of Lewis acids as catalysts were
screened under standard conditions (Scheme 3.3) and the reaction was
monitored by HPLC.
S
Br
COOH
(3b)
NaOH / NaHSO3
CATALYST
S
SO3Na
(4a)
COONa
reflux. pH 7.5- 7.7
HCl / KClS
SO3K
COOH
(5a)
100 °C
Scheme 3.3 Ullmann condensation with sodium bisulphite with different
catalysts
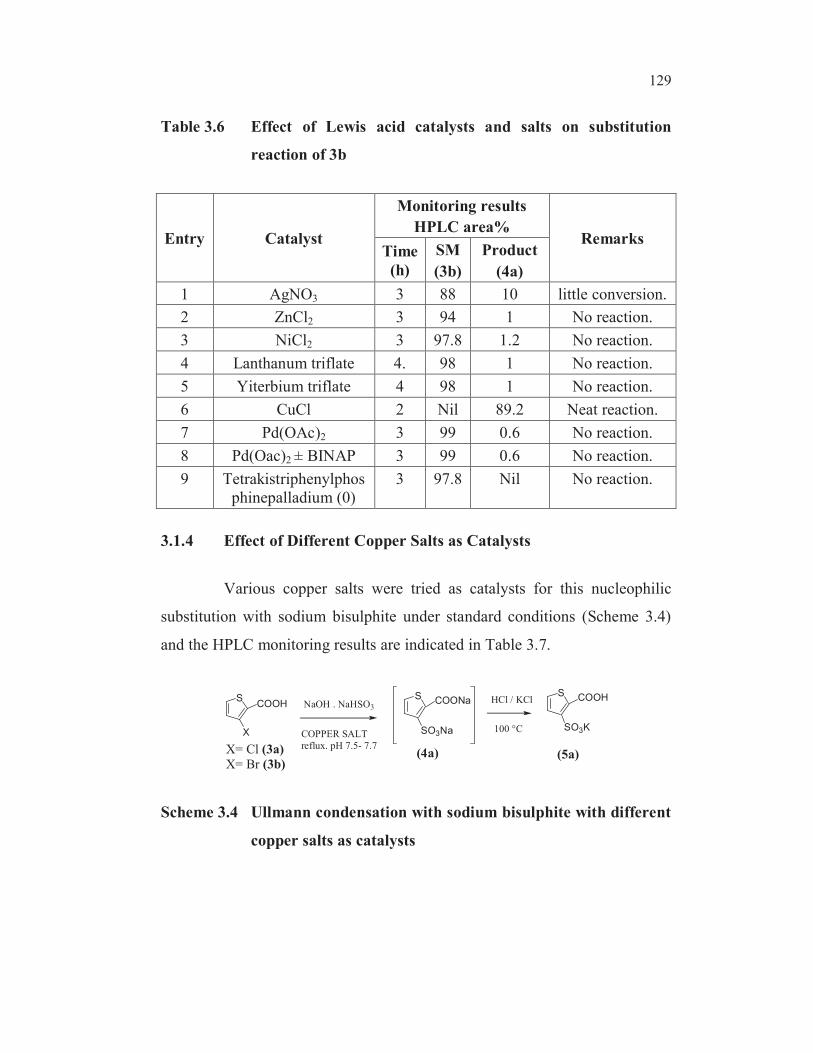
Of the several Lewis acids and salts examined, viz. silver nitrate
(entry 1, Table 3.6), zinc chloride (entry 2, Table 3.6), nickel chloride (entry3,
Table 3.6), lanthanum triflate (entry 4, Table 3.6), ytterbium triflate (entry 5,
Table 3.6), copper (I) chloride (entry 6, Table 3.6), palladium (II) acetate
(entry 7, Table 3.6), palladium (II) acetate with ligand ± 2, 2’-bis
(diphenylphosphino)-1, 1’-binaphthyl (BINAP) (entry 8, Table 3.6) and
tetrakistriphenylphosphinepalladium(0) (entry 9, Table 3.6), only cuprous
chloride was found to exhibit the best catalytic activity in effecting the
nucleophilic substitution.
129
Table 3.6 Effect of Lewis acid catalysts and salts on substitution
reaction of 3b
Entry Catalyst
Monitoring results
HPLC area%Remarks
Time
(h)
SM
(3b)
Product
(4a)
1 AgNO3 3 88 10 little conversion.
2 ZnCl2 3 94 1 No reaction.
3 NiCl2 3 97.8 1.2 No reaction.
4 Lanthanum triflate 4. 98 1 No reaction.
5 Yiterbium triflate 4 98 1 No reaction.
6 CuCl 2 Nil 89.2 Neat reaction.
7 Pd(OAc)2 3 99 0.6 No reaction.
8 Pd(Oac)2 ± BINAP 3 99 0.6 No reaction.
9 Tetrakistriphenylphos
phinepalladium (0)
3 97.8 Nil No reaction.
3.1.4 Effect of Different Copper Salts as Catalysts
Various copper salts were tried as catalysts for this nucleophilic
substitution with sodium bisulphite under standard conditions (Scheme 3.4)
and the HPLC monitoring results are indicated in Table 3.7.
S
X
COOH
X= Cl (3a)
X= Br (3b)
COPPER SALT
reflux. pH 7.5- 7.7
S
SO3Na
(4a)
COONa HCl / KClS
SO3K
COOH
(5a)
100 °C
NaOH . NaHSO3
Scheme 3.4 Ullmann condensation with sodium bisulphite with different
copper salts as catalysts
130
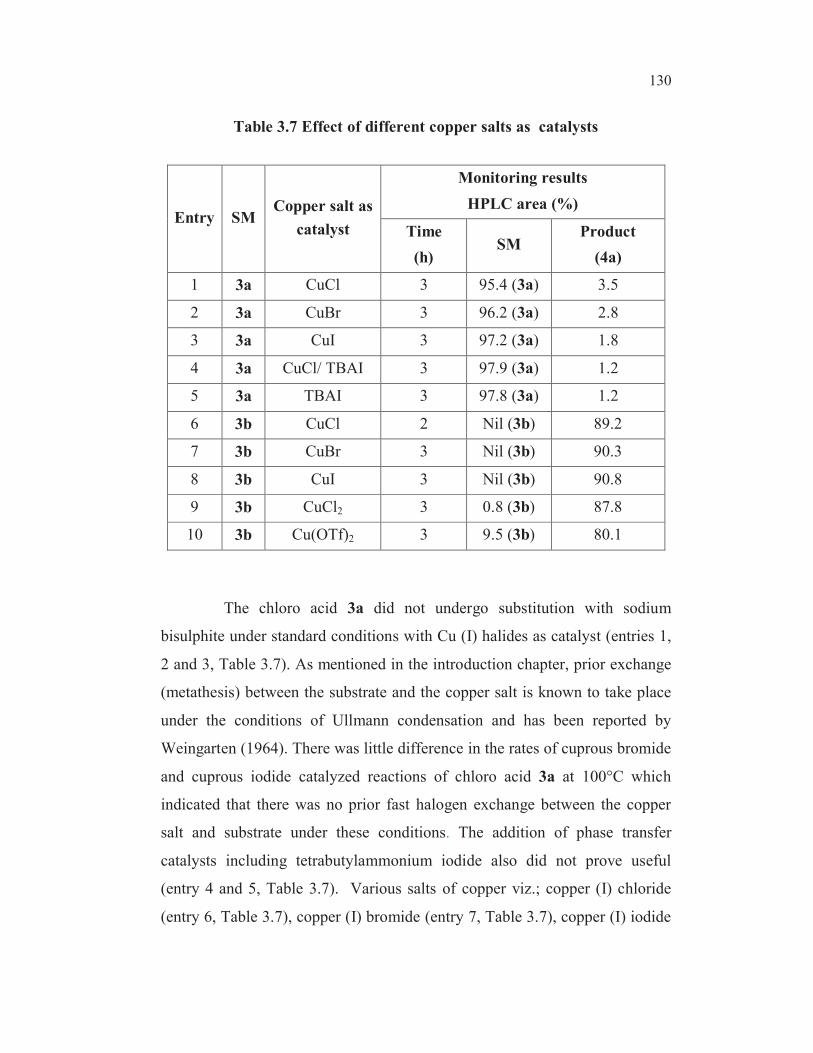
Table 3.7 Effect of different copper salts as catalysts
Entry SMCopper salt as
catalyst
Monitoring results
HPLC area (%)
Time
(h)SM
Product
(4a)
1 3a CuCl 3 95.4 (3a) 3.5
2 3a CuBr 3 96.2 (3a) 2.8
3 3a CuI 3 97.2 (3a) 1.8
4 3a CuCl/ TBAI 3 97.9 (3a) 1.2
5 3a TBAI 3 97.8 (3a) 1.2
6 3b CuCl 2 Nil (3b) 89.2
7 3b CuBr 3 Nil (3b) 90.3
8 3b CuI 3 Nil (3b) 90.8
9 3b CuCl2 3 0.8 (3b) 87.8
10 3b Cu(OTf)2 3 9.5 (3b) 80.1
The chloro acid 3a did not undergo substitution with sodium
bisulphite under standard conditions with Cu (I) halides as catalyst (entries 1,
2 and 3, Table 3.7). As mentioned in the introduction chapter, prior exchange
(metathesis) between the substrate and the copper salt is known to take place
under the conditions of Ullmann condensation and has been reported by
Weingarten (1964). There was little difference in the rates of cuprous bromide
and cuprous iodide catalyzed reactions of chloro acid 3a at 100°C which
indicated that there was no prior fast halogen exchange between the copper
salt and substrate under these conditions. The addition of phase transfer
catalysts including tetrabutylammonium iodide also did not prove useful
(entry 4 and 5, Table 3.7). Various salts of copper viz.; copper (I) chloride
(entry 6, Table 3.7), copper (I) bromide (entry 7, Table 3.7), copper (I) iodide

131
(entry 8, Table 3.7), copper (II) chloride (entry 9, Table 3.7) and copper (II)
triflate (entry 10, Table 3.7) were examined as catalysts for this substitution
reaction. Though all these copper salts brought about the Ullmann cross
coupling of bromo acid 3b with sodium bisulphite, cuprous chloride was
found to exhibit the best catalytic activity in effecting the cross coupling.
3.1.5 Effect of Different Oxidation States of Copper
Since Cu (II) is an oxidizing agent and sodium bisulphite is a reducing
agent, a control experiment was carried out to check the compatibility of Cu (II)
in presence of sodium bisulphite. When a solution of cupric chloride was mixed
with a solution of sodium bisulphite at room temperature , evolution of sulphur
dioxide gas was observed ( a filter paper wetted with potassium permanganate
solution turned colorless in presence of this liberated gas), indicating that Cu(II)
was getting reduced to Cu(I) in solution, probably as outlined in Scheme 3.5.
2CuCl2 + 2 NaHSO3 NaHSO4 + + NaCl + HCl +SO22 CuCl
Scheme 3.5 Cupric chloride in presence of sodium bisulphite
Nevertheless, a few experiments were conducted with CuCl2 as
catalyst under the standard conditions. The order of catalytic activity of various
oxidation states of copper, i. e. Cu (I) as cuprous chloride, Cu (II) as cupric
chloride and copper as Cu (0) were investigated under standard conditions
(Scheme 3.6).
S
Br
COOH
(3b)
1. NaOH / NaHSO3
reflux. pH 7.5- 7.7
S
SO3Na
COONa
(4a)
HCl / KClS
SO3K
COOH
(5a)
100 °C
CATALYST
Scheme 3.6 Ullmann condensation with sodium bisulphite with different
oxidation states of copper catalysts
132
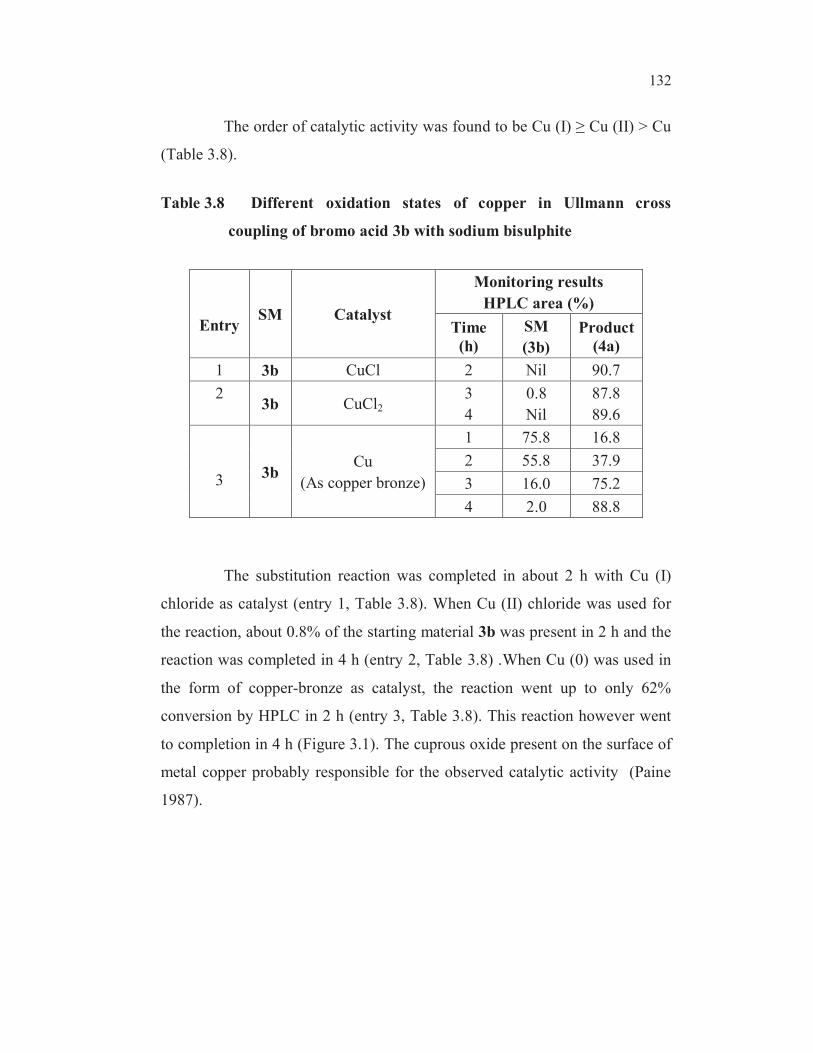
The order of catalytic activity was found to be Cu (I) Cu (II) > Cu
(Table 3.8).
Table 3.8 Different oxidation states of copper in Ullmann cross
coupling of bromo acid 3b with sodium bisulphite
EntrySM Catalyst
Monitoring results
HPLC area (%)
Time
(h)
SM
(3b)
Product
(4a)
1 3b CuCl 2 Nil 90.7
23b CuCl2
3
4
0.8
Nil
87.8
89.6
33b
Cu
(As copper bronze)
1 75.8 16.8
2 55.8 37.9
3 16.0 75.2
4 2.0 88.8
The substitution reaction was completed in about 2 h with Cu (I)
chloride as catalyst (entry 1, Table 3.8). When Cu (II) chloride was used for
the reaction, about 0.8% of the starting material 3b was present in 2 h and the
reaction was completed in 4 h (entry 2, Table 3.8) .When Cu (0) was used in
the form of copper-bronze as catalyst, the reaction went up to only 62%
conversion by HPLC in 2 h (entry 3, Table 3.8). This reaction however went
to completion in 4 h (Figure 3.1). The cuprous oxide present on the surface of
metal copper probably responsible for the observed catalytic activity (Paine
1987).
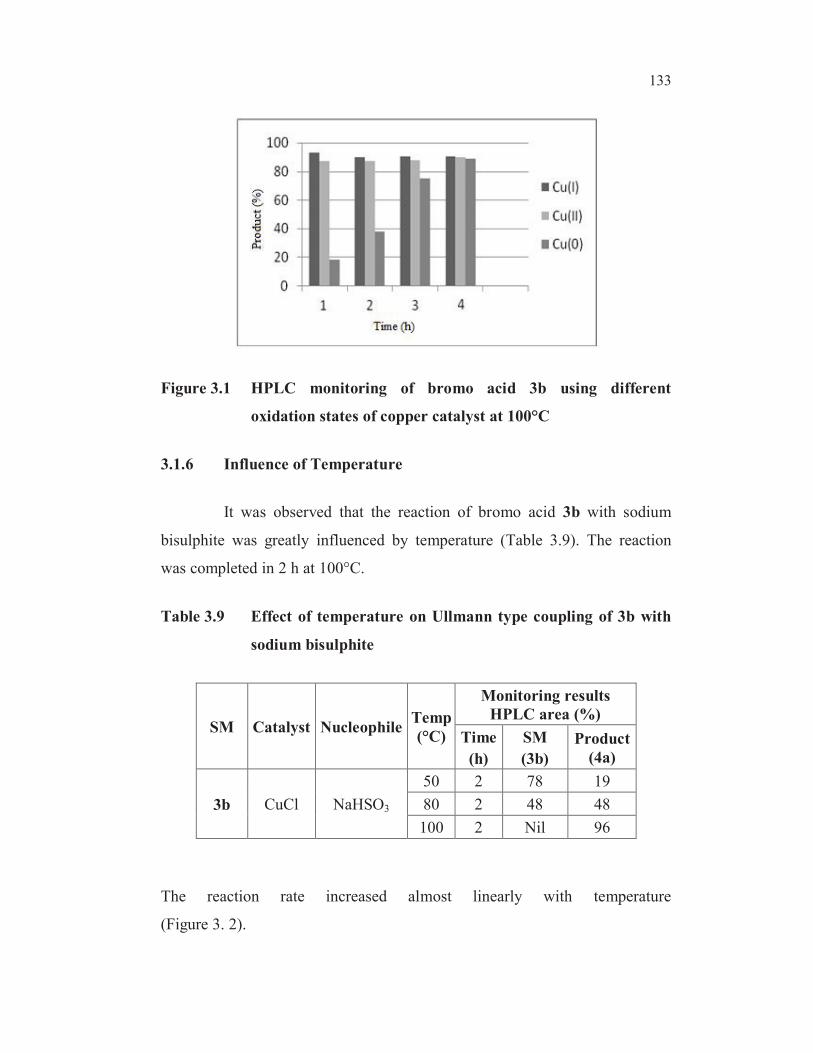
133
Figure 3.1 HPLC monitoring of bromo acid 3b using different
oxidation states of copper catalyst at 100°C
3.1.6 Influence of Temperature
It was observed that the reaction of bromo acid 3b with sodium
bisulphite was greatly influenced by temperature (Table 3.9). The reaction
was completed in 2 h at 100°C.
Table 3.9 Effect of temperature on Ullmann type coupling of 3b with
sodium bisulphite
SM Catalyst NucleophileTemp
(°C)
Monitoring results
HPLC area (%)
Time
(h)
SM
(3b)
Product
(4a)
3b CuCl NaHSO3
50 2 78 19
80 2 48 48
100 2 Nil 96
The reaction rate increased almost linearly with temperature
(Figure 3. 2).
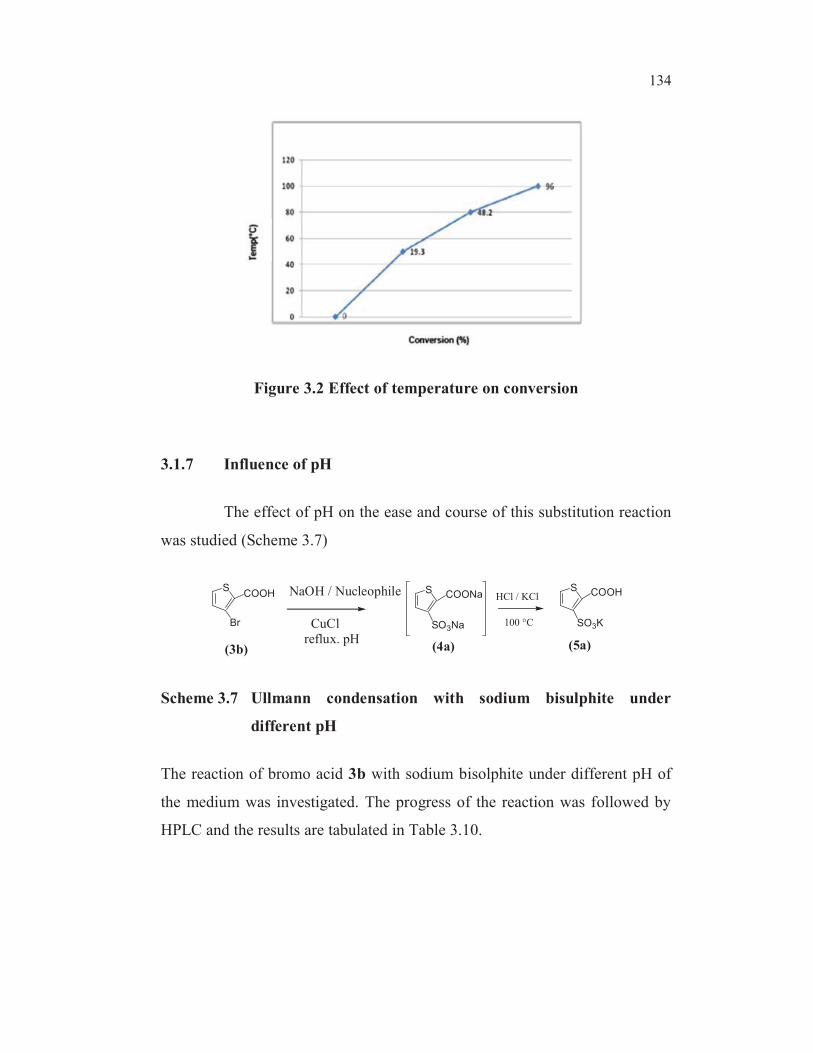
134
Figure 3.2 Effect of temperature on conversion
3.1.7 Influence of pH
The effect of pH on the ease and course of this substitution reaction
was studied (Scheme 3.7)
S
Br
COOH
(3b)
NaOH / Nucleophile
CuCl
reflux. pH
S
SO3Na
(4a)
COONa HCl / KClS
SO3K
COOH
(5a)
100 °C
Scheme 3.7 Ullmann condensation with sodium bisulphite under
different pH
The reaction of bromo acid 3b with sodium bisolphite under different pH of
the medium was investigated. The progress of the reaction was followed by
HPLC and the results are tabulated in Table 3.10.
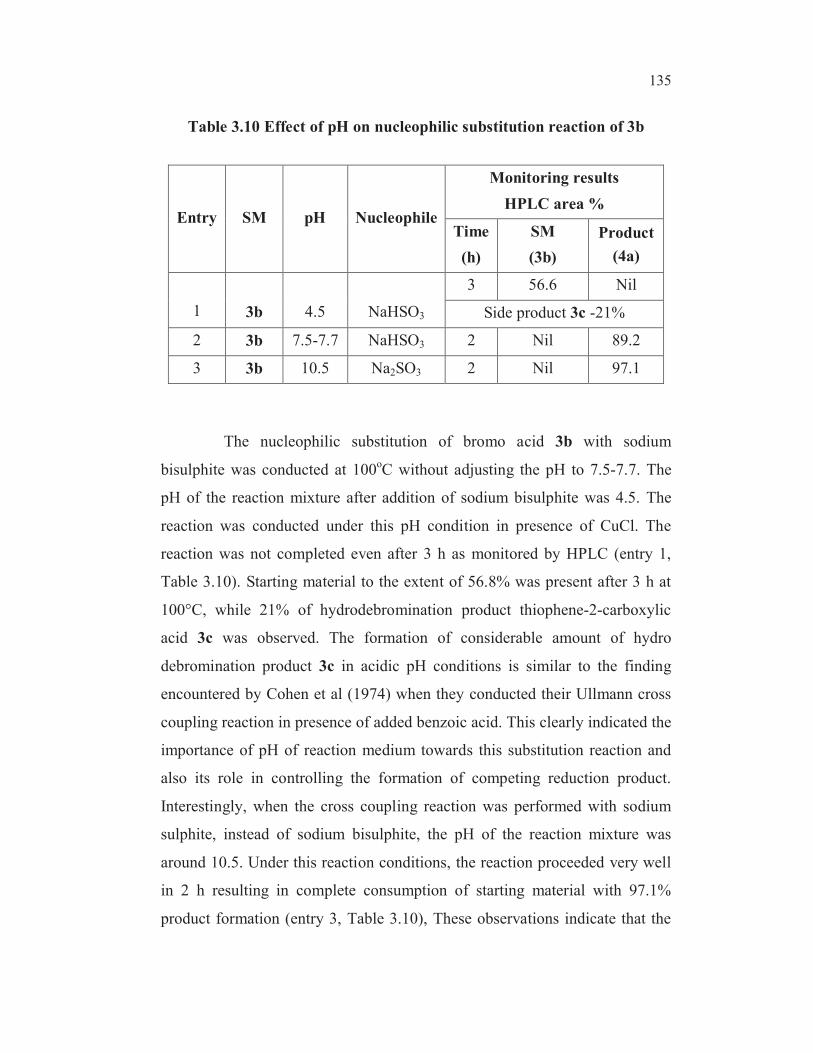
135
Table 3.10 Effect of pH on nucleophilic substitution reaction of 3b
Entry SM pH Nucleophile
Monitoring results
HPLC area %
Time
(h)
SM
(3b)
Product
(4a)
1 3b 4.5 NaHSO3
3 56.6 Nil
Side product 3c -21%
2 3b 7.5-7.7 NaHSO3 2 Nil 89.2
3 3b 10.5 Na2SO3 2 Nil 97.1
The nucleophilic substitution of bromo acid 3b with sodium
bisulphite was conducted at 100oC without adjusting the pH to 7.5-7.7. The
pH of the reaction mixture after addition of sodium bisulphite was 4.5. The
reaction was conducted under this pH condition in presence of CuCl. The
reaction was not completed even after 3 h as monitored by HPLC (entry 1,
Table 3.10). Starting material to the extent of 56.8% was present after 3 h at
100°C, while 21% of hydrodebromination product thiophene-2-carboxylic
acid 3c was observed. The formation of considerable amount of hydro
debromination product 3c in acidic pH conditions is similar to the finding
encountered by Cohen et al (1974) when they conducted their Ullmann cross
coupling reaction in presence of added benzoic acid. This clearly indicated the
importance of pH of reaction medium towards this substitution reaction and
also its role in controlling the formation of competing reduction product.
Interestingly, when the cross coupling reaction was performed with sodium
sulphite, instead of sodium bisulphite, the pH of the reaction mixture was
around 10.5. Under this reaction conditions, the reaction proceeded very well
in 2 h resulting in complete consumption of starting material with 97.1%
product formation (entry 3, Table 3.10), These observations indicate that the

