Embed Size (px)
Citation preview

Marine Genomics 10 (2013) 1–9
Contents lists available at SciVerse ScienceDirect
Marine Genomics
j ourna l homepage: www.e lsev ie r .com/ locate /margen
Chloroplast genome of one brown seaweed, Saccharina japonica(Laminariales, Phaeophyta): Its structural features andphylogenetic analyses with other photosynthetic plastids
Xiuliang Wang, Zhanru Shao, Wandong Fu, Jianting Yao, Qiuping Hu 1, Delin Duan ⁎Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China
⁎ Corresponding author. Tel./fax: +86 532 82898556E-mail address: [email protected] (D. Duan).
1 Present address: Shanghai Majorbio Bio-pharm TechnChina.
1874-7787/$ – see front matter © 2012 Elsevier B.V. Allhttp://dx.doi.org/10.1016/j.margen.2012.12.002
a b s t r a c t
a r t i c l e i n f oArticle history:Received 18 September 2012Received in revised form 19 December 2012Accepted 20 December 2012
Keywords:Photosynthetic heterokontSaccharina japonicaChloroplastPhylogenomicMolecular phylogenetics
The chloroplast genome sequence of one brown seaweed, Saccharina japonica, was fully determined. It ischaracterized by 130,584 base pairs (bp) with a large and a small single-copy region (LSC and SSC), separatedby two copies of inverted repeats (IR1 and IR2). The inverted repeat is 5015 bp long, and the sizes of SSC andLSC are 43,174 bp and 77,378 bp, respectively. The chloroplast genome of S. japonica consists of 139 protein-coding genes, 29 tRNA genes, and 3 ribosomal RNA genes. One intron was found in one tRNA-Leu gene in thechloroplast genome of S. japonica. Four types of overlapping genes were identified, ycf24 overlapped withycf16 by 4 nucleotides (nt), ftrB overlapped with ycf12 by 6 nt, rpl4 and rpl23 overlapped by 8 nt, finally,psbC overlapped with psbD by 53 nt. With two sets of concatenated plastid protein data, 40-protein datasetand 26-protein dataset, the chloroplast phylogenetic relationship among S. japonica and the other photosyntheticspecies was evaluated. We found that the chloroplast genomes of haptophyte, cryptophyte and heterokont werenot resolved into one cluster by the 40-protein datasetwith amino acid composition bias, although it was recoveredwith strong support by the 26-protein dataset.
© 2012 Elsevier B.V. All rights reserved.
1. Introduction
The photosynthetic heterokont, haptophyte and cryptophyte werepreviously postulated to have arisen from a single secondary endosym-biosis in which a non-photosynthetic eukaryote acquired a plastid byengulfing a red algal (Cavalier-Smith, 1999).
Manymolecular phylogenetic studies have been conducted to resolvethe phylogenetic relationships of these three groups, and the resultswerein a flux. Recently the photosynthetic heterokont was classified into onenew supergroup Sar, however, haptophyte and cryptophyte were listedas incertae sedis in the eukaryotes, based on nuclear genes phylogenomicanalyses to taxon broadly sampled (Adl et al., 2012). In addition to thenuclear DNA sequences, ten mitochondrial genes were used to examinethe phylogenetic relationships of the 29 taxa. The results showed thatheterokont algae formed strong sister-clusters that separated from thecryptophyte and haptophyte (Oudot-Le Secq et al., 2006). Moreover,the chloroplast genome could give important clues to the phylogeneticrelationships among the three groups (Yoon et al., 2002). For instance,an analysis of 62 plastid associated genes of 15 taxa showed that the
.
ology C., Ltd., Shanghai 201203,
rights reserved.
plastids from heterokont, dinoflagellate, haptophyte and cryptophyteare monophyletic (Sanchez-Puerta et al., 2007). However, based on theplastid protein dataset without composition bias, such phylogenetic rela-tionship was not identified for heterokont, haptophyte and cryptophyte(Khan et al., 2007).
Previous studies showed that the phylogenetic relationships amongthe three groups are hard to resolve, because the methodologies andanalysis results varied significantly using the datasets from nuclear, mi-tochondria or chloroplast (Green, 2011). Therefore, in order to resolvethe deepest branch order in the topology tree for photosyntheticheterokont, haptophyte and cryptophyte, broad sampling and newdataset are needed for phylogenetic analysis (Le Corguillé et al., 2009).
Here, we reported the sequence and structural analyses of the chloro-plast genome of Saccharina japonica from Laminariales, which is one ofthe economically important species in large scale brown seaweed aqua-culture in East Asian countries (Tseng, 2001). In comparison with othertwo available brown seaweed plastid genomes of Fucus vesiculosus andEctocarpus siliculosus (Le Corguillé et al., 2009), our data showed thatthere were higher cpDNA structural similarities between S. japonica andF. vesiculosus. With 40- and 26-plastid protein sequence datasets, we in-vestigated the phylogenetic relationships among the plastids of photo-synthetic heterokont, haptophyte and cryptophyte, and demonstratedthat the chloroplast genomes of haptophyte, cryptophyte and heterokontwere not resolved into one cluster by the 40-protein dataset, although itwas recovered with strong support by the 26-protein dataset.

Table 1Datasets used in the phylogenetic studies.
Dataset Number of amino acids Gene names Taxa that failed the amino acid composition homogeneitytest by TreePuzzle
40 proteins for 23species
7140 atpA, atpB, atpE, atpF, atpH, petA, petB, petD, petG, psaA, psaB,psaC, psaJ, psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ,psbK, psbL, psbN, psbT, rpl2, rpl14, rpl16, rpl20, rps2, rps4, rps7,rps8, rps11, rps12, rps14, rps19, ycf3, ycf4
Cyanidium caldarium, Cyanidioschyzon merolae, Fucusvesiculosus, Nephroselmis olivacea, Vaucheria litorea,Prochlorococcus marinus, Synechocystis sp.
