Embed Size (px)
Citation preview

Chapter 3
Synthesis of Varenicline

Chapter 3 110
3.1. INTRODUCTION
In 1604, King James referred to smoking as „a branch of the sin of
drunkenness, which is the root of all sins‟. Since its introduction in the
16th century, the use of tobacco has become widespread, and survived
considerable official disfavor.1 The clinical sequence of cigarette smoking
represents an unparalleled burden to public health. The use of tobacco
through cigarette smoking is the leading avoidable cause of death and
kills nearly four million people annually.2-4 According to the World Health
Organization (W H O), ten million smokers will die annually worldwide by
the year 2030.
3.1.1. Nicotine
Nicotine 1 (Figure 1) is an alkaloid was isolated from tobacco
leaves of Nicotiana tabacum in 1828 by Posselt and Reimanbasic. The
research on „nicotine effects in the brain and body‟ showed that,
although tobacco contains thousands of chemicals, the most active
ingredient that acts in the brain and produces addiction is nicotine and
the addiction produced by nicotine is very powerful and is at least as
strong as addictions to other drugs such as cocaine and heroin.
Figure 1. Nicotine

Chapter 3 111
3.1.2. nAChRs
The neuronal nicotinic acetylcholine receptors (nAChRs) are a
family of ligand-gated ion channels, which are expressed throughout the
central and peripheral nervous systems.5,6 nAChRs are made up from
pentameric assemblies of and β subunits. In mammals nine (2–10)
and three β subunits (β2–β4) have been identified to date, which combine
to form functional channels. Most commonly, functional channels are
made up of a single type of and a single type of β subunit, however,
there is evidence that „triplet‟ channels containing two different types of
and a single type of β subunit, or two types of β and one type of α
subunit exist.7-10 nAChR subunit composition has an important impact
on pharmacological sensitivity, nAChR dysfunction leads to a number of
neurological diseases, like schizophrenia, Alzheimer‟s and Parkinson‟s
diseases and also nAChRs play an important role in the response to
pain.11-13 As a result nAChRs have become important therapeutic targets.
3.1.3. How nicotine acts
When smoked, nicotine is readily absorbed through the alveolar
surface of the lungs and rapidly reaches circulation and the brain within
11 seconds after inhalation.14 Nicotine affects many neurotransmitters,
but dopamine 2 (Figure 2) seems to be most responsible for the major
addictive properties of nicotine. Nicotine directly stimulates the
acetylcholine receptors on dopamine-containing neurons. The
stimulation of these acetylcholine receptors is responsible for the

Chapter 3 112
overflow of dopamine in the reward centers of the brain, resulting in
extracellular dopamine within the nucleus accumbens and an increased
firing of dopaminergic neurons.15 Even, many subtypes of nAChRs are
associated with nicotine addiction, 4β2 subtype are thought to play a
principal role.16,17 Unlike acetylcholine 3 (Figure 2), which is degraded
quickly by acetylcholinesterase, nicotine remains active at the 4β2
receptor sites for a longer time. Nicotine stimulation at the acetylcholine
receptors causes receptor up-regulation. This up-regulation desensitizes
acetylcholine receptors, resulting in physical dependence, tolerance, and
withdrawal symptoms, thereby adding to the tendency for nicotine
addiction.17 Smokers who try to quit experience symptoms of withdrawal,
such as depression, insomnia, irritability, anxiety, difficulty
concentrating, restlessness, weight gain, increased appetite, and
decreased heart rate. The withdrawal symptoms strengthen the desire to
smoke, perpetuating the dependency cycle.18
Figure 2. Dopamine and Acetylcholine
3.1.4. Smoking cessation interventions
Addiction to nicotine is a chronic, relapsing and in many cases
enduring problem. To improve smoking cessation rates, more effective
treatments are needed and presently there are two types of treatment

Chapter 3 113
options are available. They are nor pharmacological and pharmacological
treatments. Among them pharmacological treatments (NRT) are more
effective.
3.1.4.1. Nicotine replacement therapy (NRT)
NRT was the first successful pharmacological intervention for
nicotine dependency and it refers to the administration of nicotine to
substitute that obtained from tobacco and provides the smoker with
lower, relatively safer dose of nicotine that is reported to satisfy
withdrawal symptoms. NRT allows the smoker to develop coping
strategies for the behavioral aspects of their addiction with the
physiologic component being addressed.19,20
The main mode of action of NRT is thought to be the stimulation of
nicotine receptors in the ventral tegmental area of the brain and the
consequent release of dopamine in the nucleus accumbens.21,22 Nicotine
replacement as an aid to smoking cessation, providing that steady-state
levels of nicotine can prevent a smoker from experiencing intense
withdrawal while not providing the reinforcing peaks achieved with
smoking.23
NRT is supplied in several in the forms of patch, gum (polacrilex),
inhaler, nasal spray and lozenges. The faster acting formulation nicotine
gum, nasal spray or inhaler, appears to be helpful in satiating the
positive effects of nicotine intake through smoking and reduce acute
craving, while the slow acting transdermal nicotine patch supplies low

Chapter 3 114
but constant levels of nicotine, which, depending on the concentration,
can ease nicotine withdrawal symptoms.24,25
3.1.4.2. Other pharmaceutical agents: Several other drugs including
antidepressants (Bupropion 4, Imipramine 5, Doxepin 6, Moclobemide
7), antihypertensives (Clonidine 8), anxiolytics (Buspirone 9, Diazepam
10, Meprobamate 11 and b-blockers), and sensory replacement therapy
(silver acetate) have been used in smoking cessation (Figure 3).
Figure 3. Pharmaceutical agents used in the smoking cessation.