136
actual nucleophile may be sulphite ion even in the case of reaction with
sodium bisulphite, when the reaction was conducted at pH 7.5-7.7 with
sodium bisulphite.
3.1.8 Optimal Catalyst Loading
Next, the nucleophilic substitution of bromo acid 3b with sodium
bisulphite was studied with respect to the catalyst loading (Scheme 3.8) under
standard conditions.
S
Br
COOH
(3b)
NaOH / NaHSO3
CATALYST
reflux. pH 7.5- 7.7
S
SO3Na
(4a)
COONa HCl / KClS
SO3K
COOH
(5a)
100 °C
Scheme 3.8 Ullmann condensation with sodium bisulphite under
different loading of CuCl
The reaction was monitored by HPLC and the results are shown in
Table 3.11. The optimal catalyst loading was found to be 0.1 mol with respect
to the substrate. Higher loadings resulted in the formation of more of
hydrodebromination product 3c viz., thiophene-2-carboxylic acid, though
complete conversion of the starting material 3b was observed even at the end
of 0.5 h (entry 2 and 3, Table 3.11). It was observed that excess catalyst make
the substitution reaction faster but the selectivity was reduced due to
competing side reactions such as the formation of more thiphene-2-
carboxylic acid .
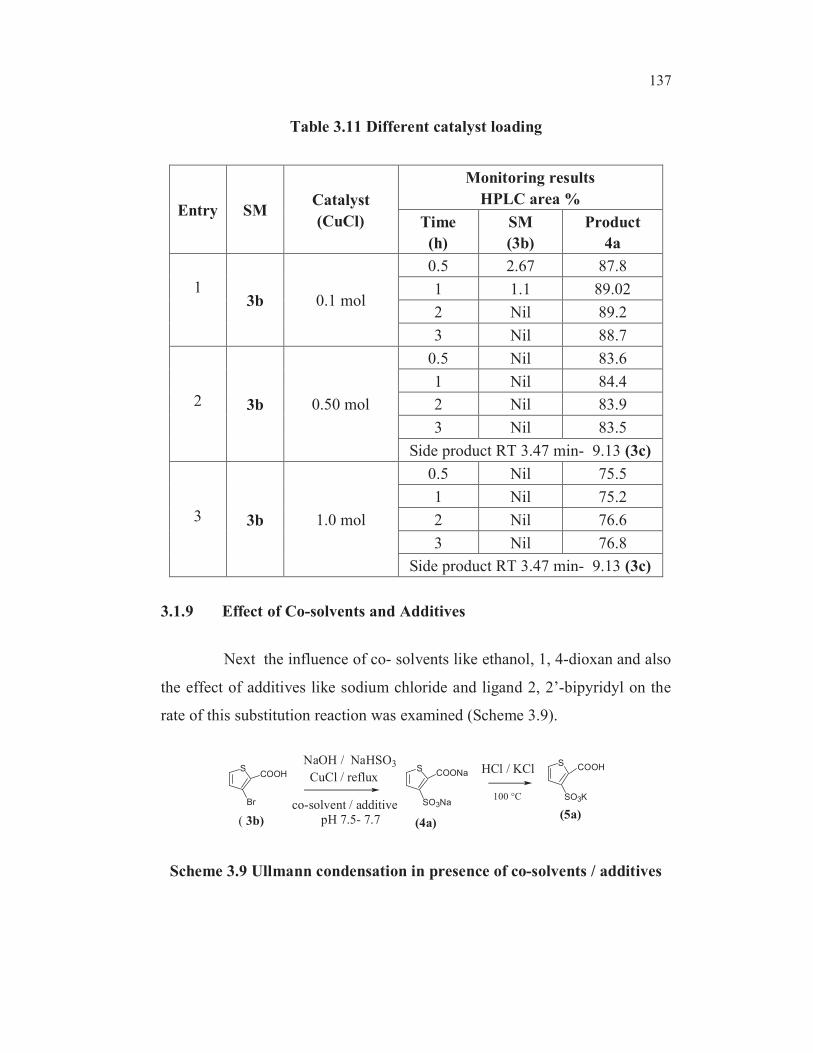
137
Table 3.11 Different catalyst loading
Entry SMCatalyst
(CuCl)
Monitoring results
HPLC area %
Time
(h)
SM
(3b)
Product
4a
13b 0.1 mol
0.5 2.67 87.8
1 1.1 89.02
2 Nil 89.2
3 Nil 88.7
2 3b 0.50 mol
0.5 Nil 83.6
1 Nil 84.4
2 Nil 83.9
3 Nil 83.5
Side product RT 3.47 min- 9.13 (3c)
3 3b 1.0 mol
0.5 Nil 75.5
1 Nil 75.2
2 Nil 76.6
3 Nil 76.8
Side product RT 3.47 min- 9.13 (3c)
3.1.9 Effect of Co-solvents and Additives
Next the influence of co- solvents like ethanol, 1, 4-dioxan and also
the effect of additives like sodium chloride and ligand 2, 2’-bipyridyl on the
rate of this substitution reaction was examined (Scheme 3.9).
S
Br
COOH
( 3b)
NaOH / NaHSO3
CuCl / reflux
co-solvent / additive
pH 7.5- 7.7
S
SO3Na
(4a)
COONa HCl / KClS
SO3K
COOH
(5a)
100 °C
Scheme 3.9 Ullmann condensation in presence of co-solvents / additives
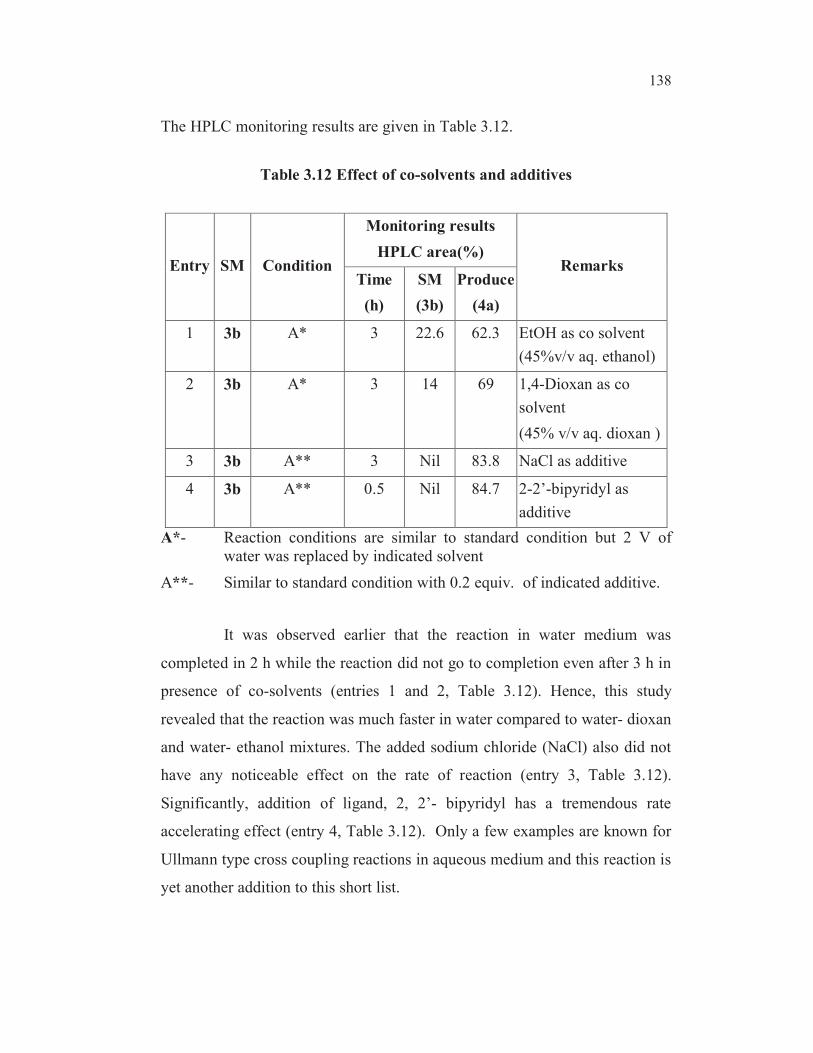
138
The HPLC monitoring results are given in Table 3.12.
Table 3.12 Effect of co-solvents and additives
Entry SM Condition
Monitoring results
HPLC area(%)Remarks
Time
(h)
SM
(3b)
Produce
(4a)
1 3b A* 3 22.6 62.3 EtOH as co solvent
(45%v/v aq. ethanol)
2 3b A* 3 14 69 1,4-Dioxan as co
solvent
(45% v/v aq. dioxan )
3 3b A** 3 Nil 83.8 NaCl as additive
4 3b A** 0.5 Nil 84.7 2-2’-bipyridyl as
additive
A*- Reaction conditions are similar to standard condition but 2 V of
water was replaced by indicated solvent
A**- Similar to standard condition with 0.2 equiv. of indicated additive.
It was observed earlier that the reaction in water medium was
completed in 2 h while the reaction did not go to completion even after 3 h in
presence of co-solvents (entries 1 and 2, Table 3.12). Hence, this study
revealed that the reaction was much faster in water compared to water- dioxan
and water- ethanol mixtures. The added sodium chloride (NaCl) also did not
have any noticeable effect on the rate of reaction (entry 3, Table 3.12).
Significantly, addition of ligand, 2, 2’- bipyridyl has a tremendous rate
accelerating effect (entry 4, Table 3.12). Only a few examples are known for
Ullmann type cross coupling reactions in aqueous medium and this reaction is
yet another addition to this short list.

139
3.1.10 Microwave Conditions
The copper mediated nucleophilic substitution of 3a and 3b with
sodium bisulphite was also performed under similar microwave conditions
(Scheme 3.10) using the same molar proportions of reagents.
S
X
COOH
X= Cl (3a)
X= Br (3b)
CuCl/ MicrowaveS
SO3Na
(4a)
COONa HCl / KClS
SO3K
COOH
(5a)
100 °C
pH 7.5- 7.7
NaOH . NaHSO3
Scheme 3.10 Ullmann condensation with sodium bisulphite under
microwave
It was observed that under microwave condition also the bromo acid
3b was more reactive than chloro acid 3a (Table 3.13).
Table 3.13 Monitoring results under microwave conditions.
Entry SM
Monitoring results
HPLC area(%)Remarks
Time
(min.)SM
Product
(4a)
1 3a 295.6
(3a)2.9
Microwave conditions, 80°C,
and 100W- No reaction
2 3b 24.9
(3b)68.8
Microwave conditions 80°C,
100W, Product formed. Better
conversion compared
to 3a

140
3.1.11 Effect of Concentration of Sodium Bisulphite
The Ullmann cross coupling of 3-bromothiophene-2-carboxylic acid
3b was studied with respect to different molar proportions of sodium
bisulphite with respect to the bromo acid 3b under standard conditions.
(Scheme 3.11)
S
Br
COOH
( 3b)
NaOH / Nucleophile
CuCl / reflux
pH 7.5- 7.7
S
SO3Na
(4a)
COOH HCl / KClS
SO3K
COOH
(5a)
100 °C
Scheme 3.11 Ullmann condensation with different concentrations of
sodium bisulphite
The reaction was monitored by HPLC (Table 3.14). Though there
was no appreciable difference between 1.15 equiv (entry 1, Table 3.14) and
1.5 equiv of sodium bisulphite (entry 2, Table 3.14), the optimal quantity of
the nucleophile would be 1.15 equiv. with respect to the substrate from the
raw material perspective. With increase in concentration of the nucleophile
(entry 3, Table 3.14), the selectivity was not affected though there was
sluggishness in the initial rate of reaction.
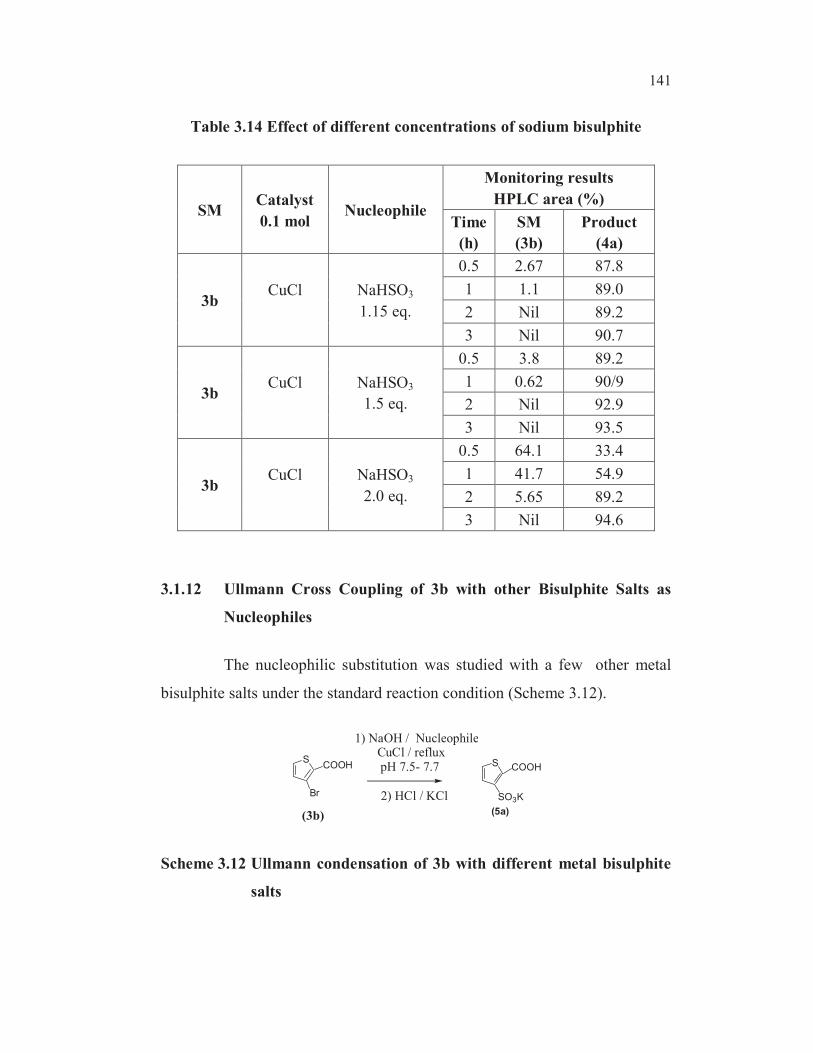
141
Table 3.14 Effect of different concentrations of sodium bisulphite
SMCatalyst
0.1 molNucleophile
Monitoring results
HPLC area (%)
Time
(h)
SM
(3b)
Product
(4a)
3bCuCl NaHSO3
1.15 eq.
0.5 2.67 87.8
1 1.1 89.0
2 Nil 89.2
3 Nil 90.7
3bCuCl NaHSO3
1.5 eq.
0.5 3.8 89.2
1 0.62 90/9
2 Nil 92.9
3 Nil 93.5
3bCuCl NaHSO3
2.0 eq.
0.5 64.1 33.4
1 41.7 54.9
2 5.65 89.2
3 Nil 94.6
3.1.12 Ullmann Cross Coupling of 3b with other Bisulphite Salts as
Nucleophiles
The nucleophilic substitution was studied with a few other metal
bisulphite salts under the standard reaction condition (Scheme 3.12).
S
Br
COOH
(3b)
1) NaOH / Nucleophile CuCl / reflux pH 7.5- 7.7
2) HCl / KCl
S
SO3K
COOH
(5a)
Scheme 3.12 Ullmann condensation of 3b with different metal bisulphite
salts
142
The substitution reaction was monitored by HPLC (Table 3.15).
Table 3.15 Reaction of 3b with different nucleophiles
EntrySM Nucleophile
Monitoring results
HPLC area (%)
Time
(h)
SM
(3b)
Product
(5a)
1 3b Calcium bisulphite 3 18.1 22.3
2 3b Cerium bisulphite 3 11.9 53.3
3 3b Sodium sulphite (pH 10.5) 2 Nil 97.1
4 3b Sodium sulphite (pH 7.5-7.7) 3 7.1 86.2
The substitution reaction of 3b was slow when calcium bisulphite
(entry 1, Table 3.15) or cerium bisulphite (entry 2, Table 3.15) was used as
nucleophile under standard condition. More of side product viz., thiophene-2-
carboxylic acid 3c was formed in these cases. Interestingly, when the cross
coupling reaction was performed with same equivalents of sodium sulphite
(pH of the reaction mixture was around 10.5) instead of sodium bisulphite,
the reaction proceeded very well in 2 h resulting complete consumption of
starting material with 97.1% product formation as monitored by HPLC
(entry 3, Table 3.15). When the same reaction with sodium sulphite was
performed at pH 7.5-7.7, by adjusting the pH from 10.5 using dilute HCl, it
was observed that even after 3 h, the reaction was incomplete and 7.1% of the
starting material 3b remained unreacted. Probably the addition of acid to
adjust the pH would have quenched some quantity of sodium sulphite
resulting in depletion of the nucleophile. These observations indicate that the
actual nucleophile may be the sulphite ion even in the case of the reaction
with sodium bisulphite as the reaction was done at pH 7.5-7.7 and that its
concentration might had been low at this pH.

143
3.1.13 Ullmann Condensation without Nucleophile
The Ullmann cross coupling of 3b was conducted without sodium
bisulphite under standard conditions (Scheme 3.13) to see the outcome. The
results are shown in Table 3.13.
S
Br
COOH
( 3b)
1. NaOH / CuCl
pH 7.5- 7.7S
H
(3c)
COOH
2. HCl / KCl
+
S
OH
COOH
(5e)
Scheme 3.13 Ullmann condensation without nucleophile
In this event, the reaction resulted in the formation of more of
hydrodebromination product 3c in addition to unreacted starting material
(entry1, Table 3.16). Formation of hydrodebromination 3c, as the major
product under alkaline condition is noteworthy. The dehalogenation reaction
was more pronounced under drastic conditions viz., 140-143°C under
autoclave conditions (entry 2, Table 3.16). Another side product at RT 3.775
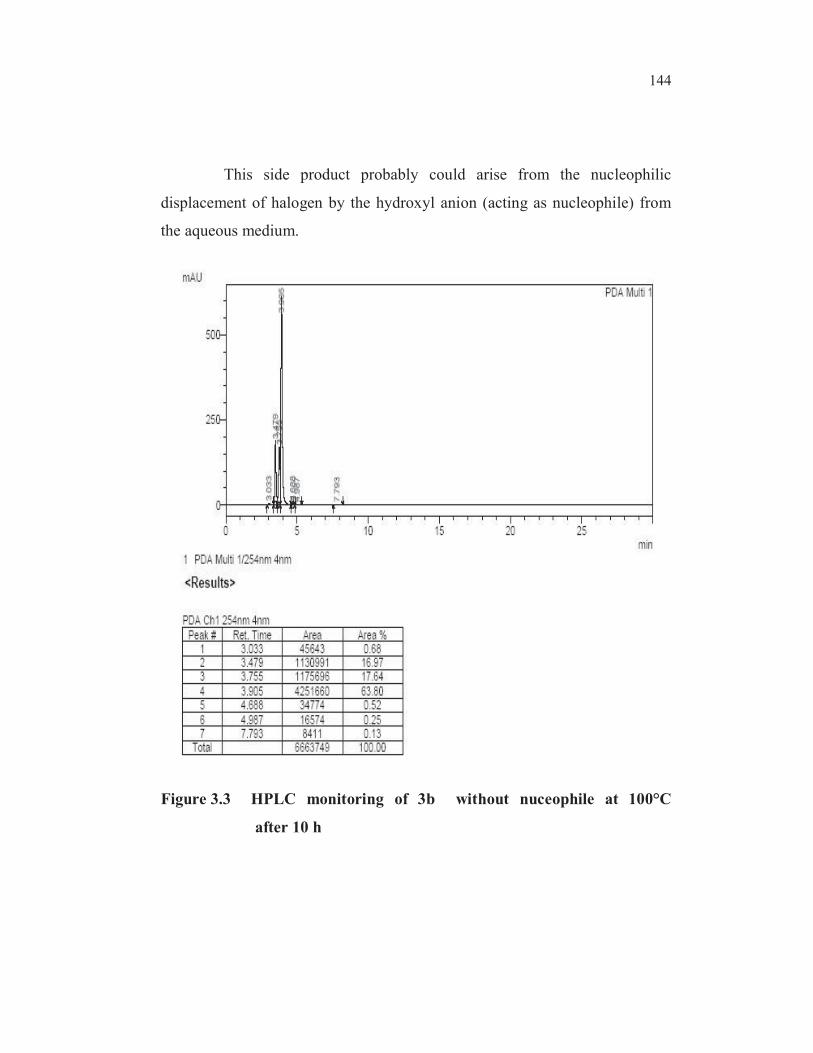
min was also observed in the HPLC trace of the monitoring sample
(Figure 3.3).
Table 3.16 Ullmann cross coupling of 3b without nuceophile
EntryCatalyst
10 mole%
Temp
( C)
Monitoring results
HPLC area (%)
Time
(h)
SM
(3b)(3c)
1CuCl 100
10 63.8 16.7
(RT 3.479 min)
2CuCl 140- 143
16 48.1 34.4
(RT 3.476 min)
144
This side product probably could arise from the nucleophilic
displacement of halogen by the hydroxyl anion (acting as nucleophile) from
the aqueous medium.
Figure 3.3 HPLC monitoring of 3b without nuceophile at 100°C
after 10 h
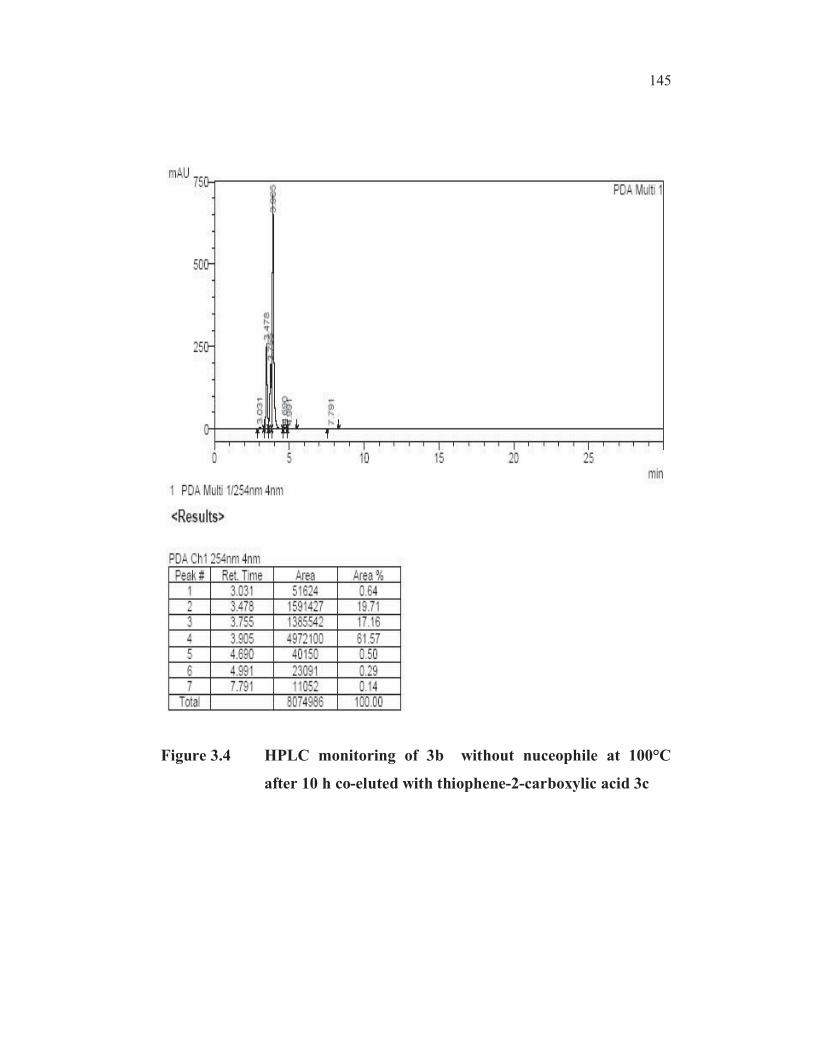
145
Figure 3.4 HPLC monitoring of 3b without nuceophile at 100°C
after 10 h co-eluted with thiophene-2-carboxylic acid 3c

