Embed Size (px)
Citation preview

ARTICLES
412 nature materials | VOL 4 | MAY 2005 | www.nature.com/naturematerials
Chiral selectivity in the charge-transfer bleaching of single-walled carbon-nanotube spectra
MICHAEL J. O’CONNELL, EZRA E. EIBERGEN AND STEPHEN K. DOORN*Chemistry Division, C-ACS, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA*e-mail: [email protected]
Published online: 10 April 2005; doi 10.1038/nmat1367
Chiral selective reactivity and redox chemistry of carbon
nanotubes are two emerging fi elds of nanoscience.
These areas hold strong promise for producing methods
for isolating nanotubes into pure samples of a single
electronic type, and for reversible doping of nanotubes
for electronics applications. Here, we study the selective
reactivity of single-walled carbon nanotubes with organic
acceptor molecules. We observe spectral bleaching of
the nanotube electronic transitions consistent with an
electron-transfer reaction occurring from the nanotubes
to the organic acceptors. The reaction kinetics are found
to have a strong chiral dependence, with rates being
slowest for large-bandgap species and increasing for
smaller-bandgap nanotubes. The chiral-dependent
kinetics can be tuned to effectively freeze the reacted
spectra at a fi xed chiral distribution. Such tunable redox
chemistry may be important for future applications
in reversible non-covalent modifi cation of nanotube
electronic properties and in chiral selective separations.
Single-walled carbon nanotubes (SWNTs) are one-dimensional conductors that can be metallic or semiconducting depending on their chirality1. Current methods of nanotube synthesis produce
a wide range of nanotube chiralities. Although advances in SWNT synthesis have yielded very narrow diameter distributions2–4, no method yet produces one type of nanotube (that is, just semiconductors or metallics). The range of nanotubes currently produced also have different bandgaps and chemical reactivity. These differences can allow chiral selective chemistry.
Examples of chiral selective chemistry include demonstration of diameter and electronic-type selectivity in nanotube osmylation5 and ozonolysis reactions6. Mechanistic studies suggest that, in addition to diameter-dependent pyramidalization and π-orbital misalignment effects, charge transfer can be an important component of chiral selective reactivity. Charge transfer is also a determining factor in the preferential reactivity of diazonium reagents towards metallic nanotube types7. The role that shifts in charge density and charge transfer plays in chiral selective reactions underscores the importance of understanding nanotube redox chemistry, an emerging fi eld that has included a number of spectro-electrochemical studies of nanotube redox behaviour8–12. Chiral selectivity in pH-dependent absorbance, Raman and fl uorescence spectra13,14 is thought to originate in bandgap-dependent electron-transfer reactions. Studies of SWNT electron-transfer reactions with small-molecule inorganic redox reagents demonstrate an increase in the SWNT reduction potential with increasing bandgap15.
In addition to fundamental interest in the origins of chiral selective reactivity, such reactions can be an important route to the separation of nanotube types. Several recent papers demonstrate routes to separation of metallics from semiconductors and rely on chiral selective chemisorption or physisorption7,16,17. Solution-phase manipulation of SWNTs to isolate nanotubes with desired electronic properties will not work for all SWNT synthesis methods, especially for nanotubes longer than a micrometre. Advances in synthesis now allow the growth of ultra-long SWNTs on a surface by chemical vapour deposition (CVD)18,19. Nanotubes with lengths of the order of a centimetre are not readily put into suspension and are often grown with desired placement and geometry. Nanotubes
nmat1367-print.indd 412nmat1367-print.indd 412 12/4/05 1:39:14 pm12/4/05 1:39:14 pm
Nature Publishing Group© 2005

ARTICLES
nature materials | VOL 4 | MAY 2005 | www.nature.com/naturematerials 413
synthesized by these methods cannot be manipulated by solution-based separation methods. Although methods for selectively eliminating metallic nanotubes on CVD-grown samples have been developed20,21, new methods are needed to control their properties for electronics applications. Chirality-selective redox chemistry may provide that control.
We describe a method for selectively and reversibly doping SWNTs, thereby controlling their electronic properties through redox reactions with small organic electron-acceptor molecules. The selectivity stems from the varying reactivity of each nanotube type based on differences in bandgap and electronic properties that depend on their diameter and chirality. Redox doping by means of electron transfer between an acceptor molecule and an electron donor (the nanotube) results in bleaching of nanotube fl uorescence and absorbance spectra. Chiral selectivity in the redox reaction is shown to result from bandgap dependence in the electron-transfer kinetics.
REDOX-INDUCED SPECTRAL BLEACHING.
We studied the reactivity of HiPco nanotubes, suspended22 in sodium dodecyl sulphate (SDS) solution, with a number of organic charge-transfer molecules, including 7,7,8,8-tetracyanoquinodimethane (TCNQ), 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (TFTCNQ), mordant yellow 10 (MY) and 4-amino-1,1-azobenzene-3,4-disulphonic acid (AB). The azobenzene compounds MY and AB were favoured for this study, as they are considerably more water-soluble than TCNQ and TFTCNQ. Nanotube spectral changes induced by reaction with these small molecules were monitored using fluorescence (excited at 785 nm) and absorption spectroscopy (see Methods). Initial studies focused on spectral behaviour in aqueous solutions under ambient oxygen conditions. In addition to the organic acceptor molecules, other species present in the aqueous suspension of nanotubes were found to play an important role in the bleaching and recovery of SWNT spectra. TritonX-405, poly(ethylene glycol)-co-poly(propylene glycol) and SDS were screened to determine the best surfactant for this study. SDS-suspended nanotubes were found to require much less organic acceptor than the non-ionic surfactants (about
a tenth as much) to effect the observed spectral changes. The difference may arise either from competing reaction of the acceptors with the non-ionic surfactants23, or through greater stabilization of formation of the nanotube–acceptor complex when it reacts in the presence of the ionic SDS15. Because of the improved reactivity observed between the nanotubes and organic acceptors, we chose SDS for the remainder of these studies.
