Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1993 by The American Society for Biochemistry and Molecular Biology, Inc. Vol. 268, No. 2, Issue of January 15, pp. 1462-1469,1993
Printed in U.S.A.
Characterization of the Forward and Reverse Integration Reactions of the Moloney Murine Leukemia Virus Integrase Protein Purified from Escherichia coli*
(Received for publication, May 19, 1992)
Colleen B. Jonsson, George A. Donzella, and Monica J. RothS From the Department of Biochemistry, University of Medicine and Dentistry of New Jersey, Robert Wood Johnson Medical School, Piscataway, New Jersey 08854
The forward and reverse reactions for integration were characterized for the Moloney murine leukemia virus integrase (M-MuLV IN) protein. The M-MuLV IN was recombinantly produced in Escherichia coli, and was purified to greater than 90% homogeneity by a one-step affinity purification scheme. M-MuLV IN was highly active for integration as measured by in vitro cleavage and strand transfer assays. Further- more, the integration of a model viral substrate into X concatamers by IN correctly produced the flanking 4- base pair duplications characteristic of M-MuLV IN. The reverse reaction of integration, disintegration, was also catalyzed by the recombinant M-MuLV IN. Two products were generated, a 3”recessed long ter- minal repeat and a ligated target DNA, from a model integration-intermediate substrate in the presence of M-MuLV IN. The requirements and optimal conditions for maximal integration and disintegration activity for M-MuLV IN were determined. The forward and re- verse reactions required different concentrations of manganese ion and reductant. Salt was also titrated for the forward and reverse reactions. Sodium chloride inhibited integration, but had little affect on disinte- gration. Low concentrations of potassium chloride en- hanced integration, but had no affect on disintegration. The dinucleotide cleavage, strand transfer, and the disintegration reactions each had a unique pH profile of activity.
Retroviral integration is a multi-step biochemical process that requires only one enzyme, the viral encoded integrase (IN),’ and the reverse transcribed retroviral DNA (for review, see Refs. 1-4). The ends of the linear double stranded viral DNA contain a long terminal repeat (LTR) that is necessary for integration (5). I n uivo studies of the Moloney murine
leukemia virus (M-MuLV) integration pathway show that only the conserved terminal 13 bp of each viral LTR is required for recognition as a substrate for integration by IN. The most conserved feature of retroviral termini is the dinu- cleotide sequence, 5‘. . . .CA..3‘, located two nucleotides from the 3’-end. The terminal two nucleotides 3’ to the CA at the end of each LTR are removed by IN prior to integration in uivo (6), and in uitro (7-9). The 3’-ends of each recessed strand subsequently undergo a strand transfer reaction to the 5‘-end of the host DNA (10-15). Integration of M-MuLV results in a gap of four nucleotides on each host DNA strand. The repair of the gap, presumably by the host cell, generates a direct duplication of the host DNA flanking the proviral DNA. Cleavage of the terminal two nucleotides is proposed to occur by a single nucleophilic attack (16, 17), and strand transfer by a one-step transesterification (17) in the absence of an exogenous energy source.
Recent studies of the HIV-1 integrase protein have shown the integration reaction is reversible (18). The reverse reac- tion of integration, or disintegration, for HIV-1 IN generates two major products as revealed upon incubation with an oligonucleotide substrate that resembles an intermediate structure for one processed LTR end (..CA-3’) joined to a host target DNA. The two enzymatic activities of HIV-1 IN disintegration are an endonuclease activity at the LTR-host DNA junction producing a 3”recessed LTR, and a ligation activity of a nick between the 3’- and 5”junction of the host DNA.
The M-MuLV IN is transcribed and translated as a gag-pol fusion protein (1). The pol portion of the peptide is processed during virion assembly to form a 14-kDa protease, a 77-kDa reverse transcriptase, and the 46-kDa IN protein. The inte- grase protein among related retroviruses share sequence ho- mology, yet the processing of the pol polypeptide varies. The human immunodeficiency virus (HIV-1 and HIV-2) (9, 19)
* This work was supported by National Science Foundation Grant and the avian sarcoma-leukosis virus (20) integrases have
DMB-9105091. The costs of publication of this article were defrayed both been expressed and purified from Esche- in part by the payment of page charges. This article must therefore richia Coli expression systems. These Proteins are active for be herebv marked “aduertisenent” in accordance with 18 U.S.C. cleavage and strand transfer reactions characteristic of the Section y734 solely to indicate this fact. integration process. M-MuLV IN purified from a Baculouirus
correspondence should be addressed. Tel.: 908-463-5048; Fax: 908- -? Scholar of the Leukemia Society of America, Inc. To whom expression system is also active for the integration reactions
(8). M-MuLV IN protein has been expressed and purified 1 The abbreviations used are: IN, integrase; “ M u ~ V , Moloney from E. coli previously, but it retained only nonspecific DNA
murine leukemia virus; HIV-1, human immunodeficiency virus type binding activity (21). We Present a One-steP affinity Purifi- 1; HIV-2, human immunodeficiency virus type 2; LTR, long terminal cation of M-MuLV IN that was expressed in E. coli. Histidine repeat; DTT, dithiothreitol; bp, base pair; Ni*+-NTA, nickel nitrilo- residues were introduced at the carboxyl terminus of IN, triacetate; HEPES, 4-(2-hydrox~eth~l)-l-~i~erazinediethanesu~f~~~~ which for selective retention on a nickel affinity resin. acid Mes, 4-morpholineethanesulfonic acid; PIPES, 1,4-piperazine-
lammonio]-l-propane-1,3-diol; IPTG, isopropyl-l-thio-P-D-ga]acto- integration, and the disintegration reaction. Disintegration pyranoside; PAGE, polyacrylamide gel electrophoresis. differed from integration in its requirements for maximal
463-4783.
diethanesulfonic acid; CHAPS, 3-[(3-~holamidopropyl)dimethy- The purified M“uLV integrase was active for the
1462

Purification and Characterization of M-MuLV Integrase 1463
activity. The integration and disintegration reactions for the M-MuLV integrase were characterized and the distinguishing features of each reaction are presented.
EXPERIMENTAL PROCEDURES
Materials-Crude [y-3ZP]ATP was purchased from ICN (Irvine, CA); [ c ~ - ~ ~ S ] ~ A T P and X packaging extracts were purchased from Amersham. Urea was purchased from Bio-Rad.
Construction of a M-MuLV IN Expression Vector-An NdeI site was incorporated 5’ of the region encoding IN by a Taq Polymerase (Perkin Elmer, Norwalk, CT) catalyzed polymerase chain reaction (Biocycler, BIOS, New Haven, CT) using the primer 5”GGCATAT- GATAGAAAATTCATCACCC-3’ and a 3’-end primer of sequence (5’-GGATCCAGTACTGACCCCTCTG-3’) on the template plasmid p150-18 (21). The 1260-bp product of the polymerase chain reaction amplification was gel isolated, and then digested with NdeI and HindIII. The resulting 290-bp NdeI-Hind111 fragment was gel isolated using glass powder (22). The 3’ terminus of IN was isolated from pRC65 (Dr. Robert Craigie, National Institutes of Health), a subclone of pMK556 (8) in which a BamHI linker was bridged to the ScaI site at position 6322 of M-MuLV. pRC65 was digested with HindIII and BamHI and the 970-bp fragment isolated. The two IN encoding fragments were directionally cloned into pETl lC (Novagene, Madi- son, WI) digested with NdeI and BamHI, producing pETIN. The region between NdeI and HindIII resulting from the polymerase chain reaction amplification was sequenced and no nucleotide substitutions were detected. The histidine tagged IN expression vector, pETINH1, was made by partial digestion of pETIN with HpaI and subsequent
dines (5’-ACATCACCATCACCATCACTAGTA-3’, 5”TACTAGT- ligation of two complimentary oligonucleotides encoding six histi-
GATGGTGATGGTGATGT-3’). The HpaI site was destroyed upon successful ligation of the oligonucleotides and thus facilitated rapid screening of transformants. The orientation of the insertion was determined by dideoxy sequencing (23).