146
Figure 3.5 HPLC monitoring of 3b without nucleophile at 140°C after
16 h
3.1.14 Isomeric Bromothiophenecarboxylic Acids
For the first time, the Ullmann type cross coupling reactions of
isomeric bromothiophene carboxylic acids, viz., 3-bromothiophene-2-
carboxylic acid 3b, 4-bromothiophene-2-carboxylic acid 3d, 5-
bromothiophene -2-carboxylic acid 3e and 2-bromothiophene -3-carboxylic
acid 3f with sodium bisulphite was studied under the standard condition in an
effort to understand the mechanism of the substitution reaction viz., the
147
relative importance of chelation and electronic effects like mesomeric
interactions (Scheme 3.14). The results are shown in Table 3.17.
S
1) NaOH / NaHSO3
CuCl / reflux
pH 7.5- 7.7S
2) HCl / KClBr
HOOC
2-COOH and 3-Br (3b)
2-COOH and 4-Br (3d)2-COOH and 5-Br (3e)
3-COOH and 2-Br (3f)(SM))
SO3K
2-COOH and 3-SO3K (5a)
2-COOH and 4-SO3K (5b)
2-COOH and 5-SO3K (5c)
3-COOH and 2-SO3K (5d)
(Product)
HOOC
Scheme 3.14 Ullmann condensation with isomeric bromothio
phenecarboxylic acids
Table 3.17 Ullmann cross coupling reaction of isomeric
bromothiophenecarboxylic acids with sodium bisulphite
Entry SM Structure
Monitoring results
HPLC area (%)Remarks
Time
(h)SM Product
1 3b SCOOH
Br
2 Nil (3b) 89.2 (5a) High
Conversion.
2 3d SCOOH
Br
3 73.6(3d) 22.4 (5b) Low
conversion
3 3e SCOOH
Br 321.1(3e) 73.6 (5c)
Faster than 3d
reaction.
16Nil (3e) 84.6 (5c)
Reaction at
140°C
4 3f SBr
COOH
0.5 2.1 (3f) 77.7 (5d) Very little SM
after 0.5 h.

148
The reaction of 4-bromo acid 3d with sodium bisulphite was significantly
slower compared to other isomers and only 22% conversion was observed
after 3 h under standard conditions (entry 2, Table 3.17). The conversion did
not improve even on drastic conditions, viz. heating in an autoclave 140°C. In
comparison, the 5- bromo acid 3e exhibited a better reactivity leading to 73%
conversion in 3 h under standard conditions. Complete conversion in this case
was observed at 140°C in an autoclave (entry 3, Table 3.17). In the case of 3f,
the reaction was fast and almost complete in 0.5 h (entry 4, Table 3.17). These
findings are similar to the results obtained by Tim et al (2000) in the case of
Pd catalyzed amination of electron deficient halothiophenes. The authors
prepared a series of functionalized aminothiophenes by palladium catalyzed
amination reactions. The authors observed that the reaction proceeded well in
these cases where the halide is adjacent to an electron withdrawing group
(Scheme 3.15). Thus the reactivity exhibited by 3-bromothiophene-2
carboxylic acid and 5-bromothiophene-2-carboxylic acid bring about the
importance of both the chelation and mesomeric interactions.
Between the two bromo acids, 3b (entry 1, Table 3.17) and 3f
(entry 4, Table 3.17), 3f displayed higher reactivity but at the same time led to
more hydrodebromination product 3h in comparison to that of 3b. The results
of this study indicated the following order of reactivity: 3f > 3b > 3e >> 3d.
This is an important finding towards understanding the mechanism of the
reaction. The mono potassium salt 5c obtained from 3e is not known in
literature. This salt was isolated and characterized by spectral data and HRMS
analysis (Section 2. 6.3). The reactivity of 3b and 3f parallels the observation
of Menno et al (1992) in the Ullmann coupling of 3-bromthiophene and 2-
bromothiophene with alkoxides.

149
SCOOMe
Br
SCOOMe
NHBu(Yield 94%) Methyl-3-n-butylaminothiophe
ne-2 carboxylate
+
n-butylamine1.2 equiv
2 equiv.Cs2CO3
5 mol% Pd(dba)2
Toluene/reflux
BuNH2
SCOOMe
SCOOMe
(Yield 76%%)Methyl-5-bromothiophene-2 carboxylate ( 1.0 equiv.)
+
N-(methyl) benzylamine, 1.2 equiv
2 equiv.Cs2CO3
5 mol% Pd(dba)2
Toluene/reflux
PhNHMe
Ph(Me)NBr
Methyl-4-N-(methyl)benzylamino-thiophene-2 carboxylate
SCN
Br
(Yield 86%)
n-butylamine1.2 equiv
2 equiv.Cs2CO3
5 mol% Pd(dba)2
Toluene/reflux
BuNH2+
3-bromo-2-cyanothiophene (1 equiv)
SCN
NHBu
3-n-butylamino-2-cyanot hiophene
SCN
(Yield 0%)
2 equiv.Cs2CO3
5 mol% Pd(dba)2
or 5 mol%Pd(OAc)2
Toluene/reflux+
4-bromo-2-cyanothiophene (1 equiv)
SCN
Br
PhNHMePh(Me)N
Methyl-5-N-(methyl)benzylamino-thiophene-2 carboxylate
Methyl-3-bromothiophene-2 carboxylate ( 1.0 equiv.)
N-(methyl) benzylamine, 1.2 equiv
Scheme 3.15 Pd catalyzed amination of electron deficient halothiophenes
They observed that the reaction in the case of 3-bromothiophne
afforded the expected ethers and no reduction product was observed whereas
the reaction in the case of 2 bromothiophene gave rise to more reduction
product. The following path way may be envisaged for the copper mediated
reduction process at C2 as shown in Scheme 3.16
S
Br
O Cu
HH
H
S
Br
CuOCH3S
H
+ CuBr + HCHO
2-bromothiophene
Scheme 3.16 Copper mediated reduction process

150
The delocalization of lone pairs of electrons on the sulphur atom
supports this path way for hydrodehalogenation. Probably the following
mechanistic pathway (Scheme 3.17) would account for a greater hydro
debromination in the case of 2-bromothiophene.
SBr
CuOCH3S
Br
Cu O
H
HH S
H + HCHO
2-bromothiophene
CuBr
Scheme 3.17 Possible pathway of reduction in 2-bromothiophene
3.1.15 Effect of Radical Scavengers
The copper mediated nucleophilic substitution of bromo acid 3b
with sodium bisulphite under standard condition was performed in the
presence of radical scavengers (Scheme 3.18).like molecular oxygen, 2, 2, 6,
6-tetramethylpiperidinyl-1-oxy (TEMPO), para di-nitrobenzene etc
(Bowman, et al 1987). The results are shown in Table 3.18
S
Br
COOH
(3b)
NaOH / NaHSO3
CuCl / T°CpH 7.5- 7.7
S
SO3K
(5a)
COOHS
SO3Na
(4a)
COOHHCl / KCl
Radical Scavenger
Scheme 3.18 Ullmann condensation in presence of radical scavengers
151
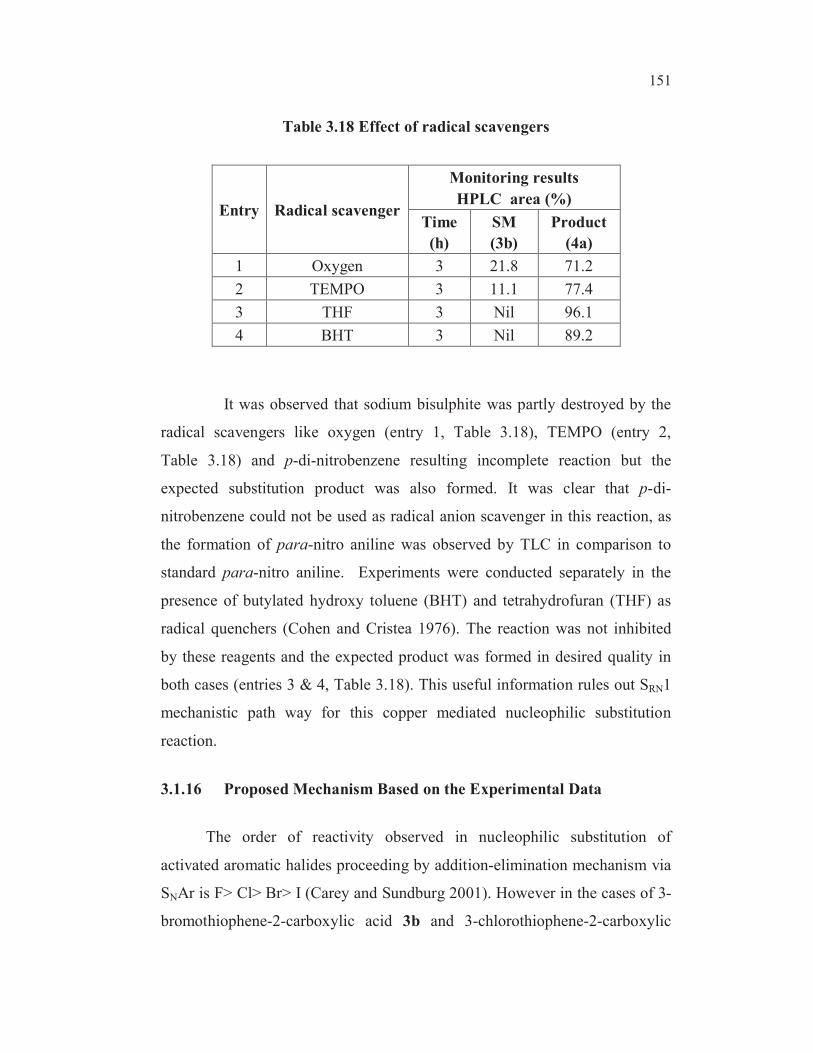
Table 3.18 Effect of radical scavengers
Entry Radical scavenger
Monitoring results
HPLC area (%)
Time
(h)
SM
(3b)
Product
(4a)
1 Oxygen 3 21.8 71.2
2 TEMPO 3 11.1 77.4
3 THF 3 Nil 96.1
4 BHT 3 Nil 89.2
It was observed that sodium bisulphite was partly destroyed by the
radical scavengers like oxygen (entry 1, Table 3.18), TEMPO (entry 2,
Table 3.18) and p-di-nitrobenzene resulting incomplete reaction but the
expected substitution product was also formed. It was clear that p-di-
nitrobenzene could not be used as radical anion scavenger in this reaction, as
the formation of para-nitro aniline was observed by TLC in comparison to
standard para-nitro aniline. Experiments were conducted separately in the
presence of butylated hydroxy toluene (BHT) and tetrahydrofuran (THF) as
radical quenchers (Cohen and Cristea 1976). The reaction was not inhibited
by these reagents and the expected product was formed in desired quality in
both cases (entries 3 & 4, Table 3.18). This useful information rules out SRN1
mechanistic path way for this copper mediated nucleophilic substitution
reaction.
3.1.16 Proposed Mechanism Based on the Experimental Data
The order of reactivity observed in nucleophilic substitution of
activated aromatic halides proceeding by addition-elimination mechanism via
SNAr is F> Cl> Br> I (Carey and Sundburg 2001). However in the cases of 3-
bromothiophene-2-carboxylic acid 3b and 3-chlorothiophene-2-carboxylic

152
acid 3a, which are activated hetero aromatic halides, a reversal in reactivity
was observed when they are subjected to Ullmann cross coupling reactions
with sodium bisulphite. The bromo acid 3b displayed a much higher reactivity
compared to the chloro acid 3a despite the patent literature recommending
chloro acid 3a over bromo acid 3b for this nucleophilic substitution reaction.
The ease of reduction of C-X bond in aromatic system under electrochemical
conditions has been found to parallel the leaving group ability (Gibson and
Spitzmesser 2003). In the case of aromatic compounds and the same aromatic
moiety, there is a rough correlation to electron transfer reactions (Denney, D.
E. and Denney, D. J. 1991 and Denney et al 1993). The ease of reductions
revealed by the reduction potential in liquid ammonia is PhI > PhBr > PhSPh
> PhCl > PhF > PhOPh (Amatore et al 1985) coincides with the reactivity
order determined under photo-initiation. Experiments carried out with pairs
of PhX in liquid ammonia under irradiation towards CH2COBut ions indicated
the following order of reactivity .PhCl / PhF= 29, PhBr / PhCl= 450 and PhI /
PhBr= 8.3. Therefore the increase in reactivity from PhF to PhI is almost
100000 (Amatore et al 1981). It was of interest to find out whether one could
expect a similar parallel between the cathodic reduction potential of C-X bond
and the ease of Ullmann cross coupling. This has been advanced as an
evidence in favour of the mechanism in the case of SRN1 reactions (Gibson
and Spitzmesser 2003). Since both the reduction of the C-X bond and
oxidative addition of a metal/metal ion into a C-X bond involve addition of 2
electrons, it was of interest to find out whether any correlation could be
observed between the reduction potentials and ease of the Ullmann cross
coupling of the various halothiophene carboxylic acids. More importantly,
whether the observed higher reactivity of 2-bromobenzoic acid 8a over 3-
bromothiophene-2-carboxylic acid 3b in the Ullmann cross coupling reaction
with sodium bisulphite could be correlated with the reduction potential of
these two bromo derivatives. So far no such study has been reported in
literature. In fact a comparative study of the electrochemical reduction of
153
various halothiophenecarboxylic acids or halobenzoic acids has not been
reported so far in literature. With a view to get some information regarding
this, the cathodic reduction potential of C- X bonds in the molecules of
interest were measured using cyclic voltammetry and the results are shown in
Table 3.19
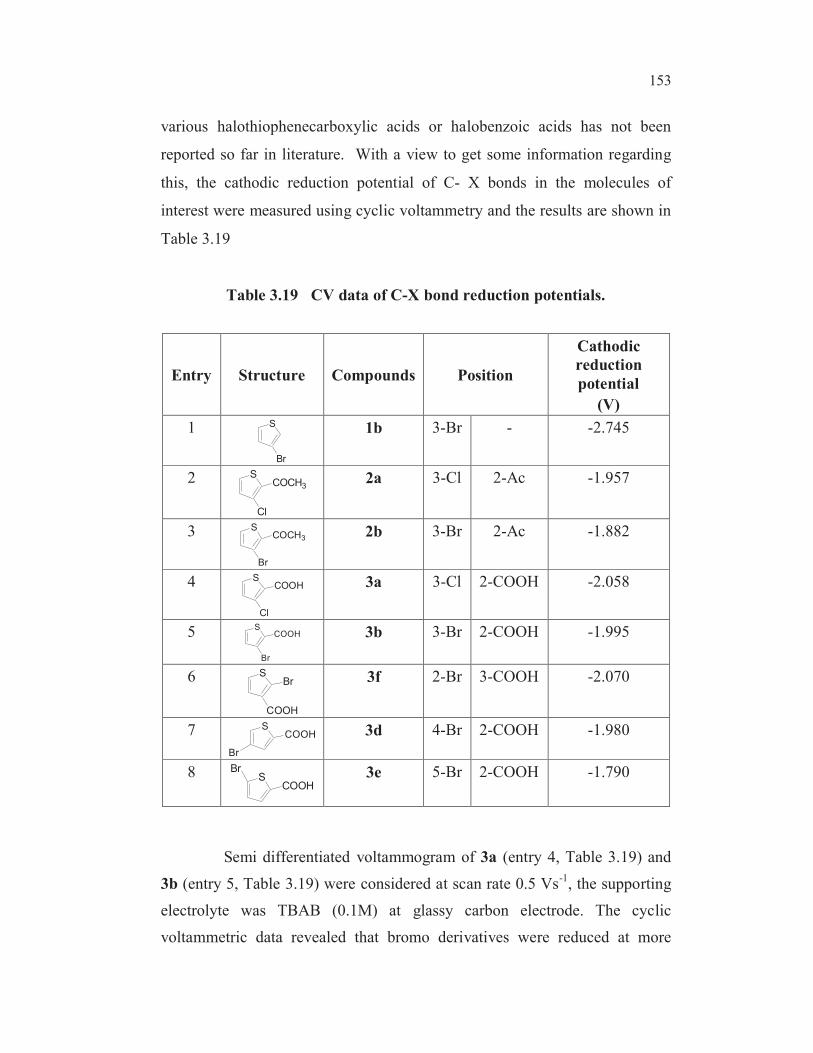
Table 3.19 CV data of C-X bond reduction potentials.
Entry Structure Compounds Position
Cathodic
reduction
potential
(V)
1 S
Br
1b 3-Br - -2.745
2 S
Cl
COCH32a 3-Cl 2-Ac -1.957
3 S
Br
COCH32b 3-Br 2-Ac -1.882
4 S
Cl
COOH 3a 3-Cl 2-COOH -2.058
5 S
Br
COOH 3b 3-Br 2-COOH -1.995
6 S
COOH
Br 3f 2-Br 3-COOH -2.070
7 SCOOH
Br
3d 4-Br 2-COOH -1.980
8 SCOOH
Br 3e 5-Br 2-COOH -1.790
Semi differentiated voltammogram of 3a (entry 4, Table 3.19) and
3b (entry 5, Table 3.19) were considered at scan rate 0.5 Vs-1
, the supporting
electrolyte was TBAB (0.1M) at glassy carbon electrode. The cyclic
voltammetric data revealed that bromo derivatives were reduced at more
154
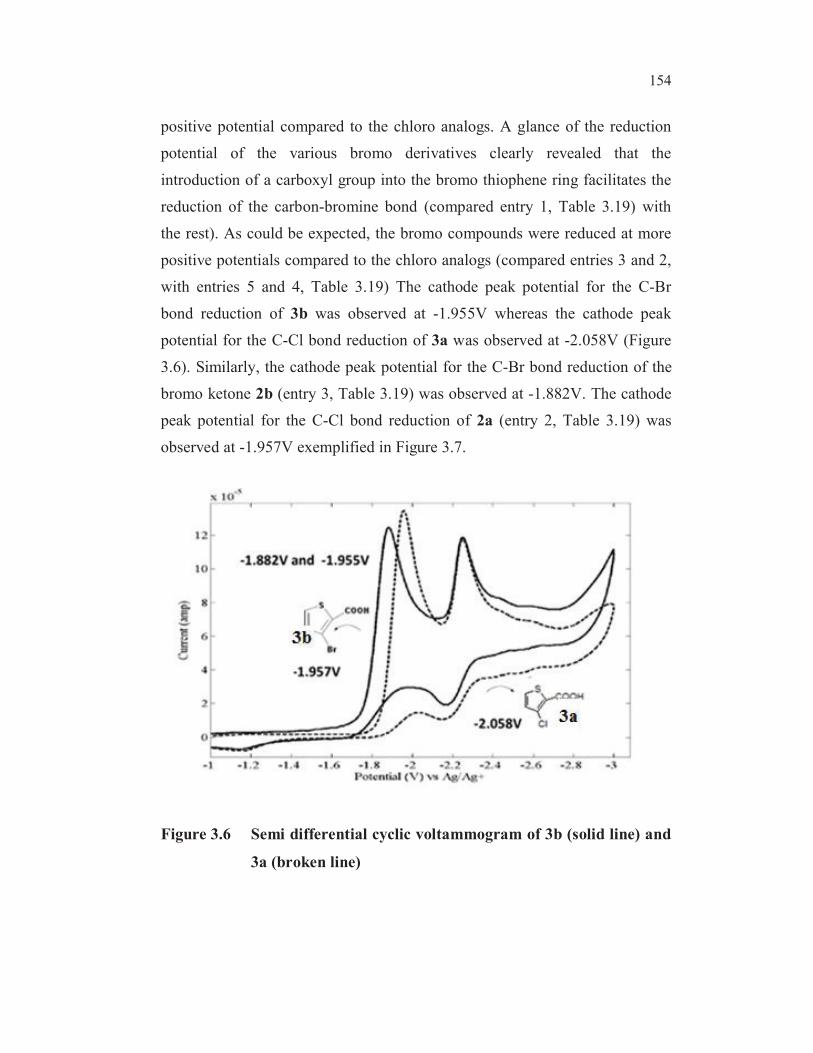
positive potential compared to the chloro analogs. A glance of the reduction
potential of the various bromo derivatives clearly revealed that the
introduction of a carboxyl group into the bromo thiophene ring facilitates the
reduction of the carbon-bromine bond (compared entry 1, Table 3.19) with
the rest). As could be expected, the bromo compounds were reduced at more
positive potentials compared to the chloro analogs (compared entries 3 and 2,
with entries 5 and 4, Table 3.19) The cathode peak potential for the C-Br
bond reduction of 3b was observed at -1.955V whereas the cathode peak
potential for the C-Cl bond reduction of 3a was observed at -2.058V (Figure
3.6). Similarly, the cathode peak potential for the C-Br bond reduction of the
bromo ketone 2b (entry 3, Table 3.19) was observed at -1.882V. The cathode
peak potential for the C-Cl bond reduction of 2a (entry 2, Table 3.19) was
observed at -1.957V exemplified in Figure 3.7.
Figure 3.6 Semi differential cyclic voltammogram of 3b (solid line) and
3a (broken line)
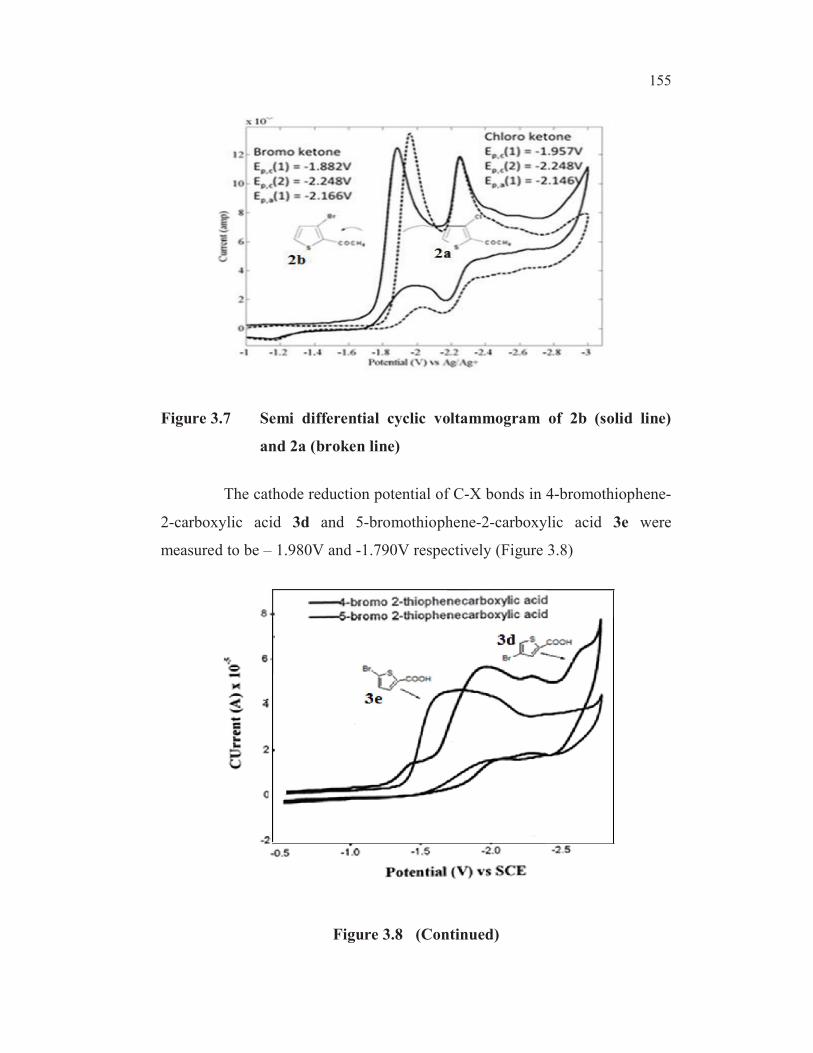
155
Figure 3.7 Semi differential cyclic voltammogram of 2b (solid line)
and 2a (broken line)
The cathode reduction potential of C-X bonds in 4-bromothiophene-
2-carboxylic acid 3d and 5-bromothiophene-2-carboxylic acid 3e were
measured to be – 1.980V and -1.790V respectively (Figure 3.8)
Figure 3.8 (Continued)

156
Figure 3.8 Semi differential cyclic voltammogram of 3d, 3e and 3f
The halo ketones undergo reduction more readily compared to the
halo acids, as one would anticipate (entry 2 vs entry 4, Table 3.19 and entry 3
vs entry 5, Table 3.19). The 2-bromothiophen-3-carboxylic acid 3f is reduced
at more negative potential (-2.070V) compared to the 3-bromo isomer 3b
(-1.995V). This could be attributed to the alpha hetero atom effect, viz.,
electron repulsion between the lone pair of electrons on the sulphur and the
developing negative charge on the C2. This parallels the ease of reduction of
bromothiophenes mediated by metals. It is known that the metal mediated
reduction of 2, 3, 4- tribromothiophenes leads first to 2,3-dibromo, then to 3-
bromo and finally to 2-bromothiophene (Gronowitz and Raznikiewicz 1973).
The reduction potential of 5-bromothioophene-2-carboxylic acid ( -1.790V) is
found to be lowest among all the bromothiophenes studied ( entry 8,
Table 3.19). This could be due to the mesomeric interaction involving
extended conjugation. One more factor may contribute for the easier
reduction of carbon-bromine bond in the case of this compound. In the case
of 3-bromothiophene-2-carboxylic acid 3b, the dipole repulsion between the
two functionalities may make the reduction process more difficult (alpha
haloketone effect and anomeric effect) whereas in the case of 5-
bromothiophene-2-carboxylic acid 3e, such a dipole repulsion does not exist.