Chapter 3 115
3.2. Discovery of varenicline16, 29
The natural product (–)-cytisine 13, found in numerous plant
species, was reported in 1994 to be a partial agonist of 4β2 nAChR and
antagonize the receptor response to its endogenous neurotransmitter,
acetylchloline. In the 1960s an early smoking cessation trial with (–)-
cytisine failed to exhibit efficacy, perhaps due to poor absorption and
limited brain penetration. And (–)-cytisine 13 was chosen as a starting
point for the SAR studies at Pfizer research center.
The initial SAR studies discovered that 4β2 nAChR binding
affinity was considerably decreased with changes at position N-9 and C-5
(Figure 4), but maintained or improved with substitution at C-3 of (–)-
cytisine. These results led them to explore pyridine replacements based
on 14. However this series of analogues exhibited a weaker affinity and
lower efficacy partial agonist profile relative to (–)-cytisine.
To find compounds with promising in vitro and in vivo profiles, the
Pfizer team searched for alternative template. The amazing similarity
between substructures of (–)-cytisine 13 and morphine 18 was
recognized, where their [3.3.1] bycyclic skeletons differ only by the
position of nitrogen atom (cf. 14 vs 15). In the 1970s, it was found that

Chapter 3 116
the simplified analogue 15 had morphine-like antinociceptive activity, as
did the modified derivative 16. However, the N-positional isomer 17
lacked antinociceptive activity. Interestingly, 3,5-bicyclic aryl piperidine
18 displayed in vivo pharmacology similar to natural nicotinic agents. On
the basis of the resemblance between 15 and 16, both antinociceptive
compounds, it is speculated that 14 and 17 might share a similar
nicotinic pharmacology. It turned out to be true that 17 is a nicotinic
antagonist.
Figure 4. Related substructures of (-)-Cytisine and Morphine
Potent natural nicotinic agonists, such as epibatidine, 19 (–)-
nicotine, 1 (+)-anatoxin , 20 and (-) cytisine, contain electron-deficient
π-systems. Electron-withdrawing substituents therefore were introduced

Chapter 3 117
on the position 1, 2 and/or 3 of 16 (Figure 5). From ortho-
dinitroderivative 21, other types of derivatives were prepared, including
quinoxalines, benzimidazoles, benzoxazoles, benzothiazoles. In vitro
activity of these analogues is sensitive to structural modification,
showing a dependence on both steric and electronic nature of
substituents. Furthermore they exhibit a range of partial agonist activity
and potency in vivo.
Figure 5. Bycyclic Aryl Piperdines and Other Analogue Derivatives
Among these analogues, varenicline 22 (quinoxaline R = H)
displays high affinity, selectivity, and a desirable in vitro partial agonist

Chapter 3 118
profile. It also has low affinity for other receptors, channels, uptake sites
and second messengers. Varenicline is partial agonist alone and fully
block nicotine‟s effect in vivo.
3.2.1. Pharmacology of varenicline
It has been reported that, In vitro varenicline 22 produces 68% of
the response seen by the binding of nicotine 1 to 4β2 receptors in In
vitro. In vivo studies have found the dopamine 2 response to varenicline
to be 32–60% of the response to nicotine. With this partial agonist–
antagonist profile, the varenicline molecule competitively inhibits
nicotine, thereby blocking the effects of nicotine at the 4β2 receptor site.
Therefore, varenicline alleviates the symptoms of nicotine craving and
withdrawal through its agonist activity while inhibiting the effects of
repeated nicotine exposure by its antagonist activity.

Chapter 3 119
3.3. Pfizer’s Approach to varenicline.16
3.3.1. Retrosynthesis
Scheme 1. Retrosynthesis of varenicline.
3.3.2. Synthesis of intermediate 2716b
They have followed two different approaches for intermediate 27
from 30, which have been prepared according to reported procedure.
Approach I
Osmium-catalyzed cis-dydroxylation of 30 produced 29. Diol 29
was conveniently converted to 28 in a two step procedure. Oxidative
cleavage of 29 with NaIO4 gave aldehyde 33 and subsequently treated
with benzyl amine in presence of NaBH(OAc)3 to get 28. The benzyl
protection was removed in presence of Pd(OH)2 to get 27 (Scheme 2).

Chapter 3 120
Scheme 2. Reagents and conditions: (a) OsO4, NMO, aq. acetone, 89%;
(b) NaIO4, aq. DCE; (c) BnNH2, Na(OAc)3BH, DCE, 82-86% for two steps;
(d) H2, Pd(OH)2, HCl:MeOH, 88-95%.
Approach II
To form methoxyhydroperoxide glycol 34, a stream of ozone was passed
through a solution of 30 in methanol at -78 ºC. Once 34 had formed
completely, it was reduced to 35 with 5% Pt/C. Benzyl amine and formic
acid was added and the hydrogenolysis was resumed to carry out the
reductive amination to afford 28. The benzyl group was removed by
Pearlman catalyst to get 27 (Scheme 3).