Protein Purification-Two liters of E. coli HMS174DE3 (Nova- gene) cells that harbored either pETl lC or pETINHl were grown to 0.7-1.0 Am units, induced with 1 mM isopropyl-1-thio-@-D-galacto- pyranoside (IPTG), and grown an additional 3 h prior to harvesting cells. The cells were solubilized in 50 ml of Buffer A (10 mM Tris base, 0.1 M Na2HPO4, 0.1% Nonidet P-40, 10 mM @-mercaptoethanol, 100 mM NaC1, 10% glycerol, 4 M urea)/pH 7.8, with slow shaking for 1 h a t room temperature. The slurry was centrifuged a t 12,000 X g for 10 min, and the supernatant slowly applied to a 1-ml nickel nitrilotriacetate affinity column (Ni*’-NTA, Qiagen, Chatsworth, CA) pre-equilibrated with Buffer A/pH 7.8, at 4 “C. All manipulations from this point were performed a t 4 “C. The column was washed with 10 column volumes of Buffer A/pH 7.8, followed by 10 column volumes of Buffer A/pH 6.3, to remove the majority of background E. coli proteins prior to elution of IN. These steps were followed by 10 column volumes of Buffer A/pH 5.9, and 10 column volumes of Buffer A/pH 4.5. IN protein eluted after 1 column volume of Buffer A/pH 4.5. Protein concentration was measured by the method of Bradford (24) (Bio-Rad).
An alternative protocol, purification B, was developed which sub- stituted the @-mercaptoethanol in Buffer A with dithiothreitol, des- ignated Buffer B. A 10-ml Ni2+-NTA column was developed as above with stepwise pH elutions after the application of crude extracts of p E T l l C or pETINH1. The majority of the E. coli proteins eluted in the first wash with Buffer B/pH 7.8. IN protein eluted throughout the first 6 column volumes of the Buffer B/pH 6.3 elution step, and the first four fractions of the Buffer B/pH 5.9 elution step. No IN protein was eluted in the Buffer B/pH 4.5 step. In all purifications, column fractions were slowly renatured by a 24-h step dialysis from 3 to 0 M urea in 2 liters of 20 mM HEPES, pH 7.4,l mM dithiothreitol (DTT), 20% glycerol, 0.1% Nonidet P-40, 400 mM monopotassium glutamate over 4 days. Fractions were frozen in liquid nitrogen and stored at -70 “C. A specific activity of 2.2 units/mg was calculated for the pH 5.9 fraction purified in purification B. One unit is defined as 1 pmol of U5 substrate converted to autointegrants/min.
SDS-PAGE, Silver Stain, and Western Analysis-Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western analysis with polyclonal antibodies made to IN were performed as previously described (25). Silver stain plus (Bio-Rad) was used as specified by the manufacturer.
Phosphorlmager Analysis-PhosphorImager screens were exposed to denaturing polyacrylamide gels containing integration or disinte- gration reaction products for 30 min to 1 h and scanned with a
Molecular Dynamics PhosphorImager (Sunnyvale, CA.). The scanned gels were quantitated with ImageQuant 3.15 software (Molecular Dynamics).
Oligonucleotide Substrate Preparation-DNA oligonucleotides were made on an Applied Biosystems Model 380B DNA Synthesizer by the UMDNJ Biochemistry Department DNA synthesis facility. Oli- gonucleotides were purified on 20% denaturing polyacrylamide gels, and eluted from the gel overnight in 0.5 M ammonium acetate, 10 mM magnesium acetate a t 37 “C. Synthetic U5 LTR substrates for cleav- age and strand transfer were prepared by end labeling the 5’-end of oligonucleotides 2783 (5”GTCAGCGGGGGTCTTTCATT) or 2784 (5”GTCAGCGGGGGTCTTTCA) with [y3*P]ATP using T4 poly- nucleotide kinase. Reactions were stopped with 33 mM EDTA, and heat inactivated for 5 min a t 90 “C. The labeled strands were then hybridized with a 5-fold molar excess of 2785 (5”AATGAAA- GACCCCCGCTGAC) in 100 mM NaCl to produce blunt (2783/2785) or precleaved (2784/2785) substrates for integration assays. Unincor- porated label was removed by G-25 Quick Spin columns (Boehringer Mannheim).
The disintegration substrate (Y-oligomer) was prepared by labeling the 5’-end (as above) of 3152 (5”CAGCAACGCAAGCTTG) and hybridizing with a 5-fold molar excess of 3154 (5”TAGTCAGCGG-
GCCCAAGCTTGCGTTGCTG), and 3192 (5”AATGAAAGACCC- CGCTGA) in 100 mM NaCl at 80 “C for 20 min, 70 “C for 30 min, 60 “C for 15 min, 55 “C for 15 min, 50 “C for 15 min, 40 “C for 20 min, and 30 “C for 15 min. The Y-oligomer was gel purified on a 15% nondenaturing polyacrylamide gel as described by Chow et al. (18).
Integration and Disintegration Assays-Standard integration re- actions (15 pl) contained 20 mM PIPES, pH 6.8,20 mM DTT, 20 mM manganese chloride (MnCL), 1 pmol of the U5 blunt or precleaved substrates, and 1.3-8.7 pmol of IN. Ten pl of stop buffer (20 mM EDTA, 95% formamide, 0.5% bromphenol blue, and xylene cyanol) was added after reactions were incubated a t 30 “C for 1 h. Titrations of pH were performed with 20 mM sodium acetate/acetic acid buffer from pH 3.6 to 5.6, 20 mM MES from pH 5.8 to 6.4, 20 mM PIPES from pH 6.6 to 7.0, and 20 mM HEPES from pH 7.2 to 8.0. Disinte- gration reactions were performed at 30 “C for 1 h in a 15-pl volume containing 1 pmol of substrate, 20 mM PIPES, pH 6.8, 1 mM DTT, 50 mM NaC1,50 mM MnCIz, 7 mM CHAPS, 0.05% Nonidet P-40, and 1-5 pmol of IN protein. The reactions were terminated as described by Chow et al. (18). X-ray film (Kodak X-Omat AR, Eastman-Kodak Co.) was exposed to dried gels at -70 “C.
Biological Integration Assay-Integration of Mini-Mo (NdeI line- arized vector, pMK471) into Xbs1sbs’9 DNA by M-MuLV IN was performed essentially as described by Fujiwara and Craigie (12). Briefly, standard reactions were preincubated on ice for 1 h and then incubated for 30 min at 30 “C in a 50-111 final volume containing 20 mM HEPES, pH 7.4, 10 mM MnCI2, 10 mM DTT, 0.2% Nonidet P- 40, 5 mM MgCI2, 120 mM monopotassium glutamate, 10% dimethyl sulfoxide, 5% polyethylene glycol, 0.5 pg of pMK471 linearized with NdeI, 1.5 pg of X concatamer, 1 pg of RNase A, 120 ng of HU protein, and 17.2 pmol of IN. Integration reactions were packaged as specified by Amersham.