Addition of AB to a nanotube solution results in a rapid depletion of the nanotube fluorescence spectrum, dependent on bandgap and nanotube diameter (Fig. 1). Time-dependent spectra of the semiconducting nanotubes show that intensity loss starts with the tubes of lowest energy bandgap and largest diameter, and progresses to the smallest-diameter/largest-bandgap semiconductors with time at a given concentration. After 5 min (Fig. 1 inset) the addition of 1 μmol of AB bleached the fluorescence of nearly all the nanotubes, leaving only the tubes with the highest-energy bandgap. Addition of more AB resulted in a complete loss of nanotube fluorescence. Similar spectral changes were observed when monitoring the nanotube absorption spectra during the reactions (see Supplementary Information). These results indicate that loss of fluorescence intensity occurs as a direct result of a decrease in transition strength (bleaching) and not through excited-state energy transfer (quenching). Degassed samples demonstrated more complex behaviour under these conditions and will be discussed later. Reaction of nanotubes with the other organic acceptor molecules used in this study resulted in similar spectral behaviour, but with differing bleaching kinetics and effectiveness.
Zheng and Diner15 have studied the reaction of DNA-wrapped nanotubes with the small inorganic oxidizing agent K2IrCl6. They observed changes in the fi rst van Hove absorbance spectra of the nanotubes similar to our spectral results: addition of oxidizer bleaches absorbance, with small-bandgap nanotubes being affected fi rst. The authors attribute this behaviour to electron transfer from the nanotube to the metal complex15. Similar redox reactivity is likely to be responsible for our observations.
Figure 2 shows the relative reduction potentials (determined by cyclic voltammetry) of the organic acceptor molecules used in our study, compared with the expected positions for a number of
900
900
1,000 1,100
1,100
1,200 1,300
1,300
1,400 1,500
1,500
1,600 1,700
1,700
Wavelength (nm)
11
0.8
0.8
0.6
0.6
0.4
0.4
0.2
0.2
0
0
Rela
tive
inte
nsity
Rela
tive
inte
nsity
Wavelength (nm)
OriginalFinal
Figure 1 Time-dependent redox bleaching of fl uorescence spectra. Spectra (with excitation at 785 nm) were taken every 15 seconds after the addition of 1 μmol of AB to 1 ml of 10 mg l–1 SWNTs suspended in 1% SDS/D2O. Long-wavelength emission is found to decay more rapidly than that at short wavelengths. The inset shows initial and fi nal spectra.
1.15 1.05 0.95 0.85 0.751.25
Transition energy (eV)
–1.75
–1.25
–0.75
–0.25
0.25
075
1.25
1.75
Pote
ntia
l (V
Vs. N
HE)
8,3 6,57,5 10,2
9,412,1 11,3 10,5
9,7 10,6 9,812,5
Fermi energy
MYAB
TCNQ
TFTCNQ
Figure 2 Relative potential energies. Plot of relative potential energies of the valence band, conduction band and Fermi levels of specifi c nanotube chiralities in relation to reduction potentials (determined by cyclic voltammetry) for the organic acceptor molecules used in this study. The nanotube fl uorescence spectrum (785 nm excitation) is superimposed. The red arrow indicates the electron transfer from the SWNT to the acceptor.
nmat1367-print.indd 413nmat1367-print.indd 413 12/4/05 1:39:21 pm12/4/05 1:39:21 pm
Nature Publishing Group© 2005
ARTICLES
414 nature materials | VOL 4 | MAY 2005 | www.nature.com/naturematerials
the nanotube chiralities observed in our fl uorescence spectra. The reduction potentials and Fermi levels for the different chiralities are referenced to the measured potential of the (6,5) nanotube obtained by Zheng and Diner15. By performing a one-electron redox titration (with K2IrCl6 as the oxidant) of a (6,5) enriched nanotube sample, they obtained its reduction potential of 0.800 mV relative to normal hydrogen electrode (NHE). In Fig. 2, the relative positions of the Fermi levels for each chirality are modelled after the results of Okazaki et al.11, for which the nanotube work function was found to vary linearly with inverse nanotube diameter. This translates to a near-linear dependence of the Fermi level on bandgap energy in our observed spectral region. As seen in Fig. 2, the change in potential between the nanotubes and the organic acceptors clearly favours an electron transfer from the nanotubes to the electron acceptors (as indicated by the downward arrow). Thus a spectral bleaching based on electron transfer is likely.
Given the difference of about 0.3 V in reduction potential found for AB and MY, differences in bleaching behaviour for the two acceptors are expected. As shown in Fig. 2, a transition in the effectiveness of AB to act as an electron acceptor should occur at a bandgap of ~1.2 eV. Nanotubes with larger bandgaps should not be oxidized by AB. As seen in Figs 1 and 3, effective bleaching of nanotube fl uorescence experiences a transition between the (6,5) and (7,5) chiralities. Signifi cant bleaching with AB occurs only for bandgaps less than or equal to that of the (7,5) nanotube. Any further bleaching of the (6,5) and (8,3) transitions at higher AB concentrations may instead result from an excited-state quenching reaction. In contrast, with MY as an acceptor, this transition in the effectiveness of bleaching should occur at a bandgap of ~1.1 eV. Our experimental results for MY demonstrate that effective bleaching occurs for nanotubes with bandgaps less than about 1.15 eV (transitions between the (10,2) and (9,4) chiralities), in good agreement with the model depicted in Fig. 2.