Chapter 3 121
Scheme 3. Reagents and condition: (a) Ozone, MeOH, -78 °C; (b) H2,
Pt/C, MeOH, 0 °C; (c) BnNH2, HCO2H, H2, Pt/C, MeOH; (d) TsOH, H2,
Pd(OH)2/C, MeOH, 40 °C, 28% overall yield.
3.3.3. Conversion of 27 to varenicline16a
Compound 27 was converted to trifluoroacetamide 26, vicinal dinitration
with nitronium triflate (CF3SO2-NO2+) yielded 25. Reduction of 25 to the
corresponding diaminophenylbisulfite and condensation with glyoxal
afforded 23. Deprotection of trifluoroacetamide completed the synthesis
of 22 (Scheme 4).
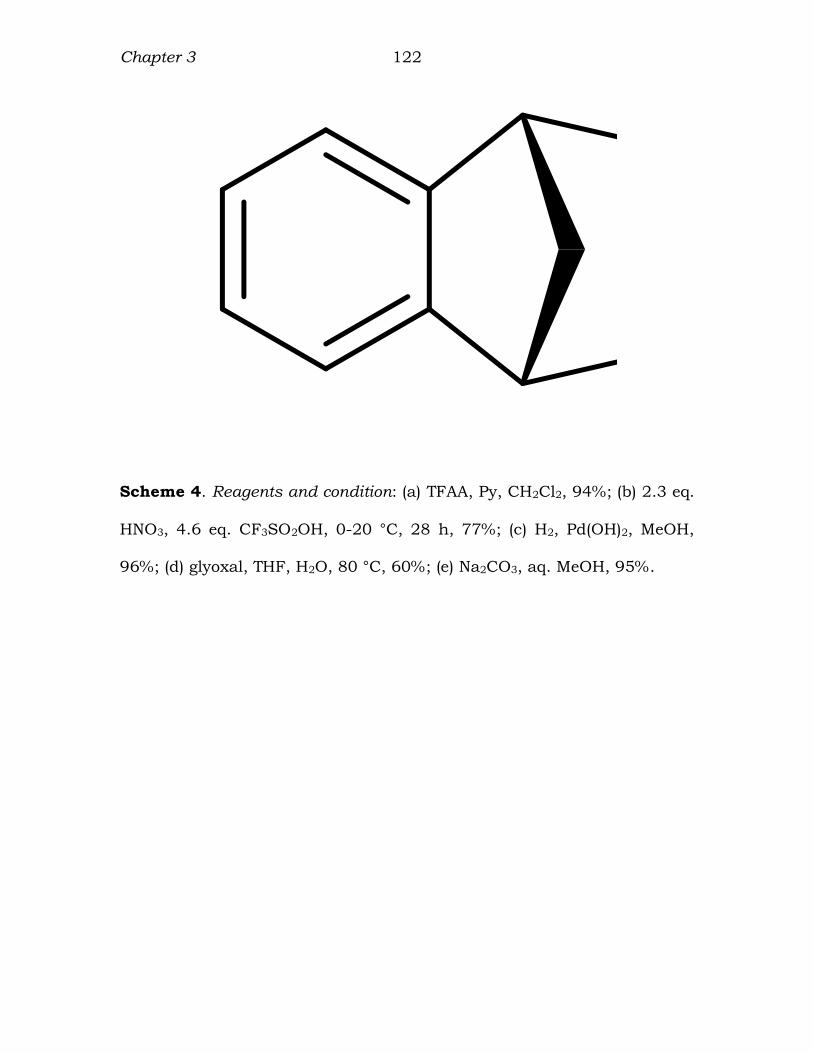
Chapter 3 122
Scheme 4. Reagents and condition: (a) TFAA, Py, CH2Cl2, 94%; (b) 2.3 eq.
HNO3, 4.6 eq. CF3SO2OH, 0-20 °C, 28 h, 77%; (c) H2, Pd(OH)2, MeOH,
96%; (d) glyoxal, THF, H2O, 80 °C, 60%; (e) Na2CO3, aq. MeOH, 95%.

Chapter 3 123
3.4. PRESENT WORK
We have developed two new and efficient approaches to varenicline
using Diels-Alder reaction and reductive amination as key steps.
3.4.1. Our Approach I
3.4.1.1. Retrosynthesis of Varenicline
As depicted in scheme 5, retrosynthetically we envisioned varenicline 22
from PBM protected varenicline 36. Compound 36 could be obtained
from 37 by means of oxidative cleavage followed by reductive amination
with 4-methoxybenzylamine. The Diels-Alder adduct 38, which was
obtained from tetra bromide 40 and norbornadiene 39 could serve as
precursor to 37.
Scheme 5. Retrosynthesis of varenicline

Chapter 3 124
3.4.1.2. Results and Discussions
As per the synthetic plan (scheme 6), our synthesis began with the
conversion of 2, 3-dimethylpyrazine 41 to its known analogue 2,3-bis-
(dibromomethyl)pyrazine31 40. Followed by NaI-mediated Diels–Alder32,33
reaction between 2,3-bis (dibromomethyl)pyrazine 40 and commercially
available norbornadiene 39 to furnish the adduct 38. Olefin 38 was
converted34 to its corresponding diol 37 with OsO4 in the presence of
NMO. Oxidative cleavage35 of the diol 37 with NaIO4 provided an
intermediate dialdehyde. Subsequent reductive amination with 4-
methoxy benzyl amine gave compound PMB-protected varenicline 36.
Finally PMB group in 36 was removed by Pd/C in the presence of
ammonium formate in methanol to yield varenicline 22.
Scheme 6. Reagents and conditions: (a) NBS, CCl4, hν, 16 h, 65%; (b)
NaI, DMF, 60 ºC, 30 min, 46%; (c) NMO, OsO4, acetone/water/tBuOH,
rt., 16 h, 85%; (d) NaIO4, CH2Cl2, silica gel, rt., 30 min; (e) PMBNH2,

Chapter 3 125
Na(CN)BH3, MeOH, AcOH, 0 ºC to rt., 2 h, 62% for two steps; (f) Pd/C,
HCO2NH4, MeOH, reflux for 30 min, rt., 24 h, 64%.
3.4.2. Our Approach II
After successful synthesis of varenicline using approach I, we
thought of developing general approach to its pyridine analogues. Our
interest in Diels-Alder strategy allowed us to develop another new
methodology to varenicline. This methodology was further demonstrated
by applying to its pyridine analogue.
3.4.2.1. Retrosynthesis of Varenicline
As depicted in scheme 7, retrosynthetically we envisioned
varenicline 22 from benzyl protected varenicline 42. This could be
obtained from 43 by Diels-Alder reaction with 2, 3-bis (dibromomethyl)
pyrazine 40. Compound 43 can be obtained from dihydroxy compound
44 by oxidative cleavage followed by reductive amination with benzyl
amine. And compound 44 could be prepared from norbornadiene 39 by
osmium tetraoxide cis-hydroxylation

Chapter 3 126
Scheme 7. Retrosynthesis of varenicline
3.4.2.2. Results and Discussions
As per the synthetic plan (scheme 8), our synthesis began with the
cis-hydroxylation36,37 of commercially available norbornadiene 39 with
OsO4 in presence of NMO to obtain 44 in 55% isolated yield. Oxidative
cleavage35 of the diol 44 with NaIO4 provided an intermediate dialdehyde.
Subsequent reductive amination with benzyl amine gave crucial
intermediate 43. The NaI mediated diels-alder32,33 reaction between 43
and 2,3-bis- (dibromomethyl)pyrazine 40 gave benzyl-protected
varenicline 42. Finally benzyl group in 42 was removed by Pd/C in the
presence of ammonium formate in methanol to yield varenicline 22.