GGTCTTTCAGGCTGCAGGTCGAC), 3153 (5”GTCGACCTGCA-
RESULTS
Expression of M-MuLV IN Protein in E. coli-The scheme for expression of M-MuLV IN protein is shown in Fig. 1. The 3’ terminus of the pol gene encoding IN was cloned under the control of the T7lac promoter of the E. coli expression vector, pETl lC (Fig. 1) (26). Previous work in our laboratory found that the carboxyl-terminal 28 amino acid residues of M- MuLV IN are not necessary for activity in uiuo (27). The DNA sequence corresponding to 6 histidine residues was introduced at the carboxyl region, replacing the DNA se- quence of the terminal 5 amino acids of wild type IN. Two to five percent of the total protein produced in E. coli, HMS174DE3/pETINHl, was IN, of which, approximately 10% was solubilized by 4 M urea. The length of induction with IPTG was limited to 3 h to avoid proteolysis of IN.
Purification of M-MuLV IN by Nickel Nitrilotriacetate Af- finity Chromatography-M-MuLV IN with 6 histidines at the carboxyl terminus was extracted from HMS1’74DE31 pETINHl with 4 M urea (Buffer A/pH 7.8) and applied to a
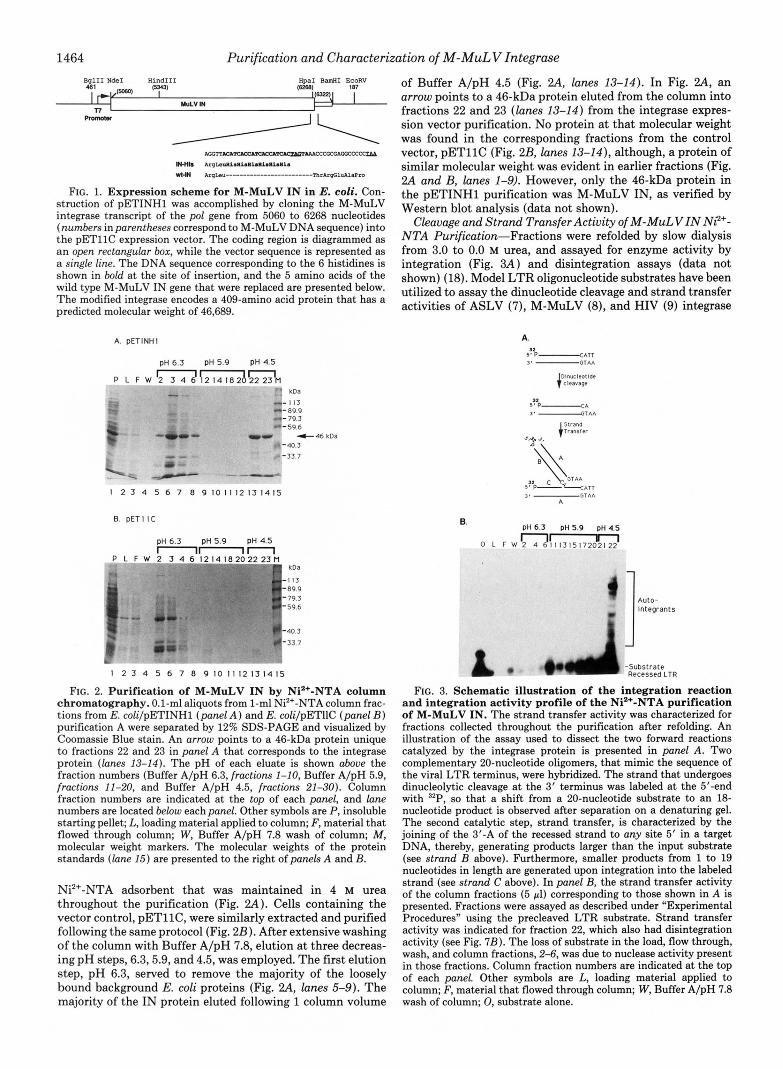
1464 Purification and Characterization of M-MuLV Integrase BglII N d e I Hind111 161
HpaI BaraHI EcoRV 1 87
Romolr TI
I I
/\ A G G m A u ~ A M c c c s x A "
.eM. ""U.U. wt-nl ArgUu""""-""mr~luu*P~
FIG. 1. Expression scheme for M-MuLV IN in E. coli. Con- struction of pETINHl was accomplished by cloning the M-MuLV integrase transcript of the pol gene from 5060 to 6268 nucleotides (numbers in parentheses correspond to M-MuLV DNA sequence) into the pETllC expression vector. The coding region is diagrammed as a n open rectangular box, while the vector sequence is represented as a single line. The DNA sequence corresponding to the 6 histidines is shown in bold at the site of insertion, and the 5 amino acids of the wild type M-MuLV IN gene that were replaced are presented below. The modified integrase encodes a 409-amino acid protein that has a predicted molecular weight of 46,689.
A. PETINHI
pH 6.3 pH 5.9 pH 4.5 "-
kDa
89.9 I13
59.6 79.3
6 4 6 koa 40.3 33.7
1 2 3 4 5 6 7 8 9 1011 1213 1415
E. pETl IC
pH 6.3 pH 5.9 pH 4.5 "n
P L F W 2 3 4 6 1 2 1 4 1 S 2 0 2 2 2 3 M '.*q .\" e kDa
I 89.9 I I 3
59.6 79.3
40.3
33.7 4 L
I 2 3 4 5 6 7 8 9 IO 1112131415
FIG. 2. Purification of M-MuLV IN by Ni2+-NTA column chromatography. 0.1-ml aliquots from 1-ml Ni*+-NTA column frac- tions from E. coli/pETINHl (panel A ) and E. coli/pETIIC (panel B ) purification A were separated by 12% SDS-PAGE and visualized by Coomassie Blue stain. An arrow points to a 46-kDa protein unique to fractions 22 and 23 in panel A that corresponds to the integrase protein (lanes 13-14). The pH of each eluate is shown above the fraction numbers (Buffer A/pH 6.3, fractions 1-10, Buffer A/pH 5.9, fractions 11-20, and Buffer A/pH 4.5, fractions 21-30). Column fraction numbers are indicated at the top of each panel, and lane numbers are located below each panel. Other symbols are P, insoluble starting pellet; L, loading material applied to column; F, material that flowed through column; W, Buffer A/pH 7.8 wash of column; M, molecular weight markers. The molecular weights of the protein standards (lane 15) are presented to the right of panels A and B.