26 proteins for 21species
5746 atpA, atpB, atpH, petB, petD, petG, psaA, psaB, psaC, psbA, psbB,psbC, psbD, psbE, psbF, psbL, psbN, psbI, rpl14, rpl2, rpl20, rps11,rps12, rps14, rps19, ycf3
None
2 X. Wang et al. / Marine Genomics 10 (2013) 1–9
2. Materials and methods
2.1. Algal material and chloroplast DNA purification
S. japonica sporophytes raised from gametophyte cultures weremaintained at the Key Laboratory of Experimental Marine Biology, In-stitute of Chinese Academy of Sciences (Wang et al., 2004). Gameto-phyte culture and sporophyte cultivation were performed accordingto the previously described method (Li et al., 2007). The chloroplastDNA was purified from fresh juvenile sporophytes according to thereported method (Fu et al., 2008).
2.2. Chloroplast genome sequencing, assembling and annotation
The shotgun libraries and PCR strategies were used to generate thecomplete chloroplast genome sequence of S. japonica. The shotgun li-braries were constructed with fragments (1.6–4 kb) of nebulized, puri-fied plastid DNA cloned into pUC19 (Fermentas). Sequencing reactionswere performed and analyzed on ABI3730xl automated sequencers.
In total, 2216 fragments were sequenced with average fragmentlength of 969 base pairs. The chloroplast genome sequences were assem-bled with the Phred–Phrap–Consed package (Gordon et al., 1998). Theinitial assembly of random clone sequences produced four large contigs.Contigswereunited by reverse sequencing of selected clones. Themissingsequences in the gapswere generated bymultiple PCR reactionswith ref-erence to the reported E. siliculosus and F. vesiculosus chloroplast genomesequences (Le Corguillé et al., 2009). The whole sequencewas verified bysequencing PCR fragments randomly selected based on the assembled ge-nome sequence. All sequences of selectedPCR fragmentswere identical tothe original sequences from the assembled chloroplast genome. Everybase of the chloroplast genome has minimum quality with Phred valueof at least 20 and was confirmed in both directions by a minimum ofthree reads. The final genome coverage was approximately sixteen-foldwith 2216 reads used for the genome assembling. The genome sequencehas beendeposited inGenBankwith the accessionnumber JQ405663. TheS. japonica chloroplast physical map of the circular genome was drawnusing GenomVx (http://wolfe.gen.tcd.ie/GenomeVx/).
Protein-coding and ribosomal RNA genes were identified by BLASTsearches (Altschul et al., 1997) of the nonredundant databases at theNational Center for Biotechnology Information. The tRNA genes werefound using tRNAscan-SE (Lowe and Eddy, 1997).
2.3. Phylogenetic analysis
The phylogenetic analysis was performed with DNA sequencesfrom the chloroplast genome of S. japonica, E. siliculosus, F. vesiculosus,Vaucheria litorea (Xanthophyte),Heterosigma akashiwo (Raphidophyte)and the pinnate diatom Fistulifera sp. strain JPCC DA0580 plus the 15algal sequences and two reference Cyanobacterium genomes (Khan etal., 2007; Le Corguillé et al., 2009). The phylogenetic analyses wereperformed with a total of two Cyanobacteria and 21 chloroplast ge-nomes, including four complete genomes from red algae, twelve from
heterokont, haptophyte and cryptophyte species, four from green spe-cies and one from glaucophytic species.
Two datasets were constructed for phylogenetic analysis (Table 1).Thefirst dataset included40 chloroplast protein-coding sequences select-ed from 44 plastid-encoded proteins (Martin et al., 2002; Khan et al.,2007; LeCorguillé et al., 2009). The seconddataset used26protein codingsequences from 21 out of 23 species excluding V. litorea and Cyanidiumcaldarum. The amino acid composition bias of the two datasets was in-vestigated using Tree-Puzzle (Strimmer and von Haeseler, 1996). Theconcatenated protein sequences were aligned with ClustalX (Larkin etal., 2007) andMEGA5 (Tamura et al., 2011). Each alignmentwas furtheroptimized using GBlocks under stringent settings (Castresana, 2000).Phylogenetic analyses of the aligned concatenated protein data werecarried out on 7140 and 5746 amino acids with unambiguous positionsfrom the respective 40- and 26-protein datasets (Table 1).
Maximum likelihood (ML) analysis for constructing phylogenetictrees was performed using PhyML (Guindon and Gascuel, 2003) andMEGA5 programs (Tamura et al., 2011). PhyML was used under modelcpREV with a gamma distribution approximated by 5 categories tomodel site rate heterogeneity.MEGA5was performed under the selectedmodel cpREV with a gamma distribution approximated by 5 categoriesto model site rate heterogeneity.
The neighbor-joining (NJ) analysis was performed under the modelJTT with a gamma distribution approximated by 5 categories to modelsite rate heterogeneity using MEGA5 (Tamura et al., 2011).
For both the ML and NJ analyses, a statistical support for individualbranch was investigated by 100 bootstrapping replicates.
Bayesian analyses were performed using MrBayes 3.2 (Huelsenbeckand Rouquist, 2001). MrBayes was performed using the cpREV modelfor amino acid sequence evolution, including four γ-distributed rate cat-egories and invariant sites under the co-variationmodel. Two runswithfour Markov chains each were run for 100,000–300,000 generationsuntil the average standard deviation of split frequencies was below0.01 and sampled every hundredth generation with a burn-in periodthat corresponded to 25% of samples.
CONSEL (Shimodaira and Hasegawa, 2001) was used to statistical-ly test the topologies of trees with approximately unbiased (AU) andShimodaira–Hasegawa (SH) analyses. Site likelihoods for each phylo-genetic tree were calculated using Tree-Puzzle (Strimmer and vonHaeseler, 1996).
3. Results
3.1. Features of S. japonica chloroplast genome
The general features of S. japonica chloroplast genome (cpDNA) werecomparedwith the chloroplast genomes of E. siliculosus and F. vesiculosus.All the three chloroplast genomes are divided into large (LSC) and smallsingle copy (SSC) regions separated by two inverted repeats (IRs)(Fig. 1; Table 2). In S. japonica and F. vesiculosus, the cpDNA IR con-tains two ribosomal operons encoding the 16S, 23S and 5S rRNAand two tRNAs, however in E. siliculosus, it contains additional coding

Fig. 1. Chloroplast genomemap of S. japonica. Genes on the outside of the circles are transcribed clockwise, whereas those on the inside counter clockwise. Annotated genes are accordingto the functional categories shown in the legend and the tRNA genes are indicated by a single-letter code of the corresponding amino-acid. Abbreviation: IR, inverted repeat.