Chapter 3 127
Scheme 8. Reagents and conditions: (a) OsO4, NMO, acetone, water,
tBuOH, tBuOOH, rt., 2 h, 55%; (b) NaIO4, CH2Cl2, silica gel, rt., 30 min;
(e) BnNH2, Na(CN)BH3, MeOH, rt., 2 h, 75% for two steps; (d) NaI, DMF,
60 °C, 30 min, 65%; (e) Pd/C, HCO2NH4, MeOH, reflux for 30 min, rt., 24
h, 85%.
3.4.3. Pyridine analogue
After the successful synthesis of varenicline using approach II, we
have applied the same methodology to pyridine analogue 48 of
varenicline in order to generalize its applicability to substitute the
pyrazine moiety of varenicline with pyridine, benzene and substitutions
on these rings.
As depicted in scheme 9, our synthesis began with the smooth
conversion of 2, 3-dimethyl pyridine 45 to its tetrabromide analogue 46
using NBS in presence of catalytic amount of benzoyl peroxide. As per
our expectation the NaI mediated Diels-Alder reaction between 43 and
2,3-bis-(dibromomethyl)pyridine 46 gave benzyl-protected varenicline 47.

Chapter 3 128
Finally benzyl group was removed by Pd/C in the presence of ammonium
formate in methanol to yield pyridine analogue 48.
Scheme 9. Reagents and conditions: (a) NBS, CCl4, hν, 16 h, 55%; (b)
NaI, DMF, 60 °C, 30 min, 65%; (c) Pd/C, HCO2NH4, MeOH, reflux for 30
min, rt., 24 h, 75%.
3.5. CONLUSIONS
In conclusion we have developed two new and efficient synthetic
routes to varenicline (approach I and II). Total synthesis of varenicline
has been achieved in a total of six steps with 10% overall yield. And this
methodology can be applied to pyrazine and pyridine analogues of
varenicline. This procedure can be scaled up for commercial use.

Chapter 3 129
3.6. EXPERIMENTAL SECTION
General procedure remains same as mentioned in Chapter 2
experimental section.
2,3-bis(dibromomethyl)pyrazine (40).
A solution of 2,3-dimethyl pyrazine 41 (15 g, 138.9 mmol) and N-bromo
succinamide (105 g, 695 mmol) in CCl4 (1 lit.) containing cat. amount
Benzoly peroxide stirred under photo light (160 W) for 14 hrs. After
completion of starting material reaction mixture was cooled to rt, the
solid was filtered and washed with CCl4. The organic layer was
evaporated and the solid washed with CH2Cl2 to afford 40. Yield: 40 g
(65%); white solid; mp 168-170 0C.
1H NMR (400 MHz, DMSO-d6): δ = 8.44 (s, 2H), 7.81 (s, 2H).
ESI-MS: m/z = 420 [M + H]+.
6,9-Dihydro-6, 9-methano-benzo[g]quinoxaline (38).
Sodium iodide was added in small portions over a period of 2 min to a
solution of the 2, 3-bis (dibromomethyl) pyrazine 40 (2.0 g, 4.71 mmol)

Chapter 3 130
and norbornadiene 39 (2.16 g, 23.58 mmol) in dry DMF (23 mL) at 60 °C.
The mixture was stirred for further 30 min. The reaction mixture was
cooled to rt, diluted with EtOAc (46 mL), and passed through a small pad
of Celite. The filtrate was washed with 10% sodium thiosulfate (3 × 20
mL), water (2 × 20 mL), and brine (1 × 20 mL). The organic layer was
dried (MgSO4) and concentrated in vacuum and the residue was purified
by flash column chromatography (silica gel, EtOAc–hexane, 10:90) to
afford 38. Yield: 420 mg (46%); white solid; mp 120-123 °C.
IR (K Br): 2519, 1708, 1649, 1215, 1182 cm-1.
1H NMR (400 MHz; DMSO-d6): δ = 8.70 (s, 2H), 7.78 (s, 2H), 6.78 (t, J =
1.7 Hz, 2H), 4.08 (t, J = 1.7 Hz, 2H), 2.45–2.43 (m, 1H), 2.32–2.30 (m,
1H).
13C NMR (50 MHz; CDCl3): δ = 153.5, 143.2, 142.5, 142.1, 120.3, 66.3,
49.6.
ESI-MS: m/z = 195 [M + H]+.
HRMS (ESI) : m/z [M + H]+ calcd for C13H11N2: 195.0922; found:
195.0931.
6,7,8,9-Tetrahydro-6,9-methano-benzo[g]quinoxaline-7,8-diol (37).
To a solution of 6, 9-Dihydro-6,9-methano-benzo[g]quinoxaline 38 (5 g,
25.7 mmol) and N-methylmorpholine N-oxide (6.1 g, 51.4 mmol) in a

Chapter 3 131
mixture of acetone (50 mL), water (25 mL) and t-BuOH (19 mL) was added
OsO4 in t-BuOH ( 1%, 6.5 mL) and the mixture was stirred at room
temperature for 16 h. Sodium sulfite (40 mg, 0.03 mmol) and florisil (6
g) was added to the reaction mixture and insoluble materials were
filtered through celite. The filtrate was basified with 6 N NaOH and
extracted with CH2Cl2 (6 × 100 mL), dried (MgSO4). After removal of the
solvent the residue was triturated with CH2Cl2 to afford pure 37. Yield:
5.2 g (90%); white solid; mp 143-145 0C.
IR (neat): 3124 (br), 2890, 1483, 1359, 1074, 956 cm–1.
1H NMR (400 MHz, DMSO-d6): δ = 8.83 (s, 2H), 7.85 (s, 2H), 5.11 (s, 2H),
3.69 (s, 2H), 3.36 (s, 2H), 2.32 (d, J = 9.4 Hz, 1H), 1.85 (dt, J = 1.6, 3.2
Hz, 1H).
13C NMR (50 MHz, DMSO-d6): δ = 148.5, 144.1, 142.3, 120.9, 70.5, 50.3,
41.8.
ESI-MS: m/z = 229 [M + H]+.
ESI-HRMS: m/z [M + H]+ calcd for C13H13N2O4: 229.0977; found:
229.0986.
PMB protected Varenicline (36).
A 0.65 M aq soln of NaIO4 (3.5 mL, 2.14 mmol) was added dropwise to a
vigorously stirred suspension of chromatography-grade silica gel (3.5 g)

