Embed Size (px)
Citation preview

Universität Ulm
Institut für Virologie
Leiter: Prof. Dr. med. Th. Mertens
Charakterisierung des Tegumentproteins
pUL24 des Humanen Zytomegalievirus und
seiner Beteiligung am Zelltropismus des Virus
Dissertation zur Erlangung
des Doktorgrades der Humanbiologie (Dr. biol. hum.)
der Medizinischen Fakultät der Universität Ulm
vorgelegt von
Robert-Elmar Pretsch
aus Günzburg
2006

II
Amtierender Dekan: Prof. Dr. Klaus-Michael Debatin 1. Berichterstatter: Prof. Dr. Thomas Mertens 2. Berichterstatter: Prof. Dr. Barbara Spellerberg Tag der Promotion: 17.11.2006

Widmung III
Meiner geliebten Frau Annette und meiner neugeborenen Tochter Lena
Es gibt nichts ergreifenderes im Leben
als einem kleinen Menschen das erste Mal
die Hand zu reichen und zu spüren,
dass wir seine Wurzeln im Baum des Lebens sind,
die ihm Halt und Geborgenheit geben.

Inhaltsverzeichnis IV
Inhaltsverzeichnis:
Abkürzungsverzeichnis ................................................................VI
1 Einleitung .................................................................................. 1
1.1 Das Humane Zytomegalievirus (HCMV)............................................... 1
1.2 Tegument ............................................................................................... 5
1.3 US22 Genfamilie .................................................................................... 6
1.4 Endothelzelltropismus des HCMV........................................................ 6
1.5 Ziel der Arbeit ........................................................................................ 8
2 Material und Methoden ............................................................ 9
2.1 Biologisches Material............................................................................ 9
2.2 Radiochemikalien ................................................................................ 11
2.3 Nukleinsäuren...................................................................................... 11
2.4 Enzyme................................................................................................. 15
2.5 Standardlösungen und Puffer ............................................................ 15
2.6 Molekularbiologische Standardmethoden ........................................ 17
2.7 Reagenzsysteme ................................................................................. 18
2.8 DNA Methoden..................................................................................... 18
2.9 Sawano-Mutagenese [Sawano et al., 2000] ....................................... 21
2.10 redαβ-Rekombination.......................................................................... 23
2.11 Ko-Immunpräzipitation........................................................................ 26
2.12 RNA ligase mediated rapid amplification of cDNA ends.................. 27
2.13 RT-PCR ................................................................................................. 28
2.14 Prokaryote Expression und Immunisierung von Kaninchen........... 29
2.15 Immunfluoreszenzanalyse .................................................................. 30
2.16 Zellkulturverfahren .............................................................................. 31
2.17 Retrovirale Transduktion .................................................................... 34
2.18 Luziferaseassay................................................................................... 35
3 Ergebnisse .............................................................................. 36
3.1 Prokaryote Expression von MBP-pUL24 ........................................... 36
3.2 Charakterisierung einer pUL24/ppUL32 Interaktion ......................... 39
3.3 Funktionelle Analyse von pUL24 mittels Luziferaseassay .............. 42

Inhaltsverzeichnis V
3.4 Generierung und Charakterisierung einer UL24-Deletionsmutante im Hintergrund des endotheliotropen HCMV-Stammes TB40E ............ 45
3.5 Charakterisierung der UL24-/UL25-Genregion mittels rapid amplification of cDNA ends (RACE)................................................... 51
3.6 Herstellung und Charakterisierung von Punktmutanten ................. 52
3.7 Transkomplementation durch retroviralen Gentransfer .................. 57
3.8 Einfluss auf Epithelzelltropismus ...................................................... 59
3.9 Einfluss auf Makrophagentropismus................................................. 61
3.10 Verringerte Infektiösität in Endothelzellen durch Zentrifugation kompensierbar ..................................................................................... 62
3.11 Beteiligung von pUL24 an Entry-Prozessen des HCMV in Endothelzellen ..................................................................................... 64
4 Diskussion .............................................................................. 67
4.1 Herstellung von Antikörpern und Lokalisation von pUL24.............. 67
4.2 Interaktion von pUL24 und ppUL32 ................................................... 68
4.3 Funktionelle Analyse von pUL24........................................................ 69
4.4 Bedeutung von UL24 für den Endothelzelltropismus ...................... 70
4.5 Bedeutung von UL24 für Epithelzell- und Makrophagentropismus 72
4.6 Mechanismus des Endothelzelltropismus ........................................ 73
5 Zusammenfassung................................................................. 76
6 Literaturverzeichnis ............................................................... 77

Abkürzungsverzeichnis VI
Abkürzungsverzeichnis AIDS aquired immunodeficiency syndrome
AK Antikörper
BAC bacterial artifical chromosome
BES N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid
BSA bovine serum albumin
CPE cytopathic effect
DMSO Dimethylsulfoxid
dpi day(s) post infection
DTT 1,4-dithio-DL-threitol
E early
EC(s) Endothelzelle(n)
EBV Epstein-Barr Virus
EDTA Ethylendiamintetraessigsäure
FCS fetal calf serum
FRT Flp recombination target
HCMV Humanes Zytomegalievirus
HEPES [4-(2-Hydroxyethyl)-piperazino]-ethansulfonsäure
HFF human foreskin fibroblast
HHV humanes Herpesvirus
HIV human immunodeficiency virus
hpi hour(s) post infectionem
HRP horseraddish peroxidase
HSV Herpes simplex Virus
HUVEC Human Umbillical Vein Endothelial Cell
IE immediate early
IP Immunpräzipitation
kB Kilobasen
kDa Kilodalton
L late
LB Luria-Bertani-Medium
LTR long terminal repeat
MOI multiplicity of infection
MCMV murines Zytomegalievirus
MCP major capsid protein
MEM minimal essential medium

Abkürzungsverzeichnis VII
NIEP noninfectious enveloped particle
OD optische Dichte
ORF open reading frame
PBS Phosphat-gepufferte Saline
PMSF Phenylmethylsulfonylfluorid
pp Phosphoprotein
PVDF Polyvinyliden Difluorid
rpm rounds per minute
RT Reverse Transkriptase
SDS-PAGE sodium dodecyl sulfate - Polyacrylamidgelelektrophorese
SSC salted sodium citrate
UL unique long
US unique short
WT, wt Wildtyp
Einleitung 1
1 Einleitung
1.1 Das Humane Zytomegalievirus (HCMV)
1.1.1 Taxonomie
Das Humane Zytomegalievirus wird nach der Klassifikation des Internationalen
Komitees für Taxonomie in die Familie der Herpesviren eingeordnet [Roizman et
al., 1981, Smith 1956]. Zu dieser Familie gehören über 100 Vertreter, von denen
acht als humane Herpesviren bezeichnet werden (Tabelle 1). Alle Vertreter dieser
Virusfamilie zeichnen sich durch Gemeinsamkeiten im Partikelaufbau und ihren
biologischen Eigenschaften aus.
HCMV ist, wie alle Herpesviren, ein komplexes Virus und gehört zur Unterfamile
der β-Herpesvirinae. Deren Mitglieder zeichnen sich durch einen hohen
Verwandtschaftsgrad im Hinblick auf ihre Genomorganisation, ein begrenztes
Wirtsspektrum als auch einen relativ langen Replikationszyklus aus [Roizman et
al., 2001].
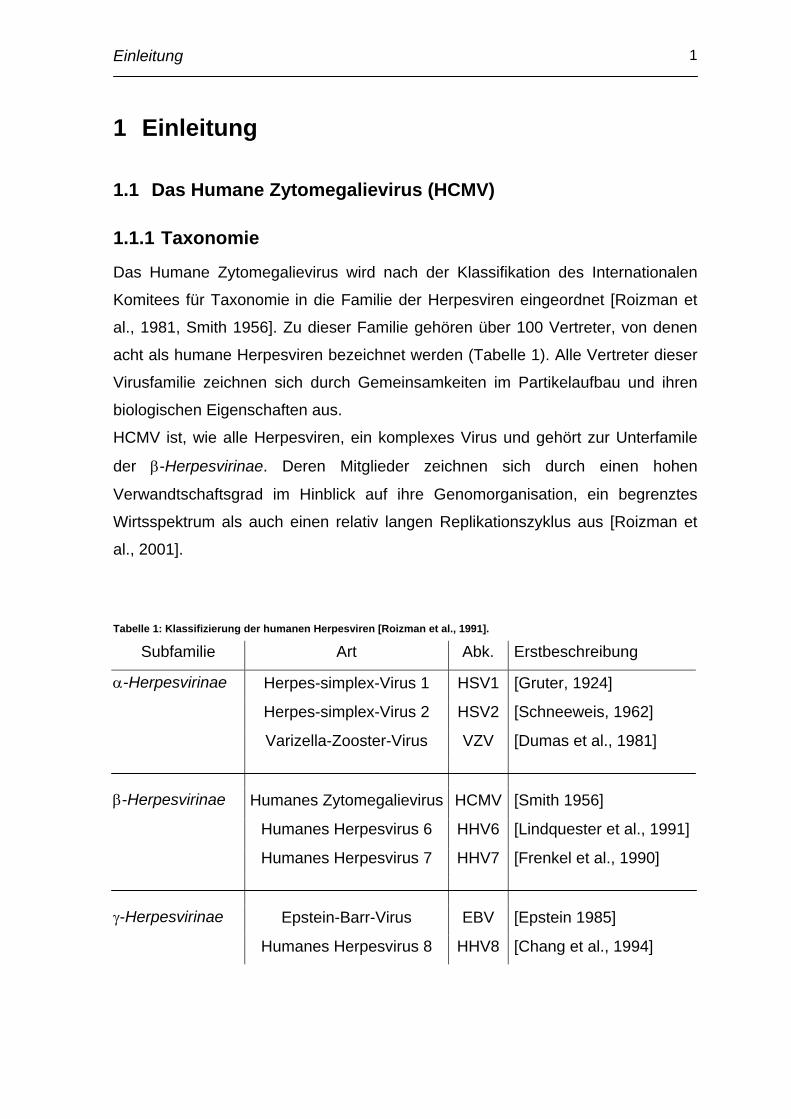
Tabelle 1: Klassifizierung der humanen Herpesviren [Roizman et al., 1991].
Subfamilie Art Abk. Erstbeschreibung
α-Herpesvirinae Herpes-simplex-Virus 1 HSV1 [Gruter, 1924]
Herpes-simplex-Virus 2 HSV2 [Schneeweis, 1962]
Varizella-Zooster-Virus VZV [Dumas et al., 1981]
β-Herpesvirinae Humanes Zytomegalievirus HCMV [Smith 1956]
Humanes Herpesvirus 6 HHV6 [Lindquester et al., 1991]
Humanes Herpesvirus 7 HHV7 [Frenkel et al., 1990]
γ-Herpesvirinae Epstein-Barr-Virus EBV [Epstein 1985]
Humanes Herpesvirus 8 HHV8 [Chang et al., 1994]

Einleitung 2
1.1.2 Morphologie
Das reife HCMV-Partikel hat einen Durchmesser von ungefähr 150-200 nm und
setzt sich aus einem 100 nm großen ikosaedrischen Kapsid, welches das Genom
enthält, einer im Vergleich zu HSV großen Tegument-Komponente und einer das
Partikel umgebenden Hülle zusammen. Diese besteht aus einer zellulären
Doppellipidmembran mit eingelagerten viralen Glykoproteinen (Abb. 1).
HCMV-infizierte Zellen produzieren drei verschiedene Partikeltypen: Die oben
beschriebenen reifen, infektiösen Virionen, sowie sogenannte NIEPs (non
infectious enveloped particles) und Dense Bodies (DBs). NIEPs setzen sich aus
denselben viralen Proteinen wie infektiöse Partikel zusammen und verfügen
ebenfalls über ein Kapsid, welches jedoch kein elektonendichtes, virales Genom
enthält. Somit können sie elektronenmikroskopisch von Virionen unterschieden
werden [Mocarski et al., 2001]. Bei den DBs handelt es sich um nicht
replikationsfähige, fusionskompetente, umhüllte Partikel, die zu ca. 60% (ca. 15%
bei Virionen) aus dem Tegumentprotein pp65 (ppUL83) bestehen. Der Anteil
anderer Proteine, wie z. B. ppUL32, ppUL82, pUL48, pUL80, pUL86 nimmt um
Faktor 5-10 ab [Varnum et al., 2004]. Die zahlenmäßigen Verhältnisse der drei
Abb. 1: Struktur eines HCMV Virions [Reddehase 2006]

Einleitung 3
Partikeltypen in Zellkultur sind abhängig vom verwendeten Virusstamm und von
der eingesetzten MOI (multiplicity of infection).
Das HCMV verfügt über ein lineares, doppelsträngiges DNA-Genom einer Größe
von zirka 235.000 Basenpaaren und gehört somit zu den humanpathogenen Viren
mit der höchsten Kodierungskapazität. Das Genom enthält mehr als 200 offene
Leserahmen (ORFs). Von vielen der ORFs konnte bisher noch nicht gezeigt
werden, dass sie tatsächlich für virale Proteine kodieren. Eine hohe Konservierung
in fünf sequenzierten, klinischen Isolaten gilt aber als deutlicher Hinweis darauf
[Dolan et al., 2004, Murphy et al., 2003].
1.1.3 Replikationszyklus
Durch ihre Oberflächenproteine adsorbieren Herpesviren an bestimmte Strukturen
der Zelloberfläche. Seit geraumer Zeit ist bekannt, dass HCMV über eine
Interaktion von Virionen mit Heparansulfat Proteoglykanen auf der Zelloberfläche
die Infektion initiiert [Compton et al., 1993]. Außerdem gibt es Hinweise darauf,
dass das Virus über das virale Glykoprotein gB (pUL55) an den zellulären EGF-
Rezeptor bindet [Wang et al., 2003]. Ebenso konnte eine Beteiligung von
Integrinen am Eintritt des Virus in die Zelle gezeigt werden [Feire et al., 2004].
Nach der Bindung des Viruspartikels an die Zelloberfläche kommt es zur Fusion
des Virus mit der Zelle. Hierbei wird das Nukleokapsid ins Zytoplasma der Zelle
freigesetzt. Die Kapside lagern sich an Mikrotubuli an und werden zu den
Kernporen transportiert, durch die das Genom in das Nukleoplasma gelangt
[Dolan et al., 2004, Murphy et al., 2003].
Nach der Infektion werden die viralen ORFs in einer organisierten und regulierten
Kaskade exprimiert. Man unterscheidet wie bei allen Herpesviren drei operational
definierte Replikationsphasen: Die sehr frühe (immediate early, IE) Phase, die
frühe (early, E) Phase und die späte (late, L) Phase [Honess et al., 1975, Stinski
1978]. In der IE-Phase kommt es bereits nach wenigen Stunden zur Expression
hauptsächlich regulatorischer Proteine wie z.B. des IE2-Proteins, dem wohl die
größte Bedeutung zukommt, da es essentiell für die Expression der frühen Gene
ist [Marchini et al., 2001, White et al., 2004]. Die Genprodukte aus der nun
folgenden frühen Phase haben unterschiedlichste Funktionen und sind
beispielsweise an der Replikation des viralen Genoms beteiligt (DNA-Polymerase
pUL54 und Proteinkinase pUL97). Gene der L-Phase kodieren überwiegend für

Einleitung 4
Strukturproteine, welche das Kapsid oder das Tegument bilden oder als
Glcoproteine in die Virushülle integriert werden.
Der gesamte Replikationszyklus läuft wie schon unter 1.1.1 erwähnt
vergleichsweise langsam ab und dauert zwischen 48 und 72 Stunden bis die
neuen, reifen Viruspartikel freigesetzt werden. Hierbei fusioniert das mit DNA
beladene virale Kapsid mit dem Zellkern und wird anschließend im Zytoplasma
tegumentiert. Die Umhüllung des tegumentierten Kapids findet bei der Knospung
in das Transgolgi-Netzwerk statt. Das Vesikel, welches nun das umhüllte Partikel
enthält, wird zur Plasmamembran transportiert, wo Vesikel und Membran unter
Freisetzung der Viruspartikel fusionieren [Mettenleiter 2004].
Alternativ zur Infektion von Zellen, bei der freie Viruspartikel mit Bestandteilen der
Zelloberfläche interagieren, können HCMV auch in Zellen gelangen, welche nicht
über diese Bestandteile verfügen. Die Kapside können durch Verschmelzen der
Membranen von infizierter zu nichtinfizierter Zelle weitergegeben werden. Man
spricht hier von einer Übertragung von Zelle zu Zelle (cell to cell spread) [Silva et
al., 2005].
1.1.4 Medizinische Relevanz
Seit fast einem Jahrhundert werden klinische Erkrankungen mit HCMV-Infektionen
in Verbindung gebracht. Zunächst wurde in der Literatur eine klinische Relevanz
des Virus bei prä- oder perinatalen Infektionen beschrieben. Diese können zu
ernsthaften Schädigungen von Embryonen oder Neugeborenen führen. Nach der
Einführung von Gewebe- und Organtransplantationen wurden die Mediziner
darauf aufmerksam, dass HCMV das am häufigsten vorkommende
opportunistische Pathogen bei immunsupprimierten Patienten darstellt [Ho 1991,
Rubin 2001]. Die klinische Manifestation einer HCMV-Infektion variiert hierbei in
Abhängigkeit von der zugrundeliegenden Erkrankung und vom Grad der
Immunsuppression, wie z. B. das Auftreten von Retinitis und Gastroenteritis bei
AIDS-Patienten oder einer Pneumonie bei Knochenmarktransplantierten. Die
Beteiligung von HCMV als Risikofaktor bei Vaskulopathien und Atherosklerose
wird gegenwärtig diskutiert. Es wird angenommen, dass eine veränderte
Migrationsfähigkeit infizierter vaskulärer Zellen pathogene Veränderungen der
Gefäßwand verstärken [Reinhardt et al., 2002, Streblow et al., 2001].

Einleitung 5
Das Virus kann über Speichel, Muttermilch oder Urin, aber auch Cervixsekret und
Sperma infizierter Personen ausgeschieden werden. Letztere machen den
Sexualverkehr zu einem wichtigen Übertragungsweg [Pass 2001]. Eine
Weitergabe ist auch durch Organtransplantation möglich.
Je nach sozioökonomischem Status kann die Serumprävalenz Jugendlicher bei
40-90% liegen. In der Regel laufen Primärinfektionen bei Immungesunden
inapparent ab; nur in ca. 5% der Fälle kommt es zu mononukleoseähnlichen
Beschwerden. Bei der intrauterinen Übertragung kommt es abhängig von
Hygienestandards und Durchseuchungsgrad der entsprechenden
Bevölkerungsgruppe in bis zu 0,1% aller Fälle zur Geburt von geschädigten
Kindern.
1.2 Tegument
Das Tegument ist eine zwischen Hüllmembran und Kapsid gelegene Schicht, die
aus mehr als 20 verschiedenen Proteinen [Gibson 1996]. Noch vor kurzem ging
man davon aus, dass das Tegument eine willkürliche Ansammlung viraler Proteine
sei. Die Struktur des HCMV-Teguments ist im Vergleich zu HSV1 und
Pseudorabies Virus (PrV) noch weitgehend unbekannt. Inzwischen zeigte sich die
Bedeutung dieser Struktur für die virale Replikation und als Aktivatoren viraler
Promotoren [Liu et al., 1992, Romanowski et al., 1997]. Es konnten außerdem
mehrere Tegumentprotein-Komplexe identifiziert werden [Bechtel et al., 2002,
Schierling et al., 2004], was darauf hinweist, dass es sich um eine gut organisierte
und strukturierte Schicht handelt. Die Hauptbestandteile des Teguments in reifen
Viruspartikeln bilden ppUL83 (pp65), ppUL32 (pp150) und ppUL82 (pp71)
[Varnum et al., 2004]. Letzteres wurde als erster Partikelbestandteil mit
transaktivierender Wirkung beschrieben [Liu et al., 1992]. Neben weiteren
transaktivierenden Tegumentproteinen wie ppUL35 und pUL69 wird es mit dem
Virion in die infizierte Zelle aufgenommen und in den Kern transportiert [Baldick,
Jr. et al., 1997, Liu et al., 2002, Winkler et al., 1996].
Durch die Charakterisierung von HCMV-Deletionsmutanten wurde gezeigt, dass
die Gene UL35, UL69 und UL82, zum Teil MOI-abhängig, sehr wichtig für die
Vermehrung des Virus sind [Bresnahan et al., 2000, Dunn et al., 2003, Hayashi et
al., 2000, Schierling et al., 2005]. Für das in HCMV essentielle ppUL32 [Dunn et

Einleitung 6
al., 2003] konnte gezeigt werden, dass es mit seinem Aminoterminus an Kapside
bindet [Baxter et al., 2001]. Weiterhin konnte dieses Protein in unserem Labor
mittels Yeast Two-Hybrid Analyse als Interaktor von pUL24 identifiziert werden.
1.3 US22 Genfamilie
Die US22 Genfamilie wurde zuerst bei HCMV beschrieben [Chee et al., 1990] und
beinhaltet 12 Mitglieder [Ménard et al., 2003], u.a. pUL24, die alle über
mindestens eins von vier konservierten Aminosäuresequenzmotiven verfügen
[Efstathiou et al., 1988, Kouzarides et al., 1988]. Bei den bisher untersuchten
Genprodukten handelte es sich überwiegend um Tegumentproteine [Adair et al.,
2002, Varnum et al., 2004]. Andere beta-Herpesviren kodieren für eine jeweils
identische Anzahl an US22 Genprodukten, was auf eine bedeutende Rolle dieser
Genfamilie für den Replikationszyklus der Viren hindeutet [Gompels et al., 1995,
Nicholas 1996, Megaw et al., 1998, Rawlinson et al., 1996, Vink et al., 2000, Bahr
et al., 2001].
Die Mitglieder der US22 Genfamilie haben unterschiedlichste Funktionen. HCMV
pUL36, pIRS1 und pTRS1 fungieren beispielsweise als transkriptionelle
Transaktivatoren des HCMV-Haupt-IE-Promotors [Colberg-Poley et al., 1992,
Stasiak et al., 1992]. In HHV6 bewirkten U16 und U3, die Homologe zu UL36 und
UL24, in transienten Transfektionsstudien eine Transaktivierung des HIV-1 LTR
[Geng et al., 1992, Mori et al., 1998]. pUL36 wurde zusätzlich eine
antiapoptotische Wirkung nachgewiesen [Skaletskaya et al., 2001]. Für MCMV
m139, m140 und m141 wurde gezeigt, dass sie für das Viruswachstum in
Makrophagen in vivo [Hanson et al., 2001] und in vitro [Ménard et al., 2003] von
großer Bedeutung sind. Bisher konnte keinem HCMV-kodierten Mitglied der US22
Genfamilie eine Beteiligung am Zelltropismus des Virus nachgewiesen werden.
1.4 Endothelzelltropismus des HCMV
Es wird angenommen, dass die Verbreitung des HCMV über den Blutstrom eine
wichtige Rolle bei der vertikalen Übertragung von der Mutter zum Kind und bei der
Organmanifestation im Verlauf einer akuten systemischen Infektion
immunsupprimierter Patienten spielt. Neben polymorphnukleären und

Einleitung 7
mononukleären Leukozyten wurden vaskuläre Endothelzellen (EC) als Zielzellen
des Virus in vivo identifiziert, die an der Ausbreitung von HCMV über den
Blutkreislauf beteiligt sein könnten [Sinzger et al., 1995]. Im Verlauf einer akuten
HCMV-Infektion kann sich das Virus im gesamten Organismus verbreiten und
anschließend praktisch jedes Organ infizieren [Bissinger et al., 2002]. Infizierte
Endothelzellen wurden hierbei in verschiedensten Geweben nachgewiesen.
Offensichtlich können sich ECs von der Gefäßwand ablösen und in den Blutstrom
eintreten, wo sie als zirkulierende, zytomegalische, infizierte Zellen nachgewiesen
werden konnten [Grefte et al., 1993].
Man unterscheidet zwischen schwach und hochgradig endotheliotropen
Virusstämmen. Bei ersten erfolgreichen Infektionen von human umbilical vein
endothelial cells (HUVECs) mit HCMV wurde deutlich, dass patientennahe
Virusisolate diesen Zelltyp effizienter infizieren als Laborstämme wie z.B. AD169
oder Towne [Ho et al., 1984], deren Infektionsrate unter normalen Bedingungen
auf wenige Prozent begrenzt ist. HCMV-Stämme, die direkt nach ihrer Isolierung
auf ECs propagiert wurden, wie z.B. TB40/E oder VHL/E [Sinzger et al., 1999],
werden als hochgradig endotheliotrop bezeichnet. Die schwach endotheliotropen
Isolate haben anscheinend bereits einen Defekt bei oder nach der Aufnahme der
Partikel. Die gegenwärtige Hypothese lautet, dass der Transport der viralen
Nukleokapside und damit auch des viralen Erbgutes zum Zellkern beeinträchtigt
ist, wodurch eine produktive Infektion unterbunden wird [Sinzger et al., 1999]. Auf
molekularer Ebene konnte bisher die Region UL128-131 als Determinante für EC-
Tropismus identifiziert werden. Jedes der Gene dieses Lokus ist per se notwendig
für ein effizientes Viruswachstum in EC [Hahn et al., 2004]. Im besonderen
zellkulturadaptierte Stämme besitzen Mutationen in den ORFs UL128, UL130 und
UL131A [Dolan et al., 2004].

Einleitung 8
1.5 Ziel der Arbeit
Die gegenwärtig zur Behandlung des Humanen Zytomegalievirus verwendeten
Medikamente wie Ganciclovir/Valganciclovir, Cidofovir, Foscarnet und das
Antisense-Oligonukleotid Fomivirsen haben mit Ausnahme des Letztgenannten
alle die virale DNA-Polymerase als Ziel. Berichte von immer häufigerem Auftreten
klinisch relevanter, resistenter HCMV zeigen, dass dringend neue Substanzen und
Mechanismen zur erfolgreichen Behandlung infizierter Personen identifiziert
werden müssen. Hier könnten Medikamente, die andere virale Proteine oder
deren Interaktionen blockieren einen neuen Ansatz darstellen. Die in
vorangegangenen Arbeiten identifizierte Interaktion der viralen Tegumentproteine
ppUL32 und pUL24 sollte dazu näher charakterisiert werden.
Die Fähigkeit zur Infektion von Endothel- und Epithelzellen durch das HCMV ist im
Hinblick auf die Pathogenität des Virus von großer Bedeutung. Endothelzellen sind
als Schnittstelle zwischen Organgewebe und Blutkreislauf von großer,
strategischer Bedeutung. Sie ermöglichen eine Ausbreitung des Pathogens im
gesamten Wirtsorganismus.
Epithelzellen sind hoch empfänglich für HCMV und bereits vor Jahrzehnten konnte
Virus aus den Speicheldrüsen infizierter Personen isoliert werden. Auch im
Epithelgewebe der Lunge, des Gastrointestinal- und des Urogenitaltraktes konnte
virale Repilikation nachgewiesen werden. Daher wird Epithelzellen eine wichtige
Rolle als Eintrittspforte und für die Verbreitung des Virus zugeschrieben. Für
HCMV ist bisher die Genregion UL128-131 als eine Determinante für Endothelzell-
und vor kurzem auch für Epithelzelltropismus identifiziert worden. Die
Identifizierung neuer Tropismusdeterminanten und das Wissen um deren
Wirkungsweise bringt somit die Möglichkeit mit sich, die Pathogenität des Virus zu
manipulieren. Mitgliedern der US22-Genfamilie konnte bisher nur in MCMV eine
Rolle am Zelltropismus des Virus bescheinigt werden.
Die Herstellung verschiedener Virusmutanten und nachfolgende
Infektionsexperimente mit unterschiedlichen Zelltypen sollten durchgeführt
werden, um eine mögliche Beteiligung von pUL24 am Zelltropismus von HCMV zu
untersuchen.