3X. Wang et al. / Marine Genomics 10 (2013) 1–9
sequences for other protein genes such as rpl21, psbA, and rpl32. Thenumber of protein-coding genes, rRNA genes and tRNA genes was sim-ilarly encoded in the three chloroplast genomes. However, the petL andycf54 genes were not found in S. japonica, and similarly, the syfB andycf17 coding genes were not found in F. vesiculosus.
Overall, sequence analysis revealed that the three chloroplastgenomes described above are highly compact. Similar to that ofF. vesiculosus, one intron was found in one tRNA-Leu gene in thecpDNA of S. japonica. In contrast, there is no intron in any genes ofcpDNA of E. siliculosus. In addition, four types of overlapping geneswere identified in the cpDNA of S. japonica. The overlapping sequenceswere 4 nucleotides (nt) between ycf24 and ycf16, 6 nt between ftrB and
ycf12, 8 nt between rpl4 and rpl23, and 53 nt between psbC and psbD.Similar gene overlappingwas found in the cpDNA of E. siliculosus. Strik-ingly, except the overlapping pair between ftrB and ycf12, all other threepairs of overlapping genes appeared to be identical to that found in thecpDNA of S. japonica. Six types of gene overlapping were found in thecpDNA of F. vesiculosus. In addition to the identical four pairs found inthe cpDNA of S. japonica, the cpDNA of F. vesiculosus contained twomore pairs of overlapping genes namely the rps1 and thiS by 4 nt, andtRNA(M) and rpl19 pair by 1 nt.
The gene identity and order are highly conserved among the threebrown seaweed cpDNAs. Approximately half of each genome is coveredby two large gene clusters, namely the ribosomal protein gene cluster

Table 2General features of three brown seaweed chloroplast genomes.
Sj Es Fv
Size (bp) 130,584 139,954 124,986Inverted repeat (bp) 5015 8615 4863Small single-copy region (bp) 43,174 42,714 40,347Large single-copy region (bp) 77,378 80,010 74,913Total G+C content (%) 31.1 30.7 28.9Gene content (total) 171 174 168% Coding sequence 83.5 80 85.5Average size intergenic spacer in bp 125.5 153.8 109.2Protein-coding genes 139 144 139rRNA genes 3 3 3tRNA genes 29 27 26No. of overlapping genes 4 3 6No. of introns 1 0 1
Abbreviations: Sj, Saccharina japonica; Es, Ectocarpus siliculosus; Fv, Fucus vesiculosus.
4 X. Wang et al. / Marine Genomics 10 (2013) 1–9
and the atpA gene cluster (Stoebe and Kowallik, 1999). Comparison ofthe cpDNA of S. japonica with that of F. vesiculosus revealed that theyshared 48 genes in the ribosomal protein gene cluster ranging fromrpl9 to rns2, and 41 genes in the shared atpA gene cluster delimited byatpA and petN. The number of genes shared between the cpDNAs ofS. japonica and E. siliculosus increased. The ribosomal protein genecluster contained 59 shared genes ranging from ycf16 to rns2. Fiftyshared genes were identified in the atpA gene cluster includingtRNA(F)–petD–petB of inverted order, delimited by ycf37 and rpl35.Besides the two larger clusters, other gene clusters including 2 to17 genes also appeared to be conserved among the three brown sea-weed cpDNAs (Table 3). GRIMM server (http://nbcr.sdsc.edu/GRIMM/grimm.cgi) was used to infer the number of inversions that appearedin the three cpDNAs. It calculated a total of 39 inversions betweencpDNAs of S. japonica and F. vesiculosus, 63 inversions between cpDNAsof S. japonica and E. siliculosus, and 59 inversions between cpDNAs ofF. vesiculosus and E. siliculosus. Interestingly, the positions of the rpl20and psbA genes varied significantly among the cpDNAs of S. japonica,F. vesiculosus and E. siliculosus. In E. siliculosus, the psbA gene is locatedin the IRs and the rpl20 is located adjacent to ycf47 and tRNA(M);whereas the cpDNAs of S. japonica and F. vesiculosus, the psbA clusterswith psaC, and rpl20 are located between ilvH and rpl35.
Table 3Sj chloroplast gene clusters shared specifically with Fv and Es cpDNAs.
Compared group Shared chloroplast gene clusters
Fv ycf19–rps16–ycf65–trnQ–trnR–rps4–ilvB–ycf33–ycf39–(ycf41)psbC–psbD– (trnN)ycf16–ycf24–ycf35–trnR–trnVrps1–(ycf40)–trnH–trnTycf37–psaM–chlI–trnF–petB–petDacsF–ycf42–rpl35–rpl20–ilvH–ycf34–trnL–(trnC)–rbcL–rbcS–trnE–psaA–psaB–rps14–petG–psbKccsA–chlL–chlN–psaF–psaJ–Ljcp97–petA–tatC–atpE–atpB–ycf3–rps18–rpl33–clpC–trnK–psaC–psbAcbbX–ycf12–ftrB–psaI–psbJ–psbL–psbF–psbE–trnG–ycf4–psaL–rbcR–psbY–rpl32–trnL
Es ycf17–ycf19–rps16–ycf65–trnQ–trnR–rps4–ilvB–ycf33–ycf39–(ycf41)–psbC–psbD–trnNilvH–ycf34trnL–trnC–rbcL–rbcSpsaA–psaB–rps14petG–petK–ccsAchlL–chlN–psaF–psaJ–Ljcp97–petA–tatC–atpE–atpB–ycf3–rps18–rpl33–clpCtrnK–psaCcbbX–ycf12–ftrB–psaI–psbJ–psbL–psbF–psbEycf4–psaL–rbcR–psbYrpl32–trnL
Genes encoded on different strands are pointed in the parenthesis. Abbreviations: Sj,Saccharina japonica; Es, Ectocarpus siliculosus; Fv, Fucus vesiculosus.