157
While the results obtained from the above study did not throw much
light to understand the mechanism of the reaction, they provided new and
interesting information. Uncatalyzed pathway or Lewis acid promoted SNAr
mechanism proceeding through addition-elimination mechanism can be ruled
out on the basis of the following observations: (i) first of all, chloro analog
displayed very poor reactivity compared to the bromo analog (ii) Other Lewis
acids did not bring about coupling reaction and only Cu (I) was found to be
the effective catalyst (iii) In the absence of cuprous chloride, cross coupling
reaction was not observed (iv) high reactivity of ortho bromo carboxylic acid,
so called ‘ortho- carboxylate effect’ observed in the present case as well as
reported by James Paine (1985) and McKillop and Bruggink (1975) in related
halobenzoic acid salts. Our findings parallels the observations of Cirigotts et
al (1974) who noted that silver nitrate, zinc chloride, nickel chloride and Pd
(II) chloride were completely ineffective catalysts for condensation of 2-
bromobenzoic acid with benzoyl acetone in ethanolic sodium hydroxide. They
had observed that catalysis by some copper species was essential for this
reaction. Elimination-addition mechanism via an aryne intermediate (Terrier
1982 and Diliang Gua et al 2008) can also be ruled out as there was no scope
for formation of any hetero aryne intermediate in the case of 3b and 3f. Any
aryne intermediate generated by decarboxylative debromination cannot give
rise to the observed products viz., ortho- sulfocarboxylic acids.
Among the classes of mechanism that have been proposed in the
literature (Lindley 1984 and Nathan Kornblum 1978) there are two
conceivable ones involving (1) radical intermediates involving SRN1 pathway
and (2) oxidative addition-reduction elimination mechanism. Reactions that
proceed via SRN1 mechanism exhibit the following characteristics (i)
acceleration of reaction rate by addition of reducing agents like Sn (II), Fe
(II), Ti (III; II) (Cohen et al 1974), (ii) inhibition of the reaction by oxygen or

158
radical traps like TEMPO (Abdelouahab et al 2005) or di-t-butylniroxide
(Bowman 1982), galvanoxyl (Buegelmans et al 1982), (iii) quenching of the
reaction by added p-di-nitrobenzene or m-di-nitrobenzene (Bowman 1982)
and (iv) formation of oligomers when styrene was added (Todres 1978). Also,
there is a correlation between reduction potential and leaving group ability of
the halide (Todres 1978). The order of reactivity in SRN1 is ArI > ArBr>
ArCl.> ArF, which is opposite to that of SNAr ArF>> ArCl> ArBr> ArI
(Linley 1984, Carey and Sundburg 2001). In SRN1 reactions of aryl halides,
ortho substituent to the halide retards the rate due to steric factors while
electron withdrawing group at the para position marginally increases the rate
of the reaction (Linley 1984). When Ullmann cross coupling of bromo acid
3b with sodium bisulphite under standard condition was performed in the
presence of radical scavengers like BHT or THF (Cohen 1976), the reaction
was not inhibited and expected product was formed at the same rate and in
desired purity and yield (entry 3 and 4,Table 3.23). This observation clearly
ruled out SRN1 mechanistic pathway.
Bowman et al (1982) investigated the mechanism in the case of
CuI catalyzed reaction of 1-chloro-4-iodobenzene with phenyl thiolate which
yielded mono coupled product where as polymeric material was obtained
under SRN1 reaction conditions and this experimental observations led to the
inference that no aryl radicals are produced under copper (I) catalyzed
conditions. Further in contrast to the 3 bromothiophene-2-carboxylic acid 3b
and 5-bromothiophene-2-carboxylic acid 3e, the cuprous chloride catalyzed
cross coupling reaction of 4-bromothiophene-2-carboxylic acid 3d isomer
with sodium bisulphite was significantly very slow. The observed reactivity
3f 3b > 3e >>3d cannot be accounted by SRN1 mechanism. Also the ortho-
carboxylate effect observed in the case of 3b cannot be rationalized on the
basis of SRN1 mechanism. The experimental findings are consistent with an

159
intramolecular oxidative addition-reductive elimination mechanism operating
in the case of 3b as depicted is Scheme 3.19.
Scheme 3.19 Intramolecular oxidative addition - reductive elimination
mechanistic pathway
The intervention of oxidative addition-reductive elimination
mechanism in Ullmann condensation was first proposed by Cohen in 1974.
This mechanism was subsequently substantiated by several literature reports
(Cohen 1976, Bowman et al 1984 and Gary et al 1996) indicating Cu (I) and
Cu (III) intermediates. Oxidative addition of cuprous halides to aryl halides is
a reversible process according to literature (Cohen 1976). The well
documented copper catalyzed halogen exchange in aryl halides is a further
indication of the reversibility (Cohen 1976). Possible role of additional
ligands may be to stabilize copper (I) species, increase the solubility of the
catalytic species, to prevent aggregation of the metal and to promote the
oxidative addition to Cu (I) complex (Cohen 1976). In fact the exceptionally
high catalytic activity of copper (I) thiophene-2-carboxylate in Ullmann cross
S
B r
O
O
C u
S
N a 2S O 3
S C O O C u
S O 3N a
In tram o le cu la r o x id a t iv e
ad d itio n
R e d u c tiv ee lim in a t io n
L
C u
O
O
B r C l
S
C u
O
O
N a O 2 S O X
IIII I I
XI
IC l
S C O O
B r
S C O O
S O 3N aS C O O
B r
C u C l
N a XX = B r,C l.
N a +
N a +
N a +N a +
N a +N a +
N a +

160
coupling reactions has been ascribed to the stabilization of Cu(III) complex
formed in the oxidative addition step thereby driving the equilibrium to the
forward direction (Gary D. Allred et al 1996).
The proposed mechanism is
consistent with the observed findings.
1. The reactivity order ArBr > ArCl parallels the leaving group
ability of the halide ion.
2. Couplings are favored by electron withdrawing groups
3. Coupling did not take place in the absence of copper catalyst.
4. There is a correlation between cyclic voltametry data on
cathodic potential and the leaving group ability of the halide.
5. Free radical inhibitors like BHT or THF did not suppress the
reaction.
6. Bromo ketone 2b and the ester 3g did not exhibit high
reactivity when compared to that of bromo acid 3b towards
this copper catalyzed substitution reaction with sodium
bisulphite.
The HPLC in-process check profile of a sample of reaction mixture
of copper mediated nucleophilic substitution of 3b with sodium bisulphite
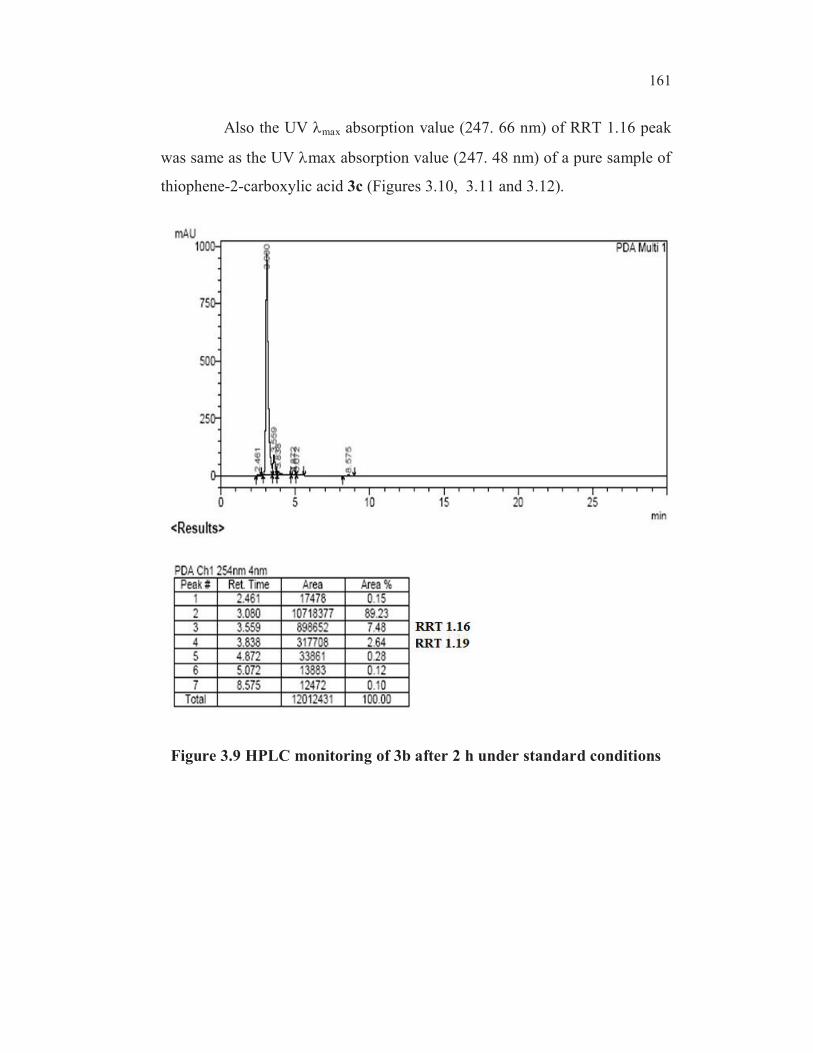
after 2 h under standard conditions is shown in Figure 3.9. There were two
impurities (side products) observed in the HPLC chromatogram. The impurity
at RT (Retention Time) 3.559 minutes corresponding to relative retention
time (RRT) 1.16 was due to the formation of thiophene-2-carboxylic acid 3c,
by hydrodebromination of 3b under reaction conditions. This was confirmed
by co-injecting a reference sample of 3c (Section 2.12) which got co-eluted
along with RRT 1.16 impurity.
161
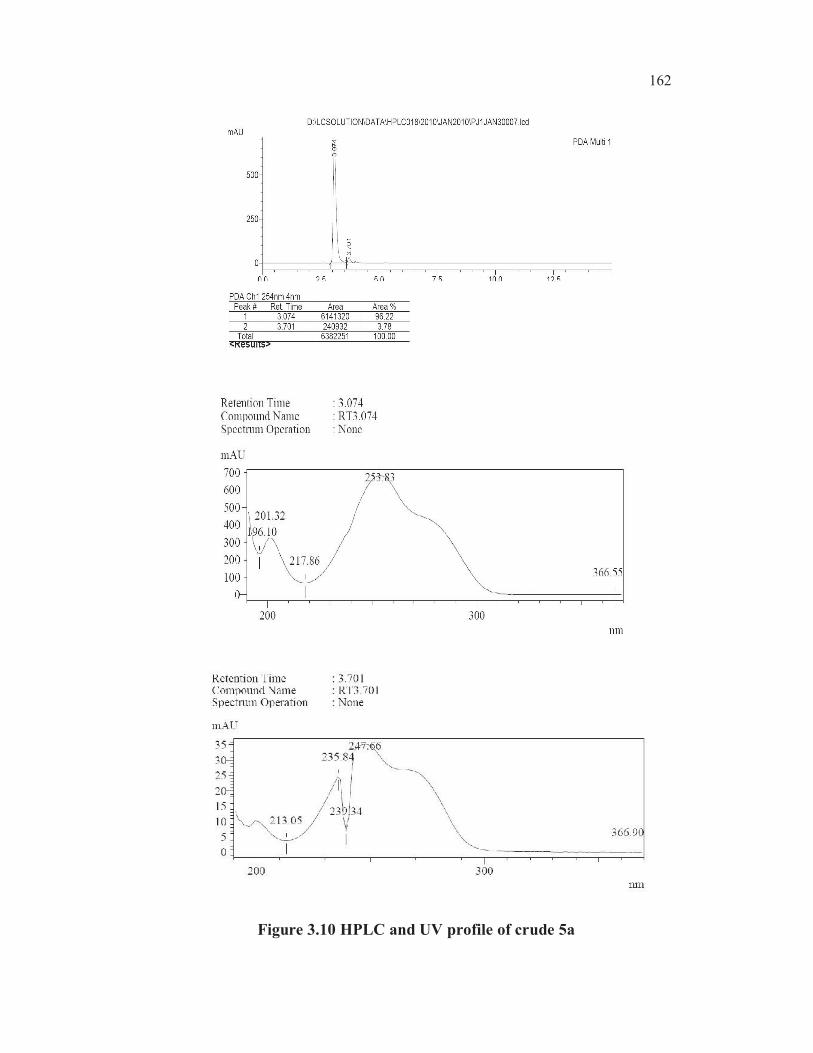
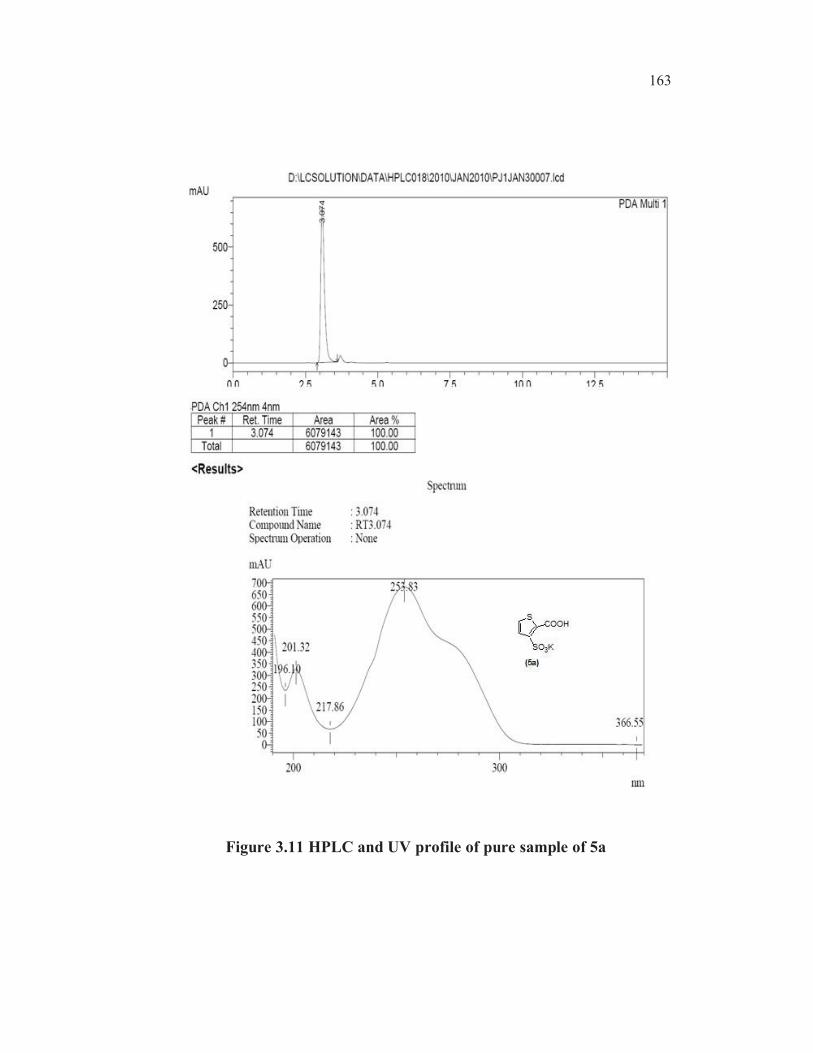
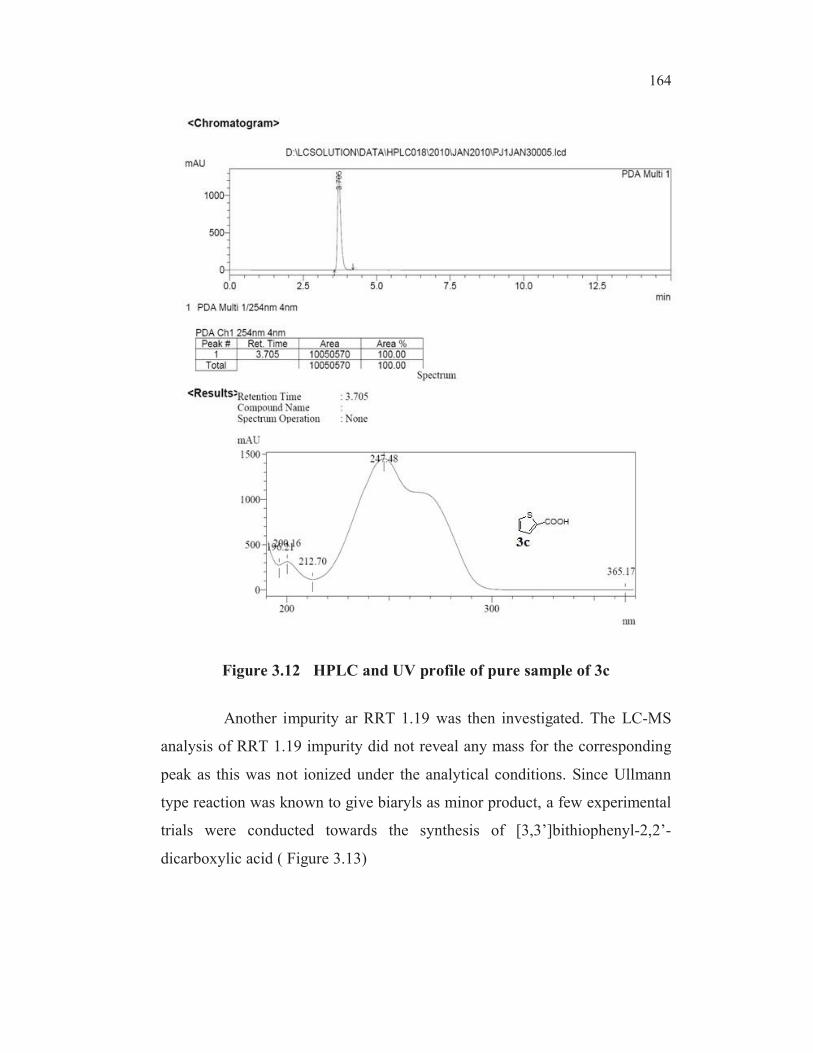
Also the UV max absorption value (247. 66 nm) of RRT 1.16 peak
was same as the UV max absorption value (247. 48 nm) of a pure sample of
thiophene-2-carboxylic acid 3c (Figures 3.10, 3.11 and 3.12).
Figure 3.9 HPLC monitoring of 3b after 2 h under standard conditions
162
Figure 3.10 HPLC and UV profile of crude 5a
163
Figure 3.11 HPLC and UV profile of pure sample of 5a
164
Figure 3.12 HPLC and UV profile of pure sample of 3c
Another impurity ar RRT 1.19 was then investigated. The LC-MS
analysis of RRT 1.19 impurity did not reveal any mass for the corresponding
peak as this was not ionized under the analytical conditions. Since Ullmann
type reaction was known to give biaryls as minor product, a few experimental
trials were conducted towards the synthesis of [3,3’]bithiophenyl-2,2’-
dicarboxylic acid ( Figure 3.13)

165
S COOH
S
COOH
Figure 3.13 Possible structure of RRT 1.19 impurity
[3,3’] Bithiophenyl-2,2’-dicarboxylic acid is known in literature
with Chemical Abstracts Service (CAS) registry number 31493-17-5
(Leardini et al 1970). As per the literature information, the synthesis of this
compound was attempted from methyl 3-bromothiophene-2-carboxylate 3g as
indicated in Scheme 3.20.
SCOOMe
Br
(3g)
Copper-Bronze
DMF, reflux
S COOMe
S
COOMe
KOH
S COOH
S
COOH
(11b)(11a)
[3,3']Bithiophenyl-2,2'-
dicarboxylic acid dimethyl ester
[3,3']Bithiophenyl-2,2'-dicarboxylic acid
3-Bromo-thiophene-2-carboxylic acid methyl ester
Scheme 3.20 Literature route of synthesis of 11b
A few experimental trials were conducted towards the synthesis of
[3,3’]bithiophenyl-2,2’-dicarboxylic acid using the literature information.
Despite limited efforts this compound could not be prepared.
Attempts to characterize the copper salt of 3b (Gary et al 1996) by a
single crystal XRD was not successful as suitable crystal of the copper salt
could not be generated. The first direct observation of Cu (I)-Cu (III) redox
steps relevant to Ullmann type coupling reaction has very recently been
provided by Alicia et al (2010). Uncertainty remains as to whether
nucleophilic substitution step precedes or follow the oxidative addition step.

166
From the experimental data generated, the intramolecular oxidative
addition-reductive elimination mechanism with -complexation of the
carboxylate copper with aromatic cloud as the driving force for promoting
oxidative addition of copper from the carboxylate salt in to the Ar-X bond
(Zheng et.al 2009) can be discounted. The authors did not reveal any support
to their proposed mechanism. The mechanism requires planarity for the -
cloud of the aromatic system and the attacking nucleophile which is
debatable. More emphasis was given to the regio-selectivity of the
substitution. These authors indicated that the substitution was more facile
with o- bromo substituent compared to o- chloro substituent (entries 1 and 23
of Table 2) of Zheng et al (2009) based on the yields obtained and suggested
aromatic -complexation of the carboxylate copper. The role of halogens was
not defined to account for this reaction rate profile. More so, this mechanism,
may not justify the rate differences between isomeric bromothiophene
carboxylic acids that were generated in this present research work. Such a
comparative study of isomeric halocarboxylic acids was not described in
detail by Zheng et al in their published work. Hence, based on the
experimental data generated in this research work, halogen assisted
complexation with ortho- carboxylate copper providing the environment for
an intramolecular oxidative addition can be justified.
3.1.17 Influence of Different EWG Substituent
With a view to get additional evidence for the operation of ortho-
carboxylate effect, the Ullmann condensation reaction with sodium bisulphite
was performed with 3- brmothiophene-2-carboxylic acid 3b, 2-acetyl-3-
bromothiophene 2b and 3-bromo-2-carbomethoxy thiophene 3g under similar
reaction conditions (Scheme 3.21).