Material und Methoden 9
2 Material und Methoden
2.1 Biologisches Material
2.1.1 Bakterien
Escherichia coli DH10B: E. coli K12 F- araD139 Δ(ara, leu)7697 ΔlacX74 galU
galK rpsL deoR φ80dlacZΔM15 endA1 nupG recA1 mcrA Δ(mrr hsdRMS mcrBC)
[Grant et al., 1990]
2.1.2 Zellen
HFF primäre humane Vorhautfibroblasten [Chang et al., 2002]
HUVEC primäre humane Endothelzellen der Nabelschnurvene
HeLa Zervixkarzinom Epithelzellen [Nelson-Rees et al., 1976]
SK-N-SH humane Neuroblastomazelllinie [Biedler et al., 1973]
293T humane embryonale Nierenzelllinie, welche das SV40 T-Antigen
exprimiert [DuBridge et al., 1987]
Phoenix Amphotrope Verpackungszelllinie zur Herstellung rekombinanter
MLV-Retroviren [Pear et al., 1993]
U373MG humane Glioblastoma/Astrozytoma Zelllinie [Ponten et al., 1968]
RPE1 Retina Pigment Epithelzellen [Bodnar et al., 1998]
2.1.3 Viren
HCMV AD169 Ein attenuierter Laborstamm, der ursprünglich von
[ROWE et al., 1956] isoliert wurde.
TB40E Patientennaher Virusstamm [Sinzger et al., 2000]
RVTB4-BAC Rekombinantes Virus aus TB4-BAC
RV TB4delUL24-BAC Rekombinantes Virus aus TB4delUL24-BAC
RV TB4repUL24-BAC Rekombinantes Virus aus TB4repUL24-BAC
RV TB4mutUL24-1.ATG Rekombinantes Virus aus TB4mutUL24-1.ATG
RV TB4mutUL24-2.ATG Rekombinantes Virus aus TB4mutUL24-2.ATG
RV TB4mutUL24-1.+2.ATG Rekombinantes Virus aus TB4mutUL24-1.+2.ATG
RVRetro-GFP Rekombinantes Virus aus p617-neo-T-EGFP-tTA3

Material und Methoden 10
RVRetro-UL24 Rekombinantes Virus aus pMLV-Retro-tTA3-DEST
Myc-UL24
2.1.4 Antikörper
2.1.4.1 Seren
αUL35 Antiserum: eingesetzt in 1:200 (Immunfluoreszenz) bzw. 1:5000 (Western
Blot) Verdünnungen [Liu et al., 2002]
αUL69 Antiserum: eingesetzt in 1:2000 (Immunfluoreszenz) bzw. 1:5000 (Western
Blot) Verdünnungen [Winkler et al., 1994]
αIE1/2 Antiserum: eingesetzt im Western Blot in einer 1:2000 Verdünnung [Gebert
et al., 1997]
αUL24 Antiserum: eingesetzt in einer 1:50 Verdünnung (Immunfluoreszenz, Ko-
Immunpräzipitation) [R. Pretsch, nicht publiziert]
2.1.4.2 monoklonale Antikörper
Zellkulturüberstände: unverdünnt eingesetzt
69-66 monoklonaler Antikörper aus Maus gegen pUL69 [W. Britt, nicht
publiziert]
CMV 355 monoklonaler Antikörper aus Maus gegen ppUL82 [Nowak et al.,
1984]
12CA5 monoklonaler Antikörper aus Maus gegen HA-tag [Niman et al.,
1983]
9E10 monoklonaler Antikörper aus Maus gegen Myc-tag [Evan et al., 1985]
63-27 monoklonaler Antikörper aus Maus gegen IE1 [Plachter et al., 1993]
aUL24 monoklonaler Antikörper aus Maus gegen UL24 [Adair et al., 2002]
XP-1 monoklonaler Antikörper aus Maus gegen pp150, 1:2000 verdünnt im
Western Blot eingesetzt [Scholl et al., 1998]
αUL25 monoklonaler Antikörper aus Maus (Ascites) 1:200 (Western Blot)
[Battista et al., 1999]

Material und Methoden 11
2.1.4.3 kommerzielle Antikörper
I.E.A. [clone E13] monoklonaler Antikörper aus Maus gegen HCMV-immediate
early antigen [Argene, Vailhes, Frankreich]
AAC10 monoklonaler Antikörper aus Maus gegen das HCMV late-
Protein ppUL83 [DAKO, Glostrup, Dänemark]
CCH2 monoklonaler Antikörper aus Maus gegen das HCMV early-
Protein pUL44 [DAKO, Glostrup, Dänemark]
Vimentin monoklonaler Antikörper aus Maus gegen Vimentin (Ab-1)
[Oncogene, Boston]
2.1.4.4 Sekundäre Antikörper
Ziege-anti-Maus, Cy3-gekoppelt, polyklonal Dianova, Hamburg
Ziege-anti-Kaninchen, Cy3-gekoppelt, polyklonal Dianova, Hamburg
Ziege-anti-Maus, Alexa 488-gekoppelt, polyklonal Invitrogen, Karlsruhe
Ziege-anti-Kaninchen, Alexa 488-gekoppelt, polyklonal Invitrogen, Karlsruhe
Ziege-anti-Maus, HRP-gekoppelt, polyklonal Pierce, Rockford, IL, USA
Ziege-anti-Kaninchen, HRP-gekoppelt, polyklonal Pierce, Rockford, IL, USA
2.2 Radiochemikalien
[α32P]dCTP 10 mCi/ml Amersham, Braunschweig
2.3 Nukleinsäuren
2.3.1 Oligonukloetide
Die Oligonukleotide wurden von MWG Biotech (Ebersberg), Sigma-ARK
(Darmstadt) und biomers.net (Ulm) bezogen.
UL24rt5’ CGGGAACGGAACTGCGTCTA
UL24rt3’ TGCTGACGTCCTTTGGGGCA
Aktin4 AGCCATGTACGTAGCCATCCAGGCT
Aktin5 GGATGTCAACGTCACACTTCATGATGG

Material und Methoden 12
UL25 5’myc Eco TATGAATTCCACCATGGAACAGAAACTCATCTCTGAAGA-
GGATCTGATGTCGTCGCGGCGTCGCAG
IE2 5’ CAACACCAATCGCTCTCTTG
IE2 3’ CGCATCCACCTCACTCTTC
UL24 5’RACE ip CGAATTCTGCGCACGTAAGCTTCCAGACAGCCCAG
UL24 5’RACE op ATCTCCTGCAGCACTAGGTT
UL25 5’RACE ip CGCGGTACCCGTTTCTTTTCGCTCGGTTTCTTGGC
UL25 5’RACE op TTCTGCTACTGTTACTGGTGGTGCGA
UL23 5’RACE ip GCTCGAGAAAGTTGTAGGTGCAGATGCTGCCGACG
UL23 5’RACE op AATTCAGTGATGCTCTCGGC
UL24-5’ CGCGGATCCTACATGGAGGAGACCCGG
UL24-3’OK GCGAATTCAACGGTGCTGACGTCC
UL24-3’-KO GTCGGTGTTTTATGCCCCAAGCAGCGTCGTCGTCACTC-
GTGGCGTCACAGACAAGGACGACGACGACAAGTAA
UL24-5’-KO CGACATCCTCTCGTCTGCTGAGGTGCGTTCGGCTCAGT-
TGCTTACCTGCACGTCGTGGAATGCCTTCGAATTC
UL24-rev CATCCTCTCGTCTGCTGAGGTGCGTTCGGCTCAGTTGC-
TTACCTGCATACATGGAGGAGACCCGGGCGGG
UL24-rev-dATG CATCCTCTCGTCTGCTGAGGTGCGTTCGGCTCAGTTGC-
TTACCTGCATACGCGTAGGAGACCCGGGCGGGACGTT
UL24del-3’ GCGAGTCAAACGGGAAGTAG
UL24del-5’ AAATCACCGTCGAGGGTTCG
UL24-M59A ACTGACTCGGCGCGTCACGCGTCAGATCTGGCTAGCCT-
GGCG
Myc-UL24-5' ATAGAATTCCACCATGGAACAGAAACTCATCTCTGAAGA-
GGATCTGATGGAGGAGACCCGGGCGGG
UL24-3’-XhoI ATACTCGAGTCAACGGTGCTGACGTCCTTTGG

Material und Methoden 13
2.3.2 Plasmide
pBlueKan: Von pBluescript KS II abgeleiteter Klonierungsvektor, bei dem die
Ampizillinresistenz durch das Resistenzgen für Kanamycin ersetzt wurde
[Schierling et al., 2005].
pCDNA3: Eukaryoter Expressionsvektor mit CMV-Promotor, Apr und Neor
(Invitrogen GmbH, Karlsruhe).
pENTR1a: Klonierungsvektor, codiert für ccdB (negativer Selektionsmarker), Knr
(Invitrogen GmbH, Karlsruhe)
pIRES2-EGFP: Eukaryoter Expressionsvektor, bei dem EGFP über eine IRES
koexprimiert wird, Knr [Invitrogen GmbH, Karlsruhe]
pFastBac-CMV: Transfervektor zur Herstellung rekombinanter Baculoviren, Apr,
Gmr [GIBCO, BRL]
pBabepuro: Vektor für retroviralen Gentransfer, Puromycinr [Morgenstern et al.,
1990]
pLNCX2: Vektor für retroviralen Gentransfer, Apr, Neor [Clontech]
pHM287: Luziferase-Reporterkonstrukt mit dem HCMV IE1/2 Enhancer (-1228 bis
+53) [Winkler et al., 1996]
pCB6: Eukaryoter Expressionsvektor mit CMV-Promotor, Apr und Neor [Brewer
1994] pp150-CB6: eukaryotes Expressionsplasmid; enthält den UL32-ORF unter
Kontrolle des HCMV IE1/2 Enhancer/Promotors [B. Plachter, Mainz]
pCDNA3-IE2: Eukaryotes Expressionsplasmid, das für IE2 kodiert [Schierling et
al., 2004]
pCDNA-UL69: Eukaryotes Expressionsplasmid, das für UL69 kodiert [Schierling
et al., 2004]
pSL-FRT: Trägt Knr flankiert von minimalen FRT-Sequenzen [Wagner et al., 1999]
pKD46: Ein low copy Plasmid mit Apr und temperatursensitivem Ori (30°C);
Enthält γ- β- und exo-Gene des Red Recombinase-System [Datsenko et al., 2000].
pCP20: Enthält den Leserahmen für die pSC-FLP-Rekombinase, repliziert und
induziert die FLP-Synthese temperaturabhängig (30°C) [Cherepanov et al., 1995].
pBRep-cre: Codiert für die Cre-Rekombinase [Hobom et al., 2000]
pCM1015: Cosmid; enthält das HindIII-Fragment des HCMV-AD169, welches die
Leserahmen UL11 bis UL37 umfasst [Fleckenstein et al., 1982].

Material und Methoden 14
pBK-UL24- full: Enthält den Leserahmen UL24 des HCMV-AD169, jedoch ohne
Start- und Stopcodon [R. Pretsch, Diplomarbeit] .
pcDNA3-Myc-N-UL24: Enthält den UL24-Leserahmen aus pBK-UL24-full, der
nach EcoRI/BamHI-Verdau in pcDNA3-Myc-N ligiert wurde.
pCMV71: Enthält den Leserahmen HCMV Towne UL82 [Liu et al., 1992]
pSL-FRT-TB4-mycUL24: Trägt mycUL24 und Knr flankiert von minimalen FRT-
Sequenzen
pSL-FRT-TB4-mycUL24 mut 2.ATG: Über Sawano-Mutagenese unter
Verwendung des Primers UL24-M59A aus pSL-FRT-TB4-mycUL24 hergestellt.
pTST101: Prokaryotes Expressionsplasmid; enthält ein Fusionsgen aus EGFP
und Maltose Bindeprotein (MBP) [G. Muth, Tübingen]. pTST-UL24: Der UL24-ORF wurde über BamHI/HindIII in pTST101 kloniert.
p617-neo-T-EGFP-tTA3 [Kühnel et al., 2004]
pMLV-Retro-tTA3-DEST: EGFP in p617-neo-T-EGFP-tTA3 wurde durch
pBabepuro Myc-UL24 TB1 : Primer UL24 5' mit Myc-Tag und UL24-3'OK EcoRI
aus TB1-BAC DNA amplifiziert, mit EcoRI gespalten und in die EcoRI-Schnittstelle
von pBabepuro ligiert.
pENTR-mycUL24-TB1: Das Myc-UL24-TB1-EcoRI Fragment aus pBabepuro
Myc-UL24 TB1 wurde über EcoRI in pENTR1a einkloniert. pMLV-Retro-tTA3-DEST Myc-UL24: Myc-UL24 aus pENTR-mycUL24-TB1 wurde
über die Gateway-Klonierungstechnik in pMLV-Retro-tTA3-DEST gesetzt
pFastBac-CMV Myc-UL24 TB4: pIRES2-EGFP Myc-UL24: Myc-UL24 wurde aus pFastBac-CMV Myc-UL24 TB4
über EcoRI in pIRES2-EGFP kloniert.
pLNCX2 Myc-UL24: Myc-UL24 wurde aus pIRES2-EGFP Myc-UL24 über EcoRI
in pLNCX2 kloniert.
HIV luc: HIV1 LTR Luziferase-Konstrukt [Winkler et al., 1994] SV40 luc: SV40 enhancer/promoter Luziferase-Konstrukt [Christian Aepinus]

Material und Methoden 15
2.3.3 BACs
TB4wtBAC [Hahn et al., 2003]
TB4delUL24 Mittels recET-abhängiger Rekombination wurde UL24 aus
TB4-BAC deletiert.
TB4repUL24 Der UL24 ORF wurde durch Rekombination in die
Deletionsmutante wieder hergestellt.
TB4mut1.ATG Der UL24 ORF wurde durch einen mutierten ORF ersetzt.
TB4mut2.ATG Der UL24 ORF wurde durch einen mutierten ORF ersetzt.
TB4mut1.+2.ATG Der UL24 ORF wurde durch einen mutierten ORF ersetzt.
2.3.4 Sonstige Nukleinsäuren
Größenstandard GeneRuler 1 kB DNA ladder (Fermentas, St. Leon-Rot)
2.4 Enzyme
Die verwendeten Restriktionsenzyme wurden von MBI Fermentas (St.Leon-Rot)
und New England Biolabs (Schwalbach) bezogen.
Das Expand High Fidelity System stammt von der Firma Roche (Mannheim),
Klenow-Fragment wurde von MBI-Fermentas (St.Leon-Rot) bezogen. RNaseA und
Proteinase K stammen von Roche (Mannheim), Mungbean Exonuclease, T4-DNA-
Ligase und T4 Polynukleotidkinase wurden von New England Biolabs
(Schwalbach) bezogen.
2.5 Standardlösungen und Puffer
Alle Medien und Lösungen wurden, soweit nicht anders beschrieben, mit bidest
H2O hergestellt.
Denaturierungslösung für Southern Blot: 1,5 M NaCl, 0,5 M NaOH
Transferpuffer für Southern Blot: 1,5 M NaCl, 0,25 M NaOH
20x SSC: 3 M NaCl, 0,3 M Trinatriumcitrat pH 7.0
50x Denhardt: 1 g Ficoll, 1 g Polyvinylpyrrolidon, 1 g BSA, ad 100 ml bidest H2O,
sterilfiltriert.

Material und Methoden 16
Prä-/hybridisierungslösung für Southern Blot: 50 % Formamid, 5xSSC, 1x
Denhardt, 0,05 M Phosphat, Hefe-RNA ca. 50 mg
10% APS-Lösung: 1 g APS in 8 ml bidest H2O gelöst, ad 10ml bidest H2O.
1x Laemmlipuffer: 2 M Glycin, 0,25 M Tris/HCl (pH 8,5), 0,5 % SDS (v/v)
5x SDS-Probenpuffer: 200 mM Tris, 5 mM EDTA, 1 M Sucrose, 0,1 %
Bromphenolblau (w/v) ,1 mM DTT, pH 8,8 mit HCl eingestellt.
Coomassie-Färbelösung: 2,5 g Coomassie Brilliant Blue R-250, 450 ml Methanol,
100 ml Eisessig, ad 1 l bidest H2O.
Coomassie-Entfärbelösung: 450 ml Methanol, 100 ml Essigsäure 99,9%, ad 1 l
bidest H2O
Transferpuffer für Westernblots: 25 mM Tris, 0,1 % SDS (v/v), 1,5 % Glycin (v/v),
20 % Methanol (v/v), pH 8,3 mit HCl eingestellt.
Ponceau S-Färbelösung: 0,5% Ponceau S (w/v), 1% Trichloressigsäure (v/v)
Blockierungslösung für Western Blot: 1x PBS, 5% Milchpulver (MP) (w/v)
Waschlösung für Western Blot: 1x PBS, 1% MP (w/v), 0,1% NP40 (v/v)
1x PBS (phosphat-buffered saline): 8 g NaCl, 0,2 g KCl, 2,68 g Na2HPO4-7H2O,
0,24 g KH2PO4, mit HCl auf pH 7,4 eingestellt, ad 1 l bidest H2O, autoklaviert.
0,01 M PBS: 150 mM NaCl, 1 mM KH2PO4, mit NaOH auf pH 7,8 eingestellt.
0,01 M PBS / EDTA: 0,01 M PBS, 1% EDTA Stammlösung (v/v)
0,01 M PBS / BSA: 0,01 M PBS, 1% BSA (v/v)
40xTAE Agarosegelelektrophoresepuffer: 1,6 M Tris, 1,6 M NaAcetat x 3 H2O,
0,04 M EDTA x 2H2O, pH 7,2 mit Eisessig eingestellt.
10x Agarosegel-Probenpuffer: 50 mM Tris (pH 7,6), 60 % Glycerol (v/v), 0,25 %
Bromphenolblau (w/v), 0,25 % Xylencyanol (w/v)
Puffer 1-3 für modifizierte Alkali-Lyse:
Puffer 1: 50 mM Glucose, 10 mM EDTA, 25 mM Tris/HCl pH 8,0
Puffer 2: 0,2 M NaOH, 1 % SDS (v/v)
Puffer 3: 3 M K-Acetat pH 4,8
2x BES (N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid): 50mM BES, 280
mM NaCl, 1,5mM Na2HPO4, pH 6,95
10x PCR-Puffer: 100 mM Tris pH 9,0, 500 mM KCl, 15 mM MgCl2
LB-Medium: 10 g Bactotrypton, 5 g Hefeextrakt, 8 g NaCl, pH 7,5 mit NaOH
eingestellt., ad 1 l bidest H2O, autoklaviert. LB-Agarplatten enthielten zusätzlich
1,5 % Agar (w/v) und die entsprechenden Antibiotika.

Material und Methoden 17
MEM und DMEM-komplett: 500 ml MEM oder DMEM (Gibco BRL, Eggenstein)
wurden jeweils mit, 10 % FCS (v/v), 1% Penicillin/Streptomycin (v/v), 1% Glutamin
(v/v), 1% nicht-essentielle Aminosäuremix.
Sucrose-Phosphatpuffer: 74,62 g Sucrose, 1,218 g K2HPO4, 0,52 g KH2PO4, ad 1 l
bidest H2O, autoklaviert.
KoIP-Lysepuffer: 50 mM Tris/HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 0,5 % NP-
40, 1x Protease Inhibitormix (Roche, Mannheim, Germany)
2 x Lysispuffer für Luziferase-Assays: 62,5 ml 100 mM Tris / H3PO4, 0,5 ml 1M
DTT, 2,5 ml Triton X-100, 0,1732 g CDTA und 25 ml Glycerol mischen, pH 7,8 mit
NaOH einstellen; H2O ad 125 ml.
Reaktionspuffer für Luziferase-Assay: 100 mM Kaliumphosphat pH 7,8, 15 mM
MgSO4, 5 mM ATP
Substratpuffer für Luziferase-Assay: Reaktionspuffer plus 1 mM D-Luziferin,
(Roche, Mannheim)
2x HBS: 8,2 g NaCl, 5,95 g Hepes, 105 mg Na2HPO4•2 H2O, 1 g Dextrose
(Glucose), 370 mg KCl, H2O ad 450 ml, pH 7,05, H2O ad 500 ml.
MBP-Elutionspuffer: 20 mM Maltose
MBP-Waschpuffer: 20 mM Tris-HCl (pH 7.4) , 0.2 M NaCl ,1 mM EDTA
2.6 Molekularbiologische Standardmethoden
Folgende Methoden gelten als Standardmethoden und werden daher im
Methodenteil dieser Arbeit nicht genauer beschrieben:
• Kleine Plasmidpräparation [Zagursky et al., 1985]
• Ethanol-Präzipitation von Nukleinsäuren
• Phenol-Chloroform-Extraktion
• Bestimmung von DNA-Konzentrationen mit dem Photometer
• Restriktionsspaltungen
• Ligationen
• Dephosphorylierungsreaktionen mit SAP oder CIP
• Auffüllreaktion mit Klenowfragment oder Mung-bean Nuklease für
Klonierungen mit glatten Enden
• Agaroregelelektrophorese [Sambrook et al., 1989]

Material und Methoden 18
2.7 Reagenzsysteme
- große Plasmidpräparation mit dem Jetstar Plasmid Maxi Kit 20 (Genomed,
Bad Oyenhausen)
- große BAC-Präparation mit dem Nucleobond AX500 Maxi Kit (Marchery-
Nagel, Düren)
- nicht-radioaktive Sequenzierung von DNA wurde mit einem
Sequenziergerät 310 Genetic Analyzer (Perkin-Elmer, Norwalk, USA) der
Firma ABI (Weiterstadt) durchgeführt. Der verwendete Sequenzier-Kit
(BigDye Termination Kitb 1.0) stammte von der Firma ABI.
- die Herstellung radioaktiver DNA-Sonden für Southern Blot Analysen wurde
mit Hilfe von Ready-to-go™ DNA Labeling Beads (Amersham Pharmacia,
Piscataway, NJ, USA) durchgeführt. Die Abtrennung radioaktiver
Nukleotide erfolgte über Sephadex G50 Fine Quick Spin™ Säulen (Roche,
Mannheim).
- die Aufreinigung von PCR-Produkten und DNA-Fragmenten nach
Restriktionsspaltungen erfolgte mittels GFX PCR DANN and Gel Band
Purification Kit (Amersham Biosciences, Buckinghamshire).
- Super Signal West Dura Extended Duration Substrate (Pierce, Rockford, IL)
- Reaktionen der Reversen Transkription mit cDNA first strand synthesis Kit
(MBI Fermentas, St. Leon-Rot)
2.8 DNA Methoden
2.8.1 PCR
Polymerase Kettenreaktionen wurden nach [Mullis et al., 1986] durchgeführt.
Annealingtemperatur (X) und Elongationszeit (Y, 1 min/kB) waren
anwendungsabhängig variabel.
2.8.1.1 High Fidelity-PCR
Die PCRs zur Klonierung von HCMV ORFs wurde mit Expand High Fidelity-Taq-
DNA-Polymerase (Roche), einem Gemisch aus Taq-DNA-Polymerase und Pwo-
DNA-Polymerase, durchgeführt.

Material und Methoden 19
Reaktionsbedingungen:
5 min 94°C
30 s 94°C
30 s X°C
Y s 72°C
7 min 72°C
∞ 4°C
30 Zyklen
Reaktionsmix:
50ng -1 µg Template-DNA
100 pm forward primer
100 pm reverse primer
1 µl dNTPs (10 mM)
5 µl 10x Expand High Fidelity -Puffer
0,5 µl Expand High Fidelity-Taq
1,25 µl DMSO
ad 50 µl H2O
2.8.1.2 Touchdown-PCR
Diese PCR wurde zur Herstellung der Fragmente für die Klonierung der
Virusmutanten in einem Geneamp PCR System 9700 (Perkin Elmer, Norwalk,
USA) durchgeführt, wobei das gleiche Pipettierschema wie unter 2.8.1.1
verwendet worden ist. Folgende Reaktionsbedingungen wurden verwendet:
2 min 94°C
30 s 94°C
30 s 72°C
3 min 72°C
7 min 72°C
∞ 4°C
jeweils nach 2 Zyklen schrittweise Herabsetzung
der Annealingtemperatur um 1°C bis auf 61°C,
dann 12 weitereZyklen.

Material und Methoden 20
2.8.1.3 Kolonie-PCR
Bei der Kolonie-PCR wurde DNA direkt aus Koloniematerial amplifiziert und der
Reaktionsmix nach folgendem Schema hergestellt:
Reaktionsmix :
Eine Bakterienkolonie
10 pm 5’-Primer Reaktionsbedingungen:
5 min 94°C
30 s 94°C
30 s X°C
X s 72°C
7 min 72°C
35 Zyklen
10 pm 3’-Primer
1 µl dNTPs (20 mM)
5 µl 10x PCR-Puffer
2 µl Taq-DNA-Polymerase [Pluthero 1993]
1,25 µl DMSO
ad 50 µl H2O
2.8.2 Präparation von BAC-DNA
Bei der Isolierung von BAC-DNA wurde versucht, eine Fragmentierung der DNA
durch Scherkräfte zu vermeiden. Deshalb wurde auf kräftiges Schütteln zugunsten
von mehrmaligem Invertieren verzichtet, und alle weiteren Schritte, in denen BAC-
DNA pipettiert wurde, wurden mit abgeschnittenen Pipettenspitzen durchgeführt.
Für Mini-Präparationen wurden 5 ml Übernachtkultur 5 min bei 3500 rpm
zentrifugiert, das Pellet in 300 µl Puffer P1 (Qiagen) aufgenommen, 300 ml Puffer
P2 (Qiagen) zugegeben, 2-3mal invertiert und 4 min bei Raumtemperatur
inkubiert. Nach Zugabe von 300 µl Puffer N3 (Qiagen) ruhte der Reaktionsmix 10
min auf Eis, bevor er 5 min bei 14000 rpm und 4°C zentrifugiert wurde, um das
Zellmaterial abzutrennen. Der Überstand wurde in ein 1,5 ml Reaktionsgefäß
überführt, wobei abgeschnittene Pipettenspitzen verwendet wurden, um die DNA
so wenig wie möglich Scherkräften auszusetzen. Die DNA wurde mit 600 µl
Isopropanol versetzt, 15 min bei 14000 rpm und RT gefällt, der Überstand
abgekippt und das Pellet nach Zugabe von 100 µl 70% EtOH wiederum 15 min bei
14000 rpm gewaschen. Der Überstand wurde verworfen, das Pellet 10 min an der
Luft getrocknet, in 50 µl Wasser aufgenommen und für 1 h bei Raumtemperatur
gelöst.

Material und Methoden 21
Um eine größere Menge an BAC-DNA zu erhalten, wurden BAC-Maxi-
Präparationen mit dem Nucleobond-BAC-Maxi-Kit (Machery-Nagel) durchgeführt.
Die Präparation erfolgte nach dem von der Herstellerfirma mitgelieferten Protokoll.
2.8.3 Southern Blot - DNA-DNA-Hybridisierung [Southern 1975]
Für die Hybridisierung wurde BAC-DNA mindestens 4 h mit Restriktionsenzymen
gespalten und anschließend über Nacht bei 30 V in einem 0,6%-igen Agarosegel
aufgetrennt. Danach wurde das Gel für 20 min in 0,25 M HCl geschwenkt, was zu
DNA-Doppelstrangbrüchen führt und den Transfer der DNA erleichtert. Nachdem
das Gel mit demin H2O gespült wurde, folgte eine 30minütige Inkubation in 1,5 M
NaCl, 0,5 M NaOH. Anschließend wurde das Gel 15 min in Transferpuffer (1,5 M
NaCl, 0,25 M NaOH) equilibriert und ein Kapillarblot aufgebaut (Sambrook et al,
1989).
Der DNA-Transfer erfolgte über Nacht. Am nächsten Tag wurde die Membran kurz
in 2x SSC gewaschen und die DNA durch 2 h Inkubation bei 80°C im
Hybridisierungsofen (Robbins Scientific Corp.) kovalent an die Membran
gebunden.
2.9 Sawano-Mutagenese [Sawano et al., 2000]
Zunächst wurde das Oligonukleotid UL24-M59A in einer Kinasereaktion nach
folgendem Ansatz phosphoryliert:
Oligo 100 pmol/ul 15 ul
10 mM ATP 2 ul
10x PNK Puffer 2 ul
PNK 1 ul
Die Reaktion fand über 2 h bei 37°C statt.

Material und Methoden 22
Die Mutagenese des Plasmids pSL-FRT-TB4-mycUL24 selbst fand in einer
zweistufigen PCR-Reaktion statt:
Reaktionsansatz:
50 ng pSL-FRT-TB4-mycUL24
14 pmol UL24-M59A (phosphoryliert)
1,25 µl dNTP (10mM)
2,5 µl 10x Pfu Buffer
2,5 U Pfu Polymerase
2,5 µl 10x Taq Ligase Buffer
20 U Taq Ligase
Reaktionsbedingungen 1:
5 min 65°C
2 min 95°C
30s 95°C
30s 55°C 18 Zyklen
10 min 65°C
7 min 75°C
je Ansatz 1 ul (20 U) DpnI zugeben, mischen;
Reaktionsbedingungen 2:
1 h 37°C
30 s 95°C
30 s 95°C 2 Zyklen
1 min 55°C
10 min 70°C
2 µl des Reaktionsansatzes nach Ablauf beider Reaktionen wurden in kompetente
DH10B transformiert, auf Selektionsplatten inkubiert (37°C) und positive Klone
mittels Sequenzanalyse identifiziert.

Material und Methoden 23
2.10 redαβ-Rekombination
Die gerichtete, PCR-basierte Mutagenese eines HCMV-BAC erfolgt in der Regel
ohne Klonierung viraler Sequenzen und damit unabhängig von geeigneten
Schnittstellen innerhalb des zu modifizierenden Genoms. Mutationen bestimmter
Gene erfolgen über homologe Rekombination in E. coli mit Hilfe des redαβ-
Rekombinase-Systems des Phagen λ, welches in den Bakterien induziert werden
kann. Das zu mutierende Gen wird durch einen Selektionsmarker (Kanamycin)
ersetzt, welcher von FRT-Sequenzen flankiert wird und in einem zweiten Schritt
mit Hilfe der FLP-Rekombinase wieder aus dem BAC entfernt werden kann
[Wagner et al., 1999]. Abb. 2 stellt das Prinzip der ET-Klonierung schematisch dar.
Abb 2: SchematischeDarstellung des dreiteiligenPrinzips der RecET-Klonierungnach Wagner (1999). FRT-
Sequenzen (rote Balken),
homologe Sequenzen zwischen
Pimern und BAC (blaue und
grüne Linien) und BAC- bzw.
Plasmid–Sequenzen (schwarze
Linien) wurden dargestellt.

Material und Methoden 24
2.10.1 Amplifikation von PCR-Fragmenten für die redαβ-
Rekombination
Zunächst wurde mittels Touchdown-PCR (siehe 2.8.1.2) das von FRT-Sequenzen
flankierte Kanamycinresistenzgen durch die Primer UL24-3’-KO und UL24-5’-KO
aus pSL-FRT [Wagner et al., 1999] amplifiziert (Abb. 2.1). Um die UL24 TB4
Wildtypsituation wiederherzustellen, wurde die Primerkombination UL24-rev/UL24-
5’-KO und pSL-FRT-TB4-mycUL24 als Template verwendet. Für die Mutation des
ersten Startkodons wurde pSL-FRT-TB4-mycUL24 mit den Primern UL24-rev-
dATG and UL24-5’-KO amplifiziert. Zur Herstellung von BACs bei denen das
zweite oder das erste und zweite Startkodon mutiert waren, wurde pSL-FRT-TB4-
mycUL24-M59A als Matrize und die Primerpaare UL24-rev/UL24-5’-KO oder
UL24-rev-dATG/UL24-5’-KO verwendet. Die Primer wurden so gewählt, dass sie
gleichzeitig homologe Sequenzbereiche zur HCMV-AD169-Sequenz [Chee et al.,
1990] von ca. 50 bp 5‘ bzw. 3‘ von UL24 besaßen. Das entstandene PCR-Produkt
wurde aufgereinigt (PCR Purification-Kit, Qiagen, Hilden), für 1 h bei 37°C mit dem
Restriktionsenzym DpnI verdaut, um Reste der eingesetzten Template-DNA zu
entfernen (DpnI spaltet nur methylierte DNA und somit nicht das PCR-Fragment)
und nach Ethanolpräzipitation in 10 µl sterilem Wasser aufgenommen.
2.10.2 Herstellung kompetenter E. coli DH10B [TB4-BAC, pKD46]
Das Plasmid pKD46 [Datsenko et al., 2000] wurde in E. coli DH10B [TB4-BAC]
transformiert. 250 ml LB (mit 25 µg/ml Cm und 100 µg/ml Ap) wurden 1:100 mit
Vorkultur angeimpft. Nach Wachstum der Kultur bei 30°C und 200 rpm bis zu einer
OD600 von 0,1-0,15 wurde Arabinose bis zu einer Endkonzentration von 0,1%
zugegeben, um das redαβ-Rekombinasesystem zu induzieren. Nach Erreichen
einer OD600 von 0,4-0,6 wurden die Bakterien für 15 min auf Eis inkubiert, 10 min
bei 7000 rpm und 4°C zentrifugiert (Beckman J2-21, Beckman, München) und das
Pellet dreimal mit 10%iger Glycerinlösung gewaschen (10 min bei 7000 rpm, 4°C).
Nach Abkippen des Überstandes wurden die Bakterien im Restvolumen
resuspendiert, in Aliquots à 60 µl aufgeteilt, in flüssigem Stickstoff schockgefroren
und bei –70°C bis zur weiteren Verwendung gelagert.