Chapter 3 132
in CH2Cl2 (59 mL). After addition of 37 (350 mg, 1.53 mmol) in CH2Cl2
(78 mL) to the resulting flaky soln, stirring was continued for another 30
min and then the mixture was passed through a filter pad onto a small
amount of Na2SO4. The retained silica gel was washed with CH2Cl2 (20
mL) and the washings were pooled with filtrate. Removal of the solvent
left the dialdehyde as a solid reddish residue, which was dissolved in
methanol (8 mL). PMB amine (210 mg, 1.53 mmol) in methanol (3 mL)
followed by AcOH (9 µL, 0.15 mmol) was added to the above-mentioned
soln at 0 ºC and after stirring at rt for 10 min, Na(CN)BH3 (145 mg, 2.3
mmol) was added at 0 ºC and stirred at rt for 2 h. Methanol was
evaporated and the residue was dissolved in water (15 mL) and extracted
with EtOAc (3 × 15 mL). The combined organic layers were washed with
water (2 × 15 mL) and brine (1 ×15 mL), dried (MgSO4), and concentrated
under reduced pressure. The crude product was purified by flash
chromatography (silica gel, EtOAc– hexane, 15:85) to afford 36. Yield:
315 mg (62%); brown-colored thick liquid.
IR (Neat): 2947, 2789, 1512, 1244, 1031 cm–1.
1H NMR (400 MHz; CDCl3): δ = 8.76 (s, 2H), 7.77 (s, 2H), 6.76 (d, J = 8.8
Hz, 2H), 6.65 (d, J = 8.8 Hz, 2H), 3.72 (s, 3H), 3.41 (s, 2H), 3.35 (br s,
2H), 2.80 (d, J = 10.7 Hz, 2H), 2.54 (d, J = 10.3 Hz, 2H), 2.34–2.30 (m,
1H), 1.85 (d, J = 10.7 Hz, 1H).
13C NMR (50 MHz; CDCl3): δ = 158.4, 150.9, 143.4, 143.3, 130.0, 129.4,
120.4, 113.4, 60.9, 57.2, 55.1, 43.1, 41.2.

Chapter 3 133
ESI-MS: m/z = 332 [M + H]+.
HRMS (ESI) : m/z [M + H]+ calcd for C21H22N3O: 332.1763; found:
332.1775.
Varenicline (22).
To a solution of 36 (50 mg, 0.15 mmol) in methanol (2 mL) was added
10% Pd/C (125 mg, 2.5 equiv, wt/wt), followed by HCO2NH4 (100 mg, 2
equiv, wt/wt), and heated to reflux for 30 min. The reaction mixture was
cooled to rt, and allowed to stand for 24 h. It was filtered through a small
pad of Celite and was washed with methanol (10 mL). The filtrate was
evaporated under reduced pressure and purified by flash
chromatography (silica gel, MeOH–CHCl3, 2:98) to afford 22. Yield: 20 mg
(64%); cream-colored solid; mp 137–139 ºC
IR (K Br): 3342, 2949, 2924, 2852, 1473, 1354 cm-1.
1H NMR (400 MHz; CDCl3): δ = 8.75 (s, 2H), 7.83 (s, 2H), 3.25 (bs, 2H),
3.15 (d, J = 13.0 Hz, 2H), 2.92 (d, J = 13.0 Hz, 2H), 2.48 (m, 1H), 2.09 (d,
J = 8.8 Hz, 1H), 1.82 (br s, 1H).
13C NMR (50 MHz; CDCl3): δ= 149.6, 143.5, 143.6, 121.7, 50.5, 43.1,
42.2.
ESI-MS: m/z = 212 [M + H]+.

Chapter 3 134
HRMS (ESI) : m/z [M + H]+ calcd for C13H14N3: 212.1188; found:
212.1196.
Bicyclo[2.2.1]hept-5-ene-2,3-diol (44).
To a solution of norbornadiene 39 (5 g, 54.3 mmol) and N-
methylmorpholine N-oxide (12.7 g, 108.6 mmol) in a mixture of acetone
(65 mL), water (6.5 mL) and t-BuOH (25 mL) was added 1% OsO4 in t-
BuOH (0.14 g, 0.54 mmol) followed by catalytic amount of t-BuOOH and
the mixture was stirred at room temperature for 2 h. The reaction
mixture was quenched with saturated sodium bisulfite and extracted
with CH2Cl2 (3 × 100 mL). The combined organic extracts were washed
with water (2 × 50 mL), brine (1 × 50 mL), dried (MgSO4) and evaporated
under reduced pressure. The crude product was purified by flash column
chromatography (silica gel, EtOAc–hexane, 10:90) to afford 44. Yield: 3.8
g (55%); white solid; mp 104-106 °C.
IR (KBr): 3408, 3242, 2981, 1078 cm–1.
1H NMR (400 MHz, CDCl3): δ = 6.04 (d, J = 1.5 Hz, 2H), 3.71 (d, J = 1.5
Hz, 2H), 2.94 (br, 2H, -OH), 2.70 (d, J = 1.5 Hz, 2H), 1.89 (d, J = 9.5 Hz,
1H), 1.63 (d, J = 9.5 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ = 136.3, 68.7, 47.8, 42.1.

Chapter 3 135
ESI-MS: m/z = 127 [M + H]+.
3-Benzyl-3-aza-bicyclo[3.2.1]oct-6-ene (43).
A 0.65 M aq soln of NaIO4 (41 mL, 26.6 mmol) was added drop-wise to a
vigorously stirred suspension of chromatography-grade silica gel (26 g) in
CH2Cl2 (410 mL). After addition of 44 (2.4 g, 19.0 mmol) in CH2Cl2 (65
mL) to the resulting flaky soln, stirring was continued for another 30 min
and then the mixture was passed through a filter pad onto a small
amount of Na2SO4. The retained silica gel was washed with CH2Cl2 (100
mL) and the washings were pooled with filtrate. Removal of the solvent
left the dialdehyde as a solid reddish residue, which was dissolved in
methanol (38 mL). Benzyl amine (2.0 g, 19.0 mmol) in methanol (38 mL)
followed by AcOH (100 µL, 1.9 mmol) was added to the above-mentioned
soln at 0 ºC and after stirring at rt for 10 min, Na(CN)BH3 (1.80g, 28.56
mmol) was added at 0 ºC and stirred at rt for 2 h. Methanol was
evaporated and the residue was dissolved in water (40 mL) and extracted
with EtOAc (3 × 50 mL). The combined organic layers were washed with
water (2 × 50 mL) and brine (1 × 50 mL), dried (MgSO4), and
concentrated under reduced pressure. The crude product was purified by
flash chromatography (silica gel, EtOAc– hexane, 20:80) to afford 43.
Yield: 2.85 g (75%); colorless liquid.