167
S
Br
EWG
NaOH / aq.EtOH
CuCl / reflux S
SO3Na
EWG
EWG = COCH3 (2b)
EWG = COOH (3b)
EWG-= COOMe (3g)
HCl / KCl S
SO3K
EWG
EWG = COCH3 (4b)
EWG = COOH (4a)
EWG-= COOMe (4c)
EWG = COOH (5a)
Scheme 3.21 Ullmann condensation with different EWG at 2-position
Under the same reaction conditions stated below, only bromo acid
3b (entry 1, Table 3.20) was found to be reactive while 3-bromo-2-
acetylthiophene 2b (entry 2, Table 3.20) and 3-bromo-2-carbomethoxy
thiophene 3g (entry 3, Table 3.20) were ineffective and did not undergo any
reaction, revealing the rate acceleration was solely due to the ortho-
carboxylate coordination effect operating in the case of 3b.
Table 3.20 Effect of different electron withdrawing groups
Entry SM EWG
Monitoring results
HPLC area (%)Remarks
Time
(h)SM Product
1 3b -COOH 3 22.6 (3b) 62.3 (4a) Slow reaction
2 2b -COCH3 3 96.1 (2b) Nil (4b) No reaction
3 3g -COOCH3 3 98.2 (3g) Nil (4c) No reaction
The reaction was performed under aqueous ethnolic conditions in
order to account for solubility. The substrates were taken in aqueous alcohol
(4 ml of 50% aq. ethanol/g of substrate) along with 1.15 equiv. of sodium
bisulphite as 40% aq. solution and 10 mole % cuprous chloride as catalyst.

168
The reaction mass was heated to 100°C and samples were drawn at regular
intervals and analyzed by HPLC.
3.1.18 Ullmann Condensation of 3b with other Nucleophiles
With a view to find out if the observed high reactivity exhibited by
the bromo acid 3b towards sodium bisulphite is a special case and restricted
only to sodium bisulphite, experiments were carried out with other
nucleophiles under standard conditions. The reaction of the bromo acid 3b
was investigated with a few phenols (Scheme 3.22) and amines (Scheme
3.23) as nucleophiles.
SCOOH
Br
Reflux 100C
aq. NaOH/ CuClphenol
No reaction
aq. NaOH/ CuCl
m-cresol No reaction(3b)
Reflux 100C
Scheme 3.22 Ullmann condensation of 3b with phenoxides under
standard conditions
The nucleophilic substitution reaction of 3b with phenol as
nucleophile under standard conditions did not take place as monitored by
TLC (mobile phase 40% chloroform in methanol). At the end of 3 h of reflux
under standard conditions, the isolated product showed only starting material
by1H NMR. The same trend was observed when m-cresol was used as
nucleophile for the substitution of 3b under standard conditions.
The substitution reaction of 3b with cyclohexyl amine did not take
place under standard conditions (Scheme 3.23) as monitored by TLC (mobile
phase 30% ethyl acetate in hexane). HPLC in process check analysis at the

169
end of 4 h indicated the presence of the starting material 77.4 area% in
addition to side products at RRT 1.16 (3c- 4.3 area%) and RRT 1.19 (16.5
area%)
SCOOH
Br
(3b)
aq. NaOH / CuClCyclohexyl amine
aq. NaOH / CuClBenzyl amine
No reaction
No reaction
Reflux 100oC
Reflux 100oC
Scheme 3.23 Ullmann condensation of 3b with amine nucleophiles under
standard conditions
The substitution reaction of 3b with benzyl amine as nucleophile
under standard conditions did not take place as monitored by TLC (Mobile
phase 30% ethyl acetate in hexane). Though it is difficult to predict the reason
at this juncture with limited studies, it appears that the copper mediated
nucleophilic substitution of 3b was specific to sodium bisulphite under these
reaction conditions (may be due to higher nucleophilicity coupled with
solubility factor in the case of sodium sulphite) as the reaction did not occur
with phenol and amine nucleophiles under the same standard reaction
conditions.
3.1.19 Practical Application of the Present Work
A scalable, industrial process for the preparation of methyl ester 7,
which is a key intermediate for synthesis of API’s like Tenoxicam (Dieter
et al 1987) and Sitaxsentan sodium (Raju et al 1997) was envisaged from this
research work based on the successful scalable process established for
monopotassium salt of 3-sulphothiophene-2-carboxylic acid 5a (Table 3.3)
from 3-bromothiophene-2-carboxylic acid 3b. The key pharma intermediate

170
viz. 2-carbomethoxythiophene-3-sulphonyl chloride 7 was obtained in two
steps from 5a (Scheme 3.24) as solid with m. p. 61-63°C (lit. value of 60-
62°C). The structure of the ester 7 was confirmed by HPLC, IR, NMR and
Mass spectra (Section 2.7).
SCOOH
SO3K
POCl3 / PCl5S
COCl
SO2Cl
(5a) (6)
MeOH
RefluxReflux
SCOOCH3
SO3Cl
(7)
Scheme 3.24 Reaction scheme for the synthesis of methyl ester 7
The process for the preparation of, ester 7 was scaled up
successfully .The product ester 7 was obtained with consistent yield and
purity from the monopotassium salt 5a (Table 3.21). Thus, a scalable,
industrial process for the preparation of methyl ester 7, which is a key
intermediate for synthesis of API’s has been realized.
Table 3.21 Consistent yield and purity of ester 7
EntryInput of 5a
(Kg)
Output 7
(Kg)
Purity
HPLC area(%)
1 100 73 99.9
2 100 73.5 99.9
3 100 72 99.9
3.2 IMPURITY FORMATION DURING SCALE UP OF 5a
3.2.1 Observation of an Impurity
The mono potassium salt of 3-sulphothiophene-2-carboxylic acid 5a
was prepared as given in section 2.5, starting from bromo acid 3b. The ester 7

171
was made in laboratory according to the literature procedure (Dieter, et al
1987) and the product was characterized completely by spectral data (Section
2.7). The process was considered for kilo-lab trials. During the scale up of the
process, unexpectedly, an impurity formation in the product was observed. In
one of the kilo-lab scale up batches, while converting the bis-acid chloride 6
to the ester 7 (Scheme 3.21), this impurity formation was noticed. This
impurity could not be removed by usual crystallization methods. Various
instrumental techniques including HPLC, NMR and Mass analysis were
carried out to investigate and understand the nature of the impurity. This
impurity eluted at a relative retention time (RRT) of 2.82 in HPLC. The1H-
NMR spectrum of the crude product from this scale up batch exhibited all the
signals expected for the ester 7. However, it also showed some additional
weak signals at ð-3.94 corresponding to methyl protons of another methyl
ester group and weak signals in the aromatic region at 7.3-7.4 (Figures 3.14
and 3.15).
Figure 3.141H NMR spectrum of impure sample of 3-chlorosulphonyl
thiphene-2-carboxylic acid methyl ester 7 in CDCl3

172
Figure 3.15 Expanded1H-NMR spectrum of the impure ester 7
3.2.2 Identification and Synthesis of Impurity
GC-MS analysis of this impure ester 7 indicated the presence of an
additional compound with a mass peak corresponding to a mass of 346. The
GC-MS spectrum revealed that this impurity could be a dimer type impurity
as the base peak was observed at 173, which is exactly half the value of 346
(Figure 3.16).
Figure 3.16 GC-MS spectrum of impure ester 7

173
A close scrutiny of the GC-MS data and1H NMR spectral data, coupled with
chemistry expertise, revealed that this impurity is likely to be bis-[2-
methoxycarbonyl-3-thienyl]-disulphide 11c. (Figure 3.17)
SS
S
MeOOCS
COOMe
11c
Figure 3.17 Proposed structure of impurity
The literature survey (Corral et al 1985) indicated that this disulphide is a
known compound and is prepared by the reduction of ester 7 using Zinc/Con.
HCl (Scheme 3.25).
SCOOMe
SO2Cl
7
SS
S
MeOOCS
COOMe
11c
Zn/HCl
Scheme 3.25 Preparation of bis-[2-methoxycarbonyl-3-thienyl]-disulphide
Accordingly, disulphide 11c was synthesised and characterized by
HPLC, NMR and Mass spectra (Section 2.11). The HPLC retention time (RT)
of the authentic disulphide 11c, under the same analytical conditions matched
with that of the impurity observed in scale up lot RT 12.27 (RRT 2.82). The
proton NMR spectral signals (Figure 3.18) also correspondingly matched with
that of extra signals seen in the NMR spectrum of the impure product. The
mass spectrum of synthesised disulphide 11c indicated a molecular mass of
346 with a base peak at 173. The impurity isolated by column
chromatography from the impure ester matched well in spectral data with that
of the synthesised disulphide. A sample of disulphide 11c co-injected with
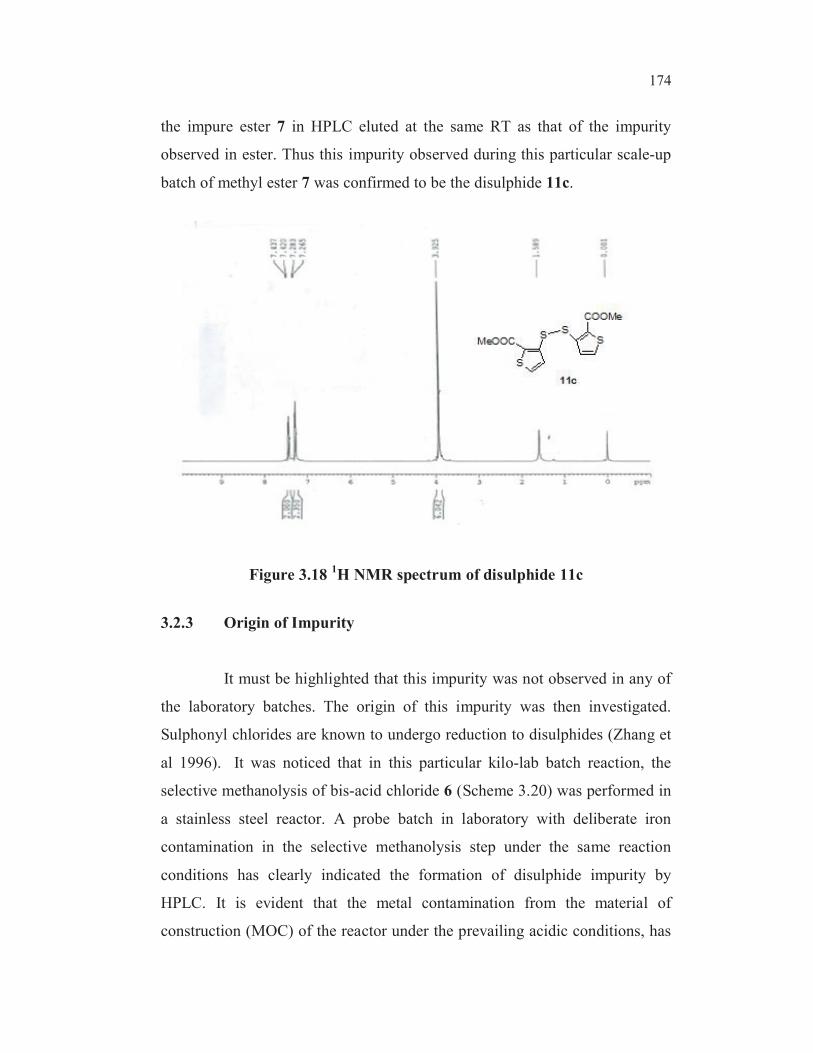
174
the impure ester 7 in HPLC eluted at the same RT as that of the impurity
observed in ester. Thus this impurity observed during this particular scale-up
batch of methyl ester 7 was confirmed to be the disulphide 11c.
Figure 3.181H NMR spectrum of disulphide 11c
3.2.3 Origin of Impurity
It must be highlighted that this impurity was not observed in any of
the laboratory batches. The origin of this impurity was then investigated.
Sulphonyl chlorides are known to undergo reduction to disulphides (Zhang et
al 1996). It was noticed that in this particular kilo-lab batch reaction, the
selective methanolysis of bis-acid chloride 6 (Scheme 3.20) was performed in
a stainless steel reactor. A probe batch in laboratory with deliberate iron
contamination in the selective methanolysis step under the same reaction
conditions has clearly indicated the formation of disulphide impurity by
HPLC. It is evident that the metal contamination from the material of
construction (MOC) of the reactor under the prevailing acidic conditions, has

175
led to the reduction of methyl ester 7 resulting in formation the disulphide
impurity 11c. The knowledge derived from successful investigation of the
impurity originating from MOC of the reactor, during the preparation of the
ester 7 from the acid chloride 6 has thus paved way for a successful and
consistent process of very high quality material of ester 7, which is a key
pharmaceutical intermediate.
3.3 SYNTHESIS OF NEW ARYL ETHERS
The Ullmann cross coupling of aryl halides and heteroaryl halides
with phenoxides is one of the popular methods for the synthesis of diaryl
ethers and heteroaryl ethers (Marcoux et al 1997 and Torroca et al 2001) and
has been extensively investigated. This reaction continues to receive wide
attention (Cristau et al 2004). In this context, it was of interest to study the
Ullmann type cross coupling of 1a, 2b and 3b with a few phenoxides
(Scheme 3.26).
X = Br, R = H (1b)
X = Cl, R = COCH3 ( 2a )
X = Br, R = COCH3 (2b)
R = R1= H (10a)
R = COCH3, R1 = H (10b)
R = COCH3, R1 = CH3 (10c)
Pyridine / CuCl
118oC
S
R
X
S
O
R
OH
NaH/R1
R1
Scheme 3.26 Preparation of aryl ethers
3.3.1 Synthesis of 2-Acetyl-3-phenoxy thiophene and 2-Acetyl-3-(m-
tolyloxy) thiophene
Experiments were conducted in order to investigate the
reactivity pattern between chloro ketone 2a and bromo ketone 2b towards
nucleophilic substitution with phenol (Scheme 3.24). The results are shown in
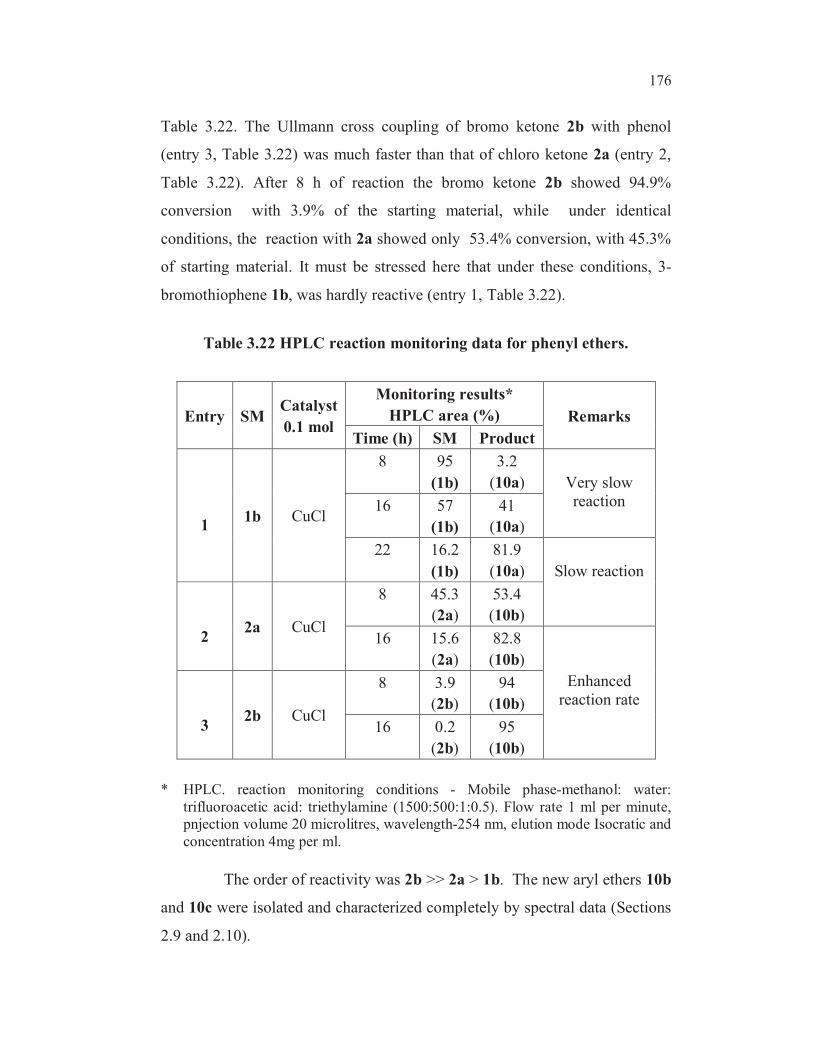
176
Table 3.22. The Ullmann cross coupling of bromo ketone 2b with phenol
(entry 3, Table 3.22) was much faster than that of chloro ketone 2a (entry 2,
Table 3.22). After 8 h of reaction the bromo ketone 2b showed 94.9%
conversion with 3.9% of the starting material, while under identical
conditions, the reaction with 2a showed only 53.4% conversion, with 45.3%
of starting material. It must be stressed here that under these conditions, 3-
bromothiophene 1b, was hardly reactive (entry 1, Table 3.22).
Table 3.22 HPLC reaction monitoring data for phenyl ethers.
Entry SMCatalyst
0.1 mol
Monitoring results*
HPLC area (%) Remarks
Time (h) SM Product
11b CuCl
8 95
(1b)
3.2
(10a) Very slow
reaction16 57
(1b)
41
(10a)
22 16.2
(1b)
81.9
(10a) Slow reaction
22a CuCl
8 45.3
(2a)
53.4
(10b)
16 15.6
(2a)
82.8
(10b)
Enhanced
reaction rate
32b CuCl
8 3.9
(2b)
94
(10b)
16 0.2
(2b)
95
(10b)
* HPLC. reaction monitoring conditions - Mobile phase-methanol: water:
trifluoroacetic acid: triethylamine (1500:500:1:0.5). Flow rate 1 ml per minute,
pnjection volume 20 microlitres, wavelength-254 nm, elution mode Isocratic and
concentration 4mg per ml.
The order of reactivity was 2b >> 2a > 1b. The new aryl ethers 10b
and 10c were isolated and characterized completely by spectral data (Sections
2.9 and 2.10).

177
3.4 COMPARISON BETWEEN HALOBENZOIC ACIDS AND
HALOTHIOPHENECARBOXYLIC ACIDS IN ULLMANN
CONDENSATION
Halothiophenes are relatively inert to nucleophilic substitution while
that are conjugatively substituted with electron withdrawing groups are
comparatively more reactive to nucleophilic substitution (Kassmi, 1992).
SRN1 reactions occur with halogen derivatives of thiophene but less readily
compared to phenyl halides (Bunnet and Bernhard 1976, Miller 1968, Salo
Gronowitz 1991 and Bunnett 1983). The nucleophilic substitution of
unactivated halides are effected under harsh conditions compared to activated
halides. For the first time, a comparitive study on the Ullmann type
nucleophilic substitution with reference to ortho- carboxylic acid effect in 3-
halothiophene-2-carboxylic acids and 2-halobenzoic acids with sodium
bisulphite was undertaken. This study was also extended to a few other
nucleophiles like amines and phenols.
3.4.1 Ullmann Cross Coupling with Isomeric Bromobenzoic Acids
The cuprous chloride catalyzed Ullmann cross coupling of
halobenzoic acids with sodium bisulphite under the standard condition
(Scheme 3.27) was investigated and the experimental results are given in
Table 3.23. The o- bromobenzoic acid underwent very facile Ullmann type
coupling with sodium bisulphite under standard conditions (entry 1, Table
3.23). The meta isomer 8b (entry 2, Table 3.23) and para isomer 8c (entry 3,
Table 3.23) did not undergo substitution under standard conditions. Similarly
among the three isomeric chloro benzoic acids 8d, 8e and 8f (entries 4, 5 and
6, Table 3.23), only ortho chloro isomer 8d displayed some reactivity with
very low (23.% ) conversion compared to 8a (>94% conversion) and in the
case of 8e and 8f there was hardly any reaction.
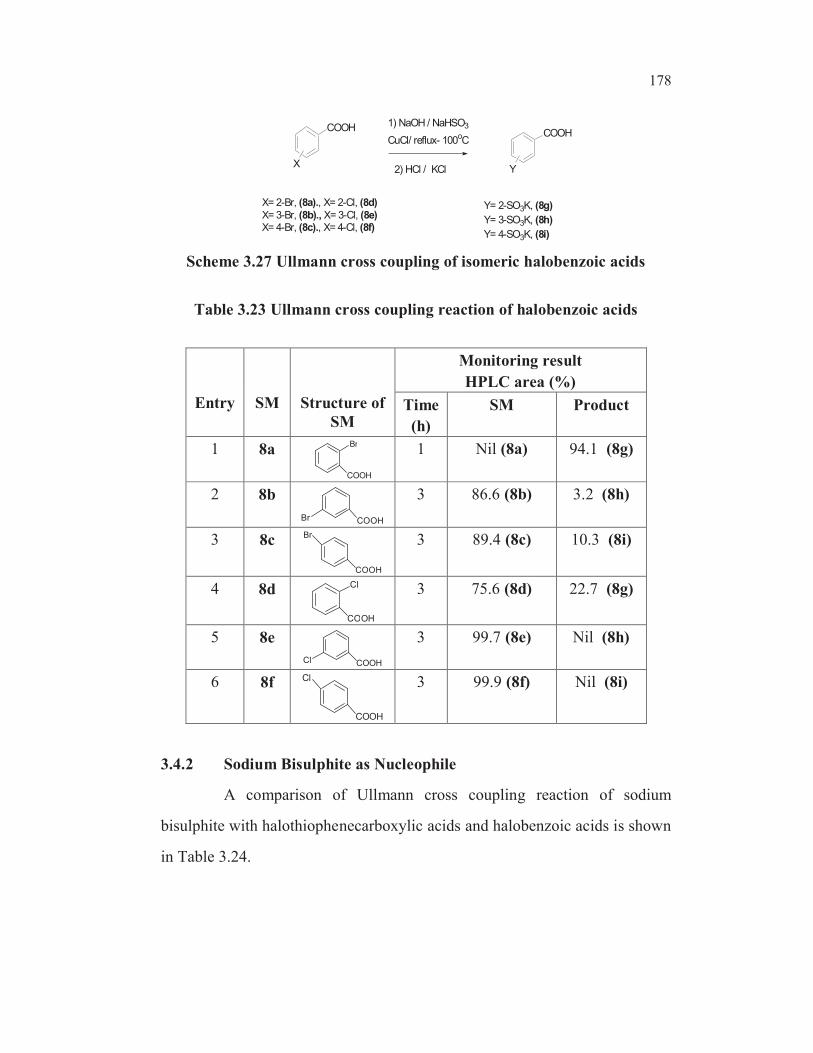
178
1) NaOH / NaHSO3
CuCl/ reflux- 100oC
X= 2-Br, (8a)., X= 2-Cl, (8d)
X= 3-Br, (8b)., X= 3-Cl, (8e)X= 4-Br, (8c)., X= 4-Cl, (8f)
Y= 2-SO3K, (8g)
Y= 3-SO3K, (8h)
Y= 4-SO3K, (8i)
2) HCl / KCl
COOH
X
COOH
Y
Scheme 3.27 Ullmann cross coupling of isomeric halobenzoic acids
Table 3.23 Ullmann cross coupling reaction of halobenzoic acids
Entry SM Structure of
SM
Monitoring result
HPLC area (%)
Time
(h)
SM Product
1 8aBr
COOH
1 Nil (8a) 94.1 (8g)
2 8b
COOHBr
3 86.6 (8b) 3.2 (8h)
3 8c
COOH
Br 3 89.4 (8c) 10.3 (8i)
4 8d Cl
COOH
3 75.6 (8d) 22.7 (8g)
5 8e
COOHCl
3 99.7 (8e) Nil (8h)
6 8f
COOH
Cl 3 99.9 (8f) Nil (8i)
3.4.2 Sodium Bisulphite as Nucleophile
A comparison of Ullmann cross coupling reaction of sodium
bisulphite with halothiophenecarboxylic acids and halobenzoic acids is shown
in Table 3.24.
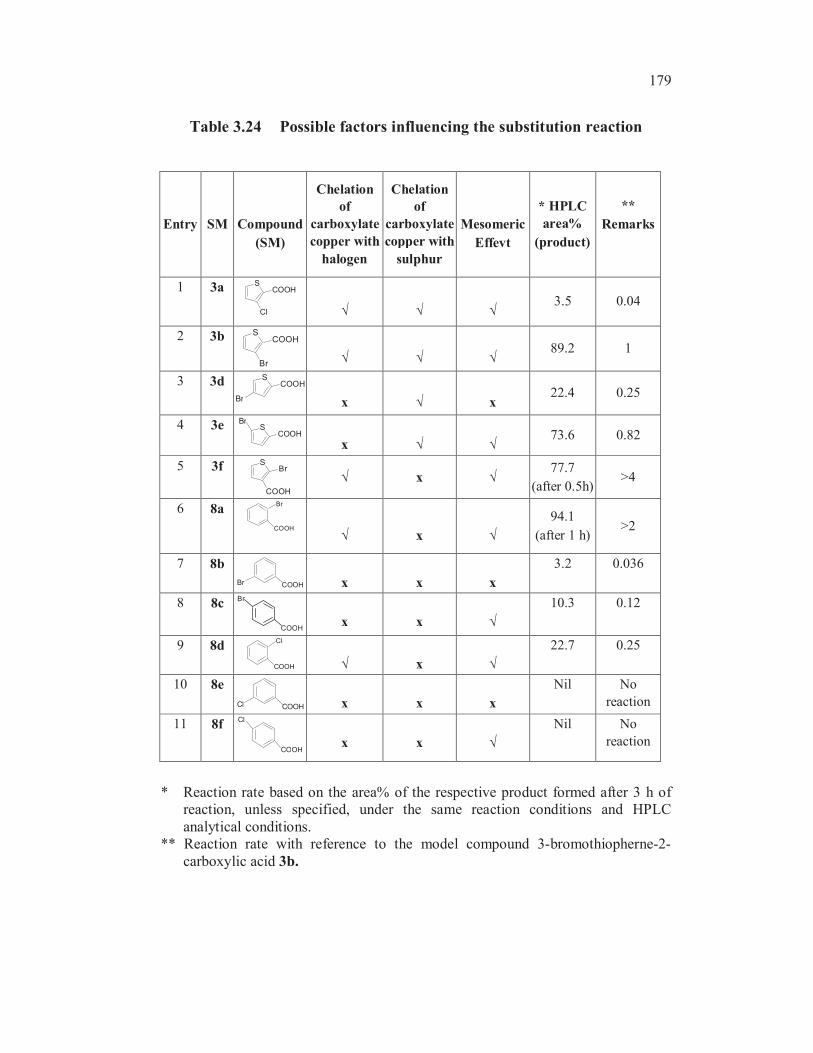
179
Table 3.24 Possible factors influencing the substitution reaction
Entry SM Compound
(SM)
Chelation
of
carboxylate
copper with
halogen
Chelation
of
carboxylate
copper with
sulphur
Mesomeric
Effevt
* HPLC
area%
(product)
**
Remarks
1 3a S
Cl
COOH
3.5 0.04
2 3b S
Br
COOH
89.2 1
3 3d SCOOH
Br x x22.4 0.25
4 3e SCOOH
Br
x73.6 0.82
5 3f S
COOH
Br
x77.7
(after 0.5h)>4
6 8aBr
COOH
x
94.1
(after 1 h)>2
7 8b
COOHBr x x x
3.2 0.036
8 8c
COOH
Br
x x
10.3 0.12
9 8dCl
COOH x
22.7 0.25
10 8e
COOHCl x x x
Nil No
reaction
11 8f
COOH
Cl
x x
Nil No
reaction
* Reaction rate based on the area% of the respective product formed after 3 h of
reaction, unless specified, under the same reaction conditions and HPLC
analytical conditions.
** Reaction rate with reference to the model compound 3-bromothiopherne-2-
carboxylic acid 3b.
180
Interestingly, the Ullmann cross coupling of sodium bisulphite with 2
bromobenzoic acid 8a (entry 1, Table 3.25), was faster (> 2 folds) compared
to that of 3-bromothiophene-2-carboxylic acid 3b.
Table 3.25 Comparison of Ullmann cross coupling of 8a and 3b with
sodium bisulphite under standard conditions
Entry SM Structure of SM
Monitoring result
HPLC area (%)
Time
(h)SM Product
1 8aBr
COOH
1 Nil
(8a)
94.1
(8g)
2 3b S
Br
COOH 2 Nil
(3b)
89.2
(5a)
The reaction in the case of acid 8a completed in 1h (entry 1, Table
3.25) as compared to 2 h in the case of 3b (entry 2, Table 3.25). It is to be
noted that the resulting product 8g has been described in literature (Anthony
1979, Kim 2010) from ortho chlorobenzoic acid 8d via Ullmann cross
coupling reaction with sodium sulphite under very harsh conditions viz., at
175°C, under pressure in an autoclave for 24 h (Scheme 3.28) and
surprisingly, there are no reports in the literature for the preparation of 8g
from bromobenzoic acid 8a.
COOH
Cl
NaOH,CuSO4.5H2O
H2O, Na2SO3 175°C, 24 h.,
6-7 Kg/cm2
pressure COOH
SO3K
(8d)2) HCl / KCl
(8g)
Scheme 3.28 Preparation of 8g reported in literature