Material und Methoden 25
2.10.3 Einbau der Resistenzkassette ins BAC-Genom
2-5 µl eines in 2.10.1 hergestellten PCR-Fragmentes wurden zu 60 µl
elektrokompetenten E. coli DH10B [TB4-BAC, pKD46] gegeben (siehe 2.10.2) und
sofort elektroporiert (2,5 kV, 200 Ω, 25 µF). Anschließend wurde 1 ml LB
dazugegeben und der Ansatz 1,5-2 h bei 37°C inkubiert. Nach kurzem
Abzentrifugieren wurden die Zellen in 150 µl LB resuspendiert, auf Platten mit 25
µg/ml Kn und 25 µg/ml Cm ausgestrichen und über Nacht bei 37°C im Brutschrank
inkubiert. Bei diesem Schritt werden Klone mit homologer Rekombination des
Amplifikats mit dem BAC (Abb. 2.2) selektioniert und pKD46 geht aufgrund seines
temperatursensitiven Replikationsorigin verloren. Die erfolgreiche Rekombination
wurde mittels Kolonie-PCR, BAC-DNA-Minipräparation mit anschließendem
Restriktionsverdau und Southern Blot-Analyse kontrolliert.
2.10.4 Entfernung der Resistenzkassette
Um die Kanamycinkassette zu entfernen, wurden Bakterien, welche das jeweilige,
mutierte BAC inklusive Kn-Resistenz enthielten mit pCP20 transformiert. Die von
dem Plasmid codierte FLP-Rekombinase schneidet die Kanamycinkassette über
die FRT-Sequenzen aus dem BAC heraus (Abb. 2.3). 1/10 des
Transformationsansatzes wurde auf LB-Agarplatten mit Chloramphenicol
(25µg/ml) und Ampicillin (100µg/ml) ausplattiert und über Nacht bei 30°C im
Brutschrank inkubiert. Es wurden fünf Kolonien gepickt, auf einer LB-Agarplatte
mit Chloramphenicol (25µg/ml) ausgestrichen und für 24 h bei 43°C inkubiert. Bei
diesem Schritt repliziert pCP20 nicht und geht deshalb verloren. 10 Kolonien von
dieser Platte wurden parallel auf Platten mit Cm, Cm und Ap oder Cm und Kn
ausgestrichen, um den Verlust der Kanamycinkassette und von pCP20 zu
überprüfen. Bakterienklone, die nur auf Cm-Platten wuchsen, wurden mittels PCR,
Restriktionsverdau, Southern Blot Analyse und Sequenzanalyse der modifizierten
Bereiche analysiert.

Material und Methoden 26
2.11 Ko-Immunpräzipitation
2.11.1 Ko-Immunpräzipitation aus transfizierten 293T Zellen
293T Zellen wurden in 6-Loch Zellkulturplatten bis zu einer Konfluenz von 70 %
kultiviert. In einem 1,5 ml Eppendorf-Reaktionsgefäß wurden je 2 μg pro zu
transfizierendem Plasmid mit sterilem H2O auf ein Endvolumen von 225 μl
verdünnt. Dann wurden 25 μl 2,5 M CaCl2 zugegeben, die Lösungen kurz
gevortext und 250 μl 2 x HBS pH 7,12 (mit sofortigem Vortexen für ca. 10 sec)
zugegeben. Das entstandene Präzipitat wurde anschließend auf die Zellen
getropft. Die Zellen wurden über Nacht inkubiert, am nächsten Tag dreimal
vorsichtig mit warmen 0,01 M PBS gewaschen und mit frischem DMEM
überschichtet. Für die Ko-Immunpräzipitation wurden die Zellen nach weiteren 24
h geerntet. Dazu wurden sie zweimal vorsichtig mit 0,01 M PBS gewaschen und
anschließend in 1 ml 0,01 M PBS vom Plattenboden abgespült. Die Zellen wurden
5 min bei 2300 × g sedimentiert, der Überstand verworfen, das Pellet in 500 μl
Lysepuffer (50 mM Tris/HCl pH 8,0, 150 mM NaCl, 5 mM EDTA, 0,5 % NP-40 plus
einmal Protease-Inhibitor-Mix, Roche, Mannheim) aufgenommen und unter
mehrmaligem Vortexen für 20 min auf Eis inkubiert. Die Zelltrümmer wurden bei
16000 × g (10 min, 4 °C) abgetrennt. Vom Überstand wurden 80 μl als
Lysatkontrolle verwendet und mit 20 μl 5 x SDS-Probenpuffer 10 min bei 95 °C
denaturiert. Der restliche Überstand wurde in ein neues Reaktionsgefäß überführt,
mit 300 μl Lysepuffer aufgefüllt und mit dem für die Präzipitation verwendeten
Antikörper versetzt. Es wurden 100 μl Hybridomüberstand bzw. 1 μl der
polyklonalen Seren verwendet. Die Immunpräzipitation erfolgte für 1,5 h bei 4 °C
unter ständigem Mischen im Überkopf-Taumler. Anschließend wurden zu jedem
Ansatz 40 μl Protein A-Sepharose (25 mg/ml in Lysepuffer) zugegeben und
weitere 2 h inkubiert. Die Sepharose wurde sedimentiert (5 min, 2000 × g, 4 °C)
und dreimal mit Lysepuffer gewaschen. Der letzte Zentrifugationsschritt erfolgte
bei 16000 × g, der Überstand wurde komplett entfernt, das Sediment in 10 μl 2 x
SDS-Probenpuffer aufgenommen und 10 min bei 95 °C denaturiert. Die
Auftrennung der Proteine erfolgte in einer 10 %igen SDS-PAGE mit
anschließendem Nachweis der ko-präzipitierten Proteine im Western Blot.

Material und Methoden 27
2.11.2 Ko-Immunpräzipitation aus infizierten HFF Zellen
HFF Zellen wurden in einer 10 cm Zellkulturschale ausgesät und mit HCMV
TB40E mit einer MOI von 2 infiziert. 96 h nach Infektion wurden die Zellen
trypsiniert, sedimentiert und zweimal mit 0,01 M PBS gewaschen. Die weiteren
Schritte erfolgten wie unter 2.11.1 beschrieben, die Zelllyse erfolgte jedoch in
einem Volumen von 300 μl.
2.12 RNA ligase mediated rapid amplification of cDNA ends
Bei dieser Methode handelt es sich um eine Weiterentwicklung der herkömmlichen
RACE, die eine alleinige Amplifikation von Volllänge-mRNA erlaubt. Die
Experimente wurden mit dem FirstChoiceTM RLM RACE Kit von Ambion
durchgeführt.
5’-Race:
In einem ersten Schritt wurden 10 µg gesamtzelluläre RNA mit Kälberdarm-
Phosphatase behandelt, um freie 5’-Phospate degradierter mRNA-, rRNA-, tRNA-
oder DNA-Stücke zu entfernen (Abb. 3.1). Komplette mRNAs sind durch ihre 5’-
cap-Struktur geschützt. Die nachfolgende TAP-(tobacco acid pyrophosphatase)
Behandlung führte zur Abspaltung des 5’-caps der Volllänge-mRNA, wobei ein 5’-
Monophosphat stehen blieb (Abb. 3.2). Dieses wird für die Ligation eines
Adapteroligonukleotids benötigt (Abb. 3.3).
Über eine anschließende Reverse Transkription erhielt man cDNAs mit einem
definierten 5’-Ende (Abb. 3.4). Durch die Verwendung von genspezifischen
Primern konnten die 5’-Regionen mittels PCR und nested PCR (Abb. 3.5)
amplifiziert, kloniert und sequenziert werden.
3’-RACE:
Über eine RT-Reaktion unter Verwendung eines Oligo-dT-Adaptermoleküls erhielt
man cDNAs mit definierter 3’-Sequenz (Abb. 3.6). Durch die Verwendung von
genspezifischen Primern konnten die 3’-Regionen mittels PCR und nested PCR
(Abb. 3.7) amplifiziert, kloniert und sequenziert werden.

Material und Methoden 28
Abb. 3: Schematische Darstellungeiner RLM-RACE. Homologe
Sequenzen zwischen Primer und
mRNA (schwarze Linie) wurden als
schwarze Balken dargestellt.
2.13 RT-PCR
Die RT-PCR dient als Methode, um eine Aussage über die Expression von viralen
als auch zellulären Genen auf mRNA-Ebene treffen zu können. Aus HFF-Zellen
wurde mit TRIZOL-Reagenz (Invitrogen, Karlsruhe) nach Angaben des Herstellers
gesamtzelluläre RNA isoliert. Diese wurde in einem ersten Schritt mit RNase-freier
DNase verdaut (MBI Fermentas, St. Leon-Rot; 30 min bei 37°C). Anschließend
wurde die mRNA in cDNA umgeschieben, welche später in der PCR als Matrize
dienen sollte. Pro Ansatz wurden 0,5 µg RNA, 1 µl Oligo d(T)18-Primer und DEPC-
H2O ad 11 µl für 5 min bei 70°C inkubiert und danach sofort auf Eis gekühlt. Nach
Zugabe von 4 µl 5x-Puffer (Fermentas), 3 µl dNTPs und 1 µl RNase-Inhibitor-Mix
wurden die Reaktionsgefäße weitere 5 min bei 37°C inkubiert. Im Anschluss daran
wurde 1 µl Reverse Transkriptase (200 u) hinzugefügt. Nach 1 h Inkubation bei
42°C, wurde das Enzym durch 10minütiges Erhitzen auf 70°C inaktiviert und sofort
auf Eis gestellt. Die Amplifikation erfolgte mit genspezifischen Primerpaaren,
wobei folgende Bedingungen verwendet wurden:

Material und Methoden 29
Reaktionsmix: Reaktionsbedingungen:
5 min 94°C
30 s 94°C
30 s 60°C 35 Zyklen
30 s 72°C
7 min 72°C
∞ 4°C
1 µl cDNA aus der RT-Reaktion
10 pm 5’-Primer
10 pm 3’-Primer
1 µl dNTPs (10 mM)
5 µl 10x PCR-Puffer
2 µl Taq-Polymerase
ad 50 µl H2O
Primerbindungstemperatur und Elongationszeit waren so gewählt, dass alle
Amplifikationen unter den selben Bedingungen ablaufen konnten. Die Auswertung
der RT-PCR-Reaktionen fand mittels 2%iger Agarosegelelektrophorese statt.
2.14 Prokaryote Expression und Immunisierung von Kaninchen
4x 500ml LB(Amp) wurden mit je 100ml Übernachtkultur DH10B pTST-UL24
inokuliert und bei 37°C bis zu einer OD578=0,5 wachsen gelassen. Nach der
Induktion des rhamnoseabhängigen Promotors mit 10 ml 10% Rhamnose je
Kolben wurden die Zellen bei Raumtemperatur bis zu einer OD578 von 2,0 weiter
wachsen gelassen. Die Bakterien wurden sedimentiert (10 min bei 5000 g) und
das Pellet in 10 ml Columnbuffer aufgenommen. Das Gesamtvolumen von zirka
25 ml wurde in einer Frenchpress (SLM Aminco, Schwäbisch Gmündt) bei einem
Druck 1500 bar, 4x aufgeschlossen, 75min bei 7000 g und 4°C zentrifugiert und
der Überstand mit 1 ml Amylose–Matrix (New England Biolabs, Ipswich, MA) für 3
h bei 4°C im Überkopftaumler geschüttelt. Nach drei Waschschritten mit
Columnbuffer (5 min, 1000 g) wurde das an die Matrix gebundene, rekombinante
Fusionsprotein in drei Schritten mit zweimal je 1 ml Elutionspuffer (20mM Maltose)
bzw. 1ml 0,1% SDS eluiert. Die Fraktionen wurden in einem Coomassieblau
gefärbten 10%igen SDS-Gel analysiert. Die anschließende Immunisierung von
Kaninchen wurde nach dem Schema in Abbildung 4 durchgeführt.

Material und Methoden 30
Abb. 4: SchematischeDarstellung derProbenaufbereitung fürdie Immunisierung von
Kaninchen.
2.15 Immunfluoreszenzanalyse
Für Immunfluoreszenzuntersuchungen wurden eukaryote Zellen in 24-Loch-
Platten auf Deckgläsern kultiviert und anschließend transfiziert bzw. infiziert. Zum
Nachweis von viralen Proteinen mittels spezifischer Antikörper, wurden indirekte
Immunfluoreszenzanalysen durchgeführt. Die Zellen wurden hierzu zweimal mit
0,01 M PBS gewaschen und mit 250 μl 4 % Paraformaldehyd überschichtet. Nach
10 min Inkubation bei Raumtemperatur wurden die Zellen erneut dreimal
gewaschen und entweder zur weiteren Lagerung bei 4 °C mit 300 μl 0,01 M PBS
überschichtet, oder aber bei sofortiger Verwendung für 2 min mit 0,1 % Triton-
X/0,01 M PBS permeabilisiert., und anschließend 30 min bis 1 h bei 37°C mit
einem entsprechend verdünnten Primärantikörper inkubiert. Polyklonale Antiseren
wurden 1:50 bis 1:5000 in 0,01 M PBS mit 1% BSA verdünnt, monoklonale
Hybridomzellüberstande wurden unverdünnt verwendet. Danach wurden die
Zellen dreimal mit 0,01 M PBS gewaschen und mit einem Fluorophor-gekoppelten,
1:200 bis 1:1000 in 0,01 M PBS/BSA verdünnten Sekundär-Antikörper für weitere
30 min bis 1 h bei 37°C inkubiert. Zur Gegenfärbung wurde der Lösung mit dem
Sekundärantikörper DAPI (1:1000 Verdünnung der Stocklösung) zugegeben.
Nach zweimaligem Waschen mit 0,01 M PBS wurden die Deckgläser kopfüber mit
einem Tropfen Vectashield Mounting Medium (Vector, Burlingame, CA, USA) auf
einen Objektträger gegeben. Die Zellen konnten nun bei UV-Licht unter dem
Fluoreszenzmikroskop betrachtet werden. Aufnahmen zur Untersuchung der
Lokalisation rekombinant exprimierter Proteine wurden mit Hilfe konfokaler

Material und Methoden 31
Mikroskopie (Leica TCS laser scanning system (Bensheim) unter Verwendung der
Leica LCS Software) durchgeführt.
2.16 Zellkulturverfahren
Arbeiten mit eukaryoten Zellen wurden stets mit sterilen Materialien an einer
sterilen Arbeitsbank durchgeführt.
2.16.1 Kultivierung der Zellen
Eukaryote Zellen wurden bei 37°C, 96% Luftfeuchtigkeit und 5% CO2-Gehalt in
einem Inkubator (Heraeus, Hanau) kultiviert. 293T- und Phoenix-Zellen wurden in
Kulturflaschen mit DMEM (mit 1% Glutamin, 1% Pen/Strep und 10% FCS),
Fibroblasten, U373MG-, HeLa- und SK-N-SH-Zellen mit MEM (mit 1% Glutamin,
1% Pen/Strep und 10% FCS) angezogen. RPE1 (HAM’s F12 : D-MEM = 1:1 mit
1% Glutamin, 1% Pen/Strep, 10% FCS und 0,348% NaHCO3), HSGE (humane
Speicheldrüsen-Epithelzellen, McCoy´s 5a, 10% FCS, 1% Pen/Strep, 1%
Glutamin, 1% AS), HUVEC (EBM mit 5% FCS, 0,4% BBE (bovine brain extract),
0,1% Hydrokortison, 0,1% GA-1000, 0,1% hEGF), Makrophagen (RPMI mit 1%
Glutamin, 1% Pen/Strep und 10% FCS). Bei den 293T-, Phoenix- und HeLa-Zellen
wurde darauf geachtet, dass sie nie eine höhere Zelldichte als 75% erreichen, da
sie sich ansonsten nicht mehr effizient genug transfizieren lassen.
Zur Subkultivierung wurden adhärent wachsenden Zellen zunächst zweimal mit
0,01 M PBS/EDTA gewaschen und anschließend mit 1 ml 0,01 M PBS/EDTA und
1 ml 0,25%igem Trypsin/EDTA vom Boden der Kulturflasche gelöst (für 5 min bei
37°C), mit frischem serumhaltigen Medium heruntergespült und anschließend auf
neue Zellkulturgefäße verteilt.
2.16.2 Infektion von Zellen
Die Zellen wurden je nach Versuchsansatz in festgelegter Anzahl in
entsprechenden Zellkulturschalen ausgebracht und für 24 h im Brutschrank
inkubiert. Am nächsten Tag wurden die Zellen nach einem Medienwechsel mit der
benötigten Infektiosität (plaque forming units pfu/ml) pro Infektionsansatz infiziert.

Material und Methoden 32
Mock-infizierte Zellen wurden zur Kontrolle genau wie die zu infizierenden Zellen
behandelt. Anstelle der Viruslösung wurde das entsprechende Volumen eines
Gemisches aus MEM-komplett und Sucrosephosphat auf die Zellen gegeben.
Dies entspricht der Situation bei den zu infizierenden Zellen, da das Virus in
50%iger Sucrosephosphatlösung eingefroren war. Es folgte eine einstündige
Inkubation (37°C, 5% CO2, 96%Luftfeuchtigkeit), wobei die Zellkulturgefäße im
Abstand von 15 min geschwenkt wurden. Anschließend wurde der Virusüberstand
wieder von den Zellen abgenommen, durch frisches serumhaltiges Medium ersetzt
und die Zellen bis zum jeweiligen Erntezeitpunkt weiter kultiviert.
2.16.3 Herstellung und Titration von Virusstocks
Zur Herstellung zellfreier Virusstocks wurden 106 HFF in einer 25 cm2
Zellkulturflasche ausgesät und nach 24 h mit einer MOI von 0,1 infiziert. Nachdem
diese Kultur zu 90 % infiziert war (nach ungefähr 5 Tagen), wurden die Zellen
trypsiniert und zusammen mit ca. 4 x 107 nicht infizierten HFF Zellen auf
insgesamt zehn 175 cm2 Zellkulturflaschen ausgebracht. Diese wurden so lange
kultiviert, bis die Infektion zur Lyse der Zellen führte. Der Kulturüberstand wurde in
einem sterilen Gefäß gesammelt, die restlichen adhärenten Zellen mit einem
Zellschaber vom Flaschenboden entfernt und die gesamte Kultur vereinigt. Nach
Zentrifugation für 5 min bei 2500 × g wurde der Überstand in ein weiteres steriles
Gefäß gegeben und das Zellpellet in ca. 3 ml MEM resuspendiert. Diese Zellen
wurden dann auf Eis in einem Homogenisator aufgeschlossen (30 Hübe pro
Aufschluss), die Zelltrümmer sedimentiert (10 min, 2500 × g) und der Überstand
mit den bereits geernteten Überständen vereinigt. Das Virus wurde nun in einer
Beckmann Ultrazentrifuge mit dem SW28 Rotor für 1 h bei 22000 rpm (4 °C)
pelletiert. Nach Abkippen des Überstands wurde das Viruspellet über Nacht bei 4
°C in 250 μl Sucrosephosphat gelöst. Die vollständig resuspendierten
Viruspräparationen wurden vereinigt und als Aliquots von 250-500 μl bei -80 °C
gelagert.
Für die Titration der Virusstocks wurden 1,6 x 104 HFF pro Loch in einer 96-Loch-
Platte ausgebracht. Am darauffolgenden Tag wurden Verdünnungsreihen (1:10)
der Stocks hergestellt und je 100 μl dieser Verdünnungen auf die Zellen pipettiert.
Um den statistischen Fehler gering zu halten, wurden die Infektionen pro

Material und Methoden 33
Verdünnung im Dreifach-Ansatz durchgeführt. Nach Zentrifugation der 96-Loch-
Platte für zweimal 15 min bei 900 × g wurden die Zellen für vier Tage im
Brutschrank inkubiert. Der Nachweis infizierter Zellen erfolgte durch die Detektion
von viralem E-Antigen mittels Immunoperoxidasefärbung. Hierzu wurde der
Zellkulturüberstand entfernt, die Zellen zweimal mit 0,01 M PBS gewaschen und
mit eiskaltem Methanol für 10 min bei –20 °C fixiert. Nach dem Entfernen des
Methanols wurden die Zellen für mindestens 10 min bei RT getrocknet. Zur
Rehydrierung wurden die Zellen anschließend zweimal mit 0,01 M PBS
gewaschen und dann für 30 min bei RT mit einem HCMV E-Ag Antikörper (CCH2,
DAKO, Glostrup, Dänemark) in einer Verdünnung von 1:50 in 0,01 M PBS/1 %
BSA inkubiert. Nach dreimaligen Waschen mit PBS erfolgte die Inkubation mit
einem horseradish-peroxidase (HRP)-gekoppelten anti-Maus Sekundärantikörper
(DAKO, Glostrup, Dänemark; 1:100 in 0,01 M PBS / 1 % BSA für 30 min, RT). Die
Zellen wurden abermals mit 0,01 M PBS gewaschen und die Antigen-
Antikörperkomplexe nach einer Färbereaktion mit AEC-Lösung (DAKO, Glostrup,
Dänemark) detektiert. Die Reaktion wurde durch Waschen mit 0,01 M PBS
gestoppt und die gefärbten Zellkerne gezählt.
2.16.4 Wachstumskinetik
Um das Replikationsverhalten von Virus vergleichen zu können, wurden „Ein-
Schritt-Wachstumskurven“ bestimmt. Ca. 5x104 HFF bzw. HUVEC pro Loch
wurden auf 24-Loch-Platten verteilt und in einem Endvolumen von 500 µl MEM-
komplett für 1 h infiziert. Parallel dazu wurde eine Titration des Inokulums
durchgeführt, um zu gewährleisten, dass initial vergleichbare Mengen an
Infektiosität eingesetzt wurden. Nach Entfernen des Virus und zweimaligem
Waschen mit 0,01 M PBS wurde der Überstand abgenommen, mit dem gleichen
Volumen Sucrosephosphat bei –70°C weggefroren (Zeitpunkt t0) und durch
frisches Medium (im Fall der HUVECs EBM) ersetzt. Ebenso wurde zu den
weiteren Erntezeitpunkten verfahren.
Im Anschluss wurde eine Titration durchgeführt, um die Anzahl der plaque forming
units über den Zeitverlauf bestimmen und somit das Replikationsvermögen der
Viren vergleichen zu können.

Material und Methoden 34
2.17 Retrovirale Transduktion
Amphotrope Phönix Helferzellen [Pear et al., 1993] die in 6 Loch Platten kultiviert
wurden, wurden durch Kalzium-Phosphat-Präzipitation mit 5 µg der retroviralen
Vektoren p617-neo-T-EGFP-tTA3 oder pMLV-Retro-tTA3-DEST Myc-UL24
transfiziert (Abb. 5.1). Am Folgetag wurde das Medium gewechselt und die Zellen
bei 32°C inkubiert. Nach weiteren 24 h wurde der mit Retroviren angereicherte
Überstand geerntet und die Zelltrümmer pelletiert (5 min, 1500 rpm ). HFF wurden
mit 1 ml frischem Virusüberstand infiziert und für 24 h bei 32°C inkubiert (Abb.
5.2). Die Infektion wurde hierbei durch einen 45minütigen Zentrifugationsschritt bei
1800 rpm verstärkt. Die transduzierten HFF wurden nach einem Waschschritt mit
TB4delUL24-BAC überinfiziert (MOI=1, Abb. 5.3). Die Überstände der infizierten
HFF wurden 5 dpi geerntet um eine erfolgreiche Transkomplementation in HFF
und HUVEC testen zu können. HFF und HUVEC wurden mit jeweils 1 ml
Überstand infiziert (Abb 5.4) und infizierte Zellen durch Immunfluoreszenzanalyse
unter Verwendung eines monoklonalen Antikörpers (E13, Argene) gegen das
HCMV IE1-Protein nachgewiesen (Abb 5.5). Zur Bestätigung einer erfolgreichen
Transduktion wurden die infizierten Zellen ebenfalls geerntet, und über RT-PCR
auf die Expression von UL24-mRNA Transkripten untersucht.
Abb. 5: Schematische Darstellung zur Vorgehensweise bei Retroviraler Transduktion.