Ni2+-NTA adsorbent that was maintained in 4 M urea throughout the purification (Fig. 2A). Cells containing the vector control, pETllC, were similarly extracted and purified following the same protocol (Fig. 2B). After extensive washing of the column with Buffer A/pH 7.8, elution at three decreas- ing pH steps, 6.3,5.9, and 4.5, was employed. The first elution step, pH 6.3, served to remove the majority of the loosely bound background E. coli proteins (Fig. ?.A, lanes 5-9). The majority of the IN protein eluted following 1 column volume
of Buffer A/pH 4.5 (Fig. 2A, lanes 13-14). In Fig. 2A, an arrow points to a 46-kDa protein eluted from the column into fractions 22 and 23 (lanes 13-14) from the integrase expres- sion vector purification. No protein at that molecular weight was found in the corresponding fractions from the control vector, pETllC (Fig. 2B, lanes 13-14), although, a protein of similar molecular weight was evident in earlier fractions (Fig. 2A and B, lanes 1-9). However, only the 46-kDa protein in the pETINHl purification was M-MuLV IN, as verified by Western blot analysis (data not shown).
Cleavage and Strand Transfer Activity of M-MuLVIN N?+- NTA Purification-Fractions were refolded by slow dialysis from 3.0 to 0.0 M urea, and assayed for enzyme activity by integration (Fig. 3A) and disintegration assays (data not shown) (18). Model LTR oligonucleotide substrates have been utilized to assay the dinucleotide cleavage and strand transfer activities of ASLV (7), M-MuLV (8), and HIV (9) integrase
A.
6. pH 6.3 DH 5.9 pH 4.5
Auto- tntegrants
ubstrate ecessedLTR
FIG. 3. Schematic illustration of the integration reaction and integration activity profile of the Ni2+-NTA purification of M-MuLV IN. The strand transfer activity was characterized for fractions collected throughout the purification after refolding. An illustration of the assay used to dissect the two forward reactions catalyzed by the integrase protein is presented in panel A. Two complementary 20-nucleotide oligomers, that mimic the sequence of the viral LTR terminus, were hybridized. The strand that undergoes dinucleolytic cleavage at the 3' terminus was labeled at the 5'-end with 32P, so that a shift from a 20-nucleotide substrate to an 18- nucleotide product is observed after separation on a denaturing gel. The second catalytic step, strand transfer, is characterized by the joining of the 3'-A of the recessed strand to any site 5' in a target DNA, thereby, generating products larger than the input substrate (see strand B above). Furthermore, smaller products from 1 to 19 nucleotides in length are generated upon integration into the labeled strand (see strand C above). In panel B, the strand transfer activity of the column fractions (5 pl) corresponding to those shown in A is presented. Fractions were assayed as described under "Experimental Procedures" using the precleaved LTR substrate. Strand transfer activity was indicated for fraction 22, which also had disintegration activity (see Fig. 7B). The loss of substrate in the load, flow through, wash, and column fractions, 2-6, was due to nuclease activity present in those fractions. Column fraction numbers are indicated at the top of each panel. Other symbols are L, loading material applied to column; F, material that flowed through column; W, Buffer A/pH 7.8 wash of column; 0, substrate alone.

Purification and Characterization of M-MuLV Integrase 1465
proteins. In our assays, the terminal 20-bp sequence of the M-MuLV U5 LTR also served as a model oligonucleotide substrate to analyze the 3"dinucleotide cleavage activity of IN, the first catalytic step in the integration reaction (Fig. 3A). The model substrate used for the strand transfer reaction is identical in size and sequence to the U5 blunt-ended sub- strate, except for the absence of the two terminal nucleotides proximal to the CA-3'-dinucleotide. Strand transfer activity is thus characterized by the autointegration of the recessed substrate in the presence of IN.
The column profile of the integration activity for the pETINHl purification presented in Fig. 2A is shown in Fig 3B. The substrate was 32P-labeled at the 5'-end of the recessed strand. Strand transfer products (autointegrants) were de- tected in fractions 22 comprising the pH 4.5 step. The 46-kDa protein in fraction 22 (Fig. 2, panel A ) copurified with the integration activity, and was also active for disintegration (see below). Fraction 23 was also active in the integration and disintegration assays (data not shown).
Silver Stain and Western Blot Analysis-The purity of the IN fraction obtained from the pH 4.5 elution step was ana- lyzed by silver staining and Western blot analysis (Fig. 4). In Fig. 4A, a silver stain analysis of fraction 22 from pETllC (lane 1) and pETINHl (lane 2) purifications is shown. No predominant protein species was observed in the control fraction, pETllC (lane 1 ). A major band corresponding to a protein with a molecular mass of 46 kDa was observed only in the pETINHl fraction, Fig. 4A (lane 2). Analysis of a
A. 6. 1 2 1 2 3
koa koa
.-"-I13 y - * ,
1-1 I S "899 ! -97 .-s6
"793
.I C 4 6 k D a "596
"45 %-"I
.L -33 7
-29
"
- . ~-
C. 0.
1 2 .I 2 -3 P"! koa koa - 116 - 97 n-116
-66 3- :: ". ". -45 - 6 4 4 6 k D a
- 29 0 - 29 - - FIG. 4. Silver stain and Western analysis of M-MuLV IN
purified from E. coZi/pETINHl by Ni2+-NTA column chro- matography. In panels A and B, a 0.1-ml aliquot of the peak IN fraction (12.5 pg) and the corresponding pETllC fraction from purification scheme A (described under "Experimental Procedures") were separated by 12% SDS-PAGE and subjected to silver stain and Western analysis. Panel A shows a silver stained SDS-PAGE of fraction number 22 from the vector control, pETllC (lane I ) , and IN from the pH 4.5 elution (lane 2). Panel B presents a Western blot analysis of fraction number 22 for the vector control, pETllC (lane 1 ), and pETINHl (lane 2) purifications from the pH 4.5 elution. In panels C and D, the peak eluates of IN from purification scheme B for pH steps, pH 5.9 and 6.3, were subjected to silver stain and Western blot analysis. Panel C is a silver stained SDS-PAGE of the pH 6.3 (lane I ) and 5.9 (lane 2 ) eluates from pETINHl purifications. Panel D is a Western blot analysis of the pH 6.3 (lane 1 ) and 5.9 (lane 2) eluates from a pETINHl purification. Prestained molecular weight markers (Sigma) were run in lane 3 of panels B and D. The molecular weight for each protein standard is presented to the right of each panel.
subsequent purification of integrase on a 17.5% SDS-PAGE gel revealed a 29-kDa protein as a minor contaminant (data not shown). Similarly, a Western blot analysis of fractions 22 from pETllC and pETINHl purifications only detected one major band at 46 kDa that reacted with antibodies raised to M-MuLV integrase (Fig. 4B, lane 2), and no detectable bands in the control parent vector (Fig. 4B, lane I ) . Minor back- ground bands observed at the dye front also reacted to the I'd- MuLV IN antibody and are presumably proteolytic fragments of IN (Fig. 4B, lane 2).
Purification of M-MuLV IN by Nickel Nitrilotriacetate Af- finity Chromatography in the Presence of DTT-Substitution of P-mercaptoethanol in Buffer A with dithiothreitol, desig- nated Buffer B, provided an alternate outcome in the purifi- cation (purification B). The column was developed as above with stepwise pH elutions of column applied material. The binding affinity of the resin is decreased in the presence of DTT? Therefore, the E. coli proteins elute at pH 7.8, while the IN protein eluted at the Buffer B/pH 6.3 and the Buffer B/pH 5.9 elution steps (data not shown). No IN protein was eluted during the Buffer B/pH 4.5 step. Silver stain analysis of the peak fractions containing IN from the pH 6.3 and pH 5.9 elution steps indicated that a 46-kDa protein was the major component (Fig. 4C). In Fig. 40, a Western blot analy- sis of the fractions from pH 6.3 (lane I ) and 5.9 (lane 2) demonstrated that the 46-kDa protein (indicated by an arrow) reacted with antibody raised to M-MuLV integrase. A com- parison of the integration and disintegration activity of the pH 5.9 fraction to the pH 4.5 fraction found a similar range of specific activity, while the pH 6.3 fraction had a slightly reduced activity (see below).