181
The higher reactivity of 8a than that of 3b in Ullmann cross
coupling reaction with sodium bisulphite is an important finding towards
understanding the mechanism of this nucleophilic substitution. The reactivity
difference between 8a and 3b could be due to the possibility for formation of
one more intramolecular chelated complex involving copper thiophene-2-
carboxylate moiety with the sulphur atom in the thiophene ring as shown in
Figure 3.19.
SO
O
Cu
Br
S
O
Br Cu
O
example a example b
Figure 3.19 Possible chelations of carboxylate copper
An intramolecular oxidative addition-reductive elimination
mechanism involving o- copper carboxylate chelate (Figure 3.19 example a)
was proposed for the copper mediated nucleophilic substitution reaction of 3b
with sodium bisulphite based on the experimental data which is discussed in
detail under section 3.1.16. A possible competitive intramolecular chelation of
carboxylate copper with sulphur (Figure 3.19 example b) reduces the
possibility of the formation of required copper complex, thus resulting in a
slower reaction rate in the case of 3b compared to 8a where the scope for this
sort of competitive coordination does not exist. It is to be noted that
intramolecular oxidative addition of Cu+ to the C-Br bond is not possible in
the cases of 3d and 3e as there is no carboxylic group present ortho to the
bromine, unlike in the cases of 3b and 8a. The oxidative addition of Cu(I) in
the cases of 3d and 3e must involve only intermolecular process. Hence the
substitution reactions in the case of 3d and 3e are significantly slower
compared to 3b and 8a. A comparison of Ullmann condensation of 8b and

182
3d with sodium bisulphite under standard conditions is shown in Table 3.26,
while that of 8c and 3e is shown in Table 3.27.
Table 3.26 Comparison of Ullmann cross coupling of 8b and 3d with
sodium bisulphite under standard conditions
Entry SM Structure of
SM
Monitoring result
HPLC area (%)
Time
(h)
SM Product
1 8b
COOHBr
3 86.6
(8b)
3.2
(8h)
2 3d SCOOH
Br
3 73.6
(3d)
22.4
(5b)
Table 3.27 Comparison of Ullmann cross coupling of 8c and 3e with
sodium bisulphite under standard conditions
Entry SM Structure of
SM
Monitoring result
HPLC area (%)
Time
(h)
SM Product
1 8c
COOH
Br 3 89.4 (8c) 10.3 (8i)
23e
SCOOH
Br
3 21.1 (3e) 73.6 (5c)
Evidently, the comparatively higher reactivity of 3e than that of 3d
is due to the extended mesomeric interaction exerted by the carboxyl group
(Figure 3.20) which facilitates the oxidative addition of Cu+ to the Br-C bond.
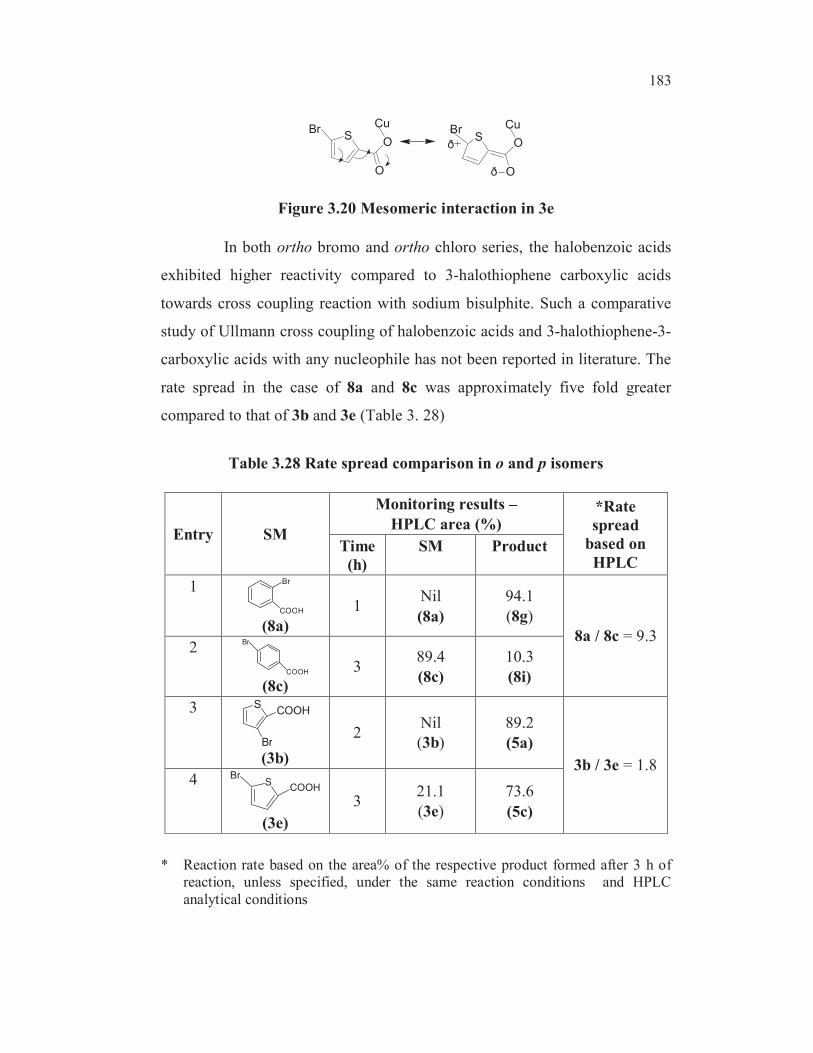
183
SO
O
CuBr
SO
O
CuBr
ð
ð
Figure 3.20 Mesomeric interaction in 3e
In both ortho bromo and ortho chloro series, the halobenzoic acids
exhibited higher reactivity compared to 3-halothiophene carboxylic acids
towards cross coupling reaction with sodium bisulphite. Such a comparative
study of Ullmann cross coupling of halobenzoic acids and 3-halothiophene-3-
carboxylic acids with any nucleophile has not been reported in literature. The
rate spread in the case of 8a and 8c was approximately five fold greater
compared to that of 3b and 3e (Table 3. 28)
Table 3.28 Rate spread comparison in o and p isomers
Entry SM
Monitoring results –
HPLC area (%)*Rate
spread
based on
HPLCTime
(h)
SM Product
1Br
COOH
(8a)
1Nil
(8a)
94.1
(8g)
8a / 8c = 9.32
COOH
Br
(8c)
389.4
(8c)
10.3
(8i)
3 S
Br
COOH
(3b)
2Nil
(3b)
89.2
(5a)
3b / 3e = 1.84 S
COOH
Br
(3e)
321.1
(3e)
73.6
(5c)
* Reaction rate based on the area% of the respective product formed after 3 h of
reaction, unless specified, under the same reaction conditions and HPLC
analytical conditions

184
The bromides exhibited higher reactivity compared to the chlorides
both in the halobenzoic acids and halothiophenecarboxylic acids which
parallels the order of reactivity observed in Pd mediated cross coupling
reactions reported in the literature (Rina Singh and George Just 1989 and
Toshihiko 1980) as shown in Schemes 3.29 and 3.30.
O2N
X
+H C5H11
1-heptyne
2 mol% of Pd(PPh3)4
3 mol% Cu2Br2
N2 atmosphere
90°C
Boiling triethylamine
O2N
C5H11
When X= Cl, the reaction completed in 1440 minwhen X= Br, the reaction was over in < 3 minutes
4-halonitrobenzene
Scheme 3.29 Pd catalyzed ethynylation
X
+
HS 8 mol% Pd(PPh3)4
DMSO (20 ml)t-BuONa- 4 mmol, 100°C
S
Halo benzene2 mmol
Phenylthiol2 mmol
Diphenyl sulphide
When X= Cl, there was no reaction in 4 h.When X= Br, 82% of the product is obtained in 4 h.
Scheme 3.30 Pd catalyzed nucleophilic substitution
Although chloro acids 3a and 8d did not undergo Ullmann
condensation appreciably with sodium bisulphite under standard conditions
compared to bromo acids 3b and 8a, it is noteworthy to compare their relative
rates of Ullmann condensation reaction under standard conditions
(Table 3.29).
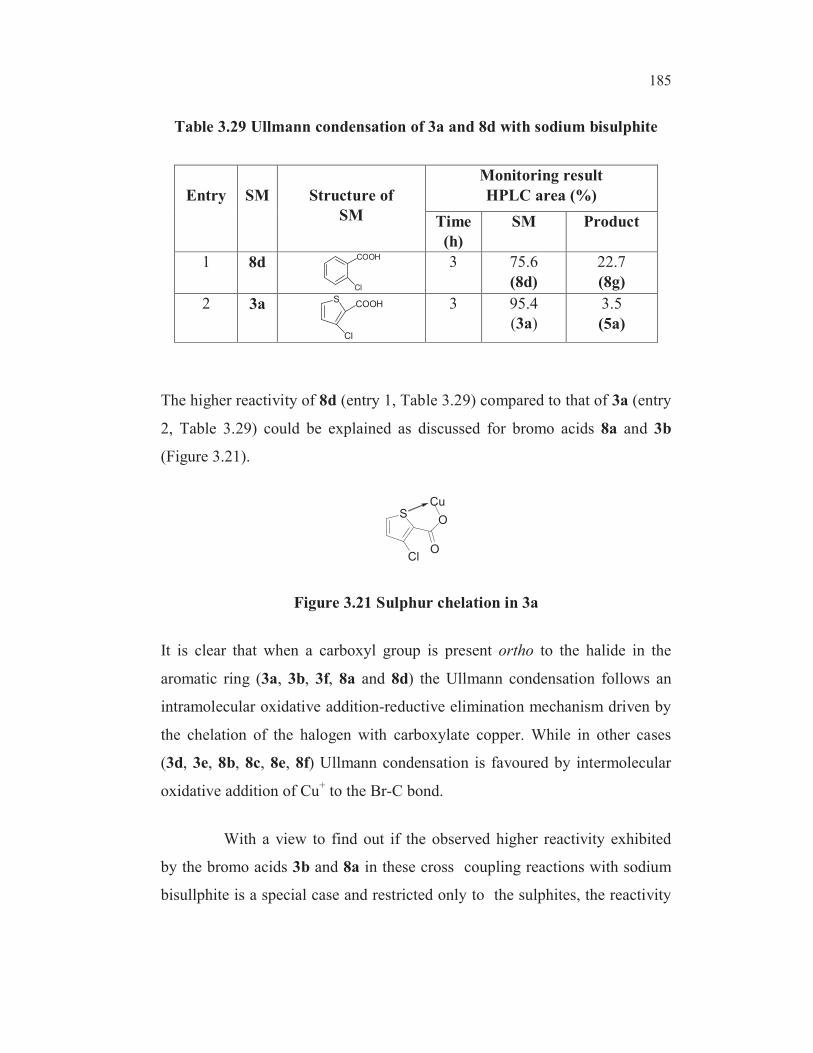
185
Table 3.29 Ullmann condensation of 3a and 8d with sodium bisulphite
Entry SM Structure of
SM
Monitoring result
HPLC area (%)
Time
(h)
SM Product
1 8d
Cl
COOH 3 75.6
(8d)
22.7
(8g)
2 3a SCOOH
Cl
3 95.4
(3a)
3.5
(5a)
The higher reactivity of 8d (entry 1, Table 3.29) compared to that of 3a (entry
2, Table 3.29) could be explained as discussed for bromo acids 8a and 3b
(Figure 3.21).
SO
O
Cu
Cl
Figure 3.21 Sulphur chelation in 3a
It is clear that when a carboxyl group is present ortho to the halide in the
aromatic ring (3a, 3b, 3f, 8a and 8d) the Ullmann condensation follows an
intramolecular oxidative addition-reductive elimination mechanism driven by
the chelation of the halogen with carboxylate copper. While in other cases
(3d, 3e, 8b, 8c, 8e, 8f) Ullmann condensation is favoured by intermolecular
oxidative addition of Cu+ to the Br-C bond.
With a view to find out if the observed higher reactivity exhibited
by the bromo acids 3b and 8a in these cross coupling reactions with sodium
bisullphite is a special case and restricted only to the sulphites, the reactivity

186
of bromo acids 3b and 8a were investigated with a few amines and phenols
and thereby compare their reactivity with these nucleophiles.
3.4.3 Amines as Nucleophiles
The Ullmann type cross coupling of aryl halides and heteroaryl
halides with amine nucleophiles is one of the popular methods for the
synthesis of aryl and heteroaryl substituted amines (Cohen and Tirpak 1975,
Sugaya et al 1994, Rottger et al 2007) and has been extensively investigated.
This reaction continues to receive wide attention (Cresteu et al 2004). In this
context, it is of interest to study the copper mediated cross coupling of 3b, 8a
with a few amine nucleophiles (cyclohexyl amine and benzyl amine) under
aqueous conditions and compare the relative reactivity under the same
reaction conditions (Scheme 3.31).
(3b)
+1 equiv. aq. NaOH
CuCl / refluxS
Br
COOH
(8a)
S
NHR
COOH
Br
COOH
+1 equiv. aq. NaOH
CuCl / reflux
NHR
COOH
R-NH2
R-NH2
R = cyclohexyl (12c)
R = benzyl (12d)
R = cyclohexyl (12a)
R = benzyl (12b)
Scheme 3.31 Ullmann condensation with amine nucleophiles
The Ullmann condensation reactions of 8a and 3b with cyclohexyl
amine were performed under standard conditions and the reactions were
monitored by HPLC. The results are tabulated in Table 3.30.
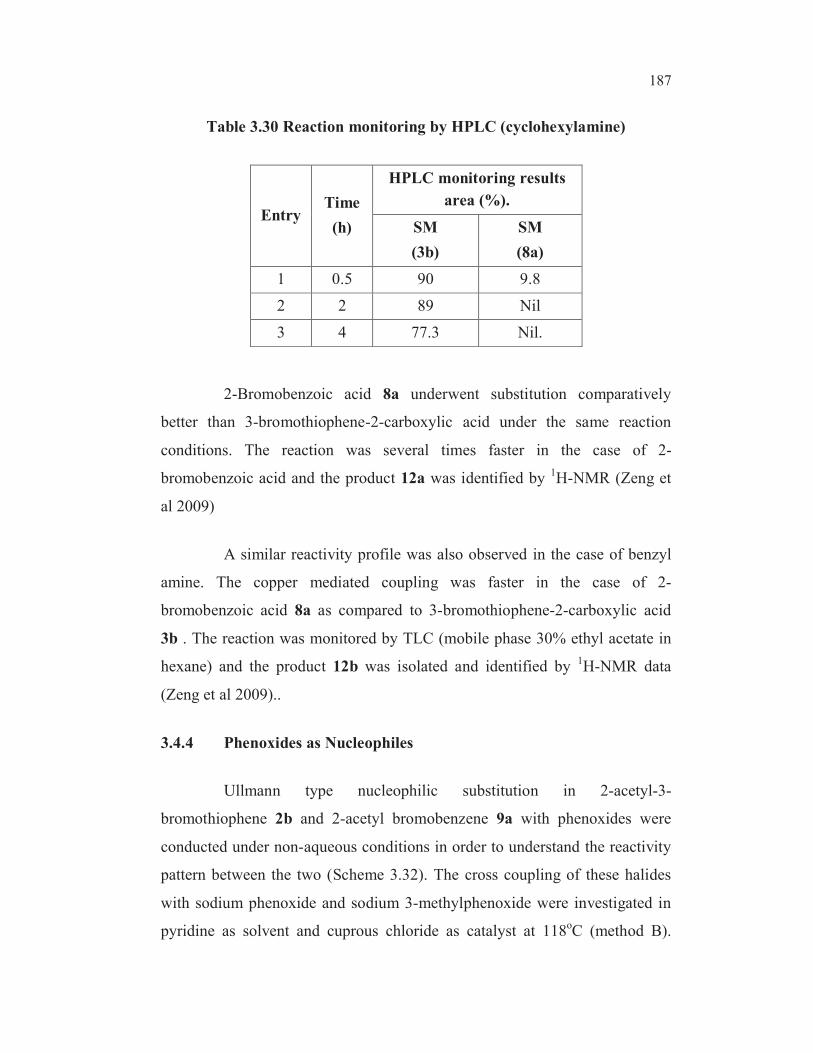
187
Table 3.30 Reaction monitoring by HPLC (cyclohexylamine)
EntryTime
(h)
HPLC monitoring results
area (%).
SM
(3b)
SM
(8a)
1 0.5 90 9.8
2 2 89 Nil
3 4 77.3 Nil.
2-Bromobenzoic acid 8a underwent substitution comparatively
better than 3-bromothiophene-2-carboxylic acid under the same reaction
conditions. The reaction was several times faster in the case of 2-
bromobenzoic acid and the product 12a was identified by1H-NMR (Zeng et
al 2009)
A similar reactivity profile was also observed in the case of benzyl
amine. The copper mediated coupling was faster in the case of 2-
bromobenzoic acid 8a as compared to 3-bromothiophene-2-carboxylic acid
3b . The reaction was monitored by TLC (mobile phase 30% ethyl acetate in
hexane) and the product 12b was isolated and identified by1H-NMR data
(Zeng et al 2009)..
3.4.4 Phenoxides as Nucleophiles
Ullmann type nucleophilic substitution in 2-acetyl-3-
bromothiophene 2b and 2-acetyl bromobenzene 9a with phenoxides were
conducted under non-aqueous conditions in order to understand the reactivity
pattern between the two (Scheme 3.32). The cross coupling of these halides
with sodium phenoxide and sodium 3-methylphenoxide were investigated in
pyridine as solvent and cuprous chloride as catalyst at 118oC (method B).