Further support for the electron-transfer mechanism was obtained by studying the effect of the addition of organic reducing agents to the already oxidized nanotube samples. Addition of the electron-donating compound B-nicotinamide adenine dinucleotide (reduced, NADH) to the quenched nanotube suspensions resulted in complete recovery of all spectra. Figure 3 shows the original fl uorescence spectrum taken before the addition of AB, the spectrum after addition of AB (resulting in spectral bleaching) and nearly
complete recovery after addition of NADH. The recovery is consistent with electron transfer from NADH to the oxidized nanotube. The recovery can also be performed stepwise through titration with the NADH solution. Another organic recovery agent, tetrathiafulvalene (TTF), was found to give partial recovery of the spectrum when added to a sample quenched with any of the organic acceptors investigated. Stoichiometric recovery was not possible, owing to the low water solubility of TTF. These recovery results demonstrate the facile reversibility of the nanotube redox reactivity.
As implied in Fig. 2 and suggested previously11,13,15, the electron transfer is expected to occur from the top of the
850 1,050 1,250 1,450 1,650Wavelength (nm)
1
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0
Rela
tive
inte
nsity
OriginalQuenchedRecovered
Figure 3 Recovery of nanotube fl uorescence. Nanotube fl uorescence spectra (785 nm excitation) showing unreacted spectrum (blue line), spectrum after addition of 1 μmol of AB to 1 ml 10 mg l–1 SWNTs suspended in 1% SDS/D2O (black line), and recovered spectrum after addition of excess NADH (red line).
Abso
rban
ce
1.5
1.3
1.1
0.9
0.7
0.5500 700 900 1,100 1,300 1,500
Wavelength (nm)
Figure 4 Nanotube fi rst and second van Hove absorbance spectra. Absorbance spectra of unreacted (red line) and bleached nanotubes (black line) after addition of 0.5 μmol of AB to 1 mL of 14 mg l–1 SWNT in 1% SDS/D2O. Spectra show both the fi rst van Hove transitions (wavelengths longer than 900 nm) and the second (wavelengths shorter than 900 nm), and demonstrate redshifting of the second van Hove spectra.
150 200 250 300
40,000
30,000
20,000
10,000
0
150 200 250 300
Raman shift (cm–1)
Rela
tive
inte
nsity
3,000
2,000
1,000
0
Figure 5 Time evolution of Raman spectra of the radial breathing mode region. Spectra (with excitation at 785 nm) were taken every 30 seconds after the addition of 1 μmol of AB to 1 ml of 10 mg l–1 SWNTs suspended in 1% SDS/D2O. Raman intensity decays on addition of AB and the relative intensities of different radial breathing modes shift. The inset shows the spectrum taken 5 min after addition of AB.
nmat1367-print.indd 414nmat1367-print.indd 414 12/4/05 1:39:23 pm12/4/05 1:39:23 pm
Nature Publishing Group© 2005
ARTICLES
nature materials | VOL 4 | MAY 2005 | www.nature.com/naturematerials 415
nanotube valence band. This suggests that only the first van Hove transitions should be strongly affected. Figure 4 shows the behaviour of the full visible/near-infrared (vis-NIR) absorbance spectrum on addition of organic oxidizer. Only the first van Hove transitions become bleached. The second van Hove transitions of the semiconductors (in the 500 to 900 nm region) remain, and this is consistent with an electron-transfer reaction occurring from the top of the valence band. The second van Hove absorbance features, however, are slightly redshifted. Similar redshifting has been observed previously when the nanotube SDS environment is exchanged for a polymer22 or other surfactants24. The redshifting has been attributed to an electronic dispersion induced by interaction of water with the nanotubes. These observations demonstrate the extreme sensitivity of nanotubes to their environment. Perturbation of the electronic structure will cause an absorbance and fluorescence redshift. The redshift results from disruption of the SDS micelle structure and/or the non-covalent interaction of the charge-transfer molecule with the nanotube sidewall. The shift therefore is consistent with formation of a charge-transfer complex.
The changes in electronic structure are also reflected in the nanotube Raman spectra (Fig. 5). Reaction with AB yields a loss of Raman intensity in the radial breathing modes. This decay results from a combination of the loss of electron density9,11 and redshifting of the resonant transitions away from the laser excitation wavelength of 785 nm. Redshifting is also manifested as a change in relative intensities of the (12,1) (at 237 cm–1) and (10,2) (at 265 cm–1) radial breathing modes (Fig. 5 inset). Whereas the (12,1) transition (at 792 nm) is shifted out of resonance, the (10,2) (at 730 nm) becomes more resonant25,26, resulting in a greater relative (10,2) intensity after nanotube oxidation. As was observed for fluorescence behaviour (Fig. 3), addition of NADH results in nearly complete recovery of the radial breathing mode intensity.