Material und Methoden 35
2.18 Luziferaseassay
U373 Zellen wurden mit 3x105 Zellen pro Loch in einer 6 Loch-Platte ausgesäht.
Am nächsten Tag wurde das Medium durch 750 μl frisches DMEM ersetzt. Die zu
transfizierende DNA (0,5 μg Reporterplasmid und je 2 μg Transaktivator-DNA)
wurde mit 8 μl DEAE-Dextran (50 mg/ml) versetzt, mit 250 μl DMEM aufgefüllt und
auf die Zellen getropft. Nach 3stündiger Inkubation im Brutschrank wurden die
Zellen zweimal mit warmem 0,01 M PBS gewaschen, mit 2 ml frischem DMEM
überschichtet und weitere 48 h kultiviert. Für die Herstellung der Zellextrakte
wurden die Zellen zweimal mit 0,01 M PBS gewaschen, mit 300 μl 1 x Lysepuffer
überschichtet und für 30 min auf einem Schüttler inkubiert. Der Überstand wurde
in Reaktionsgefäße überführt und die Zelltrümmer sedimentiert (16000 × g, 10
min, 4 °C). Für die Messung der Luziferase-Aktivität wurden 100 μl Zelllysat mit
100 μl Reaktionspuffer (100 mM Kaliumphosphat pH 7,8, 15 mM MgSO4, 5 mM
ATP) gemischt. Die Luminiszenz-Reaktion wurde durch Einspritzen von 100 μl
Substratpuffer (Reaktionspuffer plus 1 mM D-Luziferin, Roche, Mannheim) in
einem Luminometer (Lumat LB9507, Berthold, Bad Wildheim) vermessen und in
relativen Luminiszenzeinheiten (RLU) angegeben.
Ergebnisse 36
3 Ergebnisse
3.1 Prokaryote Expression von MBP-pUL24
Für die Immunisierung von Kaninchen sollte pUL24 zunächst prokaryot exprimiert
werden. Da sich in vorangegangenen Versuchen Fusionsproteine wie His-pUL24
oder GST-pUL24 gar nicht oder nur in sehr geringen Mengen herstellen ließen,
wurde pUL24 als Maltose Bindeprotein (MBP)-Fusionsprotein in E. coli DH10B in
löslicher Form exprimiert. Bereits im Überstand des Zelllysats konnte in einem
Coomassieblau gefärbten SDS-Gel eine distinkte Bande von ca. 75 kDa
nachgewiesen werden, die der erwarteten Molekularmasse des Fusionsproteins
entspricht. Das Fusionsprotein wurde durch anschließende Reinigungsschritte
über ein Amyloseresin weiter angereichert (Abb. 6 A). Das Fusionsprotein wurde
in mehreren Fraktionen eluiert, auf Nitrozellulose transferiert und mittels Ponceau-
Rot-Färbung nachgewiesen (Abb. 6 B). Die entsprechende Bande wurde
ausgeschnitten, in DMSO aufgelöst und für die Immunisierung eingesetzt.
Das Kaninchenserum wurde in Immunfluoreszenzanalysen an mit pcDNA-Myc-
UL24-IRES-EGFP transient transfizierten HeLa-Zellen getestet. Hierbei wurden
erfolgreich transfizierte Zellen über die Fluoreszenz des bicistronisch exprimierten
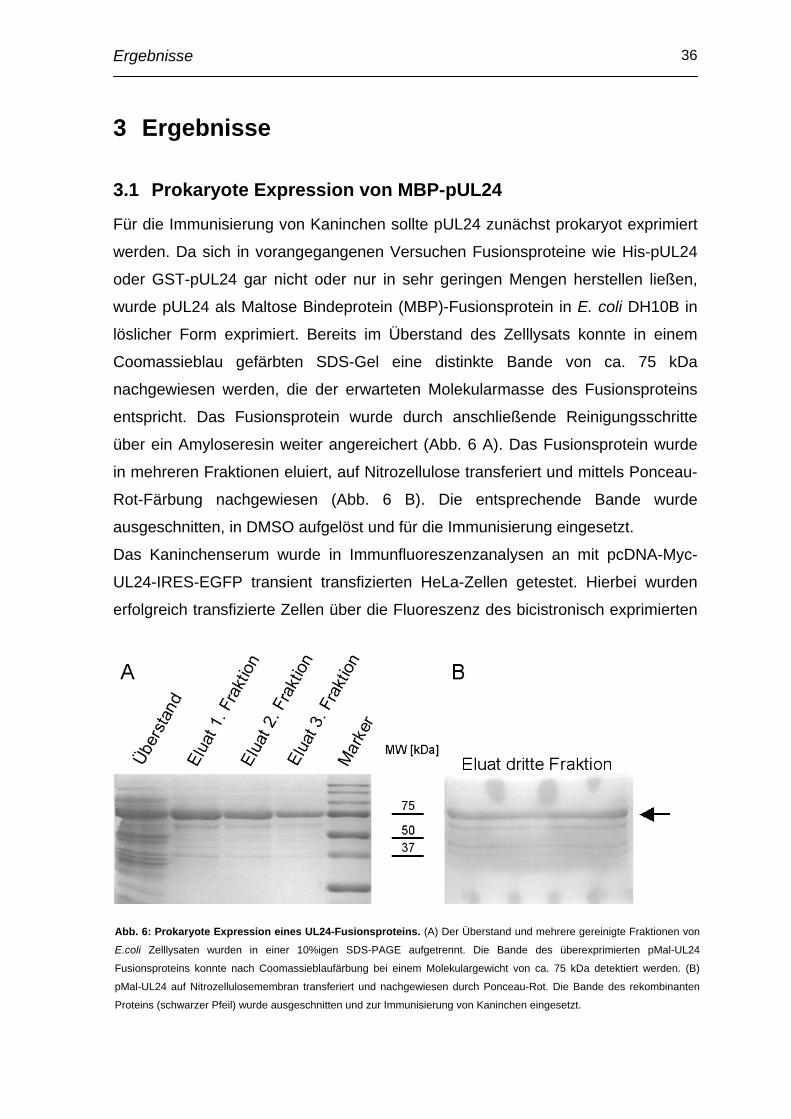
Abb. 6: Prokaryote Expression eines UL24-Fusionsproteins. (A) Der Überstand und mehrere gereinigte Fraktionen von
E.coli Zelllysaten wurden in einer 10%igen SDS-PAGE aufgetrennt. Die Bande des überexprimierten pMal-UL24
Fusionsproteins konnte nach Coomassieblaufärbung bei einem Molekulargewicht von ca. 75 kDa detektiert werden. (B)
pMal-UL24 auf Nitrozellulosemembran transferiert und nachgewiesen durch Ponceau-Rot. Die Bande des rekombinanten
Proteins (schwarzer Pfeil) wurde ausgeschnitten und zur Immunisierung von Kaninchen eingesetzt.
Ergebnisse 37
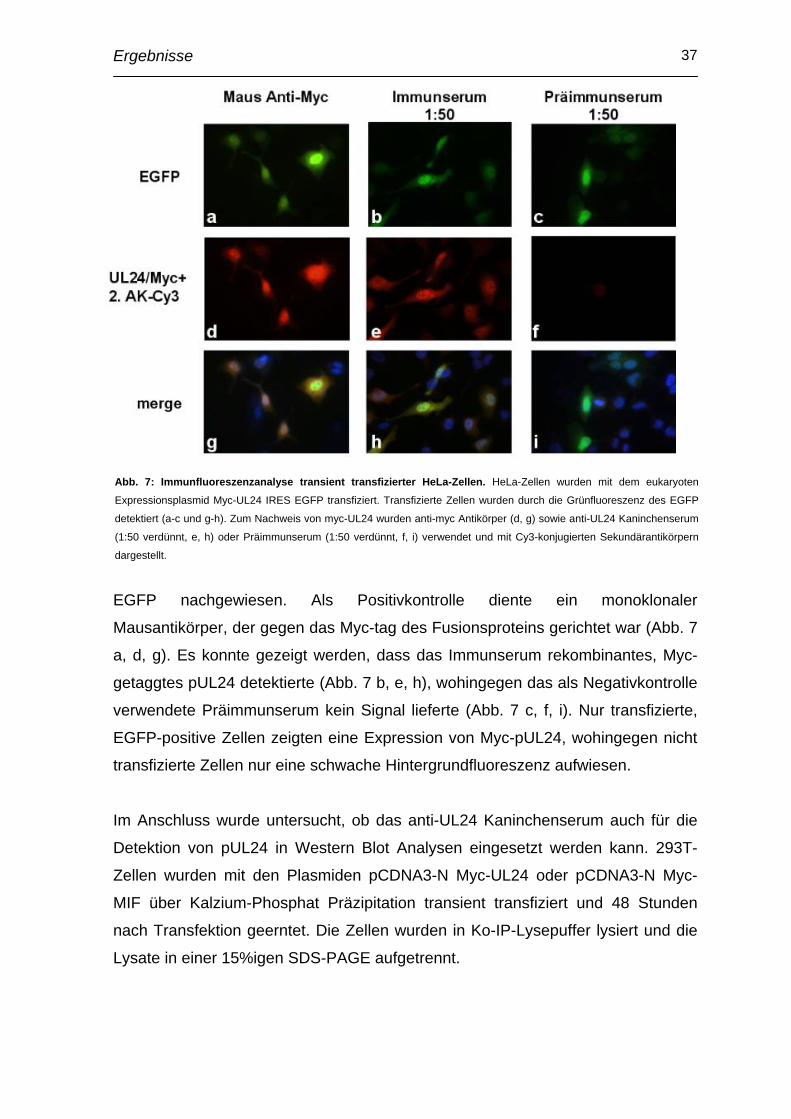
EGFP nachgewiesen. Als Positivkontrolle diente ein monoklonaler
Mausantikörper, der gegen das Myc-tag des Fusionsproteins gerichtet war (Abb. 7
a, d, g). Es konnte gezeigt werden, dass das Immunserum rekombinantes, Myc-
getaggtes pUL24 detektierte (Abb. 7 b, e, h), wohingegen das als Negativkontrolle
verwendete Präimmunserum kein Signal lieferte (Abb. 7 c, f, i). Nur transfizierte,
EGFP-positive Zellen zeigten eine Expression von Myc-pUL24, wohingegen nicht
transfizierte Zellen nur eine schwache Hintergrundfluoreszenz aufwiesen.
Abb. 7: Immunfluoreszenzanalyse transient transfizierter HeLa-Zellen. HeLa-Zellen wurden mit dem eukaryoten
Expressionsplasmid Myc-UL24 IRES EGFP transfiziert. Transfizierte Zellen wurden durch die Grünfluoreszenz des EGFP
detektiert (a-c und g-h). Zum Nachweis von myc-UL24 wurden anti-myc Antikörper (d, g) sowie anti-UL24 Kaninchenserum
(1:50 verdünnt, e, h) oder Präimmunserum (1:50 verdünnt, f, i) verwendet und mit Cy3-konjugierten Sekundärantikörpern
dargestellt.
Im Anschluss wurde untersucht, ob das anti-UL24 Kaninchenserum auch für die
Detektion von pUL24 in Western Blot Analysen eingesetzt werden kann. 293T-
Zellen wurden mit den Plasmiden pCDNA3-N Myc-UL24 oder pCDNA3-N Myc-
MIF über Kalzium-Phosphat Präzipitation transient transfiziert und 48 Stunden
nach Transfektion geerntet. Die Zellen wurden in Ko-IP-Lysepuffer lysiert und die
Lysate in einer 15%igen SDS-PAGE aufgetrennt.
Ergebnisse 38
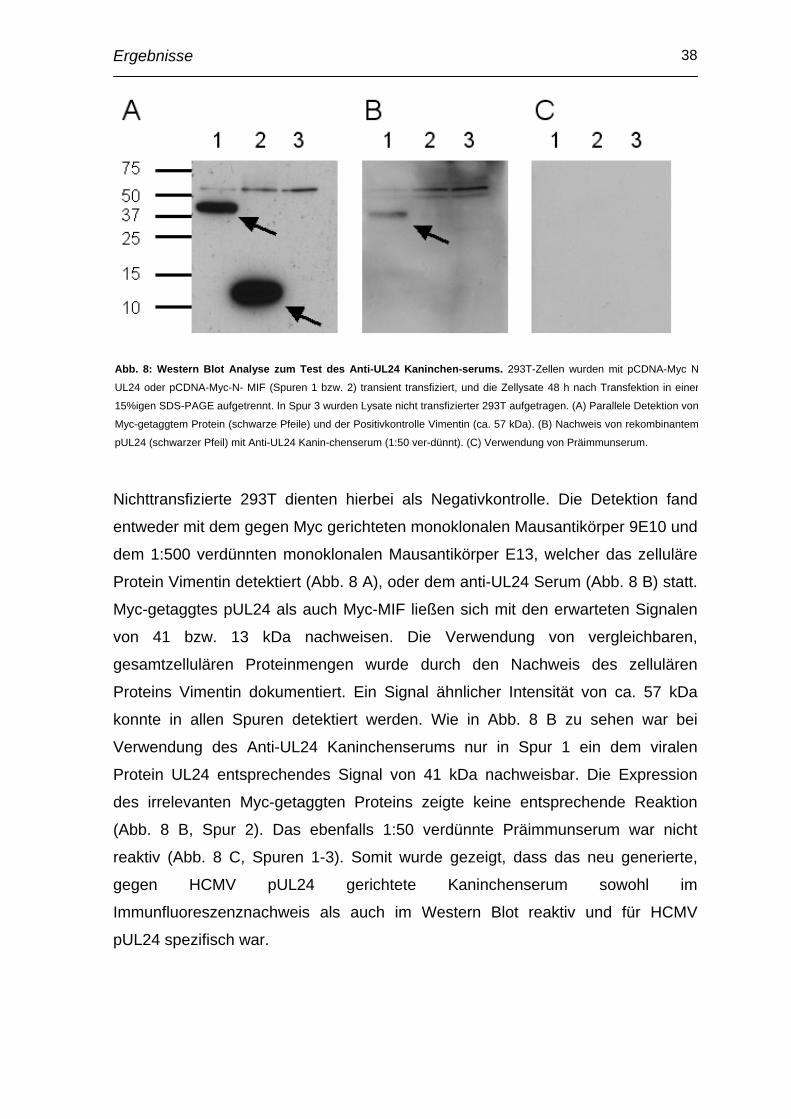
Nichttransfizierte 293T dienten hierbei als Negativkontrolle. Die Detektion fand
entweder mit dem gegen Myc gerichteten monoklonalen Mausantikörper 9E10 und
dem 1:500 verdünnten monoklonalen Mausantikörper E13, welcher das zelluläre
Protein Vimentin detektiert (Abb. 8 A), oder dem anti-UL24 Serum (Abb. 8 B) statt.
Myc-getaggtes pUL24 als auch Myc-MIF ließen sich mit den erwarteten Signalen
von 41 bzw. 13 kDa nachweisen. Die Verwendung von vergleichbaren,
gesamtzellulären Proteinmengen wurde durch den Nachweis des zellulären
Proteins Vimentin dokumentiert. Ein Signal ähnlicher Intensität von ca. 57 kDa
konnte in allen Spuren detektiert werden. Wie in Abb. 8 B zu sehen war bei
Verwendung des Anti-UL24 Kaninchenserums nur in Spur 1 ein dem viralen
Protein UL24 entsprechendes Signal von 41 kDa nachweisbar. Die Expression
des irrelevanten Myc-getaggten Proteins zeigte keine entsprechende Reaktion
(Abb. 8 B, Spur 2). Das ebenfalls 1:50 verdünnte Präimmunserum war nicht
reaktiv (Abb. 8 C, Spuren 1-3). Somit wurde gezeigt, dass das neu generierte,
gegen HCMV pUL24 gerichtete Kaninchenserum sowohl im
Immunfluoreszenznachweis als auch im Western Blot reaktiv und für HCMV
pUL24 spezifisch war.
Abb. 8: Western Blot Analyse zum Test des Anti-UL24 Kaninchen-serums. 293T-Zellen wurden mit pCDNA-Myc N
UL24 oder pCDNA-Myc-N- MIF (Spuren 1 bzw. 2) transient transfiziert, und die Zellysate 48 h nach Transfektion in einer
15%igen SDS-PAGE aufgetrennt. In Spur 3 wurden Lysate nicht transfizierter 293T aufgetragen. (A) Parallele Detektion von
Myc-getaggtem Protein (schwarze Pfeile) und der Positivkontrolle Vimentin (ca. 57 kDa). (B) Nachweis von rekombinantem
pUL24 (schwarzer Pfeil) mit Anti-UL24 Kanin-chenserum (1:50 ver-dünnt). (C) Verwendung von Präimmunserum.

Ergebnisse 39
3.2 Charakterisierung einer pUL24/ppUL32 Interaktion
Daten aus Yeast Two-Hybrid Screens (Schierling, nicht veröffentllicht) wiesen
pUL24 als Interaktor von ppUL32 aus. Um diese Daten in menschlichen Zellen zu
bestätigen, wurden 293T oder HeLa-Zellen transient mit eukaryoten
Expressionsvektoren transfiziert, welche für pUL24 oder ppUL32 kodieren oder
HFF mit HCMV infiziert und die gewonnenen Lysate für
Koimmunpräzipitationsanalysen eingesetzt.
3.2.1 Charakterisierung und Bestätigung der pUL24/ppUL32 Interaktion durch Ko-Immunpräzipitation
Die Interaktionsdaten von pUL24 und ppUL32 aus den Experimenten in Hefe
konnten nach transienter Kotransfektion in 293T-Zellen über ein Ko-
immunpräzipitationsexperiment bestätigt werden (Daten nicht gezeigt). Um
herauszufinden, welche Art von Protein-Protein Bindung der identifizierten
pUL24/ppUL32-Interaktion zugrunde liegt, wurden in diesem Experiment
verschiedene Lysepuffer mit zunehmenden Ionenkonzentrationen (150mM,
300mM und 500mM NaCl) verwendet. Dadurch sollten Interaktionen, die auf
elektrostatischen Bindungen basieren inhibiert werden. 293T-Zellen wurden mit
Abb. 9: Koimmunpräzipitation austransient transfizierten Zellen in Gegenwartansteigender Salzkonzentrationen.Angegebene Proteine wurden in 293T-Zellen
überexprimiert. (A) Die Immunpräzipitation
erfolgte mit dem monoklonalen
Mausantikörper XP1. Kopräzipitiertes pUL24
wurde mit dem spezifischen
Kaninchenantiserums detektiert. (B) In allen
Zelllysat-Kontrollen konnte pUL24
nachgewiesen werden. Zusätzlich wurde das
virale Protein IE1 nachgewiesen (Spur 4 ).

Ergebnisse 40
pCDNA3-UL24 und pCB6-UL32 bzw. pCDNA3-UL24 und pCDNA3-IE1
kotransfiziert und 48 Stunden nach Transfektion mit den Lysepuffern
unterschiedlicher Salzkonzentration aufgeschlossen. Die Lysate wurden mit dem
gegen ppUL32 gerichteten Maus-Antikörper XP1 oder im Fall der Negativkontrolle
mit dem monoklonalen Anti-IE-Mausantikörper (p63-27) inkubiert und präzipitiert.
Der Nachweis von kopräzipitiertem pUL24 erfolgte mit Anti-pUL24
Kaninchenserum. Das Protein konnte in Gegenwart von allen drei
Salzkonzentrationen nachgewiesen werden (Abb. 9, Spuren 1 bis 3), was darauf
hinweist, dass die Bindung nicht nur auf elektrostatischen Kräften beruht. Die
Negativkontrolle diente dazu, eine unspezifische Präzipitation von pUL24
auszuschließen.
Um zu zeigen, dass die Interaktion nicht nur nach Überexpression nachgewiesen
werden kann, sondern auch bei der Virusinfektion eine Rolle spielt, wurden HFF
mit HCMV TB40E infiziert (MOI=1), Zelllysate 96 hpi geerntet und mit Antiseren
gegen pUL24 (Kaninchen) oder pUL48 (Mensch) präzipitiert. Der Nachweis von
kopräzipitiertem ppUL32 erfolgte mit Hilfe des monoklonalen Maus anti-ppUL32
Antikörpers XP1. Das Protein ppUL32 konnte durch pUL24, aber nicht durch
pUL48, ein weiteres Tegumentprotein, kopräzipitiert werden (Abb. 10 A, Spuren 1
und 2). In den Lysatkontrollen (Abb. 10 B) ist zu sehen, dass ppUL32 Bestandteil
beider Präparationen ist.
In diesem Experiment konnte erstmals gezeigt werden, dass eine physikalische
Interaktion zwischen pUL24 und ppUL32 tatsächlich auch in HCMV-infizierten HFF
stattfindet und demnach auch mit hoher Wahrscheinlichkeit für das Virus bzw. eine
Infektion mit HCMV relevant ist.
Abb. 10: Ko-Immunpräzipitation aus infizierten HFF. (A)
HFF wurden mit einer MOI von 1 mit TB4wt-BAC infiziert und
96 hpi geerntet. Die Immunpräzipitation erfolgte mit anti-UL24
Kaninchenserum (Spur 1) oder einem anti-UL48
Humanserum (Spur 2). Kopräzipitiertes ppUL32 wurde im
Western Blot unter Verwendung des monoklonalen
Mausantikörpers XP1 nachgewiesen. (B) In der Lysatkontrolle
wurde in beiden Präparationen ppUL32 im Western Blot unter
Verwendung des monoklonalen Mausantikörpers XP1
nachgewiesen (Spuren 1 und 2).
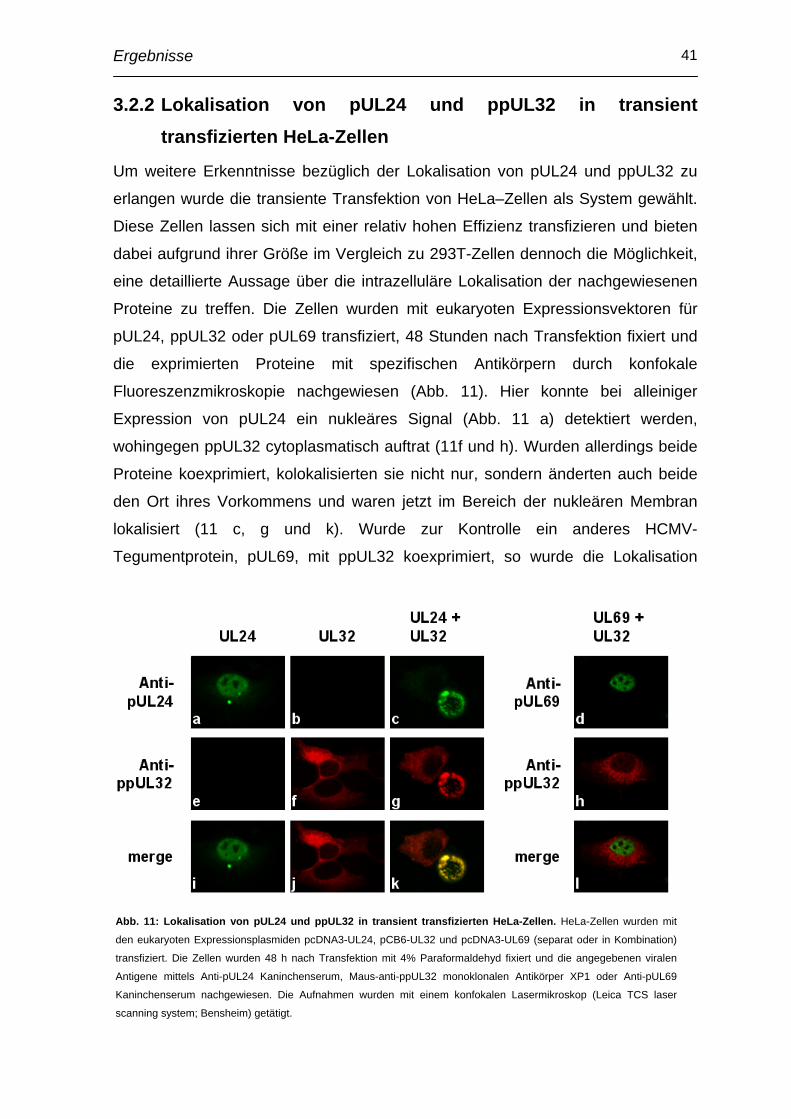
Ergebnisse 41
3.2.2 Lokalisation von pUL24 und ppUL32 in transient transfizierten HeLa-Zellen
Um weitere Erkenntnisse bezüglich der Lokalisation von pUL24 und ppUL32 zu
erlangen wurde die transiente Transfektion von HeLa–Zellen als System gewählt.
Diese Zellen lassen sich mit einer relativ hohen Effizienz transfizieren und bieten
dabei aufgrund ihrer Größe im Vergleich zu 293T-Zellen dennoch die Möglichkeit,
eine detaillierte Aussage über die intrazelluläre Lokalisation der nachgewiesenen
Proteine zu treffen. Die Zellen wurden mit eukaryoten Expressionsvektoren für
pUL24, ppUL32 oder pUL69 transfiziert, 48 Stunden nach Transfektion fixiert und
die exprimierten Proteine mit spezifischen Antikörpern durch konfokale
Fluoreszenzmikroskopie nachgewiesen (Abb. 11). Hier konnte bei alleiniger
Expression von pUL24 ein nukleäres Signal (Abb. 11 a) detektiert werden,
wohingegen ppUL32 cytoplasmatisch auftrat (11f und h). Wurden allerdings beide
Proteine koexprimiert, kolokalisierten sie nicht nur, sondern änderten auch beide
den Ort ihres Vorkommens und waren jetzt im Bereich der nukleären Membran
lokalisiert (11 c, g und k). Wurde zur Kontrolle ein anderes HCMV-
Tegumentprotein, pUL69, mit ppUL32 koexprimiert, so wurde die Lokalisation
Abb. 11: Lokalisation von pUL24 und ppUL32 in transient transfizierten HeLa-Zellen. HeLa-Zellen wurden mit
den eukaryoten Expressionsplasmiden pcDNA3-UL24, pCB6-UL32 und pcDNA3-UL69 (separat oder in Kombination)
transfiziert. Die Zellen wurden 48 h nach Transfektion mit 4% Paraformaldehyd fixiert und die angegebenen viralen
Antigene mittels Anti-pUL24 Kaninchenserum, Maus-anti-ppUL32 monoklonalen Antikörper XP1 oder Anti-pUL69
Kaninchenserum nachgewiesen. Die Aufnahmen wurden mit einem konfokalen Lasermikroskop (Leica TCS laser
scanning system; Bensheim) getätigt.

Ergebnisse 42
keines der beiden Proteine beeinflusst (11 d, h und l). Zusammenfassend kann
man sagen, dass in diesem Experiment die vorangegangenen Interaktionsdaten
bestätigt und durch die gemeinsame Lokalisation der beiden Interaktionspartner
gefestigt werden konnten. Der Umstand, dass pUL24 in Gegenwart von ppUL32
nicht mehr seine gewohnte Lokalisation im Zellkern einnimmt, ist ein Hinweis
darauf, dass eine eventuelle Beteiligung von pUL24 an nukleären Prozessen
durch die Interaktion mit ppUL32 beeinträchtigt sein könnte.
3.3 Funktionelle Analyse von pUL24 mittels Luziferaseassay
Es konnte bereits für eine Reihe von Tegumentproteinen gezeigt werden, dass es
sich bei ihnen um virale Transaktivatoren handelt (pUL82, ppUL35, pUL69
[Bresnahan et al., 2000, Liu et al., 2002, Winkler et al., 1994]). Weiterhin ändert
pUL24 in Gegenwart seines Interaktionspartners ppUL32 seine gewohnte,
nukleäre Lokalisation, wodurch eine eventuelle genregulierende Funktion von
pUL24 aufgrund der Interaktion beeinträchtigt sein könnte. Aus diesen Gründen
wurde ein Set an Experimenten durchgeführt, welches einer Beteiligung von
pUL24 an regulatorischen Prozessen nachgeht.
3.3.1 Verwendung verschiedener Promotorkonstrukte
Mit pUL36, pIRS1 und pTRS1 ist für mehrere Mitglieder der US22 Genfamilie eine
transkriptionell transaktivierende Funktion auf den HCMV IE-Promotor
beschrieben [Colberg-Poley et al., 1992, Stasiak et al., 1992]. Da auch das UL24-
Homolog aus HHV6, U3, eine bis zu fünffache Transaktivierung des HIV-1 LTR
bewirkt [Mori et al., 1998], wurde über Luziferase-Reportergenassays eine
mögliche Wirkung von pUL24 auf unterschiedliche Promotoren untersucht. U373-
Zellen wurden mit Konstrukten transfiziert, welche Luziferase unter der Kontrolle
von HIV-1-LTR, SV40- oder HCMV-IE-Promotor exprimieren. Gleichzeitig wurden
die Zellen mit Plasmiden, welche für pUL24 oder ppUL32 kodieren, kotransfiziert.
Bei allen drei verwendeten Promotor-Luziferasekonstrukten konnte in Gegenwart
von pUL24 eine 2-fache Transaktivierung beobachtet werden (Abb. 12, Spuren 1,
5 und 9), wohingegen ppUL32 allein zumindest für den HIV-1 LTR und den
HCMV-IE Promotor eine Repression bewirkte (Abb. 12, Spuren 2 und 9). Der
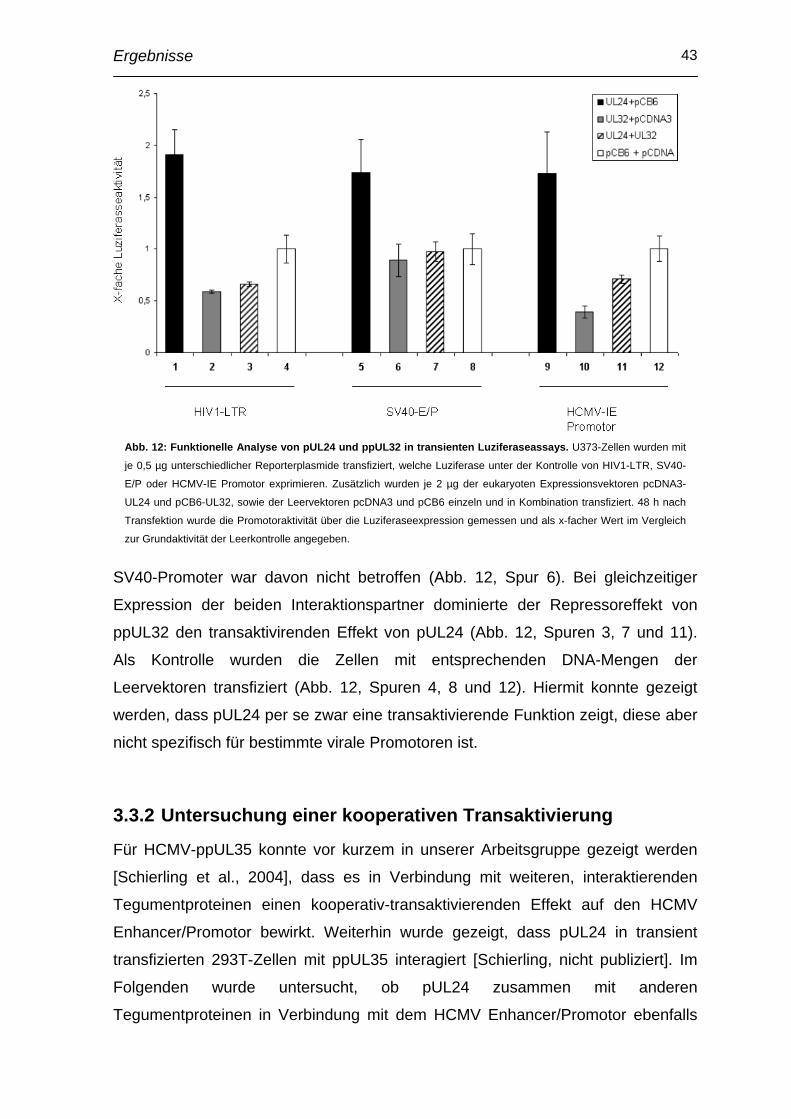
Ergebnisse 43
SV40-Promoter war davon nicht betroffen (Abb. 12, Spur 6). Bei gleichzeitiger
Expression der beiden Interaktionspartner dominierte der Repressoreffekt von
ppUL32 den transaktivirenden Effekt von pUL24 (Abb. 12, Spuren 3, 7 und 11).
Als Kontrolle wurden die Zellen mit entsprechenden DNA-Mengen der
Leervektoren transfiziert (Abb. 12, Spuren 4, 8 und 12). Hiermit konnte gezeigt
werden, dass pUL24 per se zwar eine transaktivierende Funktion zeigt, diese aber
nicht spezifisch für bestimmte virale Promotoren ist.
Abb. 12: Funktionelle Analyse von pUL24 und ppUL32 in transienten Luziferaseassays. U373-Zellen wurden mit
je 0,5 µg unterschiedlicher Reporterplasmide transfiziert, welche Luziferase unter der Kontrolle von HIV1-LTR, SV40-
E/P oder HCMV-IE Promotor exprimieren. Zusätzlich wurden je 2 µg der eukaryoten Expressionsvektoren pcDNA3-
UL24 und pCB6-UL32, sowie der Leervektoren pcDNA3 und pCB6 einzeln und in Kombination transfiziert. 48 h nach
Transfektion wurde die Promotoraktivität über die Luziferaseexpression gemessen und als x-facher Wert im Vergleich
zur Grundaktivität der Leerkontrolle angegeben.
3.3.2 Untersuchung einer kooperativen Transaktivierung
Für HCMV-ppUL35 konnte vor kurzem in unserer Arbeitsgruppe gezeigt werden
[Schierling et al., 2004], dass es in Verbindung mit weiteren, interaktierenden
Tegumentproteinen einen kooperativ-transaktivierenden Effekt auf den HCMV
Enhancer/Promotor bewirkt. Weiterhin wurde gezeigt, dass pUL24 in transient
transfizierten 293T-Zellen mit ppUL35 interagiert [Schierling, nicht publiziert]. Im
Folgenden wurde untersucht, ob pUL24 zusammen mit anderen
Tegumentproteinen in Verbindung mit dem HCMV Enhancer/Promotor ebenfalls
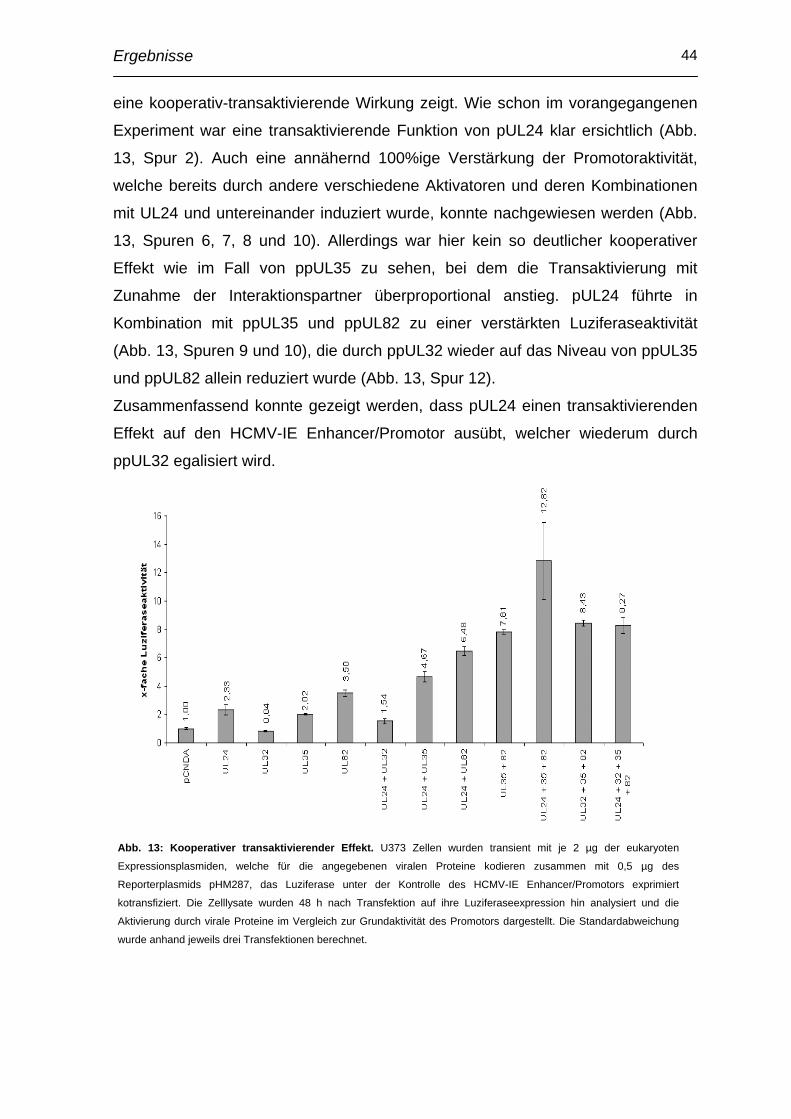
Ergebnisse 44
eine kooperativ-transaktivierende Wirkung zeigt. Wie schon im vorangegangenen
Experiment war eine transaktivierende Funktion von pUL24 klar ersichtlich (Abb.
13, Spur 2). Auch eine annähernd 100%ige Verstärkung der Promotoraktivität,
welche bereits durch andere verschiedene Aktivatoren und deren Kombinationen
mit UL24 und untereinander induziert wurde, konnte nachgewiesen werden (Abb.
13, Spuren 6, 7, 8 und 10). Allerdings war hier kein so deutlicher kooperativer
Effekt wie im Fall von ppUL35 zu sehen, bei dem die Transaktivierung mit
Zunahme der Interaktionspartner überproportional anstieg. pUL24 führte in
Kombination mit ppUL35 und ppUL82 zu einer verstärkten Luziferaseaktivität
(Abb. 13, Spuren 9 und 10), die durch ppUL32 wieder auf das Niveau von ppUL35
und ppUL82 allein reduziert wurde (Abb. 13, Spur 12).
Zusammenfassend konnte gezeigt werden, dass pUL24 einen transaktivierenden
Effekt auf den HCMV-IE Enhancer/Promotor ausübt, welcher wiederum durch
ppUL32 egalisiert wird.
Abb. 13: Kooperativer transaktivierender Effekt. U373 Zellen wurden transient mit je 2 µg der eukaryoten
Expressionsplasmiden, welche für die angegebenen viralen Proteine kodieren zusammen mit 0,5 µg des
Reporterplasmids pHM287, das Luziferase unter der Kontrolle des HCMV-IE Enhancer/Promotors exprimiert
kotransfiziert. Die Zelllysate wurden 48 h nach Transfektion auf ihre Luziferaseexpression hin analysiert und die
Aktivierung durch virale Proteine im Vergleich zur Grundaktivität des Promotors dargestellt. Die Standardabweichung
wurde anhand jeweils drei Transfektionen berechnet.