188
Method B: Phenol was dissolved in dry pyridine and one equivalent of NaH
(as 60% mineral oil suspension) was added under nitrogen atmosphere. The
starting material (0.33 equivalents) was added followed by addition of 0.33
equivalent of CuCl catalyst. The reaction mixture was heated to reflux
temperature under nitrogen and monitored by TLC.
S COCH3
Br
Ullmann cross coupling
Pyridine, CuCl, RefluxNucleophile
S COCH3
Nu
(2b)Nu = OPh (10b)Nu = m-tolyloxy (10c)
COCH3
Br
(9a)
Ullmann cross coupling
Pyridine, CuCl, RefluxNucleophile
COCH3
Nu
Nu = OPh (9b)
Nu = m-tolyloxy (9c)
Scheme 3.32 Comparitive study with phenoxides as nucleophile
The reaction of 2-acetyl bromobenzene 9a with phenoxide was
much faster compared to 2-acetyl-3-bromothiophene 2b by TLC in process
check analysis (30% ethyl acetate v/v in hexane). The reaction of 2-acetyl
bromobenzene 9a was completed in 2 h while in the case of 2-acetyl-3-
bromothiophene 2b the starting material was observed in TLC even after 8 h
of reaction. 2-Acetyldiphenyl ether 9b (Pellon et al 2003) was characterized
by NMR data. Following this procedure, new aryl ether 10b was prepared
from 2b and characterized by NMR spectral data and HRMS data (Section
2.9). A similar reactivity difference was observed between 2b and 9a, when
3-methyl phenol was used as the nucleophile under the same reaction
conditions. The reaction was monitored by TLC (mobile phase 30% ethyl
acetate v/v in hexane). The reaction of 2-acetyl bromobenzene 9a was
completed in 2 h while in the case of 2-acetyl-3-bromothiophene 2b the
starting material was observed in TLC even after 8 h of reaction and the
isolated products 10c (Section 2.10) and 9c (Pellon et al 2003) respectively,

189
were identified by NMR data. The cross coupling reactions of 3b and 8a with
p- chloro phenol were performed under aqueous conditions at 100°C using
cuprous chloride (0.1 mol) as catalyst (Scheme 3.33).
(3b)
+
Cl
OH 1 equi aq.NaOH
CuCl / refluxS
Br
COOH
(8a)
S
O
COOH
Cl
,(9d)
(10d)
Br
COOH
+
Cl
OH 1 equi aq.NaOH
CuCl / reflux
O
COOH
p-chlorophenol
p-chlorophenol
Cl
Scheme 3.33 Nucleophilic substitution with p-chlorophenol under
aqueous conditions
The reactions were monitored by HPLC using the standard
conditions (Table 3.31).
Table 3.31 HPLC monitoring of 3b and 8a with p-chlorophenol
EntryTime
(h)
HPLVC monitoring results
area (%)
SM
(3b)
SM
(8a)
1 0.5 92.7 21.1
2 2 89.3 19.9
3 4 86.4 19.3
Under aqueous conditions at 100°C, the substitution was faster in 2-
bromobenzoic acid 8a compared to 3-bromothiophene-2-carboxylic acid 3b
where there was no significant reaction. The crude product 9d obtained from
8a was isolated and identified by proton NMR (Pellon et al 2003) . Hence it
was observed that the Ullmann condensation with phenoxide was faster in 2-

190
bromobenzoic acid compared to 3-bromothiophene-2-carboxylic acid. The
findings from this study clearly revealed that ortho- carboxylate effect is
mainly responsible for the high reactivity in the case of 3-bromothiophene-2-
carboxylic acid and 2-bromobenzoic acid in Ullmann cross coupling reactions
irrespective of the nature of the nucleophile, whether it is S or N or O. It is
also clear that the cross coupling reactions are several fold faster in the case
of 2-bromobenzoic acid compared to that of 3-bromothiophene-2-carboxylic
acid. The difference in the reactivity can be rationalized as explained below.
3.4.5 Plausible Mechanism for Higher Reactivity of 8a Compared to
3b in Ullmann Condensation
A competitive intramolecular chelation involving coordination of
sulphur atom of the thiophene moiety with the carboxylate copper
(Figure 3.22) reduces the probability for the oxidative addition of the metal to
the carbon halogen bond at C3 of thiophene moiety. This type of competitive
coordination does not exist in the case of benzene system. This may be the
reason for the higher reaction rate of 2-bromothiophene-3-carboxylic acid 3f
compared to 3-bromothiophene-2-carboxylic acid 3b in nucleophilic
substitution with sodium bisulphite under standard conditions.
Figure 3.22 Coordination with sulphur atom
S
Br
OS
Br
O
OCu
O Cu

191
3.5 CORRELATION OF CYCLIC VOLTAMMETRY DATA
WITH THE PROPOSED MECHANISM
Electrochemical techniques are concerned with the interplay
between the electricity and chemistry, namely the measurements of electrical
quantities such as current, potential or charge and their relationship to
chemical parameters. Such use of electrical measurements for analytical
purposes has found a vast range of applications including environmental
monitoring, industrial quality control and bio-medical analysis (Joseph Wang,
2001). Cyclic Voltammetry (CV) is the most widely used technique for
acquiring qualitative as well as quantitative information about electrochemical
reactions (Wang et al 2012, Plana et al 2009, Iotov et al 2009, Barnes et al
2011). The potential is linearly scanned using a triangular waveform from an
initial potential to a final potential. The current is measured corresponding to
the potential scanning. The peak current corresponds to the concentration of
the electro active species present. The three electrode assembly is used in
studying them which consist of a working electrode (glassy carbon electrode),
counter electrode (platinum wire) and reference electrode (Ag/AgCl).
Normally Tetra butyl ammonium bromide is used as supporting electrode in
solvents such as acetonitrile and DMF.
The reductive cleavage of organic compounds continues to be a
fascinating topic of research on account of diverse mechanistic pathways
mediated by the solvent characteristic and magnitude of the driving force.
Among various organic substrates, aromatic halides are especially interesting
in view of the difficulty in their reduction as one passes from carbon-iodine to
carbon-fluorine bond. It is customary to anticipate that these reductions lead
to the neutral radical and the corresponding halide anions. The stabilization of
these radicals can be controlled by (i) carbon-halogen bond energy (ii)
presence of electron withdrawing group in aromatic moiety and (iii) polarity

192
of the solvent. Cyclic voltammetry is the customary technique for studying
kinetics and mechanism of electro-organic reactions. For the first time,
attempts were made to correlate the reduction potentials of the C-X bond
obtained from cyclic voltammogram of halothiophenes and halothiophene
carboxylic acids with their reactivity towards Ullmann cross coupling reaction
with bisulphite and more specifically, the ortho- carboxylate effect (Scheme
3.34) observed in the Ullmann coupling is reflected in the cathodic reduction
potentials of these substrates.
+
1. 1 equiv. aq.NaOHCuCl / reflux
S
X
COOH S
SO3K
COOH
X
COOH
+
SO3K
COOH
NaHSO3
NaHSO3
2. HCl / KCl
(5a)
(8g)
(X= Cl, Br)
(X= Cl, Br)
1. 1 equiv. aq.NaOHCuCl / reflux
2. HCl / KCl
Scheme 3.34 General scheme for Ullmann type nucleophilc substitution
It was also felt worthwhile to compare the reduction potentials of
bromothiophene-2-carboxylic acids with those of bromobenzoic acids and see
if any correlation could be found between this comparison and the reactivity
of these two aromatic systems towards Ullmann cross coupling with
nucleophiles with reference to ortho- carboxylate effect.
3.5.1 CV of thiophene and benzene halides
The cyclic voltammetry data of halothiophenes and halobenzene
compounds are indicated in Table 3.32. It was observed that 2-
bromothiophene 1d, 3-bromothiophene 1b and 3,4-dibromothiophene 1e, all
underwent reduction at more or less in the same potential region viz., -2.72 to
-2.75V (Figure 3.23, examples a, b and c). It was surprising to note that there
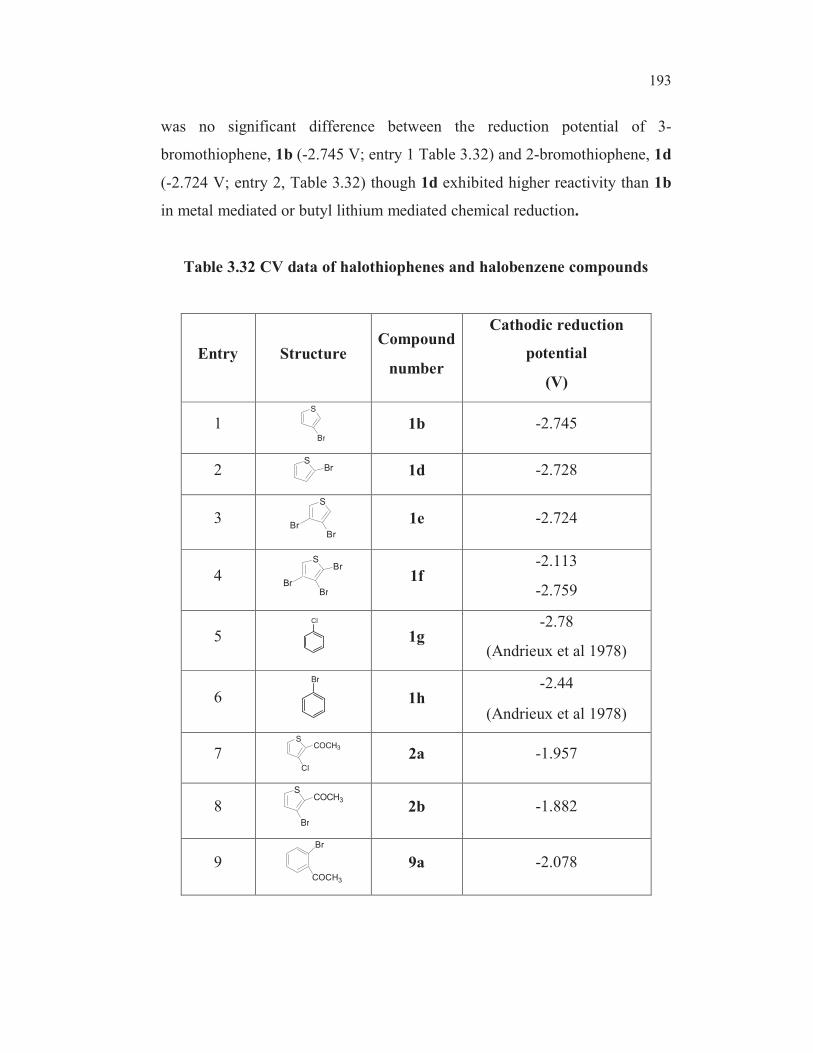
193
was no significant difference between the reduction potential of 3-
bromothiophene, 1b (-2.745 V; entry 1 Table 3.32) and 2-bromothiophene, 1d
(-2.724 V; entry 2, Table 3.32) though 1d exhibited higher reactivity than 1b
in metal mediated or butyl lithium mediated chemical reduction.
Table 3.32 CV data of halothiophenes and halobenzene compounds
Entry StructureCompound
number
Cathodic reduction
potential
(V)
1S
Br
1b -2.745
2S
Br 1d -2.728
3S
BrBr
1e -2.724
4S
BrBr
Br
1f-2.113
-2.759
5Cl
1g-2.78
(Andrieux et al 1978)
6
Br
1h-2.44
(Andrieux et al 1978)
7S
Cl
COCH3
2a -1.957
8S
Br
COCH3
2b -1.882
9
Br
COCH3
9a -2.078
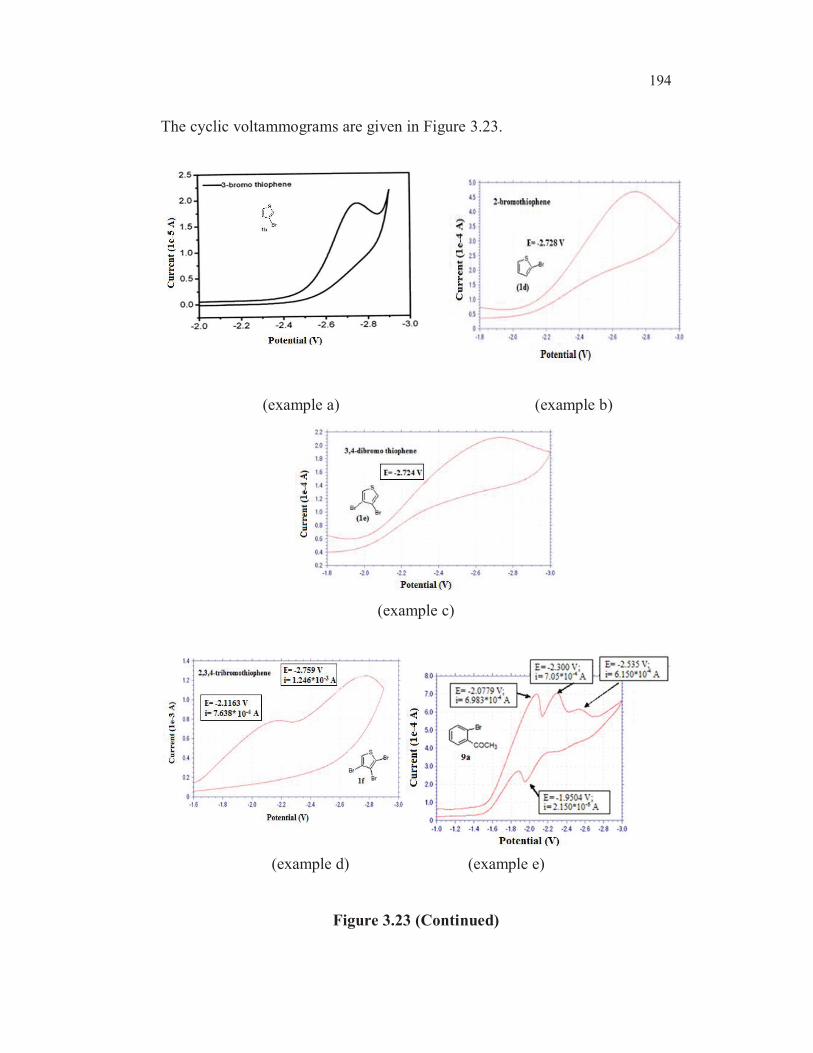
194
The cyclic voltammograms are given in Figure 3.23.
(example a) (example b)
(example c)
(example d) (example e)
Figure 3.23 (Continued)
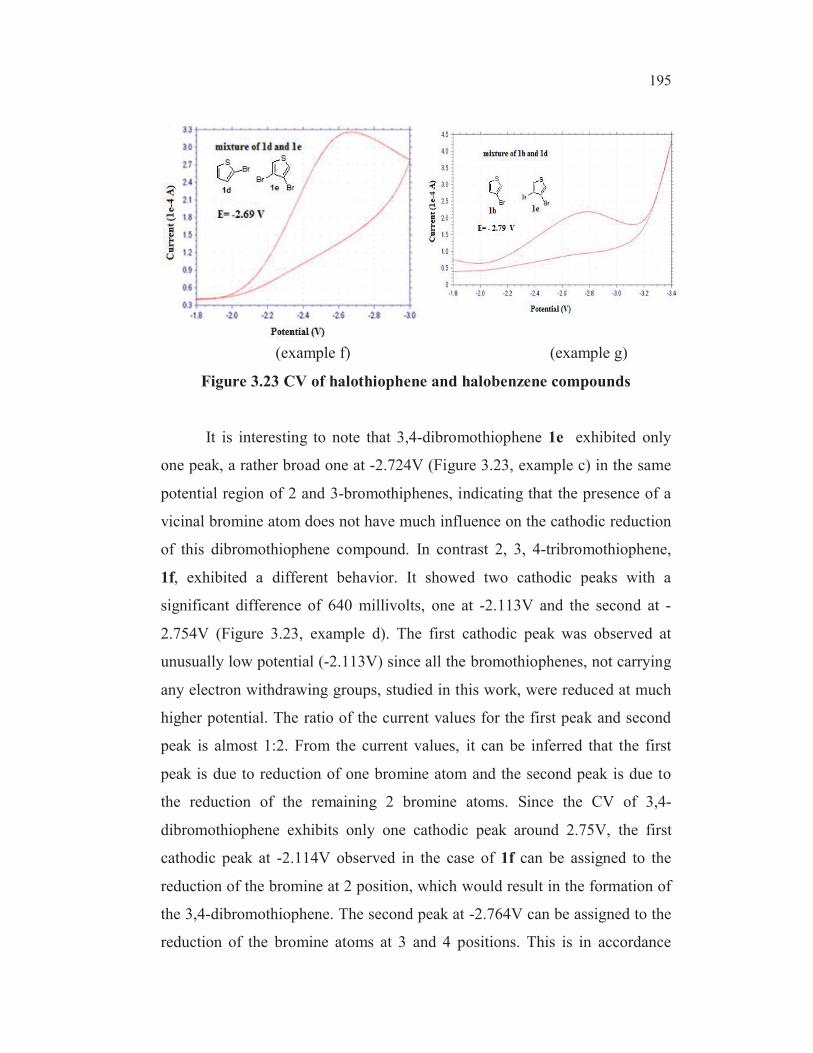
195
(example f) (example g)
Figure 3.23 CV of halothiophene and halobenzene compounds
It is interesting to note that 3,4-dibromothiophene 1e exhibited only
one peak, a rather broad one at -2.724V (Figure 3.23, example c) in the same
potential region of 2 and 3-bromothiphenes, indicating that the presence of a
vicinal bromine atom does not have much influence on the cathodic reduction
of this dibromothiophene compound. In contrast 2, 3, 4-tribromothiophene,
1f, exhibited a different behavior. It showed two cathodic peaks with a
significant difference of 640 millivolts, one at -2.113V and the second at -
2.754V (Figure 3.23, example d). The first cathodic peak was observed at
unusually low potential (-2.113V) since all the bromothiophenes, not carrying
any electron withdrawing groups, studied in this work, were reduced at much
higher potential. The ratio of the current values for the first peak and second
peak is almost 1:2. From the current values, it can be inferred that the first
peak is due to reduction of one bromine atom and the second peak is due to
the reduction of the remaining 2 bromine atoms. Since the CV of 3,4-
dibromothiophene exhibits only one cathodic peak around 2.75V, the first
cathodic peak at -2.114V observed in the case of 1f can be assigned to the
reduction of the bromine at 2 position, which would result in the formation of
the 3,4-dibromothiophene. The second peak at -2.764V can be assigned to the
reduction of the bromine atoms at 3 and 4 positions. This is in accordance
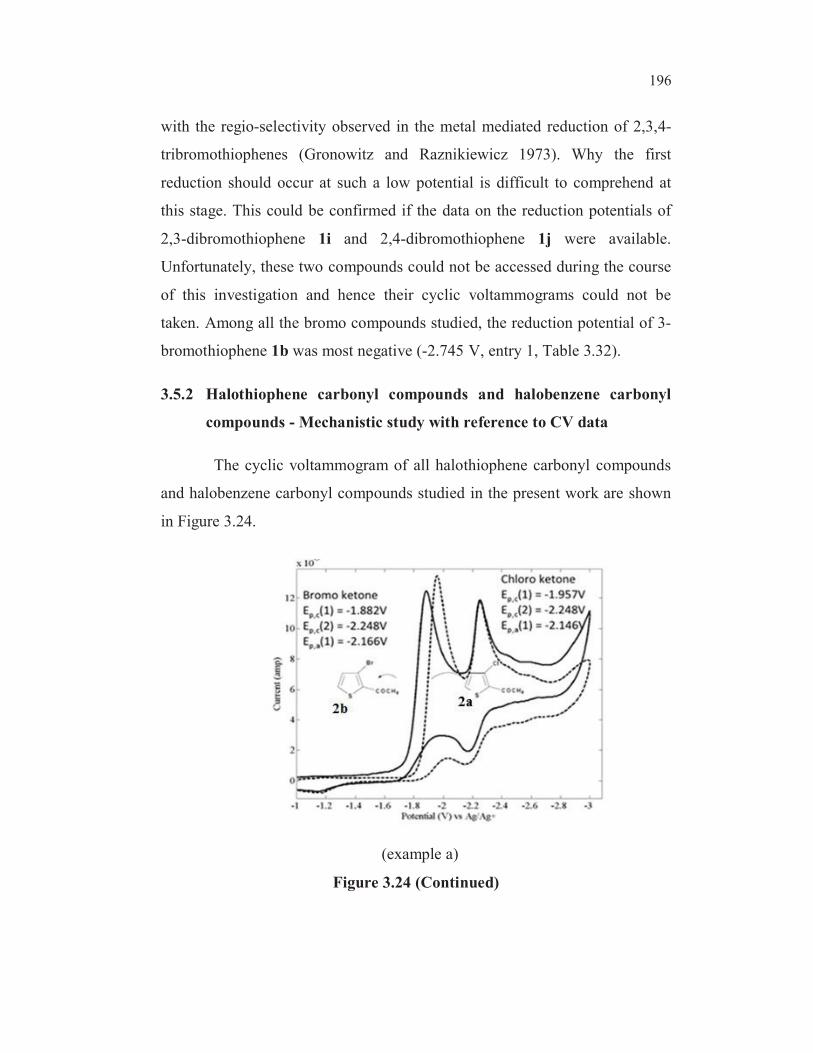
196
with the regio-selectivity observed in the metal mediated reduction of 2,3,4-
tribromothiophenes (Gronowitz and Raznikiewicz 1973). Why the first
reduction should occur at such a low potential is difficult to comprehend at
this stage. This could be confirmed if the data on the reduction potentials of
2,3-dibromothiophene 1i and 2,4-dibromothiophene 1j were available.
Unfortunately, these two compounds could not be accessed during the course
of this investigation and hence their cyclic voltammograms could not be
taken. Among all the bromo compounds studied, the reduction potential of 3-
bromothiophene 1b was most negative (-2.745 V, entry 1, Table 3.32).
3.5.2 Halothiophene carbonyl compounds and halobenzene carbonyl
compounds - Mechanistic study with reference to CV data
The cyclic voltammogram of all halothiophene carbonyl compounds
and halobenzene carbonyl compounds studied in the present work are shown
in Figure 3.24.
(example a)
Figure 3.24 (Continued)
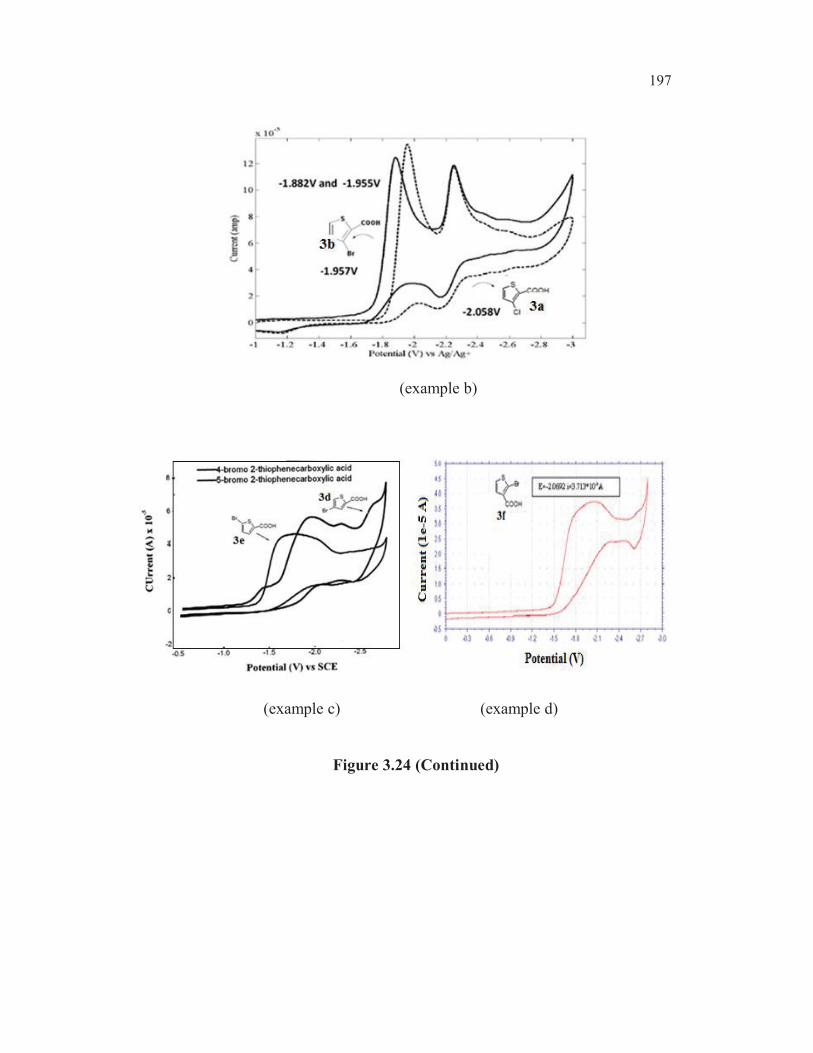
197
(example b)
(example c) (example d)
Figure 3.24 (Continued)
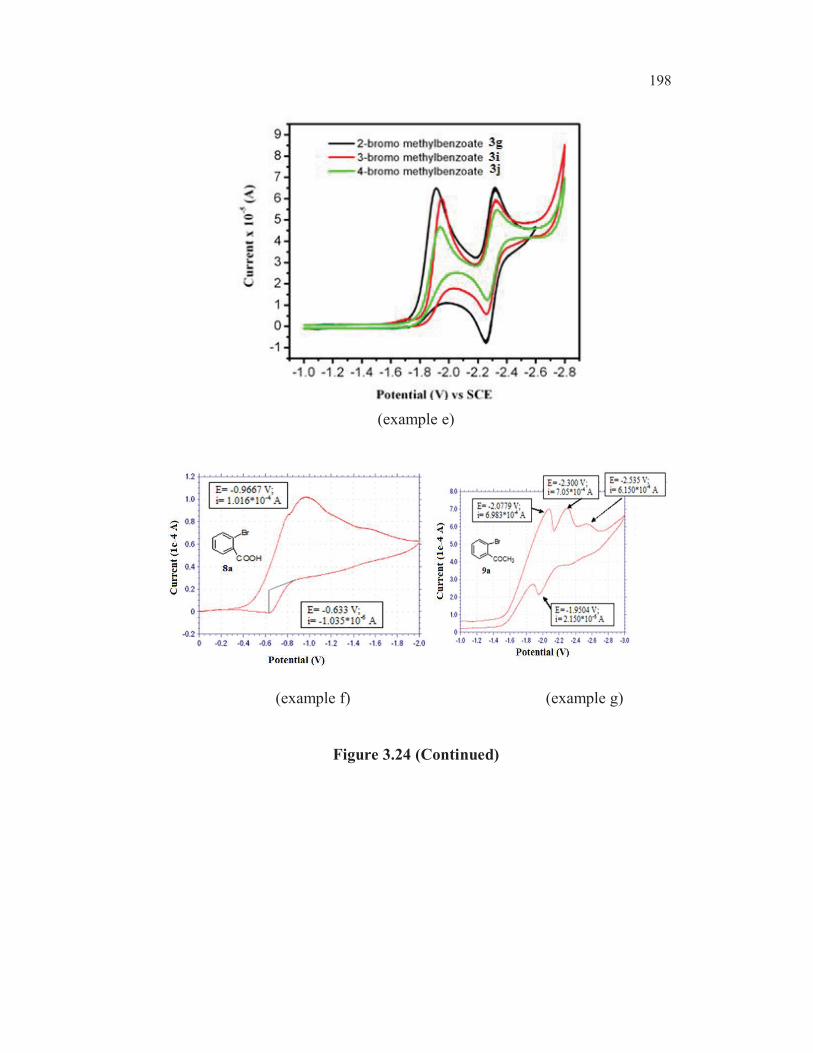
198
(example e)
(example f) (example g)
Figure 3.24 (Continued)
199
(example h) (example i)
Figure 3.24 CV of halothiophene carbonyl compounds and halobenzene
carbonyl compounds
2-aceyl-3-bromothiophene, ketone 2b and bromothiophenecarboxylic acids
3b and 3d showed two cathodic peaks, one corresponding to the reduction of
C-Br bond (first wave and less negative) and another due to the reduction of
the carbonyl group/H+ (second wave, more negative potential). This was
evident by comparing the cyclic voltammogram of 3-bromothiophene 1b
(Figure 3.23, example a) with that of 3-bromothiophene-2-carboxylic acid 3b
(Figure 3.24, example b) and also with those of 4-bromo and 5-bromo
thiophene-2-carboxylic acids 3d and 3e (Figure 3.24, example c) respectively.
The other two bromothiophene carboxylic acids 3f (Figure 3.24, example c)
and 3e (Figure 3.24, example d) showed only one broad peak.
The reduction potentials of all these compounds are listed in Table 3.33.
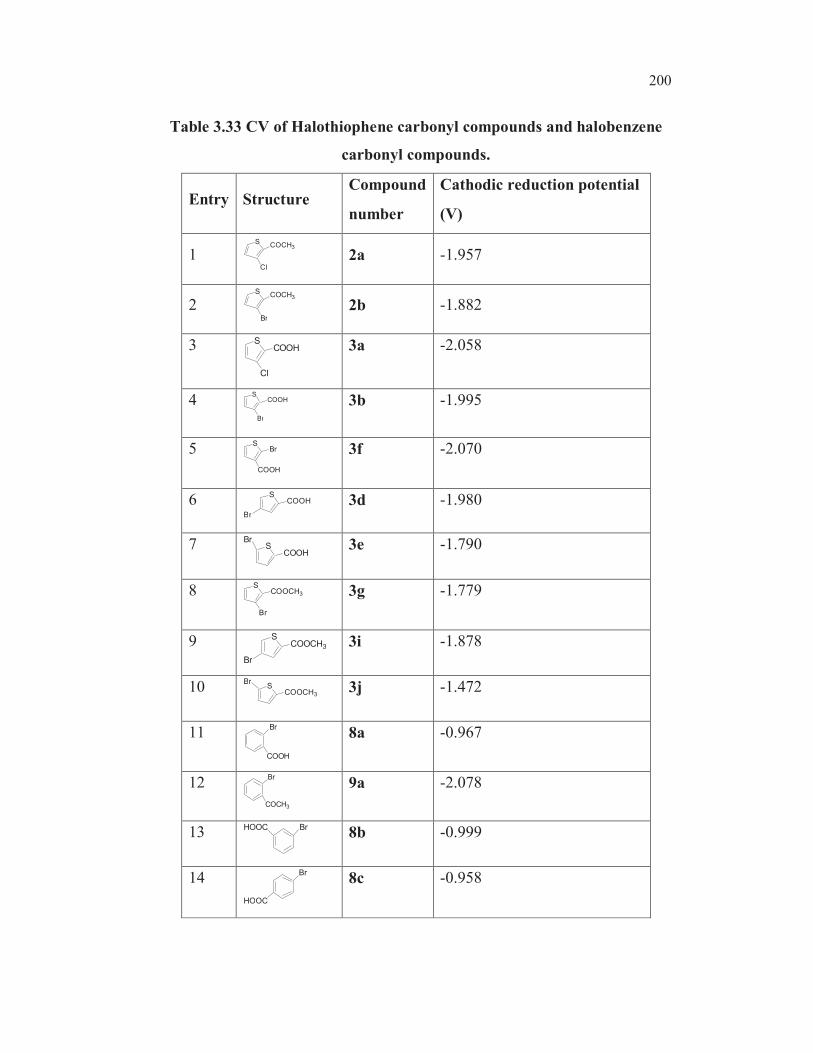
200
Table 3.33 CV of Halothiophene carbonyl compounds and halobenzene
carbonyl compounds.
Entry StructureCompound
number
Cathodic reduction potential
(V)
1S
Cl
COCH3
2a -1.957
2S
Br
COCH3
2b -1.882
3 S
Cl
COOH 3a -2.058
4S
Br
COOH 3b -1.995
5S
COOH
Br 3f -2.070
6S
COOH
Br
3d -1.980
7 SCOOH
Br 3e -1.790
8S
Br
COOCH3 3g -1.779
9 SCOOCH3
Br
3i -1.878
10 SCOOCH3
Br3j -1.472
11Br
COOH
8a -0.967
12Br
COCH3
9a -2.078
13 BrHOOC8b -0.999
14Br
HOOC
8c -0.958