Requirements for Nucleolytic Cleavage and Strand Transfer for M-MuLV IN-The affect of divalent cations, salt, reduc- ing agent, and pH on the integration activity of the M-MuLV IN purified from E. coli was examined. In Fig. 5, panels A-C, only the strand transfer activity is presented, however, similar requirements were observed for the dinucleotide cleavage event. A notable exception was found in the case of pH optimum, therefore, both activities are presented in Fig. 5, panel D. Manganese was required for integration activity, as has been demonstrated from M-MuLV IN from Baculouirus (8). The optimal concentration of manganese for the E. coli purified M-MuLV IN was from 5 to 10 mM for autointegration (Fig. 5A, lanes 3-4), and cleavage activities (data not shown). Magnesium-dependent cleavage activity has been reported for the avian sarcoma-leukosis virus IN purified from E. coli (7), although the efficiency of strand transfer for the avian sar- coma-leukosis virus IN is reduced in the presence of magne- sium as opposed to manganese. The cell-free viral systems are also able to catalyze the strand transfer reaction with mag- nesium ion (10, 12). No cleavage or strand transfer activity for M-MuLV IN was detected in the presence of 1-50 mM magnesium or 50 mM EDTA (data not shown).
The effect of NaCl on the integration activity was studied. Maximal activity was observed in the absence of NaCl (Fig. 5B, lane 2). NaCl concentration as low as 25 mM diminished strand transfer activity (Fig. 5B, lane 3 ) ) while no activity could be detected in 100 mM NaCl (Fig. 5B, lanes 5-7). Potassium chloride (5-50 mM) stimulated integration activity 4-8-fold (data not shown). These conditions differ from the integration reaction conditions reported for HIV-1, which is active for integration in the presence of NaCl and inhibited by KC1 (15, 17, 28).
Dithiothreitol (DTT) was an absolute requirement for strand transfer activity (Fig. 5C). In contrast to HIV-1 IN
* Qiagen QIAexpress Manual (1991), Chatsworth, CA.

1466 Purification and Characterization of M-MuLV Integrase A. 8. C.
mM Mn2' mM NaCl mM DTT
C ' 0 5 1015202530' c ' 0 2" 9 \@P P ' c n 0 I02030 I 2 3 4 5 6 7 8 1 2 3 4 5 6 7 1 2 3 4 5
Auto- lntcgrar
Auto- megrants
D.
Strand Transfer Actlvltles
pH profile l 0 m 0 N P O O N O O m O N O O m o N * O ~ o ~
nt H 0 1 2 3 4 5 6 7 8 91011121314151617181920212223 r i n P P P o n n n a n o O o o O c ~ i e c m
33- P"' 30- *
1 Auto- lntcgrants
zl-.
- 1 ' cleavage Product
Dlnucleotlde Cleavage Actlvltles
Q O O N P O O N P O ~ O N O O ~ O N P O ~ ~ n n o * l l n n n n n O O Q * ~ c c c c c m
0 I 2 3 4 5 6 7 8 9 1 0 1 1 1 2 1 3 1 4 ' 5 1 6 1 7 1 8 1 9 2 0 2 1 2 2 2 3
PH Droftle
+lent Cleavage Product
FIG. 5. Enzymatic analysis of M-MuLV IN integration ac- tivities. Requirements for optimal strand transfer and cleavage reactions for the E. coli M-MuLV IN protein were examined under various reaction conditions as described under "Experimental Pro- cedures." The oligonucleotide substrate assayed in the absence of protein is presented in lane I of eachpanel. In paneld, strand transfer activity of the M-MuLV IN (0.64 pg/14 pmol IN) was titrated with varying concentrations of MnC12: 0 (lane 2), 5 (lane 3 ) , 10 (lane 4), 15 (lane 5), 20 (lane 6), 25 ( l a n e 7), 30 ( l a n e 8) mM. In panel B, varying concentrations of NaCl were added to the standard reaction for strand transfer activity: 0 (lane 2), 25 (lane 3 ) , 50 (lane 4), 100 (lane 5), 125 (lane 6), and 150 (lane 7) mM. Panel C shows the effect of increasing DTT in the strand transfer reaction for the M-MuLV IN protein (0.64 pg/14 pmol IN); 0 (lane 2), 10 (lane 3 ) , 20 (lane 4), and 30 (lane 5) mM. In panel D, the affect of pH on the cleavage and strand transfer reactions is presented for the M-MuLV IN protein (0.64 pg) using the U5 blunt substrate, 2783/2785. The pH of each reaction is indicated above each lane number in the panel. In the top
(9), 8-mercaptoethanol did not substitute for DTT in the integration reaction buffer at equimolar concentrations (data not shown). Approximately 10-fold higher concentrations of 8-mercaptoethanol was needed to produce autointegrants at a similar level.
The optimal pH for strand transfer and cleavage activities was examined with the 20-bp U5 blunt-ended substrate. The optimal pH for maximal strand transfer activity was 20 mM MES, pH 6.2 (Fig. 50, lane 14), with detectable activity at pH values as low as 5.6 and as high as pH 7.6 (Fig 50, lanes 11, 20). A dramatic decrease in the dinucleotide cleavage activity of the terminal two nucleotides was observed at pH 6.0 (Fig. 50, lane 13) and below. The optimal pH for maximal cleavage activity ranged from pH 6.2 to 7.6 (Fig. 50 , lane 14- 21 ); a higher and broader optimal pH range than observed for the strand transfer reaction. Basal levels of cleavage activity were noted from pH 5.4 to 6.0. No strand transfer or cleavage activity was observed from pH 3.6 to 5.4. M-MuLV viral particles containing integrase similarly display decreased strand transfer activity at higher pH (10). The pattern of autointegrants was nonrandom throughout these experiments as depicted throughout the Fig. 5.
Strand Transfer Actiuity of the pH 6.3, 5.9, and 4.5 Rena- tured Eluates-Using the optimal conditions defined above, strand transfer activity of the IN eluates from pH steps 6.3, 5.9, and 4.5, isolated by the two purification schemes, were each titrated with increasing M-MuLV IN protein. A titration of the pH 6.3 fraction (purification B) is shown in Fig. 6A. The strand transfer activity of the pH 6.3 IN eluate increased over a range of 10-70 pmol of M-MuLV IN protein (1 pmol of substrate) (Fig. 6A, lanes 4-6); after which the activity remained constant (Fig. 6A, lanes 6-7). Below a ratio of 1O:l (1N:substrate) autointegrants were not observed (lanes 2-3). The IN protein isolated from the pH 5.9 step had greater integration activity than the pH 6.3 fraction. The strand transfer activity of the pH 5.9 eluate was detected over a 60- fold range of IN (0.25-30 pmol) to 1 pmol of the recessed substrate showing a maximal activity at 5 pmol of IN (data not shown). Under optimal conditions, 20% of the precleaved strand transfer substrate was converted to product, as deter- mined by PhosphorImager analysis. Using 1 pmol of the precleaved substrate and 5 pmol of IN, the reaction was followed over 2 h (Fig. 6B). The time course of the reaction was linear for at least the first 10 min. Analysis of the integration properties of the pH 4.5 fraction from purification A yielded levels of activities similar to the pH 5.9 fraction (data not shown).