Ergebnisse 45
3.4 Generierung und Charakterisierung einer UL24-Deletionsmutante im Hintergrund des endotheliotropen HCMV-Stammes TB40E
Nachdem für einige Mitglieder der US22-Genfamilie auch eine Rolle für den
Zelltropismus von CMV gezeigt werden konnte und bei vorangegangenen
Experimenten mit einer UL24-Deletionsmutante im an Fibroblasten adaptierten
Laborstamm AD169 bei der Infektion von HFF keinerlei Defekt auftrat (R. Pretsch,
Diplomarbeit), wurde für die Generierung eines UL24-Deletionsvirus das TB4-BAC
gewählt, welchem der hochgradig endotheliotrope HCMV-Stamm TB40E zugrunde
liegt. Der Vorteil der Verwendung diesen BACs ist es, dass von ihm abstammende
rekombinante Viren immer noch über die Fähigkeit verfügen, unterschiedlichste
Zelltypen infizieren zu können.
3.4.1 Deletion und Reinsertion des UL24-ORF in TB4-BAC mittels ET-Klonierung
Die Entfernung des kompletten UL24-Leserahmens aus dem HCMV-Genom
erfolgte mittels der Rec-ET-Rekombination durch homologe Rekombination in E.
coli (Abb. 14). Dazu wurde das UL24-Gen zunächst durch eine
Kanamycinresistenz-Kassette ersetzt. Der Marker wurde in einem anschließenden
Schritt über FLP-vermittelte Rekombination zweier FRT-Sequenzen, welche das
Resistenzgen umgeben, wieder aus dem BAC entfernt, so dass die
Deletionsmutante TB4delUL24-BAC unter Verbleib einer 80 Basenpaare
umfassenden Insertion (eine FRT-Sequenz und zwei Bindestellen für die zur
Amplifikation der Markerkassette verwendeten Oligonukleotide) resultierte. Eine
Revertante wurde durch Wiedereinsetzen von UL24 hergestellt. Hierfür wurde
UL24 zusammen mit dem Kanamycingen aus pSL-FRT-TB4-mycUL24 amplifiziert,
in TB4delUL24 einrekombiniert und der Marker anschließend wieder über FLP-
vermittelte Rekombination entfernt. Das entstandene BAC wurde nachfolgend mit
TB4revUL24-BAC bezeichnet und war vom Wildtyp durch eine in der verbliebenen
FRT-Sequenz vorhandene EcoRI-Schnittstelle zu unterscheiden (Abb. 15 A). Aus
einer vergleichenden Restriktionsanalyse konnte man ersehen, dass es im Verlauf
der Rekombination nicht zu größeren Deletionen im viralen Genom gekommen ist
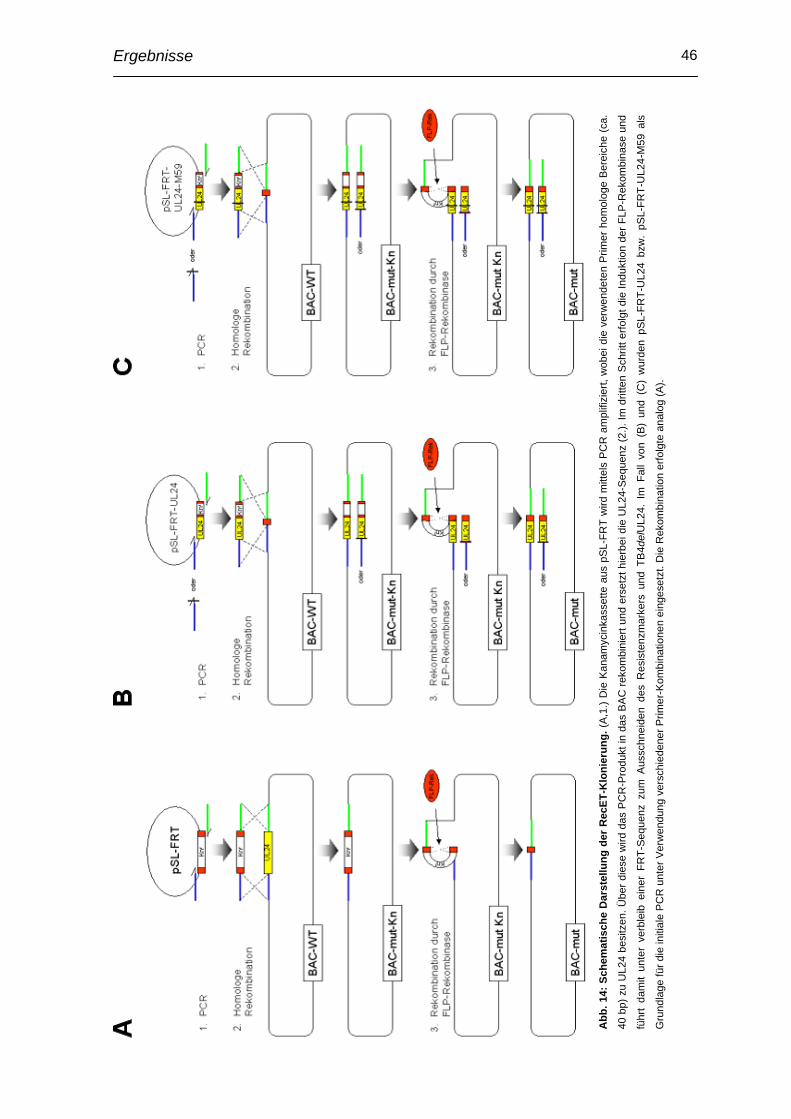
Ergebnisse 46
Abb
. 14:
Sch
emat
isch
e D
arst
ellu
ng d
er R
ecET
-Klo
nier
ung.
(A,1
.) D
ie K
anam
ycin
kass
ette
aus
pS
L-FR
T w
ird m
ittel
s P
CR
am
plifi
zier
t, w
obei
die
ver
wen
dete
n P
rimer
hom
olog
e B
erei
che
(ca.
40 b
p) z
u U
L24
besi
tzen
. Übe
r die
se w
ird d
as P
CR
-Pro
dukt
in d
as B
AC
reko
mbi
nier
t und
ers
etzt
hie
rbei
die
UL2
4-S
eque
nz (2
.). Im
drit
ten
Sch
ritt e
rfolg
t die
Indu
ktio
n de
r FLP
-Rek
ombi
nase
und
führ
t da
mit
unte
r ve
rble
ib e
iner
FR
T-S
eque
nz z
um A
ussc
hnei
den
des
Res
iste
nzm
arke
rs u
nd T
B4d
elU
L24.
Im
Fal
l vo
n (B
) un
d (C
) w
urde
n pS
L-FR
T-U
L24
bzw
. pS
L-FR
T-U
L24-
M59
als
Gru
ndla
ge fü
r die
initi
ale
PC
R u
nter
Ver
wen
dung
ver
schi
eden
er P
rime r
-Kom
bina
tione
n ei
nges
etzt
. Die
Rek
ombi
natio
n er
folg
te a
nalo
g (A
).
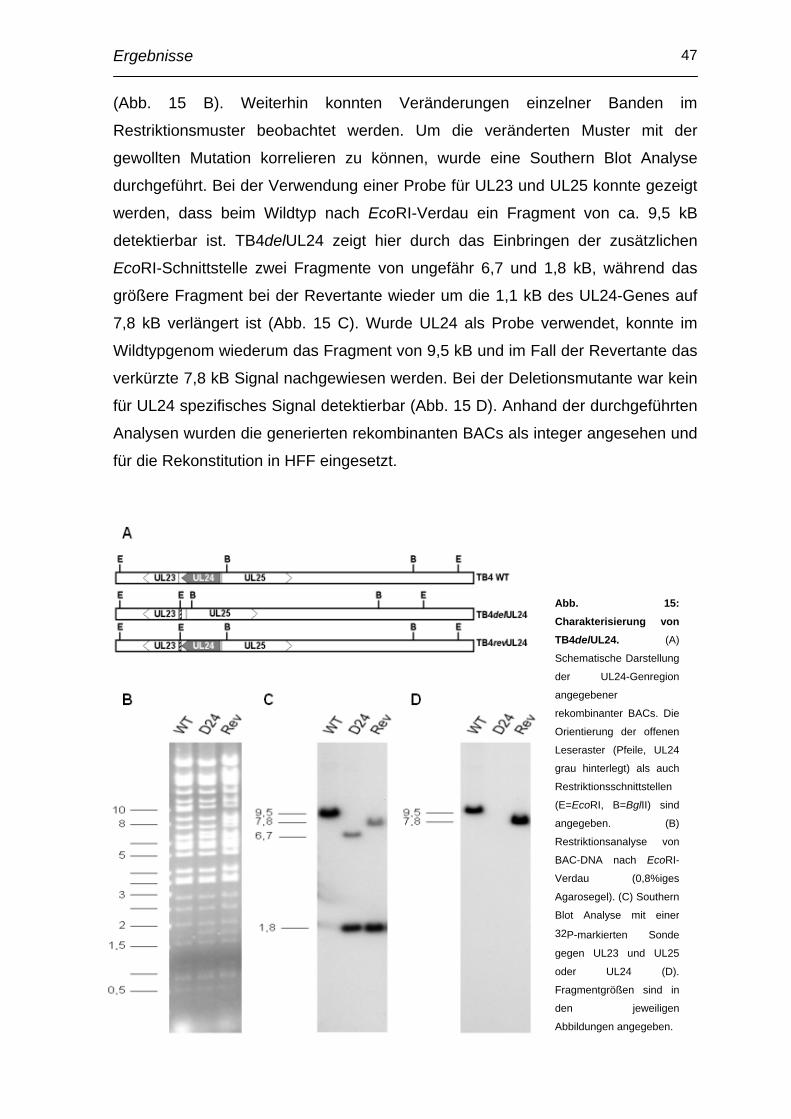
Ergebnisse 47
(Abb. 15 B). Weiterhin konnten Veränderungen einzelner Banden im
Restriktionsmuster beobachtet werden. Um die veränderten Muster mit der
gewollten Mutation korrelieren zu können, wurde eine Southern Blot Analyse
durchgeführt. Bei der Verwendung einer Probe für UL23 und UL25 konnte gezeigt
werden, dass beim Wildtyp nach EcoRI-Verdau ein Fragment von ca. 9,5 kB
detektierbar ist. TB4delUL24 zeigt hier durch das Einbringen der zusätzlichen
EcoRI-Schnittstelle zwei Fragmente von ungefähr 6,7 und 1,8 kB, während das
größere Fragment bei der Revertante wieder um die 1,1 kB des UL24-Genes auf
7,8 kB verlängert ist (Abb. 15 C). Wurde UL24 als Probe verwendet, konnte im
Wildtypgenom wiederum das Fragment von 9,5 kB und im Fall der Revertante das
verkürzte 7,8 kB Signal nachgewiesen werden. Bei der Deletionsmutante war kein
für UL24 spezifisches Signal detektierbar (Abb. 15 D). Anhand der durchgeführten
Analysen wurden die generierten rekombinanten BACs als integer angesehen und
für die Rekonstitution in HFF eingesetzt.
Abb. 15:Charakterisierung vonTB4delUL24. (A)
Schematische Darstellung
der UL24-Genregion
angegebener
rekombinanter BACs. Die
Orientierung der offenen
Leseraster (Pfeile, UL24
grau hinterlegt) als auch
Restriktionsschnittstellen
(E=EcoRI, B=BglII) sind
angegeben. (B)
Restriktionsanalyse von
BAC-DNA nach EcoRI-
Verdau (0,8%iges
Agarosegel). (C) Southern
Blot Analyse mit einer
32P-markierten Sonde
gegen UL23 und UL25
oder UL24 (D).
Fragmentgrößen sind in
den jeweiligen
Abbildungen angegeben.

Ergebnisse 48
3.4.2 Rekonstitution der Virusmutanten
Die jeweilige BAC-DNA wurde mittels Elektroporation in HFF transfiziert. Ungefähr
zehn bis 14 Tage nach Transfektion waren erste für eine HCMV-Infektion typische
zytopathische Effekte der HFF erkennbar, wobei kein merklicher Unterschied
zwischen den verschiedenen rekombinanten Viren erkennbar war. Nachdem die
Zellkulturen nahezu durchinfiziert waren, wurden die Viren expandiert und
Virusstocklösungen angelegt, welche für die Folgeexperimente verwendet wurden.
3.4.3 Einfluss auf den Endothelzelltropismus des HCMV
Vaskuläre Endothelzellen wurden als Zielzellen des Virus identifiziert, welche an
der Verbreitung des Virus im Blutstrom des Wirtsorganismus beteiligt sind. Aus
diesem Grund sind Virusmutanten, bei denen der Endothelzelltropismus des Virus
beeinträchtigt ist, von besonderem Interesse. In den nachfolgenden Experimenten
wurde untersucht, ob die Absenz des UL24-Genproduktes einen derartigen Effekt
hervorruft. Falls ja, sollte im weiteren Verlauf genauer charakterisiert werden, in
welcher Phase der viralen Replikation der Defekt auftritt.
3.4.3.1 Analyse der Replikationskinetiken von HCMV-Mutanten in HFF und HUVEC
HFF als auch HUVEC wurden mit TB4wtBAC, delUL24 oder revUL24 mit einer
MOI von 1 infiziert. Anschließend wurde das erste Mal Überstand geerntet und
somit der Zeitpunkt t0 gesetzt. Virushaltige Zellkulturüberstände wurden zu den
Zeitpunkten 1, 4, 7, 9 und 12 Tage nach Infektion (days post infection, dpi)
abgenommen und bis zum Zeitpunkt der Analyse bei –80°C gelagert. Die Anzahl
der jeweils vorhandenen infektiösen Einheiten (plaque forming units, pfu) wurde
mittels Virustitration auf HFF bestimmt. Hier konnte gezeigt werden, dass das
UL24-Deletionsvirus (Abb. 16 A, gefüllte Quadrate) in HFF im Vergleich zu Wildtyp
(offener Kreis) und Revertante (offenes Dreieck) keinen detektierbaren
Wachstumsdefekt aufweist. In den Endothelzellen dagegen konnte mehrfach
gezeigt werden, dass die von delUL24 erreichten Virustiter 12 dpi (Abb. 16 B,
schwarze Quadrate) 2-3 Logstufen unter den von WT oder Revertante erreichten
Werten lagen. Somit konnte ein ausgeprägter, zelltypspezifischer
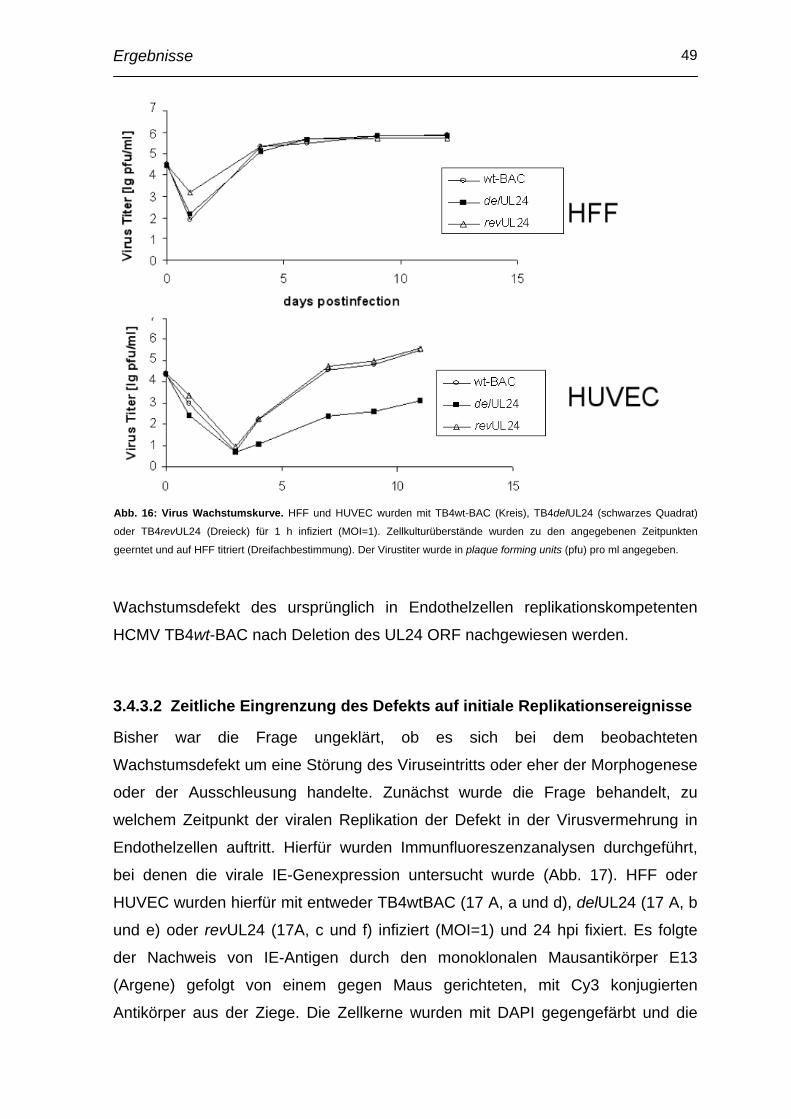
Ergebnisse 49
Wachstumsdefekt des ursprünglich in Endothelzellen replikationskompetenten
HCMV TB4wt-BAC nach Deletion des UL24 ORF nachgewiesen werden.
Abb. 16: Virus Wachstumskurve. HFF und HUVEC wurden mit TB4wt-BAC (Kreis), TB4delUL24 (schwarzes Quadrat)
oder TB4revUL24 (Dreieck) für 1 h infiziert (MOI=1). Zellkulturüberstände wurden zu den angegebenen Zeitpunkten
geerntet und auf HFF titriert (Dreifachbestimmung). Der Virustiter wurde in plaque forming units (pfu) pro ml angegeben.
3.4.3.2 Zeitliche Eingrenzung des Defekts auf initiale Replikationsereignisse
Bisher war die Frage ungeklärt, ob es sich bei dem beobachteten
Wachstumsdefekt um eine Störung des Viruseintritts oder eher der Morphogenese
oder der Ausschleusung handelte. Zunächst wurde die Frage behandelt, zu
welchem Zeitpunkt der viralen Replikation der Defekt in der Virusvermehrung in
Endothelzellen auftritt. Hierfür wurden Immunfluoreszenzanalysen durchgeführt,
bei denen die virale IE-Genexpression untersucht wurde (Abb. 17). HFF oder
HUVEC wurden hierfür mit entweder TB4wtBAC (17 A, a und d), delUL24 (17 A, b
und e) oder revUL24 (17A, c und f) infiziert (MOI=1) und 24 hpi fixiert. Es folgte
der Nachweis von IE-Antigen durch den monoklonalen Mausantikörper E13
(Argene) gefolgt von einem gegen Maus gerichteten, mit Cy3 konjugierten
Antikörper aus der Ziege. Die Zellkerne wurden mit DAPI gegengefärbt und die
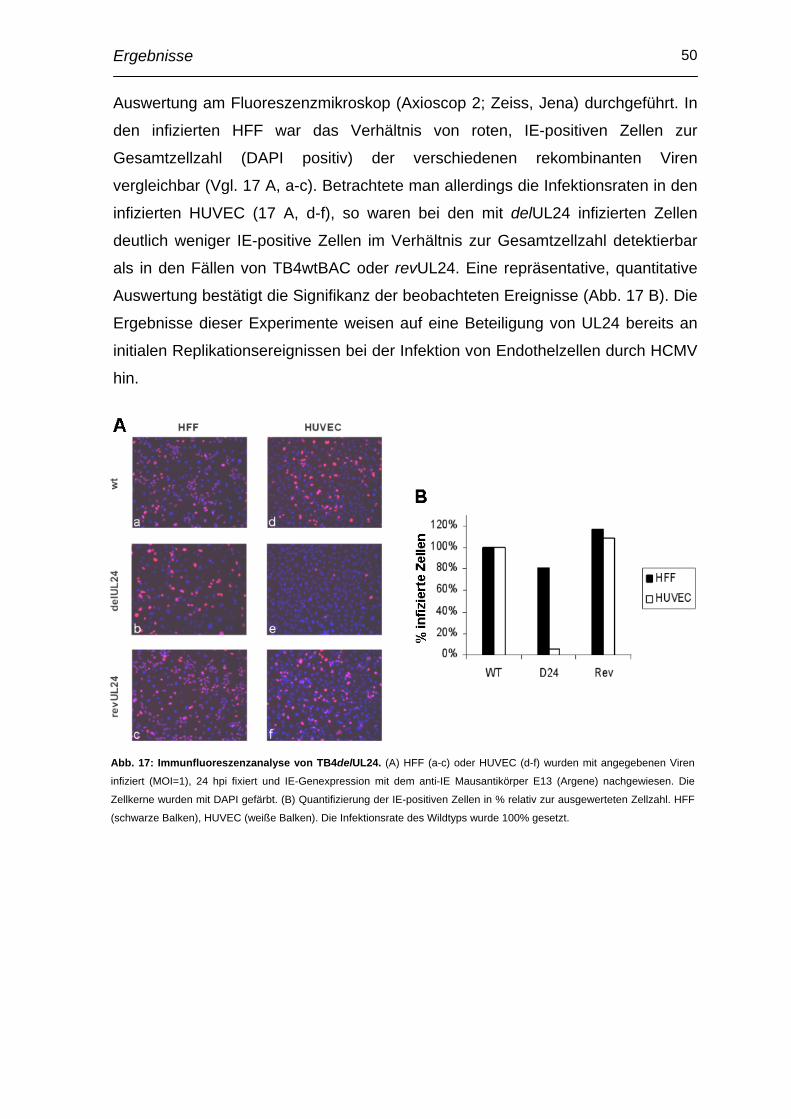
Ergebnisse 50
Auswertung am Fluoreszenzmikroskop (Axioscop 2; Zeiss, Jena) durchgeführt. In
den infizierten HFF war das Verhältnis von roten, IE-positiven Zellen zur
Gesamtzellzahl (DAPI positiv) der verschiedenen rekombinanten Viren
vergleichbar (Vgl. 17 A, a-c). Betrachtete man allerdings die Infektionsraten in den
infizierten HUVEC (17 A, d-f), so waren bei den mit delUL24 infizierten Zellen
deutlich weniger IE-positive Zellen im Verhältnis zur Gesamtzellzahl detektierbar
als in den Fällen von TB4wtBAC oder revUL24. Eine repräsentative, quantitative
Auswertung bestätigt die Signifikanz der beobachteten Ereignisse (Abb. 17 B). Die
Ergebnisse dieser Experimente weisen auf eine Beteiligung von UL24 bereits an
initialen Replikationsereignissen bei der Infektion von Endothelzellen durch HCMV
hin.
Abb. 17: Immunfluoreszenzanalyse von TB4delUL24. (A) HFF (a-c) oder HUVEC (d-f) wurden mit angegebenen Viren
infiziert (MOI=1), 24 hpi fixiert und IE-Genexpression mit dem anti-IE Mausantikörper E13 (Argene) nachgewiesen. Die
Zellkerne wurden mit DAPI gefärbt. (B) Quantifizierung der IE-positiven Zellen in % relativ zur ausgewerteten Zellzahl. HFF
(schwarze Balken), HUVEC (weiße Balken). Die Infektionsrate des Wildtyps wurde 100% gesetzt.
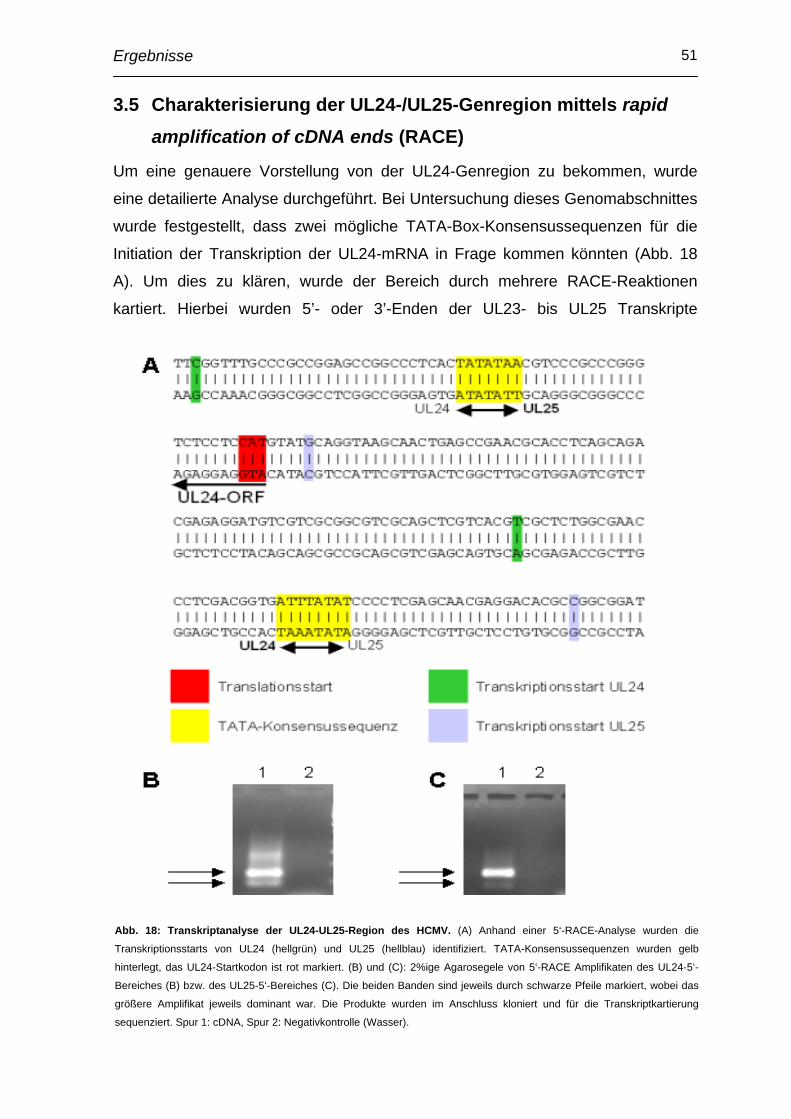
Ergebnisse 51
3.5 Charakterisierung der UL24-/UL25-Genregion mittels rapid amplification of cDNA ends (RACE)
Um eine genauere Vorstellung von der UL24-Genregion zu bekommen, wurde
eine detailierte Analyse durchgeführt. Bei Untersuchung dieses Genomabschnittes
wurde festgestellt, dass zwei mögliche TATA-Box-Konsensussequenzen für die
Initiation der Transkription der UL24-mRNA in Frage kommen könnten (Abb. 18
A). Um dies zu klären, wurde der Bereich durch mehrere RACE-Reaktionen
kartiert. Hierbei wurden 5’- oder 3’-Enden der UL23- bis UL25 Transkripte
Abb. 18: Transkriptanalyse der UL24-UL25-Region des HCMV. (A) Anhand einer 5‘-RACE-Analyse wurden die
Transkriptionsstarts von UL24 (hellgrün) und UL25 (hellblau) identifiziert. TATA-Konsensussequenzen wurden gelb
hinterlegt, das UL24-Startkodon ist rot markiert. (B) und (C): 2%ige Agarosegele von 5‘-RACE Amplifikaten des UL24-5‘-
Bereiches (B) bzw. des UL25-5‘-Bereiches (C). Die beiden Banden sind jeweils durch schwarze Pfeile markiert, wobei das
größere Amplifikat jeweils dominant war. Die Produkte wurden im Anschluss kloniert und für die Transkriptkartierung
sequenziert. Spur 1: cDNA, Spur 2: Negativkontrolle (Wasser).

Ergebnisse 52
amplifiziert, teilweise kloniert und anschließend sequenziert. Die
Transkriptionsstarts für UL24 bzw. UL25 liegen bei –57 und +78 (UL24) bzw. bei
+5 und +133 (UL25) jeweils vom UL24-Translationsstart aus gesehen. Es konnte
gezeigt werden, dass beide erwähnten Initiationsstellen genutzt werden und die
längeren, bevorzugten Transkriptformen (Abb. 18 B und C) von UL24 und UL25
überlappen. UL23 verfügt über einen separaten Transkriptionsstart, wobei die
UL24-mRNA koterminal mit dem UL23-Transkript endet. Diese Ergebnisse führen
zu dem Schluss, dass das Entfernen des kompletten UL24-Leserahmens, und
somit auch des UL25-Promoters, das benachbarte Gen funktionell ausschalten
könnte.
3.6 Herstellung und Charakterisierung von Punktmutanten
Durch das Entfernen eines kompletten Leserahmens könnten auch benachbarte
beeinflusst werden. Aus diesem Grund wurden Mutanten von TB4-BAC generiert,
bei denen nicht der komplette UL24-Leserahmen entfernt, sondern nur das erste,
ein potentielles zweites oder beide Startkodons durch Alanine ersetzt wurden.
Hierbei wurde untersucht, ob tatsächlich allein die mangelnde Expression an
pUL24 für die bisher beobachteten Effekte verantwortlich ist.
3.6.1 Herstellung von Punktmutanten
Aus pSL-FRT-TB4-mycUL24 wurde über eine Mutagenese-PCR das Konstrukt
pSL-FRT-TB4-mycUL24-M59A generiert, bei dem das zweite ATG-Kodon (an
Position 59) unter Entstehung einer neue BglII Schnittstelle in ein GCG
umgewandelt wurde. pSL-FRT-TB4-mycUL24 und pSL-FRT-TB4-mycUL24-M59A
wurden anschließend als Matrizen zur Herstellung der für die Rekombination
benötigten PCR-Produkte verwendet (Tabelle 2). Die entstandenen BACs wurden
durch differentiellen Restriktionsverdau und anschließende Southern Blot Analyse
kontrolliert. Die Restriktionsmuster stimmten erwartungsgemäß weitgehend mit
den unter 3.4.1 beschriebenen Mustern des TB4wtBAC überein.