201
The reduction potentials of the C-Br bond of bromothiophene
carboxylic acids were more positive compared to those of bromothiophenes
by as much as 750 millivolts or more (entries 4, 5, 6 and 7, Table 3.33
compared with entries 1,2 and 3, Table 3.32). Even when the bromine and
COOH are not conjugated as in the case of the acid 3d the C-Br bond
reduction occurs remarkably at a lower potential viz., - 1.980V (entry 6, Table
3.33) compared to 2/3-bromothiophene (-2.74V). Surprisingly there is a
marginal difference of 15 millivolts only between the reduction potentials of
acid 3b (-1.995V) and 3d (-1.980V) in spite of the fact that in the case of acid
3b, the bromine atom is conjugated with the -COOH group. When the
bromine is conjugated with the -COOH group as in the case of acid 3e but not
vicinally located, the reduction potential is further shifted to more positive
direction with a significant difference of 205 millivolts (-1.79V, entry 7,
Table 3.33). One may advance the following tentative explanation as to why
the 2 or 3-bromo acids (3f and 3b) is reduced at more negative potential
compared to the 5- bromo acid 3e. While resonance is present in both the
compounds 3b/3f and 3e, the developing negative charge on the carbon will
experience dipole-dipole repulsion in the case of 3b/3f due to the adjoining
CO group rendering the reduction process energetically more difficult. In the
case of 5-bromoacid 3e, the C-Br bond and -COOH are farther away from
each other and there is no dipole-dipole repulsion due to the developing
negative charge. -Haloketones and anomeric effects are well known
examples for the effect of dipole repulsion in influencing the physical /
chemical properties of a molecule (Carey and Sundburg 2003). The easier
reduction of 5-bromoacid 3e compared to that of the 4-bromoacid 3d can be
attributed to mesomeric interaction. Resonance involving the bromine and the
carboxyl in the case of 5-bromoacid 3e would facilitate the reduction of C-Br
bond whereas in 3d, the bromine is not in conjugation with the COOH group.

202
In the case of bromo and the corresponding chloro thiophene carbonyl
compounds, the bromo isomers were reduced at a more positive potential
compared to the corresponding chloro isomers as would be expected (Figure
3.24, examples a and b). This trend parallels their reactivity in the Ullmann
cross coupling reaction. The bromo ketone 2b and bromo ester 3g are reduced
at more positive potential when compared to the corresponding bromo acid 3b
(entries 2, 8 and 4, Table 3.33). It is interesting to note that while bromo
ketone 2b (entry 1, Table 3.33) is reduced at a more positive potential
compared to bromo acid 3b (entry 4, Table 3.33), the trend is reversed in
corresponding benzene system i.e. bromo acid 8a (entry 11, Table 3.33) is
reduced at a more positive potential compared to bromo ketone 9a (entry 12,
Table 3.33).
The order of ease of reduction of C-Br bond in bromothiophene
carboxylic acids as revealed by cyclic voltammetry data is 3e>3d=3b>3f. 2,
3, 4, 5-tetrabromothiophene 1k could be regio-selectively reduced stepwise
by zinc or n-butyl lithium to 2, 3, 4-tribromothiophene, 1f then to 3, 4-
dibromothiophene, 1e and then to 3-bromothiophene 3b (Gronowitz and
Raznikiewicz 1973). In fact this property has been used for the synthesis of 3-
bromothiophene. However this trend is not reflected in the case of Ullmann
cross coupling reactions which exhibited the following order of reactivity
3f>3b>3e>3d even though the reduction potentials of 2-bromo acid, 3f (-
2.070V) and 3-bromo acid 3b (-1.995 V) were very close but more negative
compared to that of 5-bromo acid 3e (-1.79V). The 2-bromo acid 3f is more
reactive compared to 3-bromo acid 3b. The observed order of reactivity
towards Ullmann cross coupling reaction is in contrast to the reduction
potentials which follows the order 3e>3d=3b>3f. Thus it is clear that factors
other than ease of reduction of the C-Br play a significant role in determining
the reactivity in Ullmann cross coupling reactions. The higher reactivity
exhibited by the bromo acids 3b and 3f in the Ullmann cross coupling with

203
sodium bisulphite is attributed to chelation and the availability of an
intramolecular oxidative addition pathway in these compounds (Figure 3.25).
Figure 3.25 Chelation supporting the oxidative addition
The lower reactivity of the acid 3b compared to that of 3f may be due
to the chelation of the copper carboxylate with sulphur atom in the case of 3b
thus reducing the probability for the oxidative addition of copper in to C-Br
bond. This sort of chelation is not possible in the case of 2-bromothiophene-3
carboxylic acid 3f and hence its enhanced reactivity. The higher reactivity of
3f over 3e may be due to the effect of chelation of Cu+ with the thiophene
ring sulphur. One may speculate the following explanation to account for the
higher reactivity of 3e over 3d. The chelation of Cu+ with thiophene ring
sulphur atom will bring down the charge density on sulphur and the effect of
this will be transmitted to the ring. The carbon C2 nearer to S atom will
experience this effect more than C3 or C4 of the thiophene moiety and as a
result of this lowering of electron density at this centre, the oxidative addition
(intermolecular) of Cu+ to the C-Br bond is facilitated. The present study
brings out the role and importance of chelation factor and lends support to the
proposed mechanism involving the intramolecular oxidative addition. In the
light of the interesting findings from the CV studies, it will be worthwhile to
examine the cyclic voltammetry behavior of thiophene compounds viz., 2, 3,
4, 5-tetrabromothiophene, 1k, 3-bromothiophene-4-carboxylic acid 3k and 2-
bromothiophene-4-carboxylic acid, 3l and compare the ease of reduction of
C-Br bonds in these compounds with their reactivity towards Ullmann cross
coupling reaction with sodium bisulphite.
S
Br
OS
Br
O
OCu
O Cu

204
S
Br1k
Br
Br
BrS
COOH3k
Br
S
COOH3l
Br
Figure 3.26 Thiophene compounds
In the case of 3-bromothiophene-4-carboxylic acid 3k, while
intramolecular oxidative addition can take place, there is no scope for
alternate chelation of copper carboxylate with the sulphur atom. Hence it
should exhibit high reactivity similar to that of 3f. With regard to 2-bromo-
thiophene-4-carboxylic acid, 3l, intramolecular oxidative pathway is not
possible. At the same time, chelation of Cu+
salt with ring sulphur is also not
possible. It remains to be seen how its reactivity would compare with that of
3d. The bromine atom is not conjugated with the -COOH group in both these
acids, 3k and 3l and also there is no dipole-dipole repulsion factor due to
developing negative charge in the case of the reduction of 3l. Based on this
reasoning, one could expect its reduction potential (C-Br bond) should be
lower (more positive) than that of 3k and also higher than that of 3e. Hence it
will be worthwhile to investigate the electrochemical reduction and Ullmann
cross coupling reactions of these acids.
The rate profile for Ullmann cross coupling reaction of halothiophenes
with phenol is 3-bromo-2-acetyl-thiophene (2b) > 3-chloro-2-acetyl thiophene
(2a) > 3-bromothiophene (1b). This is in accordance with the corresponding
CV data for the C-X bond reduction potentials (2b -1.882V, 2a -1.957V and
1b -2.745V).

205
3.5.3 Comparison of Thiophene and Benzene Halocarboxylic Acid
Systems in Ullmann Condensation Based on their CV Data
Nucleophilic substitutions of aromatic halides and heteroaroamtic
halides have been extensively investigated (Bunnet 1951, 1974 amd 1976,
Lindley 1984, Denney and Denney 1991, Diederich and Stang 1998, Carey
and Sundburg 2001, Gibson and Spitzmesser 2003, Laszlo and Barbara 2005,
Naohiko and Eiichi 2012). It is known that haloaromatics exhibit higher
reactivity compared to halothiophenes towards nucleophiles (Bunnet and
Bernhard 1976, Miller 1968, Salo Gronowitz 1991 and Bunnett 1983).
Different physical methods (X-ray, electron diffusion and microwave studies)
reveal a high double bond character of the C-S bond in thiophene (Figure
3.18).
SS S S S S
C2
C1
C3
C4
Figure 3.27 Canonical structures for thiophene molecule
In excessive heteroaromatics which contain a five membered ring
and contain only one heteroatom, the electron density is higher on the ring
carbon. This implies low reactivity towards nucleophiles. The C(2)-C(3) and
C(3)-C(4) bonds in thiophene molecule are respectively shorter and longer
than C-C bond of in a benzene ring and therefore have a correspondingly
higher and lower bond order. This fact causes the occurrence of different
ortho-relations corresponding to 2, 3 and 3, 4 di-substituted thiophenes. Many
kinetic, spectroscopic and reactivity data support this difference and lead to
distinguish the two molecules as hyper-ortho and hypo-ortho respectively.
Moreover, in the five membered ring of thiophene the values of internal
angles S-C1-C2 (111°28’) and C1-C2-C3 (112 °27’) are not far from those of

206
sp3 hybridized carbon atom (109°5’). This is similar to those represent the
reaction centre in the complexes for both SNAr and SEAr. At the same time
the high values of external angle makes the distance between the two ortho-
like substituents larger than in benzene derivatives, affecting both the
interactions between these substituents and their geometry with respect to the
ring. Theoretical and experimental studies indicate that thiophene is less
aromatic than benzene (resonance energy of thiophene 130 KJmol-1
and that
of benzene 151 KJmol-1
). The geometric structure and the aromaticity surely
affect the reactivity of thiophene derivatives with nucleophiles. Under
analogous reaction conditions, the reactivity of thiophene in SEAr is higher
than that of the corresponding benzene derivatives, for example, the nitration
and acetoxymercuration of thiophene are faster than benzene respectively by a
factor 850 (at 10°C) and 105 at 25C°. The piperidinodechlorination in ethanol
at 25° C of 2-chloro-3,5-dinitrothiophene is faster by a factor 500 than that of
2,4-dinitrochlorobenzene. This is in contrast to the nucleophilic substitution
occurring through addition-elimination SNAr mechanism.
SCl
NO2
O2N
Cl
NO2O2N
ethanol
ethanol
NH
NH
SN
NO2
O2N
N
NO2O2N
k thiophene / k benzene > 500
25° C
25° C
Scheme: 3.35 Piperidinodechlorination reaction
The high electron density on the ring carbon, characteristic of
excessive heteroaromatics causes a stabilization of the transition state for

207
SEAr reaction in thioiphene, where as the high electron delocalization ability
of the electron withdrawing aromatic residues favors SNAr reaction in
thiophene derivatives (Bunnet 1978). The intrinsic ability of the thiophene
ring to transit the electronic effect of the substituent is greater than the ability
of the benzene ring (a factor that can also be enhanced by hyper-ortho or
quasi-para relations and also can be enhanced by involving the transition of
electronic effects through sulphur heteroatom).
Unactivated halothiophenes are relatively inert to nucleophilic
substitution while those conjugatively substituted with electron withdrawing
groups are comparatively more reactive to nucleophilic substitution (Kassmi
1992). In unactivated systems, SRN1 reactions occur with halogen derivatives
of thiophene but less readily compared to phenyl halides (Bunnet and
Bernhard 1976, Miller 1968, Salo Gronowitz 1991, Bunnett 1983). This
order of reactivity is reversed in the case of activated substrates containing
highly electron withdrawing groups. The nucleophilic substitutions of
unactivated halides are effected under harsh conditions when compared to
activated halides.
It has been observed in literature that there is a parallel between the
reduction potentials of C-X bond of halides and the leaving group ability of
the halides (Gibson and Spitzmesser 2003). The order of reduction potential
of halobenzenes in liquid ammonia is PhI > PhBr > PhSPh > PhCl > PhF >
PhOPh (Amatore et al 1985) which coincides with the reactivity order
determined under photo initiation. Experiments carried out in liquid ammonia
under irradiation, with pair of PhX towards-CH2COBu
t, the following
reactivity order was found: PhCl / PhF= 29, PhBr / PhCl= 450. PhI / PhBr=
8.3. Therefore the increase in reactivity from PhF to PhI is almost 100000
(Amatore et al 1981). One could expect a similar parallel between the
cathodic reduction potential of C-X bond and the ease of Ullmann cross

208
coupling. With a view to get some insight into mechanism of this reaction, the
cathodic reduction potential of C- X bonds in the molecules of interest were
measured using cyclic voltammetry analysis. The CV data of aromatic halides
has been advanced as evidence in support of the mechanism in the case of
SRN1 reactions (Bunnet 1978). Since both the reduction of the C-X bond and
oxidative addition of a metal/metal ion into a C-X bond involve addition of 2
electrons, it was of interest to find out whether any correlation could be
observed between the reduction potentials and ease of the Ullmann cross
coupling of the various halothiophene carboxylic acids. More importantly, it
was our interest to check if the observed higher reactivity of 2-bromobenzoic
acid 8a over 3-bromo-thiophene-2-carboxylicacid 3b in the Ullmann cross
coupling reaction with sodium bisulphite could be correlated with the
reduction potential of these two bromo derivatives. So far no such study has
been reported in literature. In fact a comparative study of the electrochemical
reduction of various halothiophenecarboxylic acids or halobenzoic acids has
not been reported so far in literature. In view of this, cyclic voltammograms
of several halothiophene carboxylic acids and halobenzoic acids were
measured and the cathodic peak potentials are given in Table 3.33.
The Ullmann cross coupling of 2-bromobenzoic acid 8a with
sodium bisulphite was much faster compared to that of 2-chlorobenzoic acid
8d. The relative reactivity was similar to that observed in the case of 3-
bromothiphene-2-carboxlic acid 3b and 3-chloro-thiophebne-2-carboxylic
acid 3a. Harsh conditions have been employed in literature (Anthony 1979,
Kim 2010) for the Ullmann cross coupling of 2-chlorobenzoic acid 8d with
sodium bisulphite (175°C, autoclave, 7 kg/cm2 pressure for 24 h). It
interesting to note that 2-bomobenzoic acid 8a was several folds more
reactive than 3-bromothiophene-2-carboxylic acid 3b. The conversion profile
is tabulated in Table 3.25. Likewise, 5-bromo-thiophene-2-carboxylic acid 3e
was found to be more reactive than 4-bromobenzoic acid 8c (Table 3.27). One

209
could advance the following explanation to account for the observed higher
reactivity of 2- bromobenzoic acid 8a compared to that of 3-bromothiophene-
2-carboxylic acid 3b. In the case of the thiophene acid 3b, the copper
carboxylate can form two chelates (Figure 3.19) i.e. one involving ring
sulphur and another involving the bromine. Of these only one can participate
in intramolecular oxidative addition and lead to product (Figure 3.19, example
a), whereas the other copper chelate cannot (Figure 3.19, example b). Thus,
the probability for the intramolecular oxidative addition involving the copper
complex which is the crucial step in this nucleophilic substitution reaction is
reduced in 3b. In the case of 2- bromobenzoic acid 8a such a situation does
not prevail and only one chelate involving the copper carboxylate is possible
and thus the probability factor is in favour of 2-bomobenzoic acid 8a
compared to 3-bromothiophene-2-carboxylic acid 3b. In addition to the rate
accelerating ortho carboxylate effect, some other electronic factor involving
the ring sulphur may also play a role.
A comparative study of the electronchemical reduction of various
halothiophenecarboxylic acids or halobenzoic acids has not been reported so
far in literature. The cathodic reduction potential of bromobenzene 1h (-2.44
V) was less negative (difference of 290-310 millivolts) compared to that of 3-
bromothiophene 3b and 2-bromothiophene 1d ( -2.72 V and -2.75 V)
indicating the significant influence of sulphur on the ease of reduction of the
C-Br bond in bromothiophenes. Since the thiophene ring is an activated
heteroaromatic system compared to benzene (super aromatic) for electrophilic
substitution reactions and vice versa for nucleophilic substitution reactions, it
is evident that the addition of electrons to such a system will be unfavourable
compared to that of simple benzene ring. This is reflected in the cathodic
reduction potential values of bromothiophene and bromobenzene. Based on
dipole-dipole repulsion consideration discussed earlier, and based on a
comparison of the cathodic potentials of bromothiophenecarboxylic acids, one

210
would expect that 4-bromobenzoic acid 8d would exhibit a more positive
cathodic potential compared to that of 2-bromobenzoic acid, 8a. Though this
trend is observed, the difference (50 to 60 millivolts) is only marginal and not
as big as that observed in the case of thiophene acids 3b and 3e (205
millivolts). 2-bromobenzoic acid 8a is reduced only at a slightly more
negative potential (-0.967V) compared to the 4-isomer 8c (-0.958V). As a
follow up of this study, the cyclic voltammograms of 3-bromo-2- acetyl
thiophene 2b and 2-acetyl bromobenzene 9a were measured and compared
with their relativities towards Ullmann cross coupling reaction with
phenoxides as nucleophiles. It is interesting to note that a rather huge
difference (-1.111V) in the reduction potential between 2-bromobenzoic acid
8a and 2-bromoacetophenone 9a (bromo acid and bromo ketone of benzene
system) was observed compared to the difference of + 0.113V in the case of
3-bromothiophene-2-carboxylic acid and 2-acetyl-3-bromohiophene (bromo
acid and bromo ketone of thiophene system). The cathodic reduction potential
of C-X bond in 3-bromo-2- acetylthiophene 2b and 2-acetylbromobenzene 9a
are -1.882 V and -2.078V respectively. Based on these CV data, one may
expect that bromo ketone of thiophene system viz., 2b should manifest higher
reactivity compared to bromo ketone of benzene system viz., 9b towards
Ullmann cross coupling reaction. However 9b was found to be more reactive
than 2b, pointing to some role played the sulphur atom.
The present study has shown that the cathodic reduction potential
values of halobenzoic acids, haloacetophenones, halothiophene carboxylic
acids and ketones cannot be correlated in all cases to their respective
reactivity towards Ullmann cross coupling reactions, though a good
correlation has been observed in literature in the case of SRN1 reactions. The
present cyclic voltammetry investigation has led to some very interesting
findings which are worthy of further exploration.