Chapter 3 136
IR (neat): 2939, 2187, 1452, 1363 cm–1.
1H NMR (400 MHz, CDCl3): δ = 7.29-7.21 (m, 5H), 6.00 (s, 2H), 3.61 (s,
2H), 2.64 (dt, J = 3.0, 4.9 Hz, 2H), 2.52 (br, 2H), 2.14 (d, J = 10.1 Hz,
2H), 2.07-1.95 (m, 1H), 1.32 (d, J = 9.8 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ = 138.0, 132.9, 129.1, 128.0, 126.7, 61.8,
52.6, 44.0, 39.3.
ESI-MS: m/z = 200 [M + H]+.
ESI-HRMS: m/z [M + H]+ calcd for C14H18N: 200.1439; found: 200.1436.
Benzyl protected Varenicline (42).
Sodium iodide (900 mg, 6 mmol) was added in small portions over a
period of 2 min to a solution of the 2, 3-bis (dibromomethyl)pyrazine 40
(492 mg, 1.2 mmol) and 52 (200 mg, 1.0 mmol) in dry DMF (5 mL) at 60
°C. The mixture was stirred for further 60 min. The reaction mixture was
cooled to rt, diluted with EtOAc (20 mL), and washed with 10% sodium
thiosulfate (3 × 20 mL), water (2 × 20 mL), and brine (1 × 20 mL). The
organic layer was dried (MgSO4) and concentrated in vacuum and the
residue was purified by flash column chromatography (silica gel, EtOAc–
hexane, 20:80) to afford 42. Yield: 197 mg (65%); brown color liquid.
IR (neat): 2947, 2792, 1473, 1359 cm–1.

Chapter 3 137
1H NMR (400 MHz, CDCl3): δ = 8.76 (s, 2H), 7.78 (s, 2H), 7.10 (dd, J =
2.0, 5.0 Hz, 3H), 6.84-6.82 (m, 2H), 3.47 (s, 2H), 3.35 (t, J = 4.1 Hz, 2H),
2.99 (dt, J = 1.8, 3.7 Hz, 2H), 2.57 (d, J = 9.8 Hz, 2H), 2.36-2.31 (m, 1H),
1.86 (d, J = 10.7 Hz, 1H).
13C NMR (50 MHz, CDCl3): δ = 150.9, 143.2, 141.6, 138.1, 128.2, 127.9,
126.6, 120.3, 61.4, 57.3, 43.1, 41.2.
ESI-MS: m/z = 302 [M + H]+.
ESI-HRMS: m/z [M + H]+ calcd for C20H20N3: 302.1657; found: 302.1643.
2,3-Bis-dibromomethyl-pyridine (46).
A solution of 2,3-Dimethyl-pyridine (2 g, 18.7 mmol) and N-bromo
succinamide (16.6 g, 93.5 mmol ) in CCl4 (94 mL) containing benzoly
peroxide (cat) stirred under photo light (160 W) for 14 hrs. After
completion of starting material reaction mixture was cooled to rt., the
solid was filtered and washed with CCl4. The organic layer was
evaporated to afford 46. Yield: 4.1 g (58%); white solid;
IR (KBr): 3047, 3018, 1568, 1427 cm–1.
1H NMR (400 MHz, DMSO-d6): δ = 8.70 (dd, J = 1.5, 4.6 Hz, 1H), 8.26 (d,
J = 7.9 Hz, 1H), 7.77 (s, 1H), 7.76 (s, 1H), 7.58 (dd, J = 4.6, 7.9 Hz, 1H).
ESI-MS: m/z = 422 [M]+

Chapter 3 138
Compound (47).
Sodium iodide (750 mg, 5 mmol) was added in small portions over a
period of 2 min to a solution of the 2,3-bis(dibromomethyl)pyridine 46
(531 mg, 1.25 mmol) and 43 (250 mg, 1.25 mmol) in dry DMF (5 mL) at
60 °C. The mixture was stirred for further 60 min. The reaction mixture
was cooled to rt, diluted with EtOAc (20 mL), and washed with 10%
sodium thiosulfate (3 × 20 mL), water (2 × 20 mL), and brine (1 × 20 mL).
The organic layer was dried (MgSO4) and concentrated in vacuum and
the residue was purified by flash column chromatography (silica gel,
EtOAc–hexane, 20:80) to afford 47. Yield: 245 mg (65%); brown color
liquid.
IR (neat): 2945, 2792, 1452 cm–1.
1H NMR (400 MHz, CDCl3): δ = 8.83 (dd, J = 1.8, 4.4 Hz, 1H), 8.10 (dd, J
= 1.5, 8.4 Hz, 1H), 7.80 (s, 1H), 7.50 (s, 1H), 7.33 (dd, J = 4.4, 8.4 Hz,
1H), 7.10-7.09 (m, 3H), 6.85-6.83 (m, 2H), 3.47 (s, 2H), 3.30 (dd, J = 4.0,
19.4 Hz, 2H), 3.00-2.92 (m, 2H), 2.54 (d, J = 9.9 Hz, 2H), 1.82 (d, J =
10.6 Hz, 1H), 2.33-2.27 (m, 1H).
13C NMR (100 MHz, CDCl3): δ = 105.0, 148.7, 148.6, 146.5, 138.3,
135.7, 128.3, 127.9, 126.5, 120.6, 119.9, 118.6, 61.5, 57.4, 43.2, 41.3,
40.9.