Target Site Duplication Activity of M-MuLV IN from E. coli-One characteristic of the M-MuLV IN integration event is the generation of a 4-bp duplication in the target substrate (1). The fidelity of the duplication was verified for M-MuLV IN (Table I). A biological assay for integration was performed that screens for integration into X concatamers by a model viral substrate in the presence of IN (8). The substrate, Mini- MoMLV, contains an ampicillin and a tetracycline resistance gene bracketed by the recessed LTR sequence for M-MuLV. After the integration reaction, the X DNA is packaged, and
portion of the panel, a long exposure of the autoradiogram is presented to show the autointegrant products of strand transfer at the indicated pH. A shorter exposure is presented in the lower h a l f of the panel to depict the pH profile of the dinucleotide cleavage activity. An arrow specifies the expected position for specific cleavage of the 20-nucleo- tide LTR substrate to an 18-nucleotide product. In panel D, lane 0 is an 18-nucleotide size marker, and lane I is the 20-nucleotide substrate in the absence of integrase. Lane numbers are presented above each panel. Migration of nucleotide size standards is shown to the right or left of each panel.

Purification and Characterization of M-MuLV Integrase 1467
allowed to infect E. coli. Integration events are selected by growth in the presence of ampicillin and tetracyclin. The junction between the X DNA and the synthetic precleaved viral substrate, Mini-MoMLV, was sequenced for three dif- ferent integration events. Each pair of junctions contained 4 bp perfect direct repeats. These repeats establish that the E. coli IN is able to correctly process the target DNA for inte- gration.
Characterization of Disintegration Activity for the M-MuLV IN from E. coli-The structure of the substrate for disinte- gration is shown in Fig. 7A. The structure is a Y-shape comprised of four oligonucleotide strands. The DNA strands drawn as thick solid lines represent the viral LTR, the other represents the host target DNA. During the disintegration reaction, IN catalyzes the formation of two major products that have unique sizes of 19 and 30 nucleotides. The 19-
A W I N
C'&'b5%bb\b5%bb' I 2 3 4 5 6 7
l : L 5 0 100 150
Time (mln)
FIG. 6. Strand transfer activity of the pH 6.3 and 5.9 frac- tions containing M-MuLV IN. Titration of the strand transter activity for M-MuLV IN from the pH 6.3 fraction is presented in panel A: 0 (lane I ) , 0.064 (lane 2) , 0.32 (lane 3), 0.64 (lane 4 ) , 1.6 (lane 5), 3.2 (lane 6), 6.4 (lane 7) pg of protein. Panel B presents a time course for strand transfer for the pH 5.9 fraction (5 pmol) using 1 pmol of the 2784/2785 precleaved substrate. Activity measurements are based on PhosphorImager analysis of the strand transfer products as described under "Experimental Procedures."
oligonucleotide product is the result of a cleavage on the B strand between the LTR (thick solid line) after the conserved 5'..CA..3' and the host target DNA (thick stipled line). IN catalyzes the formation of the 30-nucleotide product by join- ing the 5'-end of the cleaved target portion of the B strand, 14 nucleotides (thick stipled line) with the 3'-end of the C strand, 16 nucleotides (thin solid line) of the target DNA.
The joining activity of M-MuLV IN is presented in Fig. 'IB (lane 3) using the renatured fraction 22 from the integrase purification (Fig. 2A) . An arrow points to the unique 30- nucleotide product created by the joining activity of M-MuLV IN. No such product was observed in the control, pETllC, protein fraction 22 (see Fig. 7B, lane 2). By using "P-labeled B strand, the release of the B strand could be assayed by the appearance of a 19-nucleotide product. Similar to the joining activity, this cleavage activity was unique to the integrase containing fraction and was not observed in the control frac- tion (data not shown).
The effect of divalent cations on disintegration activity was determined. In Fig. 7C, lane 5, 25 r n ~ MnC12 provided con- ditions for maximal activity, while concentrations a t 100 mM (Fig. 7C, lane 7) had significantly reduced activity. Maximal joining activity was reproducibly found a t a manganese ion concentration 2.5 to 5 times higher (Fig. 7C) than that re- quired for integration activity (Fig. 5B). Manganese was essential for disintegration activity. No joining or cleavage activity was observed with magnesium and EDTA (data not shown). NaCl was also examined in the disintegration reac- tion for M-MuLV IN. In contrast to the integration reaction for M-MuLV IN, up to 400 mM NaCl did not diminish the joining activity (data not shown). However, NaCl did not enhance the joining activity of M-MuLV IN, as with HIV-IN (18). Potassium chloride, which enhanced integration activity up to &fold, had no stimulating affect on the joining activity (data not shown).
A clear distinction in the forward and reverse reactions was observed upon a comparison of the pH profiles. Disintegration activity was detected over a wider range of pH, 4.6-8.0 (Fig. 7 0 , lane 7-23), than was found for integration (Fig. 50). Maximal disintegration activity occurred at pH 6.4, where 38% of the substrate was converted to the 30-nucleotide product (lane 15) . Disintegration activity tapered above pH 7.6 and below pH 5.4. Between pH 5.6 and 7.6 (Fig. 7 0 , lane 10-20), the yield of product ranged from 17 to 38%, as determined by PhosphorImager analysis.
DISCUSSION
A one-step affinity purification scheme was developed for the M-MuLV IN protein expressed in E. coli that was greater than 90-95% homogeneous. The M-MuLV IN protein had integration activities (Fig. 5) similar to that observed for the M-MuLV IN isolated from Baculovirus (8), and the cell-free systems using M-MuLV viral particles (10,12). The M-MuLV IN also had the disintegration activity (Fig. 7 ) recently de-
TABLE I Duplications generated by integration oiMini-MoMLV into X by M-MuLV IN
The integration of Mini-MoMLV into X in the presence of M-MuLV IN was performed as described under "Experimental Procedures." The results below describe the DNA sequence for three Mini-MoMLV:X integration junctions. An arrow is drawn over each duplication highlighted in bold. Mini-MoMLV DNA sequence is presented in italic type.