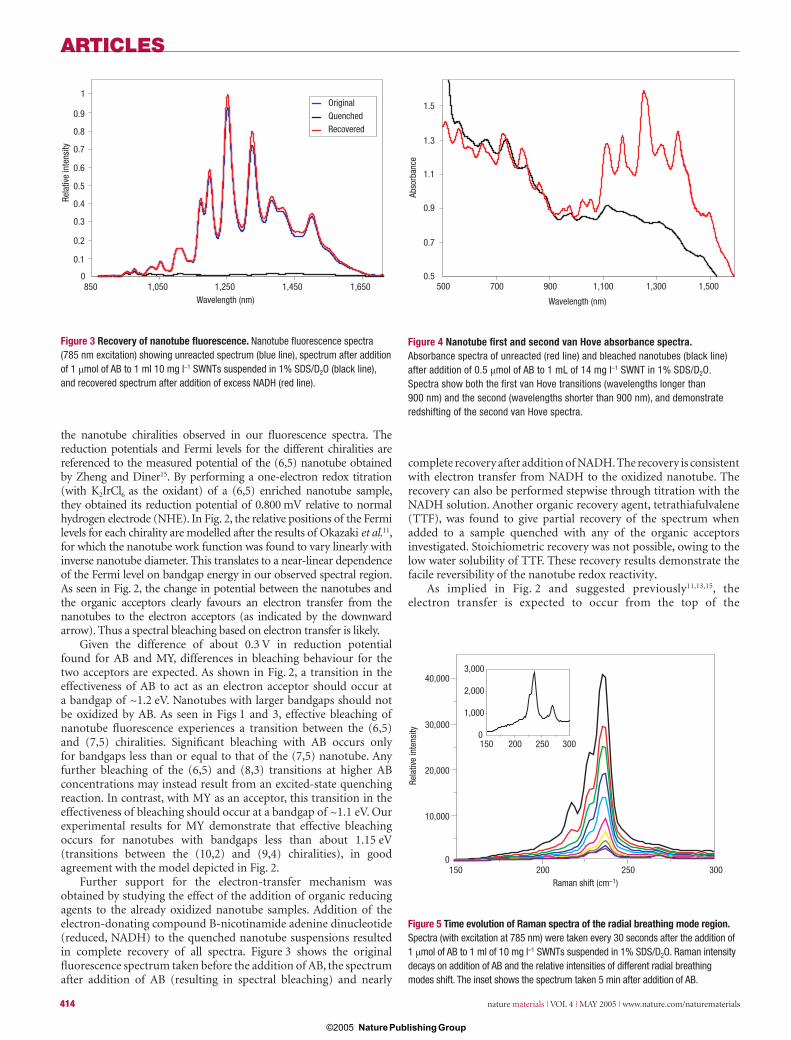
Figure 6 Chirality-dependent kinetics. Plots of relative intensity as a function of time for fl uorescence at a, 0.83 eV, b, 0.99 eV, c, 1.06 eV and d, 1.12 eV. Squares represent actual data with a pseudo fi rst-order fi t (red solid line) superimposed. e, Plot of pseudo fi rst-order rate constants as a function of bandgap energy for a range of specifi ed nanotube chiralities. Decay data were obtained from addition of 1 μmol of AB to 1 ml of 10 mg l–1 SWNTs in 1% SDS/D2O solution.
a b
c d
e
0.8 0.85 0.9 0.95 1 1.05 1.1
Bandgap energy (eV)
0.053
0.043
0.033
0.023
0.013
0.003
Rate
con
stan
t (s–1
)
12,5
10,6
9,8
9,7
10,5
11,312,1
9,4
0 50 100 150 200 250 300Time (s)
1.0
0.8
0.6
0.4
0.2
0.0
0 50 100 150 200 250 300
Time (s)
1.0
0.8
0.6
0.4
0.2
0.0
0 50 100 150 200 250 300Time (s)
1.0
0.8
0.6
0.4
0.2
0.0
0 50 100 150 200 250 300
Time (s)
1.0
0.9
0.8
0.7
0.6
0.5
0.4
Rela
tive
inte
nsity
Rela
tive
inte
nsity
Rela
tive
inte
nsity
Rela
tive
inte
nsity
CHIRAL-DEPENDENT BLEACHING KINETICS
The bandgap-dependent change in driving force for the electron transfer from the valence band of the nanotubes to acceptor molecules (illustrated in Fig. 2) suggests that there should be a chirality dependence in the rate of electron transfer. This is observed in Fig. 1, for which we noted that fl uorescence from large-diameter/small-bandgap nanotubes decays faster than for larger-bandgap nanotubes. Closer inspection of the decay behaviour (Fig. 6a–d) reveals an exponential decrease in fl uorescence intensity with time, fi tted well by a fi rst-order decay curve and indicating a process with pseudo fi rst-order kinetics. Similar results are obtained from absorbance monitoring. On the basis of estimates of 0.2 to 0.4 valence electrons available per 100 carbon atoms15, AB is present in a 500- to 1,000-fold excess compared with the number of valence electrons available for reaction. Therefore pseudo fi rst-order kinetic behaviour should be observed in this system. We also fi nd a linear change in rate constant with change in organic acceptor concentration (see Supplementary Information), as would be expected for a pseudo fi rst-order process.
The decay curves obtained for different fl uorescence wavelengths (corresponding to different nanotube chiralities) change markedly with bandgap energy. The longest-wavelength peak has the highest rate of decay, and the shortest-wavelength feature has the slowest. A plot of the pseudo fi rst-order rate constant (k) against nanotube bandgap energy (Ebg) (Fig. 6e) is nearly linear. This near-linearity is also preserved in a plot of the natural log of the rate constant against Ebg (see Supplementary Information). Such behaviour is consistent with electron-transfer rate theory, which predicts that the rate constant should depend exponentially on the electrochemical driving force (∆G), resulting in
ln(k) ∝ –ΔG ≈ nF(EA – (EF – 1/2Ebg)). (1)
nmat1367-print.indd 415nmat1367-print.indd 415 12/4/05 1:39:24 pm12/4/05 1:39:24 pm
Nature Publishing Group© 2005
ARTICLES
416 nature materials | VOL 4 | MAY 2005 | www.nature.com/naturematerials
In equation (1), n is 1 (the number of electrons), F is the Faraday constant, EA is the redox potential of the organic acceptor, and EF is the Fermi level of a given nanotube. A 10-fold change in electron-transfer rates is found across the range of nanotubes plotted in Fig. 6e. As seen in equation (1), the change in bandgap affects the reaction rates in two ways. The fi rst is through an effective change in relative position of the top of the valence band. In the absence of any other effects (that is, zero change in Fermi level with chirality), this results in a variation across the observed chiralities of only 150 mV. Although this small change may not contribute much to the large range in observed rates, it may be responsible for the slight curvature observed in the bandgap dependence. The second and more signifi cant factor is the near-linear decrease in potential of the Fermi level as bandgap decreases. As modelled in Fig. 2, this decrease gives an additional driving force of about 1 V to the electron-transfer from small-bandgap nanotubes compared with the largest-bandgap tubes. Moreover, we note that the near-linear dependence of the Fermi level on bandgap translates directly to the observed near-linearity in the reaction rates.