3.2. Calculation of amino acid composition bias of 26- and40-protein datasets
For the investigation of whether there is any amino acid com-position bias in the 26- and 40-protein datasets, our analysesshowed that the amino acid composition of the concatenated pro-tein sequences from Cyanidium caldarium, Cyanidioschyzon merolae,F. vesiculosus, Nephroselmis olivacea, V. litorea, Prochlorococcus marinusand Synechocystis sp. failed to the expected frequency distribution atP=0.05 (Table 1). In contrast, no biased amino acid composition wasfound for the 21 species in the 26 concatenated protein dataset (Table 1).
3.3. Phylogenetic analyses on the 40 and 26 concatenated protein datasets
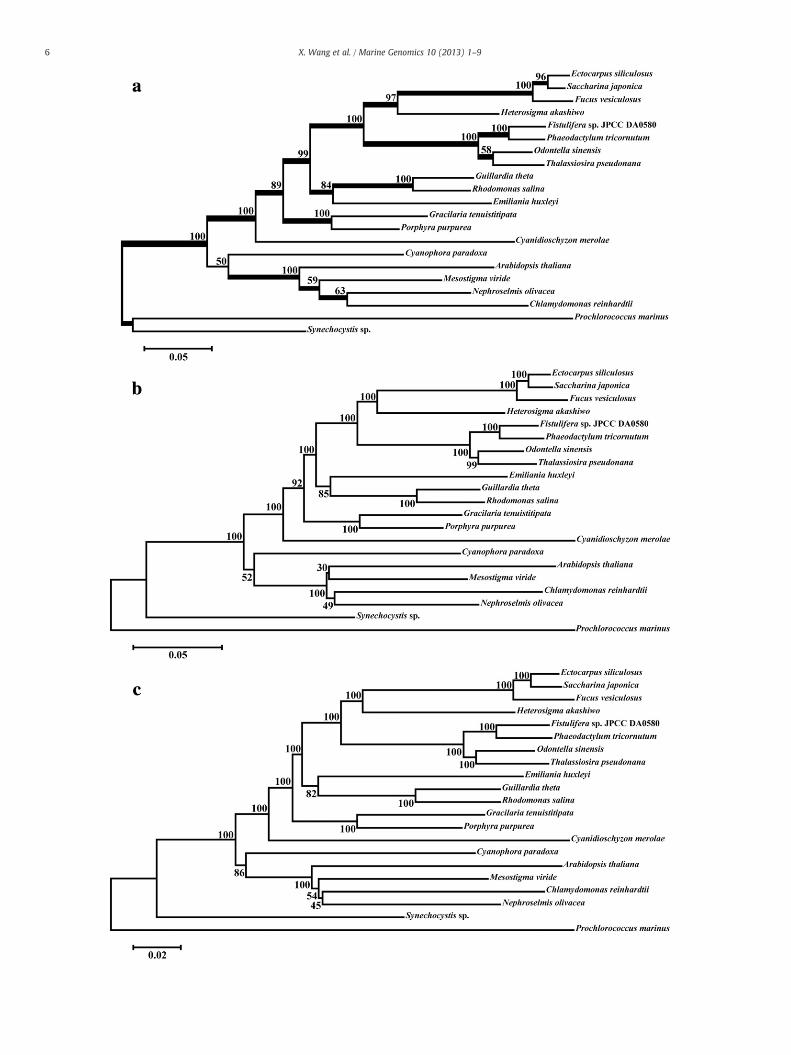
The phylogenetic analyses of the 40-protein dataset were conductedwith several programs (Fig. 2). The PhyML, MrBayes and MEGA(NJ)analyses yielded the same tree topology (Fig. 2a). S. japonica was clus-tered together with E. siliculosus. This was strongly supported by thedata from all four analyses. Moreover, the chloroplast monophylies foreach group were recovered with a strong or medium support, such asbrown seaweed, heterokont (three species of brown seaweed, oneXanthophyte species, one Raphidophyte species and four diatom spe-cies branched off first), cryptophyte and haptophyte, Florideophyteand Bangiophyte, Cynidiales, and green algae where Chlamydomonasreinhartii and N. olivacea clustered together, and Arabidopsis thalianaclosed to Mesostigma viride. Also, Cyanidiales always emerged at thebase of a strongly or moderately supported clade that includes theFlorideophyceae and Bangiophyceae, together with the cryptophyteand haptophyte, and heterokont. However, the plastid monophyly ofheterokont, cryptophyte and haptophyte was not significantly supportedby these analyses except in the MrBayes analysis with support value of100. The support values for this cluster were 74, 48 and 68 from the anal-yses of PhyML, MEGA(NJ) and MEGA(ML), respectively. The glaucophyteCyanophora paradoxa grouped outside the clade of green algae whenanalyzed with the PhyML, MrBayes and MEGA(NJ). It grouped outside abig cluster including all species except Synechocystis sp. and P. marinuswhenwithMEGA(ML) analysis (Fig. 2b). Overall, in all analyses, the clus-ter of Florideophyceae and Bangiophyceae grouped together outside theclade of heterokont, haptophyte and cryptophyte.
The chloroplast phylogenetic relationships were further examinedwith phylogenetic analyses of the 26-protein dataset for the 21 species(Fig. 3). PhyML and MrBayes analyses gave the same tree of topologies(Fig. 3a). Themonophyly of the heterokont chloroplastswas consistentlyrecoveredwith a strong support in all four analyses (Fig. 3). Interestingly,the monophyly of heterokont, haptophyte and cryptophyte chloroplastswas identified with significant support values by all four methods basedon the 26 concatenated protein dataset, and the support values to theanalyses of PhyML, MEGA(NJ), MEGA(ML) and MrBayes were 99, 100,100 and 100, respectively.
3.4. AU and SH tests
Based on the 26-protein dataset, the chloroplast relationships amongthe three brown algae, S. japonica, F. vesiculosus and E. siliculosus, wereevaluated by theAUand SH tests (Fig. 4). The data showed that S. japonicais closely grouped with E. siliculosus with F. vesiculosus outside. Thishypothetic topology receivedmore support than the other two hypothe-ses where S. japonica was closed to F. vesiculosus with E. siliculosus out-side, or F. vesiculosus clustered to E. siliculosus, with S. japonica branchedoff first.