1 . 5' - TGGTTACGTCTGCAAC. . . . .MINI "0. . . . GTTGCATCACGATAGA-3' + - + -+ "j +
2. 5"GGACACAAMGACAAC ..... MINI- MO.. . . GnACACTATTACAAA-3'
3. 5"TGATGACGGACCACAC ..... MINI- MO.... C~CACGACAGGCCCG-3'
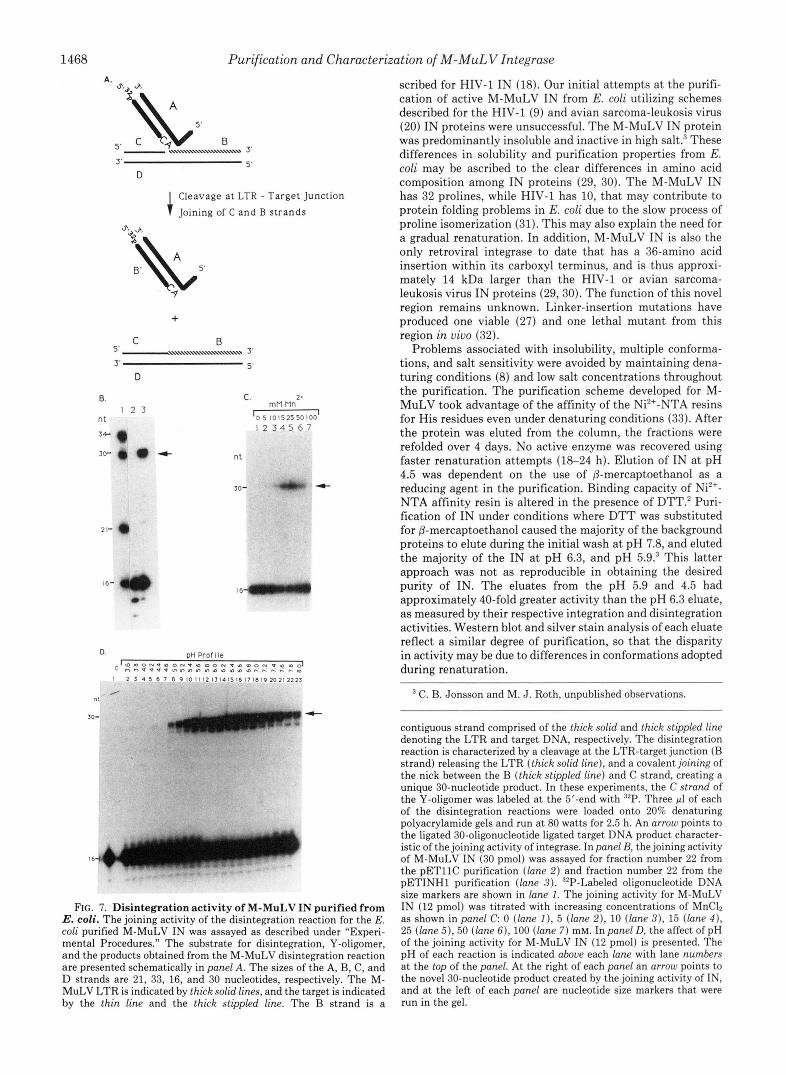
1468 Purification and Characterization of M-MuLV Integrase A
3' E. 2
D
Cleavage at LTR - Target Junction omng of C and B strands
nt 1 2 3
34- q 3 0 - 0 a - nt
30-
mM Mn 2.
'0510152550100 '
1 2 3 4 5 6 7
c
4
FIG. 7. Disintegration activity of M-MuLV IN purified from E. coli. The joining activity of the disintegration reaction for the E. coli purified M-MuLV IN was assayed as described under "Experi- mental Procedures." The substrate for disintegration, Y-oligomer, and the products obtained from the M-MuLV disintegration reaction are presented schematically in panel A. The sizes of the A, B, C, and D strands are 21, 33, 16, and 30 nucleotides, respectively. The M- MuLV LTR is indicated by thick solid lines, and the target is indicated by the thin line and the thick stippled line. The B strand is a
scribed for HIV-1 IN (18). Our initial attempts at the purifi- cation of active M-MuLV IN from E. coli utilizing schemes described for the HIV-1 (9) and avian sarcoma-leukosis virus (20) IN proteins were unsuccessful. The M-MuLV IN protein was predominantly insoluble and inactive in high salt." These differences in solubility and purification properties from E. coli may be ascribed to the clear differences in amino acid composition among IN proteins (29, 30). The M-MuLV IN has 32 prolines, while HIV-1 has 10, that may contribute to protein folding problems in E. coli due to the slow process of proline isomerization (31). This may also explain the need for a gradual renaturation. In addition, M-MuLV IN is also the only retroviral integrase to date that has a 36-amino acid insertion within its carboxyl terminus, and is thus approxi- mately 14 kDa larger than the HIV-1 or avian sarcoma- leukosis virus IN proteins (29, 30). The function of this novel region remains unknown. Linker-insertion mutations have produced one viable (27) and one lethal mutant from this region in uiuo (32).
Problems associated with insolubility, multiple conforma- tions, and salt sensitivity were avoided by maintaining dena- turing conditions (8) and low salt concentrations throughout the purification. The purification scheme developed for M- MuLV took advantage of the affinity of the Ni2+-NTA resins for His residues even under denaturing conditions (33). After the protein was eluted from the column, the fractions were refolded over 4 days. No active enzyme was recovered using faster renaturation attempts (18-24 h). Elution of IN at pH 4.5 was dependent on the use of (3-mercaptoethanol as a reducing agent in the purification. Binding capacity of Ni2+- NTA affinity resin is altered in the presence of DTT.2 Puri- fication of IN under conditions where DTT was substituted for (3-mercaptoethanol caused the majority of the background proteins to elute during the initial wash at pH 7.8, and eluted the majority of the IN at pH 6.3, and pH 5.9.3 This latter approach was not as reproducible in obtaining the desired purity of IN. The eluates from the pH 5.9 and 4.5 had approximately 40-fold greater activity than the pH 6.3 eluate, as measured by their respective integration and disintegration activities. Western blot and silver stain analysis of each eluate reflect a similar degree of purification, so that the disparity in activity may be due to differences in conformations adopted during renaturation.
C . B. Jonsson and M. J. Roth, unpublished observations.
contiguous strand comprised of the thick solid and thick stippled line denoting the LTR and target DNA, respectively. The disintegration reaction is characterized by a cleavage a t the LTR-target junction (B strand) releasing the LTR (thick solid l i n e ) , and a covalent joining of the nick between the B (thick stippled line) and C strand, creating a unique 30-nucleotide product. In these experiments, the C strand of the Y-oligomer was labeled a t the 5'-end with "P. Three pl of each of the disintegration reactions were loaded onto 20% denaturing polyacrylamide gels and run a t 80 watts for 2.5 h. An arrow points to the ligated 30-oligonucleotide ligated target DNA product character- istic of the joining activity of integrase. In panel B, the joining activity of M-MuLV IN (30 pmol) was assayed for fraction number 22 from the pETllC purification (lane 2) and fraction number 22 from the pETINHl purification (lane 3) . 32P-Labeled oligonucleotide DNA size markers are shown in lane 1. The joining activity for M-MuLV IN (12 pmol) was titrated with increasing concentrations of MnCh as shown in panel C 0 (lane I), 5 (lane 2), 10 (lane 3 ) , 15 (lane 4 ) , 25 (lane 5), 50 (lane 6), 100 (lane 7) mM. In panel D, the affect of pH of the joining activity for M-MuLV IN (12 pmol) is presented. The pH of each reaction is indicated above each lane with lane numbers at the top of the panel. At the right of each panel an arrow points to the novel 30-nucleotide product created by the joining activity of IN, and at the left of each panel are nucleotide size markers that were run in the gel.