Ergebnisse 53
Tabelle 2: Primer-Template-Kombinationen für UL24-Punktmutanten
Primerkombination: Template: Mutante:
UL24-rev-dATG + UL24-3’-KO pSL-FRT-TB4-mycUL24 mut1. ATG
UL24-rev + UL24-3’-KO pSL-FRT-TB4-mycUL24-M59A mut2. ATG
UL24-rev-dATG + UL24-3’-KO pSL-FRT-TB4-mycUL24-M59A mut1.+2. ATG
Wie man in Abb. 19 sehen kann, war im Fall der M59A Mutation (Spuren 5 und 6)
nach BglII-Verdau bei Hybridisierung mit einer UL24-Probe ein Signal von ca. 0,4
kB durch die zusätzlich eingebrachte Schnittstelle detektierbar. Zuletzt wurden der
relevante Bereich der hergestellten Punktmutanten sequenziert und die
Mutationen der gewünschten Stellen bestätigt (Daten nicht gezeigt). Nachdem die
Integrität der BACs durch oben genannte Analysen getestet war, wurden die
DNAs für die Rekonstitution der Viren in HFF transformiert.
Abb. 19:Charakterisierung derUL24-Punktmutanten. (A) Schematische
Darstellung der UL24-
Genregion angegebener
rekombinanter BACs. Die
Orientierung der offenen
Leseraster (Pfeile, UL24
grau hinterlegt) als auch
Restriktionsschnittstellen
(E=EcoRI, B=BglII) sind
angegeben. (B)
Restriktionsanalyse von
BAC-DNA nach BglII-
Verdau (0,8%iges
Agarosegel). (C)
Southern Blot Analyse mit
einer 32P-markierten
Sonde gegen UL23 und
UL25 oder (D) UL24.
Fragmentgrößen in kb
sind in den jeweiligen
Abbildungen angegeben.
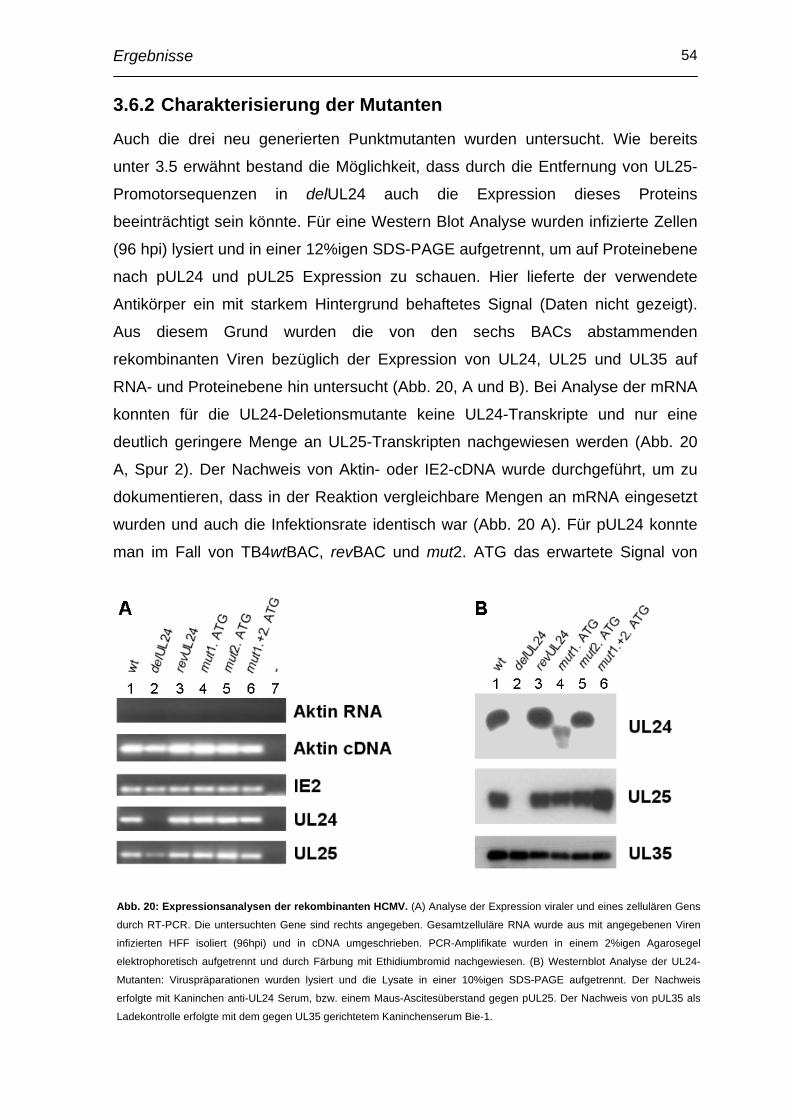
Ergebnisse 54
3.6.2 Charakterisierung der Mutanten
Auch die drei neu generierten Punktmutanten wurden untersucht. Wie bereits
unter 3.5 erwähnt bestand die Möglichkeit, dass durch die Entfernung von UL25-
Promotorsequenzen in delUL24 auch die Expression dieses Proteins
beeinträchtigt sein könnte. Für eine Western Blot Analyse wurden infizierte Zellen
(96 hpi) lysiert und in einer 12%igen SDS-PAGE aufgetrennt, um auf Proteinebene
nach pUL24 und pUL25 Expression zu schauen. Hier lieferte der verwendete
Antikörper ein mit starkem Hintergrund behaftetes Signal (Daten nicht gezeigt).
Aus diesem Grund wurden die von den sechs BACs abstammenden
rekombinanten Viren bezüglich der Expression von UL24, UL25 und UL35 auf
RNA- und Proteinebene hin untersucht (Abb. 20, A und B). Bei Analyse der mRNA
konnten für die UL24-Deletionsmutante keine UL24-Transkripte und nur eine
deutlich geringere Menge an UL25-Transkripten nachgewiesen werden (Abb. 20
A, Spur 2). Der Nachweis von Aktin- oder IE2-cDNA wurde durchgeführt, um zu
dokumentieren, dass in der Reaktion vergleichbare Mengen an mRNA eingesetzt
wurden und auch die Infektionsrate identisch war (Abb. 20 A). Für pUL24 konnte
man im Fall von TB4wtBAC, revBAC und mut2. ATG das erwartete Signal von
Abb. 20: Expressionsanalysen der rekombinanten HCMV. (A) Analyse der Expression viraler und eines zellulären Gens
durch RT-PCR. Die untersuchten Gene sind rechts angegeben. Gesamtzelluläre RNA wurde aus mit angegebenen Viren
infizierten HFF isoliert (96hpi) und in cDNA umgeschrieben. PCR-Amplifikate wurden in einem 2%igen Agarosegel
elektrophoretisch aufgetrennt und durch Färbung mit Ethidiumbromid nachgewiesen. (B) Westernblot Analyse der UL24-
Mutanten: Viruspräparationen wurden lysiert und die Lysate in einer 10%igen SDS-PAGE aufgetrennt. Der Nachweis
erfolgte mit Kaninchen anti-UL24 Serum, bzw. einem Maus-Ascitesüberstand gegen pUL25. Der Nachweis von pUL35 als
Ladekontrolle erfolgte mit dem gegen UL35 gerichtetem Kaninchenserum Bie-1.
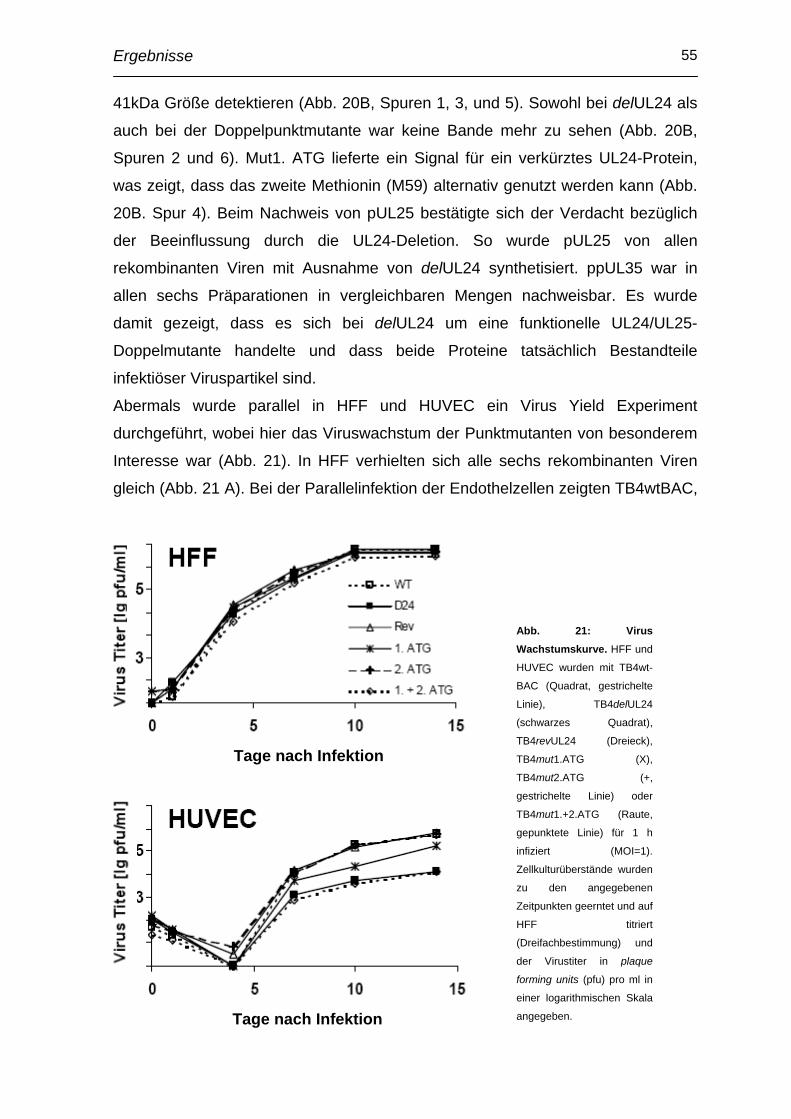
Ergebnisse 55
41kDa Größe detektieren (Abb. 20B, Spuren 1, 3, und 5). Sowohl bei delUL24 als
auch bei der Doppelpunktmutante war keine Bande mehr zu sehen (Abb. 20B,
Spuren 2 und 6). Mut1. ATG lieferte ein Signal für ein verkürztes UL24-Protein,
was zeigt, dass das zweite Methionin (M59) alternativ genutzt werden kann (Abb.
20B. Spur 4). Beim Nachweis von pUL25 bestätigte sich der Verdacht bezüglich
der Beeinflussung durch die UL24-Deletion. So wurde pUL25 von allen
rekombinanten Viren mit Ausnahme von delUL24 synthetisiert. ppUL35 war in
allen sechs Präparationen in vergleichbaren Mengen nachweisbar. Es wurde
damit gezeigt, dass es sich bei delUL24 um eine funktionelle UL24/UL25-
Doppelmutante handelte und dass beide Proteine tatsächlich Bestandteile
infektiöser Viruspartikel sind.
Abermals wurde parallel in HFF und HUVEC ein Virus Yield Experiment
durchgeführt, wobei hier das Viruswachstum der Punktmutanten von besonderem
Interesse war (Abb. 21). In HFF verhielten sich alle sechs rekombinanten Viren
gleich (Abb. 21 A). Bei der Parallelinfektion der Endothelzellen zeigten TB4wtBAC,
Tage nach Infektion
Tage nach Infektion
Abb. 21: VirusWachstumskurve. HFF und
HUVEC wurden mit TB4wt-
BAC (Quadrat, gestrichelte
Linie), TB4delUL24
(schwarzes Quadrat),
TB4revUL24 (Dreieck),
TB4mut1.ATG (X),
TB4mut2.ATG (+,
gestrichelte Linie) oder
TB4mut1.+2.ATG (Raute,
gepunktete Linie) für 1 h
infiziert (MOI=1).
Zellkulturüberstände wurden
zu den angegebenen
Zeitpunkten geerntet und auf
HFF titriert
(Dreifachbestimmung) und
der Virustiter in plaque
forming units (pfu) pro ml in
einer logarithmischen Skala
angegeben.
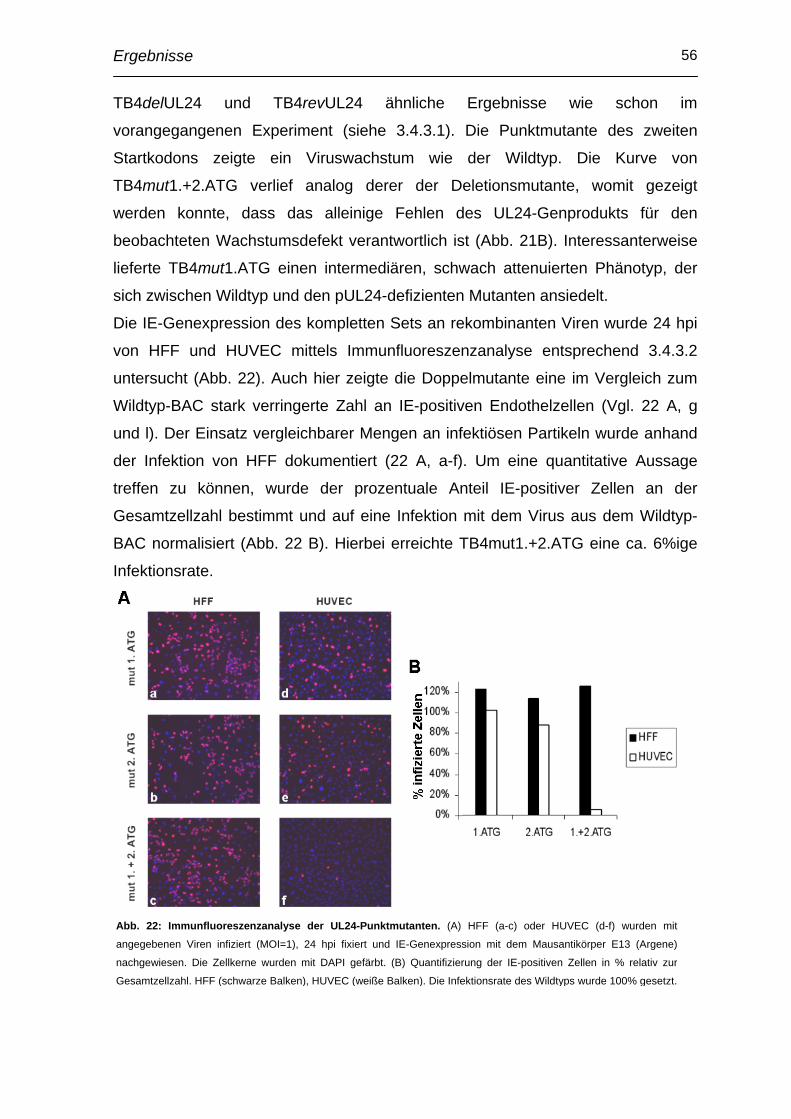
Ergebnisse 56
TB4delUL24 und TB4revUL24 ähnliche Ergebnisse wie schon im
vorangegangenen Experiment (siehe 3.4.3.1). Die Punktmutante des zweiten
Startkodons zeigte ein Viruswachstum wie der Wildtyp. Die Kurve von
TB4mut1.+2.ATG verlief analog derer der Deletionsmutante, womit gezeigt
werden konnte, dass das alleinige Fehlen des UL24-Genprodukts für den
beobachteten Wachstumsdefekt verantwortlich ist (Abb. 21B). Interessanterweise
lieferte TB4mut1.ATG einen intermediären, schwach attenuierten Phänotyp, der
sich zwischen Wildtyp und den pUL24-defizienten Mutanten ansiedelt.
Die IE-Genexpression des kompletten Sets an rekombinanten Viren wurde 24 hpi
von HFF und HUVEC mittels Immunfluoreszenzanalyse entsprechend 3.4.3.2
untersucht (Abb. 22). Auch hier zeigte die Doppelmutante eine im Vergleich zum
Wildtyp-BAC stark verringerte Zahl an IE-positiven Endothelzellen (Vgl. 22 A, g
und l). Der Einsatz vergleichbarer Mengen an infektiösen Partikeln wurde anhand
der Infektion von HFF dokumentiert (22 A, a-f). Um eine quantitative Aussage
treffen zu können, wurde der prozentuale Anteil IE-positiver Zellen an der
Gesamtzellzahl bestimmt und auf eine Infektion mit dem Virus aus dem Wildtyp-
BAC normalisiert (Abb. 22 B). Hierbei erreichte TB4mut1.+2.ATG eine ca. 6%ige
Infektionsrate.
Abb. 22: Immunfluoreszenzanalyse der UL24-Punktmutanten. (A) HFF (a-c) oder HUVEC (d-f) wurden mit
en Viren infiziert (MOI=1), 24 hpi fixiert und IE-Genexpression mit dem Mausantikörper E13 (Argene)
wiesen. Die Zellkerne wurden mit DAPI gefärbt. (B) Quantifizierung der IE-positiven Zellen in % relativ zu
angegeben
nachge r
Gesamtzellzahl. HFF (schwarze Balken), HUVEC (weiße Balken). Die Infektionsrate des Wildtyps wurde 100% gesetzt.

Ergebnisse 57
3.7 Transkomplementation durch retroviralen Gentransfer
Um mit einem alternativen Ansatz sicherzustellen, dass allein die Absenz des
UL24-Genproduktes für den initial beobachteten Effekt auf den Zelltropismus des
Virus verantwortlich ist, wurde versucht, den Defekt des UL24-Deletionsvirus
TB4delUL24 durch Transkomplementation von GFP oder pUL24 in HFF zu
kompensieren. Da sich die primären Fibroblasten nur mit sehr geringer Effizienz
transfizieren lassen, pUL24 aber in möglichst vielen mit TB4delUL24 infizierten
Zellen vorhanden sein sollte, wurde auf die weitaus effizientere Methode der
retroviralen Transduktion zurückgegriffen. HFF wurden mit rekombinanten
Retroviren transduziert, welche als Kontrolle GFP oder UL24 exprimieren sollten.
48 Stunden später wurden die Zellen mit TB4delUL24 überinfiziert. Die
entsprechenden Überstände wurden wiederum fünf Tage später verwendet, um
parallel HFF und HUVEC zu infizieren. Wie in Abbildung 23 zu sehen, zeigten die
Viren in den Fibroblasten vergleichbare Infektionsraten, womit gewährleistet war,
dass die Virustiter der verwendeten Überstände vergleichbar waren. Durch die in
trans Expression von pUL24 gelang es, den Anteil an IE-positiven HUVEC wieder
auf Wildtyp-Niveau anzuheben (Abb. 23 B, d und f). Die Expression von GFP
zeigte keinerlei Auswirkung auf den ursprünglichen Phänotyp von TB4delUL24 in
Endothelzellen (Abb. 23 B, e).
Um zu demonstrieren, dass die mit dem UL24-Retovirus transduzierten HFF im
Gegensatz zu den Kontrollzellen auch tatsächlich UL24 exprimieren, wurde aus
den Zellen der jeweiligen Ansätze gesamtzelluläre RNA isoliert und cDNA
generiert. In der folgenden PCR wurde gezeigt, dass UL24-spezifische cDNA
nachweisbar war, und zwar nur in den mit UL24-Retrovirus transduzierten Zellen
(Abb. 23 C, Spuren 1 und 2). Eine DNA-Kontamination der RNA-Proben wurde
durch eine Aktin-PCR bei Verwendung von RNA als Template ausgeschlossen
(Abb. 23 C, Spuren 1 und 2). Die erfolgreiche Generierung von vergleichbaren
Mengen an cDNA wurde durch eine Aktin-PCR unter Vorgabe der verschiedenen
präparierten cDNAs gewährleistet (Abb. 23 C, Spuren 1 und 2).
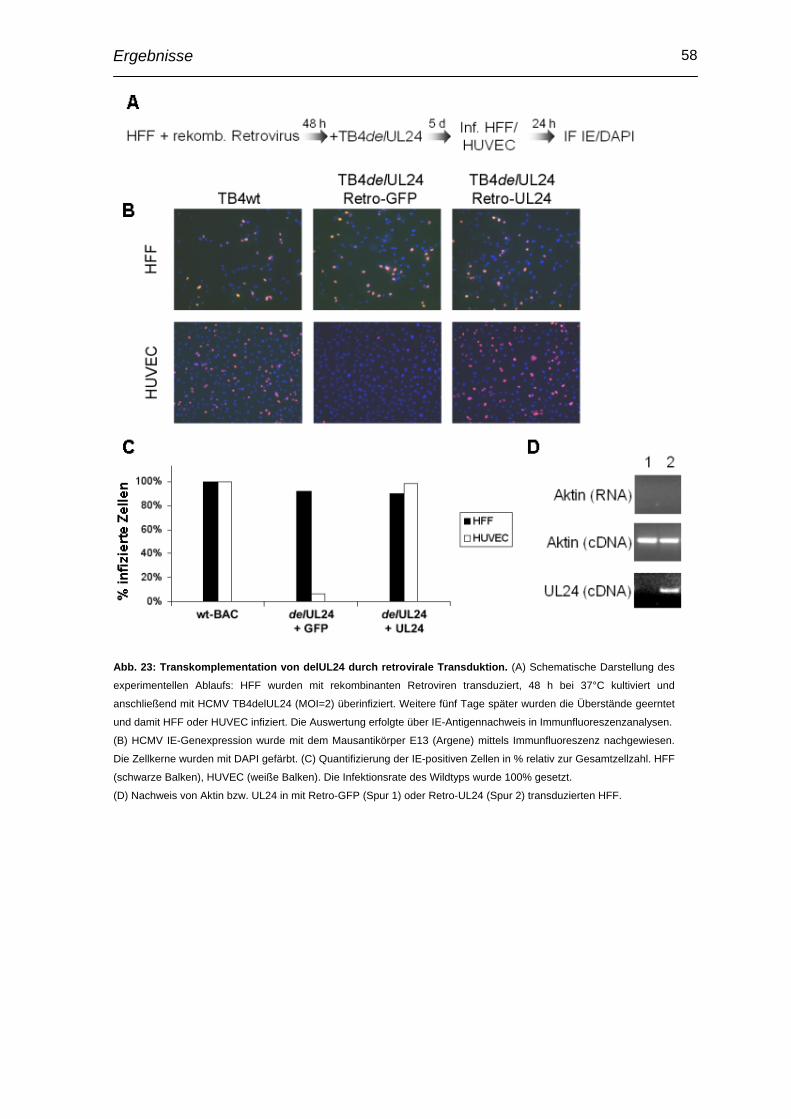
Ergebnisse 58
(D) Nachweis von Aktin bzw. UL24 in mit Retro-GFP (Spur 1) oder Retro-UL24 (Spur 2) transduzierten HFF.
(B) HCMV IE-Genexpression wurde mit dem Mausantikörper E13 (Argene) mittels Immunfluoreszenz nachgewiesen.
Die Zellkerne wurden mit DAPI gefärbt. (C) Quantifizierung der IE-positiven Zellen in % relativ zur Gesamtzellzahl. HFF
(schwarze Balken), HUVEC (weiße Balken). Die Infektionsrate des Wildtyps wurde 100% gesetzt.
Abb. 23: Transkomplementation von delUL24 durch retrovirale Transduktion. (A) Schematische Darstellung des
experimentellen Ablaufs: HFF wurden mit rekombinanten Retroviren transduziert, 48 h bei 37°C kultiviert und
anschließend mit HCMV TB4delUL24 (MOI=2) überinfiziert. Weitere fünf Tage später wurden die Überstände geerntet
und damit HFF oder HUVEC infiziert. Die Auswertung erfolgte über IE-Antigennachweis in Immunfluoreszenzanalysen.

Ergebnisse 59
3.8 Einfluss auf Epithelzelltropismus
Nachdem UL24 eine Rolle beim Endothelzeltropismus von HCMV spielt, wurde im
Folgenden untersucht, ob auch die Infektion weiterer Zelltypen durch das Virus
beeinträchtigt wird. Nachdem in kürzlich veröffentlichten Studien [Wang et al.,
2005a] gezeigt wurde, dass das UL131 Gen, welches ebenfalls eine Determinante
für Endothelzelltropismus darstellt, auch für den Epithelzelltropismus des Virus
mitverantwortlich ist, sollte im Rahmen dieser Arbeit ein möglicher Einfluss von
pUL24 auf die Infektion von ekotodermalen Zellen wie Epithelzellen oder Zellen
neuronalen Ursprungs untersucht werden. In beiden Fällen handelt es sich um für
das Virus pathogenetisch relevante Zelltypen: Epithelzellen unterschiedlichster
Gewebe (Muttermilchkanal, Speicheldrüse, Niere usw.) stellen für HCMV
vermutlich eine der Hauptstätten für die Virusproduktion dar [Britt et al., 1996] und
ermöglichen es dem infektiösen Virus somit über Muttermilch, Speichel, Urin usw.
verbreitet zu werden. Zellen mit neuronalem Hintergrund stellen für das HCMV,
wie bei anderen Herpesviren (HSV-1, VZV) möglicherweise einen Ort dar, an dem
es sicher vor Angriffen des Immunsystems im Wirtskörper verweilen kann (Ort der
Latenz).
HeLa-Zellen oder RPE-1-Zellen stellvertretend für Epithelzellen und SK-N-SH oder
MGU373-Zellen als Zellen neuronaler Abstammung wurden mit TB4wt-BAC,
delUL24 oder revUL24 inkubiert und infizierte Zellen 24 hpi mit einem gegen
HCMV IE gerichteten Antikörper in der Immunfluoreszenzanalyse nachgewiesen
(Abb. 24 A). Es wurde gezeigt, dass die Deletion des UL24-Leserasters keine
Auswirkung auf die Fähigkeit des Virus hat, Zellen des zentralen Nervensystems
zu infizieren (Abb. 24 A, i, j). Bei den Epithelzellen waren wie schon bei den
Endothelzellen deutlich weniger IE-positive Zellen im Vergleich zu WT oder
Revertante zu sehen (Abb. 24 A, g, h). Im Rahmen der quantitativen Auswertung
wurden Infektionsraten von ca. 13% (RPE1) bzw. 2% (HeLa) im Vergleich zur
Wildtypinfektion gemessen (Abb. 24 B).
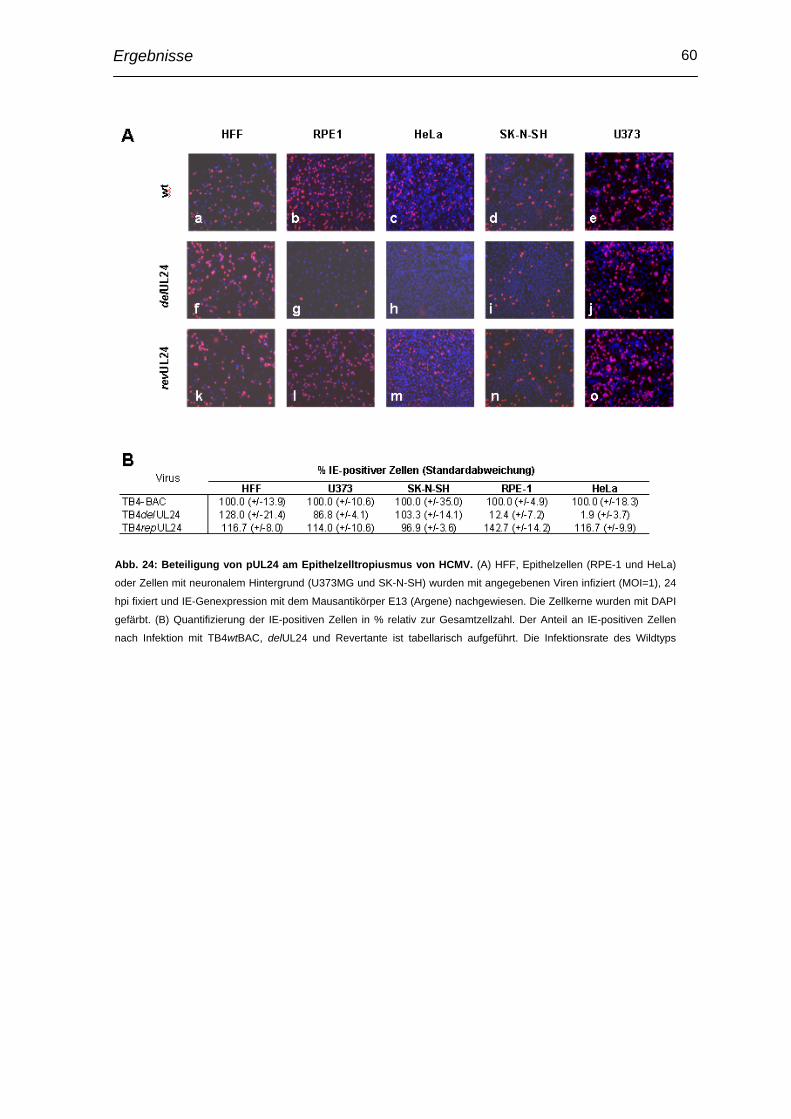
Ergebnisse 60
Abb. 24: Beteiligung von pUL24 am Epithelzelltropiusmus von HCMV. (A) HFF, Epithelzellen (RPE-1 und HeLa)
oder Zellen mit neuronalem Hintergrund (U373MG und SK-N-SH) wurden mit angegebenen Viren infiziert (MOI=1), 24
hpi fixiert und IE-Genexpression mit dem Mausantikörper E13 (Argene) nachgewiesen. Die Zellkerne wurden mit DAPI
gefärbt. (B) Quantifizierung der IE-positiven Zellen in % relativ zur Gesamtzellzahl. Der Anteil an IE-positiven Zellen
nach Infektion mit TB4wtBAC, delUL24 und Revertante ist tabellarisch aufgeführt. Die Infektionsrate des Wildtyps

Ergebnisse 61
3.9 Einfluss auf Makrophagentropismus
Makrophagen als Teil der unspezifischen Immunantwort ein äußerst interessanter
Zelltyp im Bezug auf virale Infektionen. Im Rahmen von Entzündungen oder der
zellulären Immunantwort sind sie häufig stimulierenden oder aktivierenden
Substanzen ausgesetzt, wodurch die Zellen eine Reihe intrinsischer oder
extrinsischer antiviraler Faktoren produzieren. An diese feindliche Umgebung
muss sich das Virus adaptieren.
Verschiedenste Faktoren können am Makrophagentropismus von HCMV beteiligt
sein. Der Grad der Differenzierung von Monozyten bis hin zu aktivierten
Makrophagen beispielsweise kann sich auf die Suszeptibilität der Zellen auswirken
[Soderberg-Naucler et al., 1997] aber auch genetische Faktoren auf Seiten des
Virus spielen eine Rolle. Für das MCMV wurden bereits mehrere Gene
beschrieben, deren Mutation die virale Replikation in Fibroblasten nicht beeinflusst
aber eine deutliche Reduktion des Viruswachstums in Makrophagen bewirkt.
Darunter auch die US22-Gene M36 [Ménard et al., 2003, Skaletskaya et al., 2001]
und M139-M141 [Ménard et al., 2003]. Die HCMV Endothelzelldeterminanten
UL128-UL131 scheinen ebenfalls den Makrophagenzelltropismus zu beeinflussen
[Hahn et al., 2004] und es wird vermutet, dass der Tropismus von HCMV in
Endothelzellen und Makrophagen einem ähnlichen, wenn nicht dem gleichen
Tropismus unterliegt [Sinzger et al., 2006]. Da für pUL24 eine Beteiligung am
Endothelzelltropismus des HCMV gezeigt werden konnte, wurde nun die
Bedeutung des Proteins bezüglich des Makrophagentropismus untersucht.
HFF (Abb. 25, a-c) und humane, aus primären Monozyten differenzierte
Makrophagen (Abb. 25, d-f) wurden mit TB4wt-BAC, delUL24 oder revUL24
infiziert und 24 hpi per Immunfluoreszenzanalyse auf HCMV IE-Genexpression hin
untersucht. Für das UL24-Deletionsvirus konnte bei vergleichbarer Infektiosität in
HFF eine stark verminderte Anzahl an IE-positiven Makrophagen im Vergleich zu
Wildtyp und Revertante detektiert werden (Abb. 25, e).