4. Discussion
In this study, we determined the complete chloroplast genomesequences of one brown seaweed, S. japonica. Our sequence analysisshowed that the cpDNAs of S. japonica and F. vesiculosus contain an
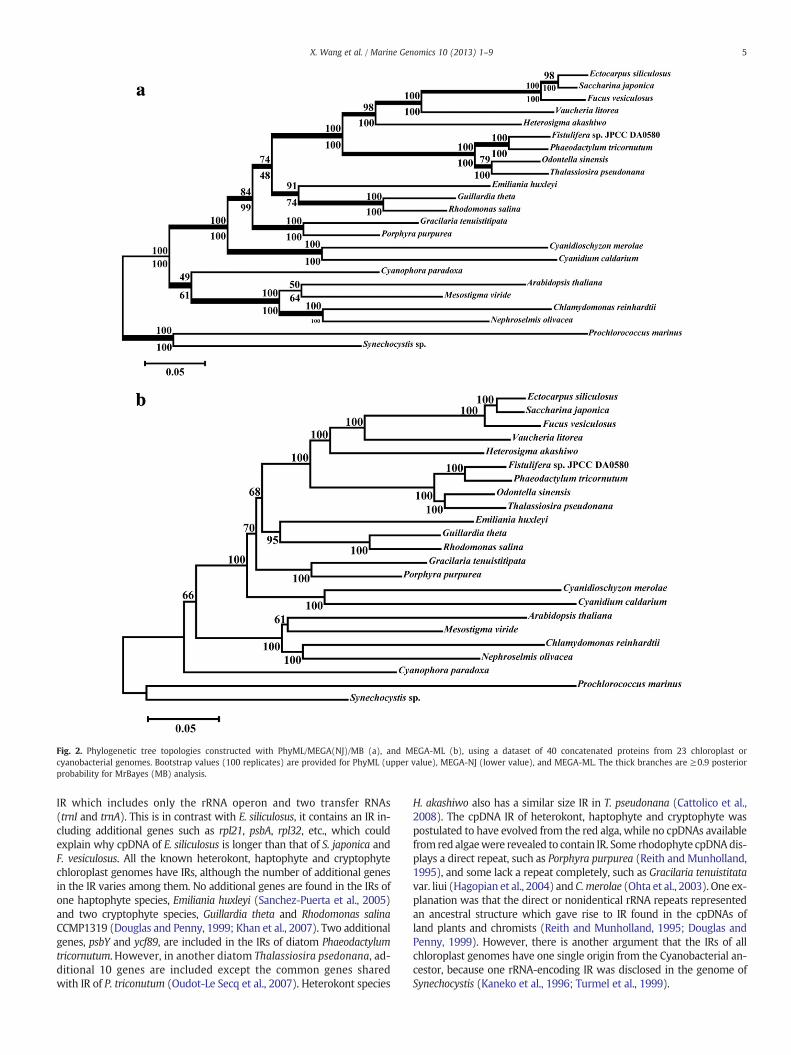
Fig. 2. Phylogenetic tree topologies constructed with PhyML/MEGA(NJ)/MB (a), and MEGA-ML (b), using a dataset of 40 concatenated proteins from 23 chloroplast orcyanobacterial genomes. Bootstrap values (100 replicates) are provided for PhyML (upper value), MEGA-NJ (lower value), and MEGA-ML. The thick branches are ≥0.9 posteriorprobability for MrBayes (MB) analysis.
5X. Wang et al. / Marine Genomics 10 (2013) 1–9
IR which includes only the rRNA operon and two transfer RNAs(trnI and trnA). This is in contrast with E. siliculosus, it contains an IR in-cluding additional genes such as rpl21, psbA, rpl32, etc., which couldexplain why cpDNA of E. siliculosus is longer than that of S. japonica andF. vesiculosus. All the known heterokont, haptophyte and cryptophytechloroplast genomes have IRs, although the number of additional genesin the IR varies among them. No additional genes are found in the IRs ofone haptophyte species, Emiliania huxleyi (Sanchez-Puerta et al., 2005)and two cryptophyte species, Guillardia theta and Rhodomonas salinaCCMP1319 (Douglas and Penny, 1999; Khan et al., 2007). Two additionalgenes, psbY and ycf89, are included in the IRs of diatom Phaeodactylumtricornutum. However, in another diatom Thalassiosira psedonana, ad-ditional 10 genes are included except the common genes sharedwith IR of P. triconutum (Oudot-Le Secq et al., 2007). Heterokont species
H. akashiwo also has a similar size IR in T. pseudonana (Cattolico et al.,2008). The cpDNA IR of heterokont, haptophyte and cryptophyte waspostulated to have evolved from the red alga, while no cpDNAs availablefrom red algaewere revealed to contain IR. Some rhodophyte cpDNAdis-plays a direct repeat, such as Porphyra purpurea (Reith and Munholland,1995), and some lack a repeat completely, such as Gracilaria tenuistitatavar. liui (Hagopian et al., 2004) and C. merolae (Ohta et al., 2003). One ex-planation was that the direct or nonidentical rRNA repeats representedan ancestral structure which gave rise to IR found in the cpDNAs ofland plants and chromists (Reith and Munholland, 1995; Douglas andPenny, 1999). However, there is another argument that the IRs of allchloroplast genomes have one single origin from the Cyanobacterial an-cestor, because one rRNA-encoding IR was disclosed in the genome ofSynechocystis (Kaneko et al., 1996; Turmel et al., 1999).

Dataset Tree topology (AU/SH) a b c
26 proteins 0.953/0.974 0.001/0.004 0.052/0.066
a
b
c
Fig. 4. AU and SH tests of phylogenetic relationships among three brown seaweeds.