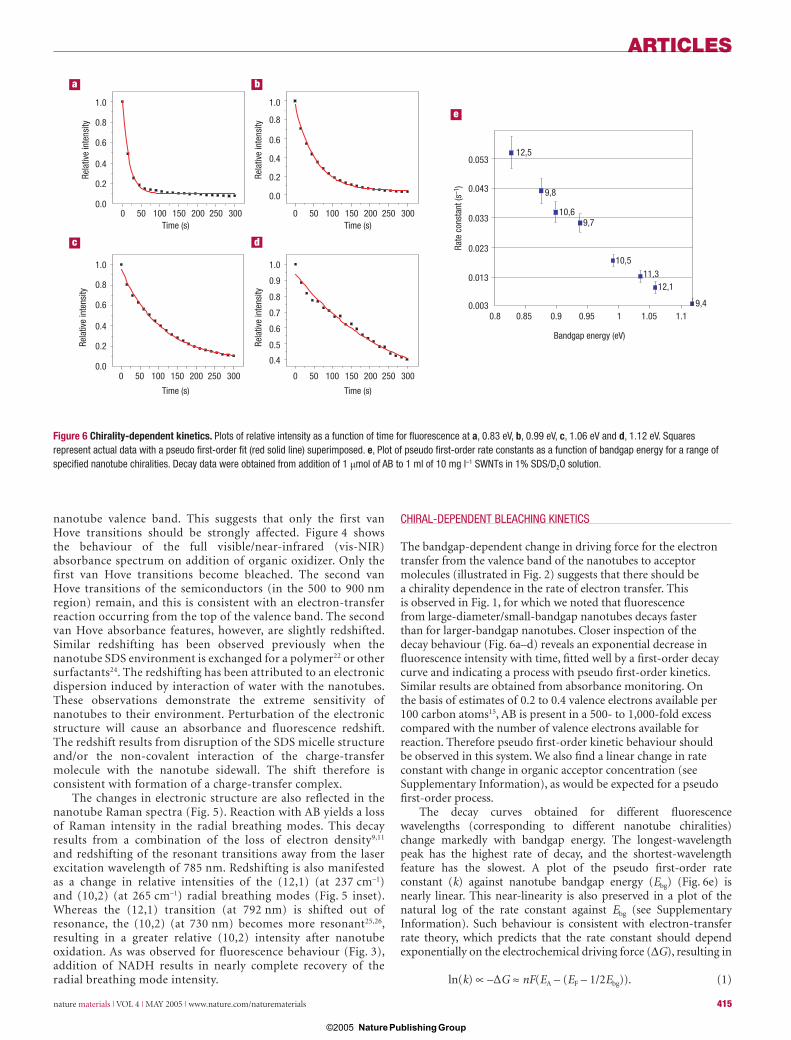
The pronounced difference in observed bleaching kinetics as a function of nanotube chirality can provide a high degree of control over the fi nal distribution of chiralities affected by these redox reactions. The fi nal distribution can be controlled through changes in relative nanotube and organic acceptor concentrations, which tune the pseudo fi rst-order rate constants for reaction. As a result, the extinction of SWNT NIR absorbance and fl uorescence features could be effectively ‘frozen’ at a particular chiral distribution by addition of an appropriate acceptor concentration. Bleaching of additional species resumed if more was added. Figure 7a shows the NIR absorbance spectrum of SWNTs bleached from the original spectrum and frozen with the addition of 0.1 μmol of AB to 1 ml of 14 mg l–1 SWNT. Under these conditions, the spectral distribution remained stable one hour after AB addition. A second addition of 0.1 μmol of AB shifted the stable distribution further, to larger-bandgap chiralities. A comparison of relative spectral intensities before and after bleaching (Fig. 7b) reveals the fractional change in chiralities. Thus, tuned redox reactivity results in a spectral enrichment of larger-bandgap nanotubes. Coupling such chemistry to separation processes may be an effective method for purifi cation of nanotubes by chirality.
The absolute positions of the Fermi levels shown in Fig. 2 are referenced to the valence band position of the (6,5) nanotube of Zheng and Diner15 (with the reduction potential corresponding to the position of the valence band) and as a result differ signifi cantly from values originally stated in ref. 11. These differences may be due to solvation effects found for the DNA-wrapped and SDS-suspended nanotubes, effects that may not be present at the electrode surface in the work of Okazaki et al.11. Also, because the Fermi level dependence in ref. 11 was determined over a narrower range of nanotube diameters than we observe and model in Figs 2 and 6, linear extrapolation over the full chirality range may not be appropriate. This may be a further contributing factor to our observed minor nonlinearity. Additionally, Zheng and Diner15 estimate a smaller change (50 mV) in driving force as bandgap is modulated from the (6,5) to the (12,1) nanotube chirality, much less than implied by Fig. 2. This suggests that the Fermi level dependence for solubilized nanotubes may be less steep
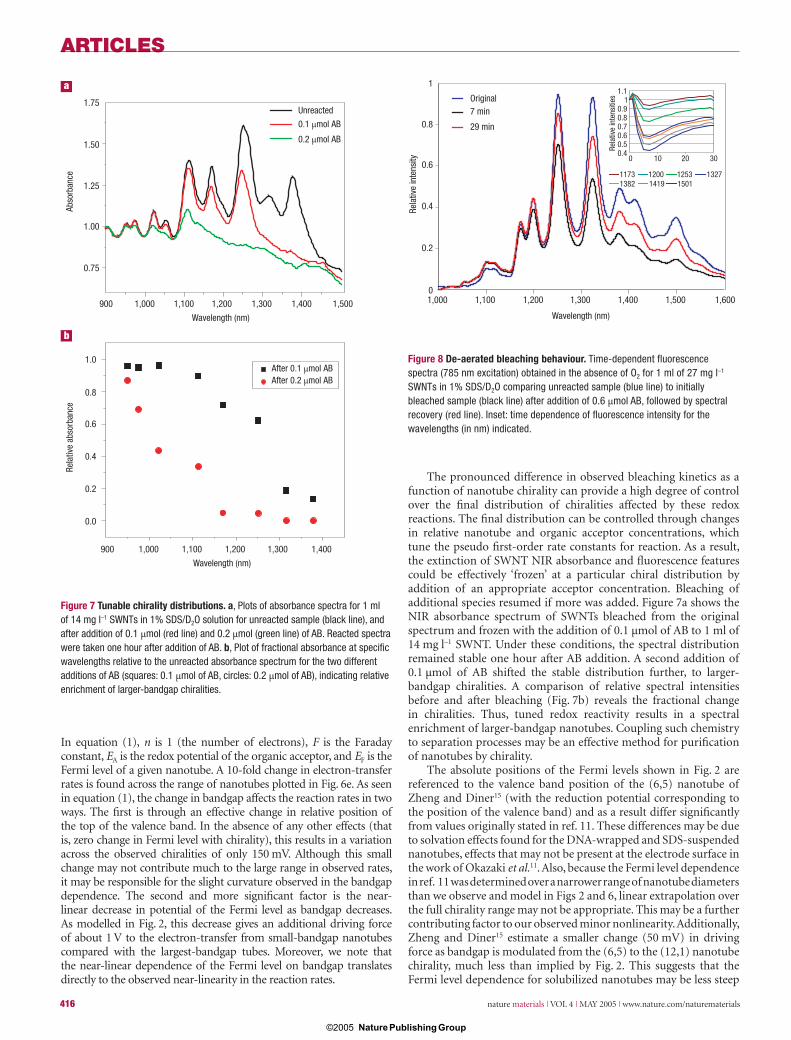
Figure 8 De-aerated bleaching behaviour. Time-dependent fl uorescence spectra (785 nm excitation) obtained in the absence of O2 for 1 ml of 27 mg l–1 SWNTs in 1% SDS/D2O comparing unreacted sample (blue line) to initially bleached sample (black line) after addition of 0.6 μmol AB, followed by spectral recovery (red line). Inset: time dependence of fl uorescence intensity for the wavelengths (in nm) indicated.