Ergebnisse 62
Abb. 25: Beteiligungvon pUL24 amMakrophagentropismus von HCMV. HFF (a-c)
oder Makrophagen (d-f)
wurden mit angegebenen
Viren infiziert (MOI=1), 24
hpi fixiert und IE-
Genexpression mit dem
Mausantikörper E13
(Argene) nachgewiesen.
Die Zellkerne wurden mit
DAPI gefärbt.
3.10 Verringerte Infektiösität in Endothelzellen durch Zentrifugation kompensierbar
Es ist beschrieben, dass durch eine niedrigtourige Zentrifugation von HCMV auf
Wirtszellen ein deutlich höherer Grad an Infektion erreicht werden kann [Hudson
et al., 1976, Hudson 1988]. Um zu überprüfen, ob der in Endothelzellen
beobachtete Phänotyp durch eine Inokulation des Virus unter Zentrifugation
beinflusst wird, wurden HFF und HUVEC parallel mit oder ohne
Zentrifugationsschritt infiziert. Hierbei konnte festgestellt werden, dass die
Infektionsrate von TB4delUL24 oder TB4mut1.+2.ATG in Endothelzellen durch
das Aufzentrifugieren deutlich erhöht werden konnte (Abb. 26, n und t bzw. r und
x). In Fibroblasten blieb die Infektionsrate mit oder ohne Zentrifugation nahezu
gleich (Abb 26, a-l). Im Rahmen dieses Experimentes konnte gezeigt werden,
dass der in der initialen Phase der viralen Replikation auftretende Defekt der
Deletionsmutante zumindest teilweise durch einen niedrigtourigen
Zentrifugationsschritt kompensiert werden kann.
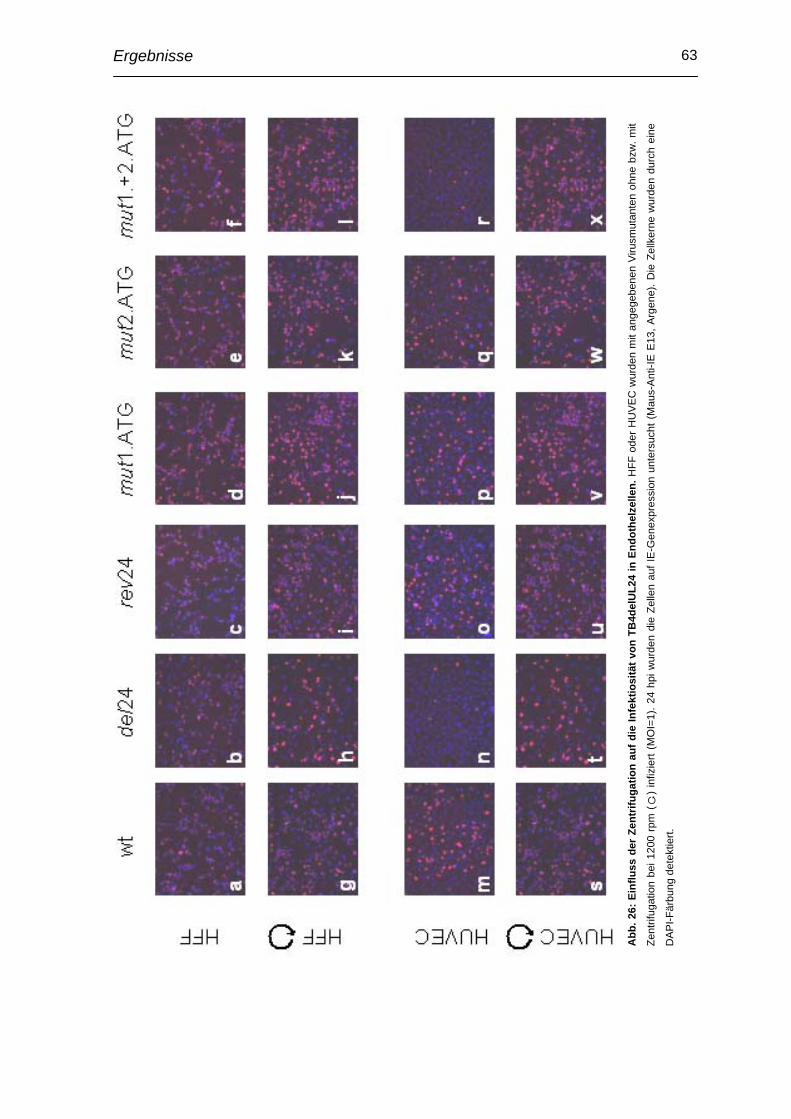
Ergebnisse 63
Abb
. 26:
Ein
fluss
der
Zen
trifu
gatio
n au
f die
Infe
ktio
sitä
t von
TB
4del
UL2
4 in
End
othe
lzel
len.
HFF
ode
r HU
VE
C w
urde
n m
it an
gege
bene
n V
irusm
utan
ten
ohne
bzw
. mit
Zent
rifug
atio
n be
i 120
0 rp
m (
)
infiz
iert
(MO
I=1)
. 24
hpi w
urde
n di
e Ze
llen
auf I
E-G
enex
pres
sion
unt
ersu
cht (
Mau
s-A
nti-I
E E
13, A
rgen
e). D
ie Z
ellk
erne
wur
den
durc
h ei
ne
DA
PI-F
ärbu
ng d
etek
tiert.

Ergebnisse 64
3.11 Beteiligung von pUL24 an Entry-Prozessen des HCMV in Endothelzellen
Die Aufnahme von HCMV erfolgt vermutlich abhängig vom Zelltyp auf
unterschiedlichen Wegen. Der in Fibroblasten überwiegend beschriebene
Mechanismus ist die Fusion des Virus mit der zytoplasmatischen Zellmembran;
aber auch die Aufnahme von Viruspartikeln über Endozytose wird gegenwärtig
besonders in Endothelzellen diskutiert.
3.11.1 Aufnahme von HCMV TB4-BAC in Endothelzellen über Endosomen
Viele Viren, die über Endozytose internalisiert werden, benötigen einen niedrigen
pH-Wert innerhalb der Endosomen, um deren Fusion mit den viralen
Glykoproteinen zu fördern. Lysosomotrope Agenzien wie zum Beispiel NH4Cl oder
Chloroquine verhindern eine Ansäuerung der Endosomen dadurch, dass sie den
endosomalen pH-Wert puffern. Es konnte bereits mehrfach gezeigt werden, dass
diese Substanzen Infektionen von Viren, welche einen niedrigen endosomalen pH-
Wert benötigen, inhibieren. Um zu testen, ob auch HCMV für die Infektion einen
niedrigen endosomalen pH-Wert benötigt, wurden HFF und HUVEC während der
frühen Phase der Infektion durch HCMV TB4-BAC gar nicht, mit NH4Cl oder mit
Chloroquine behandelt (Abb. 27). Als Toxizitätskontrolle wurden die jeweiligen
Substanzen in parallelen Ansätzen erst 4 Stunden nach Infektion, d.h. nach Ablauf
der relevanten Infektionsschritte, zu den Zellen gegeben (Abb. 27 A, i-l). Der Anteil
der IE-positiven Zellen an der Gesamtzellzahl wurde 24 hpi mittels
Immunfluoreszenz bestimmt. Bei den Fibroblasten konnte keine Abhängigkeit von
lysosomotrophen Substanzen festgestellt werden (Abb. 27 A, e und g),
wohingegen die Endothelzellen eine deutliche Reduktion an IE-positiven Zellen
aufwiesen (Abb. 27 A, f und h). Ein zytotoxischer Effekt der verwendeten Drogen
war nicht zu erkennen (Abb. 27 A, i-l). Durch dieses Experiment wurde gezeigt,
dass von HCMV-TB4 zelltypabhängig unterschiedliche Aufnahmewege verwendet
werden und in Endothelzellen der pH-abhängige, endosomale Weg favorisiert
wird. Die quantitative Analyse der Daten ist in Abb. 27 B dargestellt. Insbesondere
der Einsatz von NH4Cl führte in HUVEC zu einer Reduktion der Anzahl IE-positiver
Zellen um mehr als 90%.
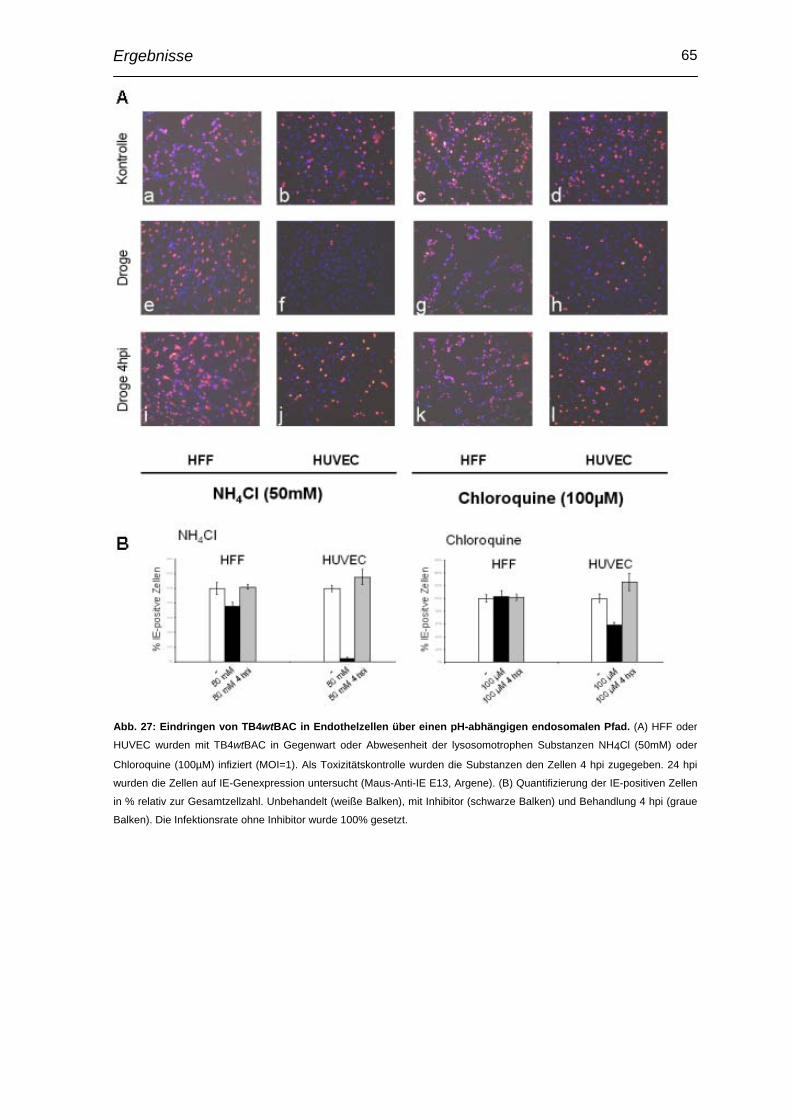
Ergebnisse 65
Abb. 27: Eindringen von TB4wtBAC in Endothelzellen über einen pH-abhängigen endosomalen Pfad. (A) HFF oder
HUVEC wurden mit TB4wtBAC in Gegenwart oder Abwesenheit der lysosomotrophen Substanzen NH4Cl (50mM) oder
Chloroquine (100µM) infiziert (MOI=1). Als Toxizitätskontrolle wurden die Substanzen den Zellen 4 hpi zugegeben. 24 hpi
wurden die Zellen auf IE-Genexpression untersucht (Maus-Anti-IE E13, Argene). (B) Quantifizierung der IE-positiven Zellen
in % relativ zur Gesamtzellzahl. Unbehandelt (weiße Balken), mit Inhibitor (schwarze Balken) und Behandlung 4 hpi (graue
Balken). Die Infektionsrate ohne Inhibitor wurde 100% gesetzt.
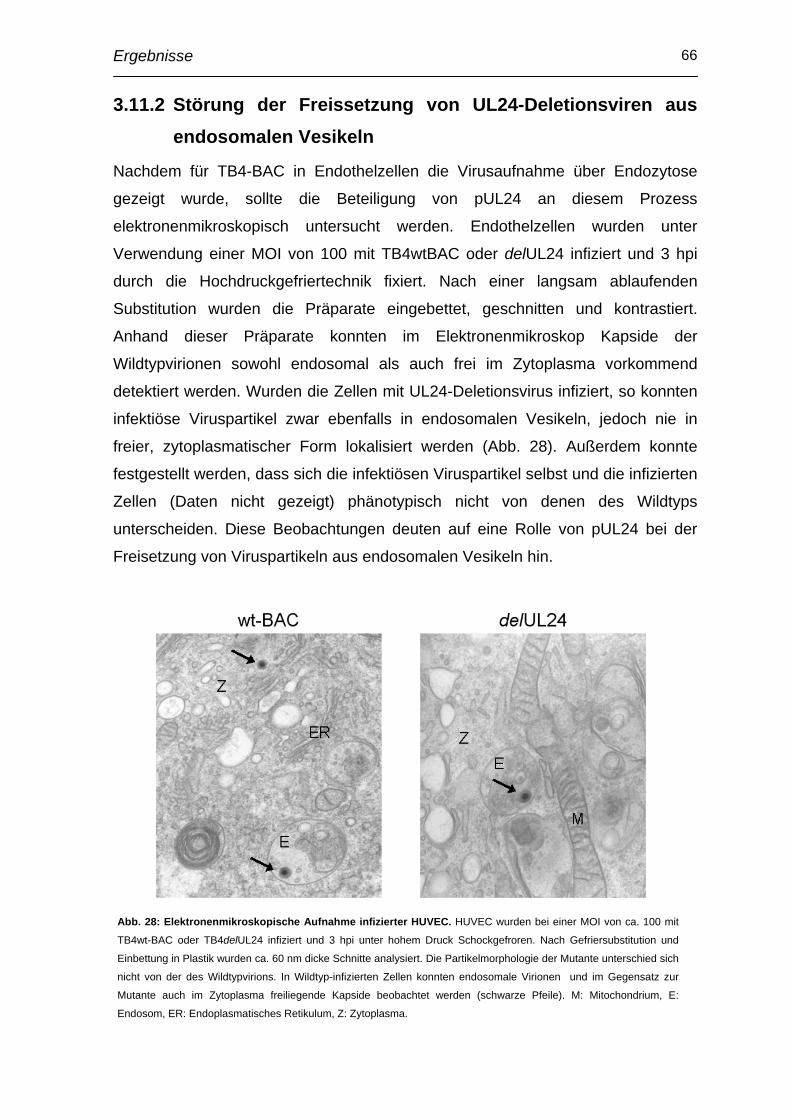
Ergebnisse 66
3.11.2 Störung der Freissetzung von UL24-Deletionsviren aus endosomalen Vesikeln
Nachdem für TB4-BAC in Endothelzellen die Virusaufnahme über Endozytose
gezeigt wurde, sollte die Beteiligung von pUL24 an diesem Prozess
elektronenmikroskopisch untersucht werden. Endothelzellen wurden unter
Verwendung einer MOI von 100 mit TB4wtBAC oder delUL24 infiziert und 3 hpi
durch die Hochdruckgefriertechnik fixiert. Nach einer langsam ablaufenden
Substitution wurden die Präparate eingebettet, geschnitten und kontrastiert.
Anhand dieser Präparate konnten im Elektronenmikroskop Kapside der
Wildtypvirionen sowohl endosomal als auch frei im Zytoplasma vorkommend
detektiert werden. Wurden die Zellen mit UL24-Deletionsvirus infiziert, so konnten
infektiöse Viruspartikel zwar ebenfalls in endosomalen Vesikeln, jedoch nie in
freier, zytoplasmatischer Form lokalisiert werden (Abb. 28). Außerdem konnte
festgestellt werden, dass sich die infektiösen Viruspartikel selbst und die infizierten
Zellen (Daten nicht gezeigt) phänotypisch nicht von denen des Wildtyps
unterscheiden. Diese Beobachtungen deuten auf eine Rolle von pUL24 bei der
Freisetzung von Viruspartikeln aus endosomalen Vesikeln hin.
Abb. 28: Elektronenmikroskopische Aufnahme infizierter HUVEC. HUVEC wurden bei einer MOI von ca. 100 mit
TB4wt-BAC oder TB4delUL24 infiziert und 3 hpi unter hohem Druck Schockgefroren. Nach Gefriersubstitution und
Einbettung in Plastik wurden ca. 60 nm dicke Schnitte analysiert. Die Partikelmorphologie der Mutante unterschied sich
nicht von der des Wildtypvirions. In Wildtyp-infizierten Zellen konnten endosomale Virionen und im Gegensatz zur
Mutante auch im Zytoplasma freiliegende Kapside beobachtet werden (schwarze Pfeile). M: Mitochondrium, E:
Endosom, ER: Endoplasmatisches Retikulum, Z: Zytoplasma.

Diskussion 67
4 Diskussion
Das Humane Zytomegalievirus besteht wie alle Herpesviren aus einem
Nukleokapsid, welches von der Tegumentschicht umgeben ist und durch die virale
Hüllmembran von der Umwelt nach außen abgegrenzt ist. Das Tegument setzt
sich aus 20-25 Virion-assoziierten Proteinen zusammen [Mocarski und Courcelle,
2001], von denen viele phosphoryliert sind und deren Funktionen gegenwärtig
noch nicht im Detail bekannt sind. Eines der am häufigsten vorkommenden
Tegumentproteine ist ppUL32. Bei letzterem handelt es sich um ein für die
Vermehrung des HCMV essentielles Protein, welches mit seinem Aminoterminus
an das virale Kapsid bindet [Baxter et al., 2001]. Analysen mittels Yeast Two-
Hybrid System in unserer Arbeitsgruppe identifizierten pUL24 als
Interaktionspartner des N-Terminus von ppUL32. Bei pUL24 handelt es sich um
einen Vertreter der US22-Genfamilie, die für Betaherpesviren spezifisch ist.
Andere Vertreter dieser Genfamilie sind wichtig für die Genregulation [Mori et al.,
1998, Romanowski et al., 1997], die Modulation des programmierten Zelltod
(Apoptose) [McCormick et al., 2003, Skaletskaya et al., 2001] und den
Zelltropismus von CMV [Hanson et al., 2001, Ménard et al., 2003, Xiao et al.,
2000]. Aus den oben genannten Gründen sollte die funktionelle Bedeutung von
pUL24 im Rahmen dieser Doktorarbeit geklärt werden.
4.1 Herstellung von Antikörpern und Lokalisation von pUL24
Zunächst wurde ein polyklonales Antiserum gegen prokaryot exprimiertes pUL24
hergestellt. Nachdem erste Expressionsversuche von pUL24 als His6- oder GST-
getaggtes Protein gescheitert waren, wurde das Maltose-Bindeprotein (MBP) als
Fusionspartner gewählt. Das MBP fungiert als eine Art Chaperon, welches die
Löslichkeit des Zielproteins erhöht und aufgrund seiner hohen Affinität zu Maltose
eine Aufreinigung des Fusionsproteins in einer Ein-Schritt-Reaktion ermöglicht [di
Guan et al., 1988, Maina et al., 1988, Kellermann et al., 1982]. Die Fusion aus
Maltose-Bindeprotein und Volllänge-pUL24 konnte in E.coli überexprimiert und für
die Immunisierung von Kaninchen eingesetzt werden. Das gewonnene Serum
zeigte im Gegensatz zum Präimmunserum sowohl in der Immunfluoreszenz-

Diskussion 68
Analyse als auch im Western Blot eine spezifische Reaktion mit pUL24. Für
pUL24 wurde eine Molekularmasse von 40 kDa gemessen, was mit der
berechneten Masse gut übereinstimmt.
Die Lokalisation von pUL24 in transfizierten 293T- und HeLa-Zellen war
überwiegend nukleär, was auf eine Beteiligung von pUL24 an nukleären
Prozessen wie z. B. Genregulation hinweist. Dies steht im Gegensatz zur
veröffentlichten zytoplasmatischen Lokalisation von pUL24 in Fibroblasten [Adair
et al., 2002]. In dieser Publikation wurde allerdings die Lokalisation nach Infektion
der Zellen mit HCMV bestimmt. Möglicherweise kann die unterschiedliche
Lokalisation durch die Mitwirkung weiterer viraler Proteine erklärt werden.
Tatsächlich konnte in Folgeexperimenten eine veränderte Lokalisation durch
Koexpression von ppUL32 beobachtet werden (siehe unten).
4.2 Interaktion von pUL24 und ppUL32
Die initial beobachtete Interaktion zwischen pUL24 und ppUL32 wurde durch Ko-
Immunpräzipitationsexperimente in transient transfizierten Zellen bestätigt. Dies
war ein weiterer Hinweis für eine physische Interaktion zwischen den beiden
Proteinen. Weitere Interaktionen [K. Schierling, Dissertation] konnten in diesem
System bestätigt werden, was zu der Annahme führt, dass pUL24 zusammen mit
ppUL35, ppUL32 und ppUL82 einen Proteinkomplex bildet (Abb. 29). Allerdings
wurden die viralen Proteine in diesem System stark überexprimiert, so dass
möglicherweise Interaktionen detektiert werden, welche nicht die natürliche
Situation wiederspiegeln. Um Gewissheit über eine tatsächliche Interaktion unter
Abb. 29: Schematische Darstellung eines möglichen pUL24/ppUL32-Proteinkomplexes. Rosa Ovale: Tegumentproteine;
rote Linien: direkte, nachgewiesene Interaktionen zwischen den Tegumentproteinen. Blaues Hexagon: Viruskapsid mit viraler
DNA.

Diskussion 69
Infektionsbedingungen zu erlangen, wurden Ko-Immunpräzipitationen aus mit
HCMV TB40E infizierten HFF durchgeführt. Die Interaktion zwischen pUL24 und
ppUL32 konnte auch hier bestätigt werden. Die Kontrollpräzipitation gegen pUL48
zeigte, dass die Interaktion jedoch unabhängig von einem weiteren vor kurzem
identifizierten Proteinkomplex, bestehend aus pUL47, pUL48, pUL69 und pUL86,
dem Haupt-Kapsidprotein (MCP) [Bechtel et al., 2002] zu sein scheint. Viele
Tegumentproteine sind früh im viralen Replikationszyklus im Zellkern anzutreffen
[Bechtel et al., 2002, Hensel et al., 1995, Liu et al., 2002, Winkler et al., 1996] und
werden erst später ins Zytoplasma transportiert, wo sie in Kompartimenten
akkumulieren, welche für die Tegumentierung verantwortlich sind [Sanchez et al.,
2000]. Im Rahmen von Kolokalisationsexperimenten wurde gezeigt, dass pUL24
bei gleichzeitiger Expression von ppUL32 seine ursprünglich nukleäre Lokalisation
in transfizierten HeLa-Zellen ändert. Auch ppUL32, welches normalerweise
cytoplasmatisch vorliegt, ändert in diesem Fall seine Lokalisation dahingehend,
dass beide Proteine in perinukleären Bereichen zu finden waren. Dies zeigt nicht
nur, dass beide Proteine im selben Kompartiment vorliegen, sondern erhärtet auch
die Daten einer funktionellen Interaktion. Weiterhin stimmen diese Ergebnisse mit
veröffentlichten Daten welche pUL24 in infizierten Zellen eine cytoplasmatische
Lokalisation in der Nähe des Zellkerns zuweisen überein.
4.3 Funktionelle Analyse von pUL24
Für verschiedene Tegumentproteine konnte eine transaktivierende Wirkung auf
den HCMV IE-Promoter gezeigt werden [Liu et al., 1992, Schierling et al., 2004,
Winkler et al., 1996]. Darunter auch die Mitglieder der US22-Genfamile IRS1 und
TRS1 [Romanowski et al., 1997]. Es wurde ebenfalls beobachtet, dass das
Positionshomolog von HCMV-UL24, das HHV6-U3-Protein in transienten
Luziferase-Reportergenassys eine Transaktivierung des HIV1 LTR bewirkt [Mori et
al., 1998]. Deshalb wurde in dieser Arbeit der Einfluss von pUL24 auf eine Reihe
verschiedener Promotoren (HCMV IE-Promotor, HIV1-LTR, SV40-Promotor)
getestet. Es konnte eine zweifache Aktivierung in Gegenwart von pUL24 gezeigt
werden, welche allerdings nicht promotorspezifisch war. Interessanterweise
zeigten die Zellen nach Transfektion von ppUL32 eine Repression der
Promotoraktivität, welche auch bei Kotransfektion der beiden Interaktionspartner

Diskussion 70
dominant war. Um zu klären, ob pUL24 im Rahmen des neu identifizierten
Proteinkomplexes einen kooperativen, transaktivierenden Effekt bewirkt, wie es für
ppUL35 und ppUL82 [Liu et al., 2002], ppUL69 und ppUL82 [Winkler et al., 1995]
oder ppUL35, ppUL69 und ppUL82 [Schierling et al., 2004] beschrieben wurde,
wurden pUL24, ppUL32, ppUL35 und ppUL82 in verschiedenen Kombinationen
kotransfiziert. Hierbei konnte eine Zunahme der Promotoraktivität um jeweils den
Faktor 2 durch die Expression von pUL24 beobachtet werden. Allerdings war in
keinem Fall ein überproportionaler Anstieg der Luziferasetranskription zu sehen.
Es lässt sich somit zusammenfassend sagen, dass pUL24 über eine
transaktivierende Wirkung verfügt, welche jedoch promotorunabhängig ist und
keinen kooperativen Effekt aufweist.
4.4 Bedeutung von UL24 für den Endothelzelltropismus
Um mehr über die biologische Funktion von pUL24 in Erfahrung zu bringen, wurde
eine UL24-Deletionsmutante, TB4delUL24, mittels gerichteter, RecET-vermittelter
BAC-Mutagenese [Wagner et al., 2002] generiert. Die Mutationen wurden im
bacterial artificial chromosome (BAC) TB4 durchgeführt, welches auf dem stark
endotheliothropen HCMV-Stamm TB40E [Hahn et al., 2003] basiert. Da in diesem
System alle Rekombinationsschritte in E.coli ablaufen, ist im Verlauf der
Mutagenese klonales Arbeiten und somit die anschließende Verwendung reiner
Viruspopulationen gewährleistet. Auf Grundlage von TB4delUL24 wurde durch
Wiedereinbringen des UL24-Leserahmens mit der gleichen Methode die
Virusrevertante TB4revUL24 generiert.
Bei der Infektion von Fibroblasten zeigte sich TB4delUL24 genauso effizient wie
das vom Wildtyp-BAC abstammende Virus oder die Revertante. Die Deletion hatte
allerdings eine starke Auswirkung auf die Fähigkeit des Virus eine Infektion in
Endothelzellen zu etablieren. Dieser Zelltyp verfügt mit seiner
Schnittstellenfunktion zwischen Blutkreislauf und Organgewebe über das
Potential, durch die Verbreitung von infizierten vaskulären Endothelzellen zur
Entwicklung von HCMV-assoziierten Erkrankungen beizutragen [Digel et al.,
2006]. Wir konnten zeigen, dass der Virustiter von TB4delUL24 2-3 log-Stufen
unter denen von TB4 oder TB4revUL24 lag. Diese Daten stimmen mit
zwischenzeitlich veröffentlichten Studien überein, bei denen komplette

Diskussion 71
Leserahmen von HCMV genomweit deletiert wurden [Dunn et al., 2003]. Allerdings
wurde diese Arbeit im Hintergrund des schwach endotheliotropen Stammes
Towne durchgeführt. Eine Beteiligung von pUL24 am natürlichen Zelltropismus
von HCMV bei einem hoch endotheliotropen Stamm wurde dagegen noch nicht
beschrieben. Das normale Wachstum der Revertante zeigte, dass außer der
Deletion des UL24-Leserahmens keine weiteren, ungewollten Mutationen den
beobachteten Phänotyp hervorgerufen haben.
Im Rahmen einer eingehenden Untersuchung der UL24-Genregion wurde anhand
von RACE-Analysen festgestellt, dass die Transkripte von UL24 und UL25
überlappen. Die komplette Deletion des UL24-ORF führte somit auch zu einer
funktionellen Inaktivierung des benachbarten UL25-Leserasters. So konnte in
Zellen, welche mit TB4delUL24 infiziert waren, nach RT-PCR nur eine deutlich
verminderte Menge an UL25-Transkripten nachgewiesen werden. Betrachtete
man die Expression von UL25 auf Proteinebene, so war im Fall der
Deletionsmutante kein pUL25 nachweisbar.
Offenbar beeinträchtigt die Deletion von UL24 den Promotor von UL25, da mit
dem UL24 Leserahmen auch eine TATA-Box entfernt wird, die vermutlich für das
UL25-Transkript verantwortlich ist. Aufgrund dieser Daten kann man nicht
ausschließen, dass allein die Expression von UL24 für den bei der
Deletionsmutante beobachteten Phänotyp verantwortlich ist. Um diese Frage zu
klären, wurden weitere Mutanten generiert. Hierbei wurden das erste Startkodon,
ein weiteres ATG an Position 59 oder beide ATGs von pUL24 mutiert. Die
Mutation des ersten ATG führte zur Entstehung eines verkürzten UL24-
Genprodukts, wohingegen die Mutation des M59 keinen Effekt zeigte. Allein nach
Umwandlung beider Methionine konnte kein pUL24 mehr nachgewiesen werden.
Bei allen drei Mutanten war die Expression von pUL25 nicht beeinträchtigt. In den
folgenden Virus Yield-Experimenten wurde für die Doppelmutante eine Reduktion
der Virustiter vergleichbar TB4delUL24 beobachtet. Interessanterweise lieferte
TB4mut1.ATG einen intermediären, schwach attenuierten Phänotyp.
Möglicherweise konnte das nicht voll funktionsfähige, verkürzte Protein immer
noch einen Teil seiner Aufgaben bei der Infektion von Endothelzellen erfüllen.
Um die Beteiligung von pUL25 am Zelltropismusdefekt von TB4delUL24 in einem
alternativen Ansatz auszuschließen, wurden HFF mit rekombinanten retroviralen
Vektoren transduziert und somit pUL24 exogen exprimiert. Wurde pUL24 in trans

Diskussion 72
zur Verfügung gestellt, konnte die Anzahl an HCMV-infizierten HUVEC auf
Wildtypniveau angehoben werden. Diese Daten stimmen mit Studien überein, in
denen gezeigt wurde, dass die Entfernung des UL25-Leserahmens keinen
Einfluss auf Endothel- noch Epithelzelltropismus ausübt [Dunn et al., 2003].
4.5 Bedeutung von UL24 für Epithelzell- und Makrophagentropismus
Epithelzellen spielen eine zentrale Rolle bei der Verbreitung und Persistenz im
immungesunden Wirt [Britt et al., 1996]. Zusätzlich wurden bei Patienten mit
geschwächtem Immunsystem zytopathische Effekte, die für eine CMV-infektion
typisch sind, in Epithelzellen unterschiedlichster Gewebe beschrieben [Ho 1991].
Nachdem für HCMV UL131, einer anderen molekularen Determinante des
Endothelzelltropismus von HCMV (HCMV UL128-131 [Hahn et al., 2004]),
berichtet wurde, dass es auch für Epithelzelltropismus benötigt wird [Wang et al.,
2005b], wurden im Rahmen dieser Arbeit Effekte auf die Infektion anderer
Zelltypen durch TB4delUL24 untersucht. In Immunfluoreszenzanalysen konnte wie
schon bei den Experimenten mit Endothelzellen eine deutlich verringerte Anzahl
an IE-positiven Epithelzellen am Beispiel von HeLa oder RPE1 gezeigt werden.
Kürzlich erschienene Studien haben basierend auf Mutationsanalysen des MCMV
sechs Gene identifiziert (M36, M45, M78, M139, M140 und M141 [Ménard et al.,
2003]), welche nach Mutation bzw. Deletion eine effektive Infektion von
Makrophagen nicht mehr ermöglichen. Sie alle verfügen über homologe Gene in
HCMV. Weiterhin ist es bemerkenswert, dass mit M36 und M139-M141 vier dieser
Gene wie auch HCMV-pUL24 der US22-Genfamilie angehören. Bei Infektion von
aus primären Monozyten differenzierten Makrophagen, zeigte TB4delUL24 erneut
den Phänotyp, der schon in Endothel- und Epithelzellen beobachtet werden
konnte. Weitere Experimente, ob es sich hierbei wie im Fall von M36 um einen
anti-apoptotischen Effekt [Ménard et al., 2003] handeln könnte, stehen noch aus.
Es scheint sich allerdings um einen zellübergreifenden Mechanismus zu handeln,
durch den das Tegumentprotein HCMV-pUL24 neben seiner Rolle bei der
Infektion von Endothelzellen auch einen Effekt auf die Infektion von Epithelzellen
und Makrophagen ausübt.