7X. Wang et al. / Marine Genomics 10 (2013) 1–9
Gene overlapping in the plastids has been identified for red algae,cryptophyte, haptophyte, diatoms, and Raphidophyte (Hagopian et al.,2004; Douglas and Penny, 1999; Khan et al., 2007; Oudot-Le Secq et al.,2007; Cattolico et al., 2008). The psbD–psbC overlap was reported toexist in all the sequenced cpDNAs of heterokont, haptophyte andcryptophytes. The overlaps involving atpD–atpF and rpl4–rpl23 wereconsidered to be common to heterokont and cryptophyte (Khan et al.,2007; Oudot-Le Secq et al., 2007; Cattolico et al., 2008). However, in
Fig. 3. Phylogenetic tree topologies constructed with PhyML/MB (a), MEGA-ML (b), andcyanobacterial genomes. Bootstrap values (100 replicates) are provided for PhyML, MEG(MB) analysis.
cpDNAs of S. japonica, E. siliculosus and F. vesiculosus, the overlap atpD–atpF has not been identified. The 4 bp overlap of ycf16–ycf24was foundto be common to the cpDNAs of the three brown seaweeds, and existedin the glaucophyte, C. paradoxa (Stirewalt et al., 1995), but the length ofycf16–ycf24 overlap in the cpDNAs of three diatoms Odontella sinensis, P.tricornutum and T. psedonanawas only 1 bp (Oudot-Le Secq et al., 2007).
Although the two clusters, the atpA gene cluster and the ribosomalprotein gene cluster, shared by cpDNAs of S. japonica and E. siliculosus,
MEGA-NJ (c), using a dataset of 26 concatenated proteins from 21 chloroplast orA-NJ, and MEGA-ML. The thick branches are ≥0.9 posterior probability for MrBayes

8 X. Wang et al. / Marine Genomics 10 (2013) 1–9
were longer than that shared by cpDNAs of S. japonica and F. vesiculosus,a total of 63 inversions could be required to convert the gene order ofE. siliculosus cpDNA into that of S. japonica cpDNA, whereas 39 inver-sions were needed to convert the gene order of F. vesiculosus cpDNAinto that of S. japonica cpDNA. This is mainly attributed by the highergene frequency of gene rearrangements and gene inversions existedbetween cpDNAs of S. japonica and E. siliculosus compared with that incpDNAs of S. japonica and F. vesiculosus. Previous studies showed thata total of 16 inversions were required to convert the gene orderof E. siliculosus cpDNA into that of F. vesiculosus cpDNA, while inthis study, 59 inversions accounted for the gene rearrangementsdisplayed by the Fucus/Ectocarpus cpDNA pairs. This difference mainlyresulted from that 165 genes, shared by the three typical brown seaweedcpDNAs, were used for GRIMM analysis in this present analysis. Howev-er, in the previous study, only genes conserved by 7 heterokont specieswere used for the GRIMM analysis, therefore few inversions wererecorded for gene rearrangements of Fucus/Ectocarpus cpDNA pairs (LeCorguillé et al., 2009).
Obviously, the cpDNA monophyly of three brown seaweeds andthat of heterokont was recovered with a significant statistical value(Figs. 2, 3). Morphological analyses have showed that Ectocarpaleswas the ancestral group of other brown algae, because of their creep-ing filaments with apical growth and an isomorphic, diplohaplontic lifecycle (Van den Hoek et al., 1995; Reviers and de Rousseau, 1999). TheLaminariales and Fucales were derived from Ectocarpales and repre-sented the specialized brown algae (Van den Hoek et al., 1995;Reviers and de Rousseau, 1999). The traditional phylogenetic relation-ships of brown algae were challenged by the enrichment of DNA se-quence data for molecular phylogenetic analysis. Chloroplast andnuclear genes were used to resolve evolutionary relationships of thekey species from 19 brown algae orders, and the results showedthat Ectocarpales was not one early diverging clade, nor did theFucales diverge early from other brown algae (Phillips et al., 2008).Here, even with higher cpDNA structural similarities between S.japonica and F. vesiculosus (Table 3), E. siliculosus always clusteredtogether with S. japonica, based on the phylogenetic analysis ofconcatenated amino sequences of cpDNAs. AUand SH test also stronglysupported this result (Fig. 4). It could be hypothesized that Ectocarpaleswas not the ancestor group of brown algae. Although Fucales branchedoff first here, it didn't mean that Fucales diverged early from the otherbrown algae. At present, our recovered phylogenetic relationshipsamong the three brown seaweeds are tentative. Additional studies are re-quired in the futurewhenmore data from brown algae become availablefor the plastid phylogenetic analysis.
To heterokont, with analyses of 40-protein dataset, the closest branchto brown seaweeds was the V. litorea (Xanthophytes), which also couldbe recovered by analysis of the 26-protein dataset (data not shown).This is consistent with the finding that one tRNA-Leu gene with a singleintron remained in both Xanthophyceae and Phaeophyceae plastidgenomes, which was believed to have been acquired from the ancestralcyanobacterial endosymbiont (Simon et al., 2003). Similarly, the closephylogenetic relationship between Xanthophyceae and Phaeophyceaewas also identified based on combined analyses of small and largesubunit ribosomal RNA genes (Ali et al., 2002).
Here, based on the analysis of the 26 concatenated protein dataset,the chloroplast monophyly of heterokont, haptophyte and cryptophytewas established with a strong support (Fig. 3). It has been reportedthat the sequence composition bias concealed in the dataset could leadto the reconstruction of incorrect relationships in phylogenetic analysis(Sanderson and Shaffer, 2002; Sanchez-Puerta et al., 2007). In the pres-ent analysis, the amino acid composition bias for the 26 concatenatedchloroplast protein dataset was analyzed, and none of the sequences inthe 26-protein dataset was not consistent with the expected assumptionat the level of P=0.05 (Table 1). However, in this study, the chloroplastmonophyly of the three groups was not established from phylogeneticanalysis of 40 concatenated protein dataset, which might be due to the
sequence composition bias concealed in the used dataset (Table 1). Sofar, only a few plastid phylogenetic analyses showed that the chloro-plast monophyly of heterokont, haptophyte and cryptophyte could beestablished based on a small number of slowly evolving concatenatedplastid-encoded genes or protein dataset (Janouškovec et al., 2010;Sanchez-Puerta et al., 2007; Bachvaroff et al., 2005; Yoon et al., 2002).Several studies showed that the chloroplast monophyly of heterokont,haptophyte and cryptophyte could not be recovered depending onsome methods or datasets (Iida et al., 2007; Khan et al., 2007). In thepresent analysis, we included the new chloroplast genome sequenceof S. japonica in the phylogenetic analysis. Although the chloroplastmonophyly of heterokont, haptophyte and cryptophyte could be recov-ered with the strong support using the 26 concatenated protein datasetwithout amino acid compositions bias, by four different phylogeneticanalysis methods, it was not resolved by the 40-protein dataset.