Figure 7 Tunable chirality distributions. a, Plots of absorbance spectra for 1 ml of 14 mg l–1 SWNTs in 1% SDS/D2O solution for unreacted sample (black line), and after addition of 0.1 μmol (red line) and 0.2 μmol (green line) of AB. Reacted spectra were taken one hour after addition of AB. b, Plot of fractional absorbance at specifi c wavelengths relative to the unreacted absorbance spectrum for the two different additions of AB (squares: 0.1 μmol of AB, circles: 0.2 μmol of AB), indicating relative enrichment of larger-bandgap chiralities.
900 1,000 1,100 1,200 1,300 1,400 1,500
Wavelength (nm)
900 1,000 1,100 1,200 1,300 1,400
Wavelength (nm)
After 0.1 µmol AB
0.1 µmol AB
0.2 µmol AB
After 0.2 µmol AB
1.75
1.50
1.25
1.00
0.75
Abso
rban
ce
a
b
1.0
0.8
0.6
0.4
0.2
0.0
Rela
tive
abso
rban
ce
Unreacted
1
0.8
0.6
0.4
0.2
0
Rela
tive
inte
nsity
1,000 1,100 1,200 1,300 1,400 1,500 1,600
Wavelength (nm)
Original7 min
29 min
0 10 20 30
1.11
0.90.80.70.60.50.4
Rela
tive
inte
nsiti
es
11731382 1419 1501
1200 1253 1327
nmat1367-print.indd 416nmat1367-print.indd 416 12/4/05 1:39:25 pm12/4/05 1:39:25 pm
Nature Publishing Group© 2005

ARTICLES
nature materials | VOL 4 | MAY 2005 | www.nature.com/naturematerials 417
than found in ref. 11, consistent with fi rst-principles treatments of the Fermi level27. However, a comparison of our results obtained for AB relative to MY (see previous discussion) is in agreement with the bandgap dependence of the Fermi level suggested in ref. 11. Further studies of nanotube redox reactivity may lead to a more accurate determination of EF and its chirality dependence for solubilized systems. This is the subject of ongoing work in our laboratory.
ROLE OF OXYGEN
Oxygen plays an important role in charge transfer associated with pH-induced bleaching of nanotube absorbance and fl uorescence spectra13. Spectral bleaching at low pH occurs only in the presence of oxygen13. It will be important to further assess the role of oxygen in other nanotube redox processes.
In the absence of quencher, we observe the same oxygen-dependent bleaching behaviour as reported previously13. At low pH, spectral bleaching occurs, which may be reversed by removal of O2. In our experiments in the absence of O2, solution spectra at neutral pH are monitored until an intensity maximum is reached on deoxygenation. Organic acceptor is then added with subsequent spectral monitoring of the redox reaction.
Addition of a large excess of acceptor (8 μmol) in the absence of O2 reproduces the result found with O2 present. Fluorescence intensity is bleached for an extended period (60 min) for all chiralities. With the addition of more moderate quencher levels, however, different behaviour is observed. Addition of 0.6 μmol of quencher in the absence of O2 produces an initial fl uorescence bleach that is followed by fl uorescence recovery on relatively short timescales (Fig. 8).
This behaviour can be explained in terms of a competing redox process proposed previously15, involving the O2/H2O redox couple:
4SWNT (red) + O2 + 4H+ → 4SWNT (ox)+ + 2H2O (2)
Zheng and Diner15 have shown how reaction (2) can be responsible for the pH- and O2-dependent behaviour presented in ref. 13. Removal of O2 results in a shift in equilibrium towards the reduced nanotube on the left-hand side of equation (2). We propose that this reverse reaction is responsible for the fl uorescence recovery shown in Fig. 8.