Diskussion 73
4.6 Mechanismus des Endothelzelltropismus
Zum bisherigen Zeitpunkt kann noch keine Aussage darüber getroffen werden,
welche biologischen Mechanismen einen verminderten Endothelzelltropismus
bewirken. Bei Analyse der IE-Genexpression von TB4delUL24 anhand von
Immunfluoreszenzanalysen konnte gezeigt werden, dass sich das Virus bereits in
dieser Phase der viralen Replikation deutlich von TB4 und TB4revUL24
unterschied. Diese Beobachtungen stehen im Einklang mit Veröffentlichungen,
welche sich mit HCMV-Endothelzelltropismus auseinandersetzen [Sinzger et al.,
1999].
Allgemein gibt es bezüglich des Endothelzelltropismus von HCMV deutliche
Hinweise darauf, dass zumindest teilweise genetische Faktoren verantwortlich
sind. So zeigte ein klinisches Isolat, welches über einen längeren Zeitraum auf
Fibroblasten (TB40F) propagiert wurde, einen Verlust seines Endotheltropismus,
während nach Kultivierung auf Endothelzellen (TB40E) sowohl Endothel- als auch
Fibroblastentropismus erhalten blieb [Sinzger et al., 1999]. Weiterhin zeigten diese
unterschiedlich kultivierten Virusstämme Unterschiede bei der
Restriktionsfragment-Analyse ihrer Genome [Sinzger et al., 1999].
In der Region UL128-UL131, welche für eine produktive Infektion von
Endothelzellen unverzichtbar ist [Hahn et al., 2004], wurden häufig Mutationen
gefunden. Vor allem in fibroblastenadaptierten Stämmen wurden Mutationen in
dieser Genregion beobachtet [Dolan et al., 2004]. Bemerkenswerterweise können
bereits drei Passagen eines endothelgängigen Stammes auf Fibroblasten
ausreichen, um eine Mutation in der UL128-131 Region zu induzieren, welche
einen Verlust des Endothelzelltropismus zur Folge hat [Akter et al., 2003]. Eine
vergleichende Analyse der UL24-ORFs von TB40E und TB40F wies keine
Abweichungen voneinander auf (R. Pretsch, Daten nicht gezeigt), sodass eine
Mutation in UL24 nicht für den unterschiedlichen Tropismus dieser beiden
Stämme verantwortlich ist.
Über den zellbiologischen Mechanismus, der dem HCMV-Zelltropismus zugrunde
liegt, gibt es gegenwärtig mehrere Ansichten. Die aktuellen Hypothesen beinhalten
eine differentielle Expression von antiapoptotischen Faktoren [Brune et al., 2001],
die Fähigkeit Mikrofusionen zwischen benachbarten Zellen zu induzieren um eine
Verbreitung von Zelle zu Zellen zu unterstützen [Gerna et al., 2000] oder die
Bildung unterschiedlicher Glykoprotein-Komplexe, wodurch eine Adsorption des

Diskussion 74
Virus an die Zelle erst möglich wird [Wang et al., 2005a]. Auch Unterschiede bei
der Freisetzung viraler Genome erfolgreich eingedrungener Viruspartikel in
Endothelzellen zu initialen Zeitpunkten der viralen Infektion wurden beschrieben.
So gibt es Veröffentlichungen, in denen gezeigt wurde, dass der Transport zum
Kern von Viruskapsiden niedrig endotheliotroper Stämme in EC fast komplett
blockiert ist, während sie in Fibroblasten effizient funktioniert [Sinzger et al., 2000].
Dies könnte daran liegen, dass HCMV bei EC einen alternativen Weg in die Zellen
geht, als die in Fibroblasten übliche Fusion der Viruspartikel mit der
Wirtszellmembran [Compton et al., 1992]. Ultrastrukturelle Untersuchungen haben
gezeigt, dass in EC die Aufnahme der Virionen mittels Endozytose erfolgt und eine
nachfolgende Degradation der Partikel der Grund für die niedrige Infektiosität des
Laborstammes AD169 ist [Bodaghi et al., 1999]. Weiterhin wurde beobachtet,
dass der Eintritt verschiedener endothel- und epithelgängiger HCMV über
Endozytose und anschließende pH-abhängige Fusion der Viruspartikel mit den
Endosomen stattfindet [Ryckman et al., 2006], was im Rahmen dieser Arbeit auch
für das vom Wildtyp-BAC TB4 abstammende Virus bestätigt werden konnte. In
elektronenmikroskopischen Untersuchungen konnten für TB4 Kapside sowohl in
endosomalen Vesikeln als auch frei im Zytoplasma nachgewiesen werden,
wohingegen TB4delUL24 bisher nur in Endosomen aufzufinden war. Dies führt zu
der Annahme, dass der Verlust von pUL24 möglicherweise einen Defekt bei der
Fusion des Viruspartikels mit der endosomalen Membran bewirkt. Eine neue
Methode, bei denen die Virusmembran mit zwei unterschiedlichen Farbstoffen,
grün- und rot-fluoreszierend, markiert ist, könnte helfen, diesen Aspekt zu klären.
Im markierten Viruspartikel sind die Farbstoffe eng gepackt und die
Grünfluoreszenz ist durch selfquenching und FRET (flourescent resonance energy
transfer) unterdrückt. Das Virus erscheint rot. Durch Fusion des Virus liegen die
Farbstoffe räumlich weniger dicht gepackt vor und es kommt zu einer verstärkten
Grünfluoreszenz (dequenching) [Sakai et al., 2006]. Offensichtlich findet der
Defekt nicht auf der Ebene der Genregulation statt, da die Anzahl der IE-positiven
Zellen bei einer Infektion mit TB4, TB4delUL24 oder TB4revUL24 nach
Zentrifugation des Virus auf die Zellen nahezu übereinstimmt. Um den
Mechanismus, wie genau pUL24 zur Infektion von Endothel- und Epithelzellen
oder Makrophagen beiträgt, detaillierter beschreiben zu können, bedarf es
weiterer Experimente. Aufschlussreich wäre beispielsweise eine biochemische

Diskussion 75
Markierung von Viruspartikeln, um den Weg der Viren in die Wirtszelle per Video-
Mikroskopie zu verfolgen.
Zusammenfassend kann gesagt werden, dass im Rahmen dieser Arbeit das virale
Protein HCMV-pUL24 als Interaktionspartner von HCMV-ppUL32 identifiziert
werden konnte. Die biologische Relevanz des Proteins konnte durch die
Generierung mehrerer Virusmutanten analysiert werden. Hierbei wurde gezeigt,
dass pUL24 am Zelltropismus von HCMV für Endothel-, Epithelzellen als auch
Makrophagen beteiligt ist.

Zusammenfassung 76
5 Zusammenfassung
Im Rahmen dieser Arbeit wurde eine initial beschriebene Interaktion der
Tegumentproteine pUL24 und ppUL32 des Humanen Zytomegalievirus (HCMV) in
transient transfizierten Zellen im Western Blot bestätigt, ihre Salzresistenz
dargestellt und sogar im Kontext infizierter Zellen gezeigt. Die Koexpression von
pUL24 und ppUL32 in transfizierten HeLa-Zellen führte zu einer Relokation beider
Interaktionspartner, sodass die Proteine im Bereich der Kernmembran
kolokalisierten. Da es Hinweise darauf gab, dass der benachbarte ORF UL25
durch eine komplette Deletion von UL24 ebenfalls beeinflusst wird, wurde eine
Transkriptkartierung der UL24 Genregion durchgeführt. Hierbei konnte eine
Überlappung der jeweils hauptverantworlichen Promotorbereiche beschrieben
werden. In transienten Transfektionsexperimenten konnte eine ungefähr
zweifache Transaktivierung verschiedener eukaryoter Promotoren beobachtet
werden.
Für eine effiziente Infektion von Fibroblasten wurde keine Relevanz von pUL24
festgestellt. Durch die Generierung einer HCMV-BAC (bacterial artificial
chromosome) UL24-Deletionsmutante im Hintergrund des endotheliotropen
Virusstammes TB40E und die Durchführung entsprechender
Infektionsexperimente wurde ein Einfluss des Gens auf den Zelltropismus des
Virus bestätigt. Hierzu erfolgten Studien in Endothel- und Epithelzellen. Anhand
von Punktmutanten und eines Transkomplementationsexperiments wurde pUL24
als verantwortliches Protein der Region um UL24 identifiziert. Letztlich wurde
anhand von elektonenmikroskopischer Aufnahmen gezeigt, dass sich die reifen
Viruspartikel der UL24 Deletionsmutante in endosomalen Vesikeln anhäufen und
somit nicht ins Zytoplasma der infizierten Zelle gelangen. Diese Beobachtung
zusammen mit Experimenten, welche zeigen, dass das Wildtyp-Virus bei der
Infektion der Wirtszellen den Weg über die Endosomen stark favorisiert, führen zu
der Hypothese, dass das UL24 Deletionsvirus einen Defekt bei der
Verschmelzung mit der endosomalen Membran hat. Um diese Frage zu klären
sind allerdings noch weiterführende Experimente notwendig, die über den
Rahmen dieser Arbeit hinaus gehen.

Literaturverzeichnis 77
6 Literaturverzeichnis
1. Adair, R., Douglas, E. R., Maclean, J. B., Graham, S. Y., Aitken, J. D.,
Jamieson, F. E. & Dargan, D. J. (2002) J Gen Virol 83, 1315-1324.
2. Akter, P., Cunningham, C., McSharry, B. P., Dolan, A., Addison, C.,
Dargan, D. J., Hassan-Walker, A. F., Emery, V. C., Griffiths, P. D.,
Wilkinson, G. W. et al. (2003) J. Gen. Virol. 84, 1117-1122.
3. Bahr, U. & Darai, G. (2001) J. Virol. 75, 4854-4870.
4. Baldick, C. J., Jr., Marchini, A., Patterson, C. E. & Shenk, T. (1997) J Virol
71, 4400-4408.
5. Battista, M. C., Bergamini, G., Boccuni, M. C., Campanini, F., Ripalti, A. &
Landini, M. P. (1999) J. Virol. 73, 3800-3809.
6. Baxter, M. K. & Gibson, W. (2001) J Virol 75, 6865-6873.
7. Bechtel, J. T. & Shenk, T. (2002) J. Virol. 76, 1043-1050.
8. Biedler, J. L., Helson, L. & Spengler, B. A. (1973) Cancer Res. 33, 2643-
2652.
9. Bissinger, A. L., Sinzger, C., Kaiserling, E. & Jahn, G. (2002) J. Med. Virol.
67, 200-206.
10. Bodaghi, B., Slobbe-van Drunen, M. E., Topilko, A., Perret, E., Vossen, R.
C., van Dam-Mieras, M. C., Zipeto, D., Virelizier, J. L., LeHoang, P.,
Bruggeman, C. A. et al. (1999) Invest Ophthalmol. Vis. Sci. 40, 2598-2607.
11. Bodnar, A. G., Ouellette, M., Frolkis, M., Holt, S. E., Chiu, C. P., Morin, G.
B., Harley, C. B., Shay, J. W., Lichtsteiner, S. & Wright, W. E. (1998)
Science 279, 349-352.
12. Bresnahan, W. A. & Shenk, T. E. (2000) Proc Natl Acad Sci USA 97,
14506-14511.
13. Brewer, C. B. (1994) Methods Cell Biol. 43 Pt A, 233-245.
14. Britt, W. J. & Alford, C. A. (1996) in Fields Virology, eds. Fields, B. N. &
Knipe, D. M. (Lippincott-Raven, Philadelphia), pp. 2493-2523.

Literaturverzeichnis 78
15. Brune, W., Menard, C., Heesemann, J. & Koszinowski, U. H. (2001)
Science 291, 303-305.
16. Chang, C. S., Wang, C. H., Su, I. J., Chen, Y. C. & Shen, M. C. (1994) J.
Formos. Med. Assoc. 93, 421-428.
17. Chang, H. Y., Chi, J. T., Dudoit, S., Bondre, C., van de, R. M., Botstein, D.
& Brown, P. O. (2002) Proc. Natl. Acad. Sci. U. S. A 99, 12877-12882.
18. Chee, M. S., Bankier, A. T., Beck, S., Bohni, R., Brown, C. M., Cerny, R.,
Horsnell, T., Hutchison III, C. A., Kouzarides, T., Martignetti, J. A. et al.
(1990) in Current topics in microbiology and immunology, ed. McDougall, J.
K. (Springer, Berlin, Heidelberg), pp. 126-169.
19. Cherepanov, P. P. & Wackernagel, W. (1995) Gene 158, 9-14.
20. Colberg-Poley, A. M., Santomenna, L. D., Harlow, P. P., Benfield, P. A. &
Tenney, D. J. (1992) J. Virol. 66, 95-105.
21. Compton, T., Nepomuceno, R. R. & Nowlin, D. M. (1992) Virology 191, 387-
395.
22. Compton, T., Nowlin, D. M. & Cooper, N. R. (1993) Virology 193, 834-841.
23. Datsenko, K. A. & Wanner, B. L. (2000) Proc. Natl. Acad. Sci. U. S. A 97,
6640-6645.
24. di Guan, C., Li, P., Riggs, P. D. & Inouye, H. (1988) Gene 67, 21-30.
25. Digel, M. & Sinzger, C. (2006) in Cytomegaloviruses - Molecular Biology
and Immunology, ed. Reddehase, M. (Caister Academic Press, Norfolk),
pp. 445-464.
26. Dolan, A., Cunningham, C., Hector, R. D., Hassan-Walker, A. F., Lee, L.,
Addison, C., Dargan, D. J., McGeoch, D. J., Gatherer, D., Emery, V. C. et
al. (2004) J. Gen. Virol. 85, 1301-1312.
27. Dumas, A. M., Geelen, J. L., Weststrate, M. W., Wertheim, P. & van der, N.
J. (1981) J. Virol. 39, 390-400.
28. Dunn, W., Chou, C., Li, H., Hai, R., Patterson, D., Stolc, V., Zhu, H. & Liu,
F. (2003) Proc. Natl. Acad. Sci. U. S. A 100, 14223-14228.

Literaturverzeichnis 79
29. Efstathiou, S., Gompels, U. A., Craxton, M. A., Honess, R. W. & Ward, K.
(1988) Lancet 2/9, 63-64.
30. Epstein, M. A. (1985) IARC Sci. Publ. 17-27.
31. Evan, G. I., Lewis, G. K., Ramsay, G. & Bishop, J. M. (1985) Mol. Cell Biol.
5, 3610-3616.
32. Feire, A. L., Koss, H. & Compton, T. (2004) Proc. Natl. Acad. Sci. U. S. A
101, 15470-15475.
33. Fleckenstein, B., Muller, I. & Collins, J. (1982) Gene 18, 39-46.
34. Frenkel, N., Schirmer, E. C., Wyatt, L. S., Katsafanas, G., Roffman, E.,
Danovich, R. M. & June, C. H. (1990) Proc. Natl. Acad. Sci. U. S. A 87,
748-752.
35. Gebert, S., Schmolke, S., Sorg, G., Floss, S., Plachter, B. & Stamminger, T.
(1997) J. Virol. 71, 7048-7060.
36. Geng, Y. Q., Chandran, B., Josephs, S. F. & Wood, C. (1992) J. Virol. 66,
1564-1570.
37. Gerna, G., Percivalle, E., Baldanti, F., Sozzani, S., Lanzarini, P., Genini, E.,
Lilleri, D. & Revello, M. G. (2000) J. Virol. 74, 5629-5638.
38. Gibson, W. (1996) Intervirology 39, 389-400.
39. Gompels, U. A., Nicholas, J., Lawrence, G., Jones, M., Thomson, B. J.,
Martin, M. E., Efstathiou, S., Craxton, M. & Macaulay, H. A. (1995) Virology
209, 29-51.
40. Grant, S. G., Jessee, J., Bloom, F. R. & Hanahan, D. (1990) Proc. Natl.
Acad. Sci. U. S. A 87, 4645-4649.
41. Grefte, A., van der, G. M., van, S. W. & The, T. H. (1993) J. Infect. Dis. 167,
270-277.
42. Hahn, G., Revello, M. G., Patrone, M., Percivalle, E., Campanini, G.,
Sarasini, A., Wagner, M., Gallina, A., Milanesi, G., Koszinowski, U. et al.
(2004) J. Virol. 78, 10023-10033.
43. Hahn, G., Rose, D., Wagner, M., Rhiel, S. & McVoy, M. A. (2003) Virology
307, 164-177.

Literaturverzeichnis 80
44. Hanson, L. K., Slater, J. S., Karabekian, Z., Ciocco-Schmitt, G. & Campbell,
A. E. (2001) J. Virol. 75, 6292-6302.
45. Hayashi, M. L., Blankenship, C. & Shenk, T. (2000) Proc Natl Acad Sci USA
97, 2692-2696.
46. Hensel, G., Meyer, H., Gartner, S., Brand, G. & Kern, H. F. (1995) J Gen
Virol 76 ( Pt 7), 1591-1601.
47. Ho, D. D., Rota, T. R., Andrews, C. A. & Hirsch, M. S. (1984) J. Infect. Dis.
150, 956-957.
48. Ho, M. Cytomegalovirus biology and infection. 1st(21). 1991. New York,
Plenum Press. Ref Type: Case
49. Hobom, U., Brune, W., Messerle, M., Hahn, G. & Koszinowski, U. H. (2000)
J Virol 74, 7720-7729.
50. Honess, R. W. & Roizman, B. (1975) Proc. Natl. Acad. Sci. U. S. A 72,
1276-1280.
51. Hudson, J. B. (1988) J. Virol. Methods 19, 97-108.
52. Hudson, J. B., Misra, V. & Mosmann, T. R. (1976) Virology 72, 235-243.
53. Kellermann, O. K. & Ferenci, T. (1982) Methods Enzymol. 90 Pt E, 459-
463.
54. Kouzarides, T., Bankier, A. T., Satchwell, S. C., Preddy, E. & Barrell, B. G.
(1988) Virology 165, 151-164.
55. Kühnel, F., Fritsch, C., Krause, S., Mundt, B., Wirth, T., Paul, Y., Malek, N.
P., Zender, L., Manns, M. P. & Kubicka, S. (2004) Nucleic Acids Res. 32,
e30.
56. Lindquester, G. J. & Pellett, P. E. (1991) Virology 182, 102-110.
57. Liu, B. & Stinski, M. F. (1992) J. Virol. 66, 4434-4444.
58. Liu, Y. & Biegalke, B. J. (2002) J Virol 76, 2460-2468.
59. Maina, C. V., Riggs, P. D., Grandea, A. G., III, Slatko, B. E., Moran, L. S.,
Tagliamonte, J. A., McReynolds, L. A. & Guan, C. D. (1988) Gene 74, 365-
373.
60. Marchini, A., Liu, H. & Zhu, H. (2001) J. Virol. 75, 1870-1878.

Literaturverzeichnis 81
61. McCormick, A. L., Skaletskaya, A., Barry, P. A., Mocarski, E. S. &
Goldmacher, V. S. (2003) Virology 316, 221-233.
62. Megaw, A. G., Rapaport, D., Avidor, B., Frenkel, N. & Davison, A. J. (1998)
Virology 244, 119-132.
63. Ménard, C., Wagner, M., Ruzsics, Z., Holak, K., Brune, W., Campbell, A. E.
& Koszinowski, U. H. (2003) J Virol 77, 5557-5570.
64. Mettenleiter, T. C. (2004) Virus Res. 106, 167-180.
65. Mocarski, E. S. & Courcelle, C. T. (2001) in Fields Virology, ed. Straus, S.
E. Philadelphia, Lippincott-Raven), pp. 2629-2673.
66. Morgenstern, J. P. & Land, H. (1990) Nucleic Acids Res. 18, 1068.
67. Mori, Y., Yagi, H., Shimamoto, T., Isegawa, Y., Sunagawa, T., Inagi, R.,
Kondo, K., Tano, Y. & Yamanishi, K. (1998) Virology 249, 129-139.
68. Mullis, K., Faloona, F., Scharf, S., Saiki, R., Horn, G. & Erlich, H. (1986)
Cold Spring Harb. Symp. Quant. Biol. 51 Pt 1, 263-273.
69. Murphy, E., Yu, D., Grimwood, J., Schmutz, J., Dickson, M., Jarvis, M. A.,
Hahn, G., Nelson, J. A., Myers, R. M. & Shenk, T. E. (2003) Proc. Natl.
Acad. Sci. U. S. A 100, 14976-14981.
70. Nelson-Rees, W. A. & Flandermeyer, R. R. (1976) Science 191, 96-98.
71. Nicholas, J. (1996) J. Virol. 70, 5975-5989.
72. Niman, H. L., Houghten, R. A., Walker, L. E., Reisfeld, R. A., Wilson, I. A.,
Hogle, J. M. & Lerner, R. A. (1983) Proc. Natl. Acad. Sci. U. S. A 80, 4949-
4953.
73. Nowak, B., Sullivan, C., Sarnow, P., Thomas, R., Bricout, F., Nicolas, J. C.,
Fleckenstein, B. & Levine, A. J. (1984) Virology 132, 325-338.
74. Pass, R. F. (2001) in Fields Virology, ed. Straus, S. E. Philadelphia,
Lippincott-Raven), pp. 2675-2705.
75. Pear, W. S., Nolan, G. P., Scott, M. L. & Baltimore, D. (1993) Proc. Natl.
Acad. Sci. U. S. A 90, 8392-8396.
76. Plachter, B., Britt, W., Vornhagen, R., Stamminger, T. & Jahn, G. (1993)
Virology 193, 642-652.

Literaturverzeichnis 82
77. Pluthero, F. G. (1993) Nucleic Acids Res. 21, 4850-4851.
78. Ponten, J. & Macintyre, E. H. (1968) Acta Pathol. Microbiol. Scand. 74, 465-
486.
79. Rawlinson, W. D., Farrell, H. E. & Barrell, B. G. (1996) J. Virol. 70, 8833-
8849.
80. Reddehase, M. (2006) Cytomegaloviruses - Molecular Biology and
Immunology (Caister Academic Press, Norfolk, UK).
81. Reinhardt, B., Minisini, R. & Mertens, T. (2002) Herpes. 9, 21-23.
82. Roizman, B. & Baines, J. (1991) Comp Immunol. Microbiol. Infect. Dis. 14,
63-79.
83. Roizman, B., Carmichael, L. E., Deinhardt, F., de-The, G., Nahmias, A. J.,
Plowright, W., Rapp, F., Sheldrick, P., Takahashi, M. & Wolf, K. (1981)
Intervirology 16, 201-217.
84. Roizman, B. & Pellett, P. E. (2001) in Fields Virology, ed. S.E.Straus
Philadelphia, Lippincott-Raven), pp. 2381-2397.
85. Romanowski, M. J., Garrido-Guerrero, E. & Shenk, T. (1997) J. Virol. 71,
5703-5705.
86. ROWE, W. P., HARTLEY, J. W., WATERMAN, S., TURNER, H. C. &
HUEBNER, R. J. (1956) Proc. Soc. Exp. Biol. Med. 92, 418-424.
87. Rubin, R. H. (2001) Transpl. Infect. Dis. 3 Suppl 2, 1-5.
88. Ryckman, B. J., Jarvis, M. A., Drummond, D. D., Nelson, J. A. & Johnson,
D. C. (2006) J. Virol. 80, 710-722.
89. Sakai, T., Ohuchi, M., Imai, M., Mizuno, T., Kawasaki, K., Kuroda, K. &
Yamashina, S. (2006) J. Virol. 80, 2013-2018.
90. Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989) Molecular Cloning (CSHL
Press, Cold Spring Harbor).
91. Sanchez, V., Greis, K. D., Sztul, E. & Britt, W. J. (2000) J. Virol. 74, 975-
986.
92. Sawano, A. & Miyawaki, A. (2000) Nucleic Acids Res. 28, E78.

Literaturverzeichnis 83
93. Schierling, K., Buser, C., Mertens, T. & Winkler, M. (2005) J. Virol. 79,
3084-3096.
94. Schierling, K., Stamminger, T., Mertens, T. & Winkler, M. (2004) J. Virol. 78,
9512-9523.
95. Scholl, P. R., O'Gorman, M. R., Pachman, L. M., Haut, P. & Kletzel, M.
(1998) Bone Marrow Transplant. 22, 1215-1218.
96. Silva, M. C., Schroer, J. & Shenk, T. (2005) Proc. Natl. Acad. Sci. U. S. A
102, 2081-2086.
97. Sinzger, C., Eberhardt, K., Cavignac, Y., Weinstock, C., Kessler, T., Jahn,
G. & Davignon, J. L. (2006) J Gen. Virol. 87, 1853-1862.
98. Sinzger, C., Grefte, A., Plachter, B., Gouw, A. S., The, T. H. & Jahn, G.
(1995) J Gen Virol 76, 741-750.
99. Sinzger, C., Kahl, M., Laib, K., Klingel, K., Rieger, P., Plachter, B. & Jahn,
G. (2000) J Gen Virol 81, 3021-3035.
100. Sinzger, C., Schmidt, K., Knapp, J., Kahl, M., Beck, R., Waldman, J.,
Hebart, H., Einsele, H. & Jahn, G. (1999) J Gen Virol 80, 2867-2877.
101. Skaletskaya, A., Bartle, L. M., Chittenden, T., McCormick, A. L., Mocarski,
E. S. & Goldmacher, V. S. (2001) Proc. Natl. Acad. Sci. U. S. A 98, 7829-
7834.
102. Smith, M. G. (1956) Proc. Soc. Exp. Biol. Med. 92, 424-430.
103. Soderberg-Naucler, C., Fish, K. N. & Nelson, J. A. (1997) Cell 91, 119-126.
104. Southern, E. M. (1975) J. Mol. Biol. 98, 503-517.
105. Stasiak, P. C. & Mocarski, E. S. (1992) J. Virol. 66, 1050-1058.
106. Stinski, M. F. (1978) J. Virol. 26, 686-701.
107. Streblow, D. N., Orloff, S. L. & Nelson, J. A. (2001) J Nutr 131, 2798S-
27804S.
108. Varnum, S. M., Streblow, D. N., Monroe, M. E., Smith, P., Auberry, K. J.,
Pasa-Tolic, L., Wang, D., Camp, D. G., Rodland, K., Wiley, S. et al. (2004)
J. Virol. 78, 10960-10966.
109. Vink, C., Beuken, E. & Bruggeman, C. A. (2000) J. Virol. 74, 7656-7665.

Literaturverzeichnis 84
110. Wagner, M., Jonjic, S., Koszinowski, U. H. & Messerle, M. (1999) J. Virol.
73, 7056-7060.
111. Wagner, M., Ruzsics, Z. & Koszinowski, U. H. (2002) Trends Microbiol 10,
318-324.
112. Wang, D. & Shenk, T. (2005b) J. Virol. 79, 10330-10338.
113. Wang, D. & Shenk, T. (2005a) Proc. Natl. Acad. Sci. U. S. A 102, 18153-
18158.
114. Wang, X., Huong, S. M., Chiu, M. L., Raab-Traub, N. & Huang, E. S. (2003)
Nature 424, 456-461.
115. White, E. A., Clark, C. L., Sanchez, V. & Spector, D. H. (2004) J. Virol. 78,
1817-1830.
116. Winkler, M., Rice, S. A. & Stamminger, T. (1994) J Virol 68, 3943-3954.
117. Winkler, M., Schmolke, S., Plachter, B. & Stamminger, T. (1995) Scand J
Infect Dis Suppl 99, 8-9.
118. Winkler, M. & Stamminger, T. (1996) J Virol 70, 8984-8987.
119. Xiao, J., Tong, T., Zhan, X., Haghjoo, E. & Liu, F. (2000) J. Virol. 74, 9488-
9497.
120. Zagursky, R. J., Baumeister, K., Lomax, N. & Berman, M. L. (1985) Gene
Anal Tech 2, 89-94.

Lebenslauf 85
Danksagung Zuallererst möchte ich Herrn PD Dr. Michael Winkler danken, der mich im Verlauf
meiner Doktorarbeit mit viel Geduld betreut hat und von dessen vielen Ideen ich
profitieren konnte. Herrn Prof. Dr. Thomas Mertens danke ich für viele anregende
Diskussionen während unserer Abteilungsseminare und dafür, dass er auch nach
meiner Diplomarbeit in der Abteilung Virologie Vertrauen in mich gezeigt hat.
Ich möchte mich bei Frau Prof. Dr. Barbara Spellerberg für die Übernahme des
Koreferats meiner Dissertation bedanken. Weiterhin bedanke ich mich bei Dr.
Heike Wilts, Dr. med Lutz von Müller und Dr. Barbara Reinhardt und Prof. Dr.
Detlef Michel für ihre Hilfe in manch kniffligen Fragen. Großer Dank gebührt auch
Anke Lüske und Monika Dürre, die meine Arbeiten tatkräftig unterstützt haben.
Besonders bedanken möchte ich mich bei meinen Mitdoktoranden, speziell bei
Jens Holl, den ich schon im Grundstudium kennen und im Verlauf meines
Studiums immer mehr schätzen gelernt habe.
Nicht vergessen möchte ich die „Fraktion Tischtennis“, Frank, Mike, Steffen, Jan
aus der HIV-AG und seit wenigen Monaten auch Jens von Einem.
Bei organisatorischen Fragen konnte ich mich stets getrost an Ingrid Bennett
wenden, die immer dafür gesorgt hat, das die Bürokratie nicht überhand nahm.
Natürlich bedanke ich mich bei meiner Familie, meinen Eltern und Freunden für
Ihre Unterstützung über mein ganzes Leben hinweg.
Annette, bei Dir bedanke ich mich, dass Du „in guten und in schlechten Zeiten“
immer für mich da warst, bist und auch sein wirst. Zusammen mit unserer Lena,
die dieses Jahr das Licht der Welt erblickt hat.





