Chapter 3 139
ESI-MS: m/z = 301 [M + H]+
ESI-HRMS: m/z [M + H]+ calcd for C21H21N2: 301.1705; found: 301.1701.
Pyridine analogue of varenicline (48).
To a solution of 47 (25 mg, 0.083 mmol) in methanol (2 mL) was added
10% Pd/C (63 mg, 2.5 equiv, wt/wt), followed by HCO2NH4 (50 mg, 2
equiv, wt/wt), and heated to reflux for 30 min. The reaction mixture was
cooled to rt, and allowed to stand for 24 h. It was filtered through a small
pad of Celite and was washed with methanol (10 mL). The filtrate was
evaporated under reduced pressure and purified by flash
chromatography (Silica gel, MeOH–CHCl3, 2:98) to afford 48. Yield: 18
mg (65%); brownish gummy material.
IR (neat): 2956, 2926, 1030 cm–1.
1H NMR (400 MHz, MeOH-d4): δ = 8.83 (dd, J = 1.5, 4.4 Hz, 1H), 8.38 (d,
J = 7.7 Hz, 1H), 8.00 (s, 1H), 7.94 (s, 1H), 7.54 (dd, J = 4.4, 8.0 Hz, 1H),
3.60 (d, J = 13.2 Hz, 2H), 3.49 (dt, J = 2.0, 3.8 Hz, 2H), 3.25-3.24 (m,
2H), 2.48-2.44 (m, 1H), 2.24 (d, J = 11.3 Hz, 1H).
ESI-MS: m/z = 211 [M + H]+.
ESI-HRMS: m/z [M + H]+ calcd for C14H15N2: 211.1235; found: 211.1228.

Chapter 3 140
3.7. SPECTRAL DATA (1H and 13C NMR)
1H NMR of Compound 40
1H NMR of Compound 46

Chapter 3 141
1H and 13C NMR of Compound 38

Chapter 3 142
1H and 13C NMR of Compound 37
Chapter 3 143
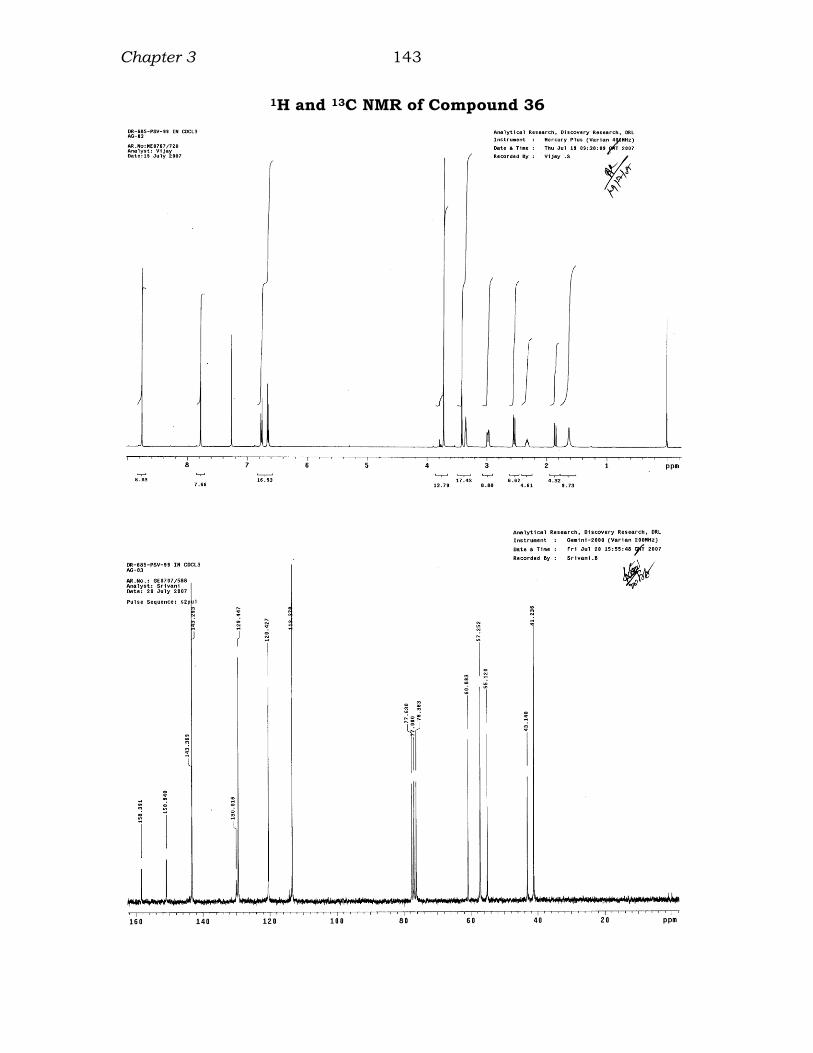
1H and 13C NMR of Compound 36

Chapter 3 144
1H and 13C NMR of Compound 22
Chapter 3 145
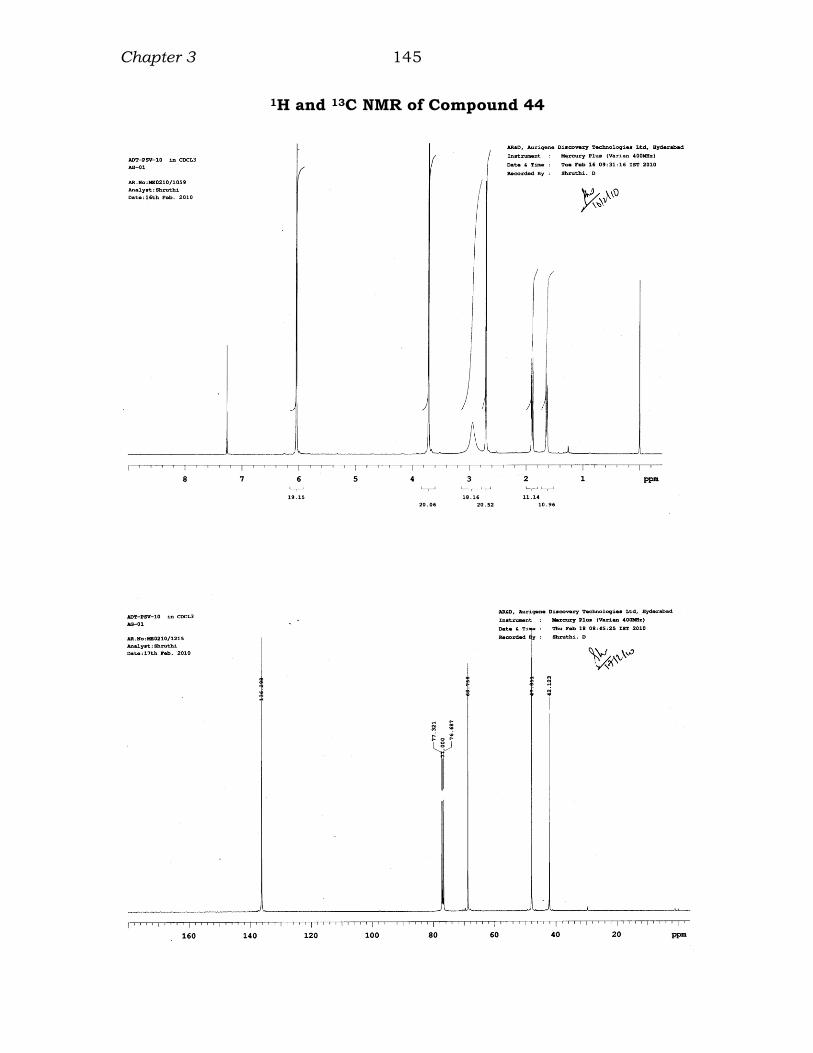
1H and 13C NMR of Compound 44