4SWNT (red) + 4A → 4SWNT+ (ox) + 4A– (red) (3a)
4SWNT+ (ox) + 2H2O → 4SWNT (red) + O2 + 4H+ (3b)
Initial fl uorescence bleaching occurs on addition of acceptor (A) (3a). In the absence of O2, the subsequent reduction of the oxidized nanotube (3b) occurs at a competitive rate to regenerate the initial spectrum. The net result of reactions (3a) and (3b) is the catalytic reduction of the acceptor by the nanotube (see Supplementary Information).
Thus, the behaviour observed in Fig. 8 provides support for the hypothesis of Zheng and Diner that coupling of the O2/H2O redox pair to nanotube oxidation and reduction can be important. The oxygen dependence of the organic-based redox chemistry mirrors the effects observed for pH dependence13,15. As was observed for K2IrCl6 oxidized nanotubes15, we find that nanotubes oxidized by organic acceptors slowly recover their spectra over a period of days in O2-saturated solutions. In the presence of O2 and at neutral pH, water acts slowly as a reducing agent15, as implied by equation (3b). In much the same way, however, that reaction (3b) is accelerated at low proton concentration15, we find that it is also aided by removal of oxygen at neutral pH (see also Supplementary Information).
CONCLUSION
Our results demonstrate a new class of electron-transfer reactions between nanotubes and organic acceptors, broadening the growing range of nanotube redox studies. Our demonstration of chiral-dependent redox kinetics has implications for potentially using solution-phase redox chemistry for pinning the absolute potential of the Fermi level, as well as for future applications in chiral selective separations. Control over the reaction kinetics by adjustment of relative concentrations and tuning the reduction potentials of acceptor species can result in selectable distributions of reacted and unreacted nanotube chiralities. Redox doping also provides a pathway towards non-covalent and reversible tuning of nanotube electronic properties. Such a capability will be important for both future nanotube sensors and electronics applications.
METHODS
CHEMICALSHiPco nanotubes (supplied by Rice University) were suspended as previously reported22 in D2O (99.9%,
Acros Organics) with 1% SDS (99%, Fisher). The following quenching and recovery agents (Aldrich)
were used as received: 4-amino-1,1-azobenzene-3,4-disulphonic acid, sodium salt (97%), mordant
yellow 10 (85%), 7,7,8,8-tetracyanoquinodimethane (TCNQ) (98%), 2,3,5,6-tetrafl uoro-7,7,8,8-
tetracyanoquinodimethane (TFTCNQ) (97%), tetrathiafulvalene (TTF) (97%), B-nicotinamide adenine
dinucleotide, reduced disodium salt hydrate (NADH) (98%).
SPECTROSCOPIC ANALYSISAbsorbance and fl uorescence spectra were taken in a stirred, quartz cuvette capped with a septum.
Additions of quenching and recovery agents were made by syringe. Absorbance measurements
were made with fi bre-optic spectrometers (Stellar Net) covering a range from 190 nm to 1,700 nm.
Fluorescence measurements with a 785-nm diode laser excitation source were made with a Fourier-
transform Raman instrument (Bruker RFS 100/S) modifi ed for NIR emission. Data were collected at
8 cm–1 resolution and averaged over 16 scans (for spectra collected at 15-s intervals) or 64 scans (for
spectra collected at 2-min intervals). Raman spectra were obtained using 15 mW of 785 nm excitation
with detection using a fi bre-optic probe head coupled to an f /1.8 imaging spectrograph (Kaiser Optical)
with 1-s integration times on a CCD (charge-coupled device) detector.
PHOTODESORPTION OF O2
Samples were fi rst placed in a round-bottom fl ask and subjected to 1 torr vacuum until bubbling
subsided. Samples were transferred by syringe to a capped cuvette purged with nitrogen, followed by
irradiation with a deuterium lamp for about 1 h. Fluorescence spectra were used to follow this process.
Organic acceptors were added by syringe once spectral intensity had maximized. The lamp was left on
for the duration of the experiment.
CYCLIC VOLTAMMETRYSamples were analysed using a CH Instruments Electrochemical Workstation. Working and counter
electrodes were platinum foil, and a silver wire was used as the reference electrode. Measurements were
made in DMF solutions, using ferrocene as a reference compound. Data were collected with scan rates
of 50, 100 and 200 mV s–1 with the results being averaged to obtain the reduction potential relative to
ferrocene. Potentials relative to NHE were obtained by adding 0.67 to the values relative to ferrocene
(see for example ref. 28). Tetrabutyl ammonium hexafl uorophosphate was used as electrolyte.
Received 16 November 2004; accepted 28 February 2004; published 10 April 2005.
References1. Saito, R., Dresselhaus, G. & Dresselhaus, M. S. Physical Properties of Carbon Nanotubes (Imperial
College Press, London, 1998).
2. Bachilo, S. M. et al. Narrow (n,m)-distribution of single-walled carbon nanotubes grown using a
solid supported catalyst. J. Am. Chem. Soc. 125, 11186–11187 (2003).
3. Maruyama, S., Miyauchi, Y., Murakami, Y. & Chiashi, S. Synthesis of single-walled carbon
nanotubes with narrow diameter-distribution from fullerene. Chem. Phys. Lett. 375, 553–559
(2003).
4. Miyauchi, Y., Chiashi, S., Murakami, Y., Hayashida, Y. & Maruyama, S. Fluorescence spectroscopy of
single-walled carbon nanotubes synthesized from alcohol. Chem. Phys. Lett. 387, 198–203 (2004).
5. Banerjee, S. & Wong, S. S. Selective metallic tube reactivity in the solution-phase osmylation of
single-walled carbon nanotubes. J. Am. Chem. Soc. 126, 2073–2081 (2004).