Acknowledgments
This work was supported by the National High Tech 863 Project(2012AA10A406), NSFC (31272660, 30500383, 40976085), Pro-ject of Achievement Transformation of Agricultural Sci. & Tech.(2011GB2491005), Projects of Key Laboratory of ExperimentalMarineBiology of CAS, Project Program of Key Laboratory of Feed Biotechnolo-gy of MOAPRC, and visiting scholarships from CAS.
References
Adl, S.M., Simpson, A.G.B., Lane, C.E., Lukeš, J., Bass, D., Bowser, S.S., Brown, M.W., Burki,F., Dunthorn, M., Hampl, V., Heiss, A., Hoppenrath, M., Lara, E., Gall, L.L., Lynn, D.H.,Mcmanus, H., Mitchell, E.A.D., Mozley-Stanridge, S.E., Parfrey, L.W., Pawlowski, J.,Rueckert, S., Shadwick, L., Schoch, C.L., Smirnov, A., Spiegel, F.W., 2012. The revisedclassification of eukaryotes. J. Eukaryot. Microbiol. 59, 429–493.
Ali, A.B., Baere, R.D., Wachter, R.D., de Peer, Y.V., 2002. Evolutionary relationshipsamong heterokont algae (the autotrophic stramenopiles) based on combined anal-ysis of small and large subunit ribosomal RNA. Protist 153, 123–132.
Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., Lipman, D.J.,1997. Gapped BLAST and PSI-BLAST: a new generation of protein database searchprograms. Nucleic Acids Res. 25, 3389–3402.
Bachvaroff, T.R., Sanchez Puerta, M.V., Delwiche, C.F., 2005. Chlorophyll c-containingplastid relationships based on analyses of a multigene data set with all fourchromalveolate lineages. Mol. Biol. Evol. 22, 1772–1782.
Castresana, J., 2000. Selection of conserved blocks from multiple alignments for theiruse in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552.
Cattolico, R.A., Jacobs, M.A., Zhou, Y., Chang, J., Duplessis, M., Lybrand, T., McKay, J., Ong,H.C., Sims, E., Rocap, G., 2008. Chloroplast genome sequencing analysis ofHeterosigmaakashiwo CCMP452 (West Atlantic) and NIES293 (West Pacific) strains. BMC Geno-mics 9, 211.
Cavalier-Smith, T., 1999. Principles of protein and lipid targeting in secondary symbiogenesis:euglenoid, dinoflagellate, and sporozoan plastid origins and the eukaryote family tree.J. Eukaryot. Microbiol. 46, 347–366.
Douglas, S.E., Penny, S.L., 1999. The plastid genome of the cryptophyte alga, Guillardiatheta: complete sequence and conserved synteny groups confirm its common ances-try with red algae. J. Mol. Evol. 48, 236–244.
Fu, W., Yao, J., Wang, X., Liu, F., Chong, Z., Duan, D., 2008. Purification of high-qualityplastid DNA from the brown alga Laminaria japonica sporophyte. Plant Mol. Biol.Report. 26, 114–120.
Gordon, D., Abajian, C., Green, P., 1998. Consed: a graphical tool for sequence finishing.Genome Res. 8, 195–202.
Green, B.R., 2011. After the primary endosymbiosis: an update on the chromalveolatehypothesis and the origins of algae with chl c. Photosynth. Res. 107, 103–115.
Guindon, S., Gascuel, O., 2003. A simple, fast and accurate algorithm to estimate largephylogenies by maximum likelihood. Syst. Biol. 52, 696–704.
Hagopian, J.C., Reis, M., Kitajima, J.P., Bhattacharya, D., Oliveira, M.C., 2004. Compara-tive analysis of the complete plastid genome sequence of the red alga Gracilariatenuistitata var. liui provides insights into the evolution of rhodoplasts and their re-lationship to other plastids. J. Mol. Evol. 59, 464–477.
Huelsenbeck, J.P., Rouquist, F., 2001. MrBayes: Bayesian inference of phylogenetictrees. Bioinformatics 17, 754–755.
Iida, K., Takishita, K., Ohshima, K., Inagaki, Y., 2007. Assessing themonophyly of chlorophyll-ccontaining plastids by multi-gene phylogenies under the unlinked model conditions.Mol. Phylogenet. Evol. 45, 227–238.
Janouškovec, J., Horák, A., Oborník, M., Lukeš, J., Keeling, P.J., 2010. A common red algalorigin of the apicomplexan, dinoflagellate, and heterokont plastids. Proc. Natl.Acad. Sci. U. S. A. 107, 10949–10954.
Kaneko, T., Sato, S., Kotani, H., Tanaka, A., Asamizu, E., Nakamura, Y., Miyajima, N., Hirosawa,M., Suqiura, M., Sasamoto, S., Kimura, T., Hosouchi, T., Matsuo, A., Muraki, A., Nakazaki,N., Naruo, K., Okumura, S., Shimpo, S., Takeuchi, C., Wada, T., Watanabe, A., Yamada,M., Yasuda, M., Tabata, S., 1996. Sequence analysis of the genome of the unicellular

9X. Wang et al. / Marine Genomics 10 (2013) 1–9
cyanobacterium Synnechocystis sp. strain PCC6803. II. Sequence determinationof the en-tire genome and assignment of potential protein-coding regions. DNA Res. 3, 109–136.
Khan, H., Parks, N., Kozera, C., Curtist, B.A., Parsons, B.J., Bowman, S., Archibald, J.M.,2007. Plastid genome sequences of the cryptophyte alga Rhodomonas salinaCCMP1319: lateral transfer of putative DNA replication machinery and a test ofchromist plastid phylogeny. Mol. Biol. Evol. 24, 1832–1842.
Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam, H.,Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., Thompson, J.D., Gibson, T.J., Higgins,D.G., 2007. ClustalW and ClustalX version 2.0. Bioinformatics 23, 2947–2948.
Le Corguillé, G., Pearson, G., Valente, M., Viegas, C., Gschloessl, B., Corre, E., Bailly, X.,Peters, A.F., Jubin, C., Vaccherie, B., Cock, J.M., Leblanc, C., 2009. Plastid genomesof two brown algae, Ectocarpus siliculosus and Fucus vesiculosus: further insightson the evolution of red-algal derived plastids. BMC Evol. Biol. 9, 253.
Li, X., Cong, Y., Yang, G., Shi, Y., Qu, S., Li, Z., Wang, G., Zhang, Z., Luo, S., Dai, H., 2007.Trait evaluation and trial cultivation of Dongfang No. 2, the hybrid of a male game-tophyte clone of Laminaria longissima (Laminarales, Phaeophyta) and a female oneof L. japonica. J. Appl. Phycol. 19, 139–151.
Lowe, T.M., Eddy, S.R., 1997. tRNAscan-SE: a program for improved detection of trans-fer RNA genes in genomic sequences. Nucleic Acids Res. 25, 955–964.
Martin, W., Ruan, T., Richly, E., Hansen, A., Cornelsen, S., Lins, T., Leister, D., Stoebe, B.,Hasegawa, M., Penny, D., 2002. Evolutionary analysis of Arabidopsis, cyanobacterial,and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterialgenes in the nucleus. Proc. Natl. Acad. Sci. U. S. A. 99, 12246–12251.
Ohta, N., Matsuzaki, M., Misumi, O., Miyagishima, S., Nozaki, H., Tanaka, K., Shin-I, T.,Kohara, Y., Kuroiwa, T., 2003. Complete sequence and analysis of the plastid genomeof the unicellular red alga Cyanidioschyzon merolae. DNA Res. 10, 67–77.
Oudot-Le Secq,M.P., Loiseau-deGoer, S., Stam,W.T., Olsen, J.L., 2006. Completemitochondrialgenomes of the three brown algae (Heterokonta: Phaeophyceae) Dictyota dichotoma,Fucus vesiculosus and Desmarestia viridis. Curr. Genet. 49, 47–58.
Oudot-Le Secq, M.P., Grimwood, J., Shapiro, H., Armbrust, E.V., Bowler, C., Green, B.R.,2007. Chloroplast genomes of the diatoms Phaeodactylum tricornutum and Thalassiosirapsedonana: comparison with other plastid genomes of the red lineage. Mol. Genet.Genomics 277, 427–439.
Phillips, N., Burrowes, R., Rousseau, F., de Reviers, B., Saunders, G.W., 2008. Resolvingevolutionary relationships among the brown algae using chloroplast and nucleargenes. J. Phycol. 44, 394–405.
Reith,M.E.,Munholland, J., 1995. Complete nucleotide sequences of the Porphyra purpureachloroplast genome. Plant Mol. Biol. Rep. 13, 333–335.
Reviers, B., de Rousseau, F., 1999. Towards a new classification of the brown algae. Prog.Phycol. Res. 13, 107–201.
Sanchez-Puerta, M.V., Bachvaroff, T.R., Delwiche, C.F., 2005. The complete plastid ge-nome sequence of the haptophyte Emiliania huxleyi: a comparison to other plastidgenomes. DNA Res. 12, 151–156.
Sanchez-Puerta,M.V., Bachvaroff, T.R., Delwiche, C.F., 2007. Sortingwheat fromchaff inmulti-gene analyses of chlorophyll c containing plastids. Mol. Phylogenet. Evol. 44, 885–897.
Sanderson, M.J., Shaffer, H.B., 2002. Troubleshooting molecular phylogenetic analyses.Annu. Rev. Ecol. Syst. 33, 49–72.
Shimodaira, H., Hasegawa, M., 2001. CONSEL: for assessing the confidence of phyloge-netic tree selection. Bioinformatics 17, 1246–1247.
Simon, D., Fewer, D., Friedl, T., Bhattacharya, D., 2003. Phlogeny and self-splicing abilityof the plastid tRNA-Leu group I intron. J. Mol. Evol. 57, 710–720.
Stirewalt, V.L., Michalowski, C.B., L ffelhardt, W., Bohnert, H.J., Bryant, D.A., 1995. Nu-cleotide sequence of the cyanelle genome from Cyanophora paradoxa. Plant Mol.Biol. Report. 13, 327–332.
Stoebe, B., Kowallik, K.V., 1999. Gene-cluster analysis in chloroplast genomics. TrendsGenet. 15, 344–347.
Strimmer, K., von Haeseler, A., 1996. Quartet puzzling: a quartet maximum likelihoodmethod for reconstructing tree topologies. Mol. Biol. Evol. 13, 964–969.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., Kumar, S., 2011. MEGA5:molecular evolutionary genetics analysis using maximum likelihood, evolution-ary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739.
Tseng, C.K., 2001. Algal biotechnology industries and research activities in China. J. Appl.Phycol. 13, 375–380.
Turmel, M., Otis, C., Lemieux, C., 1999. The complete chloroplast DNA sequence of thegreen alga Nephroselmis olivacea: insights into the architectural chloroplast genomes.Proc. Natl. Acad. Sci. U. S. A. 96, 10248–10253.
Van den Hoek, C., Mann, D.G., Jahns, H.M., 1995. Algae: An Introduction to Phycology.Cambridge University Press, United Kingdom.
Wang, X.-L., Yang, Y.-X., Cong, Y.-Z., Duan, D.-L., 2004. DNA fingerprinting of selectedLaminaria (Phaeophyta) gametophytes byRAPDmarkers. Aquaculture 238, 143–153.
Yoon, H.S., Hackett, J.D., Pinto, G., Bhattacharya, D., 2002. The single ancient origin ofchromist plastids. Proc. Natl. Acad. Sci. U. S. A. 99, 15507–15512.