Purification and Characterization of M-MuLV Integrase 1469
The pH 4.5 and 5.9 eluates containing IN (5 pmol) con- verted 20% of the recessed LTR substrate (1 pmol) to autoin- tegrants under reaction conditions for maximal a~tivity.~ Ti- tration of autointegration activity at lower ratios, 0.25:l (pmol 1N:recessed LTR), yielded 4-10% autointegrants. HIV-1 IN exhibits a similar range of dinucleotide cleavage activity at this ratio of protein to substrate (19). In HIV and M-MuLV, this ratio represents 2-6 pmol of IN for each pmol of product produced a ratio higher than expected if IN works catalyti- cally. This low conversion of substrate to product is proposed to be due to aggregation and precipitation of IN (19). Con- versely, IN could have a very low turnover or the percent of IN refolded in an active conformation is low. The necessity in uiuo for a high enzymatic turnover of the viral DNA substrate is debatable, since the estimated ratio of IN to available viral LTR ends is 25:l (I) , and once the viral DNA is integrated no suitable substrate is available for IN. Further kinetic analysis of the rate of the forward and reverse reac- tions is necessary to ascertain the stoichiometry of these reactions.
M-MuLV IN, like HIV-1 IN, catalyze the reversal of inte- gration or disintegration (18). Both enzymes recognize an intermediate (Y-oligomer) in the integration process, and catalyze the reverse reaction,. disintegration, yielding a re- cessed LTR and a ligated target DNA as the sole products (Fig. 4). The optimal MnC12 and salt requirements differed between the two retroviral integrases. Preliminary results indicate that M-MuLV differs from HIV-1 IN in its ability to cleave single stranded LTR DNA substrates ( 18).4
Several reaction parameters, pH, salt, reductant, and man- ganese ion requirements differ significantly between the for- ward and the reverse reactions of the M-MuLV IN protein. In our studies, we observed that the joining activity of the disintegration reaction for M-MuLV IN was detectable over a much broader range of pH and salt than the cleaving and joining activity for the integration reaction. The optimal concentration of the manganese ion in the M-MuLV disinte- gration reaction was 3-fold higher than for the integration reaction. Finally, DTT could be substituted at equimolar concentration with 6-mercaptoethanol in the disintegration reaction, but not in the integration reaction.
The nucleotides M-MuLV IN recognizes or requires for binding, cleavage, strand transfer, and disintegration, as well as the protein domains, and the critical residues comprising the active site are still undefined. The mechanistic basis for the differences we observed in the cation, salt, reductant, and pH requirements between the forward and reverse reaction
G. A. Donzella, C. B. Jonsson, and M. J. Roth, unpublished results.
for M-MuLV IN is also not understood at present. Further differences were noted in the reaction conditions established for HIV IN in comparison to M-MuLV IN. These differences may indicate a disparity between the specificities for the integration mechanisms, protein stabilities, or turnover of the HIV and M-MuLV integrases. Our development of a rapid purification scheme for IN that was highly active for forward and reverse integration processes should facilitate the exam- ination of some of these questions.
Acknowledgment-We thank Dr. Robert Craigie (National Insti- tutes of Health) for the plasmids, pRC65 and pMK471, bacterial strain, MK459, and HU protein.
REFERENCES 1. Varmus, H., and Swanstrom, R. (1984) in RNA Tumor Viruses (Weiss, R.,
Teich, N., Varmus, H., and Coffin J., e&) pp. 369-512, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
2. Goff, S. P., and Lohel, L. I. (1987) Blochim. Biophys. Acta 907,93-123 3. Varmus, H., and Brown, P. (1989) in Mobile DNA (Berg, D. E., and Howe,
M. M., eds) pp. 53-108, American Society of Microbiology, Washington,
4. Kulkosky, J., and Skalka, A. M. (1990) J. Acq. Immune Def. Syn. 3 , 839- D. C.
85 1 5. Coiicilli, J., and Goff, S. P. (1988) J. Mol. Biol. 199 , 47-59 6. Roth, M. J., Schwartzberg, P. L., and Goff, S. P. (1989) Cell 68, 47-54 7. Katzman, M., Katz, R. A,, Skalka, A. M., and Leis, J. (1989) J. Virol. 6 3 ,
8. Craigie, R., Fujiwara, T., and Bushman, F. (1990) Cell 62,829-837 9. Sherman, P. A., and Fyfe, J. A. (1990) Proc. Nutl. Acad. Sci. U. S. A. 8 7 ,
10. Brown, P. O., Bowerman, B., Varmus, H. E., and Bishop, J. M. (1987) Cell
11. Fujiwara, T., and Mizuuchi, K. (1988) Cell 64,497-504 12. Fujiwara, T., and Craigie, R. (1989) Proc. Natl. Acud. Sci. U. S. A. 8 6 ,
13. Katz R. A., Merkel, G., Kulkosky, J., Leis, J., and Skalka, A. M. (1990)
14. Bushman, F. D., and Craigie R. (1991) Proc. Nutl. Acad. Sci. U. S. A. 8 8 ,
15. Vink, C., van Gent, D. C., Elgersma, Y., and Plasterk, R. H. A. (1991) J. 16. Vink, C., Yeheskiely, E., van der Marel, G. A,, van Boom, J. H., and
17. Engelman, A., Mizuuchl, K., and Craigie, R. (1991) Cell 67,1211-1221 18. Chow, 9. A., Vincent, K. A,, Ellison, V., and Brown P. 0. (1992) Science
5319-5327
5119-5123
49,347-356
3065-3069
Ceil63,87-95
1339-1343
Virol. 66,4636-4644
Plasterk, R. H. A. (1991) Nucleic Acids Res. 19,6691-6698
255. 7?R-??G 19. van Gent, D. C., Elgersma, Y., Bolk, M. W. J., Vink, C., and Plasterk, R.
20. Terry, R., Soltis, D. A,, Katzman, M., Cobrink, D., Leis, J., and Skalka, A.
21. Roth, M. J., Tanese, N., and Goff, S. P. (1988) J. Mol. Biol. 2 0 3 , 131-139 22. Vogelstein, B., and Gillespie, D. (1979) Proc. Nutl. Acud. Sci. U. S. A. 7 6 ,
- - -, . - - . - - H. A. (1991) Nucleic Acids Res. 19,3821-3827
M. (1988) J. Virol. 6 2 , 2358-2365
61 5-61 9 23. Sanger, F., Nicklen, S., Coulen, A. R. (1977) Proc. Nutl. Acud. Sci. U. S. A.
24. Bradford, M. M. (1976) Anal. Biochem. 72,248-254 25. Tanese, N., Roth, M. J., and Goff, S. P. (1986) J. Virol. 69, 328-340 26. Studier, F. W., and Moffat, B. A. (1986) J. M d . Biol. 189,113-130 27. Roth, M. J. (1990) J. Virol. 6 6 , 2141-2145 28. Craigie, R., Mizuuchi, K., Bushman, F. D., and Engelman, A. (1991) Nucleic
29. Johnson, M. S., McClure, M. A., Feng, D. F., Gray, J., and Doolittle, R. F.
30. Lin, T., Quinn, T. P., Grandgenett, D., and Walsh, M. T. (1989) Proteins
31. Jaenicke. R. (1991) Biochemistrv 30.3147-3161
"_ "_ 74,5463-5467
Acids Res. 19,2729-2734
(1986) Proc. Natl. Acud. Sci. U. S. A . 8 3 , 7648-7652
Struct. Funct. Genet. 6, 156-165
32. Donehower, L. A. (1988) J. Vir& 6 2 , 3956-3964 33. Hochuli, E., Bannwarth, W., Dobeli, H., Gentz, R., and Stuber, D. (1988)
BiolTechnology 6,1321-1325





