6. Banerjee, S. & Wong, S. S. Demonstration of diameter-selective reactivity in the sidewall ozonation
of SWNTs by resonance Raman spectroscopy. Nano Lett. 4, 1445–1450 (2004).
7. Strano, M. S. et al. Electronic structure control of single-walled carbon nanotube functionalization.
Science 301, 1519–1522 (2003).
8. Kavan, L., Rapta, P. & Dunsch, L. In situ Raman and Vis-NIR spectroelectrochemistry at single-
walled carbon nanotubes. Chem. Phys. Lett. 328, 363–368 (2000).
9. Kavan, L. et al. Electrochemical tuning of electronic structure of single-walled carbon nanotubes:
in-situ Raman and vis-NIR study. J. Phys. Chem. B 105, 10764–10771 (2001).
10. Stoll, M., Rafailov, P. M., Frenzel, W. & Thomsen, C. Electrochemical and Raman measurements on
single-walled carbon nanotubes. Chem. Phys. Lett. 375, 625–631 (2003).
11. Okazaki, K., Nakato, Y. & Murakoshi, K. Absolute potential of the Fermi level of isolated single-
walled carbon nanotubes. Phys. Rev. B 68, 035434 (2003).
nmat1367-print.indd 417nmat1367-print.indd 417 12/4/05 1:39:27 pm12/4/05 1:39:27 pm
Nature Publishing Group© 2005

ARTICLES
418 nature materials | VOL 4 | MAY 2005 | www.nature.com/naturematerials
12. Corio, P., Jorio, A., Demir, N. & Dresselhaus, M. S. Spectro-electrochemical studies of single wall
carbon nanotube fi lms. Chem. Phys. Lett. 392, 396–402 (2004).
13. Strano, M. S. et al. Reversible, band-gap-selective protonation of single-walled carbon nanotubes in
solution. J. Phys. Chem. B 107, 6979–6985 (2003).
14. Weisman, R. B., Bachilo, S. M. & Tsyboulski, D. Fluorescence spectroscopy of single-walled carbon
nanotubes in aqueous suspension. Appl. Phys. A 78, 1111–1116 (2004).
15. Zheng, M. & Diner, B. A. Solution redox chemistry of carbon nanotubes. J. Am. Chem. Soc. 126,
15490–15494 (2004).
16. Chattopadhyay, D., Galeska, I. & Papadimitrakopoulos, F. A route for bulk separation of
semiconducting from metallic single-wall carbon nanotubes. J. Am. Chem. Soc. 125, 3370–3375
(2003).
17. Zheng, M. et al. Structure-based carbon nanotube sorting by sequence-dependent DNA assembly.
Science 302, 1545–1548 (2003).
18. Huang, S. M., Maynor, B, Cai, X. Y. & Liu, J. Ultralong, well-aligned single-walled carbon nanotube
architectures on surfaces. Adv. Mater. 15, 1651–1655 (2003).
19. Zheng, L. et al. Ultralong single-wall carbon nanotubes. Nature Mater. 3, 673–676 (2004).
20. Collins, P. G., Arnold, M. S. & Avouris, P. Engineering carbon nanotubes and nanotube circuits
using electrical breakdown. Science 292, 706–713 (2001).
21. An, L., Lu, C. & Liu, J. A simple chemical route to selectively eliminate metallic carbon nanotubes in
nanotube network devices. . J. Am. Chem. Soc. 126, 10520–10521 (2004).
22. O’Connell, M. J. et al. Band gap fl uorescence from individual single-walled carbon nanotubes.
Science 297, 593–596 (2002).
23. Jana, A. K., Mukhopadhyay, S. K. & Bhowmik, B. B. Absorption spectra of 7, 7, 8, 8-
tetracyanoquinodimethane in micellar solutions. Spectrochim. Acta A 57, 2687–2893 (2001).
24. Moore, V. C., Strano, M. S., Haroz, E. H., Hauge, R. H. & Smalley, R. E. Individually suspended
single-walled carbon nanotubes in various surfactants. Nano Lett. 3, 1379–1382 (2003).
25. Doorn, S. K., Heller, D. A., Barone, P. W., Usrey, M. L. & Strano, M. S. Resonant Raman excitation
profi les of individually dispersed single walled carbon nanotubes in solution. Appl. Phys. A 78,
1147–1155 (2004).
26. O’Connell, M. J., Sivaram, S. & Doorn, S. K. Near-infrared resonance Raman excitation profi le
studies of single-walled carbon nanotube intertube interactions: a direct comparison of bundled
and individually dispersed HiPco nanotubes. Phys. Rev. B 69, 235415 (2004).
27. Zhao, J., Han, J. & Lu, J. P. Work functions of pristine and alkali-metal intercalated carbon nanotubes
and bundles. Phys. Rev. B 65, 193401 (2002).
28. Lai, R. Y. & Bard, A. J. Electrogenerated chemiluminescence 71. Photophysical, electrochemical, and
electrogenerated chemiluminescent properties of selected dipyrromethene-BF2 dyes. J. Phys. Chem.
B 107, 5036 (2003).
AcknowledgementsWe thank Ming Zheng for sharing his results on K2IrCl6 oxidation of nanotubes before publication.
M.O’C. acknowledges the support of the DCI postdoctoral fellowship program. This work was
supported in part by the LANL LDRD program.
Correspondence and requests for materials should be addressed to S.K.D.
Supplementary Information accompanies the paper on www.nature.com/naturematerials.
Competing fi nancial interestsThe authors declare that they have no competing fi nancial interests.
nmat1367-print.indd 418nmat1367-print.indd 418 12/4/05 1:39:28 pm12/4/05 1:39:28 pm
Nature Publishing Group© 2005





