Chapter 3 146
1H and 13C NMR of Compound 43

Chapter 3 147
1H and 13C NMR of Compound 42

Chapter 3 148
1H and 13C NMR of Compound 47

Chapter 3 149
3.8. REFERENCES
1. Russell, M. A. Br J Med Psychol 1971, 1, 44.
2. Department of Health. Smoking kills: a white paper on tobacco:
London UK Government Report. London: The Stationery Office;
1998.
3. World Health Organization. Tobacco or health: a global status
report. Geneva: World Health Organization; 1997.
4. World Health Organization. The European report on tobacco
control policy. WHO European Ministerial Conference for a
Tobacco-Free Europe. Warsaw: World Health Organization; 2002
(February 18–19).
5. Hogg, R. C.; Raggenbass, M.; Bertrand, D. Rev Physiol Biochem
Pharmacol 2003, 147, 1–46.
6. Gotti, C.; Clementi, F. Prog Neurobiol 2004, 74, 363–96.
7. Groot-Kormelink, P. J.; Boorman, J. P.; Sivilotti, L. G. Br J
Pharmacol 2001, 134, 789–96.
8. Boorman, J. P.; Groot-Kormelink, P. J.; Sivilotti, L. G. J Physiol
2000, 529 (Part 3), 565–77.
9. Groot-Kormelink, P. J.; Luyten, W. H.; Colquhoun, D.; Sivilotti, L.
G. J Biol Chem 1998, 273, 15317–20.
10. Wang, F.; Gerzanich, V.; Wells, G. B.; Anand, R.; Peng, X.; Keyser,
K.; et al. J Biol Chem 1996, 271, 17656–65.

Chapter 3 150
11. Decker, M. W.; Meyer, M. D.; Sullivan, J. P. Expert Opin Investig
Drugs 2001, 10, 1819–30.
12. Zhang, X.; Xiao, H. S. Curr Opin Mol Ther 2005, 7, 532–7.
13. Vincler, M. Expert Opin Investig Drugs 2005, 14, 1191–8.
14. Benowitz, N. L. Prim Care. 1999, 26, 611-31.
15. George, T. P.; O‟ Malley, S. S. Trends Pharmacol Sci. 2004, 25, 42-
8.
16. (a) Coe, J. W.; Brooks, P. R.; Vetelino, M. G.; and et al. J Med
Chem. 2005, 48, 3474-7. (b) Brooks, P. R.; Caron, S.; Coe, J. W.;
and et al. Synthesis 2004, 1755-1758.
17. Harris, D. S.; Anthenelli, R. M. Curr Psychiatry Rep. 2005, 7, 344-
51.
18. Hughes, J.; Hatsukami, D. K. Tob Control. 1998, 7, 92-3.
19. Hughes, J. R. J Consult Clin Psychol 1993, 61, 751–60.
20. Lerman, C.; Patterson, F.; Berrettini, W. J Clin Oncol 2005, 23,
311–323.
21. Benowitz, N. L. Annu Rev Pharmacol Toxicol 1996, 36, 597–613.
22. Benowitz, N. L. Prim Care 1999, 26, 611–31.
23. Rennard, S. I.; Daughton, D. M. Chest 2000, 117, 360.
24. Henningfield, J. E. N Engl J Med 1995, 333, 1196–203.
25. Okuyemi, K. S.; Ahluwalia, J. S.; Harris, K. J. Arch Fam Med 2000,
9, 270–81.

Chapter 3 151
26. Hughes, J. R.; Stead, L. F.; Lancaster, T. Cochrane Database Syst
Rev 2003, CD000031.
27. Jorenby, D. E. et al. N Engl J Med 1999, 340, 685–91.
28. Ascher, J. A.; et al. J Clin Psychiatry 1995, 56, 395–401.
29. Coe, J. W.; Brooks, P. R.; Wirtz, M. C.; and et al. Bioorg. Med.
Chem. Lett. 2005, 15, 4889-4897.
30. Mehta, G.; Reddy, D. S. Chem. Commun. 1999, 21, 2193-2194.
31. Chou, T.-C.; Liao, K.-C.; Lin, J.-J. Org. Lett. 2005, 7, 4843.
32. Shepherd, M. K. J. Chem. Soc., Perkin Trans. 1 1986, 1495.
33. Diaz-Ortiz, A.; De la Hoz, A.; Moreno, A.; Prieto, P.; Leon, R.;
Herrero, M. A. Synlett 2002, 2037–2038.
34. Kobayashi, T.; Kobayashi, S. Molecules 2000, 5, 1062–1067.
35. Perez-Castro, I.; Caamano, O.; Gracia, M. D.; Fernadez, F.; Lopez,
C. Synthesis 2007, 9, 1385–1391.
36. Luthje, S.; Bornholdt, C.; Luning, U. Eur. J. Org. Chem. 2006, 909-
915.
37. Sano, H.; Sugai, S. Tetrahedron 1995, 51, 4635-4646.





























